
1 H MAS NMR spectra including TRAPDOR 29 Si MAS NMR 27 Al 3QMAS NMR 27 Al MAS NMR

NMR Studies of Inclusion Compounds
Sahar Nikkhou Aski
Stockholm University Stockholm University

ii
© Sahar Nikkhou Aski, Stockholm 2008
ISBN 978-91-7155-715-5
Printed in Sweden by US-AB, Stockholm 2008
Distributor: Department of Physical, Inorganic and Structural Chemistry
Stockholm University

iii
NMR Studies of Inclusion Compounds
Sahar Nikkhou Aski Abstract This thesis presents the application of some of the NMR methods in studying host-guest complexes, mainly in solution. The general focus of the work is on investigating the reorientational dynamics of some small molecules that are bound inside cavities of larger moieties. In the current work, these moieties belong to two groups: cryptophanes and cyclodextrins. Depending on the structure of the cavities, properties of the guest molecules and the formed complexes vary. Chloroform and dichloromethane are in slow exchange between the cage-like cavity of the cryptophanes and the solvent, on the chemical shift time scale, whereas adamantanecarboxylic acid, quinuclidine and 1,7-heptanediol in complex with cyclodextrins are examples of fast exchange. Kinetics and thermodynamics of complexation are studied by measuring exchange rates and translational self-diffusion coefficients by means of 1-dimenssional exchange spectroscopy and pulsed-field gradient (PFG) NMR methods, respectively. The association constants, calculated using the above information, give estimates of the thermodynamic stability of the complexes. Carbon-13 spin relaxation data were obtained using conventional relaxation experiments, such as inversion recovery and dynamic NOE, and in some cases HSQC-type (Hetereonuclear Single Quantum Correlation Spectroscopy) experiments. Motional parameters for the free and bound guest, and the host molecules were extracted using different motional models, such as Lipari-Szabo, axially symmetric rigid body, and Clore models. Comparing the overall correlation times and the order parameters of the free and bound guest with the overall correlation time of the host molecule, one can estimate the degree of the motional restriction, brought by the complexation, and the coupling between the motion of the bound guest and the reorientation of the host molecule. In one case, the guest motions were also investigated inside the cavities of a solid host material.

iv

v
Contents
List of papers......................................................................................................vii
Preface ................................................................................................................ 1
1- Supramolecular structures............................................................................. 2 1.1 Classification ......................................................................................................2
1.1.1 Complex or clathrate.......................................................................................3 1.1.2 Interactions .....................................................................................................4 1.1.3 Host and guest types ......................................................................................6
1.2 Selectivity..............................................................................................................21 1.3 Application ............................................................................................................22
2- NMR spectroscopy....................................................................................... 24 2.1 - Spin Hamiltonians ...............................................................................................24 2.2 Relaxation - A very brief introduction.....................................................................27
2.2.1 Relaxation mechanisms................................................................................27 2.2.2 Spectral density functions .............................................................................29 2.2.3 Relaxation parameters..................................................................................31 2.2.4 Experimental methods ..................................................................................34
2.3 Chemical exchange..........................................................................................39 2.4 Translational diffusion ...........................................................................................41
3- Dynamics of cyclodextrins and cryptophanes studied by NMR................ 44 3.1 Stoichiometry and binding constant.......................................................................44 3.2 Nuclear magnetic relaxation..................................................................................46
4- Discussion of the papers ............................................................................. 48 4.1 Papers I-II .............................................................................................................49 4.2 Papers III-V ...........................................................................................................50
Acknowledgment .............................................................................................. 53
References ........................................................................................................ 54

vi

vii
List of papers
This thesis is based on the following publications and manuscripts. I. Exchange kinetics and 13C NMR relaxation studies of inclusion complexes of dichloromethane and some cryptophanes S. Nikkhou Aski, A.Y.H. Lo, T. Brotin, J.-P. Dutasta, M. Edén and J. Kowalewski, Journal of Physical Chemistry C, in press Reproduced with kind permission from American Chemical Society ©2008 II. Inclusion complexes of cryptophane–E with dichloromethane and chloroform: A thermodynamic and kinetic study using the 1D- EXSY NMR method S. Nikkhou Aski, Z. Takacs and J. Kowalewski Magnetic Resonance in Chemistry, in press Reproduced with kind permission from John Wiley & Sons Limited ©2008 III. Reorientational dynamics of adamantanecarboxylic acid in complex with β-cyclodextrin Z. Tosner, S. Nikkhou Aski and J. Kowalewski Journal of Inclusion Phenomena and Macrocyclic Chemistry 55 59-70 (2006) Reproduced With kind permission from Science+Bussiness Media IV. Quinuclidine compelx with α-cyclodextrin: a diffusion and 13C NMR relaxation study. S. Nikkhou Aski and J. Kowalewski Magnetic Resonance in Chemistry 46 261-267, (2008) Reproduced with kind permission from John Wiley & Sons Limited ©2008 V. Interaction between α-cyclodextrin and 1,7-heptanediol. An NMR study of diffusion and carbon-13 relaxation S. Nikkhou Aski, Z. Takacs and J. Kowalewski Manuscript

viii
The following articles are not included in the thesis. - The effect of pendant-arm modification and ring size on the dynamics of cyclic polyamines J. Wyrwal, G. Schroeder, J. Kowalewski and S. Nikkhou Aski Journal of Molecular Structure 274-279, (2006) - Cross-correlated and conventional dipolar carbon-13 relaxation in methylene groups in small, symmetric molecules L. Ghalebani, P. Bernatowicz, S. Nikkhou Aski and J. Kowalewski Concepts in Magnetic Resonance Part A 30A 100-115, (2007) - Extensive NMRD studies of Ni(II) salt solutions in water and water- grycerol mixtures J. Kowalewski, A. Egorov, D. Kruk, A. Laaksonen, S Nikkhou
Aski, G. Parigi and P.-O. Westlund Submitted

ix

1
Preface
The research work described in this dissertation is the result of my PhD study at the division of physical chemistry during the period 2004 - 2008. The thesis is mainly centered on the issue how the motional properties of small molecules change in complexation with somewhat larger moiety named host molecules in liquids. Since the discovery of the naturally occurring supramolecules, there has been an intense interest in studying them. During the past decades, researchers have been doing great amounts of work and investigation in synthetic chemistry to approach the artistic way that nature has designed the functional aggregations of molecules1.
The central approach used in this work is NMR nuclear spin relaxation, in particular, relaxation of the 13C spin. As complementary tools, it is also taken advantage of some other techniques such as diffusion measurements and kinetic studies using NMR to partly cover the kinetics and thermodynamics of the chemical exchange going on in the systems under study. I would like, however, to emphasize that the most looked forward to aspect of the work was to investigate the effect of complexation on the motion rather than the thermodynamics of inclusion. The main body of the summary is split into two parts: an account of NMR spectroscopy theory and methods, and the systems undergone investigation. The thesis starts with an explanation of inclusion phenomena and the systems chosen to be studied. To maintain briefness, this part is limited mainly to two classes of host molecules. In the following chapter I present the outlines of nuclear magnetic resonance spectroscopy and nuclear spin relaxation. In the same chapter, the experimental approaches employed to obtain the relaxation parameters of the components of the systems is reviewed. In the next part, kinetics and thermodynamics of the complexation and their significance in the applicability of the relaxation study of the system is discussed. There is at last a final conclusion and discussion chapter. This chapter covers the concluding remarks of the papers on which this thesis is set up.

2
1- Supramolecular structures
Molecular recognition2 is the study of a very interesting group of compounds in which the components are held together by non-covalent bonds3. Although non-covalent bonds are of much weaker strength than covalent analogues, the constructed assemblies can be quite complicated. Recognition may occur in all phases of gas, liquid, solid and even interface. Molecular assemblies are formed in well-defined conditions and their stability is under thermodynamic control. A very general classification divides supramolecules into two main groups4. One class comprises of self-assemblies of molecular units of the same size providing compounds that can accept smaller molecules in the formed spaces within them. The other class includes a group of large molecules that are organized prior to inclusion in such a way that they can provide enclosed spaces or binding sites for accommodation of smaller components. However, some consider these classifications as just different nomenclatures given by Cram (host–guest chemistry) and Lehn (supramolecular chemistry)5 with the common feature of non-covalent bonding.
The first report of this type of research dates back to the 1950s when Cramer introduced the inclusion complexes of cyclodextrins6. The field was further developed by the vast research of Cram and co-workers on molecular containers7. Cyclodextrins and some other biologically important molecules, such as carbohydrates in general, nucleotides, steroids, and oligopeptides all occur in nature. After a while scientists began to synthesize8 similar functional systems, highly analogous to the naturally existing ones. This activity started with the synthesis of hollow calixarenes by Collet and Cram in the early 1980s9, 10. The main motive was to produce host molecules with controlled cavity size and shape11-13.
1.1 Classification
Over the years, since the discovery of the first host molecule, the number of structures that are identified or newly synthesized has been growing quickly, resulting in various nomenclatures and classifications. There have been several attempts to sort this class of compounds into different families. However, there are always grey areas where different groups and definitions

3
overlap. According to the topology, type, application and interactions involved, they may be divided into different classes14.
1.1.1 Complex or clathrate
A very fundamental classification assigns supramolecular systems to one of two groups. At one extreme, as mentioned above, there are pre-organized lattice of molecules, called clathrates, which retain smaller compounds by steric barriers. Some examples are urea and graphite15, illustrated in figures 1.1a and b. These compounds normally exist in solid phase and decompose upon dissolution. At the other extreme, there are complexes that are basically coordinated systems of host and guest, which do not lose their structures when dissolved. Crown ethers and cryptophanes are among the typical examples of this category (see figures 1c and d). There are of course other types of aggregations that fall in between these two limiting situations and usually take a hybrid name of both mentioned categories.
a) b)
N
N
N
N
N
NN
N
NN
N
N
NN
N N
NN
NN
N
N
NNN
NN
NN
N
N
NN
NN

4
c) d)
O
O
O
O
O
O
O
O
O
CN N
N
+
H3COH3 H3
H3
H3CO H3COOC
CH2Cl2
O OO
OO
O O O
Figure 1.1. Schematic representation of a) Graphite and b) Urea clathrates, and c) 27-crown-9 and d) cryptophane-A complexes.
1.1.2 Interactions
Another criterion for defining supramolecular systems is based on the forces operating between the components. Strong and specific recognition takes place by means of involving several non-covalent bonds depending on the structures and functions. For instance, “biologically important guest molecules” have different hydrogen bonding sites. In order to form hydrogen bonds, host and guest molecules must be perfectly aligned. Consequently, the complexation produces directed molecular building blocks. On the other hand, while the organization and selectivity are provided by hydrogen bonding, other types of interactions such as hydrophobic effects enhance the complexation16.
Table 1.1 Strength of covalent bonds in comparison with several non-covalent interactions.
Type of interaction Strength kJ.mol-1
covalent bond Coulomb
ion–dipole hydrogen bond dipole–dipole
cation–π π–π
van der Waals forces hydrophobic effects
metal–ligand
350–950 250
50–200 10–65 5–50 5–80 0–50 <5
Hardly assessable 0–400

5
Compared to the strength of covalent bonds, covering the range of 350 kJmol-1 for a single bond to 942 kJmol-1 for the triple bond in N2, interactions involved in molecular recognition are of weaker magnitude1. The strength of covalent and non-covalent bonds is presented in Table 1.117.
Electrostatic forces, such as ion–ion, ion–dipole and dipole–dipole are based on Columbic attraction. Strong bindings are achieved when these types of interactions are involved in attaching the guest to the host. Another feature of electrostatic forces, in the case of dipoles, is that suitable alignment is of great importance to high binding efficiency17.
Hydrogen bonding is a very common interaction in biological systems. Many proteins retain their shape by means of hydrogen bonding and the double helix structure of DNA is stabilized by hydrogen bonds. They have the same directional nature as exists in the case of two dipoles. In fact, the hydrogen bonding relation can be considered as an attraction between two dipoles18.
Cation–π interaction is also known to be relevant in structural biology. As an example, one can consider the association of K+ with benzene19 and the binding of acetylcholine20 in biological systems.
π–π Stacking forces act between aromatic rings of molecules containing them. The relative ring positions may be “face to face” or “face to edge”, of which the latter one is weaker17.
Van der Waals forces refer to the attraction between induced dipoles of different species or with non-polar molecules. Small organic molecules are usually included loosely into cavities or within crystalline lattices by means of van der Waals interactions18.
The expulsion of non-polar weakly dissolved molecules by solvent molecules, particularly water, is called hydrophobic effect. The attraction between the water molecules in their hydrogen-bonded network is so strong that non-polar organic molecules are forced out of the network. Inclusion of organic molecules in cyclodextrins is a well-known example of hydrophobic effects in supramolecular chemistry. Hydrophobic effects result in favorable free-energy changes in solution. The presence of water inside the hydrophobic cavities of host molecules such as cyclodextrins is energy expensive. Upon guest incorporation, water molecules are driven out of the cavity. These water molecules are stabilized by joining to the pool of solvent molecules. It is also more entropy-favored when the organic guest molecules are replaced with the water molecules in the bulk solvent by complexing to the host molecules17.
An important factor influencing the host–guest interactions in solution is the solvent. As discussed above, for example hydrophobic effects arise from solvent–solvent or solvent–components relationships. The solvent can modulate the thermodynamic stability of the complex to a great extent. Moreover, the dielectric constant of the solvent is defined by the polarity of solvent molecules, and consequently controls the electrostatic interactions

6
between components in solution. This can particularly be very crucial in ion recognition. Sometimes the solvent can be a strong competitor in hydrogen–bonded host–guest systems if the individual solvent molecules are good hydrogen bond donors and acceptors. In such cases, guest or host molecules are preferentially solvated rather than complexed18.
1.1.3 Host and guest types
Guest molecules can be in the forms of cation, anion or neutral molecules. One way of classification is according to complexation ability of different host molecules with each group of the mentioned guest molecules.
1.1.3.1 Cation binding hosts
Cation guests are bound to hosts through electrostatic ion–dipole interactions. In many cases hydrogen bonds enhance the complexation even more. The cations can be of either metal or nonmetal type. There are many examples of cation binding, furnished particularly by the life sciences, and among synthetic host molecules. In the B12 vitamin, a corrin macrocycle binds cobalt ion and iron is complexed with porphyrin macrocycles in haem groups. In MRI, Gd3+complexes are used as contrast agents, and coordination of platinum to DNA hinders the growth of cancerous cells21. Some of the general groups of host molecules that are able to bind cations, such as crown ethers, cryptands, spherands, calixerenes and siderophores are discussed briefly below.
Crown ethers, considered as the simplest macrocyclic ligands, were first introduced by Charles Pedersen in 1967. Their structure is composed of ether oxygen atoms linked together via organic linkers such as methylene groups. When complexing transition metals, the oxygens can be replaced by some softer donors such as sulfur and nitrogen atoms. There is an ion–dipole attraction between the cation and the oxygen donor atoms of the rings. The most stable complexes form when there is an optimal fit between the cationic radius and the cavity size. This optimal spatial fit concept results in selective complexation of crown ethers with certain cations. In figure 1.2, two crown ethers of different cavity diameters are presented, which form complexes selectively with two different cations22.

