
Noble, A., Roesner, S., & Aggarwal, V. K. (2016). Short...
76
Noble, A., Roesner, S., & Aggarwal, V. K. (2016). Short Enantioselective Total Synthesis of Tatanan A and 3-epi-Tatanan A Using Assembly-Line Synthesis. Angewandte Chemie - International Edition, 55(51), 15920- 15924. https://doi.org/10.1002/anie.201609598 Publisher's PDF, also known as Version of record License (if available): CC BY Link to published version (if available): 10.1002/anie.201609598 Link to publication record in Explore Bristol Research PDF-document This is the final published version of the article (version of record). It first appeared online via Wiley at http://doi.org/10.1002/anie.201609598 . Please refer to any applicable terms of use of the publisher. University of Bristol - Explore Bristol Research General rights This document is made available in accordance with publisher policies. Please cite only the published version using the reference above. Full terms of use are available: http://www.bristol.ac.uk/pure/about/ebr-terms
Transcript of Noble, A., Roesner, S., & Aggarwal, V. K. (2016). Short...

Noble, A., Roesner, S., & Aggarwal, V. K. (2016). Short EnantioselectiveTotal Synthesis of Tatanan A and 3-epi-Tatanan A Using Assembly-LineSynthesis. Angewandte Chemie - International Edition, 55(51), 15920-15924. https://doi.org/10.1002/anie.201609598
Publisher's PDF, also known as Version of record
License (if available):CC BY
Link to published version (if available):10.1002/anie.201609598
Link to publication record in Explore Bristol ResearchPDF-document
This is the final published version of the article (version of record). It first appeared online via Wiley athttp://doi.org/10.1002/anie.201609598 . Please refer to any applicable terms of use of the publisher.
University of Bristol - Explore Bristol ResearchGeneral rights
This document is made available in accordance with publisher policies. Please cite only the publishedversion using the reference above. Full terms of use are available:http://www.bristol.ac.uk/pure/about/ebr-terms

Supporting Information
Short Enantioselective Total Synthesis of Tatanan A and 3-epi-Tatanan A Using Assembly-Line SynthesisAdam Noble, Stefan Roesner, and Varinder K. Aggarwal*
anie_201609598_sm_miscellaneous_information.pdf

S1
Supplementary Information for
Short, Enantioselective Total Synthesis of Tatatan A, and 3-epi-Tatanan A, Using Assembly-Line Synthesis
Adam Noble, Stefan Roesner, and Varinder K. Aggarwal*
School of Chemistry, University of Bristol
Cantock’s Close, Bristol, BS8 1TS (UK)
Contents
1. General Information ................................................................................................................................... 2
2. Materials and Reagents .............................................................................................................................. 3
3. Synthesis of 2,4,5-Trimethoxyphenyl Boronic Acid Derivatives .............................................................. 4
4. Synthesis of Benzoate Esters and Carbamates ........................................................................................... 7
5. Synthesis of Neopentyl Glycol Boronic Ester 10 ..................................................................................... 12 6. Ligand Studies in Lithiation-Borylations with Primary Benzylic TIB-ester 16 ...................................... 14
7. End Game Studies: Stereospecific Olefinations Using Suzuki and Zweifel Reactions ........................... 19
8. Synthesis of 3-epi-Tatanan A ................................................................................................................... 27
9. Synthesis of Racemic Pinacol Boronic Ester 17 ...................................................................................... 30
10. Optimization of the Diastereoselective Matteson Homologation .......................................................... 32
11. Synthesis of Tatanan A .......................................................................................................................... 35
12. Comparison of NMR Spectra of Natural and Synthetic Tatanan A ....................................................... 41
13. 1H and 13C NMR spectra ........................................................................................................................ 43

S2
1. General Information
All air and water-sensitive reactions were carried out in flame-dried glassware under a nitrogen atmosphere using standard Schlenk manifold technique. Cryogenic temperatures were achieved used the following cold baths: acetone/CO2 (–78 °C); MeOH/N2 (–95 °C).
Analytical thin-layer chromatography (TLC) was performed using aluminium backed plates pre-coated with Merck Silica Gel 60 F254. Compounds were visualized under UV light or by staining with aqueous basic potassium permanganate or an ethanolic solution of phosphomolybdic acid. Flash column chromatography (FCC) was carried out using silica gel LC60A-40 (63 µm).
Chiral HPLC was performed on a HP Agilent 1100 with a Chiralpak IB column (4.6 mm × 250 mm, 5 µm) fitted with a guard (4 mm × 10 mm) and monitored by DAD (Diode Array Detector). Chiral supercritical fluid chromatography (SFC) was performed on a Thar SFC investigator using a Daicel Chiralpak IB column (4.6 mm × 250 mm, 5 µm).
1H, 13C and 11B NMR spectra were acquired at various field strengths, as indicated, using Jeol ECS 300 MHz, Jeol ECS 400 MHz, Varian VNMR 400 MHz, Varian VNMR 500 MHz, and Bruker Cryo 500 MHz Fourier transform spectrometers. Chemical shifts (δ) are given in parts per million (ppm) and referenced to CDCl3 (7.27 ppm), methanol-d4 (3.31 ppm) or DMSO-d6 (2.50 ppm). Coupling constants (J) are given in Hertz (Hz) and refer to apparent multiplicities (s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, quin = quintet, sex = sextet, hep = heptet, m = multiplet, dd = doublet of doublets, etc.). The 1H NMR spectra are reported as follows: chemical shift (multiplicity, coupling constants, number of protons).
High resolution mass spectra (HRMS) were recorded on a VG Analytical Autospec by Electron Ionization (EI) or Chemical Ionization (CI) or on a Bruker Daltonics Apex IV by Electrospray Ionization (ESI).
Infra-red (IR) spectra were recorded on a Perkin Elmer Spectrum One FT-IR as a thin film. Selected absorption maxima (νmax) are reported in wavenumbers (cm–1).
Melting points were recorded in degrees Celsius (°C), using a Kofler hot-stage microscope apparatus and are reported uncorrected.
Microwave reactions were carried out in a Biotage Initiator EXP EU microwave synthesizer
Optical rotation ([α]DT ) was measured on a Bellingham and Stanley Ltd. ADP220 polarimeter and is quoted
in (° ml)(g dm)–1.

S3
2. Materials and Reagents
All reagents were used as received unless otherwise stated. Bulk solutions were evaporated under reduced pressure using a Büchi rotary evaporator. All anhydrous solvents were commercially supplied or dried by passing through a modified Grubbs system of alumina columns, manufactured by Anhydrous Engineering. Petroleum ether refers to the fraction collected between 40 ºC and 60 °C. nBuLi (1.6 M in hexanes) and sBuLi (1.3 M in 92:8 cyclohexane/hexane) were purchased from Acros. The molarity of organolithium solutions was regularly determined by titration using N-benzyl benzamide as an indicator.1 TMEDA was dried over CaH2 before distillation and stored in a Young’s tube under N2. Diisopropylamine and 2,2,6,6-tetramethylpiperidine were dried over NaOH before distillation and stored in a Young’s tube under N2. (+)-Sparteine was obtained as the free base (BOC sciences), distilled over CaH2 or NaOH and stored in a Young’s tube under N2. The free base of sparteine readily absorbs atmospheric carbon dioxide (CO2) and should be stored in a Young’s tube under inert atmosphere at −20 °C. Lithium diisopropylamide (LDA) and lithium 2,2,6,6-tetramethylpiperide (LiTMP) solutions were freshly prepared from the corresponding amine and n-BuLi (1.6 M in hexanes) immediately before use.
The following compounds were prepared according to literature procedure and all spectroscopic data matched those previously reported: Ethyl 2,4,6-triisopropylbenzoate (EtOTIB, S1),2 (R)-1-(trimethylstannyl)ethyl 2,4,6-triisopropylbenzoate (S2),2 vinyl diisopropylcarbamate (27),3 2-ethyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane [EtB(pin)],4 2-isopropyl-5,5-dimethyl-1,3,2-dioxaborinane [iPrB(neo)],4 (4S,4′S)-2,2′-(3-pentylidene)bis(4-tert-butyloxazoline) [(S,S)-tBu-BOX],5 (4S,4′S)-2,2′-(3-pentylidene)bis(4-isopropyloxazoline) [(S,S)-iPr-BOX, L*],5 Buchwald precatalyst (tBu3P-Pd-G3),6 and 1,1-bis[(pinacolato)boryl]ethane (S3).7
1 A. F. Burchat, J. M. Chong, N. Nielsen, J. Organomet. Chem. 1997, 542, 281 – 283. 2 M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey, V. K. Aggarwal, Nature 2014, 513, 183 − 188. 3 N. J. Webb, S. P. Marsden, S. A. Raw, Org. Lett. 2014, 16, 4718 – 4721. 4 A. P. Pulis, D. J. Blair, E. Torres, V. K. Aggarwal, J. Am. Chem. Soc. 2013, 135, 16054 – 16057. 5 M. Li, A. Hawkins, D. M. Barber, P. Bultinck, W. Herrebout, D. J. Dixon, Chem. Commun. 2013, 49, 5265 – 5267. 6 N. C. Bruno, M. T. Tudge, S. L. Buchwald, Chem. Sci. 2013, 4, 916 – 920. 7 Z.-Q. Zhang, C.-T. Yang, L.-J. Liang, B. Xiao, X. Lu,; J.-H. Liu, Y.-Y. Sun, T. B. Marder, Y. Fu, Org. Lett., 2014, 16, 6342 – 6345.
S4
3. Synthesis of 2,4,5-Trimethoxyphenyl Boronic Acid Derivatives
1-Iodo-2,4,5-trimethoxybenzene (12)
Prepared following a modified literature procedure.8 N-Iodosuccinimide (12.4 g, 55.0 mmol, 1.1 equiv) was added to a solution of 1,2,4-trimethoxybenzene (11) (8.41 g, 50.0mmol, 1.0 equiv) in anhydrous MeCN (150 mL) and stirred 16 h at ambient temperature. The solvent was removed in vacuo and the residue was dissolved in Et2O (100 mL). The ethereal solution was washed with saturated aqueous Na2S2O3 solution (100 mL) and water (100 mL), dried over MgSO4, filtered and concentrated in vacuo to give the title compound (14.7 g, 49.9 mmol, >99%) as an off-white solid, which required no further purification.
1H NMR (400 MHz, CDCl3) δH 7.22 (s, 1H), 6.52 (s, 1H), 3.90 (s, 3H), 3.86 (s, 3H), 3.84 (s, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 153.0, 150.2, 144.2, 121.9, 97.8, 73.0, 57.3, 56.7, 56.1 ppm.
All spectroscopic data are consistent with those previously reported in the literature.9
(2,4,5-Trimethoxyphenyl)boronic acid (29)
n-Butyllithium (8.13 mL, 1.60 M solution in hexanes, 13.0 mmol, 1.3 equiv) was added dropwise to a solution of 1-iodo-2,4,5-trimethoxybenzene (12) (2.94 g, 10.0 mmol, 1.0 equiv) in anhydrous THF (25 mL) at −78 °C. After stirring for 30 min triisopropyl borate (2.05 mL, 15.0 mmol, 1.5 equiv) was added slowly and the mixture was stirred for 4 h at −78 °C before allowing to warm to room temperature and stirring for a 8 M. C. Carreño, J. L. Ruano, G. Sanz, M. A. Toledo, A. Urbano, Tetrahedron Lett. 1996, 37, 4081 − 4084. 9 S. Huenig, R. Bau, M. Kemmer, H. Meixner, T. Metzenthin, K. Peters, K. Singer, J. Gulbis, Eur. J. Org. Chem. 1998, 335 − 348.
O
OMeMeO
MeI
12
O
OMeMeO
Me
11
NIS
>99%
O
OMeMeO
MeB(OH)2
31
(i) nBuLi(ii) B(OiPr)3
(iii) HCl
90%
O
OMeMeO
MeB
13
MgSO4
>99%
HO OH
O
O
(i) nBuLi(ii) iPrOB(pin)
89%
O
OMeMeO
MeB
26
O
O
O
OMeMeO
MeI
12
O
OMeMeO
Me
11
NIS (1.1 equiv)
MeCN, r.t, 16 h
>99%
O
OMeMeO
MeI
12
O
OMeMeO
MeB(OH)2
29
(i) nBuLi (1.3 equiv)THF, –78 ºC
(ii) B(OiPr)3 (1.5 equiv)(iii) HCl
90%

S5
further 16 h. Saturated aqueous NH4Cl solution (20 mL) was added and the mixture was acidified with 3 M HCl to pH 5. The organic solvent was removed under reduced pressure. The crude solids were collected by vacuum filtration, washed with water and dried in vacuo to afford the title compound (1.90 g, 8.98 mmol, 90%) as a pale green solid, which required no further purification.
1H NMR (300 MHz, CDCl3) δH 7.32 (s, 1H), 6.53 (s, 1H), 5.82 (s, 2H), 3.94 (s, 3H), 3.92 (s, 3H), 3.89 (s, 3H) ppm.
11B NMR (96 MHz, CDCl3) δB 28.1 (br s) ppm.
All spectroscopic data are consistent with those previously reported in the literature.10
5,5-Dimethyl-2-(2,4,5-trimethoxyphenyl)-1,3,2-dioxaborinane (13)
Boronic acid 29 (1.74 g, 8.20 mmol, 1.0 equiv) was dissolved in Et2O (25 mL), neopentyl glycol (855 mg, 8.20 mmol, 1.0 equiv) was added and the mixture was stirred at room temperature for 16 h. Flame-dried MgSO4 (2.96 g, 24.6 mmol, 3.0 equiv) was added and the reaction mixture was stirred for an additional 2 h at room temperature. The ethereal solution was filtered through a plug of anhydrous MgSO4 and the solids were washed with Et2O. The filtrate was concentrated in vacuo and dried under high vacuum to the title compound (2.30 g, 8.20 mmol, >99%) as a pale green solid, which required no further purification.
Mpt: 61‒63 °C (Et2O).
1H NMR (400 MHz, CDCl3) δH 7.21 (s, 1H), 6.49 (s, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.82 (s, 3H), 3.78 (s, 4H), 1.03 (s, 6H) ppm.
13C NMR (101 MHz, CDCl3) δC 159.8, 151.9, 143.0, 118.6, 97.7, 72.6, 57.4, 56.5, 56.0, 31.9, 22.1 ppm.
11B NMR (96 MHz, CDCl3) δB 25.8 (br s) ppm.
IR (νmax/cm–1, neat): 2961, 1603, 1264, 1199, 1160, 1030.
HRMS (CI+) calcd. for C19H31O5BNa [M+Na]+ 373.2160, found 373.2164.
10 a) M. J. Burns, I. J. S. Fairlamb, A. R. Kapdi, P. Sehnal, R. J. K. Taylor, Org. Lett. 2007, 9, 5397 – 5400; b) G. Wang, F. Wang, D. Cao, Y. Liu, R. Zhang, H. Ye, X. Li, L. He, Z. Yang, L. Ma, A. Peng, M. Xiang, Y. Wei, L. Chen, Bioorg. Med. Chem. Lett. 2014, 24, 3158 – 3163.
O
OMeMeO
MeB(OH)2
29
O
OMeMeO
MeB
13
(1.0 equiv)
MgSO4, Et2O
>99%
HO OH
O
O

S6
4,4,5,5-Tetramethyl-2-(2,4,5-trimethoxyphenyl)-1,3,2-dioxaborolane (26)
n-Butyllithium (6.74 mL, 1.60 M solution in hexanes, 10.8 mmol, 1.1 equiv) was added dropwise to a solution of 1-iodo-2,4,5-trimethoxybenzene (12) (2.88 g, 9.80 mmol, 1.0 equiv) in anhydrous THF (20 mL) at −78 °C. After stirring for 30 min 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2.74 g, 14.7 mmol, 1.5 equiv) was added slowly and the mixture was stirred for 1 h at −78 °C. Water (10 mL) was added carefully and the mixture was allowed to warm to room temperature. The reaction mixture was extracted with CH2Cl2 (3 × 15 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo to afford the title compound (2.57 g, 8.73 mmol, 89%) as an off-white solid, which required no further purification. Recrystallization from pentane/EtOAc gave a white solid.
Mpt: 106‒107 °C (pentane/EtOAc).
1H NMR (400 MHz, CDCl3) δH 7.20 (s, 1H), 6.50 (s, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.83 (s, 3H), 1.35 (s, 12H) ppm.
13C NMR (101 MHz, CDCl3) δC 160.2, 152.4, 142.8, 119.0, 108.4 (br), 97.5, 83.1, 57.3, 56.4, 55.7, 24.8 ppm.
11B NMR (96 MHz, CDCl3) δB 28.9 (br s) ppm.
IR (νmax/cm–1, neat): 2965, 1605, 1453, 1344, 1206, 1143, 1032.
HRMS (CI+) calcd. for C15H24O5B [M+H]+ 295.1717, found 295.1718.
(i) nBuLi (1.1 equiv)THF, –78 ºC
(ii) iPrOB(pin) (1.5 equiv)
89%
O
OMeMeO
MeB
26
O
OO
OMeMeO
MeI
12

S7
4. Synthesis of Benzoate Esters and Carbamates
Propyl 2,4,6-triisopropylbenzoate (S4)
Prepared following a modified literature procedure.2 A mixture of 2,4,6-triisopropylbenzoic acid (12.4 g, 50.0 mmol, 1.00 equiv), 1-bromopropane (22.7 mL, 250 mmol, 5.00 equiv), tetrabutylammonium bisulfate (1.36 g, 4.00 mmol, 8.00 mol%), sodium hydroxide (6.20 g, 155 mmol, 3.10 equiv), CHCl3 (250 mL) and H2O (100 mL) were stirred vigorously for 67 h. The organic phase was separated and the aqueous extracted with CH2Cl2 (3 × 80 mL). The combined organic phases were washed with brine (300 mL), dried (MgSO4), filtered, and concentrated in vacuo. To the residue was added pentane (100 mL), the insoluble salts were removed by filtration and washed with pentane (50 mL). The filtrate was concentrated in vacuo to give the title compound (14.5 g, 49.9 mmol, >99%) as a colourless oil that required no further purification.
1H NMR (400 MHz, CDCl3): δH 7.02 (s, 2H), 4.28 (t, J = 6.7 Hz, 2H), 2.90 (hept, J = 6.7, 1H), 2.87 (hept, J = 6.7, 2H), 1.77 (sex, J = 7.2 Hz, 2H), 1.27 (d, J = 6.8 Hz, 12H), 1.26 (d, J = 7.0 Hz, 6H), 1.01 (t, J = 7.4 Hz, 3H) ppm.
13C NMR (101 MHz, CDCl3): δC 171.2, 150.2, 144.9, 130.9, 121.0, 66.8, 34.6, 31.6, 24.3, 24.1, 22.2, 10.7 ppm.
All spectroscopic data are consistent with those previously reported in the literature.11
(R)-1-(Trimethylstannyl)propyl 2,4,6-triisopropylbenzoate (S5)
Prepared using a modified literature procedure.2 An oven-dried Schlenk tube was evacuated and refilled with nitrogen (× 3) before the addition of propyl 2,4,6-triisopropylbenzoate (S4) (2.11 g, 7.26 mmol, 1.00 equiv) and (+)-sparteine (2.17 mL, 9.44 mmol, 1.30 equiv) and anhydrous Et2O (36 mL, 0.20 M). The solution was cooled to –78 °C before the addition of sBuLi (1.3 M in hexane/cyclohexane, 7.26 mL, 9.44 mmol, 1.30 equiv) dropwise over 10 min (colour change: colourless to dark brown). The reaction mixture was stirred at –78 °C for 4 h. Me3SnCl (1.0 M in hexanes, 9.44 mL, 9.44 mmol, 1.30 equiv) was added dropwise to the reaction mixture over 5 min (colour change: dark brown to yellow). The reaction was stirred at –78 °C for 60 min before being warmed to room temperature and stirred for a further 60 min. The reaction was quenched by the addition of 5% aqueous H3PO4 (40 mL) and stirred for 20 min before separation of the layers. The organic layer was washed with 5% aqueous H3PO4
(3 × 20 mL). The combined aqueous layers
11 P. Beak, L. G. Carter, J. Org. Chem. 1981, 46, 2363 – 2373.
O
O
= OTIB
S4
OH
O
Br+
Bu4NHSO4 (8 mol%)NaOH (3.1 equiv)
CHCl3/H2O, r.t
>99%(5.0 equiv)
S5S4
(i) (+)-sparteine (1.3 equiv)sBuLi (1.3 equiv)
Et2O, –78 ºC
(ii) Me3SnCl (1.3 equiv)
>99%, 91% ee
OTIBMe3Sn OTIB
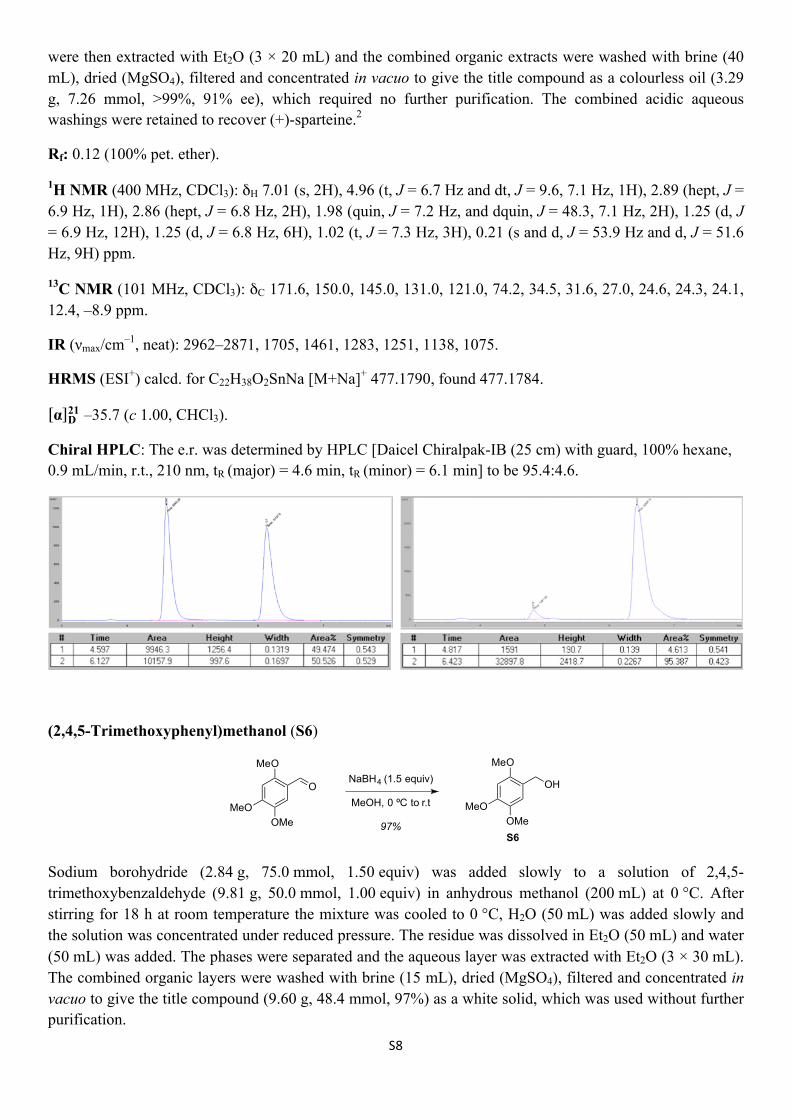
S8
were then extracted with Et2O (3 × 20 mL) and the combined organic extracts were washed with brine (40 mL), dried (MgSO4), filtered and concentrated in vacuo to give the title compound as a colourless oil (3.29 g, 7.26 mmol, >99%, 91% ee), which required no further purification. The combined acidic aqueous washings were retained to recover (+)-sparteine.2
Rf: 0.12 (100% pet. ether).
1H NMR (400 MHz, CDCl3): δH 7.01 (s, 2H), 4.96 (t, J = 6.7 Hz and dt, J = 9.6, 7.1 Hz, 1H), 2.89 (hept, J = 6.9 Hz, 1H), 2.86 (hept, J = 6.8 Hz, 2H), 1.98 (quin, J = 7.2 Hz, and dquin, J = 48.3, 7.1 Hz, 2H), 1.25 (d, J = 6.9 Hz, 12H), 1.25 (d, J = 6.8 Hz, 6H), 1.02 (t, J = 7.3 Hz, 3H), 0.21 (s and d, J = 53.9 Hz and d, J = 51.6 Hz, 9H) ppm.
13C NMR (101 MHz, CDCl3): δC 171.6, 150.0, 145.0, 131.0, 121.0, 74.2, 34.5, 31.6, 27.0, 24.6, 24.3, 24.1, 12.4, –8.9 ppm.
IR (νmax/cm–1, neat): 2962–2871, 1705, 1461, 1283, 1251, 1138, 1075.
HRMS (ESI+) calcd. for C22H38O2SnNa [M+Na]+ 477.1790, found 477.1784.
α D21 –35.7 (c 1.00, CHCl3).
Chiral HPLC: The e.r. was determined by HPLC [Daicel Chiralpak-IB (25 cm) with guard, 100% hexane, 0.9 mL/min, r.t., 210 nm, tR (major) = 4.6 min, tR (minor) = 6.1 min] to be 95.4:4.6.
(2,4,5-Trimethoxyphenyl)methanol (S6)
Sodium borohydride (2.84 g, 75.0 mmol, 1.50 equiv) was added slowly to a solution of 2,4,5-trimethoxybenzaldehyde (9.81 g, 50.0 mmol, 1.00 equiv) in anhydrous methanol (200 mL) at 0 °C. After stirring for 18 h at room temperature the mixture was cooled to 0 °C, H2O (50 mL) was added slowly and the solution was concentrated under reduced pressure. The residue was dissolved in Et2O (50 mL) and water (50 mL) was added. The phases were separated and the aqueous layer was extracted with Et2O (3 × 30 mL). The combined organic layers were washed with brine (15 mL), dried (MgSO4), filtered and concentrated in vacuo to give the title compound (9.60 g, 48.4 mmol, 97%) as a white solid, which was used without further purification.
O
OMeMeO
Me
S6
O
OMeMeO
MeNaBH4 (1.5 equiv)
MeOH, 0 ºC to r.t
97%
O OH

