
Neuromuscular Engineering 1 Neuromuscular Engineering & Technology IsoCOG.

New genetic therapies for inherited neuromuscular diseases
Francesco MuntoniDubowitz Neuromuscular Centre
UCL Institute of Child Health& Great Ormond Street Hospital
London, UK

Francesco Muntoni: disclosuresDuchenne trials• CI of AON clinical trials with AVI / Sarepta. • PI of Prosensa / Biomarin sponsored AON trials• PI of PTC phase II and III sponsored trials. • PI of Pfizer phase II clinical trial (myostatin inhibition)• CI of Summit Phase I and II trials. • CI of Esperare Phase I trial
Spinal Muscular Atrophy trials
• PI of Trophos SMA III trial• PI of Ionis/ Biogen AON Phase III study
• Other• Avexis, Biogen, Sarepta, Pfizer, PTC, Roche, Summit, Wave ad-hoc SAB

Dramatic impact of genetic therapies for Duchenne muscular dystrophy (DMD)and Spinal Muscular Atrophy (SMA)
Dealing with mutant RNA. Splice-switching antisense oligonucleotides for:
- exon skipping for DMD deletions- exon inclusion in SMA
Replacing genes with gene therapy
- AAV gene therapy for DMD- AAV gene therapy for SMA type I

Topics discussed
• Why antisense oligomers for DMD and SMA?
• Emerging data from AAV gene therapy
• Implications for clinical practice

• Most common muscular dystrophy of childhood
• X-linked, 1 in 3,500 male births• 26,000 new cases each year, ~ 100 in UK• De novo mutations hamper genetic
efforts to reduce incidence• Economic cost: at least $ 120,000 per
year
The two most common neuromuscular diseases in children
DMD SMA
• Most common lethal genetic disorder of childhood
• Autosomal recessive, 3 different levels of severity
• Cumulative incidence: 1 in 6,500 -1,10.000 births
• Heterozygous carrier frequency: 1:37 – 1: 60 in Caucasian populations

• Onset in first years of life, however mean age of diagnosis 4.5 years
• Progressive weakness affecting legs> arms• Inability to run, progressive difficulties
raising from the floor, going upstairs• Grossly elevated creatine kinase (CK)• Loss of ambulation 9.513.5 years (steroids)
Main clinical features
DMD SMA 1• Onset by 6 months of age• Diagnostic delay of ~2.5 months• Sitting posture never acquired• Severe respiratory muscle weakness• Feeding difficulties• Mean age of survival 9 months

• Lack of sarcolemmal protein dystrophin• Protects muscle from contraction
induced injury• Absent dystrophin progressive
muscle destruction• Muscle is replaced by connective tissue
and fat
PathogenesisDMD SMA
• Depletion of SMN protein• Ubiquitously expressed protein
necessary for survival of motorneurons
• Expressed in the cytoplasm, in nuclear structures GEMS, and in axons

DMD genetics• 65% out-of-frame DMD deletions• 10% out-of-frame duplications• 10-15%% nonsense mutations• Remaining: small mutations
Becker MD genetics
In framedeletions

Antisense oligonucleotides to
induce exon skipping 52
Antisense oligonucleotides: to modify splicing of the DMD gene
Skipping exon 51: 15% of all DMD boys carrying deletions

Ann Neurol. 2013;74:637-47.
2’Omethyl AON(Prosensa/GSK /Biomarin): drisapersen
Morpholino PMO AON(Sarepta)eteplirsen
AON exon 51 skipping therapy progress2007 2008 2009 2010 2011 2012 2013 2014 2015 2016 2017 2018
Clinical Trials

Muscle biopsy: Baseline vs. Week 180 Patient 01015: Eteplirsen-Treated Week 180Patient 01015: Baseline
Mendell et al, 2015
Morpholino (PMO) AON

