
nbn se uu diva-3388162617/FULLTEXT01.pdf · &rpsuhkhqvlyh 6xppdulhv ri 8ssvdod 'lvvhuwdwlrqv iurp...
68
Transcript of nbn se uu diva-3388162617/FULLTEXT01.pdf · &rpsuhkhqvlyh 6xppdulhv ri 8ssvdod 'lvvhuwdwlrqv iurp...


Dissertation for the Degree of Doctor of Philosophy (Faculty of Pharmacy) in Pharmaceutics presented at Uppsala University in 2003
ABSTRACT
Tavelin, S., 2003. New Approaches to Studies of Paracellular Drug Transport in Intestinal Epithelial Cell Monolayers. Acta Universitatis Upsaliensis. Comprehensive Summaries of Uppsala Dissertations from the Faculty of Pharmacy 285. 66 pp. Uppsala.ISBN 91-554-5582-4.
Studies of intestinal drug permeability have traditionally been performed in the colon-derived Caco-2 cell model. However, the paracellular permeability of these cell monolayers resembles that of the colon rather than that of the small intestine, which is the major site of drug absorption following oral administration. One aim of this thesis was therefore to develop a new cell culture model that mimics the permeability of the small intestine. 2/4/A1 cells are conditionally immortalized with a temperature sensitive mutant of SV40T. These cells proliferate and form multilayers at 33°C. At cultivation temperatures of 37–39°C, they stop proliferating and form monolayers. 2/4/A1 cells cultivated on permeable supports expressed functional tight junctions. The barrier properties of the tight junctions such as transepithelial electrical resistance and permeability to hydrophilic paracellular markers resembled those of the human small intestine in vivo. These cells lacked functional expression of drug transport proteins and can therefore be used as a model to study passive drug permeability unbiased by active transport. The permeability to diverse sets of drugs in 2/4/A1 was comparable to that of the human jejunum for both incompletely and completely absorbed drugs, and the prediction of human intestinal permeability was better in 2/4/A1 than in Caco-2 for incompletely absorbed drugs. The small intestinal-like paracellular permeability of 2/4/A1 thus enables better predictions of drug permeability in the small intestine than does Caco-2.
The paracellular route and its importance for intestinal drug permeability was also the focus of the second part of this thesis, in which a new principle for tight junction modulation was developed, based on the primary structure of the extracellular tight junction protein occludin. Peptides corresponding to the N-terminus of the first extracellular loop increased the permeability of the tight junctions, but lacked apical effect on the paracellular permeability. This problem was solved by conjugation of one peptide to a lipoamino acid, resulting in two diastereomers with different effects. The L-isomer had a sustained apical effect, while that of the D-isomer was transient. In conclusion, conjugated occludin peptides constitute a new class of tight junction modulators that can enhance the paracellular permeability.
Staffan Tavelin, Department of Pharmacy, Uppsala Biomedical Centre, Box 580,SE-751 23 Uppsala, Sweden
© Staffan Tavelin 2003
ISSN 0282-7484 ISBN 91-554-5582-4 Printed in Sweden by Eklundshofs Grafiska, Uppsala 2003

CONTENTS
1 PAPERS DISCUSSED 5
2 ABBREVIATIONS 6
3 INTRODUCTION 7
3.1 Drug absorption from the gastro-intestinal tract 7
3.2 Mechanisms of intestinal drug permeability 8
3.2.1 Passive transcellular transport 10
3.2.2 Active transport 11
3.2.3 Paracellular transport 12
3.3 In vitro methods for studying drug permeability 12
3.3.1 Cultured cells 13
3.3.2 Artificial membranes 15
3.4 Methods for modulating drug absorption 16
3.4.1 Composition and regulation of the tight junctions 17
3.4.2 Physiological considerations 19
3.4.3 Methods for modulating paracellular transport 20
4 AIMS OF THE THESIS 22
5 MATERIAL AND METHODS 23
5.1 Drugs and radiolabelled markers 23
5.2 Occludin peptides 23
5.3 Cell culture 23
5.3.1 2/4/A1 23
5.3.2 Caco-2 24
5.4 Hexadecane membranes 24
5.5 Microscopy 24
5.5.1 Immunofluorescence 24
5.5.2 Electron microscopy 25
5.6 Establishment of 2/4/A1-Bcl-2 clones 25
5.7 RT- PCR 25
5.8 Cell attachment 25
5.9 Analysis of toxicity and cell death 25
3

5.9.1 DNA staining 25
5.9.2 Dehydrogenase activity 26
5.9.3 Lactate dehydrogenase leakage 26
5.10 Cell monolayer integrity and drug transport studies 26
5.10.1 Electrophysiological measurements 26
5.10.2 Drug permeability studies 26
5.11 Calculations 27
5.12 Statistics 29
6 RESULTS AND DISCUSSION 30
6.1 Characterization of 2/4/A1 (Papers I–III) 30
6.1.1 Expression of SV40 large T antigen (Paper I) 30
6.1.2 Cell attachment and extracellular matrix (Paper I) 31
6.1.3 Monolayer formation (Papers I and II) 32
6.1.4 Development of tight and adherence junctions (Papers I and III) 32
6.1.5 Transepithelial electrical resistance (Papers I and II) 34
6.1.6 Expression of drug transport proteins (Paper II) 35
6.1.7 Cell death (Papers I and II) 36
6.2 Transport of drugs and hydrophilic markers (Papers I – IV) 37
6.2.1 Hydrophilic markers and average pore radius (Papers I and III) 37
6.2.2 Rapidly absorbed drugs (Paper I) 39
6.2.3 Poorly and intermediately absorbed drugs (Papers I and III) 40
6.2.4 Actively transported drugs (Papers I and II) 41
6.2.5 pH dependent permeability (Paper IV) 42
6.3 Modulation of tight junction permeability (Paper V) 44
7 SUMMARY AND CONCLUSIONS 48
8 ACKNOWLEDGEMENTS 50
9 REFERENCES 52
4

1 PAPERS DISCUSSED
This thesis includes the following papers, which will be referred to in the text by their Roman numerals:
I. Tavelin, S., Milovic, V., Ocklind, G., Olsson, S., and Artursson, P. A Conditionally Immortalized Epithelial Cell Line for Studies of Intestinal Drug Transport, J. Pharmacol. Exp. Ther. 1999, 290, 1212–1221.
II. Tavelin, S., Taipalensuu, J., Hallböök, F., Vellonen, K., Moore, V., and Artursson, P. An Improved Cell Culture Model Based on 2/4/A1 Cell Monolayers for Studies of Intestinal Drug Transport. Characterization of Transport Routes, Pharm. Res. 2003, 20, 373–381.
III. Tavelin, S., Taipalensuu, J., Söderberg, L., Morrison, R., Chong, S., and Artursson, P. Prediction of the Oral Absorption of Low Permeability Drugs Using Small Intestinal-like 2/4/A1 Cell Monolayers, Pharm. Res. 2003, 20, 397–405.
IV. Tavelin, S., Nagahara, N., and Artursson, P. The Contribution of the Paracellular Route to The pH-Dependent Epithelial Permeability to Cationic Drugs. In manuscript.
V. Tavelin, S., Hashimoto, K., Malkinson, J., Lazorova, L., Toth, I., and Artursson, P. A New Principle for Tight Junction Modulation Based on Occludin Peptides. Submitted to Mol. Pharmacol.
Reprints were made with permission from the journals.
5

2 ABBREVIATIONS
a-b apical-to-basolateral
ABC ATP-binding cassette
b-a basolateral-to-apical
EDTA ethylene diamine tetraacetate sodium
EHS Engelbreth-Holm-Swarm
FA absorbed fraction after oral administration to humans
FCS foetal calf serum
FITC fluorescein isothiocyanate
GI gastrointestinal
HBSS Hank’s balanced salt solution
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
LDH lactate dehydrogenase
MAGUK membrane-associated guanylate kinase
MDR multidrug resistance
MRP multidrug resistance associated protein
Papp apparent permeability coefficient
PBS phosphate-buffered saline
Pc permeability coefficient of the cell monolayer
PDZ peptide motif of the MAGUK proteins
PEG 4000 polyethylene glycol with an average molecular weight of 4000
P-gp P-glycoprotein
PLC phospholipase-C
RMSE root mean squared error
SD standard deviation
SV40T Simian Virus large T antigen
TER transepithelial electrical resistance
ZO zonula occludens
Å Ångström (1×10-10 m)
6

3 INTRODUCTION
The oral route is generally considered the most convenient for administration of drugs. Drug formulations intended for oral administration are preferred to their non-oral alternatives (e.g. intravenous injections) mainly for reasons such as lower production cost, better suitability for self-medication, a higher level of patient safety and better patient compliance. In order to be efficient, an orally administered drug must meet special criteria. For instance, it must be sufficiently soluble in the gastro-intestinal (GI) fluids, withstand acidic and enzymatic degradation in the GI tract and to a sufficient degree permeate the epithelium of the intestinal mucosa. Given the crucial importance of these drug properties in drug discovery, several methods have been developed to study and predict solubility and stability in and absorption from the GI tract (1-3). As a result of the introduction of high throughput pharmacological screening and combinatorial synthesis, an immense number of pharmacologically active compounds have entered the pre-clinical screening phase. Therefore the methods intended for predicting solubility and permeability have recently been modified to cope with the increasing demands for high throughput in drug discovery programmes. For instance, cell-based assays have been automated (4) and computer-based predictions of permeability and solubility have been developed (5, 6).
The work presented in the first part of this thesis deals with the development of a new cell culture model for studies of intestinal drug permeability. This cell line is particularly suitable for permeability screening since it has a permeability that is comparable to that of the human small intestine and it requires only 4–6 days of cultivation compared with several weeks (for the well-established permeability model based on Caco-2 cells).
In the development of new drugs, compounds that have poor permeability properties are generally dropped in favour of compounds with adequate permeability properties. However, in certain therapeutic areas poor permeability of the active compounds is an inherent feature. Examples include -lactam antibiotics (unless they are substrates for a transport protein) (7), peptides and proteins (8, 9). In order to deliver these drugs via the oral route, the epithelial barrier of the intestine has to be perturbed in a safe, reversible and reproducible manner. Although the use of so-called “absorption enhancers” in oral drug delivery has been exploited to a very limited extent in clinical practice, the concept still remains attractive. The new attention directed to this area in recent years is due to the mapping of the basic extracellular components of the tight junctions, occludin and the claudins (10, 11), and a better understanding of the dynamic regulation of tight junction permeability (12-20). In addition, studies have shown that bacteria regulate tight junction permeability with specific enterotoxins (21, 22).
The second part of this thesis deals with the development of a new class of tight junction modulators based on the primary structure of the transmembrane tight junction protein occludin.
3.1 Drug absorption from the gastro-intestinal tract
Drug absorption following oral administration is a fairly complex sequential series of events outlined in Figure 1. The disintegration rate is governed by properties
7

associated with the pharmaceutical formulation as well as physiological factors such as pH, gastric emptying and luminal content. The dissolution rate is dependent on the physico-chemical characteristics of the particle and the drug, as well as on luminal pH and components of the luminal fluids. Not until the drug is in solution may it permeate the intestinal epithelium. However, chemical and enzymatic degradation, as well as complex formation may limit the amount of drug in solution.
Depending on the physico-chemical properties of the drug, either the dissolution rate or the transport rate across the intestinal epithelium may be the rate-limiting step for drugs to enter the systemic circulation. In this thesis studies of drugs whose absorption is permeability-limited will be reviewed. For such a review a fundamental understanding of drug transport across the main rate-limiting barrier, the intestinal mucosa, is required. This will be discussed in the next section.
Permeability
Disintegration
DissolutionLiver extraction
Degradation/complexation
Permeability
Disintegration
DissolutionLiver extraction
Degradation/complexation
Figure 1. Schematic illustration of the sequential events during the transfer of a drug molecule from a solid dosage form in the GI tract to the systemic circulation. Dissolution and permeability across the epithelium are the two most important determinants in the absorption process. Enzymatic and chemical degradation as well as complexation in the GI tract, efflux and enzymatic degradation in the intestinal epithelium and enzymatic degradation in the liver may decrease the fraction of drug entering the systemic circulation.
3.2 Mechanisms of intestinal drug permeability
The intestinal mucosa can be considered as a system of sequential barriers to drug absorption, the outermost barrier being the mucus layer and the unstirred water layer. The gel-like structure of the mucus is thought to be a barrier to absorption of highly lipophilic drugs and some peptides because of the restricted diffusion in this matrix (23).
8

The absorptive epithelium lining the GI tract follows the folds and villi that increase the anatomical surface area of the mucosa several-fold in the small intestine (24). The villi are interspaced with crypts in which the regeneration of intestinal cells occurs. In between the crypts and the tips of the villi are the basal parts of the villi (Fig. 2). The properties which are relevant for drug absorption differ between the cells along the crypt-villus axis. This will be further discussed in section 3.2.3.
Villus tip Absorptive cells Average pore radius <6 Å
Basal part of the villus Migrating enterocytes Average pore radius 10-15 Å
Crypts Cell division Average pore radius 50-60 Å(Inaccessible to drug absorption)
Epithelial cell lining
Figure 2. Schematic illustration of the crypt-villus axis showing the properties of the different regions. The villus tip is believed to be the major site of absorption for high permeability drugs. The paracellular pathway of this region contains small pores. The paracellular pathway of the basal parts of the villus contains larger pores, and may be the site of permeation for incompletely permeable drugs. The crypts are probably not accessible for drug absorption (25).
The main purpose of the intestinal epithelium is not only to restrict access and in this way protect the body from harmful agents, but also, to allow selective absorption of nutrients and secretion of waste products and xenobiotics. The absorptive epithelium consists of several types of cells including the absorptive enterocytes and mucus secreting goblet cells, of which the enterocytes are the most abundant (26). The transport processes across absorptive epithelia are mediated through one or several of the following: passive transcellular, paracellular, active carrier-mediated and transcytosis as well as carrier-mediated efflux (Fig. 3). The transcytotic pathway has very limited capacity, and is only of relevance for the transport of small amounts of macromolecules that are excluded from the other pathways. It will therefore not be further discussed in this thesis.
9

1 2 3 4
Figure 3. Schematic illustration of the different transport routes that are relevant for drug absorption. 1. passive transcellular and 2. paracellular transport, 3. carrier-mediated efflux, and 4. carrier-mediated active transport. The membrane proteins responsible for efflux and transport may also be situated in the basolateral membrane (not shown in the figure). The transcytotic pathway may also transport drugs (not shown in the figure).
3.2.1 Passive transcellular transport
Drug transport via the passive transcellular route requires that the solute permeates the apical cell membrane. Cell membranes are made up of phospholipids arranged in bilayers that are intermingled with membrane proteins. The composition of phospholipids and proteins varies from cell type to cell type and may theoretically give rise to different permeability properties depending on the cell type. In addition, the intestinal enterocytes have a polarized cell membrane with distinct differences in membrane composition in the apical and the basolateral membrane (27). It is generally believed that the apical membrane has a lower permeability than the basolateral membrane and the former is therefore considered to be the rate-limiting barrier to passive transcellular drug transport (28, 29). One study suggests that the transcellular permeability to drugs is higher in the more distal parts of the intestine than in the small intestine (30). On the other hand, the small intestinal mucosa has an enlarged anatomical surface area due to its many folds and villi, making the small intestine the major site for passive transcellular drug absorption. However, the anatomical surface area of the intestinal epithelium is not necessarily analogous to an effective absorptive surface area (31). For example, it has been calculated that the most rapidly absorbed compound known to date, glucose, has an absorptive surface area that covers only a small part of the total anatomical surface area of the small intestine (32). By contrast, a low-permeability drug does not distribute rapidly into the cell membrane and therefore has a longer residence time in the intestinal fluids. During this time the drug will diffuse down the length of the crypt-villus axis, to eventually be absorbed over a larger absorptive surface area.
The first step in the absorption process is the permeation of the drug across the apical membrane. This step requires that the drug is sufficiently lipophilic. Therefore lipophilic drugs of moderate size are normally transported by the transcellular route. Most of the rapidly and completely absorbed drugs are absorbed by passive
10