S9
Mpt: 70‒72 °C (Et2O).
1H NMR (500 MHz, CDCl3) δH ppm 6.86 (s, 1H), 6.54 (s, 1H), 4.62 (s, 2H), 3.89 (s, 3H), 3.84 (s, 3H), 3.84 (s, 3H), 2.23 (br s, 1H) ppm.
13C NMR (126 MHz, CDCl3) δC 151.7, 149.2, 142.8, 120.6, 113.2, 97.3, 61.5, 56.6, 56.3, 56.1 ppm.
IR (νmax/cm–1, neat): 3481, 3360, 2936, 1609, 1509, 1201, 1122, 1030, 1001.
HRMS (CI+) calcd. for C10H15O4 [M+H]+ 199.0970, found 199.0961.
2,4,5-Trimethoxybenzyl 2,4,6-triisopropylbenzoate (16)
(2,4,5-Trimethoxyphenyl)methanol (S6) (2.38 g, 12.0 mmol, 1.20 equiv), 2,4,6-triisopropylbenzoyl chloride (2.67 g, 10.0 mmol, 1.00 equiv) and 4-(dimethylamino)pyridine (122 mg, 1.00 mmol, 10.0 mol%) were dissolved in anhydrous toluene (10 mL) under an inert atmosphere in a microwave vial. Et3N (2.08 mL, 15.0 mmol, 1.50 equiv) was added before the vial was sealed and heated for 4 h at 150 °C in a microwave reactor. After cooling to room temperature, the salts were removed by filtration through a plug of silica and the solids were thoroughly washed with Et2O. The filtrate was concentrated in vacuo and the residue purified by FCC (SiO2, 20% EtOAc/pentane) to afford the title compound (3.38 g, 7.89 mmol, 79%) as a white solid.
Mpt: 65‒66 °C (pentane/EtOAc).
Rf: 0.36 (20% EtOAc/pentane).
1H NMR (400 MHz, CDCl3) δH 6.98 (s, 1H), 6.98 (s, 2H), 6.53 (s, 1H), 5.35 (s, 2H), 3.91 (s, 3H), 3.84 (s, 3H), 3.83 (s, 3H), 2.87 (hept, J = 6.8 Hz, 3H), 1.23 (d, J = 6.9 Hz, 6H), 1.20 (d, J = 6.8 Hz, 12H) ppm.
13C NMR (101 MHz, CDCl3) δC 171.7, 152.7, 150.3, 150.2, 144.9, 142.9, 130.7, 120.9, 115.3, 115.2, 97.2, 62.1, 56.8, 56.4, 56.3, 34.6, 31.5, 24.2, 24.1 ppm.
IR (νmax/cm–1, neat): 2962, 1715, 1606, 1515, 1464, 1212, 1131, 1032.
HRMS (EI+) calcd. for C26H36O5 [M]+ 428.2563, found 428.2561.
O
OMeMeO
Me
16
O
OMeMeO
Me DMAP (10 mol%)Et3N (1.5 equiv)
toluene, 150 ºC, 4 h
79%
OH OTIBCl+
(1.0 equiv)S6(1.2 equiv)
O

S10
2,4,5-Trimethoxybenzyl diisopropylcarbamate (S7)
(2,4,5-Trimethoxyphenyl)methanol (S6) (1.98 g, 10.0 mmol, 1.00 equiv) and N,N-diisopropylcarbamoyl chloride (1.96 g, 12.0 mmol, 1.20 equiv) were dissolved in anhydrous toluene (10 mL) under an inert atmosphere in a microwave vial. Et3N (1.80 mL, 13.0 mmol, 1.30 equiv) was added before the vial was sealed and heated for 4 h at 150 °C in a microwave reactor. After cooling to room temperature, the salts were removed by filtration through a plug of silica and the solids were thoroughly washed with Et2O. The solvent was removed under reduced pressure and the residue was purified by FCC (SiO2, 30% EtOAc/ pentane) to afford the title compound (2.39 g, 7.34 mmol, 73%) as a white solid.
Mpt: 79‒81 °C (EtOAc/pentane).
Rf: 0.39 (30% EtOAc/pentane).
1H NMR (400 MHz, CDCl3) δH 6.91 (s, 1H), 6.52 (s, 1H), 5.11 (s, 2H), 4.06 (br m, 1H), 3.88 (s, 3H), 3.83 (br m, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 1.19 (d, J = 6.8 Hz, 12H) ppm.
13C NMR (101 MHz, CDCl3) δC 155.8, 152.0, 149.3, 142.7, 116.9, 113.7, 97.5, 61.5, 56.5, 56.4, 56.1, 45.7 (br), 20.9 (br) ppm.
IR (νmax/cm–1, neat): 2969, 1671, 1519, 1438, 1287, 1208, 1129, 1029.
HRMS (ESI+) calcd. for C17H21NNaO5 [M+Na]+ 348.1781, found 348.1770.
Chloromethyl 2,4,6-trimethylbenzoate (S8)
Prepared following a modified literature procedure.12 To an oven-dried Schlenk tube under N2 was added ZnCl2 (41 mg, 0.30 mmol, 1.0 mol%). The flask was placed under vacuum and heated with a heat gun for 2 min to fuse the ZnCl2. After allowing to cool to room temperature, the flask was backfilled with N2 before the addition of paraformaldehyde (901 mg, 30.0 mmol, 1.00 equiv) and 2,4,6-trimethylbenzoyl chloride (5.00 mL, 30.0 mmol, 1.00 equiv). The resulting suspension was stirred and heated to 80 ºC for 2.5 h to give a pale yellow solution. The reaction was allowed to cool to room temperature, resulting in a white precipitate, before purification by FCC (SiO2, 25% toluene/pet. ether) to give the title compound (3.75 g, 17.6 mmol, 59%) as a colourless oil.
Rf: 0.31 (25% toluene/pet. ether)
12 D. Rennison, O. Laita, S. Bova, M. Cavalli, B. Hopkins, D. S. Linthicum, M. B. Brimble, Bioorg. Med. Chem. 2012, 20, 3997 – 4011.
O
OMeMeO
Me
S7
O
OMeMeO
MeEt3N (1.3 equiv)
toluene, 150 ºC, 4 h
73%
OH OCbCl N+
(1.2 equiv)
O
S6
S8
(HCHO)n (1.0 equiv)ZnCl2 (1.0 mol%)
80 ºC, 2.5 h
59%
Cl
O
O
O
Cl = Cl OTMB

S11
1H NMR (500 MHz, CDCl3): δH 6.88 (s, 2H), 5.93 (s, 2H), 2.33 (s, 6H), 2.30 (s, 3H) ppm.
13C NMR (126 MHz, CDCl3): δC 167.9, 140.3, 135.9, 128.8, 128.7, 68.9, 21.2, 19.9 ppm.
IR (νmax/cm–1, neat): 2981–2864, 1612, 1440, 1748, 1242, 1161, 1056.
HRMS (EI+) calcd. for C11H13ClO2 [M]+ 212.0610, found 212.0604.
Chloromethyl 2,4,6-triisopropylbenzoate (23)
Prepared following a modified literature procedure.12 To an oven-dried vial was added anhydrous ZnCl2 (5.7 mg, 0.042 mmol, 1.0 mol%), followed by paraformaldehyde (126 mg, 4.18 mmol, 1.00 equiv) and 2,4,6-triisopropylbenzoyl chloride (1.12 g, 4.18 mmol, 1.00 equiv). The vial was purged with N2 before sealing with a screw cap. The solid mixture was heated to 90 ºC to fuse the acid chloride and the resulting heterogeneous mixture was stirred for 2.5 h. The mixture was allowed to cool to room temperature before being purified by FCC (SiO2, 1% Et2O/pet. ether) to give the title compound (440 mg, 1.48 mmol, 35%) as a white solid.
Mpt: 53–55 ºC (Et2O).
Rf: 0.34 (100% hexanes).
1H NMR (400 MHz, CDCl3): δH 7.03 (s, 2H), 5.92 (s, 2H), 2.91 (hept, J = 6.9 Hz, 1H), 2.89 (hept, J = 6.9 Hz, 2H), 1.26 (d, J = 6.9 Hz, 6H), 1.26 (d, J = 6.8 Hz, 12H) ppm.
13C NMR (126 MHz, CDCl3): δC 168.7, 151.0, 145.3, 128.3, 121.0, 68.8, 34.5, 31.3, 24.1, 23.9 ppm.
IR (νmax/cm–1, neat): 2963–2874, 1751, 1607, 1462, 1232, 1129, 1100, 1047.
HRMS (EI+) calcd. for C17H25ClO2 [M]+ 296.1543, found 296.1556.
23
(HCHO)n (1.0 equiv)ZnCl2 (1.0 mol%)
90 ºC, 2.5 h
35%
Cl
O
O
O
Cl = Cl OTIB

S12
5. Synthesis of Neopentyl Glycol Boronic Ester 10
5,5-Dimethyl-2-((2S,3R)-3-(2,4,5-trimethoxyphenyl)pentan-2-yl)-1,3,2-dioxaborinane (10)
n-Butyllithium (1.60 M in hexanes, 2.44 mL, 3.90 mmol, 1.30 equiv) was added dropwise over 10 min to a solution of (R)-1-(trimethylstannyl)propyl 2,4,6-triisopropylbenzoate (S5) (1.84 g, 4.05 mmol, 1.35 equiv) in anhydrous Et2O (15 mL) at −78 °C under an atmosphere of nitrogen and the reaction mixture was stirred for 1 h. A solution of 5,5-dimethyl-2-(2,4,5-trimethoxyphenyl)-1,3,2-dioxaborinane (13) (840 mg, 3.00 mmol, 1.00 equiv) in anhydrous Et2O (10 mL) was added dropwise at −78 °C and the mixture was stirred at –78 °C for 1 h. The reaction mixture was then removed from the dry ice bath and heated under reflux for 16 h. After cooling to room temperature the mixture was filtered through a small plug of silica and the solids were thoroughly washed with Et2O. The filtrate was concentrated under reduced pressure to give the crude boronic ester 14, which was dried under high vacuum for 2 h before being re-dissolved in anhydrous Et2O (10 mL).
In a second reaction vessel, n-butyllithium (1.60 M in hexanes, 2.25 mL, 3.60 mmol, 1.20 equiv) was added dropwise to a solution of (R)-1-(trimethylstannyl)ethyl 2,4,6-triisopropylbenzoate (S2) (1.64 g, 3.73 mmol, 1.24 equiv) in anhydrous Et2O (15 mL) at −78 °C under an atmosphere of nitrogen and the mixture was stirred for 1 h. The solution of the crude boronic ester 14 was added dropwise at –78 °C and the mixture was stirred at –78 °C for 1 h. The reaction mixture was allowed to warm to room temperature and then heated under reflux for 4 h. After cooling to room temperature the mixture was filtered through a small plug of silica and the solids were thoroughly washed with Et2O. The filtrate was concentrated in vacuo. The residue was purified by column chromatography (SiO2, 25% EtOAc/pentane + 1% Et3N) to give the title compound (572 mg, 1.63 mmol, 54%) as a white solid. The diastereomeric ratio was determined by 1H NMR spectroscopy and accounted to 95:5. The enantiomeric excess was determined to be >99.9% by chiral HPLC analysis after oxidation to the corresponding alcohol (see below).
Mpt: 66‒67 °C (pentane/EtOAc).
Rf: 0.15 (25% EtOAc/pentane + 1% Et3N).
1H NMR (500 MHz, CDCl3) δH 6.65 (s, 1H), 6.51 (s, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 3.76 (s, 3H), 3.64 (s, 4H), 2.94 (td, J = 10.8, 4.1 Hz, 1H), 1.67 – 1.62 (m, 1H), 1.55 – 1.51 (m, 1H), 1.13 (dq, J = 10.9, 7.3 Hz, 1H), 0.98 (s, 6H), 0.70 (d, J = 7.3 Hz, 3H), 0.68 (t, J = 7.3 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.8, 147.0, 143.4, 125.6, 111.5, 98.2, 72.1, 57.2, 56.6, 56.1, 42.0 (br), 31.8, 29.4, 27.2 (br), 22.1, 14.7, 12.4 ppm.
11B NMR (96 MHz, CDCl3) δB ppm 29.5 (br s).
IR (νmax/cm–1, neat): 2958, 1519, 1463, 1437, 1417, 1316, 1250, 1203, 1169, 1034.
O
OMeMeO
MeB(neo)
13
Me3Sn OTIB
S5 (91% ee, 1.35 equiv)nBuLi (1.30 equiv), Et2O, –78 ºC
(ii) ArB(neo) (13), –78 ºC to reflux
(i)
O
OMeMeO
Me
14
B(neo)
Me3Sn OTIB
S2 (>99% ee, 1.24 equiv)nBuLi (1.20 equiv), Et2O, –78 ºC
(ii) RB(neo) (14), –78 ºC to reflux
52% (2 steps),95:5 dr, >99.9% ee
(i)
O
OMeMeO
Me
10
B(neo)

S13
HRMS (ESI+) calcd. for C12H16O3I [M+H]+ 335.0144, found 335.0151.
α D21 –15.2 (c 1.05, CHCl3).
(2S,3S)-3-(2,4,5-Trimethoxyphenyl)pentan-2-ol (S9)
To a solution of neopentyl boronic ester 10 (48 mg, 0.14 mmol) in THF (3.0 mL) at 0 ºC was added 2.0 M aqueous NaOH (3.0 mL) followed by 30% aqueous H2O2 (1.5 mL). The mixture was stirred vigorously at room temperature 2 h before diluting with water (10 mL) and extracting with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by FCC (SiO2, 30% EtOAc/pet. ether) to give the title compound (33 mg, 0.13 mmol, 95%) as a colourless oil.
Mpt (racemate): 73–75 ºC (Et2O).
Rf: 0.23 (30% EtOAc/pet. ether).
1H NMR (500 MHz, CDCl3) δH 6.65 (s, 1H), 6.55 (s, 1H), 3.96 (dq, J = 6.2, 6.2 Hz, 1H, 3.89 (s, 3H), 3.84 (s, 3H), 3.80 (s, 3H), 3.00 (ddd, J = 10.7, 6.3, 4.4 Hz, 1H), 2.00 (br s, 1H), 1.89 (dqd, J = 13.5, 7.5, 4.4 Hz, 1H), 1.63 (ddq, J = 13.5, 10.8, 7.3 Hz, 1H), 1.05 (d, J = 6.2 Hz, 3H), 0.79 (t, J = 7.4 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.2, 147.8, 143.2, 121.8, 112.9, 98.1, 71.5, 56.8, 56.7, 56.0, 47.3, 22.8, 20.5, 12.3 ppm.
IR (νmax/cm–1, neat): 3421, 2962–2833, 1509, 1465, 1397, 1315, 1205, 1036.
HRMS (ESI+) calcd. for C14H22O4Na [M+Na]+ 277.1410, found 277.1408.
α D24 +19.3 (c 1.50, CHCl3).
Chiral HPLC: The e.r. was determined by HPLC [Daicel Chiralpak-IB (25 cm) with guard, 7:93 i-PrOH/hexanes, 1.0 mL/min, r.t., 210 nm, tR (major) = 9.1 min, tR (minor) = 10.2 min] to be >99.9:0.1.
For the preparation of racemic S9, see Section 9.
O
OMeMeO
Me
10
B(neo)O
OMeMeO
Me
S9
OHH2O2, NaOH
H2O/THF
95%95:5 dr, >99.9% ee
syn diastereomer (tR = 12.2 and
13.8 min)
anti diastereomer (tR = 9.1 and
10.2 min)
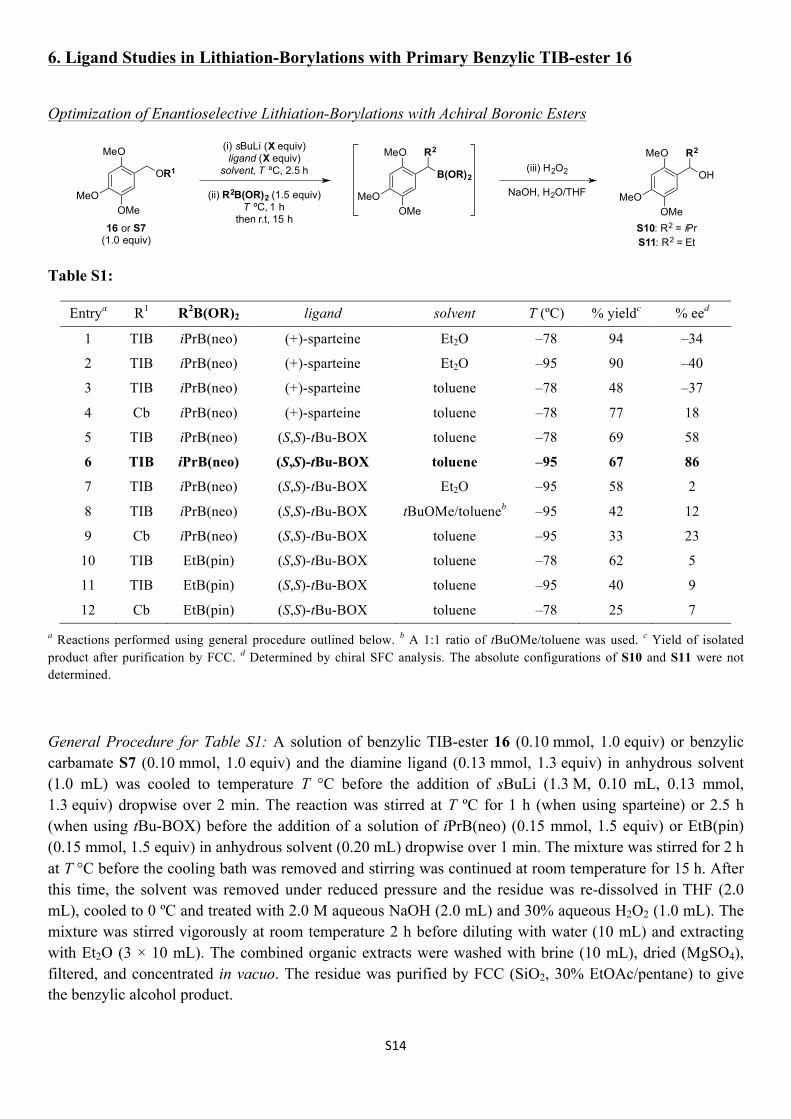
S14
6. Ligand Studies in Lithiation-Borylations with Primary Benzylic TIB-ester 16
Optimization of Enantioselective Lithiation-Borylations with Achiral Boronic Esters
Table S1:
Entrya R1 R2B(OR)2
ligand solvent T (ºC) % yieldc % eed
1 TIB iPrB(neo) (+)-sparteine Et2O –78 94 –34
2 TIB iPrB(neo) (+)-sparteine Et2O –95 90 –40
3 TIB iPrB(neo) (+)-sparteine toluene –78 48 –37
4 Cb iPrB(neo) (+)-sparteine toluene –78 77 18
5 TIB iPrB(neo) (S,S)-tBu-BOX toluene –78 69 58
6 TIB iPrB(neo) (S,S)-tBu-BOX toluene –95 67 86
7 TIB iPrB(neo) (S,S)-tBu-BOX Et2O –95 58 2
8 TIB iPrB(neo) (S,S)-tBu-BOX tBuOMe/tolueneb –95 42 12
9 Cb iPrB(neo) (S,S)-tBu-BOX toluene –95 33 23
10 TIB EtB(pin) (S,S)-tBu-BOX toluene –78 62 5
11 TIB EtB(pin) (S,S)-tBu-BOX toluene –95 40 9
12 Cb EtB(pin) (S,S)-tBu-BOX toluene –78 25 7 a Reactions performed using general procedure outlined below. b A 1:1 ratio of tBuOMe/toluene was used. c Yield of isolated product after purification by FCC. d Determined by chiral SFC analysis. The absolute configurations of S10 and S11 were not determined.
General Procedure for Table S1: A solution of benzylic TIB-ester 16 (0.10 mmol, 1.0 equiv) or benzylic carbamate S7 (0.10 mmol, 1.0 equiv) and the diamine ligand (0.13 mmol, 1.3 equiv) in anhydrous solvent (1.0 mL) was cooled to temperature T °C before the addition of sBuLi (1.3 M, 0.10 mL, 0.13 mmol, 1.3 equiv) dropwise over 2 min. The reaction was stirred at T ºC for 1 h (when using sparteine) or 2.5 h (when using tBu-BOX) before the addition of a solution of iPrB(neo) (0.15 mmol, 1.5 equiv) or EtB(pin) (0.15 mmol, 1.5 equiv) in anhydrous solvent (0.20 mL) dropwise over 1 min. The mixture was stirred for 2 h at T °C before the cooling bath was removed and stirring was continued at room temperature for 15 h. After this time, the solvent was removed under reduced pressure and the residue was re-dissolved in THF (2.0 mL), cooled to 0 ºC and treated with 2.0 M aqueous NaOH (2.0 mL) and 30% aqueous H2O2 (1.0 mL). The mixture was stirred vigorously at room temperature 2 h before diluting with water (10 mL) and extracting with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by FCC (SiO2, 30% EtOAc/pentane) to give the benzylic alcohol product.
O
OMeMeO
Me
OR1
(i) sBuLi (X equiv)ligand (X equiv)
solvent, T ºC, 2.5 h
(ii) R2B(OR)2 (1.5 equiv)T ºC, 1 h
then r.t, 15 h
O
OMeMeO
Me
B(OR)2
R2
(iii) H2O2
NaOH, H2O/THF
O
OMeMeO
Me
OH
R2
16 or S7(1.0 equiv)
S10: R2 = iPrS11: R2 = Et
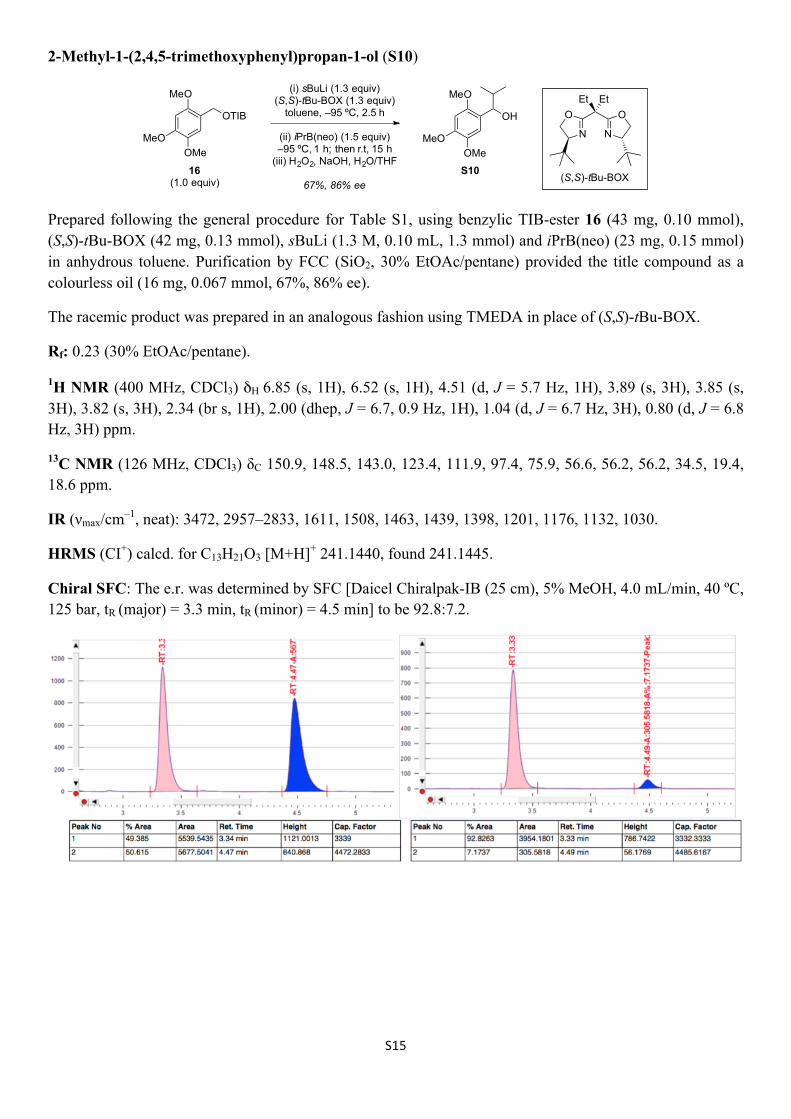
S15
2-Methyl-1-(2,4,5-trimethoxyphenyl)propan-1-ol (S10)
Prepared following the general procedure for Table S1, using benzylic TIB-ester 16 (43 mg, 0.10 mmol), (S,S)-tBu-BOX (42 mg, 0.13 mmol), sBuLi (1.3 M, 0.10 mL, 1.3 mmol) and iPrB(neo) (23 mg, 0.15 mmol) in anhydrous toluene. Purification by FCC (SiO2, 30% EtOAc/pentane) provided the title compound as a colourless oil (16 mg, 0.067 mmol, 67%, 86% ee).
The racemic product was prepared in an analogous fashion using TMEDA in place of (S,S)-tBu-BOX.
Rf: 0.23 (30% EtOAc/pentane).
1H NMR (400 MHz, CDCl3) δH 6.85 (s, 1H), 6.52 (s, 1H), 4.51 (d, J = 5.7 Hz, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.82 (s, 3H), 2.34 (br s, 1H), 2.00 (dhep, J = 6.7, 0.9 Hz, 1H), 1.04 (d, J = 6.7 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 150.9, 148.5, 143.0, 123.4, 111.9, 97.4, 75.9, 56.6, 56.2, 56.2, 34.5, 19.4, 18.6 ppm.
IR (νmax/cm–1, neat): 3472, 2957–2833, 1611, 1508, 1463, 1439, 1398, 1201, 1176, 1132, 1030.
HRMS (CI+) calcd. for C13H21O3 [M+H]+ 241.1440, found 241.1445.
Chiral SFC: The e.r. was determined by SFC [Daicel Chiralpak-IB (25 cm), 5% MeOH, 4.0 mL/min, 40 ºC, 125 bar, tR (major) = 3.3 min, tR (minor) = 4.5 min] to be 92.8:7.2.
O
OMeMeO
Me
OTIB
(i) sBuLi (1.3 equiv)(S,S)-tBu-BOX (1.3 equiv)
toluene, –95 ºC, 2.5 h
(ii) iPrB(neo) (1.5 equiv)–95 ºC, 1 h; then r.t, 15 h
(iii) H2O2, NaOH, H2O/THF
67%, 86% ee
O
OMeMeO
Me
OH
16(1.0 equiv)
S10
N N
EtEtO O
(S,S)-tBu-BOX
S16
1-(2,4,5-Trimethoxyphenyl)propan-1-ol (S11)
Prepared following the general procedure for Table S1, using benzylic TIB-ester 16 (43 mg, 0.10 mmol), (S,S)-tBu-BOX (42 mg, 0.13 mmol), sBuLi (1.3 M, 0.10 mL, 1.3 mmol) and EtB(pin) (23 mg, 0.15 mmol) in anhydrous toluene. Purification by FCC (SiO2, 30% EtOAc/pentane) provided the title compound as a colourless oil (9.0 mg, 0.040 mmol, 40%, 9% ee).
Racemic S11 was prepared as follows: To a solution of 2,4,5-trimethoxybenzaldehyde (1.96 g, 10.0 mmol, 1.0 equiv) in anhydrous toluene (30 mL) at room temperature was slowly added ethylmagnesium bromide solution (3.0 M in Et2O, 5.0 mL, 15 mmol, 1.5 equiv). The mixture was then stirred at room temperature for 20 h before cooling to 0 ºC and quenching by the slow addition of 2% aqueous NH4Cl (10 mL). The mixture was stirred for 10 min at room temperature before the layers were separated and the aqueous phase extracted with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (15 mL), dried (MgSO4), filtered and concentrated in vacuo. Purification by FCC (SiO2, 30% EtOAc/pentane) provided the title compound as a white solid (2.11 g, 9.33 mmol, 93%).
Rf: 0.18 (30% EtOAc/pentane).
Mpt (racemic): 68–69 ºC (EtOAc/pentane)
1H NMR (500 MHz, CDCl3) δH 6.89 (s, 1H), 6.52 (s, 1H), 4.78 (t, J = 6.7 Hz, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.83 (s, 3H), 2.40 (br s, 1H), 1.85‒1.72 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H) ppm.
13C NMR (101 MHz, CDCl3) δC 150.8, 148.6, 143.1, 124.1, 111.1, 97.5, 71.6, 56.6, 56.21, 56.19, 30.5, 10.4 ppm.
IR (νmax/cm–1, neat): 3417, 2954, 1659, 1609, 1512, 1450, 1402, 1208, 1029.
HRMS (CI+) calcd. for C12H19O4 [M+H]+ 227.1283, found 227.1289.
Chiral SFC: The e.r. was determined by SFC [Daicel Chiralpak-IB (25 cm), 5% MeOH, 4.0 mL/min, 40 ºC, 125 bar, tR (major) = 3.8 min, tR (minor) = 4.8 min] to be 54.5:45.5.
O
OMeMeO
Me
OTIB
(i) sBuLi (1.3 equiv)(S,S)-tBu-BOX (1.3 equiv)
toluene, –95 ºC, 2.5 h
(ii) EtB(neo) (1.5 equiv)–95 ºC, 1 h; then r.t, 15 h
(iii) H2O2, NaOH, H2O/THF
40%, 10% ee
O
OMeMeO
Me
OH
16(1.0 equiv)
S11
N N
EtEtO O
(S,S)-tBu-BOX