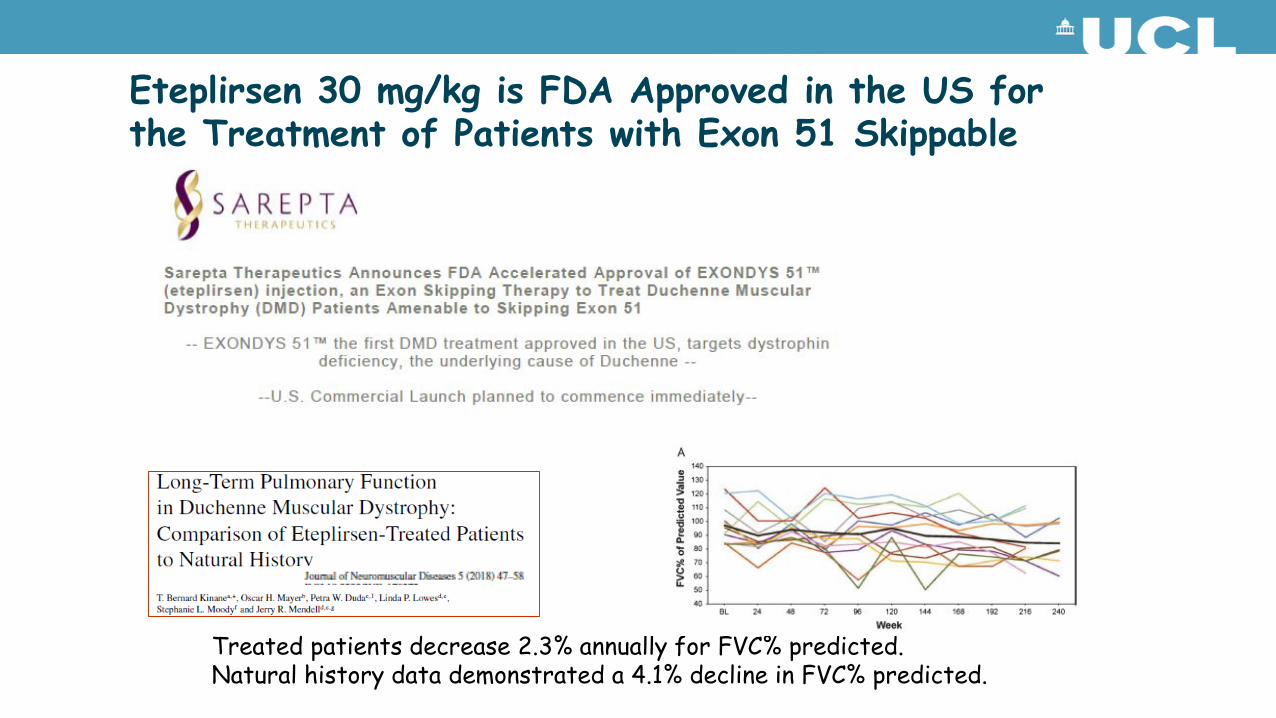
Eteplirsen 30 mg/kg is FDA Approved in the US for the Treatment of Patients with Exon 51 Skippable DMD
Treated patients decrease 2.3% annually for FVC% predicted.Natural history data demonstrated a 4.1% decline in FVC% predicted.

AON for DMD: rapidly evolving field:
1. Targeting additional exons to extend this approach to patients with other deletions
2. Next generation AONs

Targeting other DMD exons: SKIP-NMD Consortium
• Advocacy groups, research, healthcare providers and industry
• Francesco Muntoni project lead (UCL). Individual site leads are as follows:
• Institut de Myologie (Laurent Servais)• University of Newcastle upon Tyne (Volker Straub)• Università Cattolica del Sacro Cuore (Eugenio Mercuri)• Royal Holloway & Bedford New College (George Dickson)- in
vitro sequence selection & steering
Consultants for Research in Imaging & Spectroscopy