transcellular diffusion. However, if the drug is too lipophilic, in combination with other physico-chemical properties that favour a strong interaction with the cell membrane, the drug can become trapped in the apical membrane, resulting in poor bioavailability (33-35).
3.2.2 Active transport
Transport proteins embedded in the apical cell membrane actively shunt nutrients such as peptides, amino acids and sugars across the phospholipid bilayer. In order to restrict access of unwanted solutes via this pathway, these transporters display substrate specificity. Therefore, in order to utilize this pathway to increase absorption, the drug has to share some structure similarity with the normal substrate. A limited number of drugs are substrates for uptake carrier proteins. These include some cephalosporin antibiotics, cytostatics and angiotensin-converting enzyme (ACE) inhibitors that are substrates for oligopeptide transporters (36). The oligopeptide transporters have unusually broad substrate specificity (37), are abundantly expressed in the small intestine (38) and have therefore been the deliberate target for re-designing pharmaceuticals such as antiviral drugs to make them substrates for this transport protein (39, 40). Common to all absorption processes involving transport proteins is that they are saturable. Drugs that are substrates for an active transport protein can therefore display a non-linear dose-response relationship resulting in a decreasing absorbed fraction with an increasing dose. In addition, these proteins are transporters of nutrients, and therefore their capacity is likely to be influenced by food intake (41). These factors may complicate the oral delivery of drugs that are absorbed by active mechanisms.
In contrast to transport proteins acting in the absorptive direction, the active efflux proteins secrete certain drugs that are substrates for these efflux proteins. The most well-studied efflux proteins belong to the adenosine triphosphate (ATP)-binding cassette (ABC) super-family of membrane transporters. These include the multi-drug resistance 1 (MDR1; ABCB1) gene product P-gp and the multi-drug resistance protein family (MRP; ABCC) (42, 43). More recently the breast cancer related protein (BCRP, ABCG2) has been identified as potential contributor in actively limiting oral bioavailability of some drugs (44, 45). The function of the efflux proteins in the intestine may be to prevent the uptake of toxic substances and also, to eliminate such substances from the blood (46). The substrate specificity of the most well-studied efflux protein, P-gp, is broad and only a few structural features of the substrates have been identified (47-49). Examples of clinical relevance include digoxin and paclitaxel (46, 50). The substrates for the MRP family of transport proteins are more well defined and include drugs that have undergone intracellular phase 2 conjugation to derivatives of glucuronic acid, glutathione and sulfate, as well as some unconjugated drugs (43).
There are numerous examples of drugs that display significant vectorial transport in cell-based assays such as the Caco-2 model (see section 3.3.1). Whether or not these efflux proteins in the intestinal epithelium actually affect the bioavailability of orally administered drugs that are substrates is under debate (51). One concern, however, is that the lipophilic drugs generated by high throughput pharmacological screening appear to be effluxed to a greater extent by these proteins than are drugs that are
11

designed by traditional means. There is therefore a risk that drug efflux and drug-drug interactions at the level of intestinally located membrane transporters, as well as interpatient variability will be more pronounced problems in the future.
3.2.3 Paracellular transport
Drugs of small to moderate molecular weights (MWs) can permeate the intestinal epithelium through the water-filled pores between the cells. This process is known as paracellular transport, and is generally considered to be a passive process, even if this pathway appears to be selective for cationic rather than anionic and neutral drugs (52, 53). The paracellular pathway has also been shown to be saturable (54-56), by at least two separate mechanisms, one of which involves an intracellular process (56). These examples illustrate the complexity and dynamics of regulation of this pathway, which was previously considered static (see also section 3.4.1).
The drugs believed to be significantly absorbed by the paracellular route include the -receptor antagonist atenolol (57), the H2-receptor antagonists cimetidine, ranitidine
and famotidine (54, 56) and the loop-diuretic furosemide (58). These drugs are all of moderate MWs (around 200–270) and relatively hydrophilic. Paracellularly absorbed drugs are usually incompletely absorbed, since the paracellular pores cover only 0.01–0.1% of the total surface area of the intestine (59, 60) and the size-restricting gate function of the paracellular pathway significantly decreases the permeability (18). The paracellular permeability is dynamically regulated and varies both along the path of the intestine and along the crypt-villus axis (61, 62) (Fig. 2). The average pore radius of the human small intestine is 8–13 Å (63)), which will limit the paracellular permeability of drugs >4 Å and restrict those >15 Å (64). The colon is even more size-discriminating since the paracellular pathway restricts drugs <3.5 Å (65). In a recent study by Fihn et al., it was shown that the average pore radius of the most upper part of the villus of the rat small intestine contains small pores (with a radius of <6 Å) (66), while the basal part of the villus contains larger pores (with a 10–15 Å radius). Thus the paracellular pathway of the small intestine appears to be a dual-pore system, and this has also been observed in the human jejunum (67).
The rate-limiting process of paracellular drug transport is therefore the passage through the narrow pores of the tight junction complex that forms a seal at the most apical part of the intercellular space. The composition and the regulation of tight junctions are further discussed in section 3.4.1.
3.3 In vitro methods for studying drug permeability
For reasons of safety but also for reasons of cost, drug absorption studies in humans are only carried out for a limited number of well-characterized drugs. Studies of drug absorption in the intestine of the often poorly characterized drug candidates in pre-clinical studies have therefore traditionally been carried out in experimental animals. However, the introduction of combinatorial chemistry and high throughput pharmacological screening in drug discovery has significantly increased the number of compounds entering the pre-clinical phase, and this has made it impossible to assess the absorption properties of all these compounds in experimental animals. This fact has spurred the development and use of in vitro methods to assess drug
12

permeability properties in most drug discovery settings. In vitro methods for drug permeability are fairly easily adapted to automation, which further increases the capacity of these methods and at the same time reduces the costs of these otherwise cost and labour-intensive processes. Also, the insight that drug absorption across biological barriers is a complex process involving several pathways that cannot easily be delineated in experimental animals has resulted in the large interest in academic and industrial institutions in these methods (68).
A prerequisite to the successful use of in vitro methods for drug absorption studies is that the relationship between drug absorption in the human intestine in vivo and drug permeability in the in vitro model must be good (but not necessarily quantitative). Therefore, a great deal of effort has to be put into the characterization of the in vitromethod, into establishing a relationship with data obtained in humans and into ensuring that the drug permeability in the in vitro method is predictive of the drug permeability in vivo (1, 69, 70).
The methods range from the determination of partition coefficients into and transport across isotropic systems to experiments using whole tissues in situ or in vivo. The methods covered in the papers of this thesis, cultured cells and artificial membranes, are discussed below.
3.3.1 Cultured cells
In 1990, the human adenocarcinoma cell line Caco-2 was introduced as an experimental tool for mechanistic studies of intestinal transport (71-73) and a year later, it was suggested that the Caco-2 model was suitable for screening intestinal drug permeability and predicting the oral absorption potential of new drug substances (1). The Caco-2 cells were grown on permeable supports and spontaneously formed polarized monolayers that resembled that of the intestinal epithelium (Fig. 4).
Apical
BasolateralPorous filter membrane
Figure 4. Schematic illustration of the chamber used for growing Caco-2 and other cell cultures for drug transport experiments.
In many respects the Caco-2 cells are therefore functionally similar to the human small intestinal enterocyte, despite the fact that they originate from a human colorectal carcinoma (74-76). The methods that are based on cultured cells such as Caco-2 cells are, however, not only useful for drug absorption screening. It is also
13

14
possible to extract information about specific transport processes that would be difficult to obtain in more complex models such as those based on whole tissues from experimental animals. For instance, the methods enable us to investigate the relative contribution of passive transcellular and paracellular transport (58), the effect of charge on paracellular transport (52, 53) and the effect of solvent drag (52).Furthermore, cell-based assays have been used to investigate the effects of pharmaceutical excipients that may enhance passive drug transport either by enhancing the solubility of the compound e.g. (78-80) or by affecting the epithelial integrity e.g. (81-84) (see also section 3.4.3). Cell culture models have also been used extensively to study active transport mechanisms and more recently, the interplay between several transporters e.g. (85, 86), and between drug metabolism and drug transport e.g. (87, 88), and the relative contribution of passive and active transport mechanisms to the overall transport of a drug e.g. (58).
However, extensive experience in the use of Caco-2 cells has revealed some limitations with this in vitro model. For instance, the paracellular route in Caco-2 cells is tighter than that in the small intestine in vivo. While the average pore radius of the tight junctions in the human small intestine is around 8–13 Å (63), the corresponding radius in Caco-2 cells is about 5 Å (89). The lower paracellular permeability of Caco-2 cells has been attributed to the colonic origin of the cell line. Some studies suggest that the Caco-2 cell line has a paracellular permeability that is comparable to the tips of the villi of the small intestinal mucosa, and that other structural differences between the cell cultures and the intestine in vivo are the cause of the apparent lower paracellular permeability in the Caco-2 model (90, 91).
Several studies have searched alternatives to Caco-2 to circumvent the limitationsassociated with this cell line. The use of primary cultures appears attractive since these cells are not transformed. However, primary cultures are difficult to obtain, demanding to cultivate and require constant renewal since normal intestinal epithelialcells can only be maintained in culture for about 10 days (92). Most studies have therefore been carried out using spontaneously transformed or immortalized cell lines. Examples of cell cultures that have a more leaky paracellular route than Caco-2include HT29-18-C1, a monolayer forming epithelial cell line of colonic origin (93),and the small intestinal epithelial cell line, IEC-18 (94). Co-cultures of absorptive and goblet cell lines with a higher paracellular permeability have been established. However, these co-cultures express an abnormal phenotype in cell culture (95).
Another disadvantage of the Caco-2 cell line is that it requires several weeks to stabilize paracellular permeability and expression of drug transport proteins (96). Thisproblem can be circumvented, at least in part, by using cultures that differentiate faster, or by optimising the culture conditions of Caco-2 to induce faster differentiation (to less than 1 week). Alternative short-term cultures for the studies of passive drug transport include the MDCK (70) and the short-term Caco-2 cultures (97, 98). However, both MDCK and short-term Caco-2 cells express varying amounts of functional transporters such as P-gp (usually at a lower level than traditional Caco-2) (97, 98). One potential limitation with the short-term Caco-2 cells in drug transport studies may be that they can be used for only a very short time (approximately 1–2days) since their phenotype differs from one day to another and the integrity of these cultures decreases rapidly shortly after reaching maximum transepithelial electrical resistance (TER) (97).

The expression of multiple transport systems in Caco-2 is generally considered an advantage of this cell line. For instance since, the human origin of Caco-2 can provide information on human transporters in the well-differentiated enterocyte. The abundant expression of a variety of transporters in Caco-2 cells also makes it attractive to apply functional genomics tools, such as cDNA arrays in order to map the expression (99) and relative abundance of these transporters (44). Such studies have shown that while the expression of some transporters may correlate with that in vivo (44), the expression of other may differ from the in vivo situation (100). Moreover, the variety of transport systems that are expressed in Caco-2 cells may obscure the study of a specific transporter, especially if the transporter lacks a specific substrate, as in the case of most efflux transporters of the ABC transporter super-family (101). When specific membrane transporters are to be studied, simple epithelial expression systems such as those based on the kidney epithelial cell lines MDCK and LLC-PK1 are perhaps more suitable e.g. (102). However, differences in transport parameters for the human ABC transporters MDR1 and MRP2 have been observed between Caco-2 cells and the expression systems based on the MDCK cell line (103, 104). Differences between the Caco-2 and MDCK cell lines have also been reported with regard to the activity of peptide transporters (105). Since MDCK cells originate from the canine kidney epithelium, it may be speculated that the observed differences are a result of either the species difference or the different “organ origin” of the cell lines.
In summary, epithelial cell culture models are not only useful for screening absorption properties, but a vast number of studies have also shown that these models are indispensable in the elucidation of and interplay between the drug transport mechanisms. Caco-2 is the most widely used cell line for drug permeability studies, but there are alternatives that may be superior to Caco-2 for studies of specific transport routes. However, these alternative cell cultures are usually much less well characterized than the Caco-2 culture.
3.3.2 Artificial membranes
As stated in the previous section, epithelial cell cultures are well suited for studies and screening of drug permeability. However, one disadvantage of cell cultures, which is difficult to overcome, is the labour and cost-demanding maintenance of cell cultures and facilities. Also, despite efforts to automate the cell culture assays, cells are rather fragile for easy adaptation to automated processes. Therefore an alternative method has been developed for high-capacity screening of permeability properties that are based on immobilized organic solvents with or without the addition of phospholipids, or phospholipids alone prepared as vesicles or liposomes.
Experiments using vesicles or liposomes in suspension have been fairly successful in predicting intestinal drug permeability for diverse data sets of drugs (106, 107). Chromatographic methods where the stationary phase consists of immobilized phospholipids or liposomes have also been developed (108). In these methods the retention time on the column is correlated to the epithelial permeability. These methods are attractive for screening purposes since they require very little compound, are easily automated and are adaptable to diverse sets of drugs. However, the retention time of a drug does not always reflect the transport across a phospholipid bilayer. Rather, it reflects the interaction with and partitioning into the phospholipid
15

bilayer. Methods that allow the transport across a lipid membrane are therefore an attractive alternative.
A single phospholipid bilayer, a bilayer lipid membrane (BLM), can be formed by carefully placing a mixture of phospholipid in an alkane solvent over a small (0.5 mm) hole in a thin sheet of Teflon (109). However, a membrane thus formed is extremely delicate and is therefore not suitable for routine drug permeability screening. In order to circumvent the problem of the BLM being too delicate, permeable supports (similar to those on which epithelial cells are grown) are used to stabilize the membrane. Successful attempts at achieving a single stabilized lipid bilayer were reported when a mixture of egg phosphatidylcholine (PC) and cholesterol in n-decane or n-octane was added to polycarbonate filters with well-defined pores (110). It was stated that the conditions for formation of a single bilayer have to be carefully controlled to avoid formation of a “lipid plug” in the pores. This method has also been adapted to high throughput screening, for which permeable supports impregnated with a mixture of PC in an inert organic solvent were fitted in a microtiter plate (69). Drug permeability studies in this system resulted in a relationship, with fraction absorbed in the human intestine, much like that obtained with Caco-2 cells. However, the data set was selected to exclude low-MW hydrophilic drugs that might permeate the intestinal epithelium via the paracellular route. Outliers in this study were drugs known to be actively transported in vivo. Recently this approach has been taken further by using a lipid composition that corresponds to that found in the apical cell membrane in vivo (111), or simplified by removing the lipid component, thus reducing the system to consist of an organic solvent only (112). These modified systems display relationships to fraction absorbed in humans that are comparable to the relationship observed in the original paper, and in studies using Caco-2 cell monolayers. There are potential problems associated with the use of artificial membranes. For instance, in systems in which thick lipid membranes are formed drug may be retained in the membrane and result in poor mass balance and an under-estimation of the permeability coefficient of the drug. However, this can be overcome by mathematical corrections (113).
3.4 Methods for modulating drug absorption
In principle there are two strategies for modulating the absorption of a poorly permeable drug. The first involves increasing the transcellular permeability by means of a pro-drug. The second strategy is to modulate the permeability of the tight junctions and thus, increase paracellular permeability.
Research on the tight junctions of the intestinal epithelium has received much attention for three major reasons. Firstly, it has been reported that changes in the integrity of these junctions contribute to the pathology of inflammatory bowel disease e.g. (114, 115). Secondly, some bacterial toxins are known to modulate the permeability of the tight junctions (21, 116) and thirdly, it was found that by perturbing the tight junctions it is possible to increase the absorption of orally administered drugs that do not normally permeate the intestinal epithelium. Drugs that could be given orally together with a tight junction modulator include peptides and proteins (117-119), and -lactam antibiotics (7, 120, 121). Substances that are capable of modulating the permeability of the tight junctions should preferably be active
16

without irreversibly compromising the intestinal integrity and function. Several endogenous substances and hormones are known to modulate the integrity of the tight junctions (122-125). Traditionally, however, tight junction modulators used mediate the effect on the tight junctions via complex intracellular pathways, which may differ from one situation to another, jeopardizing the safety of this approach e.g. (126, 127). In order to develop safer tight junction modulators, the composition of the tight junctions have to be understood.
3.4.1 Composition and regulation of the tight junctions
The seal connecting the most apical part of the cell membrane of two neighbouring cells is known as the “tight junction complex” and consists of tight junctions and adherence junctions (128) (Fig. 5). The tight junctions are believed to be the major barrier to paracellular solute permeability, and are therefore the target of most efforts to modulate paracellular permeability. However, studies on the modulation of the adherence junction protein E-cadherin have shown that paracellular permeability can be modulated by peptides targeted also to this protein (129).
The tight junction is composed of transmembrane and cytosolic proteins that interact with each other as well as with components of the cytoskeleton. The first identified transmembrane protein that was identified is occludin (11). This approximately 65 kDa protein has four transmembrane regions and two extracellular loops of similar size. The cytoplasmatic C-terminus binds to tight junction associated proteins (ZO-1, ZO-2 and ZO-3) via PDZ binding motifs (130-132), and also directly to the actin filaments of the cytoskeleton (133). Occludin therefore provides a link between the extracellular domains of the tight junctions and is part of the regulatory elements of the junction. The first extracellular loop of occludin is unusually rich in glycine and tyrosine residues, lacks charged residues (as does also the second loop) (11) and appears to be necessary for maintaining the barrier function of the tight junctions, since over-expression of a deletion mutant lacking the N-terminus and the first extracellular loop has been reported to impair the properties of the tight junctions (16). Occludin has the capacity to form tight junction filaments when transfected into fibroblasts (which normally lack tight junctions) (134), but does not appear to be necessary for tight junction formation since an occludin knock-out mouse has been reported to have formed apparently normal tight junctions (135). The regulation of function and localization of occludin are achieved by phosphorylation of serine, threonine and tyrosine residues (135, 136). However, the phosphorylation patterns are complicated and there is ambiguity about the effects of the phosphorylation of this protein (14). Despite evidence that occludin is a constituent of the tight junctions and that its function appears to be finely tuned by intracellular regulation, its precise function remains to be elucidated.
17