S17
Ligand Effects in the Lithiation-Borylation of Chiral Boronic Esters 10 and 17
Table S2:
Entrya (OR)2 R1 ligand X equiv solvent % conv.d crude dr (syn:anti)e % yieldf purified dr
(syn:anti)e
1b neo TIB (S,S)-tBu-BOX 1.2 toluene <5 - - -
2b neo TIB (S,S)-iPr-BOX 1.5 toluene 95 92:8 72 94:6
3b neo TIB (R,R)-iPr-BOX 1.5 toluene 72 88:12 52 88:12
4b neo TIB TMEDA 1.5 toluene 77 98:2 61 98:2
5b pin TIB TMEDA 1.5 toluene 32 47:53 29 47:53
6c neo TIB (S,S)-iPr-BOX 2.0 Et2O 59 86:14 ND ND
7c neo TIB (R,R)-iPr-BOX 2.0 Et2O 63 58:42 ND ND
8c neo TIB TMEDA 2.0 Et2O 95 91:9 ND ND
9c neo Cb TMEDA 2.0 Et2O 58 88:12 ND ND a Reactions performed using general procedure outlined below. b Reactions performed using 0.2 mmol of boronic ester 10 or 17. c
Reaction performed using 0.05 mmol of boronic ester 10. d Conversion of boronic ester 10 or 17 into 15 (entries 1–5) or 19 (entries 6–9). Determined by 1H NMR analysis of the crude reaction mixture. e Determined by 1H NMR analysis. f Isolated yield after purification by FCC.
We believe the difference in the levels of selectivity between the neopentyl glycol and pinacol boronic esters is related to a change in the selectivity-determining step of the reaction. In the case of pinacol esters, boronate complex formation is reversible making 1,2-migration the stereochemistry-determining step.13 For the less sterically hindered neopentyl glycol boronic ester 10, boronate complex formation is non-reversible,14 thus making it the stereochemistry- determining step and resulting in high diastereoselectivity.
General Procedure for Table S2: To a solution of benzylic TIB-ester 16 (0.24–0.40 mmol, 1.2–2.0 equiv), or benzylic carbamate S7 (0.24–0.40 mmol, 1.2–2.0 equiv), and ligand (0.24–0.40 mmol, 1.2–2.0 equiv) in anhydrous solvent (3.0 mL) at –78 ºC was added sBuLi (1.3 M, 0.24–0.40 mmol, 1.2–2.0 equiv) dropwise over 2 min. The reaction mixture was stirred at –78 ºC for 2.5 h before adding a solution of neopentyl glycol boronic ester 10 (0.20 mmol, 1.0 equiv), or pinacol boronic ester 17 (0.20 mmol, 1.0 equiv), in anhydrous solvent (1.0 mL) dropwise over 5 min. The resulting mixture was stirred at –78 ºC for a further 1 h before
13 a) S. C. Matthew, B. W. Glasspoole, O. Eisenberger, C. M. Crudden, J. Am. Chem. Soc. 2014, 136, 5828 – 5831; b) V. Bagutski, R. M. French, V. K. Aggarwal, Angew. Chem. Int. Ed. 2010, 49, 5142 – 5145; c) B. M. Partridge, L. Chausser-Boissarie, M. Burns, A. P. Pulis, V. K. Aggarwal, Angew. Chem. Int. Ed. 2012, 51, 11795 – 11799. 14 a) S. Roesner, D. J. Blair, V. K. Aggarwal, Chem. Sci. 2015, 6, 3718 – 3723; b) V. Bagutski, R. M. French, V. K. Aggarwal, Angew. Chem. Int. Ed. 2010, 49, 5142 – 5145.
O
OMeMeO
Me
OR1
(i) sBuLi (X equiv)ligand (X equiv)
solvent, –78 ºC, 2.5 h
(ii) 10 or 17 (1.5 equiv)T ºC, 1 h
then r.t, 15 h
(iii) pinacol
NaOH, H2O/THF
when B(OR)2 = B(neo)10 or 17
(1.0 equiv)
O
OMeMeO
MeB(OR)2
16 or S7(X equiv)
O
OMeMeO
Me
B(neo)
OMe
OMeMeO
O
OMeMeO
Me
B(pin)
OMe
OMeMeO
19 15b

S18
removing from the cold bath and stirring at room temperature for 15 h (For entries 6–9, Table S2, work-up was performed at this stage and the crude neopentyl glycol boronic ester 19 analyzed by 1H NMR). The solvent was removed under reduced pressure and the residue was re-dissolved in THF (2.5 mL). Pinacol (71 mg, 0.60 mmol, 3.0 equiv) and NaOH (0.25 mL, 0.2 M in water) were added subsequently and the reaction mixture was stirred vigorously for 2 h at room temperature. Water (10 mL) and Et2O (10 mL) were added, the phases were separated, and the aqueous phase was re-extracted with Et2O (2 × 10 mL). The combined organic phases were washed with brine (20 mL), dried (Na2SO4), filtered and the solvent removed in vacuo. The crude product was analyzed by 1H NMR before being purified by FCC (2 × SiO2, 1st = 30% EtOAc/pet. ether, 2nd = 2% acetone/CH2Cl2) to give secondary pinacol boronic ester 15 as a colourless viscous oil.
4,4,5,5-Tetramethyl-2-((1S,2R,3R)-2-methyl-1,3-bis(2,4,5-trimethoxyphenyl)pentyl)-1,3,2-dioxaborolane (15b)
Prepared following the general procedure for Table S2, using 2,4,5-trimethoxybenzyl 2,4,6-triisopropylbenzoate (16) (128 mg, 0.300 mmol, 1.50 equiv), (S,S)-iPr-BOX (L*) (88 mg, 0.30 mmol, 1.5 equiv), sBuLi (1.3 M, 0.23 mL, 0.30 mmol, 1.5 equiv), neopentyl glycol boronic ester 10 (95:5 dr, 70 mg, 0.20 mmol, 1.0 equiv) and anhydrous toluene (3.0 mL + 1.0 mL). Transesterification was performed using pinacol (71 mg, 0.60 mmol, 3.0 equiv) and NaOH (0.25 mL, 0.2 M in water) in THF (2.5 mL). Purification by FCC (2 × SiO2, 1st = 30% EtOAc/pet. ether, 2nd = 2% acetone/CH2Cl2) gave the title compound (78 mg, 0.14 mmol, 72%) as a colourless viscous oil. The diastereoselectivity for the reaction was 94:6, as determined by 1H NMR. This ratio could be improved to >95:5 by careful separation of the diastereomers by FCC (SiO2, 2% acetone/CH2Cl2).
Rf: 0.29 (30% EtOAc/pet. ether), 0.30 (2% Acetone/CH2Cl2).
1H NMR (500 MHz, CDCl3) δH 6.75 (s, 1H), 6.68 (s, 1H), 6.52 (s, 1H), 6.46 (s, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.83 (s, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.67 (s, 3H), 3.06 (br s, 1H), 2.56 (d, J = 9.6 Hz, 1H), 2.34–2.24 (m, 1H), 1.88 (dqd, J = 13.2, 7.4, 5.1 Hz, 1H), 1.92–1.83 (m, 1H), 1.73–1.58 (m, 1H), 1.26 (s, 6H), 1.20 (s, 6H), 0.77 (t, J = 7.3 Hz, 3H), 0.45 (d, J = 6.9 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.8, 152.2, 147.3, 147.0, 142.87, 142.87, 124.0, 122.5, 114.6, 113.4 (br), 98.4, 97.9, 82.9, 57.0, 56.9, 56.7, 56.6, 56.14, 56.11, 43.3 (br), 39.5, 29.0 (br), 27.2 (br), 25.1, 24.7, 15.6, 12.9 ppm.
11B NMR (96 MHz, CDCl3) δB 32.2 (br s) ppm.
IR (νmax/cm–1, neat): 2961–2830, 1508, 1462, 1309, 1202, 1142, 1034.
HRMS (ESI+) calcd. for C30H45O8BNa [M+Na]+ 567.3105, found 567.3100.
α D21 +26.0 (c 0.73, CHCl3).
O
OMeMeO
Me
OTIB
(i) sBuLi (1.5 equiv)(S,S)-iPr-BOX (1.5 equiv)
toluene, –78 ºC, 2.5 hO
OMeMeO
MeB(neo)
16 (1.5 equiv)
O
OMeMeO
Me
B(pin)
OMe
OMeMeO
15b10
+(iii) pinacol (3.0 equiv)
NaOH, THF/H2O
72%, 94:6 dr
(ii) 10, –78 ºC, 1h; then r.t, 15 hN N
EtEtO O
(S,S)-iPr-BOX

S19
7. End Game Studies: Stereospecific Olefinations Using Suzuki and Zweifel Reactions
Synthesis of vinyl iodide 4 and vinyl chloride S14
1,2,4-Trimethoxy-5-(prop-1-yn-1-yl)benzene (S12)
1-Iodo-2,4,5-trimethoxybenzene (12) (294 mg, 1.00 mmol, 1.0 equiv), copper (I) iodide (38 mg, 0.20 mmol, 0.2 equiv) and bis(triphenylphosphine)palladium (II) chloride (70 mg, 0.10 mmol, 0.1 equiv) were dissolved in diethylamine (3.5 mL). The solution was sparged with an excess of propyne (balloon) for 5 min before replacing the propyne balloon with a nitrogen balloon. The reaction mixture was stirred at room temperature under N2 for 12 h. The solvent was removed in vacuo and the residue was dissolved in CH2Cl2 (20 mL) and water (20 mL) was added. The phases were separated and the aqueous layer was extracted with CH2Cl2
(2 × 15 mL). The combined organic layers were washed with brine (25 mL), dried (MgSO4), filtered through charcoal and concentrated in vacuo. The crude product was purified by FCC (SiO2, 10%–20% EtOAc/pentane) and recrystallized from Et2O to give the title compound (184 mg, 0.892 mmol, 89%) as colourless needles.
Mpt: 89‒91 °C (Et2O).
Rf: 0.25 (20% EtOAc/pentane).
1H NMR (400 MHz, CDCl3) δH 6.90 (s, 1H), 6.49 (s, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.83 (s, 3H), 2.12 (s, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 155.0, 149.6, 142.8, 116.2, 104.1, 97.2, 88.5, 75.7, 56.7, 56.4, 56.0, 4.8 ppm.
IR (νmax/cm–1, neat): 2914, 2837, 1517, 1461, 1395, 1345, 1232, 1203, 1150, 1026.
HRMS (CI+) calcd. for C12H15O3 [M+H]+ 207.1021, found 207.1022.
OMe
OMeMeO
4
II2, NaOHH2O/THF
95%, >98:2 E/Z
OMe
OMeMeO
S14
CltBuLi, THF;
C2Cl6
71%, >98:2 E/Z
OMe
OMeMeO
I
12OMe
MeO
S12
OMe
OMeMeO
S13
B(pin)Pd(PPh3)Cl2
CuI, propyne, Et2NH
89%
CuCl, P(4-MeOC6H4)3B2(pin)2, K2CO3, iPrOH, Et2O
77%, >98:2 E/Z
OMe
OMe
OMeMeO
I
12
OMe
OMeMeO
S12
Pd(PPh3)Cl2 (10 mol%)CuI (20 mol%)
propyne, Et2NH
89%

S20
(Z)-4,4,5,5-Tetramethyl-2-(1-(2,4,5-trimethoxyphenyl)prop-1-en-2-yl)-1,3,2-dioxaborolane (S13)
Following a modified literature procedure.15 1,2,4-trimethoxy-5-(prop-1-yn-1-yl)benzene (S12) (89 mg, 0.43 mmol, 1.0 equiv), bis(pinacolato)diboron (132 mg, 0.520 mmol, 1.2 equiv), tri(4-methoxyphenyl)phosphine (9.0 mg, 26 µmol, 6.0 mol%), copper (I) chloride (2.2 mg, 22 µmol, 5.0 mol%) and K2CO3 (12 mg, 86 µmol, 20 mol%) were dissolved in anhydrous Et2O (2.0 mL). i-PrOH (67 µL, 0.86 mmol, 2.0 equiv) was added and the mixture was stirred for 15 h at ambient temperature. Et2O (15 mL) and water (15 mL) were added, the phases were separated and the aqueous layer was extracted with Et2O
(2 × 15 mL). The combined organic layers were washed with brine (25 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by FCC (SiO2, 10%–20% EtOAc/pentane) to give the title compound (110 mg, 0.329 mmol, 77%) as a white solid.
Mpt: 86‒88 °C (CHCl3).
Rf: 0.28 (20% EtOAc/pentane).
1H NMR (400 MHz, CDCl3) δH 7.34 (q, J = 1.8 Hz, 1H), 6.92 (s, 1H), 6.53 (s, 1H), 3.91 (s, 3H), 3.83 (s, 3H), 3.83 (s, 3H), 1.94 (d, J = 1.8 Hz, 3H), 1.31 (s, 12H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.0, 149.1, 142.2, 137.5, 118.5, 114.2, 97.1, 83.3, 56.6, 56.3, 56.0, 24.9, 16.2 ppm.
11B NMR (96 MHz, CDCl3) δB 29.6 (br s) ppm.
IR (νmax/cm–1, neat): 2978, 1592, 1517, 1312, 1300, 1269, 1206, 1145, 1092, 1030.
HRMS (CI+) calcd. for C18H28BO5 [M+H]+ 335.2030, found 335.2019.
(E)-1-(2-Iodoprop-1-en-1-yl)-2,4,5-trimethoxybenzene (4)
Following a modified literature procedure.16 NaOH (3.0 M in water 0.620 mL, 1.86 mmol, 3.00 equiv) was added to a solution of vinyl boronic ester S13 (207 mg, 0.619 mmol, 1.00 equiv) in THF (1.2 mL). After stirring for 10 min at room temperature a solution of iodine (314 mg, 1.24 mmol, 2.00 equiv) in THF 6.2 mL) was added over 5 min and stirring was continued for 1 h. The mixture was then quenched with saturated aqueous Na2S2O4 (10 mL). The mixture was extracted with Et2O (3 × 15 mL), the combined organic layers were washed with saturated aqueous NaHCO3 solution (20 mL) and brine (20 mL), dried
15 W. Yuan, S. Ma, Org. Biomol. Chem. 2012, 10, 7266 − 7268. 16 C. Wang, T. Tobrman, Z. Xu, E. Negishi, Org. Lett. 2009, 11, 4092 – 4095.
OMe
OMeMeO
S12
OMe
OMeMeO
S13
B(pin)CuCl (5 mol%)
P(4-MeOC6H4)3 (6 mol%)
B2(pin)2 (1.2 equiv)K2CO3, iPrOH, Et2O
77%, >98:2 E/Z
OMe
OMeMeO
S13
B(pin)OMe
OMeMeO
4
II2 (2.0 equiv)
NaOH (3.0 equiv)
H2O/THF
95%, >98:2 E/Z

S21
(MgSO4), filtered and concentrated in vacuo. The residue was purified by FCC (SiO2, 10%–20% EtOAc/pentane) to afford the title compound (197 mg, 0.590 mmol, 95%, >98:2 E:Z) as a pale yellow oil [N.B. The compound should be stored at –20 ºC as significant isomerization from >98:2 to 40:60 E:Z was observed over a period of 2 days when stored at r.t.].
Rf: 0.31 (20% EtOAc/pentane).
1H NMR (500 MHz, CDCl3) δH 7.22 (q, J = 1.8 Hz, 1H), 6.73 (s, 1H), 6.50 (s, 1H), 3.90 (s, 3H), 3.83 (s, 3H), 3.81 (s, 3H), 2.58 (d, J = 1.6 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 151.2, 149.3, 142.5, 136.2, 117.9, 113.5, 97.3, 97.0, 56.7, 56.4, 56.1, 29.5 ppm.
IR (νmax/cm–1, neat): 2942, 1508, 1454, 1318, 1202, 1030.
HRMS (CI+) calcd. for C12H16O3I [M+H]+ 335.0144, found 335.0151.
(E)-1-(2-Chloroprop-1-en-1-yl)-2,4,5-trimethoxybenzene (S14)
Following a modified literature procedure.17 To a solution of vinyl iodide 4 in anhydrous THF (10.6 mL) at –78 ºC was added tBuLi (1.7 M in pentane, 0.784 mL, 1.33 mmol, 2.5 equiv) dropwise over 2 min. The resulting bright yellow solution was stirred at –78 ºC for 15 min before the dropwise addition of a solution of hexachloroethane (265 mg, 1.12 mmol, 2.1 equiv) in THF (2.1 mL) dropwise over 5 min. The mixture was stirred for 45 min at –78 ºC before saturated aqueous NH4Cl (10 mL) was added and the mixture allowed to warm to room temperature. The phases were separated and the aqueous extracted with Et2O (2 × 15 mL). The combined organic layers were washed with brine (20 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by FCC (SiO2, 20% Et2O/pet. ether) to afford the title compound (92 mg, 0.37 mmol, 71%, >98:2 E:Z) as a colourless oil.
Rf: 0.23 (20% Et2O/pet. ether).
1H NMR (500 MHz, CDCl3) δH 6.72 (s, 1H), 6.69 (s, 1H), 6.52 (s, 1H), 3.91 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H), 2.23 (s, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 151.5, 149.3, 142.6, 131.6, 123.5, 116.3, 113.7, 97.4, 56.7, 56.4, 56.1, 22.6 ppm.
IR (νmax/cm–1, neat): 2998–2834, 1513, 1464, 1439, 1397, 1319, 1215, 1206, 1127, 1080, 1035 cm‒1.
HRMS (ESI+) calcd. for C12H15O3ClNa [M+Na]+ 265.0602, found 265.0605.
17 S. Nerdinger, C. Kendall, X. Cai, R. Marchart, P. Riebel, M. R. Johnson, C.-F. Yin, N. Hénaff, L. D. Eltis, V. Snieckus, J. Org. Chem. 2007, 72, 5960 – 5967.
OMe
OMeMeO
4
IOMe
OMeMeO
S14
CltBuLi (2.5 equiv)
THF, –78 ºC;
then C2Cl6 (2.1 equiv)
71%, >98:2 E/Z

S22
Synthesis of model boronic esters S15 and S16
5,5-Dimethyl-2-(2-methyl-1-(2,4,5-trimethoxyphenyl)propyl)-1,3,2-dioxaborinane (S15)
To a solution of benzylic TIB-ester 16 (484 mg, 1.13 mmol, 1.00 equiv) and TMEDA (219 µL, 1.47 mmol, 1.30 equiv) in anhydrous toluene (5.7 mL) at –78 ºC was added sBuLi (1.3 M, 1.13 mL, 1.47 mmol, 1.30 equiv) dropwise over 2 min. The reaction was stirred at –78 ºC for 1 h before the addition of a solution of iPrB(neo) (265 mg, 1.70 mmol, 1.50 equiv) dropwise over 1 min. The mixture was stirred for 2 h at –78 ºC before the cooling bath was removed and stirring was continued at room temperature for 15 h. After this time, the solvent was removed under reduced pressure and the residue was re-dissolved in anhydrous Et2O (3.0 mL) and filtered through a plug of silica, eluting with Et2O, before concentrating in vacuo to give crude boronic ester S15 (365 mg) as a colourless oil. 1H NMR analysis showed a 1.00:0.40 ratio of S15:TIBOH, which equates to a yield of approximately 282 mg, 74%.
Neopentyl glycol boronic ester S15 was unstable to FCC and was found to decompose upon standing at ambient temperature. The product was, therefore, used without further purification.
1H NMR (400 MHz, CDCl3) δH 6.83 (s, 1H), 6.52 (s, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.78 (s, 3H), 3.59 (s, 4H), 2.26 (d, J = 10.0 Hz, 1H), 2.07–1.98 (m, 1H), 1.04 (d, J = 6.6 Hz, 3H), 0.91 (s, 6H), 0.70 (d, J = 6.6 Hz, 3H) ppm.
4,4,5,5-Tetramethyl-2-(2-methyl-1-(2,4,5-trimethoxyphenyl)propyl)-1,3,2-dioxaborolane (S16)
To a solution of benzylic TIB-ester 16 (1.29 g, 3.00 mmol, 1.00 equiv) and TMEDA (581 µL, 3.90 mmol, 1.30 equiv) in anhydrous toluene (15 mL) at –78 ºC was added sBuLi (1.3 M, 3.00 mL, 3.90 mmol, 1.30 equiv) dropwise over 2 min. The reaction was stirred at –78 ºC for 1 h before the addition of a solution of iPrB(neo) (702 mg, 4.50 mmol, 1.50 equiv) in anhydrous toluene (2.0 mL) dropwise over 1 min. The mixture was stirred for 2 h at –78 ºC before the cooling bath was removed and stirring was continued at room temperature for 15 h. After this time, the solvent was removed under reduced pressure and the residue was re-dissolved in THF (10 mL). Pinacol (1.06 g, 9.00 mmol, 3.00 equiv) and NaOH (0.2 M in water, 3.0 mL) were added subsequently and the reaction mixture was stirred for 1 h at room temperature. Water (15 mL) and Et2O (15 mL) were added, the phases were separated, and the aqueous phase was extracted with Et2O (2 × 15 mL). The combined organic phases were dried (MgSO4), filtered and the solvent was removed in vacuo. Purification by FCC (SiO2, 10%–20% EtOAc/pentane) gave the title compound (569 mg, 1.62 mmol, 54%) as a colourless oil.
O
OMeMeO
Me (i) sBuLi (1.3 equiv)TMEDA (1.3 equiv)toluene, –78 ºC, 1h
(ii) iPrB(neo) (1.5 equiv)–78 ºC, 1 h; then r.t, 15 h
~74%
OTIB
O
OMeMeO
Me
B(neo)
16 S15
O
OMeMeO
Me (i) sBuLi (1.3 equiv)TMEDA (1.3 equiv)toluene, –78 ºC, 1h
(ii) iPrB(neo) (1.5 equiv)–78 ºC, 1 h; then r.t, 15 h
(iii) pinacol, NaOH, THF/H2O
54%
OTIB
O
OMeMeO
Me
B(pin)
16 S16

S23
Rf: 0.33 (20% EtOAc/pentane).
1H NMR (400 MHz, CDCl3) δH 6.83 (s, 1H), 6.50 (s, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.77 (s, 3H), 2.40 (d, J = 9.8 Hz, 1H), 2.04 (dhept, J = 9.8, 6.5 Hz, 1H), 1.23 (s, 6H), 1.20 (s, 6H), 1.03 (d, J = 6.6 Hz, 3H), 0.73 (d, J = 6.6 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 151.7, 146.9, 142.8, 122.7, 114.0, 98.1, 82.9, 56.8, 56.4, 56.0, 32.5, 30.6, 24.8, 24.6, 23.0, 21.8 ppm.
11B NMR (96 MHz, CDCl3) δB 31.6 (br s) ppm.
IR (νmax/cm–1, neat): 2951, 1515, 1316, 1205, 1140, 1035.
HRMS (ESI+) calcd. for C19H31O4BNa [M+Na]+ 373.2160, found 373.2161.
Attempted Zweifel reaction between vinyl iodide 4 and model boronic ester S16
To a solution of vinyl iodide 4 (126 mg, 0.376 mmol, 1.20 equiv) in anhydrous THF (2.0 mL) at –78 ºC was added tBuLi (1.7 M in pentane, 0.44 mL, 0.75 mmol, 2.4 equiv) dropwise. The mixture was stirred at –78 ºC for 30 min before the dropwise addition of a solution of boronic ester S16 (114 mg, 0.313 mmol, 1.00 equiv) in anhydrous THF (1.0 mL). The mixture was stirred at the same temperature for 45 min before the addition of a solution of iodine (119 mg, 0.470 mmol, 1.5 equiv) in MeOH (2.0 mL). The mixture was stirred for 15 min at –78 ºC before the addition of a solution of sodium methoxide (51 mg, 0.94 mmol, 3.0 equiv) in MeOH (1.0 mL). The resulting mixture was allowed to room temperature and stirred for 60 min before removal of the solvents in vacuo. The residue was dissolved in Et2O (10 mL) and washed with saturated aqueous Na2S2O3. The aqueous layer was extracted with Et2O (2 × 5 mL) and the combined organic phases washed with brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo. 1H NMR and GCMS analysis of the crude product showed only unreacted starting materials 4 and S16.
The fact that vinyl iodide 4 was returned, despite complete consumption (TLC) after treatment with tBuLi and complete formation of boronate complex S18 (11B NMR), suggests that the presence of the electron-rich trimethoxyphenyl group promotes regioselective opening of the intermediate iodonium S19 to form stabilized carbocation S20, which undergoes elimination to return the starting materials (see Scheme below). This pathway dominates over the desired 1,2-metallate rearrangement to give β-iodoboronic ester S21.
(i) 4, tBuLi (2.4 equiv)THF, –78 ºC
(ii) S16, THF, –78 ºC(iii) I2 (1.5 equiv), MeOH
(iv) NaOMe (3.0 equiv), MeOH
0%
O
OMeMeO
Me
S17
O
OMeMeO
Me
B(pin) +
OMe
OMeMeO
I
4(1.2 equiv)
OMe
OMeOMeS16
(1.0 equiv)

S24
Attempted Suzuki reaction between vinyl iodide 4 and model boronic ester S15
Table S3:
Entrya conditions solvent % yieldb
1 A THF 0
2 A DME 0
3 B Et2O 0
4 B THF 0 a Reactions were performed using general procedures outlined below. b Determined by 1H NMR analysis of the crude product. DME = 1,2-dimethoxyethane.
Conditions A for Table S3, entries 1 and 2: Following a modified literature procedure.18 A vial was charged with Pd(dba)2 (2.9 mg, 5.0 µmol, 5.0 mol%), PPh3 (2.6 mg, 10 µmol, 10 mol%) and Ag2O (35 mg, 0.15 mmol, 1.5 equiv) before evacuating and backfilling with N2 three times. A solution of vinyl iodide 4 (33 mg, 0.10 mmol, 1.0 equiv) and boronic ester S15 (40 mg, 0.12 mmol, 1.2 equiv) in anhydrous THF (1.0 mL) or DME (1.0 mL) was added and the vial was sealed. The reaction was stirred and heated at 90 ºC for 16 h before allowing to cool to room temperature and filtering through a plug of silica, eluting with EtOAc, and concentrating in vacuo. The crude mixture was analyzed by 1H NMR. No desired product S17 was observed.
18 D. Imao, B. W. Glasspoole, V. S. Laberge, C. M. Crudden, J. Am. Chem. Soc. 2009, 131, 5024 – 5025.
OMe
OMeMeO
I
4 complete conversion (TLC)
OMe
OMeMeO
LiOMe
OMeMeO
B(pin)R
+Li
–
OMe
OMeMeO
B(pin)
S19
R
+ –IOMe
OMeMeO
B(pin)
S20
R–+ IOMe
OMeMeO
4
I+ RB(pin)
S16
OMe
OMeMeO
B(pin)
S21
RI OMe
OMeMeO
S17
R
tBuLi RB(pin) (S16) I2
NaOMe1,2-rearrangement
S18
Conditions A:Pd2(dba)3, PPh3, Ag2O
THF or DME, 90 ºC
Conditions B:Pd(PPh3)4, K2CO3, Ag2O
THF or Et2O, 60 ºC
0%
O
OMeMeO
Me
S17
O
OMeMeO
Me
B(neo) +
OMe
OMeMeO
I
4(1.0 equiv)
OMe
OMeOMeS15
(1.2–1.3 equiv)