16
Increase in Sarcolemma localization of newly expressed dystrophin
BASELINE BIOPSY
ON-TREATMENTBIOPSY: Wk 48
Note: Indirect immunofluorescence staining of tissue cryosections was performed using MANDYS106 and anti-laminin 2 alpha antibodies
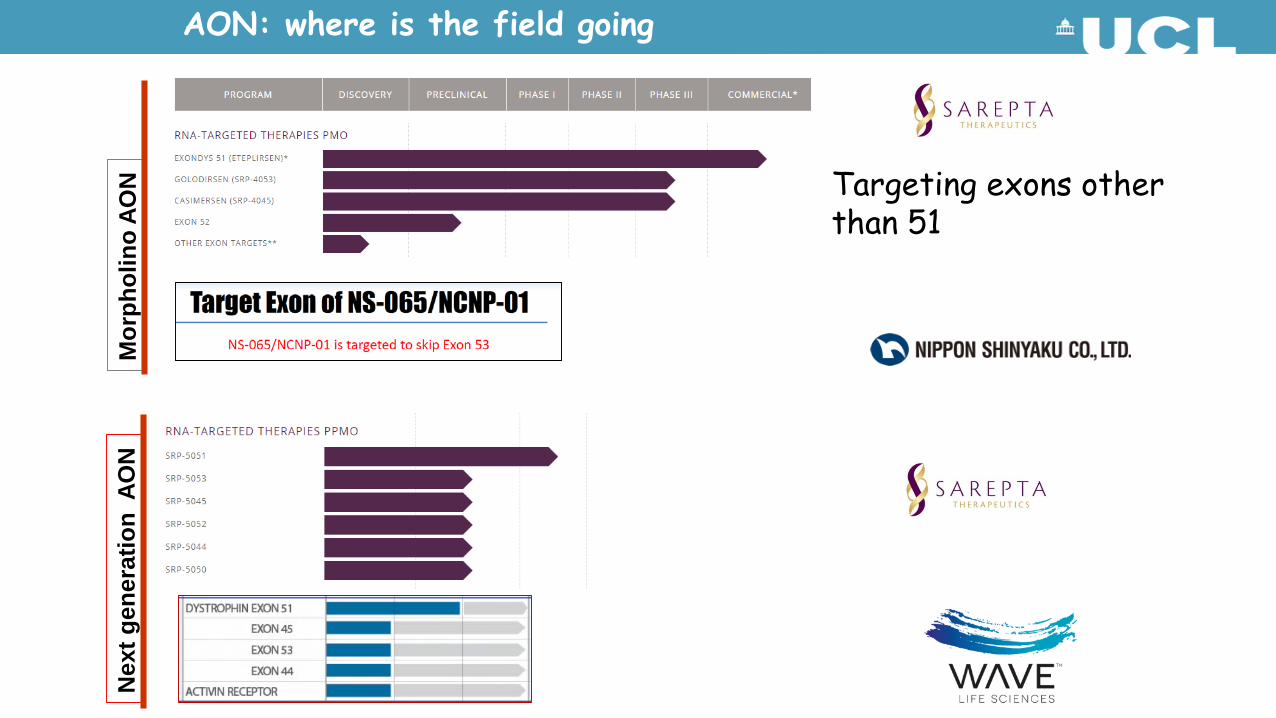
Targeting exons other than 51
Mor
phol
ino
AON
Nex
t gen
erat
ion
AO
N
AON: where is the field going
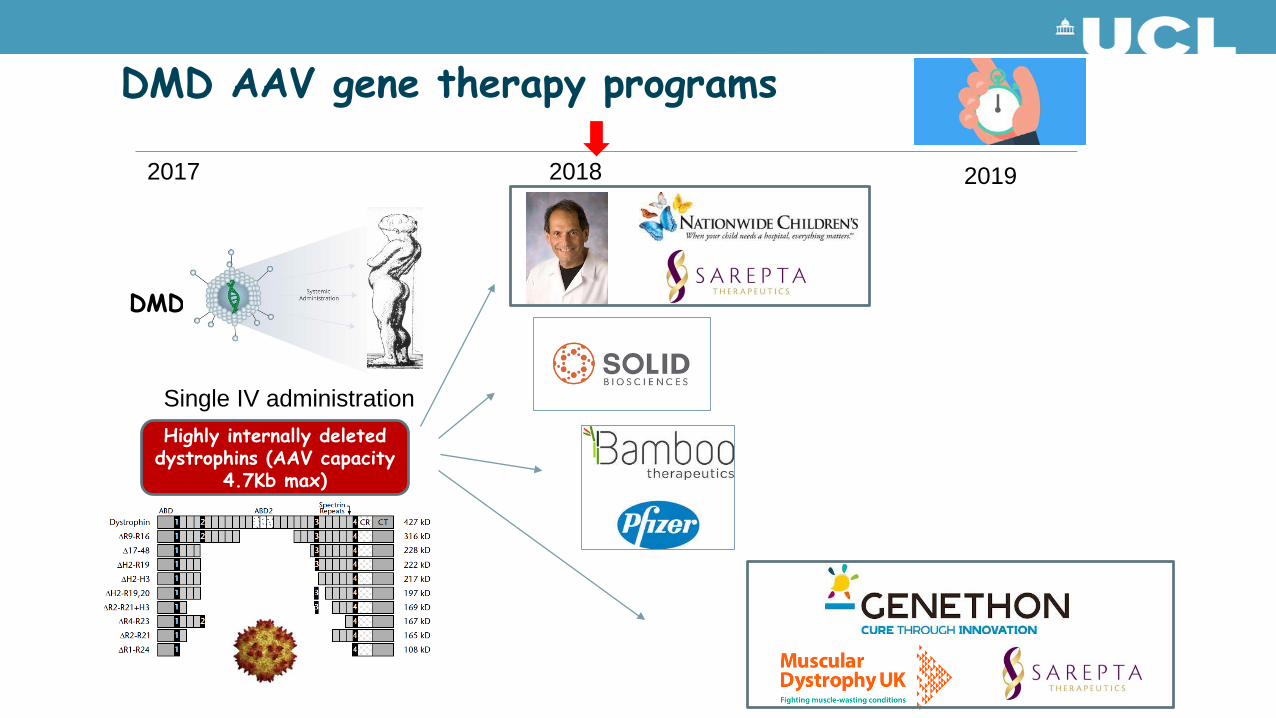
DMD
DMD AAV gene therapy programs
2017 2018 2019
Highly internally deleted dystrophins (AAV capacity
4.7Kb max)
Single IV administration