Ca2+
ZO 1 ZO 2 ZO 3
Actin filament of the cytoskeleton
Cytosolic plaque of the tight junctions
Adherence junctions
Tight junctions
Transmembrane proteins of the tight junctions e.g occludin and the claudins
Figure 5. Schematic illustration of the composition of the tight junction complex. The extracellular domain of the tight junctions consists of at least three proteins, occludin, the claudins and the junctional adhesion molecule (JAM). The cytosolic plaque of the tight junctions consists of several PDZ and non-PDZ-containing proteins. The PDZ-containing proteins (including the ZO proteins) bind to occludin and the claudins and link these to the actin filaments of the cytoskeleton. The non-PDZ-containing proteins (e.g. cingulin, symplekin and the Rab proteins; not shown in the Figure) are scaffolding proteins that are also involved in vesicular trafficking. The adherence junctions are situated just below the tight junctions, and are also linked to the cytoskeleton.
The second class of extracellular tight junction proteins that have been identified is the claudin family (10). To date 24 members of the claudin family have been identified and all members are 20–27 kDa proteins with four transmembrane domains and two extracellular loops (137). The C-terminus contains PDZ-binding motifs to ZO-1, ZO-2 and ZO-3 (138). When expressed in cells normally lacking tight junctions, the individual claudins form tight junctional strands that are more well developed than when expressing occludin (139). The claudins are therefore considered to constitute the backbone of the tight junctional strands (140). The individual claudins can interact with different claudins on the apposing cell, giving rise to a wide variety of combinations (141). Variations in the tightness and ion selectivity of various epithelia have been attributed to differential expression and ratios of the different claudins in the various tissues. For example, claudin-2 was characterized as a determinant of leaky epithelia after over-expression of this claudin was seen to change the normally tight MDCK I cells to leaky cells (142). Interestingly, this claudin is absent in the differentiated enterocyte at the tip of the villus in the intestine, but is present in cells lining the basal parts of the villi, known to be leakier (143).
Occludin and the claudins are not the only extracellular proteins. For instance, the junctional adhesion molecule (JAM) was recently identified (144). The JAM has been associated with the gate function of tight junctions (145) and also with transmigration of monocytes and dendritic cells (144, 146).
18

The cytosolic tight junction-associated proteins can be divided into two classes, PDZ and non-PDZ-containing proteins, and build the cytosolic plaque of the tight junctions (137). Examples of PDZ-containing proteins include ZO-1, ZO-2 and ZO-3 (138). The N-termini of ZO-1 and ZO-2 bind to the C-termini of occludin and the claudins, and the C-termini bind to the actin filaments of the cytoskeleton (138). In contrast to the other two ZO proteins, ZO-3 binds to the actin filaments by its N-terminal, and both the N- and the C-terminal bind to occludin (131, 133, 138). All three ZO proteins are phosphoproteins, but the effects of phosphorylation of these proteins remain unclear (137). The cytosolic tight junction proteins that lack PDZ domains include cingulin, symplekin and the Rab proteins (137). These proteins are involved in vesicular trafficking and in scaffolding the transmembrane tight junction proteins to the cytoskeleton. There are numerous other PDZ and non-PDZ-containing proteins in the cytosolic plaque of the tight junctions. These are extensively reviewed in Gonzalez-Mariscal et al. (137).
As mentioned above, many of the components of the tight junctions are regulated by phosphorylation. This implies that the tight junctions are regulated by intracellular cascades mediated by second messengers such as the intracellular concentration of Ca2+, tyrosine kinases and protein kinase C, with an altered phosphorylation pattern of the tight junction proteins as end-points in these cascades (137). However, it is difficult to distinguish the individual effects on the tight junction proteins from effects on other structures of the cell (e.g. the actin cytoskeleton). One example of a distinct action on a tight junction protein is that histamine has a specific effect on the phosphorylation of occludin, which leads to increased tight junction permeability (14).
3.4.2 Physiological considerations
Knowledge of the composition and regulation of the tight junctions is, however, not sufficient to develop a potent tight junction modulator for use in humans. Obstacles remain, such as correlation of the initial studies normally performed in cell cultures under controlled conditions to the far more complex and dynamic environment of the intestine in vivo. For one, the concentrations of the modulator and drug will be variable and difficult to predict as they are diluted in the GI fluids e.g. (147). In addition, the protective mucus layer of the mucosa in vivo may prevent the direct interaction necessary for the modulator to be effective and the pH and the different components of the GI fluids such as complexing agents and enzymes may also affect the result. Further the roles of subepthelial tissues, innervation and local blood flow cannot be predicted in cell cultures.
Also, one of the major concerns in the use of tight junction modulators in pharmaceutical products is that of toxicity. Usually acute toxicity, and damage to epithelial cells are measured and correlated to the effect of the modulator. A toxicity index (defined as the concentration that causes toxicity divided by the concentration that results in the desired tight junction modulation) can be used to define the efficacy of the modulator. Normally the toxicity index is around 2–3 in vitro (148). However, studies of in vitro acute toxicity may over-predict the toxicity of the modulator in the intestine in vivo. The reason for this is that the intestinal mucosa is under constant renewal every 3–4 days. More serious are potential systemic toxicity caused by
19

systemic absorption of the modulator, autoimmune responses and inflammatory reactions.
A third potential problem is that the effect of the modulator has to be controlled in such a way that the unwanted absorption of potentially harmful agents such as bacteria and toxins can be avoided. Therefore, the modulator has to be presented to the mucosa in close proximity to the drug to ensure that the drug is selectively absorbed and that the effect of the modulator is controlled and transient.
3.4.3 Methods for modulating paracellular transport
A large number of compounds have been identified that modulate tight junction permeability. The traditional ones include Ca2+ chelators, surfactants and cationic polymers. Most of these modulators have been studied in cell cultures such as Caco-2 cells, in order to elucidate their efficacies and mechanisms of action (148). Few studies have been carried out in vivo and even fewer modulators are used clinically e.g. (121).
Calcium ion chelators (e.g. ethylenediaminetetraacetic acid (EDTA)) were initially thought to mediate their effect by reducing extracellular Ca2+ levels and thus, affecting the Ca2+-dependent proteins of the tight junction complex. However, binding of extracellular Ca2+ also affect the intracellular levels of Ca2+, which will result in a series of intracellular events, such as activation of protein kinases and subsequent altered protein phosphorylation of the tight and adherence junctional proteins (149, 150). Therefore, Ca2+ chelators are believed to have multiple mechanisms of action on the tight junctions.
There are different classes of both natural and synthetic surfactants that are known to modulate tight junction permeability, including bile salts, anionic surfactants, medium-chain fatty acids and phosphate esters (148). Surfactants are known to interact with the cell membrane either as solubilizing agents or as agents that extract proteins and lipids from the membrane (119, 151). It is therefore likely that since surfactants are known to increase epithelial permeability, they do this by means of membrane perturbation. However, some surfactants also have distinct intracellular mechanisms of action that result in increased permeability of the tight junctions. For instance, the medium-chain fatty acid sodium caprate increases intracellular Ca2+
concentration, causes ATP depletion and triggers a phospholipase C (PLC)-mediated pathway (127). Interestingly, hexadecylphosphocholine inhibits PLC- with a concomitant increase in paracellular permeability (13). These two seemingly contradictory findings illustrate the complexity of tight junction modulation.
Chitosan is an example of a cationic polymer that has been shown to increase paracellular permeability by modulating the tight junctions. However, the efficiency of chitosan is dependent on the degree of acetylation and consequently on charge density (152). Therefore it appears that the positive charges of the polymer are directly involved in mediating the mechanism of action of this polymer. Chitosan requires direct contact with the epithelial cells in order to be effective. This fact limits the clinical use of this modulator in the intestine, since the mucus layer has been shown to reduce or abolish the efficiency of chitosan in vivo and in vitro in studies using mucus-producing cell lines (153). Another problem is that the solubility of
20

21
chitosan is low at physiologic pH since the amines of chitosan are un-ionized at a pH of around 7. However, this can be overcome by methylation of the amine groups to form permanently charged quaternary amines (83).
Recently the use of bacterial toxins as potential tight junction modulators has been investigated. For instance, the Vibrio cholerae enterotoxin zonula occludens toxin (ZOT) has been shown to increase paracellular permeability by interacting with a specific intestinal epithelial surface receptor leading to activation of the second messenger proteinkinase C, with subsequent activation of a complex intracellular cascade of events which leads to rearrangement of the actin filaments and subsequent loosening of the tight junctions (116, 154). The ZOT has also been shown to increase the absorption of insulin after oral administration to rabbits (118). Interestingly, an endogenous analogue to ZOT, zonulin, was recently identified. Zonulin interacts with the same surface receptor and has some structural features in common with ZOT. This finding suggests that intestinal epithelium regulates its paracellular permeability through specific mechanisms, for example in response to bacterial infections (155).
An alternative strategy to modulating the diffusion barrier of the tight junctions may be to directly target the tight junction modulator to the extracellular proteins of the tight junctions. It can be speculated that such direct intervention would reduce the acute toxicity of the modulator, since it does not involve an intracellular mechanism. Some bacterial toxins have as primary targets the extracellular domains of tight junction proteins. For instance, the C-terminus of Clostridium perfringens enterotoxin (CPE), which binds to the tight junction proteins claudin 3 and 4, causes an increase in paracellular permeability (21). It has also been shown that synthetic peptides can perturb the tight junction barrier. A homologous large peptide corresponding to the second extracellular loop of occludin has been shown to perturb the paracellularbarrier function (156) and peptides corresponding to the first loop of occludin have been reported to inhibit tight junction re-sealing following a Ca2+ switch (157).
Despite the potential advantages of extracellular targeting, several obstacles remain to be addressed. One is the fact that peptides targeted to the extracellular domains of tight and adherence junctions appear to be active only from the basolateral side (21,129). Whether this is an effect of easier accessibility to the junctions from the basolateral side remains unclear. Another problem is that synthetic peptides targeted to the tight junctions have a very slow onset of action (hours). One way of solving these problems may be to identify active analogues (not necessarily peptides) that can access the tight junctions from the apical side and that have a more rapid onset of action.
In conclusion, although tight junction modulators are used in clinical practice in rare cases, their mechanisms are usually non-specific and poorly understood. Thus there is a need for new approaches to tight junction modulation.

4 AIMS OF THE THESIS
The overall aims of this thesis were to develop and evaluate a new cell culture model that mimics the permeability of the small intestine and to develop a new principle for tight junction regulation. Specific aims were to:
develop a new cell culture model that better mimics the permeability of the human small intestine for studies of passive drug transport, based on the intestinal epithelial cell line 2/4/A1 which is conditionally immortalized with a temperature-sensitive mutant of the growth-promoting oncogene SV40 large T (Paper I);
improve the viability of the 2/4/A1 cell culture model and investigate different routes of drug transport in this cell line (Paper II);
characterize the paracellular route of 2/4/A1 monolayers and compare the permeabilities of incompletely absorbed oral drugs in 2/4/A1 with those in Caco-2 monolayers (Paper III);
investigate the contribution of the paracellular route to the pH-dependent permeability of cationic drugs in 2/4/A1 cell monolayers (Paper IV); and
investigate whether peptides from the extracellular loops of the tight junction protein occludin may be used as a new principle for tight junction modulation (Paper V).
22

5 MATERIAL AND METHODS
5.1 Drugs and radiolabelled markers
Acyclovir, Alprenolol-HCl, atenolol, cimetidine, digoxin, D-raffinose, fluorescein-conjugated dextran (MW 50,000), glycylsarcosine, lactulose, mannitol, metolazone, metoprolol-tartrate, mitoxantrone, Na-fluorescein, phenazone, phosphonoformic acid (foscarnet), pindolol, propranolol-HCl, sulfasalazine, sulpiride, terbutaline, verapamil and vinblastine were purchased from Sigma (St. Louis, MO). BMS 189664, BMS 187745, didanosine, metformin, nadolol, pravastatin, and lobucavir were obtained from Bristol Myers Squibb (Princeton, NJ). Alfentanil was obtained as Rapifen IV solution from Janssen-Cilag through Apoteket AB (Stockholm, Sweden). Lucifer yellow was purchased from Molecular Probes (Eugene, OR). Olsalazine was a gift from Dr. Wenche Rolfsen, Pharmacia & Upjohn (Uppsala, Sweden). [14C]-Acyclovir, [14C]-ganciclovir, [3H]-glycylsarcosine, [3H]-mitoxantrone,
14C -phosphonoformic acid and [3H]-vinblastine were obtained from Moravek Biochemicals (Brea, CA). 14C -Creatinine, 14C -mannitol, 3H -raffinose, [3H]-testosterone and 3H -vasopressin were obtained from New England Nuclear (Boston, MA). [3H]-Digoxin and [3H]-lactulose was obtained from American Radiolabeled Chemicals Inc. (St. Louis, MO). [14C]-Clodronate and unlabeled clodronate (Leiras Co., Turku, Finland) were gifts from Dr. J. Mönkkönen (University of Kuopio, Finland). 14C -PEG (MW 4,000) was obtained from Amersham Biosciences (Uppsala, Sweden). GF12918 was a gift from GlaxoWellcome (Hertfordshire, UK).
5.2 Occludin peptides
Peptide synthesis was accomplished as described previously (158). The crude peptides were purified and lyophilized to a final purity of >95%. The purified peptides were stored in a dessicator at 4˚C. Mass spectra were recorded on a Fisons VG-TOF spectrometer using matrix assisted laser desorption ionisation time-of-flight (MALDI-TOF) mass spectrometry, a VG Analytical ZAB-SE instrument using fast atom bombardment (FAB) ionisation, or a Finnigan MassLab Navigator quadrupole mass spectrometer using electrospray (ES) ionisation. The stability of the peptides (20 µg/ml) during incubation with Caco-2 cell monolayers was examined for up to 360 minutes. At 60, 120, 180, 240 and 360 minutes, samples (20 l) were removed from the apical and/or basolateral solutions. The samples were analyzed by HPLC.
5.3 Cell culture
5.3.1 2/4/A1
In paper I, the 2/4/A1 cells were cultured at 33°C in RPMI 1640 medium supplemented with 2% fetal calf serum, 10 mM HEPES, 2 mM L-glutamine, 200
g/ml geneticin, 1 mg/ml bovine serum albumin, 1 g/ml water soluble dexamethasone, equivalent to 65 ng/ml dexamethasone, 20 ng/ml epidermal growth factor, 50 ng/ml insulin-like growth factor I and ITS premixTM containing 10 g/ml insulin, 10 g/ml transferrin and 10 ng/ml selenic acid. The cells were expanded in 75 cm2 plastic cell culture flasks at 33°C, 5% CO2 and 95% humidity. The medium was changed every second day and the cells were passaged by trypsinisation at 80%
23

confluence, i.e. approximately every fourth day. Cells cultivated at passage number 30 through 43 were used. For drug transport experiments using 2/4/A1 cells, permeable supports, 0.45 m pore size, 12 mm in diameter, were coated with ECL. 2/4/A1 cells were thereafter seeded at 100,000/cm2 on the ECL coated permeable filter supports. In paper II – IV, 2/4/A1 cells were expanded as described for in paper I, with a few changes to the culture medium. The selection marker G418 was removed, the dexamethasone addition was reduced to 0.5 µg/ml, equivalent to 33.5 ng/ml and the culture medium was supplemented with 4% FCS. For functional studies, the 2/4/A1 cells were grown in RPMI 1640 supplemented with growth factors, 6% fetal calf serum (FCS) with or without G418 on permeable supports at 100,000 cells/cm2 coated with EHS matrix.
5.3.2 Caco-2
Caco-2 cells originating from a human colorectal carcinoma were obtained from American Type Culture Collection (Rockville, MD) and cultivated as described in detail previously (96). In brief, Caco-2 cells were seeded at a density of 440,000/cm2
on uncoated permeable polycarbonate filters and cultivated for 21 days. Cells at passage number 95 through 105 were used.
5.4 Hexadecane membranes
The HDM was prepare essentially as described by Whonsland et al (112), with a few modifications. 71.5 ml 5 v/v% hexadecane in hexane was added to polycarbonate filter inserts (Transwell Costar, Badhoevedorp, the Netherlands; mean pore size 0.4 mm or 3.0 mm; diameter 12 mm) (total amount of hexadecane: 3.6 ml/insert). The impregnated inserts where left at room temperature in a fume hood for at least one hour prior to drug transport studies to ensure complete evaporation of the hexane.
5.5 Microscopy
5.5.1 Immunofluorescence
Cells cultivated permeable supports were washed with PBS and fixed with 3% paraformaldehyde. The cells were then permeabilized using 0.1% Triton X-100. The F-actin distribution was visualized using direct fluorescence with rhodamine-conjugated phalloidin. Tight and adherence junction protein distributions were studied by indirect immunofluorescence using rabbit polyclonal antibody to occludin or ZO-1 and a mouse monoclonal Ig antibody to human E-cadherin respectively. The SV40 large T antigen was stained with mouse monoclonal Ig SV40 large T antigen antibody. The cells were incubated with the primary antibodies, washed with PBS and incubated with secondary FITC-labeled antibodies. The samples were washed, mounted and examined under a confocal laser scanning microscope.
24