S25
Conditions B for Table S3, entries 3 and 4: Following a modified literature procedure.13a A vial was charged with Pd(PPh3)4 (9.2 mg, 8.0 µmol, 8.0 mol%), K2CO3 (22 mg, 0.16 mmol, 1.6 equiv) and Ag2O (37 mg, 0.16 mmol, 1.6 equiv) before evacuating and backfilling with N2 three times. A solution of vinyl iodide 4 (33 mg, 0.10 mmol, 1.0 equiv) and boronic ester S15 (44 mg, 0.13 mmol, 1.3 equiv) in anhydrous Et2O (2.0 mL) or THF (2.0 mL) was added and the vial was sealed. The reaction was stirred and heated at 60 ºC for 60 h before allowing to cool to room temperature and filtering through celite, eluting with Et2O, and concentrating in vacuo. The crude mixture was analyzed by 1H NMR. No desired product S17 was observed.
Attempted stereospecific Suzuki reaction with potassium trifluoroborate S22
Potassium ((1S,2R,3R)-2-methyl-1,3-bis(2,4,5-trimethoxyphenyl)pentyl)trifluoroborate (S22)
To a solution of pinacol boronic ester 15b (79 mg, 0.15 mmol, 1.0 equiv) in MeOH (0.57 mL) at room temperature was added a solution of potassium hydrogen fluoride (102 mg, 1.31 mmol, 9.0 equiv) in H2O (0.29 mL) dropwise over 30 seconds. The resulting white suspension was stirred at room temperature for 45 min before removal of the solvents under reduced pressure. The solid was extracted with refluxing acetone (4 × 5 mL) and filtered through cotton wool before concentrating in vacuo. The resulting white solid was triturated with Et2O from hot acetone to give the title compound (47 mg, 0.087 mmol, 60%) as a white solid.
Mpt (racemate): 217–219 ºC (acetone/Et2O)
1H NMR (500 MHz, DMSO-d6) δH 7.01 (s, 1H), 6.52 (br s, 1H), 6.51 (s, 1H), 6.49 (s, 1H), 3.73 (s, 3H), 3.71 (s, 3H), 3.68 (s, 3H), 3.64 (s, 3H), 3.59 (s, 3H), 3.49 (s, 3H), 2.49–2.44 (m, 1H), 2.37–2.29 (m, 1H), 2.20 (dqd, J = 13.1, 7.4, 3.1 Hz, 1H), 2.02–1.96 (m, 1H), 1.32–1.23 (m, 1H), 0.50 (t, J = 7.2 Hz, 2H), 0.49 (d, J = 6.7 Hz, 3H) ppm.
13C NMR (126 MHz, DMSO-d6) δC 152.8, 152.3, 146.4, 145.3, 142.9, 141.5, 128.2, 127.1, 118.6, 112.7 (br), 99.1, 98.6, 56.7, 56.6, 56.4, 56.4, 55.8, 55.6, 38.3, 28.6 (br), 25.9, 17.2, 12.4 ppm.
11B NMR (96 MHz, DMSO-d6) δB 3.02 ppm.
O
OMeMeO
MeKHF2
MeOH/H2O
60%B(pin)
OMe
OMeMeO
15b>95:5 dr
O
OMeMeO
Me
BF3K
OMe
OMeMeO
S22>95:5 dr
4 or S14tBu3P-Pd-G3
K2CO3, toluene/H2O
0%
O
OMeMeO
Me OMe
OMeMeO
1
OMe
OMeOMe
O
OMeMeO
Me KHF2 (9.0 equiv)
MeOH/H2O
60%, >95:5 dr
B(pin)
OMe
OMeMeO
15b>95:5 dr
O
OMeMeO
Me
BF3K
OMe
OMeMeO
S22

S26
19F NMR (283 MHz, DMSO-d6) δF –136.8 (3F, s) ppm.
IR (νmax/cm–1, neat): 2982–2833, 1509, 1466, 1455, 1449, 1441, 1399, 1221, 1202, 1183, 1080, 1031, 993, 980, 932.
m/z (ESI+) 529.2 ([M–F3+(OMe)2]+, 100%), 513.3 ([M–F3K+Na(OMe)2]+, 26%).
Procedure for attempted stereoinvertive Suzuki cross-coupling:
Following a modified literature procedure.19 A vial was charged with a stirrer bar, potassium trifluoroborate S22 (22 mg, 42 µmol, 1.0 equiv), tBu-Pd-G3 (2.4 mg, 4.5 µmol, 10 mol%) and K2CO3 (17 mg, 0.13 mmol, 3.0 equiv). The vial was placed under vacuum and backfilled with N2 four times before the addition of a solution of vinyl chloride S14 (10 mg, 42 µmol, 1.0 equiv) [or vinyl iodide 4 (14 mg, 42 µmol, 1.0 equiv)] in degassed toluene (168 µL), followed by degassed H2O (84 µL). The vial was sealed with a screw-top cap secured with Teflon tape before being heating to 100 ºC and stirred for 24 h. After allowing to cool to ambient temperature the mixture was diluted with EtOAc (2 mL) and filtered through a plug of silica, eluting with further EtOAc (3 × 2 mL), and concentrating in vacuo.
Analysis of the crude product by 1H NMR showed that no tatanan A (1) was formed. The main side-product was the boronic acid formed by hydrolysis of the potassium trifluoroborate group of S22 (data not shown).
19 L. Li, S. Zhao, A. Joshi-Pangu, M. Diane, M. R. Biscoe, J. Am. Chem. Soc. 2014, 136, 14027 – 14030.
O
OMeMeO
Me
BF3K
OMe
OMeMeO
S22>95:5 dr
tBu3P-Pd-G3 (10 mol%)
K2CO3 (3.0 equiv)toluene/H2O100 ºC, 24 h
0%
O
OMeMeO
Me OMe
OMeMeO
1
OMe
OMeOMe
OMe
OMeMeO
X+
4 (X = I)S14 (X = Cl)
PdNH2
PtBu3OMs
tBu3P-Pd-G3

S27
8. Synthesis of 3-epi-Tatanan A
5,5'-((3R,4S,5R)-4-Methylhept-1-yne-3,5-diyl)bis(1,2,4-trimethoxybenzene) (3-epi-5)
Following a modified literature procedure.20 To a solution of boronic ester 15b (61 mg, 0.11 mmol, 1.0 equiv) and vinyl carbamate 27 (29 mg, 0.17 mmol, 1.5 equiv) in anhydrous THF (1.1 mL) at –78 ºC was added LDA (0.86 M in THF, 0.20 mL, 0.17 mmol, 1.5 equiv) dropwise over 3 min. The reaction was stirred at –78 ºC for 60 min before the reaction vessel was transferred to a –40 ºC bath and stirred for 30 min. The reaction was cooled to –78 ºC and a solution of I2 (43 mg, 0.17 mmol, 1.5 equiv) in MeOH (1.1 mL) was added dropwise over 5 min. The mixture was stirred at –78 ºC for 10 min before removing from the cold bath and stirring at room temperature for 60 min. Saturated aqueous Na2S2O3 (5 mL) was added and the product extracted into Et2O (3 × 5 mL). The combined organic extracts were washed with water (5 mL), brine (5 mL), dried (MgSO4), filtered and concentrated in vacuo. The resulting crude vinyl carbamate S23 was further dried under high-vacuum for 30 min before placing under an atmosphere of N2, dissolving in anhydrous THF (1.1 mL) and cooling to –78 ºC. LDA (0.86 M in THF, 0.78 mL, 0.67 mmol, 6.0 equiv) was added dropwise over 1 min and the resulting solution was removed from the cold bath and stirred at room temperature for 15 min. The reaction was quenched by the addition of saturated aqueous NH4Cl (5 mL) and the product extracted into Et2O (3 × 5 mL). The combined organic extracts were washed with brine (5 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by FCC (SiO2, 25% EtOAc/pet. ether) to give the title compound (46 mg, 0.10 mmol, 93%) as a colourless oil. The reaction was determined by 1H NMR analysis to proceed with 100% diastereomeric excess (de).
Rf: 0.29 (25% EtOAc/pet. ether).
20 Y. Wang, A. Noble, E. L. Myers, V. K. Aggarwal, Angew. Chem. Int. Ed. 2016, 55, 4270 – 4274.
O
OMeMeO
MeLDA, THF;
then I2, MeOH
B(pin)
OMe
OMeMeO
O
OMeMeO
Me OMe
OMeMeO
LDA, THF
93% (2 steps)100% de
O
OMeMeO
Me OMe
OMeMeO
3-epi-1OMe
O
OMeMeO
Me OMe
OMeMeO
3-epi-5S23
+ OCb
ZrCp2Cl2, AlMe3H2O, CH2Cl2;then I2, THF
62% brsm
O
OMeMeO
Me OMe
OMeMeO ArB(OH)2 (29)
Pd(PPh3)4Ag2O, THF/H2O
94%
3-epi-28
15b>95:5 dr
27
I
MeO
OMe
OCb
O
OMeMeO
MeLDA (1.5 equiv), THF
–78 ºC to –20 ºC;
then I2 (1.5 equiv)MeOH, –78 ºC to r.t
B(pin)
OMe
OMeMeO
O
OMeMeO
Me OMe
OMeMeO
LDA (6.0 equiv)
THF, –78 ºC to r.t
93% (2 steps)100% de
O
OMeMeO
Me OMe
OMeMeO
3-epi-5S23
+ OCb
27(1.5 equiv)
15b>95:5 dr
OCb

S28
1H NMR (500 MHz, CDCl3) δH 7.00 (s, 1H), 6.65 (s, 1H), 6.51 (s, 1H), 6.48 (s, 1H), 4.04 (dd, J = 7.3, 2.5 Hz, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.82 (s, 3H), 3.72 (s, 3H), 3.72 (s, 3H), 3.19 (br s, 1H), 2.25 – 2.15 (m, 1H), 2.20 (d, J = 2.5 Hz, 1H), 1.89 (dqd, J = 13.2, 7.4, 4.9 Hz, 1H), 1.71 – 1.62 (m, 1H), 0.76 (t, J = 7.3 Hz, 3H), 0.57 (d, J = 7.0 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.8, 151.1, 148.3, 147.4, 143.0, 142.8, 123.2, 120.1, 113.7, 113.5, 98.0, 97.3, 88.1, 69.1, 56.7, 56.7, 56.6, 56.4, 56.1, 56.0, 44.0, 42.0 (br), 33.7, 26.7, 14.3, 12.6 ppm.
IR (νmax/cm–1, neat): 3286, 2960–2832, 1508, 1463, 1439, 1397, 1315, 1202, 1179, 1034.
HRMS (ESI+) calcd. for C26H34O6Na [M+Na]+ 465.2248, found 465.2244.
α D24 –28.3 (c 1.00, CHCl3).
5,5'-((3S,4S,5R,E)-1-Iodo-2,4-dimethylhept-1-ene-3,5-diyl)bis(1,2,4-trimethoxybenzene) (3-epi-28)
Following a modified literature procedure.21 To a solution of zirconocene dichloride (19 mg, 0.067 mmol, 0.50 equiv) in anhydrous CH2Cl2 (1.3 mL) at 0 ºC was added trimethylaluminium (2 M in toluene, 0.40 mL, 0.80 mmol, 6.0 equiv) followed by H2O (3.6 µL, 0.20 mmol, 1.5 equiv). The mixture was allowed to warm to room temperature and stirred for 30 min to give a yellow solution, which was subsequently cooled to 0 ºC. A solution of alkyne 3-epi-5 (59 mg, 0.13 mmol, 1.0 equiv) in anhydrous CH2Cl2 (1.4 mL) was added dropwise over 1 min. The ice bath was removed and the reaction was stirred at room temperature for 16 h before cooling back to 0 ºC. A solution of I2 (170 mg, 0.67 mmol, 5.0 equiv) in THF (0.7 mL) was added via cannula and the resulting mixture stirred at 0 ºC for 20 min before allowing to warm to room temperature and stirring for a further 60 min. Saturated aqueous Na2S2O3 (5 mL) was added and the phases separated. The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by FCC (SiO2, 2% EtOAc/CH2Cl2) to give the title compound (37 mg, 0.063 mmol, 48%, 62% brsm) as a colourless oil, as well as recovered alkyne 3-epi-5 (14 mg, 0.032 mmol, 24%).
Rf: 0.45 (2% EtOAc/CH2Cl2).
1H NMR (500 MHz, CDCl3) δH 6.68 (s, 1H), 6.62 (s, 1H), 6.52 (s, 1H), 6.45 (s, 1H), 6.09 (s, 1H), 3.91 (s, 3H), 3.86 (s, 3H), 3.83 (s, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.73 (d, J = 10.1 Hz, 1H), 3.65 (s, 3H), 3.19 (br s, 1H), 2.38 (dqd, J = 10.1, 7.0, 4.5 Hz, 1H), 1.74 – 1.68 (m, 2H), 1.72 (s, 3H), 0.79 (t, J = 7.3 Hz, 3H), 0.60 (d, J = 6.9 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.9, 152.0, 149.3, 147.9, 147.4, 143.0, 142.5, 122.7, 121.7, 113.8, 112.9, 98.0, 97.5, 76.1, 56.84, 56.77, 56.67, 56.67, 56.00, 55.99, 49.2, 40.1 (br), 38.6, 26.8, 24.5, 14.3, 12.7 ppm.
IR (νmax/cm–1, neat): 2994–2831, 1609, 1508, 1463, 1396, 1315, 1204, 1179, 1036. 21 G. Jürjens, A. Kirschning, Org. Lett. 2014, 16, 3000 – 3003.
ZrCp2Cl2 (50 mol%)AlMe3 (6.0 equiv)
H2O (1.5 equiv), CH2Cl2;
then I2 (5.0 equiv), THF
62% brsm
O
OMeMeO
Me OMe
OMeMeO
3-epi-28
O
OMeMeO
Me OMe
OMeMeO
3-epi-5I

S29
HRMS (ESI+) calcd. for C27H37IO4Na [M+Na]+ 607.1527, found 607.1516.
α D24 +32.0 (c 0.77, CHCl3).
5,5',5''-((3R,4S,5R,E)-2,4-Dimethylhept-1-ene-1,3,5-triyl)tris(1,2,4-trimethoxybenzene) (3-epi-1)
A vial was charged with vinyl iodide 3-epi-28 (17 mg, 0.029 mmol, 1.0 equiv), 2,4,5-trimethoxyphenylboronic acid (29) (9.2 mg, 0.044 mmol, 1.5 equiv), Pd(PPh3)4 (3.4 mg, 2.9 µmol, 10 mol%), and Ag2O (17 mg, 0.073 mmol, 2.5 equiv). The vial was sealed with a septum and triple evacuated/N2 filled before the addition of degassed (sparged with N2 for 10 min) THF/H2O (5:1, 0.58 mL). The mixture was stirred at room temperature for 15 h before filtering through a plug of silica, eluting with EtOAc, and concentrating in vacuo. The residue was purified by preparative TLC (SiO2, 40% EtOAc/pet. ether) to give the title compound (17 mg, 0.027 mmol, 94%) as a colourless oil.
Rf: 0.40 (40% EtOAc/pet. ether).
1H NMR (500 MHz, CDCl3) δH 6.87 (s, 1H), 6.78 (s, 1H), 6.75 (s, 1H), 6.59 (s, 1H), 6.55 (s, 1H), 6.54 (s, 1H), 6.46 (s, 1H), 3.91 (s, 6H), 3.87 (s, 3H), 3.83 (s, 3H), 3.82 (s, 3H), 3.81 (s, 3H), 3.80 (s, 3H), 3.72 (s, 3H), 3.66 (s, 3H), 3.68 – 3.62 (m, 1H), 3.54 – 3.39 (m, 1H), 2.52 – 2.43 (m, 1H), 1.85 – 1.76 (m, 2H), 1.64 (s, 3H), 0.82 (t, J = 7.3 Hz, 3H), 0.59 (d, J = 6.9 Hz, 3H) ppm.
1H NMR (500 MHz, CD3OD) δH 6.88 (s, 1H), 6.79 (s, 1H), 6.77 (s, 1H), 6.67 (s, 1H), 6.65 (s, 1H), 6.57 (s, 1H), 6.55 (s, 1H), 3.86 (s, 6H), 3.81 (s, 3H), 3.81 (s, 3H), 3.76 (s, 3H), 3.75 (s, 3H), 3.74 (s, 3H), 3.73 (s, 3H), 3.65 (s, 3H), 3.65 – 3.61 (m, 1H), 3.55 – 3.46 (m, 1H), 2.51 – 2.42 (m, 1H), 1.92 – 1.73 (m, 2H), 1.57 (s, 3H), 0.80 (t, J = 7.3 Hz, 3H), 0.59 (d, J = 6.9 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 153.1, 152.2, 151.7, 148.0, 147.30, 147.28, 143.1, 142.6, 142.5, 140.2, 123.9, 123.5, 120.3, 119.0, 114.7, 114.1, 112.8, 98.02, 97.97, 97.9, 57.0, 56.76, 56.76, 56.70, 56.68, 56.4, 56.2, 56.00, 55.96, 48.2, 41.2 (br), 39.1, 26.8, 18.9, 14.3, 12.9 ppm.
13C NMR (126 MHz, CD3OD) δC 153.6, 152.6, 152.2, 148.5, 147.9, 147.8, 143.0, 142.5, 142.4, 139.7, 123.9, 123.1, 120.0, 119.1, 115.34, 115.34, 113.5, 98.18, 98.16, 98.1, 56.34, 56.26, 56.0, 55.71, 55.66, 55.6, 55.4, 55.31, 55.30, 48.0, 40.8 (br), 39.1, 26.2, 17.8, 13.5, 11.9 ppm
IR (νmax/cm–1, neat): 2996–2832, 1508, 1464, 1455, 1439, 1396, 1315, 1203, 1178, 1036.
HRMS (ESI+) calcd. for C36H48O9Na [M+Na]+ 647.3191, found 647.3201.
α D23 +47.6 (c 0.77, CHCl3).
Pd(PPh3)4 (10 mol%)Ag2O (2.5 equiv)
THF/H2O, r.t, 15 h
94%
O
OMeMeO
Me OMe
OMeMeO
3-epi-1OMe
O
OMeMeO
Me OMe
OMeMeO
3-epi-28
I
MeO
OMe
(HO)2B
OMeMeO
OMe+
29(1.5 equiv)

S30
9. Synthesis of Racemic Pinacol Boronic Ester 17
2,4,5-Trimethoxypropiophenone (Isoacoramone) (S24)
To a stirred solution of 1,4,5-trimethoxybenzene (11) (1.49 mL, 10.0 mmol, 1.00 equiv) and propionic anhydride (1.92 mL, 15.0 mmol, 1.50 equiv) in anhydrous CH2Cl2 (27 mL) at 0 ºC was added aluminium trichloride (2.20 g, 15.0 mmol, 1.50 equiv) in one portion. The reaction was removed from the ice bath and stirred at room temperature for 2 h. The reaction was quenched with 0.5 M aqueous HCl (50 mL) and the layers separated. The aqueous layer was further extracted with CH2Cl2 (2 × 20 mL) and the combined organic extracts were dried (MgSO4), filtered, and concentrated in vacuo. The resulting solid was recrystallized from Et2O/pet. ether to yield the title compound (1.86 g, 8.29 mmol, 83%) as a white solid.
Mpt: 106–108 ºC (Et2O/pet. ether) [Lit. 108–109 ºC (MeOH)].22
1H NMR (400 MHz, CDCl3) δH 7.44 (s, 1H), 6.51 (s, 1H), 3.96 (s, 3H), 3.92 (s, 3H), 3.88 (s, 3H), 3.00 (q, J = 7.2 Hz, 2H), 1.17 (t, J = 7.2 Hz, 3H) ppm.
All spectroscopic data match those reported in the literature.23
(E)-4,4,5,5-Tetramethyl-2-(3-(2,4,5-trimethoxyphenyl)pent-2-en-2-yl)-1,3,2-dioxaborolane (S25)
Prepared following a modified literature procedure.24 To a solution of 1,1-bis[(pinacolato)boryl]ethane (S3) (274 mg, 0.972 mmol 1.10 equiv) in anhydrous THF (2.2 mL) at 0 ºC was added freshly prepared LiTMP (0.4 M in THF, 2.43 mL, 0.972 mmol, 1.10 equiv) dropwise over 1 min. The mixture was stirred at 0 ºC for 5 min before the addition of solid 2,4,5-trimethoxypropiophenone (S24) (198 mg, 0.883 mmol, 1.0 equiv) in one portion. The reaction was removed from the ice bath and stirred at room temperature for 2 h before filtering through a plug of silica, eluting with Et2O, and concentrating in vacuo. The residue was purified by 22 T. Högberg, S. Bengtsson, T. de Paulis, L. Johansson, P. Ström, H. Hall, S. O. Ögren, J. Med. Chem. 1990, 33, 1155 – 1163. 23 C. H. Park, K. H. Kim, I. K. Lee, S. Y. Lee, S. U. Choi, J. H. Lee, K. R. Lee, Arch. Pharm. Res. 2011, 34, 1289 – 1296. 24 K. Endo, M. Hirokami, T. Shibata, Org. Lett. 2010, 75, 3469 – 3472.
O
OMeMeO
MeB(pin)
(±)-17
O
OMeMeO
Me
B(pin)
S25
O
OMeMeO
Me O
S24
B(pin)(pin)B
S3
+LiTMP, THF
93%, >95:5 E/Z
Pd/C, H2, MeOH
>99%, >95:5 dr
O
OMeMeO
Me O
S24
O
OMeMeO
Me
11
(EtCO)2O (1.5 equiv)AlCl3 (1.5 equiv)
DCM, 0 ºC to r.t
83%
O
OMeMeO
Me
B(pin)
S25
O
OMeMeO
Me O
S24
B(pin)(pin)B+
LiTMP (1.1 equiv)
THF, 0 ºC to r.t
93%, >95:5 E/ZS3(1.1 equiv)

S31
FCC (SiO2, 10–15% EtOAc/pet. ether) to yield the title compound (297 mg, 0.820 mmol, 93%) as a colourless oil that crystallized upon standing. The E:Z ratio of the product was determined by 1H NMR to be >95:5.
Mpt: 79–80 ºC (EtOAc).
Rf: 0.19 (10% EtOAc/pet. ether).
1H NMR (500 MHz, CDCl3) δH 6.58 (s, 1H), 6.50 (s, 1H), 3.89 (s, 3H), 3.82 (s, 3H), 3.77 (s, 3H), 2.42 (br s, 2H), 1.85 (s, 3H), 1.02 (br s, 12H), 0.88 (t, J = 7.6 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 151.4, 151.0, 148.1, 142.2, 126.0, 115.4, 98.2, 82.7, 56.9, 56.4, 56.3, 26.1, 24.5, 15.8, 12.2 ppm.
11B NMR (96 MHz, CDCl3) δB 29.4 (br s) ppm.
IR (νmax/cm–1, neat): 2975–2844, 1506, 1465, 1390, 1371, 1361, 1293, 1296, 1146, 1109, 1038.
HRMS (ESI+) calcd. for C20H31O5BNa [M+Na]+ 385.2160, found 385.2172.
4,4,5,5-Tetramethyl-2-((2S*,3R*)-3-(2,4,5-trimethoxyphenyl)pentan-2-yl)-1,3,2-dioxaborolane [(±)-17]
Prepared following a modified literature procedure.25 To a 5 mL flask was added vinyl boronic ester S25 (1.18 g, 1.25 mmol, 1.0 equiv) and MeOH (13.0 mL). The flask was purged with N2 for 5 min before adding 10% palladium on carbon (345 mg, 0.325 mmol, 10 mol%). The flask was then purged with H2 for 2 min before placing under a static atmosphere of H2. The reaction was stirred vigorously for 2 h before filtering through a plug of silica, eluting with EtOAc, and concentrating in vacuo. Purification by FCC (SiO2, 15% EtOAc/pet. ether) gave the title compound (1.18 g, 3.25 mmol, >99%) as a colourless oil. The diastereomeric ratio was determined by 1H NMR to be >95:5.
For spectroscopic data, see the synthesis of enantioenriched 17 in Section 11.
(2S*,3S*)-3-(2,4,5-Trimethoxyphenyl)pentan-2-ol [(±)-S9]
Racemic pinacol boronic ester (±)-17 (12 mg, 0.033 mmol) in THF (2.0 mL) at 0 ºC was treated with 2.0 M aqueous NaOH (2.0 mL) and 30% aqueous H2O2 (1.0 mL) using the same procedure as described in Section 5 (for the oxidation of 10) to give the title compound (8.0 mg, 0.031 mmol, 95%) as a colourless solid.
25 E. Hupe, I. Marek, P. Knochel, Org. Lett. 2002, 4, 2861 – 2863.
O
OMeMeO
MeB(pin)
(±)-17
O
OMeMeO
Me
B(pin)
S25
Pd/C (10 mol%), H2
MeOH
>99%, >95:5 dr
O
OMeMeO
Me
(±)-17
B(pin)O
OMeMeO
Me
(±)-S9
OHH2O2, NaOH
H2O/THF
95%

S32
10. Optimization of the Diastereoselective Matteson Homologation
Reaction of Boronic Esters 10 or 17 with Lithiated Chloromethyl Esters
Table S4:
Entrya B(OR)2 X base addition time /min
base (m equiv)
T ºC % SM
(10 or 17)b
% yieldb
dr (syn:anti)b
1 B(pin) OTMB (S8) 2 LDA (1.2) –78 34 45 69:31
2 B(pin) OTMB (S8) 2 LDA (1.5) –78 29 52 69:31
3 B(pin) OTMB (S8) 2 LDA (2.0) –78 <5 55 77:23
4 B(pin) OTMB (S8) 2 LiTMP (2.0) –78 <5 53 75:25
5 B(pin) OTMB (S8) 2 nBuLi (2.0) –78 8 55 71:29
6 B(pin) OTMB (S8) 30 LDA (2.0) –78 50 17 53:47
7 B(pin) OTMB (S8) 2 LDA (2.0) –95 <5 66 70:30
8 B(pin) OTMB (S8) 0.5 LDA (2.0) –95 <5 72 69:31
9 B(pin) Cl (CH2Cl2) 0.5 LDA (2.0) –95 0 52 48:52
10 B(pin) OTIB (23) 0.5 LDA (2.0) –95 8 78 77:23
11 B(neo) OTMB (S8) 0.5 LDA (2.0) –95 5 55 89:11
12 B(neo) TIB (23) 0.5 LDA (2.0) –95 10 50 90:10 a Reactions performed using general procedure outlined below. b Determined by 1H NMR using 1,3-dinitrobenzene as an internal standard. c Determined by 1H NMR. TMB = 2,4,6-trimethylbenzoyl; TIB = 2,4,6-triisopropylbenzoyl.
General Procedure for Table S4: A solution of boronic ester 10 or 17 (0.1 mmol, 1.0 equiv) and chloromethyl ester S8 or 23 or CH2Cl2 (0.22 mmol, 2.2 equiv) in anhydrous THF (1.0 mL) was cooled to temperature T ºC (–78 ºC or –95 ºC). LDA (0.86 M in THF, 0.12–0.20 mmol, 1.2–2.0 equiv) was added dropwise over 0.5–30 min and the resulting mixture stirred at T ºC for 60 min before removing from the cold bath and stirring at room temperature for 30 min. The reaction was quenched by the addition of saturated aqueous NH4Cl (5 mL) and the phases separated. The aqueous phase was extracted with Et2O (3 × 5 mL)
O
OMeMeO
Me
LDA, THF, –95 ºC
B(pin) O
OMeMeO
Me
B(pin)
Cl OTIB
23
15a17
OMe
MeOOMe
THF, –78 ºC to RT
65%, 95:5 dr
O
OMeMeO
Me OTIB
B(pin)
24a
25
OMe
OMeMeO
MgCl
O
OMeMeO
Mebase (m equiv)
THF, T ºC, 30 min;the r.t, 60 min
B(OR)2
O
OMeMeO
Me X
B(OR)2+ Cl X
CH2Cl2/23/S8(2.2 equiv)10 or 17 24: B(OR)2 = B(pin)
S26: B(OR)2 = B(neo)