Full-length
Chromosome 5
SMN1
SMN2
SMN-FL SMND7 SMN-FL
SMN1SMN2
Full-length38kD
Truncated & less stable
~10% ~90% ~100%
Gene
mRNA
Protein
SPINAL MUSCULAR ATROPHY
AONs to induce exon 7 inclusion
SMN2 Gene
1 2 2 3 4 5 6 8 SMN2 mRNA
1 2a 2b 3 4 5 6 7 8
C to T
Functional Protein
7
IONIS-SMNRx

Intratechal administration of the antisense oligonucleotide spinraza
• AONs do not cross the blood brain barrier• Intratechal administration required• After a loading phase (4 IT injection in 2 months),
the long half life of the AON enables infrequent dosing
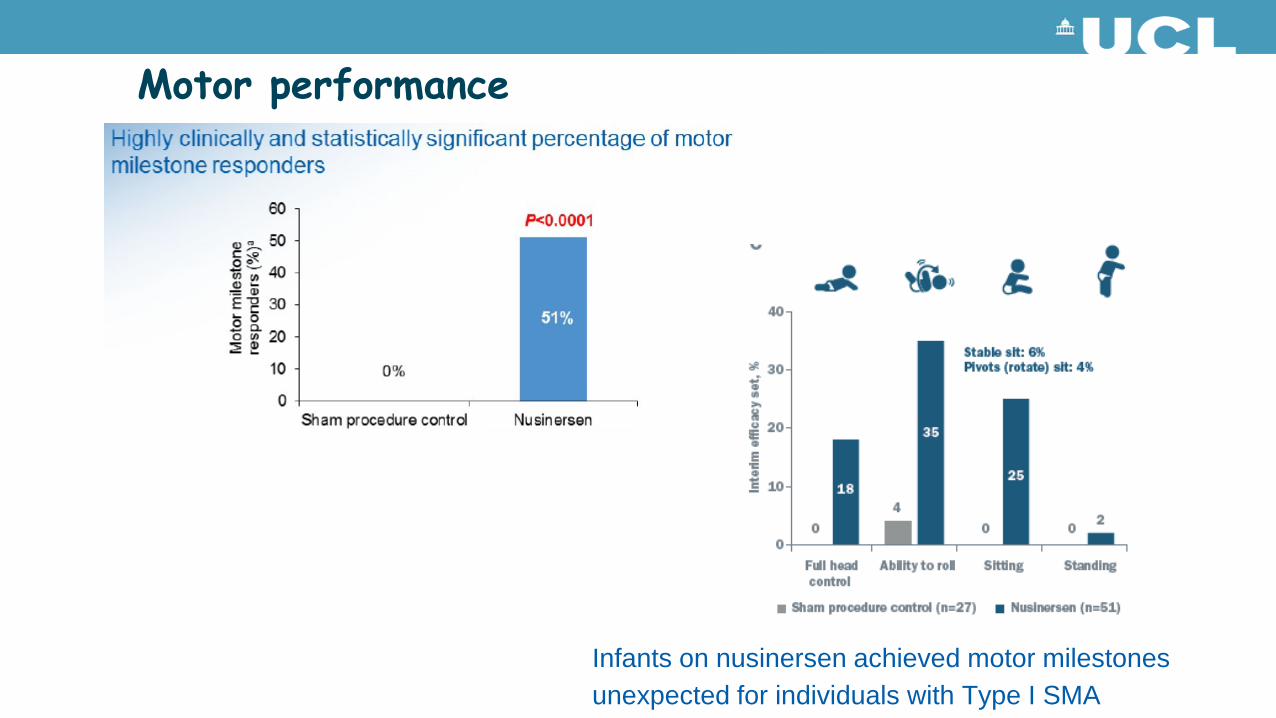
Motor performance
Infants on nusinersen achieved motor milestones unexpected for individuals with Type I SMA
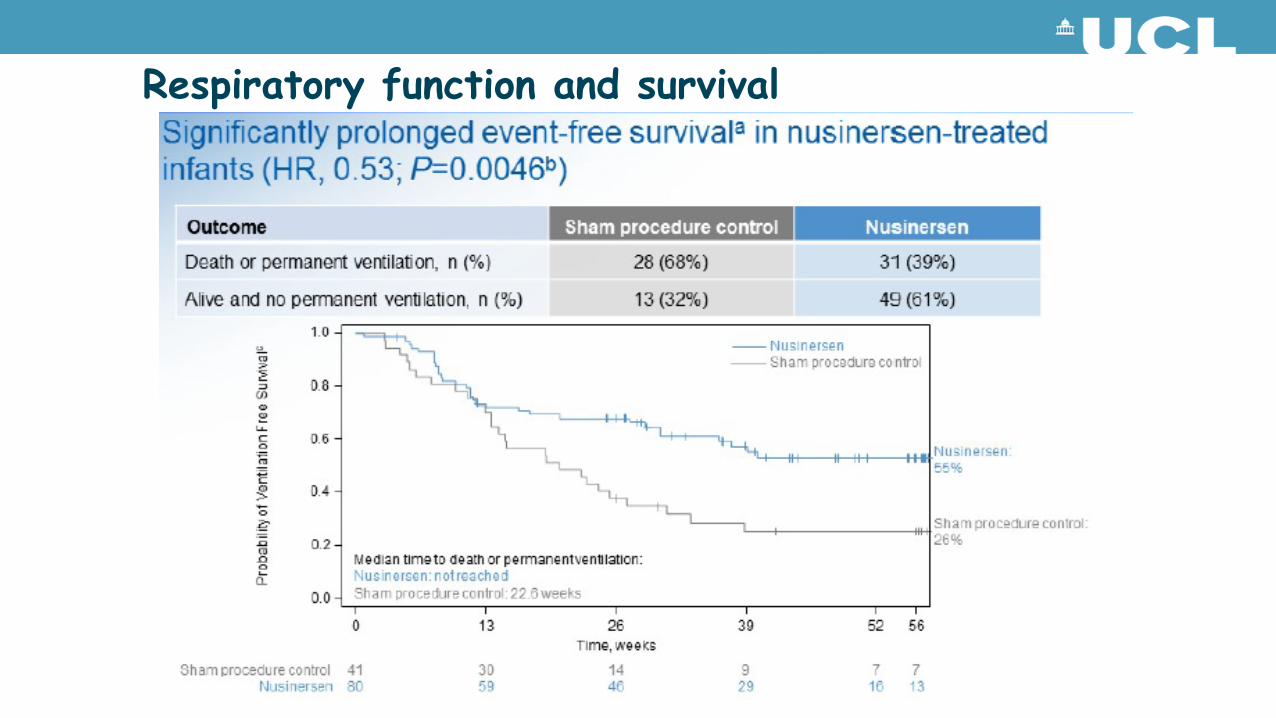
Respiratory function and survival