5.5.2 Electron microscopy
2/4/A1 and Caco-2 cells cultivated on permeable supports as described above were fixed in 2.5% glutaraldehyde, then immersed in 1% osmium tetroxide and dehydrated with ethanol. Thin sections were stained with uranyl acetate plus lead citrate and examined by transmission electron microscopy.
5.6 Establishment of 2/4/A1-Bcl-2 clones
2/4/A1 cells at 80% confluence were trypsinized and pelleted, harvested by centrifugation and washed. Two plasmids were added to each fraction; human Bcl-2 cDNA containing plasmid (159) and hygromycin B resistance plasmid (P3’SS). Electroporation was performed. After electroporation the cells were plated onto culture dishes and left overnight. Twenty-four hours after transfection, 200 U/ml hygromycin B was added to the conditioned medium. Clones were immediately isolated with cloning cylinders when they appeared. The clones were harvested by local trypsinization followed by successive expansion.
For the characterization of Bcl-2 clones, cells were harvested, washed and used for the preparation of RNA. RNA was quantified using UV absorption at 260/280 nm, and RNA integrity was checked by assessing the sharpness of ribosomal RNA bands on a native 1% agarose gel. Bcl-2 clones were investigated for Bcl-2 expression using Northern blot analysis, with a 32P-labelled full-length (1.9 Kbp) Bcl-2 cDNA-probe, and using a human Bcl-2 ELISA kit.
5.7 RT- PCR
For the characterization of transport and efflux systems, approximately 7 106 filter-grown 2/4/A1 cells were harvested using ice-cold phosphate-buffered saline solution and a cell scraper. Cells were kept on ice during the scraping procedure and subsequently recovered by centrifugation. 2/4/A1 cells and the rat ileal tissue (positive control) were homogenized, the RNA was extracted and checked for absence of contaminating genomic DNA as previously described (44). Also, PCR primer-design and reaction conditions were as previously described (44).
5.8 Cell attachment
For the cell attachment assay, 2/4/A1 cells were seeded on plastic chamber slides coated with extracellular matrix proteins and were further studied after 24 hours incubation. The plastic chamber slides were coated with collagen I, fibronectin, mouse laminin, or a mixture of entactin, collagen IV and laminin (ECL).
5.9 Analysis of toxicity and cell death
5.9.1 DNA staining
2/4/A1 cells were seeded on plastic chamber slides coated with ECL. After 24 hours, the medium containing non-attached cells was replaced with fresh medium. After four
25

days in culture, the cells were washed, fixed and permeabilized. The cells were stained with propidium iodide. The slides were examined under a fluorescence microscope. Cells containing highly condensed chromatin and irregular nuclear DNA were defined as dying (160). Cells with homogeneous DNA staining throughout a regularly shaped nucleus were considered normal.
5.9.2 Dehydrogenase activity
In paper II, the effect of G418 on cell viability was assessed by measuring the intracellular dehydrogenase activity using the MTT method (161) as described previously (162). 2/4/A1 cells were seeded in the presence of different concentrations of G418 in the culture medium and incubated at 39˚C for 48 hours before carrying out the MTT assay.
In paper V, The effect of the peptides on cell viability was assessed by measuring the intracellular dehydrogenase activity of Caco-2 cells using the MTT method as described above. Caco-2 cells were seeded in the presence of different concentrations of occludin peptide and incubated for 6 hours before carrying out the MTT assay.
5.9.3 Lactate dehydrogenase leakage
The effect of the peptides on membrane integrity was assessed by measuring the amount of lactate dehydrogenase (LDH) leakage. Caco-2 cell monolayers were incubated with peptide for 6 hours. LDH leakage into the apical and basolateral solutions was determined using an LDH release kit.
5.10 Cell monolayer integrity and drug transport studies
5.10.1 Electrophysiological measurements
The transepithelial electrical resistance (TER) of 2/4/A1 and Caco-2 cell monolayers grown on permeable supports was measured with an Endohm tissue resistance measurement chamber connected to an Evohm resistance meter. The monolayers were equilibrated for 20-25 minutes before TER measurements.
5.10.2 Drug permeability studies
All experiments were carried out at 37°C in HBSS as described previously (96). The solutions were preheated to 37°C and the experiments were performed at 37 1°C on a heating plate. The cell monolayers were washed with preheated HBSS and equilibrated in the same buffer for 20-25 minutes prior to the transport experiments. The filters were then transferred to wells containing fresh preheated HBSS. The compounds were generally dissolved in HBSS 25 mM HEPES at pH 7.4 to a final concentration of 0.1-5 mM. In paper IV, MES 10 mM (pH 5.0 – 6.5) and HEPES 25 mM (pH 7 – 8.0) was used to control the apical pH. The amount of each particular compound used was based on its solubility, detection limit, presence of saturable active transport mechanisms, and effects of the compound on monolayer integrity.
26

The drug solution was added to the donor side of the cell monolayers and the inserts were placed on a calibrated shaker and kept at 37°C throughout the experiment. At 4 – 6 regular time intervals, samples were taken from the receiver solution and replaced with an equal amount of HBSS. Radioactive samples were analyzed using a liquid scintillation counter and the unlabeled samples were analyzed using reversed-phase HPLC.
5.11 Calculations
In paper I-III and V, the apparent permeability coefficient (Papp, cm/s) was determined according to the following equation:
PappK Vr
A 1
where K is the steady state rate of change in concentration in the receiver chamber (Ct/C0) versus time (s), Ct is the concentration in the receiver compartment at the end of each time interval, C0 is the initial concentration in the apical chamber at each time interval (mole/ml), Vr is the volume of the receiver chamber (ml) and A is the surface area of the filter membrane (cm2).
In paper IV, the apparent permeability coefficients (Papp, cm/s) were calculated using the non-sink condition analysis that is general and imposes less restriction on the experimental conditions than the analysis assuming the previously used sink conditions (77, 163).
CR(t) = [M/(VD + VR)] + [CR,0 - M/(VD + VR)] × e-Papp × A × (1/VD + 1/VR) × t 2
where VD is the volume in the donor compartment, CR,0 is the drug concentration in the receiver compartment at beginning of the interval and t is the time from the start of the interval. The sampling procedure necessitates the recalculation of M and CR,0for each succeeding interval according to mass balance. Papp for model drugs were then determined by nonlinear regression, minimizing the sum of squared residuals ( (CRi,obs – CRi,calc)2), where CRi,obs is the observed receiver concentration at the end of interval i and CR,i,calc is the corresponding concentration calculated according to eq. 1.
Subsequently, the membrane permeability coefficients (Pm) of the drugs were calculated by determining Papp at two different stirring rates (V, 100 rpm and 500 rpm). Pm was calculated from the slope of the linear relationship V/Papp and V as described previously (96).
VPapp
1K
1Pc
1Pf 3
V
27

28
where Pf is the calculated permeability coefficient of the filter support and K is a constant.
The sigmoidal equation:
FA = 100
1+ Pc
Pc50
γ 4
was fitted to the drug transport data. FA is the fraction of a drug absorbed in humans after oral administration, Pc50 is the Pc at an FA value of 50% and γ is a slope factor. The equation was fitted to the data by minimizing the unweighted sum of square residuals.
The molecular hydrodynamic radii of the marker molecules were estimated from the cubic root of the molecular weight of each compound multiplied by a constant K, which was given by the ratio between the radius of mannitol (3.6 Å) and the cubic root of the molecular weight of mannitol (MW 182).
3 MWKr ⋅= 5
This approximation of molecular radii assumes that all molecules have the same hydrodynamic shape in HBSS at pH 7.4.
The average pore radii of the tight junctions in 2/4/A1 and Caco-2 cells were calculated using the pore-restricted diffusion equation, also named the “Renkin function”. This equation describes a mathematical model of the diffusion of a solute in a cylinder (89, 164):
)/( RrFDPp ⋅⋅=δε
6
where Pp is the paracellular permeability of the solute, ε is the porosity of the cell monolayer, δ is the tortuousity times the path length, D is the diffusion coefficient and F(r/R) is the Renkin equation where r is the radius of the solute and R is the radius of the cylinder:
⋅−
⋅+
⋅−⋅
−=
532
95.009.2104.211)/(R
r
R
r
R
r
R
rRrF 7
ε/δ and R are constant for any given cell monolayer. It is thus possible to get an estimate of these constants by measuring the permeability for small hydrophilic molecules and fitting the data to Equation 6.

5.12 Statistics
The results were expressed as mean values SD. One-way ANOVA was used to compare means. A 95% probability was considered significant. The root mean square error (RMSE), i.e. the standard error of regression, and the coefficient of determination (r2) were used to measure how well the sigmoidal equation fits the observations.
Rank correlation coefficients, a measure of the association between two separate rankings of n items, were obtained using Spearman’s rank coefficient according to:
)(6
1 3 nnd
rs
2
8
where d is the difference of the two ranks associated with each compound and n is the number of compounds. The value rs is always between -1 and +1. Large rs values indicate greater association. Small rs values indicate less association.
29

6 RESULTS AND DISCUSSION
6.1 Characterization of 2/4/A1 (Papers I–III)
The basic steps in establishing and characterizing an epithelial cell line as a cell model for drug transport studies include studies of cell attachment on various supports, cell proliferation, differentiation, and monolayer integrity and function. The obvious prerequisite is that the cells form intact monolayers on permeable supports. In some cases the cell line (e.g. Caco-2) secretes its own extracellular matrix, but other cell lines (e.g. 2/4/A1, see section 6.1.2) require that an extracellular matrix is added to the permeable support to promote cell attachment. The cell proliferation rate is an important factor for practical reasons. The population doubling time of the Caco-2 and 2/4/A1 cell lines are 30-35 h. These growth rates enable the necessary expansion of the cultures within a reasonable time frame. An example of a cell population doubling taking too long is that of tsFHI cells, the human analogue to 2/4/A1, which have a population doubling time of approximately 7 days (165). This makes this otherwise promising cell line unsuitable for routine drug permeability studies. In this thesis, the development of a cell culture model based on the tsFHI cells was initially considered, in order to establish a human counterpart to 2/4/A1. However, this idea had to be abandoned, due to the slow growth of the tsFHI cells. The integrity of the cell monolayers can be measured by TER. From a drug delivery point of view, a more preferable way of measuring the integrity is testing the permeability to various paracellular markers. For tight epithelial cell monolayers (e.g. Caco-2), the permeability to a low MW paracellular marker such as mannitol is an adequate measure of the integrity. For leaky epithelial cell lines such as 2/4/A1, permeability to a series of paracellular markers that span a range of MWs has to be studied at least initially, to ensure the size-discriminating capacity of the cell monolayer. Microscopic evaluation of selected epithelial markers reveals the differentiation pattern of the cell line.
The properties of the foetal rat intestinal cell line 2/4/A1 depend on culture temperature, since this cell line was conditionally immortalized by tsSV40T (166). Therefore the first part of the characterization was to study the temperature-dependent expression of this oncogene. Since the temperature dependent differential expression of SV40T influence many properties of 2/4/A1, the rest of the characterization was also carried out at 33ºC (where the oncogene is fully active), 37ºC (where the oncogene seems to be partly active) and 39ºC (where the oncogene is inactive).
6.1.1 Expression of SV40 large T antigen (Paper I)
The temperature-dependent expression of SV40T was studied by using an antibody raised against SV40T. Immunofluorescence microscopy showed that the SV40 large T antigen was expressed in an immunoreactive form to a varying degree in all cells (Fig. 6), and that the antigen was relatively evenly distributed in all cell nuclei at 33°C. At 37°C all cells still showed expression of SV40 large T but the staining was fainter, indicating a lower degree of expression or, more likely, immunoreactivity. At 39°C only some fragments of the nuclei were stained in an irregular pattern. These results indicate a gradual loss of immunoreactivity of the mutated SV40 large T
30

antigen with an increase in temperature, which is consistent with the reported temperature sensitivity of this antigen (167).
Figure 6. Immunofluorescence staining of temperature-sensitive SV40 large T antigen in 2/4/A1 cells after four days of culture at 33°C (A), 37°C (B) and 39°C (C) on ECL. The immunostaining of the antigen decreased with increasing temperature, consistent with the temperature sensitivity of the antigen. Bar=10 m
6.1.2 Cell attachment and extracellular matrix (Paper I)
When grown at 33°C, 2/4/A1 cells attached readily to cell culture-treated plastics. When the culture temperature was increased to either 37°C or 39°C, the cells also attached initially, but after approximately 2-3 days at 37°C and after 24 hours at 39°C they started to detach. One explanation for this phenomenon may be anoikis, a form of apoptosis induced by lack of proper cell attachment (168). In order to promote better cell attachment, a number of commercially available extracellular matrices were tested. At all temperatures significantly higher numbers of 2/4/A1 cells attached to fibronectin, laminin and ECL-coated supports than to the uncoated supports or to supports coated with collagen I and IV. Since preliminary results indicated that fibronectin reduced the survival of 2/4/A1 cells and since cells grown on laminin formed diffuse cell borders (data not shown) the ECL-coated supports were selected for all further studies (Fig. 7).
Figure 7. Attachment of 2/4/A1 cells to plastic (P), collagen I (C I), collagen IV (C IV), fibronectin (F), laminin (L) and ECL at 33°C (A), 37°C (B) and 39°C (C). At all temperatures attachment rates were better to fibronectin, laminin and ECL than to plastic or the collagens. *p<0.05, **p<0.01 vs plastic (calculations by one-way analysis of variance (ANOVA)). Each bar represents the mean ± standard deviation (SD) of four experiments.
31

6.1.3 Monolayer formation (Papers I and II)
Morphological analysis using light microscopy showed that the 2/4/A1 cells formed continuous multi-layers after 4 days of culture grown on ECL-coated supports at 33°C. At 37°C the cells formed monolayers within 24 hours and remained as intact monolayers for at least 10 days in culture. When seeded onto ECL at 39°C, 2/4/A1 cells initially formed monolayers comparable to those observed at 37°C. However, during the first 24 hours the monolayers of these cells developed defects and within the following 24 hours in culture, cell detachment was observed. The cell detachment increased over the next 4 days in culture, after which <50% of the surface area of the support was covered with cells.
Examination of the cell nuclei after propidium iodide staining showed that at 33°C the 2/4/A1 cells grew and divided at a high cell density, which is consistent with the formation of multi-layers observed in the light microscope. The cell nuclei of 2/4/A1 cells grown at 37°C were more evenly distributed, supporting the finding that the cells form monolayers at this temperature. The cell nuclei had a normal oval morphology at both 33° and 37°C. In contrast, many of the cells grown at 39°C displayed condensed and fragmented nuclear DNA, indicating that many cells died at this temperature (Cell death is further discussed in section 6.1.7).
6.1.4 Development of tight and adherence junctions (Papers I and III)
When the original cell culture procedure was used (Paper I), the tight junction protein ZO-1 was present in 2/4/A1 cells grown at all three temperatures (Fig. 8A-C). Its distribution was less well ordered in cells grown at 33°C than in cells grown at 37°C. At 37°C, ZO-1 formed a continuous network. By contrast, after cultivation of 2/4/A1 cells at 39°C the ZO-1 network became discontinuous, indicating that the cell-to-cell contacts had loosened. Similarly the protein E-cadherin, which is associated with adherence junctions, was present at all temperatures (Fig. 8D-F). E-cadherin formed a diffusely distributed dot-like network at 33°C. At 37°C the dot-like network was more marked and was located closer to the apical cell membrane and the cell junctions. At 39°C the E-cadherin network at the cell junctions was not as distinct as that observed at 37°C. Actin filaments (Fig. 8G-I) were found at the cell borders, but also appeared as stress fibres at 33°C. At 37°C actin had redistributed to the peri-junctional area of the terminal web. At 39°C the actin network showed a broadening of extracellular spaces and defects in the monolayer. These findings indicate that 2/4/A1 cells grown at 37°C form monolayers consisting of polarized epithelial cells with typical junctional complexes, containing ZO-1 at tight junctions and E-cadherin at areas of cell-cell contact.
32