7
Figure 1.2 Role of size in selective complexation of crown ethers with cations.
The preference for binding a cation with a certain diameter maximizes the electrostatic interaction between the two species. Most of the complexes are of 1:1 stoichiometry, although there are some examples where large crowns complex two cations simultaneously. Besides metallic cations, crown ethers can encapsulate ammonium and alkyl ammonium cations through hydrogen bonding. Therefore, a combination of electrostatic and hydrogen bonding stabilizes the complexation. The key point to this type of complexes is the complementary orientation of the oxygen atoms in the crown ether to form the hydrogen bonds17. The role of symmetry in efficient complexation is illustrated in figure 1.3. Figure 1.3 Role of symmetry in selective complexation by crown ethers.
Cryptands are three-dimensional, cage-like analogues to crown ethers. They have high affinity for complexing group 1 and 2 metal cations.
O
O
O
O
OOMe
H
MeMeH
H+
18-crown-6
MeH H
MeMeH O
O
O
O
O
+
15-crown-5
O
O
O
OO
OO
O
O O
O
O
O
K+
Cs+
Cs+
K+
diameter = 2.66 Å
Cavity diameter = 2.6 - 3.2 Å
diameter = 3.34 Å
Cavity diameter = 3.4 - 4.3 Å
18-crown-6 21-crown-7

8
Compared to crown ethers, cryptands encapsulate the cation entirely in a more selective way. This is due to their bicyclic structure and their more enhanced ionophore-like properties. Higher stability constants are therefore attained for cation complexes compared to those of analogous crown ethers. This is ascribed to favorable enthalpy and entropy changes when the cation is shielded from the solvent molecules inside the sphere of cryptand. The complexes are commonly named cryptates23. The most famous and commercially available cryptand, [2.2.2]cryptand, is shown in figure 1.4.
Figure 1.4 [2.2.2]cryptand
First discovered by Donald Cram, spherands are another group of macrocyclic cation hosts, synthesized with a preorganized structure. In contrast to crown ethers and cryptands, which have flexible structures in solution, these molecules have a convergent pocket-shape cavity for cation guests. This structure rigidity enables them to have better selectivity and bonding strength compared to crown ethers and cryptands. Higher bonding strength gives rise to slower complexation – decomplexation kinetics in these systems. However, their preorganized rigid structures limit them to binding small-size cations only, such as sodium ions. Figure 1.5 illustrates how oxygen atoms are in an octahedral configuration for binding cations24.
Figure 1.5 One of the first synthesized spherand host molecules.
OOO
OOO
ON
O O
OO
ON

9
Calixarene structure is composed of phenol parts, linked by methylene groups. They exist in four different conformations, namely, cone, partial cone, 1,3- alternate and 2,2- alternate (see figure 1.6). Depending on the polarity of the solvent, the amount of each conformation may vary in solution. In a polar solvent, for example, the cone conformation constitutes the highest amount. The reason is that the cone is the most polar of all four conformations, by having all OH groups located to one side of the molecule. In this conformation, cations can either be held by the OH groups of the lower rim or be involved in a π-cation interaction with the aromatic rings17.
Figure 1.6 Different conformations of Calixarenes molecules.
Siderophores exist both in natural and synthetic form. Their general name means “iron bearer” reflecting the fact that their ability to complex iron ions is enormous. The most frequent naturally occurring oxidation state of iron, Fe(III), is not soluble in water. This is a problem when, at physiological pH of about 7, a concentration of 10-7 mol dm-3 is needed. To tackle this difficulty, in plants, bacteria and some higher organisms the Fe3+ delivery to the cell is accomplished effectively by siderophores. Siderophores complex with iron(III) through oxygen atoms in hydroxamate or catecholate groups. Figure 1.7 shows how enterobactin, a bacterial siderophore, makes a ∆ configuration around the metal ion by catecholates18.
ROOROR OR
Cone
OR OROR
RO
Partial Cone
OR OR
OR RO
1,3-alternate
OR
OR
OR
OR
1,2-alternate

10
∆ configuration
Figure 1.7 Enterobactin makes a ∆ configuration around Fe(III).
1.1.3.2 Anion binding hosts
Anion binding is an important process that benefits different areas such as chemistry, environmental chemistry, biology and medicine. Anion reactivity as catalyst or base can be altered by binding to a host molecule. Anion pollutants and toxic byproducts can be selectively sensed and extracted by recognition processes. 70 to 75 per cent of biologically important molecules such as adenosine triphosphate(ATP) and deoxyribonucleic acid (DNA) are negatively charged18.
In extending the discussion from cation binding to anion complexation, some new aspects emerge which can be attributed to the special features of anion moieties. The new features of anions compared to cations comprise of their size, pH dependence of properties, the way they interact with solvent molecules, and their geometry. Compared to their isoelectronic cations, anions are relatively larger. This means that the receptors encapsulating them must have larger cavities. Many anions, such as carboxylates, and phosphates, exist in a narrow region of pH. Changing the pH can make them lose their negative charges. Anion solvation depends on size, charge and the pH range in which the ion exists. In general, anions have higher solvation free energies than cations of similar sizes. Anions come in different shapes and geometries. They may exhibit, e.g. spherical (F-, Cl-, Br-, I-,), linear (N3
-, CN-, SCN-, OH-), planar (NO3
-, PtCl4-), tetrahedral (PO4
3-, SO42-, MnO4
-),
O
O
O NH
Fe3+
O
O
O
NH
O
O
O
NH
ONH
O
NH
OH
OH
O
O
NH
O
O
O
OO
OH
OH
OH
OH
enterobactin

11
and octahedral (PF6-, Co(CN)6
3-) geometries, and even some complicated shapes which are common among biologically important anions17.
The main similarity between cation binding and anion recognition is that electrostatic forces again play an important role in strengthening the recognition. The simplest way of hosting an anion could be an electrostatic ion–ion interaction. There are other options, however, such as arrays of hydrogen bonding groups acting as electron pair acceptors. The directionality property of hydrogen bonds makes it possible to differentiate between anions with different geometries using specifically shaped host molecules. Another option that works in a similar way to hydrogen bonding is binding to the Lewis-acid hosts. They also can accept electron pairs into their vacant orbitals17.
As mentioned above, anion complexation is sensitive to the range of pH. This issue, however, can be used as a tool to transform a cation receptor to an anion host. A vast number of cryptand molecules that complex cations through the nitrogen bridgeheads, and secondary amine chains can bind anions through protonating the amine groups by varying the solution pH. However, only cryptands that have large cavities are suitable for this purpose. The macrotricyclic cryptand in figure 1.8 is a versatile example, which is commonly named “soccer ball” because of its perfectly spherical shape. The presence of nitrogen atoms gives it Lewis-base properties so that it can bind cations. In its tetraprotonated form it is also a good receptor for ions such as Cl-, forming hydrogen bonds from one side and strengthening the complexation from the other side by electrostatic interactions with ether oxygen atoms. In its diprotonated form it can also bind neutral molecules such as water17. Figure 1.8 The “soccer ball”-shaped cryptand complexing both cations and anions at different pH.
Two-dimensional macrocycles such as those depicted in figure 1.9, which are nitrogen analogues of the crown ethers, are the very first anion-binding hosts. However, the cavity of hexacyclen is partially filled by the NH protons and cannot accept any anion. There are some large-ring species that can accommodate large anions such as [Fe(CN)6]
4– 17.
O
ON
N
O
O
N
NO
O
12
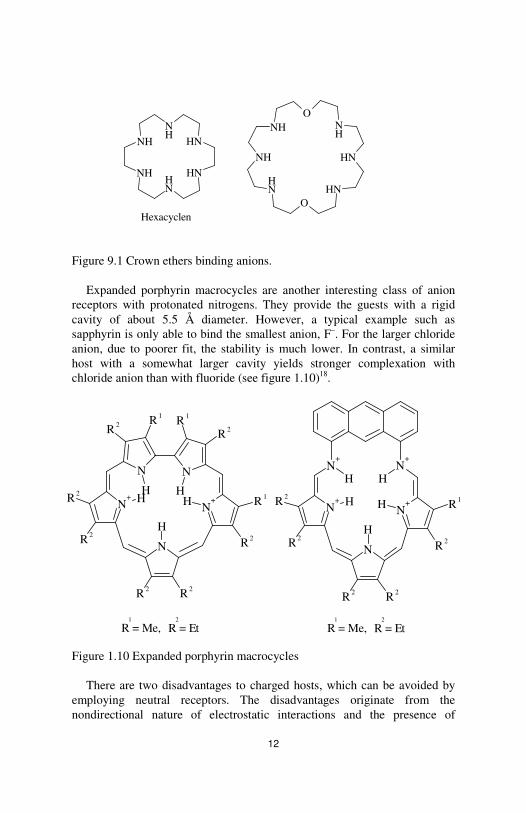
Figure 9.1 Crown ethers binding anions.
Expanded porphyrin macrocycles are another interesting class of anion receptors with protonated nitrogens. They provide the guests with a rigid cavity of about 5.5 Å diameter. However, a typical example such as sapphyrin is only able to bind the smallest anion, F–. For the larger chloride anion, due to poorer fit, the stability is much lower. In contrast, a similar host with a somewhat larger cavity yields stronger complexation with chloride anion than with fluoride (see figure 1.10)18.
Figure 1.10 Expanded porphyrin macrocycles
There are two disadvantages to charged hosts, which can be avoided by employing neutral receptors. The disadvantages originate from the nondirectional nature of electrostatic interactions and the presence of
NH
NH
NH
NH
NH
NH
NHO
NH
NH
NH
NH
ONH
Hexacyclen
N
N+
N
N+
N
R 2
R 1
R 2R 1R 1
R 2
R 2
R 2
R 2 R 2
H
H HH H
R = Me,1
R = Et2
N+
N+
N
N+
N+
R 2
R 1R 2
R 2
R 2 R 2
H H
H H
H
R = Me,1
R = Et2

13
counter-ions of the hosts. Despite strong binding by electrostatic interaction, charged hosts can bind to almost all anions with different strength of complexation, which results in reduced selectivity. The counter-ion itself could be a competitor of the target anion guest and reduces the affinity of the host for other anions. A very practical replacement for electrostatic interaction could be the hydrogen binding interactions that are at the same time directional and strong. An example is calyx[4]pyrrole, shown in figure 1.11, which has a very small cavity in which it is possible to bind small anions. In CH2Cl2, it binds fluoride with a binding constant of 1.7×104 M-1 with a high selectivity factor of 50 over chloride inclusion. Another interesting feature of this host is that it adopts different conformations when empty or bound to the anion, which is an indication of conformation rearrangement induced by complexation17. Figure 1.11 calyx[4]pyrrole binds fluoride selectively.
Another well-known group of neutral receptors is named zwitterions, which are both positively and negatively charged so that the overall charge is zero. Many anion-binding enzymes and proteins are zwitterionic. Some examples are the amino acids phenylalanine and tryptophan in which the CO2H protonates the NH2 group. This also facilitates the proteins membrane solubility18.
1.1.3.3 Hosts binding neutral molecules
In comparison to strong, permanent electrostatic interactions that hold ions to the receptors, neutral (mostly organic) molecules may be bonded with weaker interactions such as hydrogen bonds, van der Waals and hydrophobic effects. Neutral guests may be either captured in solid-state networks in the form of clathrates or be embedded within the cavity of cavitands17.
NH
NHNH
NH
calix[4]pyrrole

14
Solid state clathrates are made up of both organic and inorganic compounds. Clathrate hydrates are examples of inorganic clathrate compounds that form under very specific conditions of pressure and temperature. For example, there is no cavity in the normal ice structure, but in the presence of some hydrate-forming guests, such as Cl2, H2S and CS2, a polyhedral cavity is formed via a template reaction. This new conformation alters some of the physical properties of ice such as rigidity at different temperatures, and thermal conductivity17.
Porous aluminosilicates, known as zeolites, exist in two main categories according to the Si/Al ratio. The featured property of zeolites is their strongly built channels and cavities which are not disrupted by the guest species entering or leaving. This feature makes them favorable molecular sieves and reaction vessels with high selectivity25.
Cycloteriveratrylene (CTV) (shown in figure 1.12) is both a versatile host molecule and an important building block in two forms, bowl-shaped and saucer-shaped, used for constructing a vast range of hosts that can bind either neutral guests or anionic compounds in solid phase and in solution.
Figure 1.12 CTV building block
A wide range of small molecules, such as benzene, water, toluene and
CS2 can be included in CTV in the solid-state. CTV is predominantly a hydrogen bond acceptor. Even weak hydrogen-bond donor guests such as benzene can provide hydrogen to methoxy groups oxygens. Otherwise, in a less probable mode, the methoxy group acts as the hydrogen-bond donor to the hydrogen bond acceptor guests. These two modes of inclusion are known as α and β phase of CTV. A new clathrate phase of γ is also known, with a single example of (CTV)4.acetone. The interesting aspects of the (CTV)4 arrangement are its intracavity inclusion form and its being the non-covalent analogue of cryptophanes(see below). Buckminsterfullerene (C60) has also
O
OO
O
O OCH3
CH3
CH3

15
been shown to form an intracavity complex with CTV. Highlights of this inclusion system are a good curvature match between guest and host structures and complementation of C60 electron deficiency with electron-rich methoxy groups17.
The inclusion systems discussed above are the results of intermolecular interactions between the host and guest, so that the guest species fill the spaces left between the host molecules in their crystalline form. Hosts possessing permanent cavities can form complexes in the solid state and in solution. In this way the host can encapsulate the guest in its intrinsic curvature. These groups of hosts, termed cavitands, are molecular containers with permanently concave surfaces. They are commonly classified according to the building blocks that construct them. Besides having intrinsic curvatures, sometimes the walls of the container may be formed by aryl rings and a variety of spacers. This series of containers is named cyclophanes. The presence of aromatic rings linked by aliphatic chains provides a hydrophobic cavity for non-polar guests particularly in water. Among cyclophanes those with well-defined cavities set up by parallel aromatic rings are of special interest. The aromatic groups in the wall empower the receptor with a preorganized rigid cavity17.
Cyclodextrins(CDs) – CDs are cyclic oligosaccharides composed of six to eight D-glucopyranoside units that are linked by 1,4-glycosidic(depicted in figure 1.13) bonds. Cavitands of this group are able to bind neutral, charged and even radical guests in solid and solution phases26. The three important naturally occurring ones are α-, β-, and γ-cyclodextrins. These three members of the family contain six, seven and eight glucopyranoside units, respectively.
Figure 1.13 1,4-Glycosidic link Cyclodextrin resembles a truncated funnel with upper and lower rims,
similar to the calixarene cone. The narrower rim is composed of primary hydroxyl groups while the secondary rim is built up of -CH2OH groups. The schematic shape of CDs is presented in figure 1.1417.
OO O
OH
OH
OHOH
OH
OHO
O
1
234
5
6
2
3
4 5
6
1