S33
and the combined organics washed with brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude product was analyzed by 1H NMR and the yield of 24 or S26 determined using 1,3-dinitrobenzene as an internal standard. The diagnostic 1H NMR signals for boronic esters 24a, 24b, 24c, S26a, and S26b are as follows:
Pinacol boronic ester 24a (X = OTIB): 1H NMR (400 MHz, CDCl3) δH 4.84 (d, J = 2.2 Hz, 1H, XCHB(pin), syn diastereomer), 4.58 (d, J = 5.1 Hz, 1H, XCHB(pin), anti diastereomer) ppm.
Pinacol boronic ester 24b (X = OTMB): 1H NMR (400 MHz, CDCl3) δH 4.87 (d, J = 2.4 Hz, 1H, XCHB(pin), syn diastereomer), 4.50 (d, J = 6.0 Hz, 1H, XCHB(pin), anti diastereomer) ppm.
Pinacol boronic ester 24c (X = Cl): 1H NMR (400 MHz, CDCl3) δH 3.73 (d, J = 4.8 Hz, 1H, XCHB(pin), syn diastereomer), 3.51 (d, J = 6.4 Hz, 1H, XCHB(pin), anti diastereomer) ppm.
Neopentyl glycol boronic ester S26a (X = OTIB): 1H NMR (400 MHz, CDCl3) δH 4.76 (d, J = 2.2 Hz, 1H, XCHB(pin), syn diastereomer), 4.38 (d, J = 5.7 Hz, 1H, XCHB(pin), anti diastereomer) ppm.
Neopentyl glycol boronic ester S26b (X = OTMB): 1H NMR (400 MHz, CDCl3) δH 4.80 (d, J = 2.4 Hz, 1H, XCHB(pin), syn diastereomer), 4.30 (d, J = 6.2 Hz, 1H, XCHB(pin), anti diastereomer) ppm.
Sequential Matteson Homologation/Grignard Addition of Boronic Ester 17
Table S5:
Entrya R base n equiv % yield of 24b
dr (syn:anti) of 24c
% yield of 15ab
dr (anti:syn) of 15ac
1 TMB (S8) LDA 1.8 8 >98:2 65 63:37
2d TMB (S8) LDA 1.8 10 >98:2 68 63:37
3 TMB (S8) LiTMP 2.0 31 >98:2 53 57:43
4 TMB (S8) LDA 2.0 5 >98:2 63 73:27
5 TIB (25) LDA 2.0 16 >98:2 49 88:12
6 TIB (25) LDA 2.2 23 >98:2 54 80:20
7e TIB (25) LDA 2.0 19 21:79 64 (65) 95:5
a Reactions performed using general procedure outlined below. b Determined by 1H NMR using 1,3-dinitrobenzene as an internal standard. Number in parenthesis is the yield of isolated product after purification by FCC. c Determined by 1H NMR. d The solution of crude boronic ester 24 was added to a solution of Grignard 25 at –78 ºC. e An aqueous work-up was performed before the reaction of crude 24 with Grignard 25. TMB = 2,4,6-trimethylbenzoyl; TIB = 2,4,6-triisopropylbenzoyl.
O
OMeMeO
Me (i) base (2.0 equiv)THF, –95 ºC
30 min; the r.t, 60 minB(pin) O
OMeMeO
Me
B(pin)+ Cl OR
23 or S8(2.2 equiv) 15a17
OMe
MeOOMe
25 (n equiv)
OMe
OMeMeO
MgCl(ii)
–78 ºC, 2h; then r.t, 16 h
O
OMeMeO
Me OR
B(pin)+
24a (R = TIB)24b (R = TMB)

S34
General Procedure for Table S5: A solution of boronic ester 17 (38 mg, 0.10 mmol, 1.0 equiv) and chloromethyl TMB-ester S8 (47 mg, 0.22 mmol, 2.2 equiv), or chloromethyl TIB-ester 23 (65 mg, 0.22 mmol, 2.2 equiv), in anhydrous THF (1.0 mL) was cooled to –95 ºC before the addition of LDA (0.86 M in THF, 0.23 mL, 0.20 mmol, 2.0 equiv) dropwise over 30 s. The resulting mixture was stirred at –95 ºC for 60 min before removing from the cold bath and stirring at room temperature for 30 min. The mixture was then cooled to –78 ºC before the addition of aryl Grignard 25 (0.4 M in THF, 0.45–0.55 mL, 0.18–0.22 mmol, 1.8–2.2 equiv) dropwise over 90 s. The reaction was stirred at –78 ºC for 2 h before removing from the cold bath and stirring at room temperature for 16 h. The reaction was quenched by the addition of saturated aqueous NH4Cl (5 mL) and the phases separated. The aqueous phase was extracted with Et2O (3 × 5 mL) and the combined organics washed with brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude product was analyzed by 1H NMR and the yields of 15a and 24 determined using 1,3-dinitrobenzene as an internal standard.
Procedure for the synthesis of aryl Grignard 25 (0.4 M in THF)
To a solution of 1-iodo-2,4,5-trimethoxylbenzene (12) (159 mg, 0.541 mmol, 1.10 equiv) in anhydrous THF (0.82 mL) at 0 ºC was added isopropylmagnesium chloride (2.0 M in THF, 0.25 mL, 0.49 mmol, 1.0 equiv) dropwise over 1 min. The resulting mixture was stirred at 0 ºC for 30 min to give an approximately 0.40 M solution of (2,4,5-trimethoxyphenyl)magnesium chloride (25), which was used immediately.
iPrMgCl (1.0 equiv)
THF, 0 ºC, 30 min
25(0.4 M in THF)
OMe
OMeMeO
MgClOMe
OMeMeO
I
12(1.1 equiv)
S35
11. Synthesis of Tatanan A
4,4,5,5-Tetramethyl-2-((2S,3R)-3-(2,4,5-trimethoxyphenyl)pentan-2-yl)-1,3,2-dioxaborolane (17)
An oven-dried Schlenk tube was charged with propyl 2,4,6-triisopropylbenzoate (S4) (784 mg, 2.70 mmol, 1.35 equiv) before being evacuated and refilled with nitrogen three times. (+)-Sparteine (0.597 mL, 2.60 mmol, 1.30 equiv) and anhydrous Et2O (10 mL) were then added to the Schlenk tube. The resulting solution was cooled to –78 °C before the addition of sBuLi (1.3 M in cyclohexane/hexanes, 2.00 mL, 2.60 mmol, 1.30 equiv) dropwise over 10 min (colour change: colourless to dark brown). The reaction mixture was stirred at –78 °C for 4 h before the addition of a solution of aryl pinacol boronic ester 26 (588 mg, 2.00 mmol, 1.00 equiv) in anhydrous Et2O (6.6 mL) dropwise over 30 min. The resulting mixture was stirred at –78 ºC for 2 h before removing from the cold bath and stirring at room temperature for 20 h.
A separate oven-dried Schlenk tube was charged with ethyl 2,4,6-triisopropylbenzoate (S1) (857 mg, 3.10 mmol, 1.55 equiv) before being evacuated and refilled with nitrogen three times. (+)-Sparteine (0.689 mL, 3.00 mmol, 1.50 equiv) and anhydrous Et2O (10 mL) were then added to the Schlenk tube. The resulting solution was cooled to –78 °C before the addition of sBuLi (1.3 M in cyclohexane/hexanes, 2.31 mL, 3.00 mmol, 1.50 equiv) dropwise over 10 min (colour change: colourless to dark brown). The reaction mixture
O
OMeMeO
MeB(pin)
26
OTIBS4
(+)-sparteine, sBuLiEt2O, –78 ºC
O
OMeMeO
Me
S27
B(pin)
OTIBS1
(+)-sparteine, sBuLiEt2O, –78 ºC
74% (2 steps),94:6 dr, >99% ee
O
OMeMeO
Me
17
B(pin)
LDA, THF, –95 ºC to r.t
O
OMeMeO
Me
B(pin)
Cl OTIB
15a
OMe
MeOOMe
(i)
LDA, THF;then I2, MeOH O
OMeMeO
Me OMe
OMeMeO
LDA, THF
97% (2 steps)100% de
O
OMeMeO
Me OMe
OMeMeO
1OMe
S28
OCb
ZrCp2Cl2, AlMe3H2O, CH2Cl2;then I2, THF
58% brsm
O
OMeMeO
Me OMe
OMeMeO ArB(OH)2 (29)
Pd(PPh3)4Ag2O, THF/H2O
93%
28
(27)
I
MeO
OMe
OCb
O
OMeMeO
Me OMe
OMeMeO
5
(23)
(ii) ArMgCl (25), –78 ºC to r.t
65%, 95:5 dr[>95:5 dr after recryst.]
O
OMeMeO
MeB(pin)
26
OTIBS4 (1.35 equiv)
(+)-sparteine (1.30 equiv)sBuLi (1.30 equiv)Et2O, –78 ºC, 4 h
(ii) 26, –78 ºC to r.t, 20 h
(i)
O
OMeMeO
Me
S27
B(pin)
OTIBS1 (1.55 equiv)
(+)-sparteine (1.05 equiv)sBuLi (1.50 equiv)Et2O, –78 ºC, 4 h
(ii) S27, –78 ºC to r.t, 14 h
74% (2 steps),94:6 dr, >99% ee
(i)
O
OMeMeO
Me
17
B(pin)

S36
was stirred at –78 °C for 4 h before the addition of the contents of the first Schlenk tube (containing S27) dropwise via cannula over 40 min. The resulting mixture was stirred at –78 ºC for 2 h before removing from the cold bath and stirring at room temperature for 14 h. To the reaction was added 2 M aqueous HCl (20 mL) and the mixture stirred vigorously for 10 min before separation of the layers. The organic layer was washed with 2 M aqueous HCl (2 × 20 mL) then the combined aqueous washings were further extracted with Et2O (2 × 20 mL) [the acidic aqueous washings were retained for the recovery of (+)-sparteine].2 The combined organic layers were washed with brine (40 mL), dried (MgSO4), filtered and concentrated in vacuo to give crude boronic ester 17, which was determined to have a diastereomeric ratio of 91:9 by 1H NMR. Purification by FCC (SiO2, 5% Et2O/toluene + 1% Et3N) gave title compound (540 mg, 0.697 mmol, 74%) as a colourless oil, which crystallized upon standing. The diastereomeric ratio of the purified product was determined to be 94:6 by 1H NMR. Further enhancement to >95:5 dr was possible by FCC.
Mpt: 43–46 ºC (Et2O).
Rf: 0.18 (10% Et2O/toluene) [minor diastereomer = 0.14 (10% Et2O/toluene)].
1H NMR (500 MHz, CDCl3) δH 6.67 (s, 1H), 6.51 (s, 1H), 3.88 (s, 3H), 3.81 (s, 3H), 3.76 (s, 3H), 2.97 (td, J = 11.0, 3.8 Hz, 1H), 1.65 (dqd, J = 13.3, 7.4, 3.9 Hz, 1H), 1.54 (ddq, J = 13.3, 10.9, 7.3 Hz, 1H), 1.33 (dq, J = 11.2, 7.4 Hz, 1H), 1.29 (s, 6H), 1.28 (s, 6H), 0.74 (d, J = 7.4 Hz, 3H), 0.69 (t, J = 7.3 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.7, 147.0, 143.3, 125.1, 111.5, 98.0, 82.9, 56.9, 56.4, 56.0, 42.3, 29.1, 24.8, 24.7, 23.5, 14.3, 12.2 ppm.
11B NMR (96 MHz, CDCl3) δB 34.0 (br s) ppm.
IR (νmax/cm–1, neat): 2981–2835, 1508, 1456, 1397, 1378, 1317, 1204, 1175, 1144, 1037.
HRMS (ESI+) calcd. for C20H33BO5Na [M+Na]+ 387.2317, found 387.2327.
α D22 –9.0 (c 0.50, CHCl3).
Chiral HPLC: The e.r. was determined by HPLC [Daicel Chiralpak-IB (25 cm) with guard, 7:93 i-PrOH/hexanes, 1.0 mL/min, r.t., 210 nm, tR (major) = 9.1 min, tR (minor) = 10.2 min] to be 99.7:0.3.
syn diastereomer (tR = 12.2 and
13.8 min)
anti diastereomer (tR = 9.1 and
10.2 min)

S37
4,4,5,5-Tetramethyl-2-((1R,2R,3R)-2-methyl-1,3-bis(2,4,5-trimethoxyphenyl)pentyl)-1,3,2-dioxaborolane (15a)
A solution of boronic ester 17 (109 mg, 0.300 mmol, 1.00 equiv) and chloromethyl TIB-ester 23 (196 mg, 0.660 mmol, 2.20 equiv) in anhydrous THF (3.0 mL) was cooled to temperature –95 ºC before the addition of LDA (0.86 M in THF, 0.70 mL, 0.60 mmol, 2.0 equiv) dropwise over 45 s. The resulting mixture was stirred at –95 ºC for 60 min before removing from the cold bath and stirring at room temperature for 30 min. The reaction was quenched by the addition of saturated aqueous NH4Cl (15 mL) and the phases separated. The aqueous phase was extracted with Et2O (3 × 10 mL) and the combined organics washed with water (15 mL), brine (15 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue was further dried under high vacuum for 30 min before dissolving in anhydrous THF (3.0 mL) and cooling to –78 ºC. Aryl Grignard 25 (0.4 M in THF, 1.50 mL, 0.600 mmol, 2.0 equiv) was added dropwise over 90 s and the reaction stirred at –78 ºC for 2 h before removing from the cold bath and stirring at room temperature for 16 h. The reaction was quenched by the addition of saturated aqueous NH4Cl (15 mL) and the phases separated. The aqueous phase was extracted with Et2O (3 × 10 mL) and the combined organics washed with water (15 mL), brine (15 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by FCC (2 × SiO2, 30% EtOAc/pet. ether followed by 1% acetone/CH2Cl2) to give the title compound (105 mg, 0.193 mmol, 65%) as a colourless oil that crystallized upon standing. The diastereomeric ratio was determined by 1H NMR to be 95:5. This ratio could be improved to >95:5 by recrystallization from Et2O/pet. ether.
Boronic ester 15a could also be prepared without aqueous work-up of intermediate 24. However, this results in diminished yield and diastereoselectivity (54%, 80:20 dr, see Table S5).
Mpt: 153–155 ºC (Et2O/pet. ether). Mpt (racemate): 132–134 ºC (Et2O/pet. ether).
Rf: 0.35 (2% acetone/DCM).
1H NMR (500 MHz, CDCl3) δH 6.95 (s, 1H), 6.59 (s, 1H), 6.50 (s, 1H), 6.48 (s, 1H), 3.87 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.70 (s, 3H), 3.05 (br s, 1H), 3.02 (d, J = 6.0 Hz, 1H), 2.13–2.06 (m, 1H), 1.96 (dqd, J = 14.5, 7.2, 4.2 Hz, 1H), 1.63–1.53 (m, 1H), 1.26 (s, 6H), 1.24 (s, 6H), 0.76 (d, J = 6.9 Hz, 3H), 0.71 (t, J = 7.3 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC152.6, 151.7, 147.1, 147.0, 143.0, 142.5, 125.2, 123.1, 114.9, 112.7 (br), 98.1, 97.8, 82.8, 56.8, 56.7, 56.6, 56.4, 56.1, 56.0, 43.2 (br), 39.5, 27.7 (br), 25.8, 24.9, 24.8, 16.4, 12.1 ppm.
11B NMR (96 MHz, CDCl3) δB 32.1 (br s) ppm.
IR (νmax/cm–1, neat): 2975–2832, 1509, 1464, 1371, 1313, 1204, 1144, 1038.
HRMS (ESI+) calcd. for C30H45O8BNa [M+Na]+ 567.3105, found 567.3081.
α D24 +26.2 (c 0.89, CHCl3).
O
OMeMeO
Me
LDA (2.0 equiv)THF, –95 ºC, 30 min;
then r.t, 60 min
B(pin) O
OMeMeO
Me
B(pin)
Cl OTIB
23 (2.2 equiv)
15a17
OMe
MeOOMe
THF, –78 ºC, 2h;then r.t, 16 h
65%, 95:5 dr[>95:5 dr after recryst.]
O
OMeMeO
Me OTIB
B(pin)
24
25 (2.0 equiv)
OMe
OMeMeO
MgCl

S38
5,5'-((3S,4S,5R)-4-Methylhept-1-yne-3,5-diyl)bis(1,2,4-trimethoxybenzene) (5)
Following a modified literature procedure.20 To a solution of boronic ester 15a (132 mg, 0.242 mol, 1.00 equiv) and vinyl carbamate 27 (62 mg, 0.36 mmol, 1.5 equiv) in anhydrous THF (2.40 mL) at –78 ºC was added LDA (0.86 M in THF, 0.42 mL, 0.36 mmol, 1.5 equiv) dropwise over 3 min. The reaction was stirred at –78 ºC for 60 min before the reaction vessel was transferred to a 0 ºC bath and stirred for 30 min. The reaction was cooled to –78 ºC and a solution of I2 (92 mg, 0.36 mmol, 1.5 equiv) in MeOH (2.40 mL) was added dropwise over 5 min. The mixture was stirred at –78 ºC for 10 min before removing from the cold bath and stirring at room temperature for 60 min. Saturated aqueous Na2S2O3 (10 mL) was added and the product extracted into Et2O (3 × 10 mL). The combined organic extracts were washed with water (10 mL), brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue containing S28 was further dried under high vacuum for 30 min before placing under an atmosphere of N2, dissolving in anhydrous THF (2.40 mL) and cooling to –78 ºC. LDA (0.86 M in THF, 2.11 mL, 1.82 mmol, 6.00 equiv) was added dropwise over 1 min and the resulting solution was removed from the cold bath and stirred at room temperature for 15 min. The reaction was quenched by the addition of saturated aqueous NH4Cl (10 mL) and the product extracted into Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by FCC (SiO2, 25% EtOAc/pet. ether) to give the title compound (104 mg, 0.235 mmol, 97%) as a colourless oil. The reaction was determined by 1H NMR analysis to proceed with 100% diastereomeric excess (de)
Rf: 0.29 (25% EtOAc/pet. ether).
1H NMR (500 MHz, CDCl3) δH 7.17 (s, 1H), 6.63 (s, 1H), 6.52 (s, 1H), 6.51 (s, 1H), 4.55 (t, J = 2.9 Hz, 1H), 3.89 (s, 3H), 3.88 (s, 3H), 3.86 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H), 3.79 (s, 3H), 3.20 (br s, 1H), 2.28 (d, J = 2.6 Hz, 1H), 2.07 (dqd, J = 13.4, 7.4, 3.9 Hz, 1H), 2.01 (br s, 1H), 1.68 (br s, 1H), 0.76 (t, J = 7.3 Hz, 3H), 0.58 (d, J = 6.7 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.6 (br), 150.2, 148.2, 147.4, 143.1 (br), 142.5, 124.8 (br), 120.8, 114.3, 111.9 (br), 98.2, 97.4, 83.6, 72.2, 57.0 (br), 56.8, 56.7, 56.23, 56.22, 56.0, 41.4 (br), 40.6 (br), 33.9, 24.9 (br), 13.4, 11.8 (br) ppm.
IR (νmax/cm–1, neat): 3287, 2993–2835, 1509, 1464, 1458, 1439, 1398, 1316, 1238, 1204, 1183, 1036.
HRMS (ESI+) calcd. for C26H34O6Na [M+Na]+ 465.2248, found 465.2252.
α D22 +67.6 (c 0.88, CHCl3).
O
OMeMeO
MeLDA (1.5 equiv), THF
–78 ºC to 0 ºC;
then I2 (1.5 equiv)MeOH, –78 ºC to r.t
B(pin)
OMe
OMeMeO
O
OMeMeO
Me OMe
OMeMeO
LDA (6.0 equiv)
THF, –78 ºC to r.t
97% (2 steps)100% de
O
OMeMeO
Me OMe
OMeMeO
5S28
+ OCb
27(1.5 equiv)
15a>95:5 dr
OCb

S39
5,5'-((3R,4S,5R,E)-1-Iodo-2,4-dimethylhept-1-ene-3,5-diyl)bis(1,2,4-trimethoxybenzene) (28)
Following a modified literature procedure.21 A solution of zirconocene dichloride (23 mg, 0.079 mmol, 1.0 equiv) in anhydrous CH2Cl2 (0.80 mL) was degassed by three freeze/pump/thaw cycles before cooling to –78 ºC. Trimethylaluminium (2 M in toluene, 0.24 mL, 0.47 mmol, 6.0 equiv) was added followed by H2O (2.1 µL, 0.12 mmol, 1.5 equiv) and the reaction was transferred to a 0 ºC bath and stirred for 30 min, giving a bright yellow solution, before cooling to –78 ºC. In a separate flask, a solution of alkyne 5 (35 mg, 0.079 mmol, 1.0 equiv) in anhydrous CH2Cl2 (0.80 mL) was degassed by three freeze/pump/thaw cycles. This solution was added dropwise over 1 min to the ZrCp2Cl2/AlMe3 mixture. The cold bath was removed and the reaction was stirred at room temperature for 18 h before cooling back to –78 ºC. A solution of I2 (100 mg, 0.395 mmol, 5.00 equiv) in degassed anhydrous THF (0.40 mL) was added dropwsie over 2 min and the resulting mixture stirred at –78 ºC for 30 min, 0 ºC for 30 min, then at room temperature for 30 min. The reaction was cooled to 0 ºC and quenched by the slow [Gas evolution!] addition of saturated aqueous K2CO3 (2 mL). The phases were separated, the aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by preparative TLC (2 × SiO2, 1st = 15% EtOAc/toluene, 2nd = 0.5% EtOAc/CH2Cl2) to give the title compound (16 mg, 0.027 mmol, 35%, 58% brsm) as a colourless oil, as well as recovered alkyne 5 (14 mg, 0.031 mmol, 40%).
Rf: 0.51 (1% EtOAc/CH2Cl2), 0.40 (15% EtOAc/toluene).
1H NMR (500 MHz, CDCl3) δH 6.81 (s, 1H), 6.51 (s, 2H), 6.39 (s, 1H), 5.95 (d, J = 1.2 Hz, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.86 (s, 3H), 3.77 (s, 3H), 3.71 (s, 3H), 3.57 (d, J = 11.4 Hz, 1H), 3.44 (s, 3H), 3.04 (br s, 1H), 2.42 (dqd, J = 13.7, 6.7, 3.1 Hz, 1H), 1.79–1.62 (m, 2H), 1.62 (d, J = 1.0 Hz, 3H), 0.81 (d, J = 6.8 Hz, 3H), 0.77 (t, J = 7.3 Hz, 3H) ppm.
13C NMR (126 MHz, CDCl3) δC 152.8, 152.3, 148.1, 147.8, 147.5, 142.5, 142.0, 121.8, 121.4, 114.7, 112.2, 97.9, 96.8, 77.2, 56.9, 56.7, 56.2, 56.1, 56.0, 55.5, 50.8, 36.6, 26.8, 20.3, 14.6, 12.9 ppm. One signal (corresponding to ArCHCH2) is not observed.
IR (νmax/cm–1, neat): 2961–2832, 1508, 1463, 1455, 1316, 1203, 1179, 1037.
HRMS (ESI+) calcd. for C27H37O6INa [M+Na]+ 607.1527, found 607.1513.
α D23 +91.1 (c 0.90, CHCl3).
ZrCp2Cl2 (1.0 equiv)AlMe3 (6.0 equiv)
H2O (1.5 equiv), CH2Cl2;
then I2 (5.0 equiv), THF
58% brsm
O
OMeMeO
Me OMe
OMeMeO
28
O
OMeMeO
Me OMe
OMeMeO
5I