Figure 8. Immunofluorescence staining of ZO-l (A, B, C), E-cadherin (D, E, F) and F-actin (G, H, I) in 2/4/A1 cells four days after seeding on ECL at 33°C, 37°C and 39°C as described in Paper I. The ZO-1 distribution was visualized using an rabbit polyclonal antibody to ZO-1 protein, E-cadherin was visualized using a mouse monoclonal Ig antibody to human E-cadherin and the F-actin distribution was visualized using direct fluorescence with rhodamine-conjugated phalloidin. In contrast to the cells grown at 33°C and 39°C, cells at 37°C had a continuous network of tight and adherence junction-associated proteins. Images were obtained by confocal laser scanning microscopy. Bar=10 m.
When the modified cell culture procedure was used (Paper II), the tight junction protein occludin was expressed at the transcriptional level in equal amounts in both undifferentiated 2/4/A1 cells (grown at 33°C) and differentiated 2/4/A1 cells (grown at 39°C). By contrast, the expression of the claudins depended on the differentiation level of the 2/4/A1 cells. Claudin 1 was more abundantly expressed in differentiated 2/4/A1 cells grown at 39°C, while claudin 3 was more abundantly expressed in undifferentiated 2/4/A1 cells grown at 33°C.
Transmission electron microscopy confirmed the findings of the light and fluorescence microscopy. At 33°C, 2/4/A1 cells grew as multi-layers two to four cells thick, while at 37°C monolayers of cuboidal cells connected by tight junctions were observed (Fig. 9). Cells grown at 39°C showed the morphology of dying cells with irregular and shrunken cell nuclei and defects in the cell monolayers.
33

Figure 9. Transmission electron micrographs of tight junctions and the intercellular spaces of 2/4/A1 (A) and Caco-2 cells (B). 2/4/A1 and Caco-2 cells were cultivated on permeable supports at 37°C for 4 and 21 days respectively as described in paper I. 2/4/A1 cells form tight junctions (arrow) and relatively straight lateral spaces at 37°C but display few microvilli at the apical surface (A). Caco-2 cells form tight junctions (arrow) with tortuous lateral spaces and display a more differentiated morphology with numerous microvilli. Magnification 32,000 (A) and 11,000 (B).
In summary, these data show that 2/4/A1 cell monolayers express several components of functional tight junctions. However, the expression of perhaps the most important class of tight junction proteins, the claudins, indicates that 2/4/A1 cells preferentially express claudin 1 while the other investigated claudins are expressed to a lesser extent or not at all. It is difficult to speculate what may be the consequences of this, but recent data suggest that the expression of various claudins gives rise to special tissue-specific tightness and selectivity of the tight junctions (169).
6.1.5 Transepithelial electrical resistance (Papers I and II)
In Paper I the studies of the temperature-dependent differentiation showed that 2/4/A1 cells grown on ECL formed monolayers of viable cells only at the intermediate temperature of 37°C. Therefore, 2/4/A1 cells grown at this temperature were selected for further functional studies on the integrity and permeability of the cell monolayers. The TER in 2/4/A1 cells increased with time and reached a plateau value of 25 cm2
after 4 days in culture (Fig. 10A). The TER was maintained for at least 12 days. This TER value may seem low but lies within the range of TER values obtained in segments of the rat jejunum (30). By comparison, the TER in Caco-2 cell monolayers reached a plateau of 234 cm2 and maintained this value for at least 40 days in culture (Fig. 10B), which is in agreement with previous findings (72). The TER of confluent 2/4/A1 monolayers was therefore approximately ninefold lower than that of Caco-2 monolayers. The fact that the TER values of both 2/4/A1 and Caco-2 reach stable plateaus after 4 and 14 days, respectively, is an advantage, from a practical viewpoint, over short-term Caco-2 cultures in which the integrity is lost only a few days after reaching maximum TER. This plateau allows flexibility in planning of drug transport experiments.
34
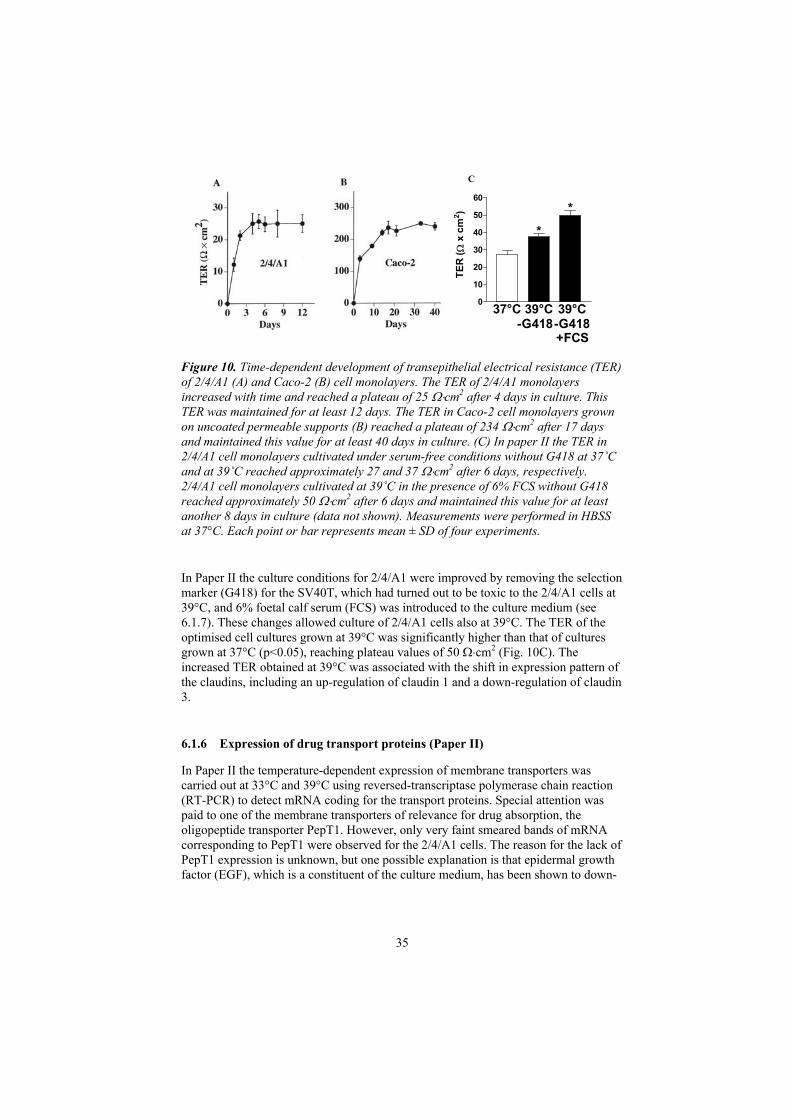
C
37°C 39°C -G418
39°C -G418 +FCS
0
10
20
30
40
50
60
TER
( x
cm
2 )
**
Figure 10. Time-dependent development of transepithelial electrical resistance (TER) of 2/4/A1 (A) and Caco-2 (B) cell monolayers. The TER of 2/4/A1 monolayers increased with time and reached a plateau of 25 cm2 after 4 days in culture. This TER was maintained for at least 12 days. The TER in Caco-2 cell monolayers grown on uncoated permeable supports (B) reached a plateau of 234 cm2 after 17 days and maintained this value for at least 40 days in culture. (C) In paper II the TER in 2/4/A1 cell monolayers cultivated under serum-free conditions without G418 at 37˚Cand at 39˚C reached approximately 27 and 37 cm2 after 6 days, respectively. 2/4/A1 cell monolayers cultivated at 39˚C in the presence of 6% FCS without G418 reached approximately 50 cm2 after 6 days and maintained this value for at least another 8 days in culture (data not shown). Measurements were performed in HBSS at 37°C. Each point or bar represents mean ± SD of four experiments.
In Paper II the culture conditions for 2/4/A1 were improved by removing the selection marker (G418) for the SV40T, which had turned out to be toxic to the 2/4/A1 cells at 39°C, and 6% foetal calf serum (FCS) was introduced to the culture medium (see 6.1.7). These changes allowed culture of 2/4/A1 cells also at 39°C. The TER of the optimised cell cultures grown at 39°C was significantly higher than that of cultures grown at 37°C (p<0.05), reaching plateau values of 50 cm2 (Fig. 10C). The increased TER obtained at 39°C was associated with the shift in expression pattern of the claudins, including an up-regulation of claudin 1 and a down-regulation of claudin 3.
6.1.6 Expression of drug transport proteins (Paper II)
In Paper II the temperature-dependent expression of membrane transporters was carried out at 33°C and 39°C using reversed-transcriptase polymerase chain reaction (RT-PCR) to detect mRNA coding for the transport proteins. Special attention was paid to one of the membrane transporters of relevance for drug absorption, the oligopeptide transporter PepT1. However, only very faint smeared bands of mRNA corresponding to PepT1 were observed for the 2/4/A1 cells. The reason for the lack of PepT1 expression is unknown, but one possible explanation is that epidermal growth factor (EGF), which is a constituent of the culture medium, has been shown to down-
35

regulate the expression of PepT1 (170). However, EGF could not be removed from the culture medium without causing loss of integrity.
In contrast to the expression of PepT1, both genes that code for P-gp in rodents, MDR1a and MDR1b, were expressed in 2/4/A1. Neither MRP2 nor MRP3 mRNA was detected in 2/4/A1 samples. RT-PCR was a valuable tool for detecting transcripts, but had to be confirmed at the functional level, since a proper expression of mRNA does not always correlate with proper function of the corresponding protein. Functional assays for PepT1, P-gp, MRP2 and BCRP are discussed in section 6.2.4.
6.1.7 Cell death (Papers I and II)
In Paper I the cells died soon after seeding to permeable supports at 39°C and the dying cells contained highly condensed chromatin and irregular nuclear DNA. However, cells grown at an intermediate temperature of 33°C and 37°C were viable. In Paper II the cause of the cell death at 39°C was confirmed to be apoptosis, as seen by the fragmentation pattern of DNA at this temperature. The finding that the cell death of 2/4/A1 cells at 39°C was caused by apoptosis agrees with observations in other cell lines immortalized by tsSV40T (171-174). Thus, cell death by apoptosis appears to be an inherent feature of these cell lines. The apoptosis in tsSV40T-expressing cell lines has been attributed to the release of the p53 tumour suppression protein upon inactivation of SV40T at 39°C (171). Despite the fact that apoptosis may have been an inherent feature of cells immortalized with tsSV40T, attempts were made to overcome apoptosis and stabilize the 2/4/A1 cells at 39°C. First, the anti-apoptotic gene Bcl-2 was over-expressed in 2/4/A1 cells.
Successful inhibition of apoptosis by over-expression of Bcl-2 in cell lines immortalized by tsSV40T had previously been shown in kidney tubule (175) and hippocampal neuronal (176) cell lines. Therefore in Paper II a plasmid-containing human Bcl-2 cDNA was introduced to 2/4/A1 cells by means of electroporation in order to inhibit the apoptosis in this cell line. Northern blots of the resulting clones showed that three out of 37 clones expressed Bcl-2 at different levels. The one clone expressing the highest levels of Bcl-2 mRNA (2/4/A1/Bcl-2) also showed expression at the protein level as seen by enzyme-linked immunosorbent assay (ELISA). Visual inspection in the confocal microscope showed that each 2/4/A1/Bcl-2 cell covered a large surface area and displayed a flat rather than cuboidal or columnar morphology at 33°C, 37°C and 39°C. The continuous 2/4/A1/Bcl-2 cell monolayers reached TER values of only 16 cm2 when grown for 4 days at 39°C.
In another attempt to overcome apoptosis, extracellular factors that induce apoptosis were identified in the serum-free cell culture medium for 2/4/A1 cells, which was used in Paper I. Thus, it was found that the toxicity of SV40T selection marker G418 turned out to be temperature-dependent. A concentration-dependent reduction of the intracellular dehydrogenase activity with an IC50 value of 18 µg/ml was observed for 2/4/A1 cells at 39°C, while G418 was non-toxic for 2/4/A1 cells at 33°C or 37°C (IC50 >400 µg/ml). For this reason G418 was removed from the cell culture medium. Serum deprivation may induce apoptosis (172); consequently, increasing amounts of FCS were added to the culture medium. At serum concentrations of >6% intact cell monolayers of fully viable cuboidal cells were obtained that survived for at least 14 days at 39°C.
36

In summary, these results show that over-expression of Bcl-2 inhibited apoptosis and allowed the formation of intact cell monolayers. However, the undesirable change in morphology and the comparably low barrier function of the 2/4/A1/Bcl-2 cells make these cells unsuitable for studies of drug transport. In contrast, the modified culture protocol allowed formation of intact 2/4/A1 cell monolayers at 39°C that were used in all further studies in Papers II-IV.
6.2 Transport of drugs and hydrophilic markers (Papers I – IV)
Once 2/4/A1 was established as and could be cultivated on permeable supports, the main focus of the first part of this thesis was to study and evaluate this cell line as an epithelial cell model for drug permeability studies. In paper I, the integrity to hydrophilic markers was studied, and a relationship between FA after oral administration to humans and Pc was established for a diverse set of passively transported drugs. With the improved cell culture procedure presented in paper II, the permeability to hydrophilic drugs was marginally reduced compared to in paper I, while the permeability to lipophilic drugs remained unchanged. The focus of paper II was to study active drug transport. In paper III, the average pore radius of 2/4/A1 was assessed by studying the permeability of a set of small hydrophilic markers. The performance of 2/4/A1 in predicting the intestinal permeability to incompletely absorbed drugs was also studied. In paper IV, by varying apical pH, the 2/4/A1 cell monolayers were used in parallel to artificial membranes and Caco-2 to disclose the contribution of the paracellular route to the observed pH dependent drug permeability.
6.2.1 Hydrophilic markers and average pore radius (Papers I and III)
In Paper I, the permeability to the low-MW markers mannitol, fluorescein and Lucifer yellow, and the high-MW markers PEG 4 000 and dextran 50 000 decreased with increasing size of the marker molecule, and was comparable to that obtained for hydrophilic markers in the human small intestine (61, 177). The permeabilities ranged from 15.5 2.09 10-6 cm/s for the smallest marker molecule mannitol (MW 182) to 0.40 0.03 10-6 cm/s for the largest marker molecule, dextran (MW 50,000), or 38-fold. By comparison, for Caco-2 monolayers, the Papp values ranged only 5-fold. Thus, the permeability of confluent 2/4/A1 monolayers to the hydrophilic marker molecules depends more strongly on molecular weight than that of Caco-2 monolayers, despite that the values were significantly higher in 2/4/A1 than in the Caco-2 monolayers (8-62 times) (Fig. 11).
37

Figure 11. Molecular weight-dependent permeability (Papp) of 2/4/A1 (A) and Caco-2 cell monolayers (B) to the hydrophilic marker molecules mannitol (M) (MW 182), fluorescein (F) (MW 376), Lucifer yellow (L) (MW 450), PEG (P) (MW 4 000) and dextran (D) (MW 50 000) at 37°C. Note the 100-fold difference in scale on the y-axis in the two figures. Each bar represents the mean ± SD of six experiments.
In Paper III the transport of low-MW hydrophilic marker molecules (MW 60–504) was studied in 2/4/A1 and Caco-2 monolayers in order to determine the average pore radius of these cell monolayers. The permeability to the paracellular marker molecules of 2/4/A1 and Caco-2 monolayers differed both qualitatively and quantitatively (Fig. 12). Although both cell models discriminated the paracellular markers in a size-dependent manner, the Pc values were 10–190-fold higher in 2/4/A1 than in Caco-2 cell monolayers and the fitted curve was more shallow in 2/4/A1 than in Caco-2 cell monolayers. The resulting average pore radii in the two cell models were 9.0 0.2 Å and 3.7 0.1 Å for 2/4/A1 and Caco-2 cell monolayers, respectively.
2 3 4 50
10
20
30
402/4/A1Average pore radius: 9.0 Å
Radius (Å)
P c x
106 (c
m/s
)
A
2 3 4 50
10
20
30
40
Caco-2Average pore radius: 3.7 Å
Radius (Å)
P c x
107 (c
m/s
)
B
2 3 4 50
10
20
30
402/4/A1Average pore radius: 9.0 Å
Radius (Å)
P c x
106 (c
m/s
)
A
2 3 4 50
10
20
30
40
Caco-2Average pore radius: 3.7 Å
Radius (Å)
P c x
107 (c
m/s
)
B
Figure 12. Determination of average pore radii in 2/4/A1 and Caco-2 cell monolayers. The permeability coefficients of urea (MW 60), creatinine (MW 113), erythritol (MW 122), mannitol (MW 182), foscarnet (MW 126), clodronate (MW 244) and lactulose (MW 382) were determined in 2/4/A1 (A) and Caco-2 (B) cell monolayers. The effective hydrodynamic radii of the molecules were determined using Equation 5. The two curves represent the best fit to Equation 6 in the respective cell model (n = 4).
38