16
Figure 1.14 Schematic presentation of CDs. From α- to γ-CD, physical and chemical properties change smoothly
except for the solubility in water which is much lower for β-CD, than for the other two and even larger cyclodextrins such as δ-CD. Several arguments have been put forward to explain this abnormal trend. One explanation is that, since the water molecule has even numbers of hydrogen bond donors and acceptors the six- and eight- fold symmetries of the two other CDs are more compatible with the solvent cage, resulting in more favorable hydration enthalpy and entropy values. Another explanation states that the intramolecular hydrogen bonding on the secondary rim of β-CD limits its interaction with solvent molecules26, 27.
As discussed above, having a rigid and deep cavity is advantageous in forming stable complexes with non-polar guests, which is based on weak interactions. For a long time it was thought that cyclodextrins with fully saturated rings could also offer such rigidity because of their highly curved surfaces. This understanding relied on X-ray studies28,29. But many experimental data, mostly acquired using solution and solid-state NMR, and theoretical investigations proved that cyclodextrins cannot be rigid moieties. One of the first experimental and theoretical proofs was the low barrier of internal rotation obtained for the ether C-O bond connecting the rigid glucopyranose rings30. This is also in agreement with the fact that CDs should be flexible to some extent to be able to include a variety of guests with different shapes. However, higher equilibrium constants are also observed for guests with saturated structures that benefit from higher degree of freedom, and therefore higher flexibility, to fit into the cavity of the CD31. CDs are obtained from biochemical degradation of starch, and they crystallize from water as hydrates with some varying amount of water captured inside their cavities. Being hydrophobic, the cavity has little interaction with these water molecules, and therefore these high-energy
13.70 Å 15.30 Å 16.90 Å
5.70 Å 7.80 Å 9.50 Å
a-CD β-CD γ-CD
13.70 Å 15.30 Å 16.90 Å
5.70 Å 7.80 Å 9.50 Å
a-CD β-CD γ-CD

17
water molecules are driven out in the process of guest binding. Upon the release of water molecules, most frequently, a 1:1 inclusion complex is formed. A common relationship of thermodynamic equilibrium may be written for the process:
1.1 1.2
where K is the stability constant of the complex.
However, it is possible that higher equilibria exist simultaneously. The important factors that drive the complexation are steric fit, release of high-energy water molecules, hydrophobic effects, van der Waals interactions, dispersive forces, dipole–dipole interactions, charge-transfer interactions, electrostatic interactions (in the case of ionic species) and hydrogen bonding. Inspecting the sizes of typical guests binding CDs, it appears that there is a proportion between the sizes of the host and the guest occupying its cavity. Several phenomena in cyclodextrin complexation could result in either favorable or unfavorable entropy and enthalpy changes. For example, release of high-energy solvent molecules and their rejoining the bulk sometimes creates enthalpy gain. Entropy loss is also obtained when two holes in the network of the solvent molecules, produced by the presence of host and free guest molecules, coalesce to one as they non-covalently bind. There are some other sources of enthalpy gain, such as hydrogen binding between the guest and hydroxyl groups of CDs and dipolar interactions. For example, when aliphatic guests of type CH3(CH2)nX bind to CD, if X is a polar group, such as –COOH or –OH, the binding is much stronger than when X is for example a methyl group. Moreover, due to presence of –OH groups in the structure of CDs, the cavity is not 100 per cent non-polar but rather semi-polar. This fact controls the favorability of the interactions operating between host, guest and solvent molecules. Consequently an enthalpy-entropy compensation process averages out the influence of different factors altering the energetics of the encapsulation. Thus the change in the association constant, K, is less than what expected from the enthalpy variation observed in experiments32.
CDs are eminently applicable in different areas, mainly because they are selective host molecules. CDs are the cheapest commercially available hosts that cover a large range of industrial applications. Having very low toxicity, even in high doses, and excellent temperature stability, 70 to 80 per cent of their production volume is devoted to food industry. They also have extensive application in pharmaceuticals industry as drug-delivery systems. They can be used as protecting agents to prevent any early metabolism of the
CD + G CD.G
[ ][ ] [ ]GCD
GCDK
.=

18
drug. CDs can modify and enhance the solubility of the drug without any need for modifying the drug itself. Another field of extensive application is analytical chemistry, particularly in chromatographic methods such as high-pressure liquid chromatography (HPLC) and gas chromatography. They are used either as part of the mobile or stationary phase to assist in the separation of closely related compounds, especially enantiomers. In the case of selecting and separating enantiomers, the process is commonly referred to with the term “chiral recognition”17. CDs can be used both as homogenous and heterogeneous catalysts. There have been several reports on catalytic activities of cyclodextrins mimicking enzymes. They are usually used to model the function of the enzymes in order to elucidate the mechanism with which the enzymes operate. However, CDs mostly influence the stereoselectivity of the reaction rather than its yield27. This vast range of applications has encouraged the synthesis of chemically and enzymatically modified cyclodextrins, so that the number of synthesized CDs has already passed 1500 derivatives33.
Cryptophanes – CTVs (shown in figure 1.12) are the building blocks of the cryptophanes. As discussed before, CTVs, with their saucer-like shape, are good anion receptors, whereas they do not have any considerable ability to include neutral guests. However, there are limited examples such as intracavity complexation of C60 and spherical carborane, o-C2B10H12. The low solution-binding ability of the CTV is sometimes ascribed to its shallowness. Deepening the CTV cavity might enhance its binding properties in solution, but a modification will be much more effective if the formed structure is a three-dimensional closed shell that can protect the guest from the solvent medium and slow down the exchange of the guest with the bulk solvent. The first series of three-dimensional hosts were synthesized by Collet34, using two CTV units facing each other and linked covalently by –(CH2)n-, -CH2-CH=CH-CH2- or –CH2-C≡C-CH2- bridges with n = 3–8. They were named cryptophanes and are usually of two distinct forms, with D3-anti or C3h-syn symmetry (see figure 1.15). The first series comprises cryptophane-A and –B, which are the anti/syn derivatives with X = -(CH2)2- bridges, and cryptophane-E and -F where X = -(CH2)3-. Afterwards came cryptophane-C and –D, with the same bridges as cryptophane-A/B but lacking methoxy groups in one hemisphere12.

19
Figure 1.15 Two forms of cryptophane derivatives.
Small, tetrahedral molecules such as methane and halogenated derivatives are the best choices of guest to fit in the cavity of the cryptophanes. In a non-polar solvent medium, a binding constant of 470 M-1 is obtained for a complex of CHCl3@cryptophane-E which, considering the absence of hydrophobic effects, is still very strong bonding. This is brought out by the large binding constant of 7700M-1 for chloroform in water with a host similar to cryptophane-E, with the difference that the methoxy groups are replaced by hydrophilic carboxyl groups17.
Cryptophanes are apparently able to discriminate among guests on the basis of van der Waals volume differences less than 5%35. For instance, as a comparison between cryptophane-E and -C, the smaller CH2Cl2 molecule is preferred by cryptophane-C where, compared to cryptophane-E, the bridges are shorter and accordingly the cavity should be of smaller size. Cryptophane-E shows selectivity for the larger chloroform molecule. However, it is worth breaking down the free energies into their enthalpy and entropy parts to get a better insight into the size-fit conception of the complexation. The results of a van t’Hoff plots for CH2Cl2 and CHCl3 binding by the above mentioned cryptophane derivatives are listed in the Table 1.217. It is clear from the Table that, although a favorable enthalpy contribution is produced when cryptophane-C binds CHCl3 but an unfavorable entropy change makes this host more selective for the smaller dichloromethane molecule. This implies that chloroform probably fits too well into the cryptophane-C cavity, so that its degree of freedom becomes
O
OO
X
O
OOO
O
X
OO O
X
OO
OO
X
O
OOO
O
X
OO O
X
O
D3-anti C3h-syn
X = -(CH2)n-, -(CH2)-CH=CH-CH2-, alkyne, C6H4
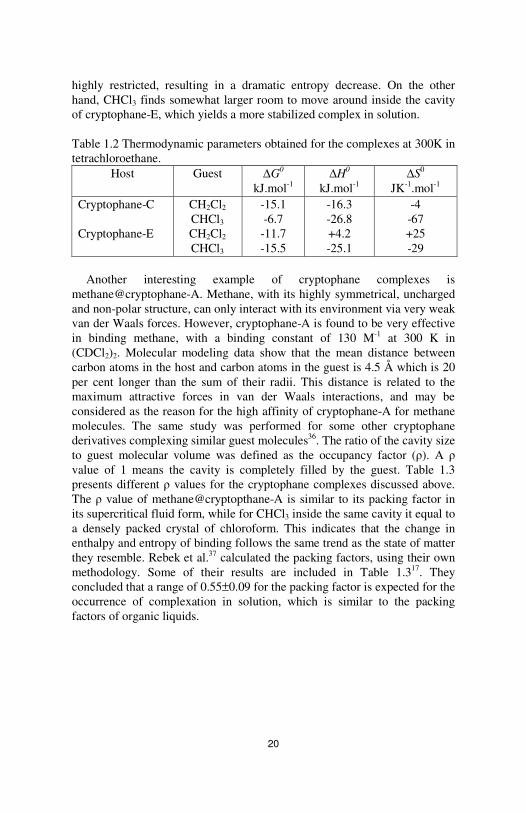
20
highly restricted, resulting in a dramatic entropy decrease. On the other hand, CHCl3 finds somewhat larger room to move around inside the cavity of cryptophane-E, which yields a more stabilized complex in solution. Table 1.2 Thermodynamic parameters obtained for the complexes at 300K in tetrachloroethane.
Host Guest ∆G0 kJ.mol-1
∆H0 kJ.mol-1
∆S0
JK-1.mol-1 Cryptophane-C Cryptophane-E
CH2Cl2 CHCl3
CH2Cl2
CHCl3
-15.1 -6.7
-11.7 -15.5
-16.3 -26.8 +4.2 -25.1
-4 -67 +25 -29
Another interesting example of cryptophane complexes is
methane@cryptophane-A. Methane, with its highly symmetrical, uncharged and non-polar structure, can only interact with its environment via very weak van der Waals forces. However, cryptophane-A is found to be very effective in binding methane, with a binding constant of 130 M-1 at 300 K in (CDCl2)2. Molecular modeling data show that the mean distance between carbon atoms in the host and carbon atoms in the guest is 4.5 Å which is 20 per cent longer than the sum of their radii. This distance is related to the maximum attractive forces in van der Waals interactions, and may be considered as the reason for the high affinity of cryptophane-A for methane molecules. The same study was performed for some other cryptophane derivatives complexing similar guest molecules36. The ratio of the cavity size to guest molecular volume was defined as the occupancy factor (ρ). A ρ value of 1 means the cavity is completely filled by the guest. Table 1.3 presents different ρ values for the cryptophane complexes discussed above. The ρ value of methane@cryptopthane-A is similar to its packing factor in its supercritical fluid form, while for CHCl3 inside the same cavity it equal to a densely packed crystal of chloroform. This indicates that the change in enthalpy and entropy of binding follows the same trend as the state of matter they resemble. Rebek et al.37 calculated the packing factors, using their own methodology. Some of their results are included in Table 1.317. They concluded that a range of 0.55±0.09 for the packing factor is expected for the occurrence of complexation in solution, which is similar to the packing factors of organic liquids.

21
Table 1.3 Packing factor for cryptophanes complexes. Guest Cryptophane-A Cryptophane-C Cryptophane-E CH4
CH2Cl2
CH2Br2 CHCl3
0.35 0.70 0.60a
0.80 0.89 0.75a
0.70
0.89
0.65 0.49a
0.81 0.61a
a Data from Rebek et al37. In accordance with the factors governing supramolecular recognition
strength in solution, cryptophane complexation is also significantly influenced by solvent effects. Early studies of cryptophane complexes were carried out in CDCl3 solvent, assuming that solvent molecules cannot pass through the effectively closed surface of the cryptophane structure. However, very low binding constants of 1–2 M-1 were obtained for guests such as dichloromethane that were similar in size to the solvent molecules. This is an indication of a significant solvation effect originating from guest encapsulation. In the case of water as solvent, this effect reinforces the binding by benefiting from the hydrophobic effects and restoring the strong hydrogen bonds between water molecules. However, the chloroform molecule is a potent competitor of similar-sized guests, resulting in reduced binding constants.
The first application of cryptophanes was the separation of enantiomers of CHFClBr, which is the simplest chiral compound. Using the intrinsic chirality of the D3-anti cryptophanes, complexation of a partially resolved sample of (±)-CHFClBr with a resolved sample of (+)-cryptpohane-C was examined by Collet et al.38. Separate signals for (+)host:(+)guest and (+)host:(-)guest were observed in NMR spectroscopy, and the optical rotation of the guest was defined. Cryptophanes are very promising synthetic hosts, and the research is going on towards the stage where their remarkable recognition properties can be used in different areas of separation, molecular delivery and sensing.
1.2 Selectivity
Discriminating among different guests in binding is called selectivity which is in fact the main goal of supramolecular chemistry. This criterion is requested both in nature and synthetic systems. The first assessment of the selectivity of a host for a particular guest is their equilibrium constant. One can therefore consider the selectivity, in thermodynamic terms, as below:

22
1.3
For example, in blood it is important that haemoglobin should selectively take up O2, from the mixture of oxygen with water, CO2 and N2
17. However, there is another kind of assessment in which one looks at the
rate of the transformation of one particular guest along a reaction path, in comparison with other guest species. In this way the kinetic selectivity of the host is examined and the guest which is transferred fastest is considered to be effectively selected by the host. This kind of selectivity is needed in processes such as supramolecular catalysis and guest sensing. Therefore in the application where the kinetics of the process is of importance the thermodynamic selectivity, i.e. high equilibrium constant, is inhibitory and is not favored17.
1.3 Application
Application of molecular recognition is progressing in two fields in parallel. Molecular biology and nanotechnology both benefit from the new horizons opening in this area. Supramolecules such as crown ethers can be used as phase transfer agents to solubilize salts in non-polar solvents. A number of organic reactions became feasible by means of these agents. Molecular recognition may be used in separation of one species from a mixture of many components. For example, in removal of pollutants such as toxic metal ions from aqueous solutions, supramolecular chemistry is of great use. As another instance, purification of C60 from C70 impurities using p-tert-butylcalix[8]arene in toluene can be mentioned. Benefiting from the shape fit of spherical C60 into the cup-shaped cavity of calixarene, the complex is precipitated from the solvent while C70 and other impurities remain in the mixture. In the next step, the complex can be transferred to a solvent such as chloroform, where the host is soluble but the guest is not, and therefore can be filtered off. Some receptors are able to report the presence of guests bound to them by some physical means. This group of hosts may be used as molecular sensors. They are usually selective to some guests and can moreover be used to estimate the concentration of the sensed molecule. The receptor can be appended to a polymer electrode and produce a response when placed in contact with the relevant guest species. Alternatively there could be a functional group in the host structure that has a special electrochemical or spectroscopic property which can be altered by including a special guest compound. Instead of silicone chips, molecular hosts may one day be used as on-off switches and logic gates. In particular, nanoscale
2
1
Guest
Guest
K
KySelectivit =

23
molecules that use light as input or output are of interest, since light has high velocity and is easily controllable using fiber optics. For example, as depicted in figure 1.16, the anthracene group is not fluorescent when, in the absence of hydrogen, photoinduced electron transfer (PET) occurs from the nitrogen group to the aromatic rings. But as soon as the nitrogen group is protonated PET is prevented and therefore emission from the anthracene unit is observed18.
Figure 1.16 A molecular ‘on-off’ switch.
In biological application of supramolecules, an important feature that attracts great attention is the catalytic influence of the hosts on the substrate. One usually wishes to gain control over regio- and stereoselectivity in catalytic reactions. In pharmaceutical research, molecular and especially chiral recognition is highly appreciated in drug design. Some examples are the application of inclusion complexes as MRI contrast and anti-cancer agents and anti-HIV products18.
Cl
N
O
Cl
NH+
O
H+
PET No PET
not fluorescent fluorescent

24
2- NMR Spectroscopy
In this chapter a number of topics in nuclear magnetic resonance spectroscopy, related to the research presented in the attached articles and drafts, are briefly discussed.
2.1 - Spin Hamiltonians
A nucleus with non-zero nuclear spin possesses a total angular momentum: .II h=ˆ 2.1
I is a dimensionless angular momentum operator. A nuclear magnetic moment, µ , is attributed to the total angular momentum: 2.2 From the sum of individual magnetic moments in an ensemble of identical nuclei, a macroscopic total magnetic moment is produced, constituting a molecular spin system39.
Through µ , the spin can interact with the magnetic fields present in its environment. Magnetic fields can be either external, such as the static Bo field or the radiofrequency B1 field, or internal due to presence of other spins within the sample. The total Hamiltonian for spin interactions can be written as: 2.3
oH consists of the time-independent interaction with Bo and all other
relevant time-independent contributions. (t)H1ˆ is the Hamiltonian that
describes the interaction of the spin with the fluctuating B1(t) field. Due to the interaction with this field, spin rotates and consequently deviates from its equilibrium position. Any interaction that can drive the spin back to its initial position is considered as a relaxation interaction, represented by (t)H R
ˆ 40. In the case of spin-1/2, the components of the time-independent
Hamiltonian are as follows:
Iγ.µ ˆˆ =
(t)H(t)HHH Rˆˆˆˆ
1 ++=o

25
2.4 The interaction of each nuclear spin with the Bo field, aligned along the z
direction of the laboratory frame, is called nuclear Zeeman interaction, and the Hamiltonian for the spin system is given by41: 2.5
γ
k is the gyromagnetic ratio and k enumerates individual spins in the molecular spin system.
The presence of electrons in the system changes the magnitude of the local field at the site of the nucleus. In an external magnetic field, electrons produce an induced magnetic field that changes the local effect of Bo, a phenomenon known as chemical shielding. The related Hamiltonian is written as41:
2.6
Ik is a vector with the components Î
k
x, Î k
y and Î k
z, and σk is the chemical
shielding tensor. Magnetic moments can also magnetically perturb each other either
through space or via the electronic structure. The Hamiltonian for the direct through space dipolar coupling of two spins41 reads:
2.7
The interaction depends on the inverse third power of the distance between the spins, rkl
-3, which is included in the coupling tensor, Dkl. In the case of indirect coupling of spins that are connected via the
electronic bonds, the interaction Hamiltonian is written as41: lklk
kk,lJ IJIH ...2ˆ π∑
>
= 2.8
Another important feature of the spin interactions is that they can be either isotropic, meaning they are the same in all directions in space, or anisotropic, i.e., they depend on the direction. If the molecule is fixed with respect to the laboratory frame, the anisotropy of interactions is important. This is actually the case in the solid state or in highly viscous systems. When the directions in space matter, we deal with the tensor-type quantities. Anisotropic interactions are therefore characterized as second-rank tensors. A second-rank Cartesian tensor (3×3 matrix) can be broken up into three irreducible tensors, which are of zero, first and second rank. In solution, on
DJσ HHHzHH ˆˆˆˆˆ +++=o
kz
k
kz I..BγH ˆˆ
o∑−=
kk
k
kσ .I.σ.BγH
o∑=ˆ
∑>
=kk,l
kklkD .I.DIH

26
the other hand, interactions become isotropic due to isotropic molecular motions. One can characterize the isotropic interactions by a scalar value, which is the zero-rank component of the interaction tensor. The zero-rank component of the tensor is the average value of the tensor. This component of the dipolar interaction tensor is zero, and the interaction thus plays no role in the appearance of liquid samples. The first rank component is also zero. However, the second rank components contribute significantly to the process of spin relaxation, which is discussed in the next section. Even though indirect dipolar interaction is basically an anisotropic interaction, only the isotropic component is taken into account because the anisotropies are usually small.
The chemical shielding tensor can be illustrated as an ellipsoid, as shown in figure 2.1a41.
Figure 2.1 a) Pictorial representation of chemical shielding tensor b) Relationship between other anisotropic interaction tensors: line N is the intersection of the XY and σxxσyy coordinate planes. Looking down from N toward origin, σzz and Z axes can rotate about N.
To obtain the magnitude of the interaction in any direction, σ(θ,φ) is
defined as: 2.9 Relative values of σxx, σyy and σzz define different powder patterns in solid-state NMR. The molecular frame of the system is defined according to the properties of this tensor. Other anisotropic interaction tensors, if any, are defined using α, β and γ Euler angles with respect to this tensor (see figure 2.1b.).
θσθφ σθφ σσ(θ,φ) zzyyxx22222 cossinsinsincos ++=
B0
θ
φ σxx σyy
σzz a)
σyyα
N
X
Z Y
b)
γ
β
σzz
σxxσyy
α
N
X
Z Y
b)
γ
β
σzz
σxx

27
2.2 Relaxation - A very brief introduction
Besides the magnetic fields discussed previously, Bo and B1, there are some other fluctuating local magnetic fields present at the sites of the nuclei. These fields are produced by the interactions of spins with one another or with their environments. These interactions act as weak perturbations of the energy levels induced for spins by Bo. Following a perturbation caused by radio-frequency field at the Larmor frequency, which causes transitions between states, spins start to relax by exchanging the absorbed energy among themselves and also releasing it to the surrounding medium; these processes are the so-called spin–spin and spin–lattice relaxation, respectively. In liquids, the relaxation process is caused by rapid and stochastic molecular motions. The influence of molecular motions on the local magnetic fields is to make them time-dependent. The most important motions are rotations which modify those (anisotropic) local magnetic fields. Due to reorientation of internuclear axes with respect to each other and the stationary field, local magnetic fields fluctuate with time. To play an active role in a relaxation process, oscillations should reach the transition frequencies of spin energy levels. For small molecules in isotropic liquids, the rotational motion may take place at rates comparable to the Larmor frequency of the nuclei42.
2.2.1 Relaxation mechanisms
Usually, all the time-dependent random interactions may contribute to relaxation. In the absence of quadruploar nuclei (I > 1/2) or paramagnetic impurities, there are two major mechanisms involved. The first one is the chemical shift anisotropy (CSA). As mentioned earlier, chemical shift originates from the shielding effect of surrounding electrons. When the distribution of electron density around the nucleus is non-spherical, the nuclear spin feels a fluctuating field when the molecule tumbles. For non-protonated carbons, such as carbonyl or olefinic carbons, this is the dominant mechanism leading to relaxation. Since CSA comes from the shielding effect, its strength is proportional to the strength of the applied magnetic field.
The other mechanism is the dipole–dipole interaction through space between pairs of spins, which is considered as the dominant mechanism for relaxation in the case of 13C-nuclei with directly bonded protons. Protons, as high natural abundance nuclei with a large gyromagnetic ratio, are particularly important. In the absence of motion, a simplified form of the local field induced by a proton spin that is close enough to the corresponding carbon can be expressed as:

28
2.10
r is the length of the relaxation vector, i.e. the axis connecting two spins, and θ defines the orientation of that axis with respect to the static magnetic field. Through molecular tumbling, the angle varies randomly, and a fluctuating field is induced at the site of the other spin (figure 2.2a). In figure 2.2b the orientation of the relaxation vector in the molecular frame is shown with spherical coordinates. θ and φ are equivalent to the α and β Euler angles, respectively. Assuming a symmetric interaction around the relaxation vector, φ is normally defined to be zero43.
a) b)
Figure. 2.2 a) The field induced by spin 1(1H) at the site of spin 2(13C). b) The relaxation vector in a spherical coordinate system.
The classical interaction energy of the carbon-13 dipole moment with Bdip is given by: 2.11 The Hamiltonian is obtained by replacing all variables with their quantum mechanical operator equivalents. Its simplified form under secular approximation is:
2.12
This approximation is applied in high magnetic fields where the Zeeman interactions are dominant. In such a case only those terms in the Hamiltonian are retained that commute with the Zeeman Hamiltonian44.
3
2
11cos3
4 r
µB
)( θ
π
µo
dip
−=
θ
φ
x
y
z
[0,π] € θ[0,2π] € φr
θ
φ
x
y
z
[0,π] € θ[0,2π] € φr
Spin1
Spin 2Bdip
rθ
Bo
Spin1
Spin 2Bdip
rθ
Bo
dipdip .BµE 2−=
213
21 ˆˆ4
12cos3ˆzz
oD II.
πr
)θ(γγµH
−−=

29
In solid-state NMR, where the rotational motion is frozen, dipolar interaction gives direct information on the geometry of the molecule. In an isotropic liquid, however, the anisotropy is averaged out: 2.13 This happens because molecular tumbling changes the orientation of the relaxation vector on a time scale that is faster than the dipolar coupling. However, this means that the ensemble averaged dipolar Hamiltonian is zero at every moment, i.e. ( ) 0ˆ =tDH whereas, in relaxation theory, we deal with the expression of the type )2(ˆ)1(ˆ tDHtDH . t1 and t2, are the time points at which ( )tH is still correlated44.
In a more general fashion, the dipolar Hamiltonian may be rewritten as a scalar product of two tensors:
2.14
One tensor introduces the spin operator functions described by second-rank irreducible spherical tensor operators, T2,m(I1,I2). Irreducibility means that the tensor has no components of ranks 0 and 1. This tensor gives information on the spin operators. The other one incorporates the spatial functions, which are second-rank spherical harmonics, Y2,m(Ω(θ)). Spherical harmonics are components of a tensor defining the direction of the relaxation vector in spherical coordinates, Ω(t) = (θ(t)φ(t)), with respect to the laboratory frame. Ω(t) represents the time-dependent orientation of the relaxation vector. In fact, the C–H vector is fixed in the molecular frame, and it is the frame that varies with time with respect to the fixed-in-space laboratory coordinate system. The two coordinate systems are associated through Euler transformation41, 45.
2.2.2 Spectral density functions
As noted above, if rotational motion of the molecule has the suitable frequency, it can stimulate transitions in the eigenstate of the spins. The probability of a transition depends on the different frequencies that are provided for the system by thermal motion of the molecules. The probability of finding the desired frequency is given by the spectral density function, J(ω). J(ω) is obtained as the Fourier transform of the correlation function, C(τ), of the spherical harmonics, Y2m(Ω):
021cos3 2 =− ) / ( θ
( ) ( ) ( )( ) ( )21
2
2 2ˆ
213
221
40ˆ II
- m ,mT,θΩ,- mYm
r
γγ
π
µtDH ∑
=−=
h

30
2.15 < > denotes an equilibrium ensemble average41.
In infrared and Raman spectroscopy, Fourier transforms of band shapes can be associated with correlation functions, while in the case of NMR spectroscopy one needs J(ω) to describe relaxation processes41,45. Correlation functions tell us how “self similar” Y2m(Ω) is after a certain time. Correlation functions examine the values of Y2m(Ω) in short time intervals, comparable to the timescale of the fluctuations, and these values tend to be similar. If one probes after a longer time, the function has already lost its memory since this is the nature of being random and fluctuating.
In the simplest case, time correlation function is proportional to a single exponential, corresponding to the reduced Lorentzian spectral density function given by44:
2.16
τc is the correlation time of rank-two spherical harmonics and ω is a frequency. The correlation time represents the duration in which the orientation of the relaxation vector has changed by a significant amount, approximately 1 radian. In such a case, equation 2.16 is valid for isotropic reorientation of a rigid body, excluding any internal motion.
Spectral density with one characteristic time is Lorentzian. In the cases where the motion of the system needs to be expressed with more characteristic times, we would deal with more complex formulae. Calculation of the correlation function is based on a physical model for the rotational motion of the molecules in liquids. According to small-step Brownian rotational diffusion theory, the correlation time is related to the rotational diffusion constant, DR:
2.17 η is the solution viscosity, kB is the Boltzmann’s constant, T is the absolute temperature and r is the radius of the sphere. This is the case, however when the relaxation vector diffuses isotropically around the x, y and z molecular coordinate axes. Generally speaking, each system can be characterized by a rotational diffusion tensor in the molecular frame. Then, there will be three non-zero principal elements in the tensor, Dx, Dy and Dz. If the molecule is a symmetric top, that is, it diffuses at the same rate around the x and y axes the number of diffusion coefficients reduces to two quantities, , and Dz = D||. D_|_ defines the tumbling of the symmetry z axis, whereas D||
( ) ( ) ( )[ ] ( )[ ] dt etΩYΩY dteτC ωJ itω
- mmitω
- ∫∫∞
∞
∗∞
∞== 22 0
( )221
2
21
c
c
τω
τ
πωJ
+=
Tk
ηπr
Dτ
BR
c 68
61 3
==
⊥== DDD yx