S40
Tatanan A (1)
A vial was charged with vinyl iodide 28 (25 mg, 0.043 mmol, 1.0 equiv), 2,4,5-trimethoxyphenylboronic acid (29) (14 mg, 0.064 mmol, 1.5 equiv), Pd(PPh3)4 (4.9 mg, 4.3 µmol, 10 mol%), and Ag2O (25 mg, 0.11 mmol, 2.5 equiv). The vial was sealed with a septum and triple evacuated/N2 filled before the addition of degassed (sparged with N2 for 10 min) THF/H2O (5:1, 0.86 mL). The mixture was stirred at room temperature for 16 h before filtering through a plug of silica, eluting with EtOAc, and concentrating in vacuo. The residue was purified by preparative TLC (SiO2, 25% EtOAc/toluene) to give the title compound (25 mg, 0.040 mmol, 93%) as a white solid. Recrystallization from hot MeOH gave colourless crystals.
Mpt: 136–138 ºC (MeOH) [Lit. 125–127 ºC (MeOH), 123–125 ºC (CD3OD)].26,27
Mpt (racemate): 122–124 ºC (MeOH).
Rf: 0.33 (40% EtOAc/pet. ether).
1H NMR (500 MHz, CD3OD) δH 6.86 (s, 1H), 6.65 (s, 1H), 6.63 (s, 1H), 6.59 (s, 1H), 6.53 (s, 1H), 6.52 (br s, 1H), 6.26 (s, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.81 (s, 3H), 3.77 (s, 3H), 3.76 (s, 3H), 3.71 (s, 3H), 3.70 (s, 6H), 3.52 (s, 3H), 3.51 (d, J = 10.5 Hz, 1H), 3.10 (br s, 1H), 2.55 (dqd, J = 10.4, 6.8, 3.1 Hz, 1H), 1.84–1.68 (m, 2H), 1.55 (s, 3H), 0.95 (d, J = 6.8 Hz, 3H), 0.75 (t, J = 7.3 Hz, 3H) ppm.
1H NMR (500 MHz, CDCl3) δH 6.89 (s, 1H), 6.64 (s, 1H), 6.54 (s, 2H), 6.48 (s, 1H), 6.43 (s, 1H), 6.34 (s, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.87 (s, 3H), 3.86 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.75 (s, 3H), 3.72 (s, 3H), 3.53 (d, J = 11.3 Hz, 1H), 3.50 (s, 3H), 3.08 (br s, 1H), 2.51 (dqd, J = 11.2, 6.9, 3.4 Hz, 1H), 1.81–1.62 (m, 2H), 1.60 (d, J = 1.3 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H), 0.79 (t, J = 7.3 Hz, 3H) ppm.
13C NMR (126 MHz, CD3OD) δC 153.2, 152.8, 152.1, 148.4, 147.79, 147.75, 142.4, 142.3, 142.0, 138.2, 123.6, 122.3, 121.6, 119.9, 115.3, 115.1, 113.8, 98.53, 98.46, 97.2, 56.4, 56.20, 56.16, 55.9, 55.5, 55.41, 55.35, 55.3, 54.7, 50.6, 40.5 (br), 36.9, 25.8, 14.4, 13.5, 11.9 ppm.
13C NMR (126 MHz, CDCl3) δC 152.8, 152.5, 151.7, 148.0, 147.3, 147.3, 142.5, 142.4, 142.0, 138.5, 123.8, 122.3, 121.8, 120.1, 114.7 (br), 114.5, 112.7, 98.5, 98.2, 96.9, 57.0, 56.7, 56.7, 56.6, 56.4, 56.2, 56.1, 56.0, 55.6, 51.0, 37.0, 26.8, 14.8, 14.5, 12.9 ppm. One signal (corresponding to ArCHCH2) is not observed.
IR (νmax/cm–1, neat): 2958–2832, 1508, 1464, 1455, 1439, 1396, 1317, 1204, 1178, 1037.
HRMS (ESI+) calcd. for C36H48O9Na [M+Na]+ 647.3191, found 647.3188.
α D24 +90.0 (c 0.10, MeOH) [Lit: α D20 +10 (c 0.1 MeOH),26 α D23 +55.1 (c 0.1 MeOH)].27
26 G. Ni, Z.-F. Shen, Y. Lu, Y.-H. Wang, Y.-B. Tang, R.-Y. Chen, Z.-Y. Hao, D.-Q. Yu, J. Org. Chem. 2011, 76, 2056 – 2061. 27 Q. Xiao, J. J. Jackson, A. Basak, J. M. Bowler, B. G. Miller, A. Zakarian, Nat. Chem. 2013, 5, 410 – 416.
Pd(PPh3)4 (10 mol%)Ag2O (2.5 equiv)
THF/H2O, r.t, 15 h
93%
O
OMeMeO
Me OMe
OMeMeO
1OMe
O
OMeMeO
Me OMe
OMeMeO
28
I
MeO
OMe
(HO)2B
OMeMeO
OMe+
29(1.5 equiv)
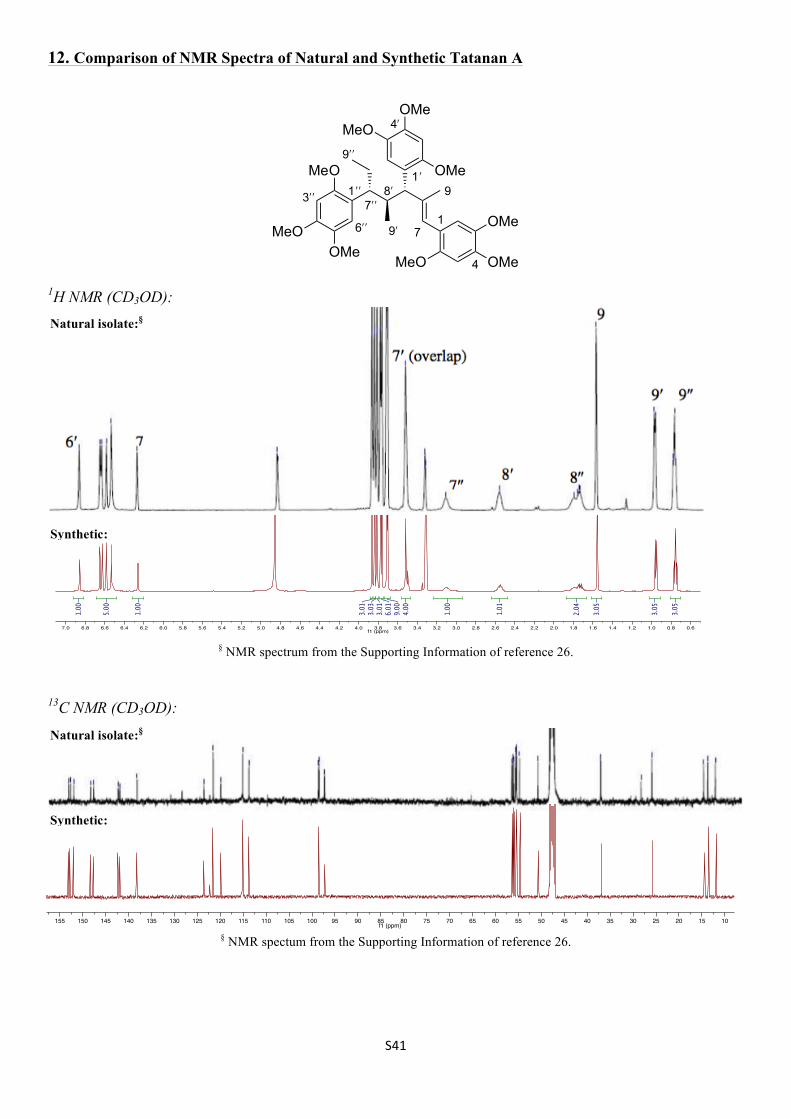
S41
12. Comparison of NMR Spectra of Natural and Synthetic Tatanan A
1H NMR (CD3OD):
§ NMR spectrum from the Supporting Information of reference 26.
13C NMR (CD3OD):
§ NMR spectum from the Supporting Information of reference 26.
O
OMeMeO
Me OMe
OMeMeO
OMeMeO
OMe
1ʹʹ
1
1ʹ
7ʹʹ
7
8ʹ
6ʹʹ
4
4ʹ
3ʹʹ
9ʹʹ
9
9ʹ
0.60.81.01.21.41.61.82.02.22.42.62.83.03.23.43.63.84.04.24.44.64.85.05.25.45.65.86.06.26.46.66.87.0f1 (ppm)
3531 AN-4-683/10
3.0
5
3.0
5
3.0
5
2.0
4
1.0
1
1.0
0
4.0
0
9.0
0
6.0
1
3.0
1
3.0
3
3.0
1
1.0
0
5.0
0
1.0
0
0.7
4
0.7
6
0.7
7
0.9
5
0.9
6
1.5
5
1.6
8
1.7
0
1.7
1
1.7
2
1.7
3
1.7
4
1.7
5
1.7
6
1.7
8
1.8
42
.52
2.5
3
2.5
4
2.5
4
2.5
5
2.5
6
2.5
6
2.5
7
2.5
8
3.1
0
3.4
9
3.5
2
3.7
0
3.7
1
3.7
6
3.7
7
3.8
1
3.8
4
3.8
6
6.2
6
6.2
6
6.5
2
6.5
3
6.5
9
6.6
3
6.6
5
6.8
6
Natural isolate:§
Synthetic:
Natural isolate:§
101520253035404550556065707580859095100105110115120125130135140145150155f1 (ppm)
3531 AN-4-683/11
11
.90
13
.51
14
.44
25
.77
36
.90
40
.50
47
.60
MeO
D
50
.64
54
.66
55
.28
55
.35
55
.41
55
.47
55
.85
56
.16
56
.20
56
.34
97
.23
98
.46
98
.53
11
3.7
6
11
5.1
4
11
5.3
2
11
9.9
2
12
1.6
3
12
2.3
1
12
3.6
3
13
8.2
3
14
1.9
8
14
2.3
1
14
2.4
1
14
7.7
5
14
7.7
9
14
8.4
0
15
2.1
0
15
2.7
9
15
3.2
1
Synthetic:

S42
1H and 13C NMR Data (CD3OD):
Number Natural Isolate26 Synthetic δH
(m, J) δC δH (m, J)a δCb
1 121.4 121.30 2 153.5 153.49 3 6.59 (s) 98.7 6.59 (s) 98.61 4 149.8 149.78 5 143.8 143.79 6 6.64 (s) 116.6 6.63 (s) 116.71 7 6.28 (s) 123.0 6.26 (s) 123.01 8 139.7 139.62 9 1.57 (s) 14.9 1.55 (s) 14.90 1' 125.1 125.01 2' 154.2 154.18 3' 6.66 (s) 100.0 6.65 (s) 99.91 4' 149.2 149.13 5' 143.7 143.70 6' 6.87 (s) 115.2 6.86 (s) 115.15 7' 3.53 (overlap) 52.0 3.51 (d, 10.5) 52.03 8' 2.56 (1H, m) 38.3 2.55 (dqd, 10.4, 6.8, 3.1) 38.29 9' 0.97 (d, 6.5) 15.8 0.95 (d, 6.8) 15.83 1'' 123.8 123.70 2'' 154.6 154.60 3'' 6.54 (s) 99.9 6.52 (s) 99.85 4'' 149.2 149.18 5'' 143.4 143.37 6'' 6.54 (s) 116.6 6.53 (s) 116.52 7'' 3.11 (m) 29.5c 3.10 (br s) 41.88 (broad)c
8'' a) 1.74 (m); b) 1.80 (m) 27.2 a) 1.73 (m); b) 1.79 (m) 27.15 9'' 0.77 (t, 6.5) 13.3 0.75 (t, 7.3) 13.29
OMe 3.53 (s) 56.1 3.52 (s) 56.05 OMe 3.71 (s) 56.7 3.70 (s) 56.66 OMe 3.71 (s) 56.8 3.70 (s) 56.74 OMe 3.71 (s) 56.8 3.71 (s) 56.80 OMe 3.77 (s) 56.9 3.76 (s) 56.86 OMe 3.78 (s) 57.3 3.77 (s) 57.24 OMe 3.82 (s) 57.6 3.81 (s) 57.54 OMe 3.84 (s) 57.6 3.84 (s) 57.58 OMe 3.87 (s) 57.8 3.86 (s) 57.73
a Referenced to CD3OD at 3.31 ppm. b Referenced to C-2'' at 154.60 ppm. c Peak misassigned in the isolation paper due to impurity in the 13C NMR spectrum. The reassigned signal at 41.9 ppm is very broad and easily missed.
O
OMeMeO
Me OMe
OMeMeO
OMeMeO
OMe
1ʹʹ
1
1ʹ
7ʹʹ
7
8ʹ
6ʹʹ
4
4ʹ
3ʹʹ
9ʹʹ
9
9ʹ

S43
13. 1H and 13C NMR spectra
1H NMR (400 MHz, CDCl3):
13C NMR (101 MHz, CDCl3):
O
OMeMeO
MeB
13
O
O

S44
1H NMR (400 MHz, CDCl3):
13C NMR (101 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
AN-3-511
12.24
3.04
3.04
3.04
1.01
1.00
1.35
3.83
3.89
3.91
6.50
7.20
7.27
O
OMeMeO
MeB
13
O
O
O
OMeMeO
MeB
26
O
O

S45
1H NMR (400 MHz, CDCl3):
13C NMR (101 MHz, CDCl3):
0102030405060708090100110120130140150160170180f1 (ppm)
AN-3-511
24
.78
55
.74
56
.40
57
.32
76
.85
cdcl3
77
.17
cdcl3
77
.49
cdcl3
83
.14
97
.54
10
8.4
3
11
9.0
0
14
2.8
2
15
2.4
3
16
0.2
1
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
an131138_AN-3-407-SM_PROTON_01
3.00
18.09
2.00
3.00
2.00
2.00
0.99
1.01
1.03
1.25
1.25
1.27
1.27
1.73
1.74
1.76
1.78
1.80
1.82
2.82
2.84
2.86
2.86
2.87
2.88
2.89
2.90
2.91
2.92
2.93
2.93
2.95
4.26
4.28
4.29
7.02
7.27
O
O
S4
O
OMeMeO
MeB
26
O
O

S46
1H NMR (400 MHz, CDCl3):
13C NMR (101 MHz, CDCl3):
-100102030405060708090100110120130140150160170180190200f1 (ppm)
an131138_AN-3-407-SM_CARBON_01
10.7
2
22.1
5
24.1
1
24.2
9
31.6
3
34.5
8
66.7
7
76.8
4 c
dcl3
77.1
6 c
dcl3
77.4
8 c
dcl3
120.9
7
130.8
6
144.8
5
150.1
6
171.1
7
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
an130904_AN-3-407_PROTON_01
9.16
3.07
18.82
2.02
3.18
1.00
2.01
0.21
1.00
1.02
1.04
1.24
1.26
1.92
1.93
1.94
1.96
1.98
1.99
2.01
2.02
2.81
2.83
2.84
2.86
2.88
2.89
2.91
2.93
2.95
4.93
4.94
4.95
4.96
4.96
4.97
4.98
4.99
7.01
O
O
S4
S5
Me3Sn OTIB

S47
1H NMR (500 MHz, CDCl3):
13C NMR (126 MHz, CDCl3):
-20-100102030405060708090100110120130140150160170180190200f1 (ppm)
an130904_AN-3-407_CARBON_01
-8.9
3
12.4
4
24.1
0
24.3
4
24.5
5
27.0
1
31.5
7
34.5
3
74.1
7
76.8
4 c
dcl3
77.1
6 c
dcl3
77.4
8 c
dcl3
120.9
5
130.9
8
144.9
9
150.0
0
171.5
7
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
sr15344_SR689a_PROTON_001
0.94
6.04
3.04
2.03
1.01
1.00
2.23
3.84
3.89
4.62
6.54
6.86
7.27
S5
Me3Sn OTIB
O
OMeMeO
Me
S6
OH

S48
1H NMR (400 MHz, CDCl3):
13C NMR (101 MHz, CDCl3):
0102030405060708090100110120130140150160170180f1 (ppm)
sr15344_SR689a_CARBON_001
56.1
3
56.2
5
56.6
0
61.4
5
76.7
8 c
dcl3
77.0
3 c
dcl3
77.2
9 c
dcl3
97.3
4
113.2
4
120.5
8
142.8
4
149.1
9
151.6
7
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
sr105174_SR1181b_PROTON_01
18.62
3.00
6.12
3.06
2.04
1.00
3.08
1.19
1.21
1.22
1.23
2.82
2.84
2.85
2.87
2.89
2.90
2.92
3.83
3.84
3.91
5.35
6.53
6.98
6.98
7.27
O
OMeMeO
Me
S6
OH
O
OMeMeO
Me
16
OTIB
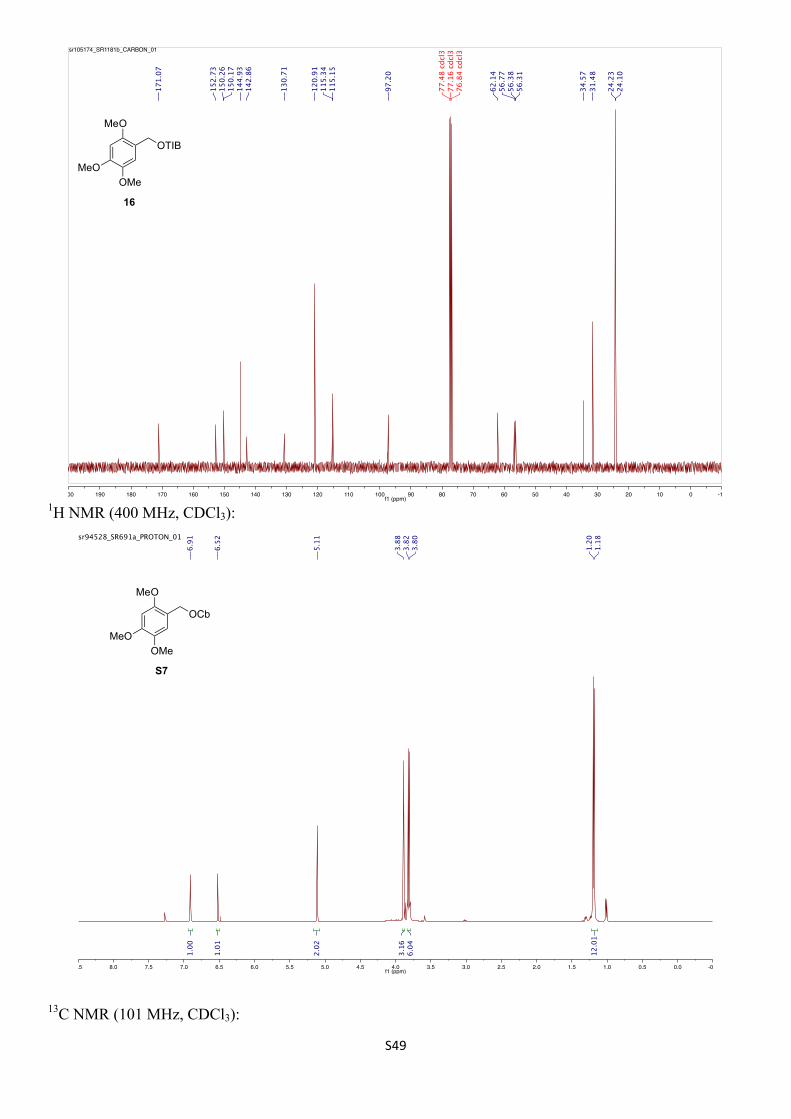
S49
1H NMR (400 MHz, CDCl3):
13C NMR (101 MHz, CDCl3):
-100102030405060708090100110120130140150160170180190200f1 (ppm)
sr105174_SR1181b_CARBON_01
24
.10
24
.23
31
.48
34
.57
56
.31
56
.38
56
.77
62
.14
76
.84
cdcl3
77
.16
cdcl3
77
.48
cdcl3
97
.20
11
5.1
5
11
5.3
4
12
0.9
1
13
0.7
1
14
2.8
6
14
4.9
3
15
0.1
7
15
0.2
6
15
2.7
3
17
1.0
7
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
sr94528_SR691a_PROTON_01
12.01
6.04
3.16
2.02
1.01
1.00
1.18
1.20
3.80
3.82
3.88
5.11
6.52
6.91
O
OMeMeO
Me
16
OTIB
O
OMeMeO
Me
S7
OCb
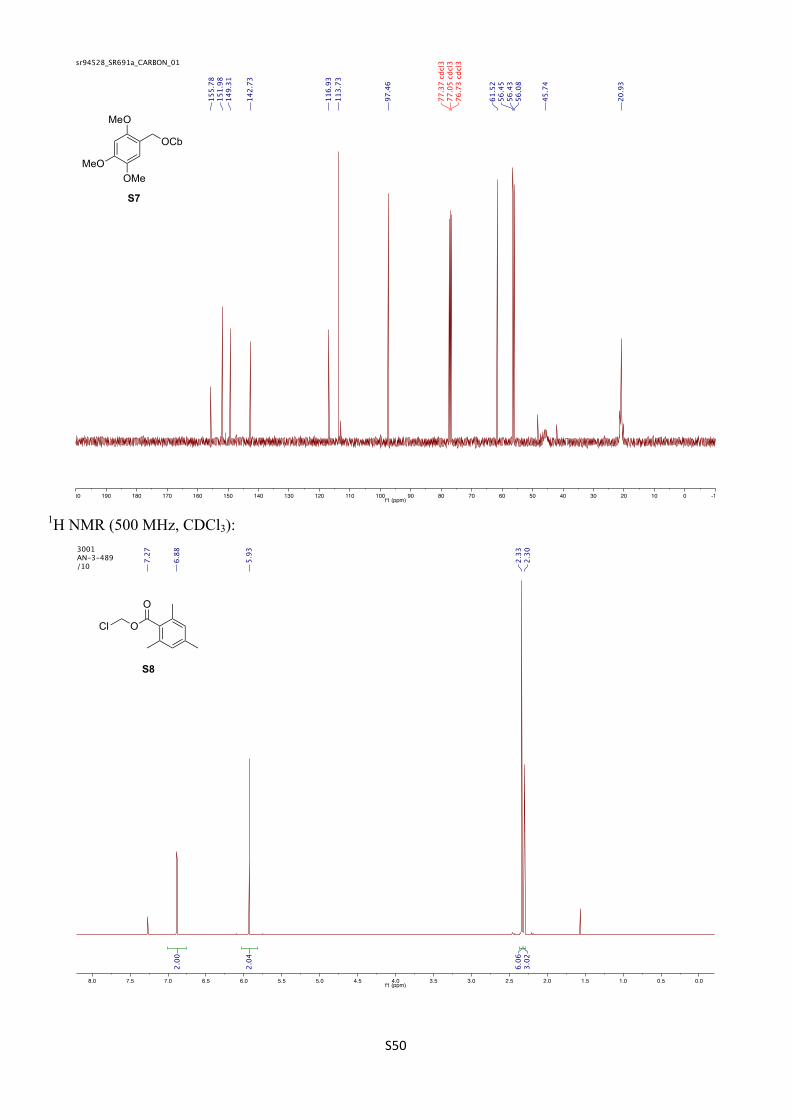
S50
1H NMR (500 MHz, CDCl3):
-100102030405060708090100110120130140150160170180190200f1 (ppm)
sr94528_SR691a_CARBON_01
20
.93
45
.74
56
.08
56
.43
56
.45
61
.52
76
.73
cdcl3
77
.05
cdcl3
77
.37
cdcl3
97
.46
11
3.7
3
11
6.9
3
14
2.7
3
14
9.3
1
15
1.9
8
15
5.7
8
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
3001
AN-3-489
/10
3.02
6.06
2.04
2.00
2.30
2.33
5.93
6.88
7.27
O
OMeMeO
Me
S7
OCb
S8
O
O
Cl

S51
13C NMR (126 MHz, CDCl3):
1H NMR (400 MHz, CDCl3):
0102030405060708090100110120130140150160170180f1 (ppm)
3001
AN-3-489
/11
19
.87
21
.19
68
.86
76
.77
CD
Cl3
77
.02
CD
Cl3
77
.28
CD
Cl3
12
8.6
5
12
8.7
5
13
5.9
0
14
0.3
2
16
7.8
7
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
an168390_AN-3-515_PROTON_01
18.48
3.03
2.00
2.02
1.25
1.25
1.26
1.27
2.84
2.86
2.87
2.89
2.91
2.93
2.94
2.96
5.92
7.03
S8
O
O
Cl
23
O
O
Cl
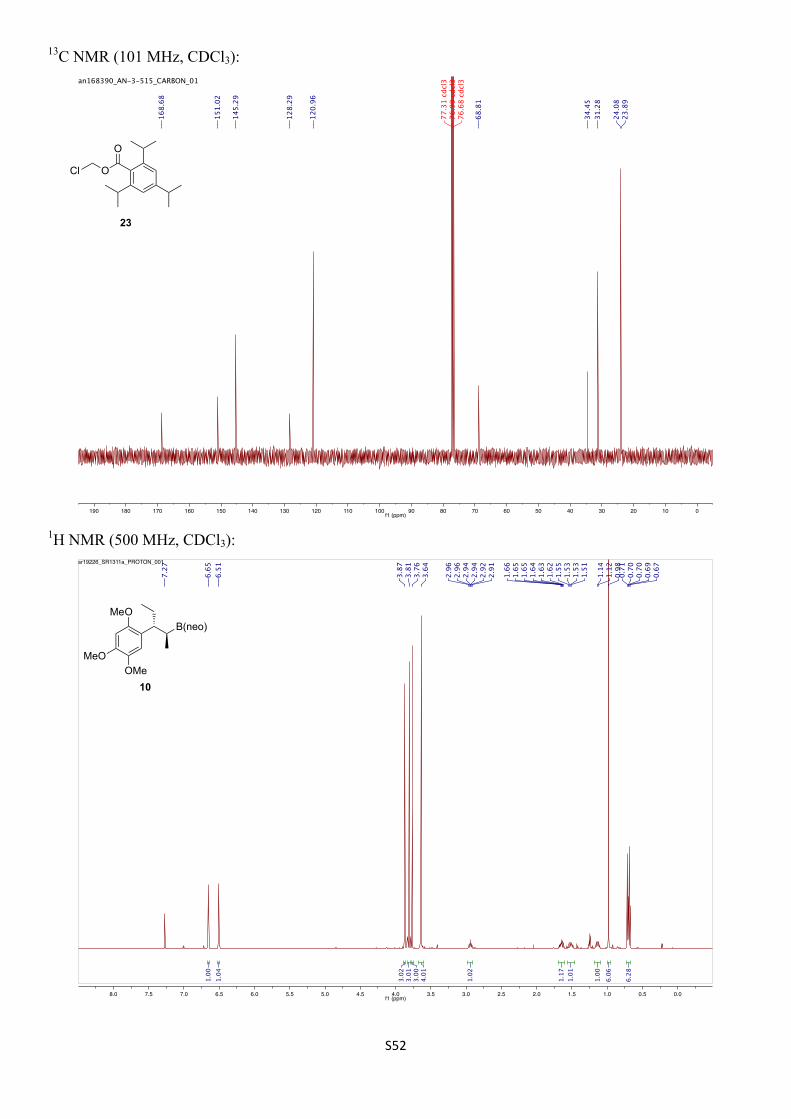
S52
13C NMR (101 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
0102030405060708090100110120130140150160170180190f1 (ppm)
an168390_AN-3-515_CARBON_01
23
.89
24
.08
31
.28
34
.45
68
.81
76
.68
cdcl3
76
.99
cdcl3
77
.31
cdcl3
12
0.9
6
12
8.2
9
14
5.2
9
15
1.0
2
16
8.6
8
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
sr19226_SR1311a_PROTON_001
6.28
6.06
1.00
1.01
1.17
1.02
4.01
3.00
3.01
3.02
1.04
1.00
0.67
0.69
0.70
0.70
0.71
0.98
1.12
1.14
1.51
1.53
1.53
1.55
1.62
1.63
1.64
1.65
1.65
1.66
2.91
2.92
2.94
2.94
2.96
2.96
3.64
3.76
3.81
3.87
6.51
6.65
7.27
O
OMeMeO
Me
10
B(neo)
23
O
O
Cl

S53
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-100102030405060708090100110120130140150160170180190200f1 (ppm)
sr19226_SR1311a_CARBON_001
12
.44
14
.68
22
.08
27
.19
29
.37
31
.81
42
.04
56
.12
56
.56
57
.18
72
.12
76
.91
cdcl3
77
.16
cdcl3
77
.41
cdcl3
98
.17
11
1.4
7
12
5.6
1
14
3.4
3
14
7.0
2
15
2.8
1
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
an1134_AN-4-607_PROTON01
3.08
3.04
1.47
1.63
1.00
3.09
3.08
3.09
1.05
1.02
1.01
0.78
0.79
0.81
1.04
1.05
1.61
1.62
1.63
1.63
1.64
1.65
1.66
1.87
1.88
1.89
1.89
2.98
2.99
2.99
3.00
3.01
3.01
3.02
3.80
3.84
3.89
3.93
3.95
3.96
3.97
3.98
6.55
6.65
7.27
O
OMeMeO
Me
10
B(neo)
O
OMeMeO
Me
S9
OH