The seemingly contradictory results obtained in Paper I and III were in fact a result of the choice of markers in the two studies. In Paper I the molecular radii of the markers spanned the average pore radius of 2/4/A1, while they were all equal to or larger than the average pore radius of Caco-2. This explains the stronger size selectivity of 2/4/A1 in Paper I. Accordingly in Paper III the molecular radii of the markers spanned the average pore radius of Caco-2, while they were all smaller than the average pore radius of 2/4/A1. This explains the stronger size selectivity of Caco-2 in Paper III. Consequently, 2/4/A1 is more selective for the molecular radius of large molecules and Caco-2 is more selective for that of smaller molecules.
6.2.2 Rapidly absorbed drugs (Paper I)
In Paper I, Pc values for a series of 17 drugs and markers were determined in 2/4/A1 cell monolayers and compared with those obtained in Caco-2 cells (Fig. 13). Out of the 17 drugs five drugs have reported FA values >90%. This cut-off value constitutes the limit between completely and incompletely absorbed drugs according the Biopharmaceutical classification system (BCS) (178). These drugs are relatively lipophilic (ClogP 0.41–2.86) and are therefore presumably absorbed via the transcellular route. Any difference between the Pc values in 2/4/A1 and in Caco-2 cells for the rapidly transported drugs would disclose differences in the phospholipid barrier between the cell lines. However, only small differences could be seen between the Pc values in the two cell lines. By fitting a sigmoidal equation to the experimental data, cut-off values could be estimated, and the resulting cut-off value for drugs with a FA >90% was identical in 2/4/A1 and Caco-2 cells (Pc >55 x 10-6 cm/s). Therefore the transcellular route in 2/4/A1 and in Caco-2 cell monolayers was considered to be comparable, and differences in Pc values were attributed to differences in either functional expression of active drug transport proteins or in the paracellular route.
Figure 13. Relationship between the absorbed fraction of structurally diverse sets of orally administered drugs and permeability coefficients obtained in 2/4/A1 monolayers (diamonds), Caco-2 monolayers (circles) (5, 179) and after in vivo perfusion of the human jejunum (triangles) (180). Sigmoidal relationships were obtained between fraction absorbed and the permeability coefficients in all models.
39

40
6.2.3 Poorly and intermediately absorbed drugs (Papers I and III)
In paper I, the remaining 12 compounds had FA values <90%. The ClogP values of these ranged from –8.09 – 4.50 and the molecular weights ranged from 113 – 504. In contrast to the subset of drugs that were completely absorbed, the Pc values for the incompletely absorbed drugs were 40 - 300 times higher in 2/4/A1 than in Caco-2(Fig. 13). The sigmoidal relationship between FA and Pc for 2/4/A1 cells is thus steeper than for Caco-2. Despite this difference, the distinction between completely and incompletely absorbed drugs was identical in the two models. Based on the observations that the permeability of the hydrophilic markers were 8 – 62 fold higher, and that TER was 9 fold lower in 2/4/A1 compared to Caco-2, the difference in permeability to the incompletely absorbed compounds was attributed to the higher permeability of the paracellular spaces in 2/4/A1. Most encouragingly, the sigmoidal relationship between FA and permeability was similar to that found after perfusion of the human jejunum in vivo. The quantitative overlap in permeability between 2/4/A1 cell monolayers and the human jejunum warranted a further examination of the properties of the 2/4/A1 cell monolayers.
In Paper III, the aim was to investigate if the 2/4/A1 cell monolayers could predict the intestinal permeability of incompletely absorbed drugs, using Caco-2 cell monolayers as a reference. In particular, drugs that have a FA of less than or equal to 80% were included in this study. This cut-off value was chosen since it constitutes the border between high and low permeability drugs in drug discovery settings (181). In total, 13 drugs were investigated in this part of the study. Eight of these drugs were categorized as sparingly absorbed (FA = 0 to 20%) and 5 drugs were categorized as intermediately absorbed (FA = 20 to 80%). These drugs were used to establish a relationship between FA and Pc in 2/4/A1 and Caco-2. The relationships were then used to predict FA for an additional set of drugs (defined as an ad hoc test set) (Fig. 14).
Figure 14. Sigmoidal relationships between FA and permeability coefficients in 2/4/A1 (A) and Caco-2 (B) cell monolayers. Open symbols represent drugs in the training set and solid symbols represent drugs in the ad hoc test set. The two curves represent the best fit to equation 4 for the training set.
The selection of these compounds was based on the observation that their FA values are particularly difficult to predict using Caco-2 cell monolayers. The average errors in the prediction of FA for the drugs in the test set were RMSEtest=15.6 and 21.1 for
-8 -7 -6 -50
25
50
75
Log Pc Caco-2 (cm/s)
FA
(%
)
-5.50 -5.25 -5.00 -4.75 -4.500
25
50
75
Log Pc 2/4/A1 (cm/s)
FA
(%
)
A B
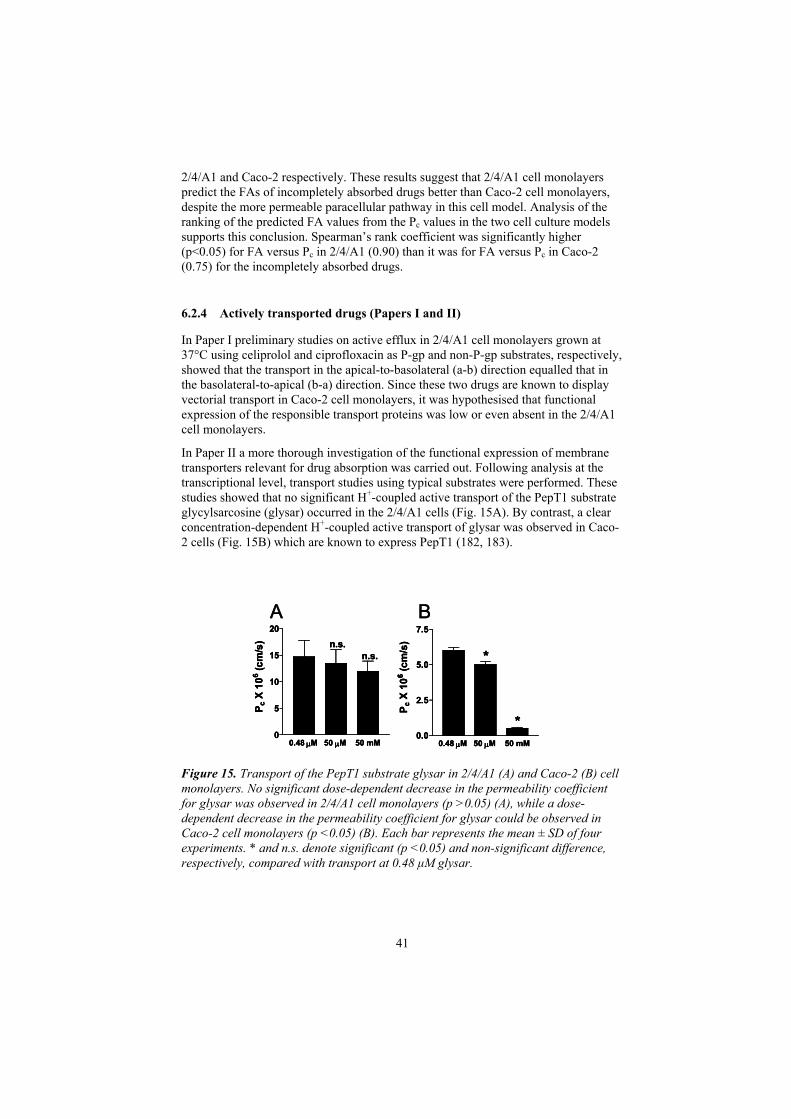
2/4/A1 and Caco-2 respectively. These results suggest that 2/4/A1 cell monolayers predict the FAs of incompletely absorbed drugs better than Caco-2 cell monolayers, despite the more permeable paracellular pathway in this cell model. Analysis of the ranking of the predicted FA values from the Pc values in the two cell culture models supports this conclusion. Spearman’s rank coefficient was significantly higher (p<0.05) for FA versus Pc in 2/4/A1 (0.90) than it was for FA versus Pc in Caco-2 (0.75) for the incompletely absorbed drugs.
6.2.4 Actively transported drugs (Papers I and II)
In Paper I preliminary studies on active efflux in 2/4/A1 cell monolayers grown at 37°C using celiprolol and ciprofloxacin as P-gp and non-P-gp substrates, respectively, showed that the transport in the apical-to-basolateral (a-b) direction equalled that in the basolateral-to-apical (b-a) direction. Since these two drugs are known to display vectorial transport in Caco-2 cell monolayers, it was hypothesised that functional expression of the responsible transport proteins was low or even absent in the 2/4/A1 cell monolayers.
In Paper II a more thorough investigation of the functional expression of membrane transporters relevant for drug absorption was carried out. Following analysis at the transcriptional level, transport studies using typical substrates were performed. These studies showed that no significant H+-coupled active transport of the PepT1 substrate glycylsarcosine (glysar) occurred in the 2/4/A1 cells (Fig. 15A). By contrast, a clear concentration-dependent H+-coupled active transport of glysar was observed in Caco-2 cells (Fig. 15B) which are known to express PepT1 (182, 183).
0.48 M 50 M 50 mM0
5
10
15
20
P c X
106 (c
m/s
) n.s.n.s.
0.48 M 50 M 50 mM0.0
2.5
5.0
7.5
P c X
106 (c
m/s
)
*
*0.48 M 50 M 50 mM
0
5
10
15
20
P c X
106 (c
m/s
) n.s.n.s.
0.48 M 50 M 50 mM0.0
2.5
5.0
7.5
P c X
106 (c
m/s
)
*
*0.48 M 50 M 50 mM
0
5
10
15
20
P c X
106 (c
m/s
) n.s.n.s.
0.48 M 50 M 50 mM0.0
2.5
5.0
7.5
P c X
106 (c
m/s
)
*
*
Figure 15. Transport of the PepT1 substrate glysar in 2/4/A1 (A) and Caco-2 (B) cell monolayers. No significant dose-dependent decrease in the permeability coefficient for glysar was observed in 2/4/A1 cell monolayers (p >0.05) (A), while a dose-dependent decrease in the permeability coefficient for glysar could be observed in Caco-2 cell monolayers (p <0.05) (B). Each bar represents the mean ± SD of four experiments. * and n.s. denote significant (p <0.05) and non-significant difference, respectively, compared with transport at 0.48 µM glysar.
41

42
Further, there was no vectorial transport in the b-a direction across 2/4/A1 cell monolayers for any of the substrates for the efflux systems investigated (P-gp, MRP2 and BCRP) (Fig. 16). In Caco-2 cell monolayers, on the other hand, all the substrates displayed several-fold higher permeability coefficients in the b-a than in the a-bdirection. Since Caco-2 cells express multiple drug efflux systems with overlapping substrate specificity, the efflux in more specific epithelial expression systems was also studied. Stable MDCK II and LLC-PK1 cell clones that over-expressed each of the efflux systems were used for this purpose. All of the typical substrates displayed higher efflux ratios (b-a/a-b) in the expression systems than in the correspondinguntransfected cell lines.
Figure 16. Permeability coefficients in the a-b (open bars) and b-a (closed bars) directions for the P-gp substrate digoxin (A), the MRP2 substrate vinblastine (B) and the BCRP substrate mitoxantrone (C) in 2/4/A1, Caco-2 (A–C), MDCK II (A and B), MDCK-MDR1 (A), MDCK-MRP2 (B), LLC-PK1 (C) and LLC-BCRP (C) cell monolayers. The P-gp inhibitor GF120918 (2 µM) was added in the studies of vinblastine transport in MDCK II and MDCK-MRP2. Each bar represents the mean ± SD of three to six experiments. * and n.s. denote a significant (p <0.05) and non-significant difference between a-b and b-a transport, respectively.
Therefore, despite 2/4/A1 cells expressing some drug transporters at the transcriptional level, the cells lacked functional expression of the investigated transport proteins. The reasons for this may be that the mRNA is not translated into protein, that the protein is not functional, or that it is not properly sorted into the apical cell membrane. This apparent drawback can, however, be advantageous in certain cases. For instance, when structure-permeability relationships are established for the passive drug transport routes, these data are not biased by active transport phenomena, which facilitates the design of the drug transport experiments.
6.2.5 pH dependent permeability (Paper IV)
The aim of Paper IV was to quantify the contribution of the paracellular route to the permeability across epithelial cell monolayers. In a previous study the ionized form of the drug was reported to have contributed significantly to the permeability in Caco-2cell monolayers at pH values where the fraction un-ionized (fu) of the drug was <0.1
Digoxin
2/4/
A1
Cac
o-2
MD
CK
MD
CK
-MD
R10
10
20
30
40
A
Pc
x 10
6 (c
m/s
)
Mitoxantrone
2/4/
A1
Cac
o-2
LL
C-P
K1
LLC
-BC
RP0
10
20
30
CVinblastine2/
4/A
1
Cac
o-2
MD
CK
MD
CK
-MR
P20
10
20
30
40
50
60
B
**
*
*
* *
**
n.s.n.s. **

43
(77). In this study the pH-dependent permeability of one rapidly transported drug, alfentanil, and one slowly transported drug, cimetidine (shown to be transported by the paracellular route in vivo), was investigated in three models of the intestinal epithelium (hexadecane-impregnated membranes (HDMs), the Caco-2 model and 2/4/A1 cells) (Fig. 17). These models vary in their permeability to slowly transported drugs, presumably owing to differences in their paracellular transport routes. Thetransport studies were carried out at apical pH values ranging from 5.0 to 8.0 (representing the pH interval of the GI). In this pH interval the fu of alfentanil varied from 0.031 to 0.969 and the fu of cimetidine, from 0.016 to 0.941.
Figure 17. Schematic illustrations of the membrane models used in Paper IV.
Sigmoidal relationships were obtained between the membrane permeability (Pm) and pH in all models, but the relationships were qualitatively different (Fig. 18). These differences became evident when the permeability to the ionized (Pmi) and the unionized (Pmu) forms of alfentanil and cimetidine were determined from the linear relationships obtained between Pm and fu. In HDM, no contribution from the ionized forms of the drugs was observed, whereas significant contribution was observed in the two cell models. The selectivity of the permeability to the unionized form over the ionized form was higher in Caco-2 than in 2/4/A1 for alfentanil (73.8 vs 18.4), as well as for cimetdine (10.1 vs 1.5). Thus the permeability properties of HDM, Caco-2 and 2/4/A1, with regard to the ionized form of at least hydrophilic drugs can be categorized as follows: HDM – no contribution, Caco-2 – small contribution, and 2/4/A1 – large contribution. A reasonable assumption is that the difference in selectivity between Caco-2 and 2/4/A1 is a consequence of the difference in the relative contribution from the paracellular pathways.
: Passive transcellular : Passive paracellular
Hexadecanemembrane Caco-2 2/4/A1
230 Ω⋅cm2 50 Ω⋅cm2
Non-paracellular Tight-paracellular Leaky-paracellular
: Passive transcellular: Passive transcellular : Passive paracellular: Passive paracellular
Hexadecanemembrane Caco-2 2/4/A1
230 Ω⋅cm2 50 Ω⋅cm2
Non-paracellular Tight-paracellular Leaky-paracellular
: Passive transcellular : Passive paracellular
Hexadecanemembrane Caco-2 2/4/A1
230 Ω⋅cm2 50 Ω⋅cm2
Non-paracellular Tight-paracellular Leaky-paracellular
: Passive transcellular: Passive transcellular : Passive paracellular: Passive paracellular
Hexadecanemembrane Caco-2 2/4/A1
230 Ω⋅cm2 50 Ω⋅cm2
Non-paracellular Tight-paracellular Leaky-paracellular

Cimetidine in Caco-2
4 5 6 7 8 90.02.55.07.5
10.0
pH
Cimetidine in 2/4/A1
4 5 6 7 8 9250
300
350
400
pH
Cimetidine in HDM
4 5 6 7 8 90.000.250.500.751.00
pH
P m (1
0-7 c
m/s
)A B C
Figure 18. Sigmoidal relationships between Pm for cimetidine and pH were obtained in all models. The values are presented as mean ± SD, n = 4.
In summary, the results of Paper IV show that 2/4/A1 cell monolayers can be used to disclose the dominant passive transport route. The tight Caco-2 cell monolayers may result in under-estimation of the paracellular route for small hydrophilic drugs.
6.3 Modulation of tight junction permeability (Paper V)
Paper V constitutes the second part of this thesis. In this paper, a new approach to tight junction modulation was taken. The aim of this study was to investigate whether peptides from the extracellular loops of the tight junction protein occludin could be used as a new principle for tight junction modulation by an extracellular mechanism. Peptides homologous to the extracellular sequences of human occludin were synthesized and tested for their ability to modulate the tight junction permeability in Caco-2 cell monolayers. First, the two peptides corresponding to the two complete extracellular loops of occludin were investigated. In the presence of the first loop peptide (OP90-135), the permeability to the paracellular marker mannitol increased approximately 3-fold after two hours. No increase in permeability could be observed after incubation with the second loop peptide (OP196-243) (Fig. 19).
44