31
describes the motion around the z axis. The spectral density function is then written as44:
2.18
If the molecule cannot be modeled as a rigid body, i.e. it enjoys internal
degrees of freedom, one should switch to the types of spectral densities where extra parameters are involved to identify the segmental motions in the system. One of the very popular ones is the model-free approach of Lipari and Szabo46, 47. In this model the overall reorientation, described by a global correlation time, τM is considered to be either isotropic or anisotropic while some part of the system is undergoing fast internal motions, characterized by a local correlation time, τe. A generalized order parameter, S, defines the degree of spatial restriction. Order parameter lies in the range of 0≤ S2 ≤1, in which lower values indicate higher freedom for internal motion. The relevant spectral density is written as44:
2.19
where τ-1 = τ e-1 + τ M
-1. If the internal motion is very slow, so that τe-1
approaches zero, equation 2.19 reduces to the same form as equation 2.16. Sometimes the relaxation data cannot be accounted for by the simple two-parameter Lipari-Szabo model. Clore et al. 48 proposed a model, originally for interpretation of 15N relaxation data in proteins, to tackle this issue. In their approach two types of local motion, slow and fast, are allowed, each characterized by an order parameter and a local correlation time. The spectral density considering and isotropic overall reorientation is: 2.20
2.2.3 Relaxation Parameters
Relaxation parameters, measured using NMR spectroscopic methods, are the macroscopic properties that provide the link to the properties at molecular
( )( )
+
−+
+=
22
2
22
2
1
1
12
1
τω
τS
τω
τS
πωJ
M
M
( ) ( )( )
( ) ( )]
ωDD
DDθ
ωDD
DDθθ
ωD
Dθ[
πωJ
||
||
||
||
224
2222
22
22
42
42sin
43
5
5sincos3
6
61cos
4
1
2
1
++
++
++
+
++
−=
⊥
⊥
⊥
⊥
⊥
⊥
( )( )
( )( )
( )
+
−+
+
−+
+=
2
22
2
2
22
2
11
1
121
's
'sf
'f
'ff
M
M
ωτ
τSS
ωτ
τS
τω
τS
πωJ

32
level. They correspond to the rates at which the populations of nuclear energy states change or the coherence of individual magnetic moments is lost. From the spectral densities one can obtain the relaxation parameters. The equations below describe the case of 13C–H dipolar interaction if the 13C and 1H are defined as S and I, respectively44. 2.21a
2.21b 2.21c
T1 and T2 are the longitudinal and transverse relaxation rates, respectively and σIS is the cross-relaxation rate. Spectral densities involve three important frequencies, ωS, ωI and zero. Assuming the simple spectral density in equation 2.16, if the molecular reorientation is in a motional regime that is called extreme narrowing, where ω2
τC2 is much less than unity, then the
spectral density function and the relaxation parameters will be independent of the magnetic field strength. At longer correlation times, the product approaches unity, i.e. ωτC ≈ 1, and 1
1−T obtains a maximum value as well. In
contrast, with J(ω=0), 12−T contains a term proportional to the correlation
time. J(ωc) describes the contribution of a single transition, implying an energy exchange with the lattice, while the two other terms represents a cooperative energy exchange of both spins with the lattice. This behavior is the origin of another important phenomenon, namely cross relaxation, which takes place only when relaxation is through the dipolar interaction mechanism. The equations can become quite complicated with more than one mechanism for the relaxation.
If CSA contributes to the relaxation rate, the equation will be the sum of the rates induced by both mechanisms:
2.22a 2.22b
( ) ( ) ( )[ ]
( ) ( ) ( )
( )
( ) ( )[ ]
SI
ISIS
SISIISIS
SII
SSIIS
SISSIIS
π r
γγµb
ωωJωωJbπ
σ
ωωJ)J(ω
ωJωωJJbπ
T
ωωJωJωωJbπ
T
4
65
]3323
21
02[5
635
0
2
212
211
h−=
−−+=
+++
+−+=
+++−=
−
−
( ) ( )SSCSA
CSADip
ωJσBγπ
T
TTT
20
11
11
11
11
5
2=
+=
−
−−−
( ) σσ σ σ σσσ/σ yyxx_|_zz||_|_|| ===−= and32
:CSA symmetricaxially For

33
Considering the complexity of the spin system and the type of information desired, one can study the relaxation phenomenon from a classical, vector-model point of view or involve more complicated quantum mechanical concepts. The simplest approach is to treat an ensemble of isolated spins, using Bloch equations. In the absence of the radio-frequency field, relaxation processes are characterized by two first-order rate constants:
2.23a
2.23b
Mz
0 is the equilibrium macroscopic magnetic moment, which is the sum over all magnetic moments of individual spins, Mz is the component along the z direction of the laboratory frame, and M+ is the observable transverse magnetization in the rotating frame (the frame rotating at the Larmor frequency), induced by the influence of the r.f. field. R1 describes the recovery of the longitudinal magnetization to thermal equilibrium, or the return of the population of the energy levels to the Boltzmann distribution. R2 describes the decay of the observable magnetization to zero. The Bloch formulation can be the basis for experimental measurements of relaxation rates, which are discussed in next section.
.
Figure 2.3 Rate constant diagram for an interacting two-spin system. For interacting spins, relaxation parameters can be obtained using
Solomon equations. When a liquid sample is placed in the strong external magnetic field of Bo, spins are distributed between different energy levels. A relaxation process takes place by transition of spins between these energy levels. Figure 2.3 shows the rate constants between the Zeeman energy
( )( )( )
( )( ) ><−=
><
><−><−=><
++ tMR
dt
tMd
MtMRdt
tMdzz
z
2
01
W0
W2W1I
W1I
αIβS
W1S
W1S
βIαS
βIβS
αIαS
W0
W2W1I
W1I
αIβS
W1S
W1S
βIαS
βIβS
αIαS

34
levels of a system of two spin-1/2 nuclei, labeled as I and S; α and β denote the eigenstates of isolated spin-1/2 nuclei44. The rate constants, W1I and W1S, determine the rates of the transitions where only one type of spin is flipped, and the other two, W0 and W2 govern the transition where both spin types are involved42. Iz and Sz components are proportional to the population differences between the eigenstates. The rate of the change of populations yields differential equations for ∆Iz and ∆Sz as below:
2.24a
2.24b where
<Kz0> is the equilibrium value of the z component of the magnetizations.
ρI and ρS are equivalent to the R1I and R1S relaxation rate constants in the Bloch terminology, and σIS is the cross-relaxation rate constant for the exchange of magnetization between the two spins42. The cross-relaxation rate is related to the phenomenon of nuclear Overhauser enhancement, see below.
2.2.4 Experimental methods
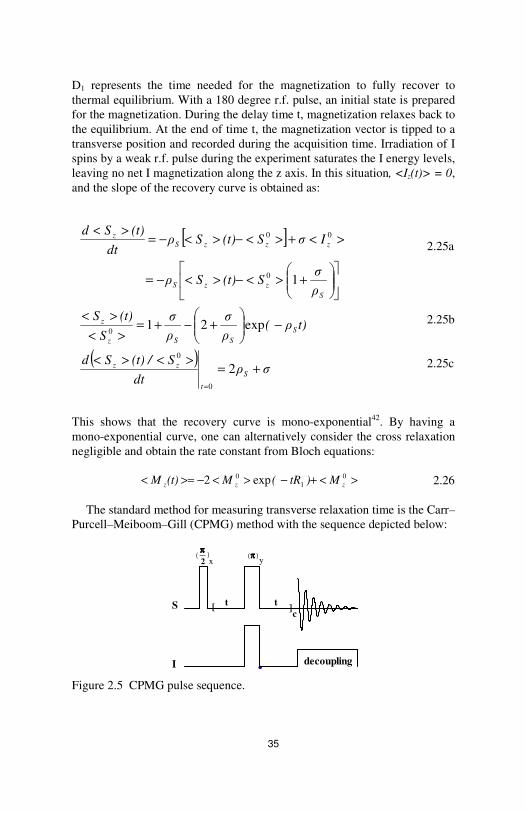
The conventional experiment to measure longitudinal relaxation time is the inversion recovery method. The pulse sequence is:
Figure 2.4 Pulse scheme of an inversion recovery experiment.
( ) I or S KKtK∆K
σ
ρ
ρ
zz
IS
S
I
=><−>=<
−=
++=
++=
0
02
21S0
21I0
WW
W2WW
W2WW
ππππ ππππ _ 2
t
I decoupling
S D1
ππππ ππππ _ 2
t
I decoupling
S D1
( )( ) ( )
( )( ) ( )t∆Iσt∆Sρ
dt
tSd
t∆Sσt∆Iρdt
tId
zISzSz
zISzIz
−−=∆
−−=∆
35
D1 represents the time needed for the magnetization to fully recover to thermal equilibrium. With a 180 degree r.f. pulse, an initial state is prepared for the magnetization. During the delay time t, magnetization relaxes back to the equilibrium. At the end of time t, the magnetization vector is tipped to a transverse position and recorded during the acquisition time. Irradiation of I spins by a weak r.f. pulse during the experiment saturates the I energy levels, leaving no net I magnetization along the z axis. In this situation, <Iz(t)> = 0, and the slope of the recovery curve is obtained as:
2.25a
2.25b 2.25c
This shows that the recovery curve is mono-exponential42. By having a mono-exponential curve, one can alternatively consider the cross relaxation negligible and obtain the rate constant from Bloch equations:
2.26 The standard method for measuring transverse relaxation time is the Carr–
Purcell–Meiboom–Gill (CPMG) method with the sequence depicted below:
Figure 2.5 CPMG pulse sequence.
><+−><−>=< 01
0 exp2 zzz M)tR(M(t)M
[ ]
( )σρ
dt
S(t) /Sd
t)ρ(ρ
σ
ρ
σ
S
(t)S
ρ
σS(t)Sρ
IσS(t)Sρdt
(t)Sd
S
t
zz
SSSz
z
SzzS
zzzSz
+=><><
−
+−+=
><
><
+><−><−=
><+><−><−=><
=
2
exp21
1
0
0
0
0
00
x ππππ(_ )
2 ( ππππ )
y
S [ t t
] c
I
decoupling

36
(t – πy –t)c is the refocusing sequence, the spin-echo, where any contribution from inhomogeneity of the magnetic field to the value of the R2 rate is eliminated. Again the R2 rate can be obtained using Bloch formalism:
2.27 However chemical exchange during the spin-echo sequence can affect the
value of the measured transverse relaxation rate 42: R2 = R2
Dip + Rex 2.28
Rex is the exchange contribution to the decay of transverse magnetization. If the exchange occurs between two equally populated sites, Rex depends on the first-order rate constant of the exchange process, kex.
Another relaxation parameter, complementary to R1 and R2 rates, is the NOE. The heteronuclear NOE is used, along with R1 and R2, to characterize molecular dynamics. Homonuclear (proton) NOE is used to determine the distance between two interacting spins in biological system, and thus the molecular structure. The cross-relaxation rate is proportional to the inverse sixth power of rSI. NOE is either characterized by σIS in Solomon equations or an enhancement factor, η. These two parameters are measured in transient and steady-state NOE experiments, respectively. In steady-state NOE measurement, irradiating the I spins for a time period much longer than the relaxation rates of both spins puts the I spins in a saturated situation. By setting d∆Sz/dt=0 and <Iz
0>=0 one obtains the steady-state I as below:
2.29a 2.29b 2.29c 1+η is the nuclear Overhauser enhancement factor. A pulse sequence for measuring NOE is shown in figure 2.649, which is called dynamic NOE. To measure the enhancement factor, the experiment is repeated twice with different values of the time interval t. In the first experiment, t is set to a negligible value, and in the second experiment it is defined to be much longer than the relaxation time of the S nuclei. Cross relaxation through dipolar coupling between I and S spins results in the NOE enhancement.
)tR(M(t)M zz 20 exp −>>=<<
( )
( )
ηγρ
γσS/S
S/IσS/S
IσSSρdt
Sd
SS
IISzz
zSzISzz
zISzzSz
ρ
+=+=
+=
=+−−=
11
1
0
0
000
00

37
Figure 2.6 Dynamic NOE pulse scheme.
Sometimes, a need of higher signal-to-noise for nuclei with small
gyromagnetic ratio, or a demand for more highly resolved spectra, has led to application of two-dimensional methods for measurement of relaxation parameters44. There is a two-dimensional analogue of the NOE experiment, called NOESY, for measuring the cross-relaxation. Since σIS is proportional, to the inverse sixth power of the interspin distance, for a quantitative measurement of the distance one should directly measure the cross relaxation rate. In this experiment, the intensity of the cross peak is under certain conditions proportional to σIS. The NOESY sequence is illustrated in figure 2.7a. During the first π/2–t1–π/2 block of the sequence, magnetization is labelled with a chemical shift and returned back to the z axis. During τM, the mixing time, magnetization transfer occurs via dipolar coupling, and is observed with the final read pulse.
Figure 2.7 a) NOESY and b) ROESY pulse sequences.
ππππ _ 2
S
I
decoupling
t
ππππ _ 2
I
ττττ m
spinlock
b)
t1 t2
ππππ _ 2
I
ττττ m
spinlock
b)
t1 t2
ππππ _ 2
I ττττ m
a) ππππ _ 2
I ττττ m
a)
t1t2
ππππ _ 2
I ττττ m
a) ππππ _ 2
I ττττ m
a)
t1t2