S54
13C NMR (126 MHz, CDCl3):
1H NMR (400 MHz, CDCl3):
0102030405060708090100110120130140150160170180f1 (ppm)
an1134_AN-4-607_CARBON01
12
.30
20
.48
22
.81
47
.29
56
.06
56
.65
56
.78
71
.53
76
.74
cdcl3
77
.00
cdcl3
77
.25
cdcl3
98
.07
11
2.8
9
12
1.7
9
14
3.1
5
14
7.8
4
15
2.1
6
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
sr103338_SR1157a_PROTON_01
3.04
3.02
1.05
0.92
3.07
3.09
3.11
1.00
1.02
1.01
0.79
0.81
1.03
1.05
1.94
1.96
1.98
2.00
2.01
2.03
2.05
2.06
2.34
3.82
3.85
3.89
4.50
4.52
6.52
6.85
7.27
O
OMeMeO
Me
OH
S10
O
OMeMeO
Me
S9
OH
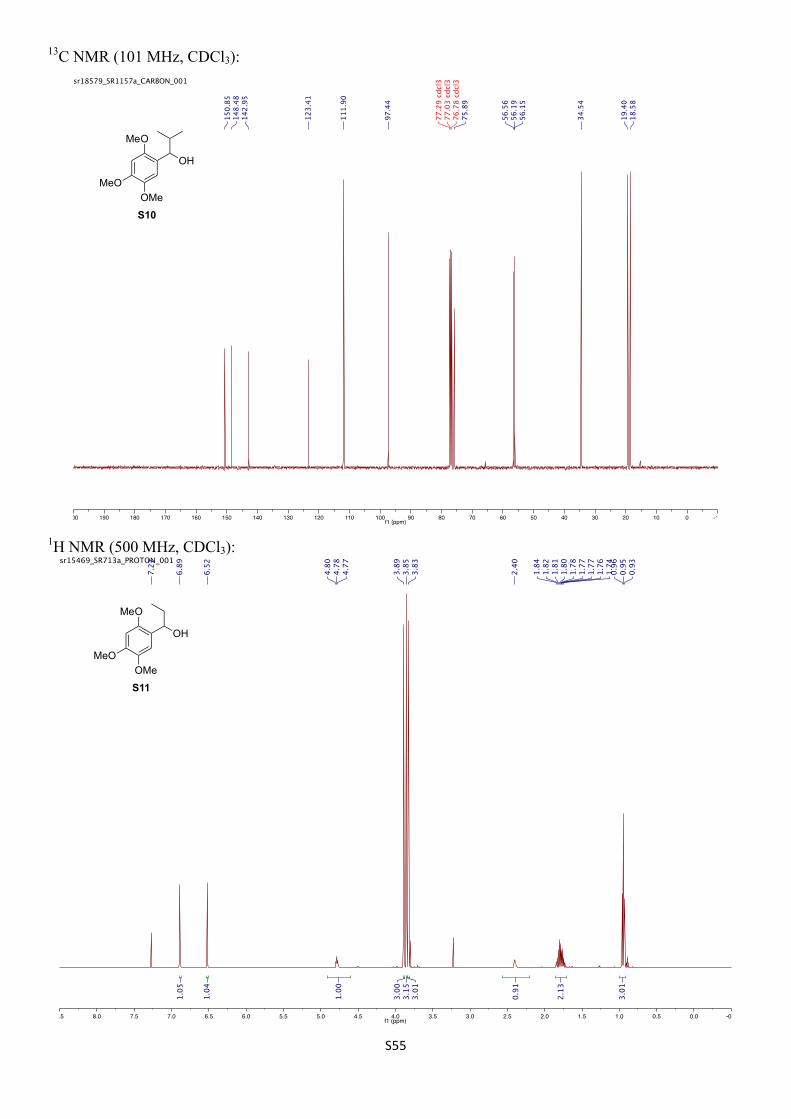
S55
13C NMR (101 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-100102030405060708090100110120130140150160170180190200f1 (ppm)
sr18579_SR1157a_CARBON_001
18.5
8
19.4
0
34.5
4
56.1
5
56.1
9
56.5
6
75.8
9
76.7
8 c
dcl3
77.0
3 c
dcl3
77.2
9 c
dcl3
97.4
4
111.9
0
123.4
1
142.9
5
148.4
8
150.8
5
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
sr15469_SR713a_PROTON_001
3.01
2.13
0.91
3.01
3.15
3.00
1.00
1.04
1.05
0.93
0.95
0.96
1.74
1.76
1.77
1.77
1.78
1.80
1.81
1.82
1.84
2.40
3.83
3.85
3.89
4.77
4.78
4.80
6.52
6.89
7.27
O
OMeMeO
Me
OH
S10
O
OMeMeO
Me
OH
S11
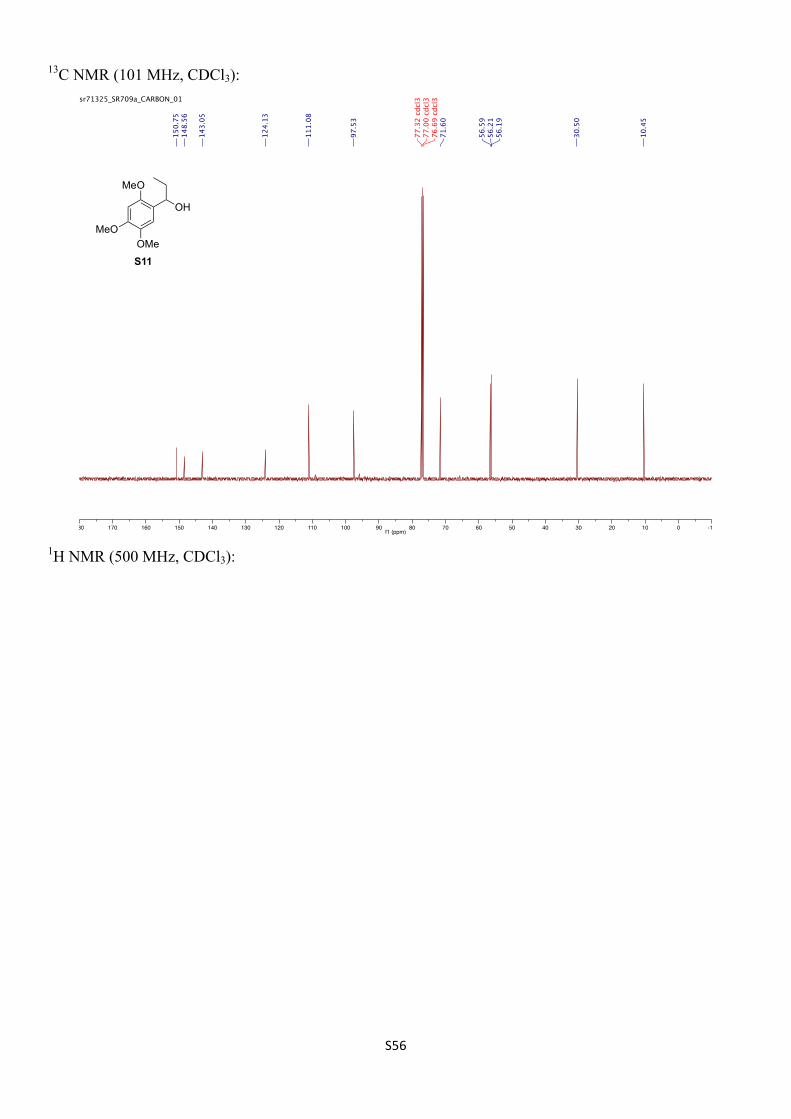
S56
13C NMR (101 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-100102030405060708090100110120130140150160170180f1 (ppm)
sr71325_SR709a_CARBON_01
10.4
5
30.5
0
56.1
9
56.2
1
56.5
9
71.6
0
76.6
9 c
dcl3
77.0
0 c
dcl3
77.3
2 c
dcl3
97.5
3
111.0
8
124.1
3
143.0
5
148.5
6
150.7
5
O
OMeMeO
Me
OH
S11

S57
13C NMR (126 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
1483_AN-3-385/10AN-3-385-single diastereomer
3.0
2
3.1
4
6.0
26.3
6
1.3
5
1.0
5
1.0
8
1.0
1
0.9
8
3.0
13.1
13.0
53.2
93.1
13.3
3
1.0
21.0
21.0
01.0
1
0.4
40.4
5
0.7
60.7
70.7
9
1.2
01.2
61.6
61.8
61.8
71.8
81.8
91.8
9
2.2
82.2
82.2
92.3
02.3
02.5
62.5
7
3.0
6
3.6
73.7
53.8
13.8
33.8
53.8
9
6.4
66.5
26.6
86.7
5
7.2
7
-100102030405060708090100110120130140150160170180f1 (ppm)
1483_AN-3-385/11
AN-3-385
12.8
6
15.5
7
24.6
5
25.1
0
27.2
4
28.9
7
39.4
7
43.3
1
56.1
1
56.1
4
56.5
6
56.7
4
56.9
1
57.0
0
76.9
1 C
DC
l3
77.1
6 C
DC
l3
77.4
1 C
DC
l3
82.9
2
97.9
1
98.3
5
113.4
2
114.6
4
122.4
8
124.0
0
142.8
7
146.9
5
147.2
8
152.1
6
152.8
4
O
OMeMeO
Me
B(pin)
OMe
OMeMeO
15b
O
OMeMeO
Me
B(pin)
OMe
OMeMeO
15b
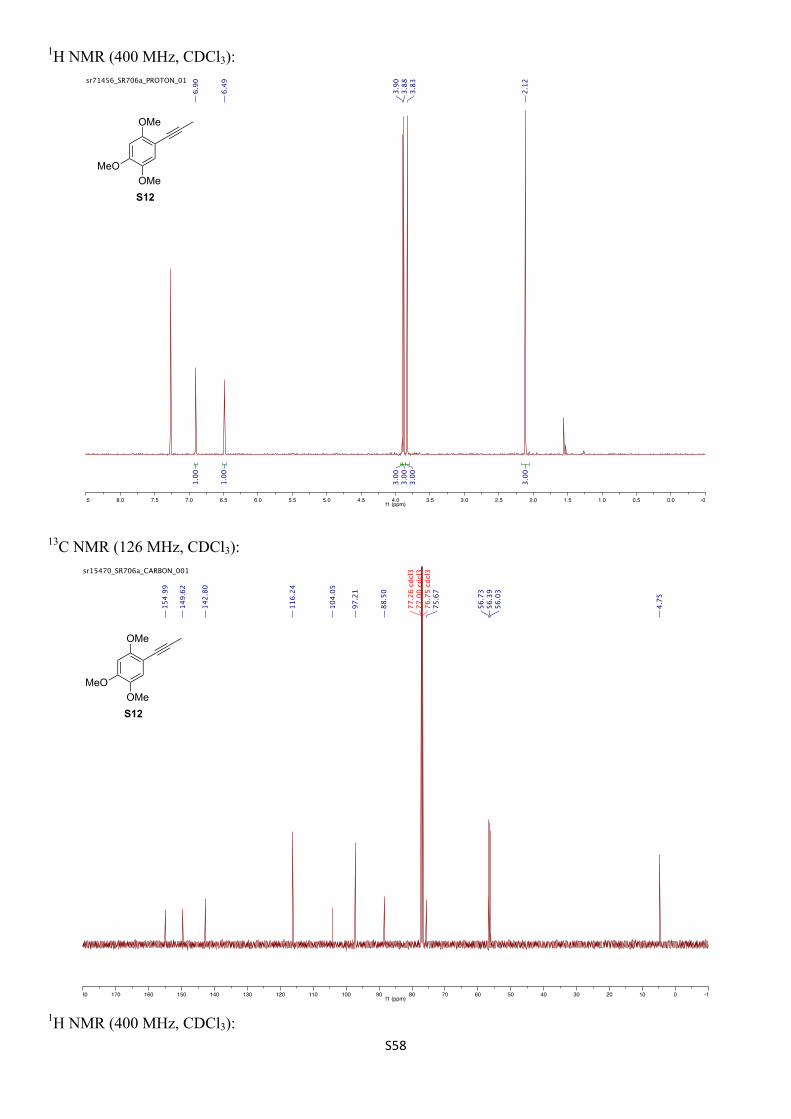
S58
1H NMR (400 MHz, CDCl3):
13C NMR (126 MHz, CDCl3):
1H NMR (400 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
sr71456_SR706a_PROTON_01
3.00
3.00
3.00
3.00
1.00
1.00
2.12
3.83
3.88
3.90
6.49
6.90
-100102030405060708090100110120130140150160170180f1 (ppm)
sr15470_SR706a_CARBON_001
4.7
5
56.0
3
56.3
9
56.7
3
75.6
7
76.7
5 c
dcl3
77.0
0 c
dcl3
77.2
6 c
dcl3
88.5
0
97.2
1
104.0
5
116.2
4
142.8
0
149.6
2
154.9
9
OMeMeO
S12
OMe
OMeMeO
S12
OMe

S59
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
sr71739_SR718a_PROTON_01
12.08
3.00
6.15
3.03
1.01
1.00
1.00
1.31
1.31
1.94
1.94
3.83
3.83
3.83
3.91
3.91
6.53
6.92
7.27
7.33
7.33
7.34
7.34
-100102030405060708090100110120130140150160170180f1 (ppm)
sr15494_SR718a_CARBON_001
16.1
6
24.8
9
56.0
2
56.3
4
56.5
7
76.7
6 c
dcl3
77.0
1 c
dcl3
77.2
6 c
dcl3
83.3
2
97.1
0
114.1
9
118.5
0
137.5
3
142.1
7
149.1
0
152.0
3
OMe
OMeMeO
S13
B(pin)
OMe
OMeMeO
S13
B(pin)
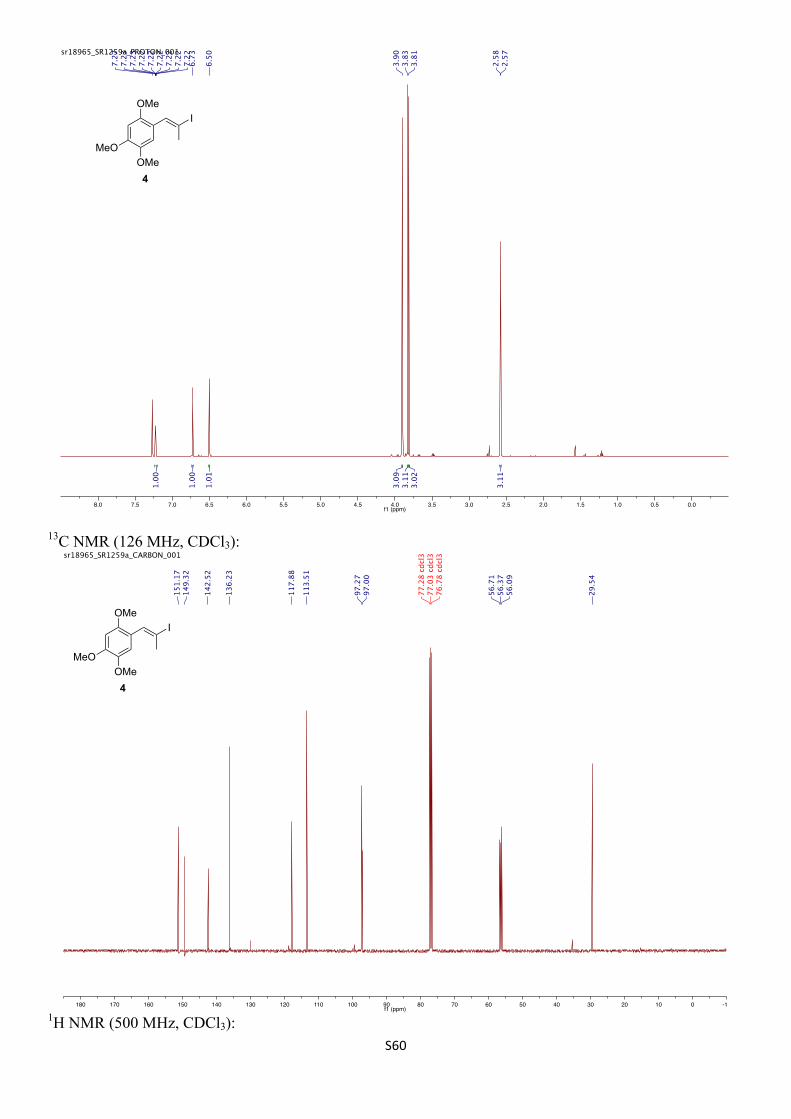
S60
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
sr18965_SR1259a_PROTON_001
3.11
3.02
3.11
3.09
1.01
1.00
1.00
2.57
2.58
3.81
3.83
3.90
6.50
6.73
7.22
7.22
7.22
7.22
7.22
7.22
7.23
7.23
7.27
-100102030405060708090100110120130140150160170180f1 (ppm)
sr18965_SR1259a_CARBON_001
29
.54
56
.09
56
.37
56
.71
76
.78
cdcl3
77
.03
cdcl3
77
.28
cdcl3
97
.00
97
.27
11
3.5
1
11
7.8
8
13
6.2
3
14
2.5
2
14
9.3
2
15
1.1
7
OMe
OMeMeO
4
I
OMe
OMeMeO
4
I

S61
13C NMR (126 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
an23686_AN-4-718_PROTON_001
3.06
3.06
3.06
3.11
1.01
0.94
1.00
2.23
3.82
3.84
3.91
6.52
6.69
6.72
7.27
-100102030405060708090100110120130140150160170180f1 (ppm)
an23686_AN-4-718_CARBON_001
22
.62
56
.10
56
.39
56
.72
76
.76
cdcl3
77
.02
cdcl3
77
.27
cdcl3
97
.39
11
3.7
2
11
6.3
0
12
3.4
9
13
1.5
6
14
2.6
1
14
9.2
6
15
1.4
6
OMe
OMeMeO
S14
Cl
OMe
OMeMeO
S14
Cl
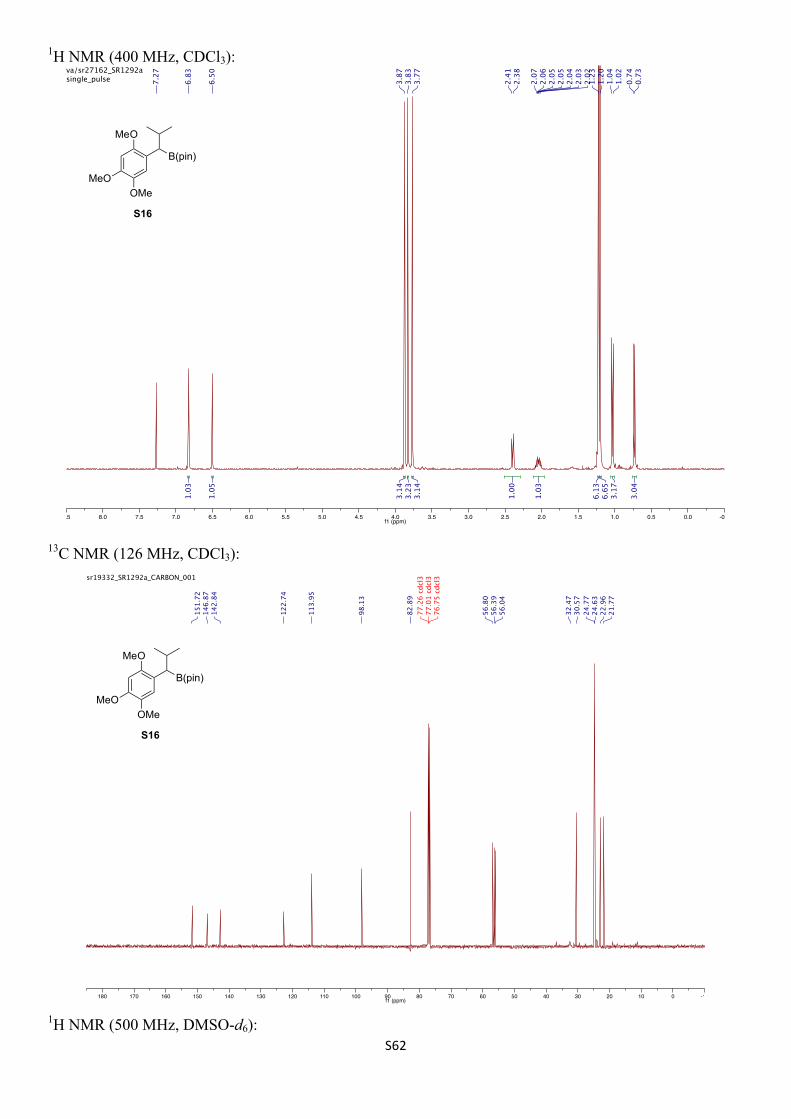
S62
1H NMR (400 MHz, CDCl3):
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, DMSO-d6):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
va/sr27162_SR1292asingle_pulse
3.04
3.17
6.65
6.13
1.03
1.00
3.14
3.23
3.14
1.05
1.03
0.73
0.74
1.02
1.04
1.20
1.23
2.02
2.03
2.04
2.05
2.05
2.06
2.07
2.38
2.41
3.77
3.83
3.87
6.50
6.83
7.27
-100102030405060708090100110120130140150160170180f1 (ppm)
sr19332_SR1292a_CARBON_001
21
.77
22
.96
24
.63
24
.77
30
.57
32
.47
56
.04
56
.39
56
.80
76
.75
cdcl3
77
.01
cdcl3
77
.26
cdcl3
82
.89
98
.13
11
3.9
5
12
2.7
4
14
2.8
4
14
6.8
7
15
1.7
2
O
OMeMeO
Me
B(pin)
S16
O
OMeMeO
Me
B(pin)
S16

S63
13C NMR (126 MHz, DMSO-d6):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
3647 AN-4-714/10
6.0
5
1.0
1
1.0
0
1.1
2
1.0
1
1.0
4
3.0
0
3.0
1
3.0
1
3.0
4
3.0
0
3.1
2
3.0
1
1.0
0
0.4
8
0.4
9
0.5
0
0.5
0
0.5
1
1.2
3
1.2
7
1.2
8
1.3
1
1.3
21.9
9
2.1
8
2.1
8
2.1
9
2.2
0
2.2
0
2.2
1
2.2
2
2.2
2
2.3
3
2.4
6
2.4
7
2.4
8
3.4
9
3.5
9
3.6
4
3.6
8
3.7
1
3.7
3
6.4
9
6.5
1
6.5
2
7.0
1
-100102030405060708090100110120130140150160170180190f1 (ppm)
3647 AN-4-714/11
12.3
8
17.2
0
25.8
5
28.5
7
38.2
7
39.1
9 D
MSO
39.3
5 D
MSO
39.5
2 D
MSO
39.6
9 D
MSO
39.8
5 D
MSO
55.5
6
55.8
2
56.3
7
56.3
7
56.6
2
56.7
1
98.6
0
99.1
4
112.6
6
118.5
5
127.0
9
128.1
5
141.4
7
142.8
7
145.2
9
146.4
3
152.3
0
152.8
4
O
OMeMeO
Me
BF3K
OMe
OMeMeO
S22
O
OMeMeO
Me
BF3K
OMe
OMeMeO
S22
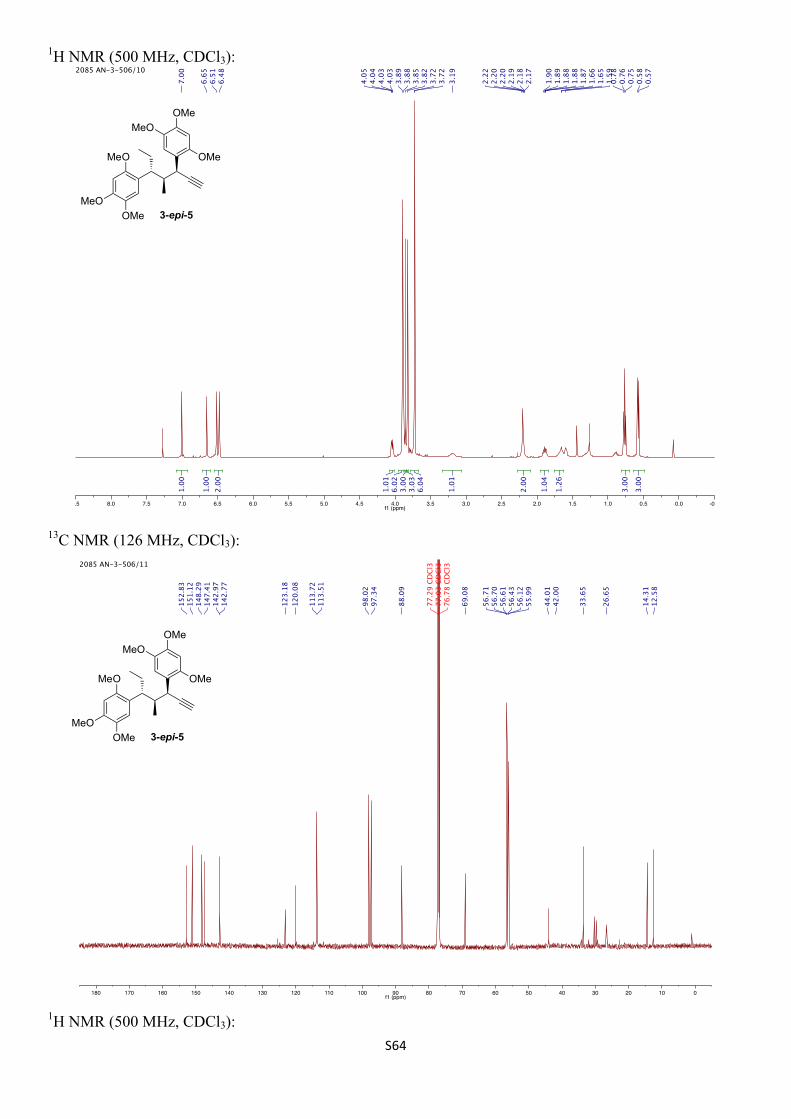
S64
1H NMR (500 MHz, CDCl3):
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
2085 AN-3-506/10
3.0
0
3.0
0
1.2
6
1.0
4
2.0
0
1.0
1
6.0
4
3.0
3
3.0
0
6.0
2
1.0
1
2.0
0
1.0
0
1.0
0
0.5
7
0.5
8
0.7
5
0.7
6
0.7
81.5
9
1.6
5
1.6
6
1.8
7
1.8
8
1.8
8
1.8
9
1.9
0
2.1
7
2.1
8
2.1
9
2.2
0
2.2
0
2.2
2
3.1
9
3.7
2
3.7
2
3.8
2
3.8
5
3.8
8
3.8
9
4.0
3
4.0
3
4.0
4
4.0
5
6.4
8
6.5
1
6.6
5
7.0
0
0102030405060708090100110120130140150160170180f1 (ppm)
2085 AN-3-506/11
12.5
8
14.3
1
26.6
5
33.6
5
42.0
0
44.0
1
55.9
9
56.1
2
56.4
3
56.6
1
56.7
0
56.7
1
69.0
8
76.7
8 C
DC
l3
77.0
3 C
DC
l3
77.2
9 C
DC
l3
88.0
9
97.3
4
98.0
2
113.5
1
113.7
2
120.0
8
123.1
8
142.7
7
142.9
7
147.4
1
148.2
9
151.1
2
152.8
3
O
OMeMeO
Me OMe
OMeMeO
3-epi-5
O
OMeMeO
Me OMe
OMeMeO
3-epi-5
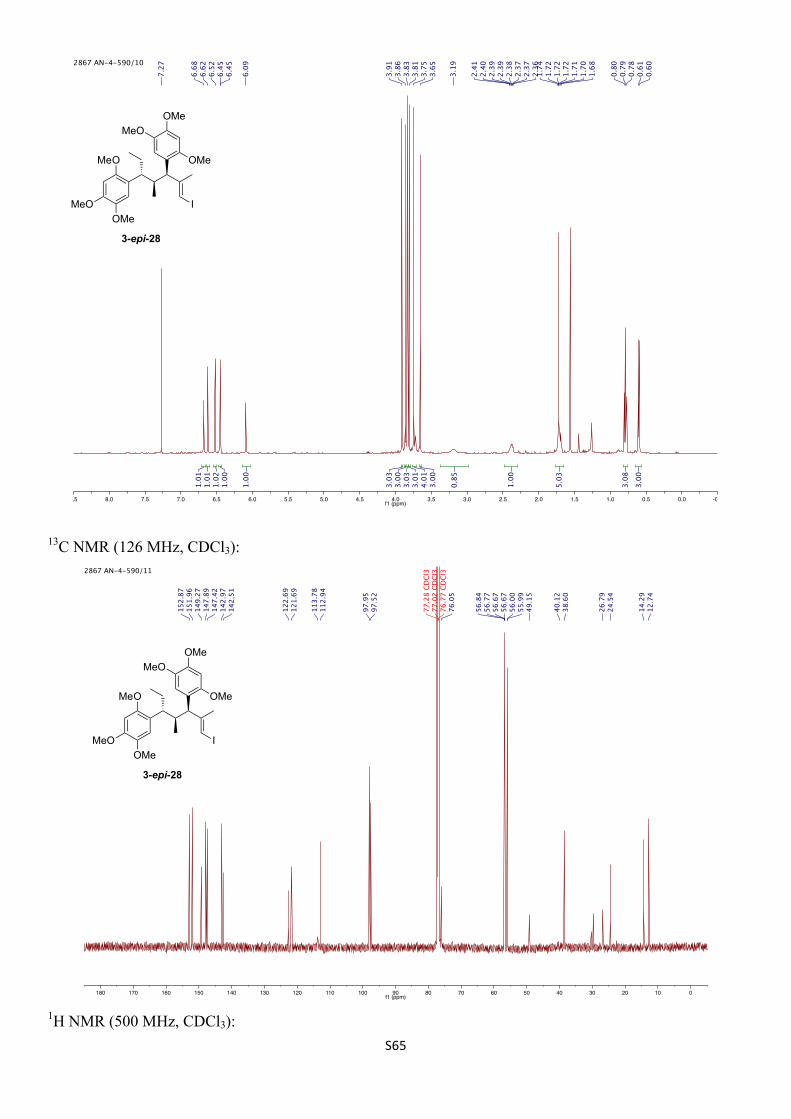
S65
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
2867 AN-4-590/10
3.0
0
3.0
8
5.0
3
1.0
0
0.8
5
3.0
0
4.0
1
3.0
1
3.0
3
3.0
0
3.0
3
1.0
0
1.0
0
1.0
2
1.0
1
1.0
1
0.6
0
0.6
1
0.7
8
0.7
9
0.8
0
1.6
8
1.7
0
1.7
1
1.7
2
1.7
2
1.7
2
1.7
42.3
6
2.3
7
2.3
7
2.3
8
2.3
9
2.3
9
2.4
0
2.4
1
3.1
9
3.6
5
3.7
5
3.8
1
3.8
3
3.8
6
3.9
1
6.0
9
6.4
5
6.4
5
6.5
2
6.6
2
6.6
8
7.2
7
0102030405060708090100110120130140150160170180f1 (ppm)
2867 AN-4-590/11
12
.74
14
.29
24
.54
26
.79
38
.60
40
.12
49
.15
55
.99
56
.00
56
.67
56
.67
56
.77
56
.84
76
.05
76
.77
CD
Cl3
77
.02
CD
Cl3
77
.28
CD
Cl3
97
.52
97
.95
11
2.9
4
11
3.7
8
12
1.6
9
12
2.6
9
14
2.5
1
14
2.9
7
14
7.4
2
14
7.8
9
14
9.2
7
15
1.9
6
15
2.8
7
O
OMeMeO
Me OMe
OMeMeO
3-epi-28
I
O
OMeMeO
Me OMe
OMeMeO
3-epi-28
I