Control OP90-135 OP196-2430
1
2
3
4
Occludin peptide
P app
x 1
07 (cm
/s)
Figure 19. After exposure to OP90-135 for two hours, the permeability to mannitol in Caco-2 cell monolayers increased 3-fold compared to control. The permeability to mannitol did not change after exposure to OP196-243. The values are presented as mean ± SD, n = 4
Next, the effects of two shorter peptides were studied. Two active peptides corresponding to the N-terminus of the first extracellular loop were identified. The 24 aa long peptide (OP90-113) was approximately 10 fold more potent than the 14 aa long peptide (OP90-103). Further truncation of the 14 aa peptide resulted in peptides that were inactive. These peptides were non-toxic as judged by the lactate dehydrogenase leakage and dehydrogenase activity assays. However, the peptides were fully active only when added to both the apical and basolateral sides of the Caco-2 cell monolayers, and the onset of action of these peptides was slow (hours).
Since tight junction modulators will be added to the apical (serosal) side of epithelial barriers in vivo, the reason for the lack of apical effect had to be identified. The stability of the occludin peptides showed that significant degradation occurred when the peptides were added to the apical but not when they were added to the basolateral side (Fig. 20). This degradation was most likely mediated by peptidases situated in the apical membrane. Another possible reason for the lack of apical effect could be that the accessibility to the tight junctions is more size-restricted from the apical side than from the basolateral side. The problem with size-restricted accessibility may have been further pronounced by the reported tendency of full-length OPs to form aggregates (134).
45

0 1 2 3 4 5 6 7405060708090
100110
OP90-103OP90-103 BL
OP90-103 AP + InhOP90-103 BL + Inh
OP90-103 AP
Time (hours)Fr
actio
n re
mai
ning
(%)
Figure 20. Degradation of OP90-103 after incubation with Caco-2 cells. After 6 hours, approximately 50% of the OP90-103 was degraded on the apical side, while no degradation could be observed for the basolateraly exposed OP90-103. The apical degradation of OP90-103 could be inhibited by a peptidase inhibitor cocktail (bestatin, 0.29 mM; diprotin A, 1 mM and captopril, 1 mM; (184)). The values are presented as mean ± SD, n = 4.
In order to increase accessibility of the OPs to the tight junctions from the apical side, a lipoamino acid derivative of the shortest active peptide, OP90–103, was synthesized. This conjugate has several potential advantages as compared with the free peptide. Firstly, the conjugation of a lipoamino acid of sufficient length (185, 186) would insert into the cell membrane and concentrate the peptide at the cell surface; secondly, it would release the active peptide through the action of brush border peptidases (i.e. at the site adjacent to the tight junctions); thirdly, it would limit degradation of the released peptide by apical peptidases (158); and fourthly, it would possibly also reduce aggregation.
The conjugation resulted in a racemic mixture of two diastereomers. After isolation of the isomers, the stability of D- and L-C14-OP90–103 added to the apical side of the Caco-2 cell monolayers was examined. The L-isomer released OP90–103 15 times faster (5.01±0.11 nmol/hr) than did the D-isomer (0.33±0.60 nmol/hr). Only intact OP90–103was released, indicating that the lipoamino acid protected the released OP90–103 from further degradation by the apical peptidases. Thus the conjugation solved the degradation problem.
Encouragingly, each of the isomers increased the permeability approximately 15-fold compared to control already 20 minutes after apical addition to Caco-2 cell monolayers. The permeability was further increased to approximately 40-fold compared to the control after 40 minutes for the L-isomer, while the effect of the D-isomer gradually returned to base line levels (Fig. 21A). The increase in permeability could be efficiently inhibited by peptidase inhibitors, indicating that release of native peptide from the conjugate was required for effect. The effect of the two isomers on TER was in close agreement with that observed for the effect on mannitol permeability with a rapid decrease in TER for both isomers after 10 minutes. The exposure of the L-isomer resulted in a further decrease that reached a plateau after approximately 20 minutes and at this time, the TER was reduced to 30% of control. The effect of the D-isomer on TER was reversible and returned to values comparable to controls within 20 minutes (Fig. 21B). C14-OP90-103 did not cause acute toxicity to Caco-2 cells at concentrations as high as 1 mM as judged by measurements of the
46

intracellular dehydrogenase activity. There was some concern that the free lipoamino acid could mediate some of the observed effect. However, the lipoamino acid presented alone or in combination with unconjugated peptide was inactive.
0 50 100 150 2000
10
20
30
40
50
Time (min)
P app
x 1
07 (cm
/s)
L-C14-OP90-103
D-C14-OP90-103Contr.
0 30 60 90 1200
100
200
300
400
500
Time (min)TE
R (
cm2 )
L-C14-OP90-103
Control
D-C14-OP90-103
A B
0 50 100 150 2000
10
20
30
40
50
Time (min)
P app
x 1
07 (cm
/s)
L-C14-OP90-103
D-C14-OP90-103Contr.
0 30 60 90 1200
100
200
300
400
500
Time (min)TE
R (
cm2 )
L-C14-OP90-103
Control
D-C14-OP90-103
0 50 100 150 2000
10
20
30
40
50
Time (min)
P app
x 1
07 (cm
/s)
L-C14-OP90-103
D-C14-OP90-103Contr.
0 30 60 90 1200
100
200
300
400
500
Time (min)TE
R (
cm2 )
L-C14-OP90-103
Control
D-C14-OP90-103
A B
Figure 21. (A) Time dependent studies showed that both L- and D-C14-OP90-103induced a rapid 15-fold increase in mannitol permeability. The activity was further increased to approximately 40-fold after 30 minutes for the L-isomer, while the effect reversed to base line levels with time for the D-isomer. (B) The effects on TER of the two isomers were in close agreement with those observed for mannitol permeability, with a rapid initial decrease in TER for both isomers followed by a return to control levels for the D-isomer while the L-isomer maintained a reduced TER for at least two hours. Values are presented as mean ± SD (n = 4).
In summary, in this study peptides originating from the first extracellular loop of occludin were shown to modulate the paracellular permeability of Caco-2 cell monolayers. A rapid apical effect was obtained after coupling of a lipoamino acid to the N-terminus of the peptide. The resulting D-isomer displayed a rapid and transient effect. The short duration of action limits unrestricted permeation of potentially toxic agents and also reduces the risk of harmful autoimmune responses. For sustained effect, the lipopeptide must rapidly release intact peptide by a mechanism mediated by apical peptidases, and this was the case only for the L-isomer. C14-OP90-103represents a prototype of a new class of tight junction modulators that act on the extracellular domains of tight junction protein occludin.
47

7 SUMMARY AND CONCLUSIONS
The results of the first part of this thesis show that the conditionally immortalized rat epithelial cell line 2/4/A1 can be used as a model epithelium for studies of passive drug transport. After 4 – 6 days on permeable supports, the integrity of the 2/4/A1 cell monolayers reaches a stable plateau that is maintained for more than a week. This plateau is reached much quicker for 2/4/A1 than for traditionally cultivated Caco-2 cell monolayers, which require approximately three weeks to stabilize. Both the passive transcellular and paracellular permeability of this cell line resembles those of the human small intestine, which will enable better quantitative predictions of intestinal drug permeability for both rapidly and slowly permeable drugs, than has been possible with Caco-2. 2/4/A1 cell monolayers lack the functional expression of several transport proteins that are relevant for intestinal drug absorption. Although an obvious disadvantage in studies of active drug transport, this feature may be an advantage in studies of structure permeability relationships for the passive transport routes. The 100-fold higher permeability for low-permeable drugs enables the use of more robust analytical techniques such as HPLC-UV since the transported amounts are larger in 2/4/A1 than in Caco-2, which often requires LC-MS analysis for low permeable drugs. Despite the approximately 100-fold higher permeability for slowly transported drugs in 2/4/A1 compared to that in Caco-2, the 2/4/A1 predicts and ranks the intestinal permeability to incompletely absorbed drugs better than does Caco-2. 2/4/A1 can also be used to study the influence of charge on the intestinal permeability.
The results of the second part of the thesis show that peptides, 14 and 24 aa in length, from the N-terminus of the first extracellular loop of occludin modulate the tight junction permeability in Caco-2 cells. The native peptides increased the tight junction permeability in a dose-dependent and non-toxic manner, but had to be added to the basolateral side of the cell monolayers. One reason for the lack of apical effect of these peptides was that they were degraded by apical peptidases. Another reason may have been that the peptides had limited access to tight junctions from the apical side. To solve the problem with the lack of apical activity, a lipoamino acid was conjugated to the N-terminus of the peptides. This conjugation inhibited degradation of the native peptide, and made the peptide active from the apical side. The two diastereomers of the conjugate had distinctly different kinetics of effect. The L-isomer rapidly released the native peptide, which resulted in a sustained effect. The D-isomer released the native peptide more slowly, resulting in a transient effect. In conclusion, the concept of conjugated peptides that act on the extracellular domains of the tight junctions constitutes a new prototype for tight junction modulation.
The specific conclusions of this thesis are:
depending on the culture conditions 2/4/A1 cells cultivated at 37ºC and 39ºC form monolayers on matrix coated permeable supports. These monolayers require 4 – 6 days to stabilize, and remain stable for at least another week.
2/4/A1 cell monolayers express the essential tight junction proteins and develop functional tight junctions.
48

2/4/A1 cell monolayers express several drug transporting membrane proteins at the transcriptional level, but there is no evidence for functional expression of these proteins in this cell line. The 2/4/A1 cell model can therefore be used for prediction of passive intestinal drug permeability.
2/4/A1 cell monolayers qualitatively and quantitatively predict the fraction absorbed after oral administration to humans for a diverse set of passively absorbed drugs. 2/4/A1 monolayers rank the intestinal permeability of incompletely absorbed drugs better than Caco-2.
2/4/A1 cell monolayers can be used to study the influence of molecular charge on the drug permeability. Studies of pH dependent drug permeability in 2/4/A1 can be performed to disclose whether the dominating passive transport route is trans- or paracellular.
Native peptides homologous to the N-terminus of the first extracellular loop of occludin modulate the tight junction permeability of Caco-2 cell monolayers in a slow and inefficient way. Conjugation of a lipoamino acid to an occludin peptide resulted in a more efficient tight junction modulator. The L-isomer of the conjugate rapidly released the native peptide, as a result of peptidase-mediated removal of the lipoamino acid. This results in a sustained increase in tight junction permeability. The D-isomer releases the native peptide more slowly, and resulted in a transient increase in tight junction permeability.
49

8 ACKNOWLEDGEMENTS
The studies in this thesis were carried out at the Department of Pharmacy, Uppsala University.
I wish to express my gratitude to the following people who contributed to this thesis:
My supervisor professor Per Artursson, for sharing your knowledge, for always aiming at high quality and for never stopping to teach me how to write papers.
Professor Göran Alderborn for providing good working facilities.
Dr. Jan Taipalensuu (co-author) for sharing your enthusiasm (and your knowledge in molecular biology).
Dr. Kei Hashimoto (co-author) for sharing your creative thinking and for pushing the occludin paper to new heights.
Naoki Nagahara (co-author) for your hard and fruitful work on the pH paper.
Dr. Vladan Milovic (co-author) for your significant contribution to the 2/4/A1 research area.
Dr. Finn Hallböök (co-author) for supervising me when I did the transfections.
Johan Gråsjö, Dr. Lucia Lazorová and Dr. Göran Ocklind (co-authors) for help and advice with mathematics and statistics, with cell culture and transport experiments, and microscopy.
Magnus Köping-Höggård, my roommate, for pleasant company and creative discussions, for your positive attitude and for your inspiring self-discipline.
Dr. Patric Stenberg and Dr. Raouf Ghaderi for most(ly) pleasant company.
Susanne Olsson for teaching me how to cultivate those tricky 2/4/A1 cells back then when I was a freshman.
Christel Bergström for providing me with the calculated descriptors and for supervising our summer students.
Ulla Wästberg-Galik for making practical matters around the writing as smooth as possible.
Eva Nises Ahlgren for your help and support.
George Farrants and Proper English for skillful linguistic assistance.
Dr. Richard Morrison, Dr. Vanessa Moore and Dr. Saeho Chong (co-authors) for stimulating discussions and valuable comments.
50

51
Kati-Sisko Vellonen, Lauri Söderberg (co-authors) and Outi Haarakangas for skillful technical assistance.
Past and present members of the cell group for being great colleagues and for trusting me with the incubators (22°C!).
All my other colleagues at Galeniken for creating the good atmosphere.
Marja-Liisa for being my love and for your support and encouragement. I especially want to thank you for handling all the practical matters in Umeå during the time I was writing this thesis, and for taking care of the cats.
Financial support from Bristol-Myers Squibb Company, AstraZeneca, The Swedish Academy of Pharmaceutical Sciences, CD Carlsons stiftelse, The Swedish Medical Research Council, Centrala försöksdjursnämnden, and Stiftelsen Forskning utan Djurförsök is gratefully acknowledged.

9 REFERENCES
1. P. Artursson and J. Karlsson. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun 175: 880-5 (1991).
2. I. Kariv, R. A. Rourick, D. B. Kassel, and T. D. Chung. Improvement of "Hit-to-Lead" Optimization by Integration of in Vitro HTS Experimental Models for Early Determination of Pharmacokinetic Properties. Comb Chem High Throughput Screen 5: 459-72 (2002).
3. C. A. Lipinski, F. Lombardo, B. W. Dominy, and P. J. Feeney. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46: 3-26 (2001).
4. P. Garberg, P. Eriksson, N. Schipper, and B. Sjöstrom. Automated absorption assessment using Caco-2 cells cultured on both sides of polycarbonate membranes. Pharm Res 16: 441-5 (1999).
5. P. Stenberg, U. Norinder, K. Luthman, and P. Artursson. Experimental and computational screening models for the prediction of intestinal drug absorption. J Med Chem 44: 1927-37 (2001).
6. C. A. Bergström, U. Norinder, K. Luthman, and P. Artursson. Experimental and computational screening models for prediction of aqueous drug solubility. Pharm Res 19: 182-8 (2002).
7. S. C. Sutton, E. L. LeCluyse, K. Engle, J. D. Pipkin, and J. A. Fix. Enhanced bioavailability of cefoxitin using palmitoylcarnitine. II. Use of directly compressed tablet formulations in the rat and dog. Pharm Res 10: 1516-20 (1993).
8. T. Uchida, Y. Toida, S. Sakakibara, Y. Miyanaga, H. Tanaka, M. Nishikata, K. Tazuya, N. Yasuda, and K. Matsuyama. Preparation and characterization of insulin-loaded acrylic hydrogels containing absorption enhancers. ChemPharm Bull (Tokyo) 49: 1261-6 (2001).
9. M. Thanou, J. C. Verhoef, P. Marbach, and H. E. Junginger. Intestinal absorption of octreotide: N-trimethyl chitosan chloride (TMC) ameliorates the permeability and absorption properties of the somatostatin analogue in vitro and in vivo. J Pharm Sci 89: 951-7 (2000).
10. M. Furuse, K. Fujita, T. Hiiragi, K. Fujimoto, and S. Tsukita. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with No sequence similarity to occludin. J Cell Biol 141: 1539-50 (1998).
11. M. Furuse, T. Hirase, M. Itoh, A. Nagafuchi, S. Yonemura, and S. Tsukita. Occludin: a novel integral membrane protein localizing at tight junctions. JCell Biol 123: 1777-88 (1993).
12. S. V. Walsh, A. M. Hopkins, J. Chen, S. Narumiya, C. A. Parkos, and A. Nusrat. Rho kinase regulates tight junction function and is necessary for tight junction assembly in polarized intestinal epithelia. Gastroenterology 121: 566-79 (2001).
52

53
13. P. D. Ward, H. Ouyang, and D. R. Thakker. Role of phospholipase C-beta inthe modulation of epithelial tight junction permeability. J Pharmacol Exp Ther304: 689-98 (2003).
14. T. Hirase, S. Kawashima, E. Y. Wong, T. Ueyama, Y. Rikitake, S. Tsukita, M. Yokoyama, and J. M. Staddon. Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and -independentmechanisms. J Biol Chem 276: 10423-31 (2001).
15. T. Y. Ma, D. Tran, N. Hoa, D. Nguyen, M. Merryfield, and A. Tarnawski. Mechanism of extracellular calcium regulation of intestinal epithelial tight junction permeability: role of cytoskeletal involvement. Microsc Res Tech 51:156-68 (2000).
16. M. S. Balda, C. Flores-Maldonado, M. Cereijido, and K. Matter. Multiple domains of occludin are involved in the regulation of paracellular permeability. J Cell Biochem 78: 85-96 (2000).
17. T. S. Jou, E. E. Schneeberger, and W. J. Nelson. Structural and functional regulation of tight junctions by RhoA and Rac1 small GTPases. J Cell Biol142: 101-15 (1998).
18. J. L. Madara. Regulation of the movement of solutes across tight junctions. Annu Rev Physiol 60: 143-59 (1998).
19. M. S. Balda, M. B. Fallon, C. M. Van Itallie, and J. M. Anderson. Structure,regulation, and pathophysiology of tight junctions in the gastrointestinal tract. Yale J Biol Med 65: 725-35; discussion 737-40 (1992).
20. S. T. Ballard, J. H. Hunter, and A. E. Taylor. Regulation of tight-junctionpermeability during nutrient absorption across the intestinal epithelium. AnnuRev Nutr 15: 35-55 (1995).
21. N. Sonoda, M. Furuse, H. Sasaki, S. Yonemura, J. Katahira, Y. Horiguchi, and S. Tsukita. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J Cell Biol 147: 195-204 (1999).
22. A. Fasano. Regulation of intercellular tight junctions by zonula occludens toxin and its eukaryotic analogue zonulin. Ann N Y Acad Sci 915: 214-22(2000).
23. A. W. Larhed, P. Artursson, J. Gråsjö, and E. Björk. Diffusion of drugs in native and purified gastrointestinal mucus. J Pharm Sci 86: 660-5 (1997).
24. J. L. Madara and J. Trier. The functional morphology of the mucosa of the small intestine, Physiology of the Gastrointestinal Tract, Raven Press, New York, 1994, pp. 1577-1622.
25. B. M. Fihn, A. Sjöqvist, and M. Jodal. Permeability of the rat small intestinal epithelium along the villus-crypt axis: effects of glucose transport. Gastroenterology 119: 1029-36 (2000).
26. T. T. Kararli. Gastrointestinal absorption of drugs. Crit Rev Ther Drug CarrierSyst 6: 39-86 (1989).