38
The problem with the NOESY experiment is that chemical exchange can also lead to cross peaks if the exchange rate is not slow compared to the mixing time. Depending on the magnitude of the overall correlation time, cross peaks from cross relaxation could have the same sign as the exchange cross peaks. Thus it is hard sometimes to distinguish between the cross peaks from exchange and cross relaxation. There is another sequence, rotating frame Overhauser effect spectroscopy (ROESY), with the advantage that the sign of the ROE cross peaks is always positive and therefore opposite in sign to the exchange cross peaks at all correlation times. In this experiment the cross relaxation between spin-locked spins using r.f. pulses is followed50, 51. The ROESY sequence is shown in figure 2.7b in which the spin-lock operates on the spins during the mixing time.
Heteronuclear single-quantum correlation spectroscopy (HSQC) is another two-dimensional method that can be used for measuring relaxation parameters, using indirect detection of the less sensitive nucleus. In these methods, magnetization transfer to more sensitive nuclei through scalar couplings results in better resolution and shorter experimental time44. A general form of the pulse sequence is presented in figure 2.852.
Figure 2.8 The general pulse scheme of the HSQC type experiments. To perform T1 measurements, the relaxation delay block is an inversion recovery. For measuring T2 or T1ρ it is replaced with a train of π pulses or spin-lock respectively. For NOE measurement, two experiments are done with one of them containing a 1200 pulses at the beginning of I channel and a long “Relaxation delay” to perform the NOE saturation. In the second experiment “Relaxation delay” is set to zero and the 1200 pulse is off. The pulse sequences contain some INEPT (insensitive nuclei enhanced by polarization transfer) blocks. In INEPT, the I spin polarization is transferred
Relaxation delay
t1
t2
Dec.
Refocused INEPT Reversed INEPT
I
S
Relaxation delay
t1
t2
Dec.
Refocused INEPT Reversed INEPT
I
S

39
to S nuclei. The enhancement ratio is ±γI / γS. The next part of the sequence is a relaxation delay. It may contain a π pulse to measure T1, a train of π pulses for T2 measurement or a spin-lock for measuring T1ρ. A reversed INEPT block returns the magnetization back to the I nuclei channel for detection.
2.3 Chemical exchange
The resonance positions of nuclei in NMR spectroscopy are sensitive to the magnetic environments. Changes in magnetic environment of the nuclei owing to some dynamic process are reflected in the NMR spectrum53. NMR provides the opportunity of studying the exchange process without perturbing the chemical equilibrium42. That is why it is called chemical exchange. One can classify chemical exchange in different groups. In these groups, spin systems can be coupled or uncoupled, and the exchange can be mutual or non-mutual. The non-mutual exchange refers to the case where two sites are not chemically equivalent. Exchange can also be either intermolecular, that is between sites in different molecules, or between different conformations of the same molecule53.
Exchange can affect the relaxation time measurements. If the spin–lattice relaxation time of a spin is different at two sites but the peaks are not resolved, the observed relaxation rate will be a weighted average of the two relaxation rates in the absence of the exchange54. If the exchange is fast enough, the curve will be single exponential.
Exchange will influence the relaxation rates even in the slow exchange. In the case of uncoupled spins, modified Bloch equations are used, which are called Bloch-McConnell if we deal with chemical reactions44. For a two-site chemical exchange, the first-order chemical reaction is described as:
The rate laws in matrix format can be formulated as42:
2.30
The modified Bloch equation can be written by introducing the matrix above:
k1
k-1
A1 A2
[ ]( )[ ]( )
[ ]( )[ ]( )
−
−=
−
−
tA
tA
kk
kk
tA
tA
dt
d
2
1
11
11
2
1

40
2.31 Here, MA(t) and MA
0 for species in the two sites, are the magnetizations at time t and at equilibrium, respectively, and RA values for 1 and 2 are the corresponding spin–lattice relaxation rates. The effect of exchange on spin–spin relaxation time is briefly discussed in the previous section.
Depending on the time scale of the exchange process, it can be measured using different techniques. In the coalescence regime, where two signals from two sites merge into one, the classical line-shape analysis is the technique of choice. This is the region where the line shape is most affected by the exchange. In the fast exchange region the signal is a single Lorentzian but it is still broadened by the exchange. However this broadening is comparable to the natural line-width and the broadening due to magnetic field inhomogeneity. Thus the contribution of exchange can be measured with a T2 experiment. In the presence of exchange, T2 is in certain situations dependent on the time intervals between the refocusing π pulses in the CPMG experiment. The equation for a two-site fast exchange is given by44:
2.32
T2 is the apparent spin–spin relaxation time, τcp presents the time intervals in the CPMG experiment, pA and pB are the populations of the two sites, kex is the exchange rate, and 2δ is the frequency difference between the signals from two sites.
Figure 2.9 Pulse scheme for the selective inversion experiment. G stands for pulsed-field gradient.
∑×=
+
−−
−−=
−
−
ii
iA
A
A
A
A
A
A
A
A
A
A
(t)A
(t)AM(t)M
M
M
R
R
(t)M
(t)M
kRk
kkR
(t)M
(t)M
dt
d
i
0
0
11
11
2
1
2
1
2
1
2
1
2
1
0
0
( )
−+∞→= −−
2tanh
21211 21
21
2cpex
cpexBAcpcp
τk
τkδ)(pp)/τ(T/τT
ππππ_ 2 ππππ
S
G
ππππSelective pulse
Mixing time
ππππ_ 2 ππππ
S
G
ππππSelective pulse
Mixing time

41
In the slow–exchange regime, excellent rate data are obtained using selective inversion experiments. The pulse sequence is depicted in figure 2.9. During the first block of the sequence, which is based on the so-called excitation sculpting procedure, one site is selectively inverted. In the second block, during the mixing time, two processes can lead to magnetization transfer. The first one is the dipolar interaction between nearby nuclei, which may lead to NOE effects, and the second one is the transfer due to exchange process. The inverted signal travels to the second site through the exchange process, and a signal is detected from the second site by the last 900 pulse.
2.4 Translational diffusion
The random Brownian motion of the molecules or ions, driven by internal thermal energy, in isotropic liquids is called diffusion55. Diffusion measurement using NMR-based techniques has some advantages over other methods. NMR can monitor the random motion of an ensemble of particles, and therefore the diffusion itself, whereas many other methods rely on detecting a concentration gradient to probe the diffusion55. NMR is also able to investigate diffusion phenomena related to formation of guest-host complexes.
According to the Stokes-Einstein equation, diffusion in the isotropic solutions is related to the size of the molecule44:
D = (kBT)/6πηr 2.33
in which D is the molecular self-diffusion coefficient, kB is the Boltzmann constant, T is the absolute temperature, η is the viscosity of the solution and r is the hydrodynamic radius of a spherical particle. The relation between the temperature and diffusion is not linear, as it appears from the equation above, since the viscosity is sensitive to the temperature. Therefore an exponential variation of the diffusion coefficient with change of the temperature may be expected:
D = D∞ e-Ea /k
BT 2.34
Ea is the activation energy of translational diffusion in the bulk solution. In water, for instance, Ea is associated with the energy needed for breaking the hydrogen bonds.
There are two main ways in NMR by which diffusion coefficients can be obtained. The first one is the analysis of T1 relaxation data to obtain the correlation time of the molecule which, as discussed previously, is related to the diffusion coefficient of the molecule56, 57. There are some limitations in

42
using the first approach, such as assumptions considered in relaxation data analysis and the need for an exact value of the hydrodynamic radius, all of which makes the second approch a better choice.
The second approach is the pulsed-field gradient (PFG) method, which directly measures translational diffusion56, 57. If in addition to the static magnetic field, there is a spatially dependent magnetic field gradient, the Larmor frequency becomes spatially dependent too. The basis of the diffusion measurement is that a well-defined field-gradient can label the position of the spin. The simplest form of the “Stejskal and Tanner” or PFG sequence is depicted in figure 2.10. The first π/2 r.f. pulse rotates the equilibrium magnetization into the x-y plane. A gradient pulse of duration δ and strength g is introduced in the middle of period τ. At the end of τ, spins experience a phase shift originating both from the main field and the gradient.
Figure 2.10 The Stejskal–Tanner pulsed-field gradient sequence.
The following π pulse at the end of the first τ period reverses the precession sign. During the second τ period, another gradient is applied, of equal magnitude and duration as the first one. If the spins have not experienced any translational motion the two gradients cancel each other and all spins are refocused. In the presence of diffusion, the phases are distributed and the echo signal is weakened. Faster diffusion results in a weaker echo signal. In this sequence, the signal is attenuated by both T2 relaxation and diffusion. Normalization of the signal with respect to the signal obtained in the absence of the applied gradients gives the attenuated signal as56: E(2τ) = exp(- γ2g2Dδ2(∆ – δ/3)) 2.35
τ τ
G
S
g
δ
∆
x)
2π
( πy
τ ττ τ
G
S
g
δ
∆
x)
2π
( πy

43
where γ is the gyromagnetic ratio, and ∆ is the separation between the gradient pulses. Typically, δ is defined to be in the range of 0–10 ms, ∆ in the range of milliseconds to seconds, and g can go up to 20 T.m-1. To measure diffusion, keeping τ constant, a series of experiments are performed with varying δ or ∆.

44
3- Dynamics of cyclodextrins and cryptophanes studied by NMR
On account of inclusion phenomena some of the physical and chemical properties of the constituting components are modified or new characterizations emerge. In the first place the guest molecule confronts a new environment where its motional freedom is restricted. Molecules can behave quite differently when restrained within a molecular container with a defined shape, volume, and chemical environment. Among different methods and techniques that are used to elucidate inclusion complexes, NMR plays a key role in determination of stoichiometry, association constants and conformations covering both domains of structure and dynamics26, 58. A host-guest system is a dynamic entity. Commonly the host is considered as the frame of reference and guest movement with respect to the frame is studied. Consequence of translational motions of the guest is the association and dissociation processes. Even though NMR is sensitive to a broad range of dynamic processes frequencies, it is necessary to have a definition of NMR time scale. Fast molecular motions tend to average out some of the NMR parameters. For example if the life times of free and bound states in a complex is in order of 10-3 or shorter the chemical shifts of two species are averaged by the exchange process26, 59. Rates of association and dissociation in cavitands such as cyclodextrins are normally fast in NMR time scale60, 61. In contrast, owing to their cage-shape cavities, cryptophanes exhibit slow exchange12.
3.1 Stoichiometry and binding constant
A primary task in studying the host-guest systems in solution is to define the stoichiometry of the complex. For cyclodextrins the most commonly observed ratios are H:G = 1:1 and 2:1. Camphor:α-CD is an example of 2:1 ratio62 while metoprolol makes an 1:1 complex with α-CD63. However some parameters such as concentration and temperature can change the stoichiometry31. In the case of having a single stoichiometry for a complex, the common method employed in NMR is the Job’s method64, 65. The

45
approach is based on the continuous variation of the host and guest proportions in a series of samples with constant total concentration of host plus guest. The sample preparation should be done in a way to cover the whole range of zero to one ratio(X) of each species concentration to the total concentration. A plot of XGuest.∆p, where p is most commonly the chemical shift, versus XHost reveals the stoichiometry.
Quantification of binding constant, K, is the next step of investigating the complexes in solution. For a 1:1 stoichiometry complex, one can estimate K providing that the equilibrium concentration (in the case of neutral moieties) of each species in equation 1.1 is in hand. When the rates of formation and decomposition are fast some of the observed NMR parameters are population weighted average. The most common observed NMR parameter for estimation of binding constant is the chemical shift. There are two main methods, graphical and curve fitting. In graphical methods a linear relationship between the chemical shift and K is sought. For example 1:1 stoichiometry is described by rectangular hyperbola and there are some proposed solutions for that66, 67. In curve fitting methods binding isotherms are calculated using the known stoichiometry and compared to the experimental data using fitting procedures68, 69.
Direct measurement of diffusion coefficient, D, is another NMR method that can be used to estimate K. It was first applied to the field of host-guest chemistry in the work of Stilbs70 in 1983 on the systems of alcohols in α- and β-cyclodextrins. It is now widely used even for large biological inclusion systems as well71, 72. Being related to the molecular size, the diffusion coefficient is a direct reporter of phenomena such as associations and aggregations. The advantage of measuring D over chemical shift is that all D parameters may be known for the system. Since the host molecule is usually much bigger than the guest it is assumed that D of host is not greatly changed due to complexation and the complexed guest has the same D as the host molecule.
When the host–guest complexation equilibrium has a slow exchange rate compared to the NMR time scale, the signals of the host and guest nuclei in the complex and free species appear at different chemical shifts. A typical approach of calculating the association constant is to extract the intensities of the signals from different species in the solution. A more precise method to extract the association constant is to measure the rates of the guest complexation and decomposition. Huber et al.73 obtained the binding constant of xenon in the cavities of two cryptophane derivatives using signal line width measurements. Brotin et al.74 studied a number of similar complexes using variable-temperature one dimensional magnetization transfer (1D-EXSY). Besides the binding constants interesting thermodynamics information is obtained 75.

46
3.2 Nuclear magnetic relaxation
As discussed in the previous chapter, relaxation parameters can provide valuable information on the structure and dynamics of a system under study. Measuring the relaxation rates due to the dipolar mechanism gives us the rotational correlation time, which characterizes overall molecular tumbling and intramolecular motions. In supramolecular complexes, the rotational correlation times of guest compounds are usually so increased that one can obtain equilibrium constants using the corresponding change of the relaxation rates32. This method was used with 81Br relaxation in cyclodextrins complexes76. 13C relaxation rates were used since 1976 to obtain correlation times for guest and host molecules in complexes by CDs. Behr discussed in his paper that, besides the thermodynamic stability and formation and dissociation kinetics, motional parameters of the species, composing a host guest system can give a wealth of information on the dynamic properties of the system77. Spin-label-induced 13C nuclear relaxation rates were used to determine guest molecule positions in α-CD with di-tert-butyl nitroxide complexes78. Proton longitudinal and transverse relaxation rates were used to determine the motion of (±)camphor guest molecules in both diastereometric complexes with α-CD 79.
Dynamics of small organic molecules complexing cryptophanes were studied in solid-state and solution using 13C and 2H80-82. In these studies effect of the cavity size on the efficiency of the complexation were investigated by calculating the motional parameters of the guest, host and the complex. Complexes of cryptophane-E with chloroform and dichloromethane in solution were investigated and the difference between the order parameter obtained for the guest molecule in each complex was discussed. It was found that these two guest molecules experience different degrees of restriction inside the same type of cavity. Moreover, they gave an estimate of the strength of dipolar coupling between the 13C and bonded protons in the guest molecules, directly using solid-state NMR methods. The 2H line shape analysis and nuclear spin relaxation studies of the same complexes were accomplished in solid-state. In the solid state, guest molecules are considered to be either encaged within the cavity or in the interstitial position, i.e. between the host molecules. It was observed that 2H spectra of encaged chloroform is broad and its correlation time is relatively long whereas the encaged dichloromethane showed a narrow line and a fast correlation time was calculated for it.
Relaxation studies were performed on other nuclei such as 129Xe. The cross relaxation effect between the proton and 129Xe nuclei was studied in complexes of Xe@cryptophanes 83. Although the relaxation time of 129Xe is hundreds of seconds, it is shortened to tens of seconds in complexing to cryptophanes or cyclodextrins.