S66
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CD3OD):
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
2964 AN-4-597/10
3.0
6
3.1
9
3.2
9
2.0
0
1.0
2
0.9
5
4.2
8
3.0
5
12.2
4
3.1
9
6.1
5
1.0
7
2.0
4
1.0
3
1.0
2
1.0
2
1.0
2
0.5
8
0.5
9
0.8
0
0.8
2
0.8
3
1.6
4
1.7
6
1.8
1
1.8
5
2.4
7
3.4
8
3.6
4
3.6
6
3.7
2
3.8
0
3.8
1
3.8
2
3.8
3
3.8
7
3.9
1
6.4
6
6.5
4
6.5
5
6.5
9
6.7
5
6.7
8
6.8
7
7.2
7
0102030405060708090100110120130140150160170180f1 (ppm)
2964 AN-4-597/11
12
.85
14
.33
18
.91
26
.81
39
.12
41
.23
48
.20
55
.96
56
.00
56
.20
56
.41
56
.68
56
.70
56
.76
56
.76
56
.98
97
.92
97
.97
98
.02
11
2.7
8
11
4.1
1
11
4.6
7
11
9.0
4
12
0.2
7
12
3.4
6
12
3.9
4
14
0.2
2
14
2.4
9
14
2.5
9
14
3.0
8
14
7.2
8
14
7.3
0
14
7.9
9
15
1.7
4
15
2.2
0
15
3.0
8
O
OMeMeO
Me OMe
OMeMeO
3-epi-1OMeMeO
OMe
O
OMeMeO
Me OMe
OMeMeO
3-epi-1OMeMeO
OMe
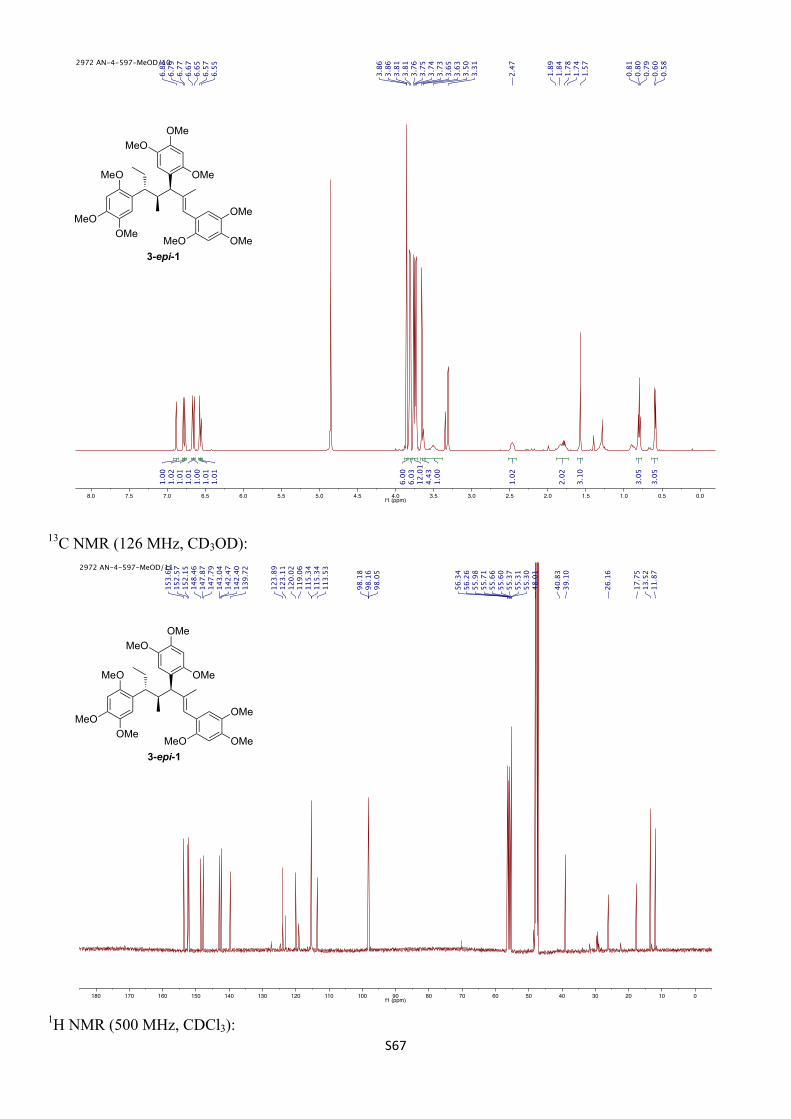
S67
13C NMR (126 MHz, CD3OD):
1H NMR (500 MHz, CDCl3):
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
2972 AN-4-597-MeOD/10
3.0
5
3.0
5
3.1
0
2.0
2
1.0
2
1.0
0
4.4
3
12
.01
6.0
3
6.0
0
1.0
1
1.0
1
1.0
0
1.0
1
1.0
1
1.0
2
1.0
0
0.5
8
0.6
0
0.7
9
0.8
0
0.8
1
1.5
7
1.7
4
1.7
8
1.8
4
1.8
9
2.4
7
3.3
1
3.5
0
3.6
3
3.6
5
3.7
3
3.7
4
3.7
5
3.7
6
3.8
1
3.8
1
3.8
6
3.8
6
6.5
5
6.5
7
6.6
5
6.6
7
6.7
7
6.7
9
6.8
8
0102030405060708090100110120130140150160170180f1 (ppm)
2972 AN-4-597-MeOD/11
11.8
7
13.5
2
17.7
5
26.1
6
39.1
0
40.8
3
48.0
1
55.3
0
55.3
1
55.3
7
55.6
0
55.6
6
55.7
1
55.9
8
56.2
6
56.3
4
98.0
5
98.1
6
98.1
8
113.5
3
115.3
4
115.3
4
119.0
6
120.0
2
123.1
1
123.8
9
139.7
2
142.4
0
142.4
7
143.0
4
147.7
9
147.8
7
148.4
6
152.1
5
152.5
7
153.6
0
O
OMeMeO
Me OMe
OMeMeO
3-epi-1OMeMeO
OMe
O
OMeMeO
Me OMe
OMeMeO
3-epi-1OMeMeO
OMe

S68
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
3661 AN-4-716/10
3.1
6
12.0
2
3.0
0
1.8
6
3.0
4
3.0
0
3.0
5
1.0
1
1.0
0
0.8
7
0.8
8
0.9
0
1.0
2
1.8
5
2.4
2
3.7
7
3.8
2
3.8
9
6.5
0
6.5
8
0102030405060708090100110120130140150160170180f1 (ppm)
3661 AN-4-716/11
12
.16
15
.78
24
.54
26
.07
56
.28
56
.39
56
.89
76
.77
CD
Cl3
77
.02
CD
Cl3
77
.27
CD
Cl3
82
.66
98
.23
11
5.4
3
12
6.0
3
14
2.2
4
14
8.1
0
15
0.9
9
15
1.4
2
O
OMeMeO
Me
B(pin)
S25
O
OMeMeO
Me
B(pin)
S25
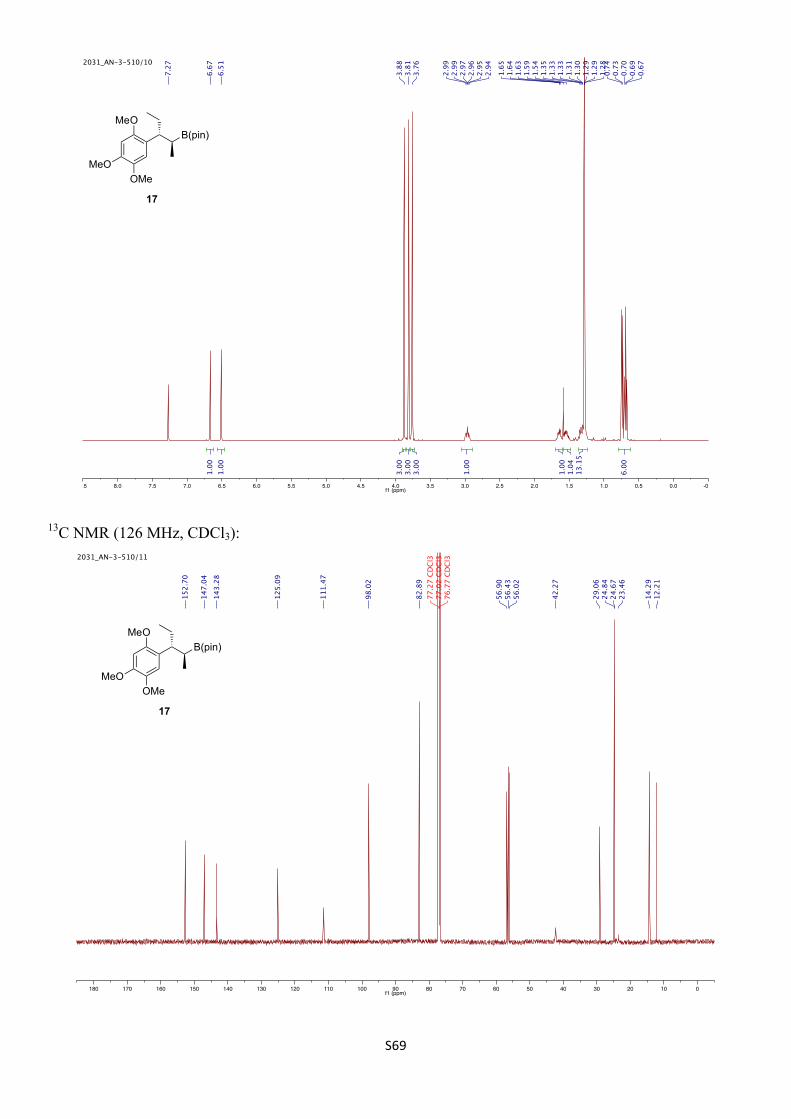
S69
13C NMR (126 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
2031_AN-3-510/10
6.00
13.15
1.04
1.00
1.00
3.00
3.00
3.00
1.00
1.00
0.67
0.69
0.70
0.73
0.74
1.28
1.29
1.29
1.30
1.31
1.33
1.33
1.35
1.54
1.59
1.63
1.64
1.65
2.94
2.95
2.96
2.97
2.99
2.99
3.76
3.81
3.88
6.51
6.67
7.27
0102030405060708090100110120130140150160170180f1 (ppm)
2031_AN-3-510/11
12.2
1
14.2
9
23.4
6
24.6
7
24.8
4
29.0
6
42.2
7
56.0
2
56.4
3
56.9
0
76.7
7 C
DC
l3
77.0
2 C
DC
l3
77.2
7 C
DC
l3
82.8
9
98.0
2
111.4
7
125.0
9
143.2
8
147.0
4
152.7
0
O
OMeMeO
MeB(pin)
17
O
OMeMeO
MeB(pin)
17
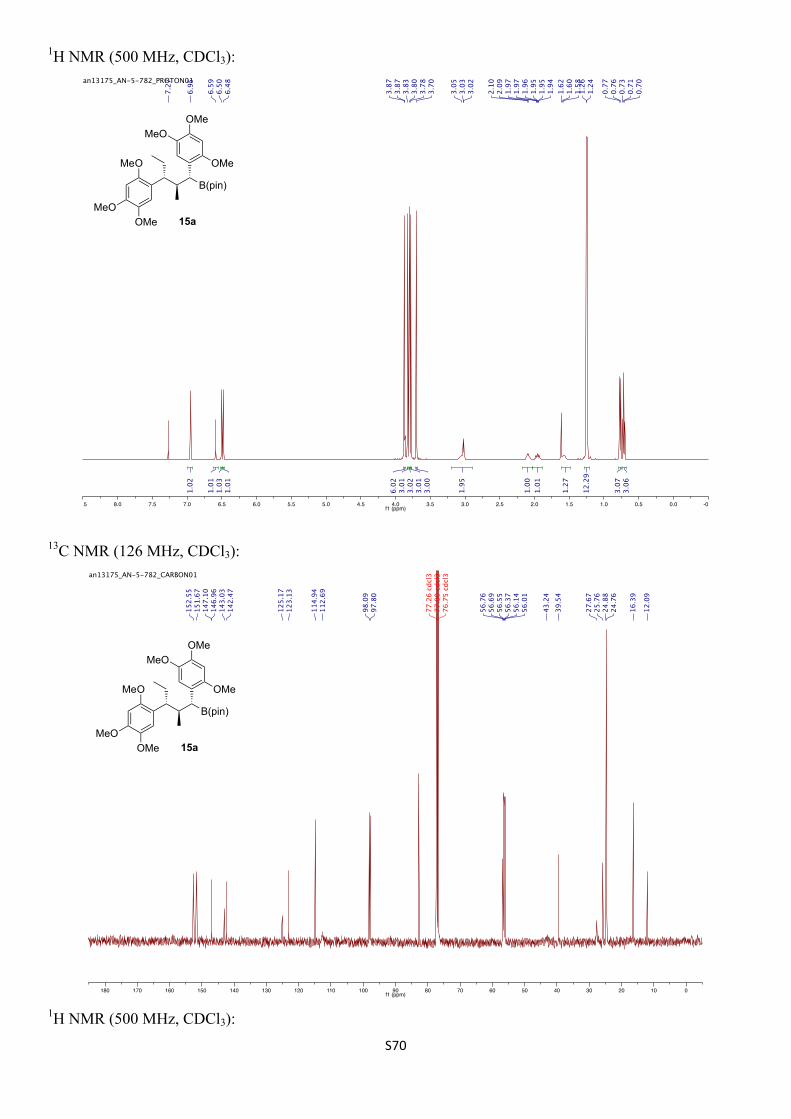
S70
1H NMR (500 MHz, CDCl3):
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
an13175_AN-5-782_PROTON01
3.06
3.07
12.29
1.27
1.01
1.00
1.95
3.00
3.01
3.02
3.01
6.02
1.01
1.03
1.01
1.02
0.70
0.71
0.73
0.76
0.77
1.24
1.26
1.58
1.60
1.62
1.94
1.95
1.95
1.96
1.97
1.97
2.09
2.10
3.02
3.03
3.05
3.70
3.78
3.80
3.83
3.87
3.87
6.48
6.50
6.59
6.95
7.27
0102030405060708090100110120130140150160170180f1 (ppm)
an13175_AN-5-782_CARBON01
12.0
9
16.3
9
24.7
6
24.8
8
25.7
6
27.6
7
39.5
4
43.2
4
56.0
1
56.1
4
56.3
7
56.5
5
56.6
9
56.7
6
76.7
5 c
dcl3
77.0
0 c
dcl3
77.2
6 c
dcl3
97.8
0
98.0
9
112.6
9
114.9
4
123.1
3
125.1
7
142.4
7
143.0
3
146.9
6
147.1
0
151.6
7
152.5
5
O
OMeMeO
Me
B(pin)
15a
OMe
MeOOMe
O
OMeMeO
Me
B(pin)
15a
OMe
MeOOMe

S71
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
3359 AN-4-667/10
3.1
3
3.1
2
1.0
2
2.0
1
1.0
2
1.0
0
3.0
4
3.0
7
3.0
2
3.1
0
6.1
3
1.0
0
2.0
0
1.0
1
1.0
0
0.5
8
0.5
9
0.7
5
0.7
6
0.7
8
1.6
82.0
5
2.0
5
2.0
6
2.0
7
2.0
7
2.0
8
2.0
9
2.1
0
2.2
7
2.2
8
2.2
8
3.2
0
3.7
9
3.8
2
3.8
4
3.8
6
3.8
8
3.8
9
4.5
5
4.5
5
4.5
6
6.5
1
6.5
2
6.6
3
7.1
7
0102030405060708090100110120130140150160170180f1 (ppm)
3359 AN-4-667/11
11
.82
13
.40
24
.91
33
.88
40
.63
41
.38
56
.01
56
.22
56
.23
56
.71
56
.83
56
.97
72
.20
76
.78
CD
Cl3
77
.03
CD
Cl3
77
.29
CD
Cl3
83
.59
97
.36
98
.22
11
1.8
8
11
4.3
0
12
0.8
2
12
4.7
7
14
2.5
3
14
3.0
9
14
7.3
8
14
8.2
3
15
0.1
8
15
2.5
7
O
OMeMeO
Me OMe
OMeMeO
5
O
OMeMeO
Me OMe
OMeMeO
5
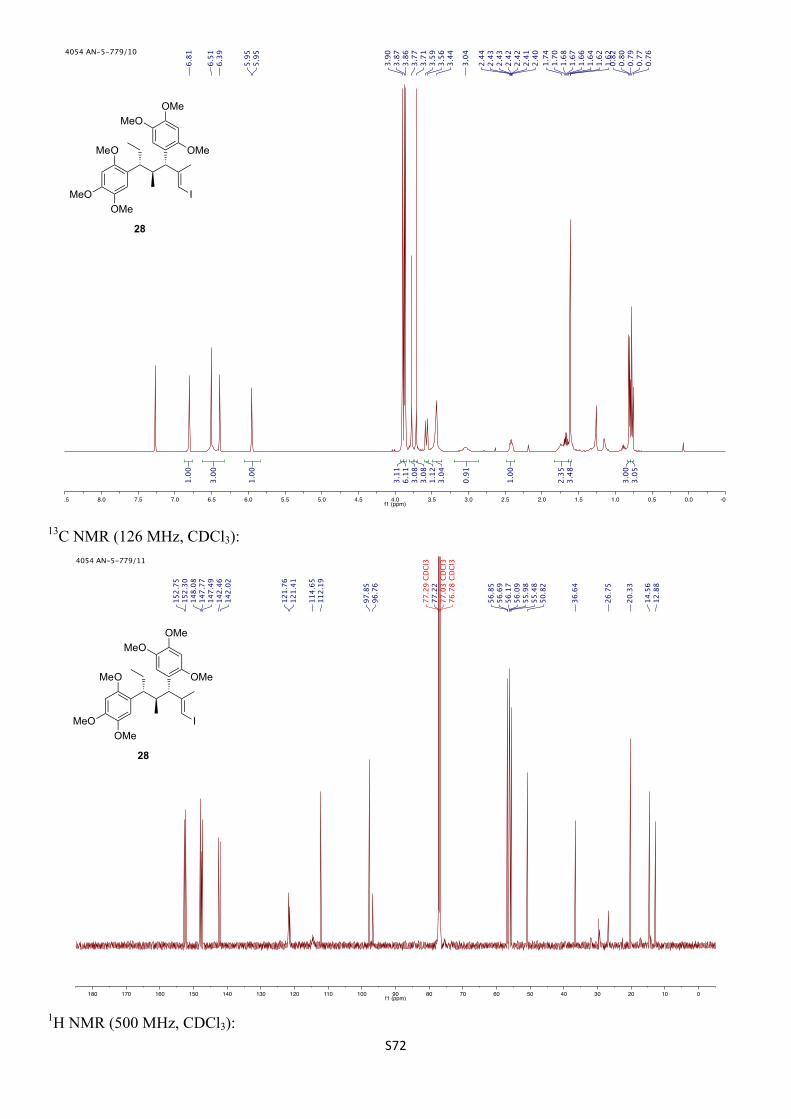
S72
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CDCl3):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
4054 AN-5-779/10
3.0
5
3.0
0
3.4
8
2.3
5
1.0
0
0.9
1
3.0
4
1.1
2
3.0
8
3.0
8
6.1
1
3.1
1
1.0
0
3.0
0
1.0
0
0.7
6
0.7
7
0.7
9
0.8
0
0.8
21.6
2
1.6
2
1.6
4
1.6
6
1.6
7
1.6
8
1.7
0
1.7
4
2.4
0
2.4
1
2.4
2
2.4
2
2.4
3
2.4
3
2.4
4
3.0
4
3.4
4
3.5
6
3.5
9
3.7
1
3.7
7
3.8
6
3.8
7
3.9
0
5.9
5
5.9
5
6.3
9
6.5
1
6.8
1
0102030405060708090100110120130140150160170180f1 (ppm)
4054 AN-5-779/11
12
.88
14
.56
20
.33
26
.75
36
.64
50
.82
55
.48
55
.98
56
.09
56
.17
56
.69
56
.85
76
.78
CD
Cl3
77
.03
CD
Cl3
77
.22
77
.29
CD
Cl3
96
.76
97
.85
11
2.1
9
11
4.6
5
12
1.4
1
12
1.7
6
14
2.0
2
14
2.4
6
14
7.4
9
14
7.7
7
14
8.0
8
15
2.3
0
15
2.7
5
O
OMeMeO
Me OMe
OMeMeO
28
I
O
OMeMeO
Me OMe
OMeMeO
28
I

S73
13C NMR (126 MHz, CDCl3):
1H NMR (500 MHz, CD3OD):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
3538 AN-4-683/10
3.0
0
3.1
7
3.0
1
2.0
7
1.0
1
0.9
6
4.0
0
3.0
0
3.0
1
6.0
0
12
.03
1.0
0
1.0
0
1.0
0
2.0
0
1.0
0
1.0
0
0.7
7
0.7
9
0.8
0
0.9
3
0.9
41
.60
1.6
0
1.7
1
1.7
3
1.7
3
1.7
3
1.7
4
1.7
6
2.4
8
2.4
9
2.5
0
2.5
0
2.5
1
2.5
2
2.5
3
3.0
8
3.5
0
3.5
2
3.5
4
3.5
5
3.7
2
3.7
5
3.7
8
3.8
0
3.8
6
3.8
7
3.8
9
3.9
1
6.3
4
6.4
3
6.4
8
6.5
4
6.6
4
6.8
9
7.2
7
0102030405060708090100110120130140150160170180f1 (ppm)
3538 AN-4-683/11
12.9
4
14.4
7
14.8
4
26.8
0
36.9
8
50.9
9
55.6
2
56.0
0
56.1
1
56.1
6
56.4
1
56.6
4
56.7
1
56.7
3
57.0
1
76.7
7 C
DC
l3
77.0
2 C
DC
l3
77.2
8 C
DC
l3
96.9
2
98.1
5
98.5
1
112.7
2
114.4
9
114.7
2
120.0
9
121.8
4
122.2
7
123.7
7
138.5
0
142.0
4
142.3
9
142.5
1
147.2
9
147.3
0
147.9
5
151.7
4
152.5
0
152.8
3
O
OMeMeO
Me OMe
OMeMeO
1OMeMeO
OMe
O
OMeMeO
Me OMe
OMeMeO
1OMeMeO
OMe

S74
13C NMR (126 MHz, CD3OD):
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
3531 AN-4-683/10
3.0
5
3.0
5
3.0
5
2.0
4
1.0
1
1.0
0
4.0
0
9.0
0
6.0
1
3.0
1
3.0
3
3.0
1
1.0
0
5.0
0
1.0
0
0.7
4
0.7
6
0.7
7
0.9
5
0.9
61.5
5
1.7
1
1.7
2
1.7
3
1.7
4
1.7
5
1.7
6
1.7
8
2.5
3
2.5
4
2.5
4
2.5
5
2.5
6
2.5
6
2.5
7
3.1
0
3.4
9
3.5
2
3.7
0
3.7
1
3.7
6
3.7
7
3.8
1
3.8
4
3.8
6
6.2
6
6.2
6
6.5
2
6.5
3
6.5
9
6.6
3
6.6
5
6.8
6
0102030405060708090100110120130140150160170180f1 (ppm)
3531 AN-4-683/11
11.9
0
13.5
1
14.4
4
25.7
7
36.9
0
40.5
047.6
0 M
eO
D
50.6
4
54.6
6
55.2
8
55.3
5
55.4
1
55.4
7
55.8
5
56.1
6
56.2
0
56.3
4
97.2
3
98.4
6
98.5
3
113.7
6
115.1
4
115.3
2
119.9
2
121.6
3
122.3
1
123.6
3
138.2
3
141.9
8
142.3
1
142.4
1
147.7
5
147.7
9
148.4
0
152.1
0
152.7
9
153.2
1
O
OMeMeO
Me OMe
OMeMeO
1OMeMeO
OMe
O
OMeMeO
Me OMe
OMeMeO
1OMeMeO
OMe










![[Finish Hash Functions; Start Asymmetric Cryptography] · 2020-04-24 · [Finish Hash Functions; Start Asymmetric Cryptography] Spring 2020 Franziska (Franzi) Roesner franzi@cs.washington.edu](https://static.fdocuments.in/doc/165x107/5ed95248f59b0f56f45f4563/finish-hash-functions-start-asymmetric-cryptography-2020-04-24-finish-hash.jpg)