AAV9 for SMA gene therapy
Jerry Mendell (Columbus, Ohio) is pursuing AAV gene therapy in infants with SMA I
Single IV injection
Two doses used:A. Cohort 1: 6.7x1013 vg/kgB. Cohort 2: 2.0x1014vg/kg
Inclusion• 9 months/6 months of age¹ or younger at
infusion• Bi-allelic SMN1 gene deletions or point
mutations• 2 copies of SMN2• Onset of disease at birth to 6 months of
age
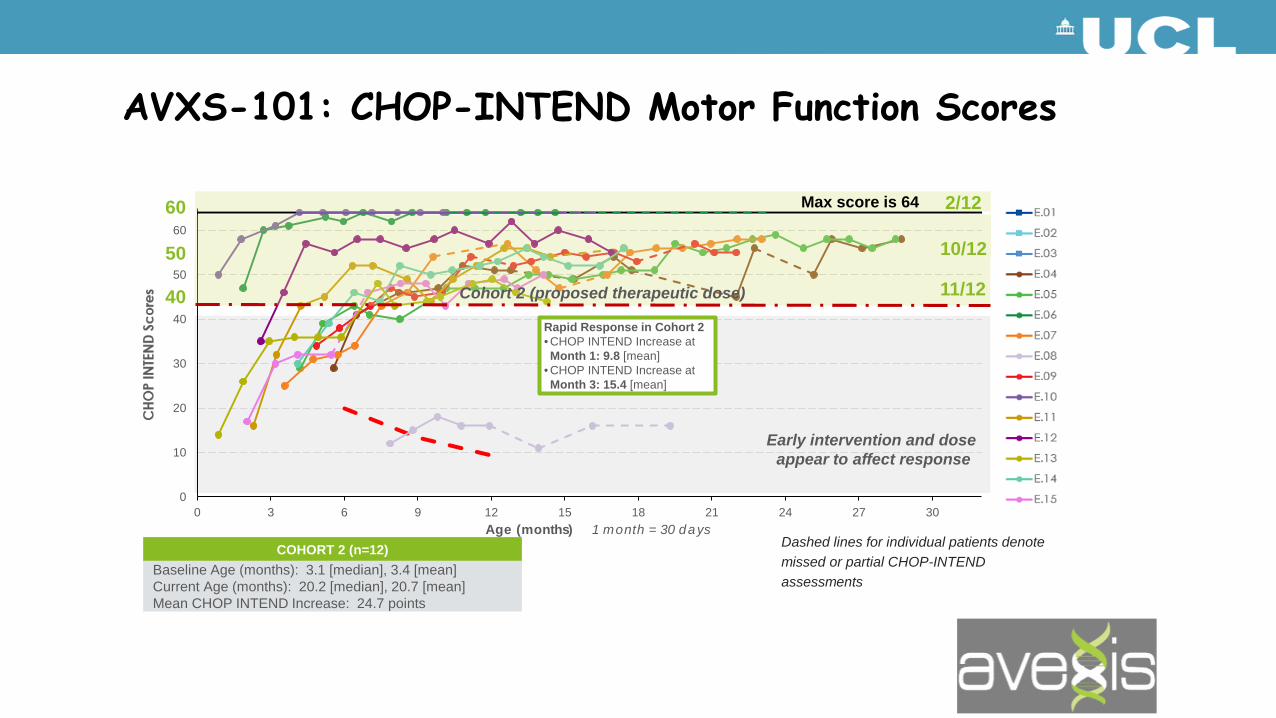
COHORT 2 (n=12)Baseline Age (months): 3.1 [median], 3.4 [mean]Current Age (months): 20.2 [median], 20.7 [mean]Mean CHOP INTEND Increase: 24.7 points
Dashed lines for individual patients denote missed or partial CHOP-INTEND assessments
Early intervention and dose appear to affect response
0
10
20
30
40
50
60
0 3 6 9 12 15 18 21 24 27 30Age (months) 1 month = 30 days
60
50
40
2/12
10/12
11/12Cohort 2 (proposed therapeutic dose)
Rapid Response in Cohort 2• CHOP INTEND Increase at Month 1: 9.8 [mean]
• CHOP INTEND Increase at Month 3: 15.4 [mean]
Max score is 64
AVXS-101: CHOP-INTEND Motor Function Scores

Cohort 2 Achieved Motor Milestones Not Seen in the Natural History of SMA Type 1

Concluding remarks (1)• Outstanding progress in the therapeutic approaches for DMD and
SMA in the last few years
• Second generation AON therapies now in clinic trials, and AAV gene therapy gaining rapid traction
• The size of therapeutic response in SMA is unprecedented, but also linked to the interval between symptom onset and initiation of treatment
• Reducing the diagnostic delay for these conditions now has very tangible implications
• Consider newborn screening

Concluding remarks (2)• While success and tolerability of these approaches encourages their
continuing development, both their long term safety, and cost/ benefit will require careful analysis
• The cost of some of these intervention is considerable, and this might preclude their access to our patients
• NHSE and NICE do not currently have a swift system to allow the assessment of emerging therapies for these devastating childhood conditions
• While cautious and measured counselling to families is imperative, there are reasons to change from pessimism to optimism for these patients, although there is a lot more work to do

AcknowledgmentClinical and research team atGOSH/ ICH
Matthew Wood, OxfordGeorge Dickson, RHUL


