47
NOEs bear structural information about interatomic distances in molecules and thus provide information about the molecular conformation. In supramolecular chemistry they can be used to probe the distances and relative orientation of the host and guest molecules in a complex. Particularly in CDs complexes, NOEs occurring between the H3 and H5 protons of the host molecule and guest protons indicates that the inclusion complex has formed32. NOE magnitude depends as well on the rotational correlation times, which characterizes overall molecular tumbling and intramolecular motions.

48
4- Discussion of the papers
This work is based on 5 papers in which the main focus of the work is on the study of the motional properties of small organic molecules complexed to larger host molecules using 13C spin relaxation measurements. Versatility of 13C relaxation measurements makes it possible to monitor even weak molecular interactions such as solvation effects. Binding to a larger moiety slows down the motion of the small molecule so that its motion falls out of the extreme narrowing regime. This allows us to collect more relaxation data by doing the measurements at different magnetic fields. The procedure is that one should adopt suitable spectral density functions that best model the motion of the molecules. Using defined spectral densities one can calculate the relaxation parameters. This is done by minimizing a chi-square function by least-square analysis. This function represents the difference between the calculated and experimental relaxation data. The calculations are performed using MATLAB 7.4.0 software. The motional parameters for the host molecule were also investigated. In all the papers it was assumed that the effect of the complexation on the motion of the host molecule is negligible. The overall correlation time of the host molecule is of special interest when compared to that of the complexed guest. Behr and Lehn77 concluded that, for supramolecualr system, in addition to thermodynamic association constants, one can define dynamic coupling coefficients. Taken as the ratio of host correlation time to that of the guest, this parameter reflects the dynamic rigidity of the system, composed of the two entities.
However, as discussed in previous chapters, the relaxation parameters are affected by the exchange going on in the systems. The stoichiometries of the systems that we chose were already studied in other research groups to be 1:1. In those cases where the data were not consistent with this stoichiometry higher ratios were examined as well, especially in the cases where the experimental conditions were somewhat different from those reported in the literatures.
Depending on the timescale of the exchange one should exclude the effect of the exchange in different ways. In fast exchange, where the exchange rates are much faster than the relaxation rates, the observed relaxation data, which are population averaged, can be resolved into the free and bound contributions if one has the information of the molar fraction of each species. A common approach is to extract the association constant of the reaction. Among several approaches, in present works, it was made use of

49
translational diffusion measurements using PFG NMR spectroscopy. In the slow exchange, the rates of the exchange are comparable to the relaxation parameters. In this regime one obtains separate relaxation data for the signals from the bound and free guest molecules but the rate contributes to the relaxation rates. This is evident by observing bi-exponential curves in the inversion recovery experiments. In this case the exact values of the forward and backward exchange rates are needed. By introducing them into modified Bloch equations one obtains the pure relaxation data in the absence of the exchange.
4.1 Papers I-II
These two papers deal with complexes of cryptophanes with dichloromethane and chloroform molecules. In the first paper complexes of dichloromethane with cryptophane-A, -223 and -233 are studied. These three molecules basically have the same structural shape with the difference that the cavity size increases from cryptophane-A to -233 by having different length of the linkers. This, on the other hand, results in a decreased symmetry of the cavity area. Despite the exchange is slow in the chemical shift timescale it turned out that it is still comparable to the scale of the relaxation time. Thus the measurement conditions were changed in the direction where the relaxation rates were dominant over the effect of the magnetization transfer due to exchange.
Relaxation parameters of dichloromethane were measured at different magnetic fields in solution for the first two hosts and in solid for the largest molecule. For the solution part, dipolar interaction was considered as the dominant relaxation mechanism for the guest whereas the host relaxation parameters show some evidence of CSA for aromatic carbons. Measuring the dipolar coupling constant using solid-state method complements the study of the three cryptophane complexes.
In Table 4.1 the order parameters obtained for dichloromethane in different cryptophanes is summarized. In this table some results from previous studies on cryptophane-E81 are also included. The order parameters of dichloromethane in different cryptophanes imply that it can gain lower motional restriction in the larger cavities. The outcome of this study was that in this series of cryptophanes the cavity size plays a central role in the complexation. Result of the solid-state study also confirms this conclusion.

50
Table 4.1 Order parameters and dipolar coupling constants for the bound dichloroemthane to cryptophanes.
Host/temperature S2 Motionally-averaged Cavity volume DCC, kHz pm3·10-6 Solution Solid-state Cryptophane-A/223K 0.46±0.06 14.2±0.9 - 95 Cryptophane-223/233K 0.20±0.03 9.4±0.7 - 102 Cryptophane-233/283K - - 8.2±1.0 117 Cryptophane-E/273K 0.02±0.003 3.2±0.3 3.3 121
The thermodynamic parameters of the complexes of dichloromethane
with cryptophane-A and -223 are presented in Table 4.2 Both complexes are enthalpy-favoured and enthropy-unfavoured, which is in fact the case in most of the inclusion complexes.
In order to involve the thermodynamic information on the complexes of cryptophane-E in the conclusion above we performed some variable temperature studies on chloroform and dichloromethane complexing cryptophane-E. In this way one can also compare the results of two different guests with different sizes and symmetry properties that interact with the same cavity. The thermodynamic quantities for all the complexes are summarized in Table 4.2. Table 4.2 Thermodynamic parameters for the cryptophane comeplexes.
Complexes ∆H0, kJ.mol-1 ∆S0, kJmol-1
CH2Cl2@ cryptophane-A -16 -24 CH2Cl2@cryptophane-223 -24 -52 CH2Cl2@cryptophane-E -26 -56 CHCl3@cryptophane-E -71 -192
A higher gain in the enthalpy of the larger guest in the largest cavity can be attributed to a better ratio of the guest to host volume.
4.2 Papers III-V
In this group of papers, container type molecules were studied as host molecules. Complexes of α-cyclodextrin with quinuclidine and 1,7-heptanediol, and β-cyclodextrin with adamantanecarboxylic acid were investigated. The interior of the cyclodextrins is hydrophobic32 and the solvent in which the complexation occurs is a mixture of deuterated water and DMSO. The first distinction between these systems and those mentioned in 4.1 is the fast kinetic of the exchange between the host cavity and the

51
solvent medium. This indicates that the complexes are less kinetically stable. One of the reasons might be the truncated cone-shape of the cyclodextrins that compared to cage-like cavity of the cryptophanes less hinders the complexation–decomposition of the guest molecule. Furthermore, depending on the interactions between guest-host, guest-solvent and guest-guest, guest molecule can penetrate with different depth into the host cavity. This is qualitatively studied by NOESY and ROESY methods. Having hydrophilic groups such as carboxylic acid group in adamantanecarboxylic acid or the hydroxyl group in diol leaves the chance of being in contact with the solvent molecules through hydrogen bonding.
Since the exchange was fast in all three complexes, translational diffusion coefficient measurements were used to estimate the association constants. Stoichiometries of all complexes were adopted from the literatures. In the case where the diffusion data did not fit to the assumed stoichiometry, other ratios of host to guest were examined. The binding constant obtained for the AdCA@β-CD complex were high at 25° and 0° C, indicating the high thermodynamic stability of this complex. In contrast, very low association constant was calculated for the quinuclidine@α-CD, and an intermediate value for the association constant of the diol@α-CD was obtained at 15° C. 13C longitudinal relaxation times and NOE enhancements were measured using conventional methods at several magnetic fields. Using the association constants, one can estimate the population of the free and bound guest molecules and the pure relaxation parameters for each group of the molecules is obtained. Reorientation of adamantanecarboxylic acid, bound to the host cavity, at 25° C is described using model-free approach, using a fixed value for overall correlation time, equal to that of host, and axially symmetric model. Comparing the rotational diffusion coefficients of AdCA in the bound and free forms shows that, due to the complexation, anisotropy of its motion is increased to a high degree, implying that, despite high association constant of the complex, the guest molecule freely rotates inside the cavity of the host molecule around its symmetry axis that coincides with that of the host. At lower temperature of 0°C it is no longer possible to model the reorientation of the bound guest using model–free formalism, and thus the Clore model, which is an extension to the model-free, is used to account for one more local motion for AdCA. It is assumed that, besides rotating around the symmetry axis, molecule is rocking to sides inside the cavity.
Owing to the low association constant of the quinuclidine@α-CD, the complex solution was prepared with a higher concentration of the host compound. Thus we decided to account for the effect of the increased viscosity compared to the sample with zero concentration of the host. Analysis of the data after viscosity correction revealed that 13C spins in quinuclidine are sensitive to the solution composition whereas the composition effect on the host spins was negligible. Therefore each sample

52
was treated separately when analysing the relaxation data. In a similar fashion, data were analysed using Lipari-Szabo and axially symmetric models, and the latter model showed that the motional anisotropy of quinuclidine is increased upon complexation with α-CD. Again, the guest molecule is free to rotate around its symmetry axis inside the cavity. Another interesting feature of the application of the axially symmetric model in both studies was that, using the perpendicular rotational correlation time of the bound guest, one can obtain an overall correlation time which is quite similar to that of the host. This is an approval of the assumption of using the overall correlation time as a fixed parameter in Lipari-Szabo analysis. This, as well, provides an estimate of the coupling effect of the complexation on the motion of the components.
In the last study a guest molecule with different geometry and flexibility features than the former guest molecules was studied in complex with α-CD. This study was accomplished semi-quantitatively since the motion of the guest molecule was rather complicated to be simply studied using common methods, explained above. The Lipari-Szabo analysis of the data showed that fixing the overall correlation time of the bound diol at the value of the host does not produce a correct trend for the calculated relaxation parameters, in comparison to the experimental ones. Thus, in a next attempt, an unconstrained Lipari-Szabo study is accomplished that yields a global correlation time much shorter than that of the host. However the trend of the calculated relaxation parameters is in agreement with the experimental data. The best fit, however, was obtained using the Clore model (see equation 2.20).

53
Acknowledgment
During the last few years, I have derived enormous personal and scientific benefit from my time spent at FOOS, specifically at physical chemistry, both from the people who work here and the environment that they have created. First and foremost, I would like to thank Jozef for his patience, continuous support and help, and the science he taught me throughout the whole period of my PhD. I am especially grateful to Arnold, Mattias, Dick, Sasha, Atto, Lena, Magnus, Zdenek, Jüri, Piotr, Peter Damberg, Leila, Torbjörn, Kristina Romare, Ann-Britt, Per-Erik and Eva for all that I have learned from them and the whole lot of help they have given me. Special thanks to Fatemeh, Katja, Andy, Zoltan, Dmytro, Alireza, Vasco, Isabel, Martin, Johan, Jonas, Ken, Adolfo, Shahriar, Emiliana, Danuta, Sasha, and rest of the friends for their friendship, help and the great times we had together. I also would like to acknowledge Jöran Karlsson and Vladislav Orekhov at the Swedish NMR center in Göteborg University, and Thierry Brotin and Jean-Pierre Dutasta at ENS Lyon for their help during the time I spent there to use their facilities. At last I would like to express my thankfulness to Shahram and my parents and siblings in Iran without whom none of this work would have been possible.

54
References
(1) Hoeben, F. J. M.; Jonkheijm, P.; Meijer, E. W.; Schenning, A. P. H. J. About Supramolecular Assemblies of Pi-Conjugated Systems. Chem.Rev. 2005, 105, 1491-1546.
(2) Cram, D. J.; Cram, J. M. Host-Guest Chemistry. Science 1974, 183, 803-809.
(3) Lehn, J.-M. Supramolecular Chemistry: Concepts and Perspectives. (Weinheim, VHC), 1995.
(4) Pons, M.; Millet, O. Dynamic NMR Studies of Supramolecular Complexes. Prog. Nucl. Magn. Reson. Spectrosc. 2001, 38, 267-324.
(5) Houk, K. N.; Leach, A. G.; Kim, S. P.; Zhang, X. Binding Affinities of Host-Guest, Protein-Ligand, and Protein-Transition-State Complexes. Angew. Chem. Int. Ed. 2003, 42, 4872-4897.
(6) Cramer, F. D. Inclusion Compounds. Revs. Pure Appl. Chem. 1955, 5, 143-164.
(7) Cram, D. J.; Cram, J. M.; Container Molecules and their Guests (Cambridge, Royal Soc. Chem.), 1997.
(8) Fyfe, M. C. T.; Stoddart, J. F. Synthetic Supramolecular Chemistry. Acc. Chem. Res. 1997, 30, 393-401.
(9) Collet, A. Cyclotriveratrylenes and Cryptophanes. Tetrahedron 1987, 43, 5725-5759.
(10) Cram, D. J. Cavitands: Organic Hosts with Enforced Cavities. Science 1983, 219, 1177-1183.
(11) Jasat, A.; Sherman, J. C. Carceplexes and Hemicarceplexes. Chem. Rev. 1999, 99, 931-967.

55
(12) Collet, A.; Dutasta, J. -P.; Lozach, B.; Canceill, J. Cyclotriveratrylenes and Cryptophanes: Their Synthesis and Applications to Host-Guest Chemistry and to Design of New Materials. Top. Curr. Chem. 1993, 165, 103-129.
(13) Warmuth, R. Inner-Phase Stabilization of Reactive Intermediates. Eur.J.Org.Chem. 2001, 423-437.
(14) Weber, E.; Josel, H. P. A Proposal for the Classification and Nomenclature of Host-Guest-Type Compounds. J.Inclusion Phenom. 1983, 1, 79-85.
(15) Davies, J. E. D. Species in Layers, Cavities and Channels (Or Trapped Species). J. Chem. Educ. 1977, 54, 536-539.
(16) Goshe, A. J.; Steele, I. M.; Ceccarelli, C.; Rheingold, A. L.; Bosnich, B. Supramolecular Recognition: On the Kinetic Lability of Thermodynamically Stable Host-Guest Association Complexes. Proc. Nal. Acad. Sci. U. S. A. 2002, 99, 4823-4829.
(17) Steed, J. W.; Atwood, J. L. Supramolecular Chemistry: A Concise Introduction. (Chichester, Wiley), 2000.
(18) Beer, P. D.; Gale, P. A.; Smith, D. K. Supramolecular Chemistry (Oxford, Oxford University), 1999.
(19) Ma, J. C.; Dougherty, D. A. The Cation-Pi Interaction. Chem.Rev. 1997, 97, 1303-1324.
(20) Sussman, J. L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic Structure of Acetylcholinesterase from Torpedo Californica: A Prototypic Acetylcholine-Binding Protein. Science 1991, 253, 872-879.
(21) Lehn, J. Supramolecular Chemistry - Scope and Perspectives Molecules, Supermolecules, and Molecular Devices (Nobel Lecture). Angew. Chem. Int. Ed. 1988, 27, 89-112.
(22) Pedersen, C. J. The Discovery of Crown Ethers (Nobel Address). Angew. Chem. 1988, 100, 1053-1059.
(23) Menon, S. K.; Hirpara, S. V.; Harikrishnan, U. Synthesis and Applications of Cryptands. Rev. Anal. Chem. 2004, 23, 233-267.

56
(24) Cram, D. J. The Design of Molecular Hosts, Guests, and their Complexes. J. Inclusion Phenom. 1988, 6, 397-413.
(25) Meier, W. M. Zeolites and Zeolite-Like Materials. Pure Appl. Chem. 1986, 58, 1323-1328.
(26) Dodziuk, H. Cyclodextrins and their Complexes. (Weinheim, Wiley), 2008.
(27) Dodziuk, H. Introduction to supramolecular chemistry. ( Boston, Kluwer Academic Publishers), 2001.
(28) Saenger, W. Cyclodextrin Inclusion Compounds in Research and Industry. Angew. Chem. 1980, 92, 343-361.
(29) Zabel, V.; Saenger, W.; Mason, S. A. Topography of Cyclodextrin Inclusion Complexes. Part 23. Neutron Diffraction Study of the Hydrogen Bonding in Beta -Cyclodextrin Undecahydrate at 120 K: From Dynamic Flip-Flops to Static Homodromic Chains. J. Am. Chem. Soc. 1986, 108, 3664-3673.
(30) Dodziuk, H.; Nowinski, K. Structure of Cyclodextrins and their Complexes. Part 2. do Cyclodextrins have a Rigid Truncated-Cone Structure? Theochem 1994, 110, 61-68.
(31) Rekharsky, M. V.; Inoue, Y. Complexation Thermodynamics of Cyclodextrins. Chem. Rev. 1998, 98, 1875-1917.
(32) Schneider, H.; Hacket, F.; Ruediger, V.; Ikeda, H. NMR Studies of Cyclodextrins and Cyclodextrin Complexes. Chem. Rev. 1998, 98, 1755-1785.
(33) Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743-1754.
(34) Gabard, J.; Collet, A. Synthesis of (D3)-Bis(Cyclotriveratrylenyl) Macrocage by Stereospecific Replication of a (C3)-Subunit. J. Chem. Soc., Chem. Comm. 1981, 1137-1139.
(35) Holman, K. T. Cryptophanes; Molecular Containers. Encyclopedia of Supramolecular Chemistry 2004, 340-348.
(36) Garel, L.; Dutasta, J.; Collet, A. Complexation of Methane and Chlorofluorocarbons by Cryptophane-A in Organic Solution. Angew. Chem. Int. Ed. 1993, 32, 1169-1171.

57
(37) Mecozzi, S.; Rebek, J.,Jr The 55% Solution: A Formula for Molecular Recognition in the Liquid State. Chem. Eur. J. 1998, 4, 1016-1022.
(38) Canceill, J.; Lacombe, L.; Collet, A. Analytical Optical Resolution of Bromochlorofluoromethane by Enantioselective Inclusion into a Tailor-made Cryptophane and Determination of its Maximum Rotation. J. Am. Chem. Soc. 1985, 107, 6993-6996.
(39) Slichter, C. P. Principles of Magnetic Resonance, with Examples from Solid State Physics. (New York, Harper & Row), 1963.
(40) Levitt, M. H. Spin Dynamics Basics of Nuclear Magnetic Resonance. (Chichester, Wiley), 2002.
(41) Smith, S. A.; Palke, W. E.; Gerig, J. T. The Hamiltonians of NMR. I. Concepts Magn. Reson. 1992, 4, 107-144.
(42) Cavanagh, J.; Fairbrother, W.; Palmer, I.,Arthur G.; Skelton, N. Protein NMR Spectroscopy: Principles and Practice. (San Diego, Academic Press), 1995.
(43) Lippens, G.; Jeener, J. The Dipolar Interaction Under all Angles. Concepts Magn. Reson. 2001, 13, 8-18.
(44) Kowalewski, J.; Mäler, L. Nuclear Spin Relaxation in Liquids: Theory, Experiments, and Applications. (New York, Taylor & Francis), 2006.
(45) Smith, S. A.; Palke, W. E.; Gerig, J. T. The Hamiltonians of NMR. Part IV: NMR Relaxation. Concepts Magn. Reson. 1994, 6, 137-162.
(46) Lipari, G.; Szabo, A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 1. Theory and Range of Validity. J. Am. Chem. Soc. 1982, 104, 4546-4559.
(47) Lipari, G.; Szabo, A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 2. Analysis of Experimental Results. J. Am. Chem. Soc. 1982, 104, 4559-4570.
(48) Clore, G. M.; Szabo, A.; Bax, A.; Kay, L. E.; Driscoll, P. C.; Gronenborn, A. M. Deviations from the Simple Two-Parameter Model-Free Approach to the Interpretation of Nitrogen-15 Nuclear Magnetic Relaxation of Proteins. J. Am. Chem. Soc. 1990, 112, 4989-4991.

58
(49) Kowalewski, J.; Ericsson, A.; Vestin, R. Determination of NOE [Nuclear Overhauser Enhancement] Factors using the Dynamic Overhauser Enhancement Technique Combined with a Nonlinear Least-Squares-Fitting Procedure. J. Magn. Reson. 1978, 31, 165-169.
(50) Bothner-By, A. A.; Stephens, R. L.; Lee, J.; Warren, C. D.; Jeanloz, R. W. Structure Determination of a Tetrasaccharide: Transient Nuclear Overhauser Effects in the Rotating Frame. J. Am. Chem. Soc. 1984, 106, 811-813.
(51) Bax, A.; Davis, D. G. Practical Aspects of Two-Dimensional Transverse NOE Spectroscopy. J. Magn. Reson. 1985, 63, 207-213.
(52) Peng, J. W.; Thanabal, V.; Wagner, G. Improved Accuracy of Heteronuclear Transverse Relaxation Time Measurements in Macromolecules: Elimination of Antiphase Contributions. J. Magn. Reson. 1991, 95, 421-427.
(53) Bain, A. D. Chemical Exchange. Mod. Magn. Reson. 2006, 1, 417-423.
(54) Zimmerman, J. R.; Brittin, W. E. Nuclear-Magnetic-Resonance Studies in Multiple-Phase Systems: Lifetime of a Water Molecule in an Adsorbing Phase on Silica Gel. J. Phys. Chem. 1957, 61, 1328-1333.
(55) Nicolay, K.; Braun, K. P. J.; de Graaf, R. A.; Dijkhuizen, R. M.; Kruiskamp, M. J. Diffusion NMR Spectroscopy. NMR Biomed. 2001, 14, 94-111.
(56) Price, W. S. Pulsed-Field Gradient Nuclear Magnetic Resonance as a Tool for Studying Translational Diffusion: Part 1. Basic Theory. Concepts Magn. Reson. 1997, 9, 299-336.
(57) Stejskal, E. O.; Tanner, J. E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288-292.
(58) Fielding, L. Determination of Association Constants (Ka) from Solution NMR Data. Tetrahedron 2000, 56, 6151-6170.
(59) Sandström, J. Dynamic NMR Spectroscopy. (New York, Academic Press), 1982.
(60) Yonemura, H.; Kasahara, M.; Saito, H.; Nakamura, H.; Matsuo, T. Spectroscopic Studies on Exchange Properties in through-Ring

59
Cyclodextrin Complexes of Carbazole-Viologen Linked Compounds: Effects of Spacer Chain Length. J. Phys. Chem. 1992, 96, 5765-5770.
(61) Berg, U.; Gustavsson, M.; Åström, N. Direct Observation and Rate of Interconversion of the 1:2 Complex between 1,4-Dimethylbicyclo[2.2.2]Octane and Alpha -Cyclodextrin by NMR. J. Am. Chem. Soc. 1995, 117, 2114-2115.
(62) Dodziuk, H.; Ejchart, A.; Lukin, O.; Vysotsky, M. O. 1H and 13C NMR and Molecular Dynamics Study of Chiral Recognition of Camphor Enantiomers by Alpha -Cyclodextrin. J. Org. Chem. 1999, 64, 1503-1507.
(63) Ikeda, Y.; Hirayama, F.; Arima, H.; Uekama, K.; Yoshitake, Y.; Harano, K. NMR Spectroscopic Characterization of metoprolol/cyclodextrin Complexes in Aqueous Solution: Cavity Size Dependency. J. Pharm. Sci. 2004, 93, 1659-1671.
(64) Job, P. Formation and Stability of Inorganic Complexes in Solution. Ann. Chim. Appl. 1928, 9, 113-203.
(65) Sahai, R.; Loper, G. L.; Lin, S. H.; Eyring, H. Composition and Formation Constant of Molecular Complexes. Proc. Nat. Acad. Sci. U.S.A. 1974, 71, 1499-1503.
(66) Mathur, R.; Becker, E. D.; Bradley, R. B.; Li, N. C. Proton Magnetic Resonance (P.M.R.) Studies of Hydrogen Bonding of Benzenethiol with several Hydrogen Acceptors. J. Phys. Chem. 1963, 67, 2190-2194.
(67) Hanna, M. W.; Ashbaugh, A. L. Nuclear Magnetic Resonance (N.M.R.) Study of Molecular Complexes of 7,7,8,8-Tetracyanoquinodimethane and Aromatic Donors. J. Phys. Chem. 1964, 68, 811-816.
(68) Hynes, M. J. EQNMR: A Computer Program for the Calculation of Stability Constants from Nuclear Magnetic Resonance Chemical Shift Data. J.Chem.Soc., Dalton Trans. 1993, 311-312.
(69) Salvatierra, D.; Diez, C.; Jaime, C. Host/guest Interactions and NMR Spectroscopy. a Computer Program for Association Constant Determination. J.Inclusion Phenom. Mol. Recognit. Chem. 1997, 27, 215-231.
(70) Rymden, R.; Carlfors, J.; Stilbs, P. Substrate Binding to Cyclodextrins in Aqueous Solution: A Multicomponent Self-Diffusion Study. J.Inclusion Phenom. 1983, 1, 159-167.

60
(71) Chien, W.; Cheng, S.; Chang, D. Determination of the Binding Constant of a Protein Kinase C Substrate, NG(28-43), to Sodium Dodecyl Sulfate Via the Diffusion Coefficient Measured by Pulsed Field Gradient Nuclear Magnetic Resonance. Anal. Biochem. 1998, 264, 211-215.
(72) Orfi, L.; Lin, M.; Larive, C. K. Measurement of SDS Micelle-Peptide Association using 1H NMR Chemical Shift Analysis and Pulsed-Field Gradient NMR Spectroscopy. Anal. Chem. 1998, 70, 1339-1345.
(73) Huber, J. G.; Dubois, L.; Desvaux, H.; Dutasta, J.; Brotin, T.; Berthault, P. NMR Study of Optically Active Monosubstituted Cryptophanes and their Interaction with Xenon. J. Phys. Chem. A 2004, 108, 9608-9615.
(74) Brotin, T.; Devic, T.; Lesage, A.; Emsley, L.; Collet, A. Synthesis of Deuterium-Labeled Cryptophane-A and Investigation of Xe-Cryptophane Complexation Dynamics by 1D-EXSY NMR Experiments. Chem. Eur. J. 2001, 7, 1561-1573.
(75) Hill, P. A.; Wei, Q.; Eckenhoff, R. G.; Dmochowski, I. J. Thermodynamics of Xenon Binding to Cryptophane in Water and Human Plasma. J. Am. Chem. Soc. 2007, 129, 9262-9263.
(76) Yamashoji, Y.; Fujiwara, M.; Matsushita, T.; Tanaka, M. Evaluation of Association between Cyclodextrins and various Inorganic Anions in Aqueous Solutions by Bromine-81 NMR Spectroscopy. Chem. Lett. 1993, 1029-1032.
(77) Behr, J. P.; Lehn, J. M. Molecular Dynamics of a-Cyclodextrin Inclusion Complexes. J. Am. Chem. Soc. 1976, 98, 1743-1747.
(78) Eastman, M. P.; Brainard, J. R.; Stewart, D.; Anderson, G.; Lloyd, W. D. Spin-Label-Induced Selective Carbon-13 Nuclear Relaxation: Structures of the Gamma -Cyclodextrin-Tempone and Alpha -Cyclodextrin-Di-Tert-Butyl Nitroxide Inclusion Complexes in Solution. Macromolecules 1989, 22, 3888-3892.
(79) Anczewski, W.; Dodziuk, H.; Ejchart, A. Manifestation of Chiral Recognition of Camphor Enantiomers by Alpha -Cyclodextrin in Longitudinal and Transverse Relaxation Rates of the Corresponding 1:2 Complexes and Determination of the Orientation of the Guest Inside the Host Capsule. Chirality 2003, 15, 654-659.
(80) Petrov, O.; Tosner, Z.; Csoeregh, I.; Kowalewski, J.; Sandström, D. Dynamics of Chloromethanes in Cryptophane-E Inclusion Complexes:

61
A 2H Solid-State NMR and X-Ray Diffraction Study. J. Phys. Chem. A 2005, 109, 4442-4451.
(81) Tosner, Z.; Lang, J.; Sandström, D.; Petrov, O.; Kowalewski, J. Dynamics of an Inclusion Complex of Dichloromethane and Cryptophane-E. J. Phys. Chem. A 2002, 106, 8870-8875.
(82) Lang, J.; Dechter, J. J.; Effemey, M.; Kowalewski, J. Dynamics of an Inclusion Complex of Chloroform and Cryptophane-E: Evidence for a Strongly Anisotropic Van Der Waals Bond. J. Am. Chem. Soc. 2001, 123, 7852-7858.
(83) Luhmer, M.; Goodson, B. M.; Song, Y.; Laws, D. D.; Kaiser, L.; Cyrier, M. C.; Pines, A. Study of Xenon Binding in Cryptophane-A using Laser-Induced NMR Polarization Enhancement. J. Am. Chem. Soc. 1999, 121, 3502-3512.

Filename: Final-jk-17August.doc Directory: C:\Documents and Settings\snikkhou.INORG\Desktop\thesis Template: C:\DOCUME~1\SNIKKH~1.INO\LOCALS~1\Temp\SU-
WordTemplate-1.dot Title: Avhandlingsmall Subject: Author: snikkhou Keywords: Comments: Creation Date: 2008-08-17 21:45:00 Change Number: 41 Last Saved On: 2008-09-22 15:59:00 Last Saved By: snikkhou Total Editing Time: 719 Minutes Last Printed On: 2008-09-22 16:00:00 As of Last Complete Printing Number of Pages: 70 Number of Words: 19 977 (approx.) Number of Characters: 105 880 (approx.)


