27. G. van Meer and K. Simons. The function of tight junctions in maintaining differences in lipid composition between the apical and the basolateral cell surface domains of MDCK cells. Embo J 5: 1455-64 (1986).
28. S. Muranishi. Absorption enhancers. Crit Rev Ther Drug Carrier Syst 7: 1-33 (1990).
29. W. L. Hayton. Rate-limiting barriers to intestinal drug absorption: a review. JPharmacokinet Biopharm 8: 321-34 (1980).
30. A. L. Ungell, S. Nylander, S. Bergstrand, A. Sjöberg, and H. Lennernäs. Membrane transport of drugs in different regions of the intestinal tract of the rat. J Pharm Sci 87: 360-6 (1998).
31. A. Strocchi and M. D. Levitt. Role of villous surface area in absorption. Science versus religion. Dig Dis Sci 38: 385-7 (1993).
32. A. L. Kawaguchi, J. C. Dunn, M. Lam, T. P. O'Connor, J. Diamond, and E. W. Fonkalsrud. Glucose uptake in dilated small intestine. J Pediatr Surg 33:1670-3 (1998).
33. D. Pal, C. Udata, and A. K. Mitra. Transport of cosalane-a highly lipophilic novel anti-HIV agent-across caco-2 cell monolayers. J Pharm Sci 89: 826-33 (2000).
34. G. A. Sawada, C. L. Barsuhn, B. S. Lutzke, M. E. Houghton, G. E. Padbury, N. F. Ho, and T. J. Raub. Increased lipophilicity and subsequent cell partitioning decrease passive transcellular diffusion of novel, highly lipophilic antioxidants. J Pharmacol Exp Ther 288: 1317-26 (1999).
35. P. Wils, A. Warnery, V. Phung-Ba, S. Legrain, and D. Scherman. High lipophilicity decreases drug transport across intestinal epithelial cells. JPharmacol Exp Ther 269: 654-8 (1994).
36. V. H. Lee. Membrane transporters. Eur J Pharm Sci 11: S41-50 (2000).
37. F. Doring, J. Will, S. Amasheh, W. Clauss, H. Ahlbrecht, and H. Daniel. Minimal molecular determinants of substrates for recognition by the intestinal peptide transporter. J Biol Chem 273: 23211-8 (1998).
38. V. Ganapathy and F. H. Leibach. Peptide transporters. Curr Opin Nephrol Hypertens 5: 395-400 (1996).
39. H. K. Han, D. M. Oh, and G. L. Amidon. Cellular uptake mechanism of amino acid ester prodrugs in Caco-2/hPEPT1 cells overexpressing a human peptide transporter. Pharm Res 15: 1382-6 (1998).
40. M. E. Ganapathy, W. Huang, H. Wang, V. Ganapathy, and F. H. Leibach. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem Biophys Res Commun 246: 470-5 (1998).
41. T. Shiraga, K. Miyamoto, H. Tanaka, H. Yamamoto, Y. Taketani, K. Morita, I. Tamai, A. Tsuji, and E. Takeda. Cellular and molecular mechanisms of dietary regulation on rat intestinal H+/Peptide transporter PepT1. Gastroenterology 116: 354-62 (1999).
54

42. Y. Tanigawara. Role of P-glycoprotein in drug disposition. Ther Drug Monit22: 137-40 (2000).
43. P. Borst, R. Evers, M. Kool, and J. Wijnholds. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst 92: 1295-302 (2000).
44. J. Taipalensuu, H. Törnblom, G. Lindberg, C. Einarsson, F. Sjöqvist, H. Melhus, P. Garberg, B. Sjöström, B. Lundgren, and P. Artursson. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelialCaco-2 cell monolayers. J Pharmacol Exp Ther 299: 164-70 (2001).
45. J. W. Jonker, J. W. Smit, R. F. Brinkhuis, M. Maliepaard, J. H. Beijnen, J. H. Schellens, and A. H. Schinkel. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst 92: 1651-6 (2000).
46. S. Drescher, H. Glaeser, T. Murdter, M. Hitzl, M. Eichelbaum, and M. F. Fromm. P-glycoprotein-mediated intestinal and biliary digoxin transport in humans. Clin Pharmacol Ther 73: 223-31 (2003).
47. T. Österberg and U. Norinder. Theoretical calculation and prediction of P-glycoprotein-interacting drugs using MolSurf parametrization and PLS statistics. Eur J Pharm Sci 10: 295-303 (2000).
48. S. Ekins, R. B. Kim, B. F. Leake, A. H. Dantzig, E. G. Schuetz, L. B. Lan, K. Yasuda, R. L. Shepard, M. A. Winter, J. D. Schuetz, J. H. Wikel, and S. A. Wrighton. Three-dimensional quantitative structure-activity relationships of inhibitors of P-glycoprotein. Mol Pharmacol 61: 964-73 (2002).
49. A. Seelig and E. Landwojtowicz. Structure-activity relationship of P-glycoprotein substrates and modifiers. Eur J Pharm Sci 12: 31-40 (2000).
50. M. M. Malingre, D. J. Richel, J. H. Beijnen, H. Rosing, F. J. Koopman, W. W. Ten Bokkel Huinink, M. E. Schot, and J. H. Schellens. Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel. J Clin Oncol 19: 1160-6 (2001).
51. W. L. Chiou, S. M. Chung, T. C. Wu, and C. Ma. A comprehensive account on the role of efflux transporters in the gastrointestinal absorption of 13 commonly used substrate drugs in humans. Int J Clin Pharmacol Ther 39: 93-101 (2001).
52. J. Karlsson, A. Ungell, J. Gråsjö, and P. Artursson. Paracellular drug transport across intestinal epithelia: influence of charge and induced water flux. Eur J Pharm Sci 9: 47-56 (1999).
53. A. Adson, T. J. Raub, P. S. Burton, C. L. Barsuhn, A. R. Hilgers, K. L. Audus, and N. F. Ho. Quantitative approaches to delineate paracellular diffusion in cultured epithelial cell monolayers. J Pharm Sci 83: 1529-36 (1994).
54. K. Lee and D. R. Thakker. Saturable transport of H2-antagonists ranitidine and famotidine across Caco-2 cell monolayers. J Pharm Sci 88: 680-7 (1999).
55

55. L. S. Gan, S. Yanni, and D. R. Thakker. Modulation of the tight junctions of the Caco-2 cell monolayers by H2-antagonists. Pharm Res 15: 53-7 (1998).
56. S. Y. Zhou, N. Piyapolrungroj, L. Pao, C. Li, G. Liu, E. Zimmermann, and D. Fleisher. Regulation of paracellular absorption of cimetidine and 5-aminosalicylate in rat intestine. Pharm Res 16: 1781-5 (1999).
57. A. Adson, P. S. Burton, T. J. Raub, C. L. Barsuhn, K. L. Audus, and N. F. Ho. Passive diffusion of weak organic electrolytes across Caco-2 cell monolayers: uncoupling the contributions of hydrodynamic, transcellular, and paracellular barriers. J Pharm Sci 84: 1197-204 (1995).
58. S. D. Flanagan, L. H. Takahashi, X. Liu, and L. Z. Benet. Contributions of saturable active secretion, passive transcellular, and paracellular diffusion to the overall transport of furosemide across adenocarcinoma (Caco-2) cells. JPharm Sci 91: 1169-77 (2002).
59. H. Nellans. Paracellular intestinal transport: modulation of absorption. AdvDrug Deliv Rev 7: 339-364 (1991).
60. J. R. Pappenheimer and K. Z. Reiss. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J Membr Biol 100: 123-36 (1987).
61. P. Artursson, A. L. Ungell, and J. E. Löfroth. Selective paracellular permeability in two models of intestinal absorption: cultured monolayers of human intestinal epithelial cells and rat intestinal segments. Pharm Res 10:1123-9 (1993).
62. J. L. Madara and M. A. Marcial. Structural correlates of intestinal tight-junction permeability. Kroc Found Ser 17: 77-100 (1984).
63. K. D. Fine, C. A. Santa Ana, J. L. Porter, and J. S. Fordtran. Effect of changing intestinal flow rate on a measurement of intestinal permeability. Gastroenterology 108: 983-9 (1995).
64. J. L. Madara and K. Dharmsathaphorn. Occluding junction structure-function relationships in a cultured epithelial monolayer. J Cell Biol 101: 2124-33 (1985).
65. B. M. Fihn and M. Jodal. Permeability of the proximal and distal rat colon crypt and surface epithelium to hydrophilic molecules. Pflugers Arch 441:656-62 (2001).
66. B. M. Fihn, A. Sjöqvist, and M. Jodal. Permeability of the rat small intestinal epithelium along the villus- crypt axis: effects of glucose transport. Gastroenterology 119: 1029-36 (2000).
67. J. D. Söderholm, G. Olaison, A. Kald, C. Tagesson, and R. Sjödahl. Absorption profiles for polyethylene glycols after regional jejunal perfusion and oral load in healthy humans. Dig Dis Sci 42: 853-7 (1997).
68. P. Artursson and R. T. Borchardt. Intestinal drug absorption and metabolism in cell cultures: Caco-2 and beyond. Pharm Res 14: 1655-8 (1997).
56

69. M. Kansy, F. Senner, and K. Gubernator. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J Med Chem 41: 1007-10 (1998).
70. J. D. Irvine, L. Takahashi, K. Lockhart, J. Cheong, J. W. Tolan, H. E. Selick, and J. R. Grove. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J Pharm Sci 88: 28-33 (1999).
71. I. J. Hidalgo, T. J. Raub, and R. T. Borchardt. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 96: 736-49 (1989).
72. P. Artursson. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorptive (Caco-2) cells. J Pharm Sci 79: 476-82 (1990).
73. A. R. Hilgers, R. A. Conradi, and P. S. Burton. Caco-2 cell monolayers as a model for drug transport across the intestinal mucosa. Pharm Res 7: 902-10 (1990).
74. J. Fogh, J. M. Fogh, and T. Orfeo. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst 59:221-6 (1977).
75. E. Grasset, M. Pinto, E. Dussaulx, A. Zweibaum, and J. F. Desjeux. Epithelial properties of human colonic carcinoma cell line Caco-2: electrical parameters. Am J Physiol 247: C260-7 (1984).
76. I. Chantret, A. Barbat, E. Dussaulx, M. G. Brattain, and A. Zweibaum. Epithelial polarity, villin expression, and enterocytic differentiation of cultured human colon carcinoma cells: a survey of twenty cell lines. Cancer Res 48:1936-42 (1988).
77. K. Palm, K. Luthman, J. Ros, J. Gråsjö, and P. Artursson. Effect of molecular charge on intestinal epithelial drug transport: pH-dependent transport of cationic drugs. J Pharmacol Exp Ther 291: 435-43 (1999).
78. P. Saha and J. H. Kou. Effect of solubilizing excipients on permeation of poorly water-soluble compounds across Caco-2 cell monolayers. Eur J Pharm Biopharm 50: 403-11 (2000).
79. B. J. Aungst, N. H. Nguyen, J. P. Bulgarelli, and K. Oates-Lenz. The influence of donor and reservoir additives on Caco-2 permeability and secretory transport of HIV protease inhibitors and other lipophilic compounds. PharmRes 17: 1175-80 (2000).
80. Y. Takahashi, H. Kondo, T. Yasuda, T. Watanabe, S. Kobayashi, and S. Yokohama. Common solubilizers to estimate the Caco-2 transport of poorly water-soluble drugs. Int J Pharm 246: 85-94 (2002).
81. D. Z. Liu, E. L. LeCluyse, and D. R. Thakker. Dodecylphosphocholine-mediated enhancement of paracellular permeability and cytotoxicity in Caco-2 cell monolayers. J Pharm Sci 88: 1161-8 (1999).
57

82. T. Lindmark, T. Nikkilä, and P. Artursson. Mechanisms of absorption enhancement by medium chain fatty acids in intestinal epithelial Caco-2 cell monolayers. J Pharmacol Exp Ther 275: 958-64 (1995).
83. A. F. Kotze, H. L. Luessen, B. J. de Leeuw, B. G. de Boer, J. C. Verhoef, and H. E. Junginger. N-trimethyl chitosan chloride as a potential absorption enhancer across mucosal surfaces: in vitro evaluation in intestinal epithelial cells (Caco-2). Pharm Res 14: 1197-202 (1997).
84. D. S. Cox, S. Raje, H. Gao, N. N. Salami, and N. D. Eddington. Enhanced permeability of molecular weight markers and poorly bioavailable compounds across Caco-2 cell monolayers using the absorption enhancer, zonula occludens toxin. Pharm Res 19: 1680-8 (2002).
85. U. Wenzel, B. Meissner, F. Doring, and H. Daniel. PEPT1-mediated uptake of dipeptides enhances the intestinal absorption of amino acids via transport system b(0,+). J Cell Physiol 186: 251-9 (2001).
86. M. Sasaki, H. Suzuki, K. Ito, T. Abe, and Y. Sugiyama. Transcellular transport of organic anions across a double-transfected Madin-Darby canine kidney II cell monolayer expressing both human organic anion-transporting polypeptide (OATP2/SLC21A6) and Multidrug resistance-associated protein 2 (MRP2/ABCC2). J Biol Chem 277: 6497-503 (2002).
87. C. L. Cummins, W. Jacobsen, and L. Z. Benet. Unmasking the dynamic interplay between intestinal P-glycoprotein and CYP3A4. J Pharmacol Exp Ther 300: 1036-45 (2002).
88. J. H. Hochman, M. Chiba, J. Nishime, M. Yamazaki, and J. H. Lin. Influence of P-glycoprotein on the transport and metabolism of indinavir in Caco-2 cells expressing cytochrome P-450 3A4. J Pharmacol Exp Ther 292: 310-8 (2000).
89. G. T. Knipp, N. F. Ho, C. L. Barsuhn, and R. T. Borchardt. Paracellular diffusion in Caco-2 cell monolayers: effect of perturbation on the transport of hydrophilic compounds that vary in charge and size. J Pharm Sci 86: 1105-10 (1997).
90. A. Collett, D. Walker, E. Sims, Y. L. He, P. Speers, J. Ayrton, M. Rowland, and G. Warhurst. Influence of morphometric factors on quantitation of paracellular permeability of intestinal epithelia in vitro. Pharm Res 14: 767-73 (1997).
91. Y. Tanaka, Y. Taki, T. Sakane, T. Nadai, H. Sezaki, and S. Yamashita. Characterization of drug transport through tight-junctional pathway in Caco-2 monolayer: comparison with isolated rat jejunum and colon. Pharm Res 12:523-8 (1995).
92. L. P. Pageot, N. Perreault, N. Basora, C. Francoeur, P. Magny, and J. F. Beaulieu. Human cell models to study small intestinal functions: recapitulation of the crypt-villus axis. Microsc Res Tech 49: 394-406 (2000).
93. A. Collett, E. Sims, D. Walker, Y. L. He, J. Ayrton, M. Rowland, and G. Warhurst. Comparison of HT29-18-C1 and Caco-2 cell lines as models for studying intestinal paracellular drug absorption. Pharm Res 13: 216-21 (1996).
58

94. E. Duizer, A. Penninks, W. Stenhuis, and J. Groten. Comparison of permeability characteristics of the human colonic Caco-2 and rat small intestinal IEC-18 cell lines. J Control Release 49: 39-49 (1997).
95. A. Wikman Larhed and P. Artursson. Co-cultures of human intestinal goblet (HT29-H) and absorbtive (Caco-2) cells for studies on epithelial transport and ntegrity. Eur J Pharm Sci 3: 171-183 (1995).
96. P. Artursson, J. Karlsson, G. Ocklind, and N. Schipper. Cell Culture Models of Epithelial Tissues - A practical Approach, Oxford University Press, New York, 1996.
97. S. Yamashita, K. Konishi, Y. Yamazaki, Y. Taki, T. Sakane, H. Sezaki, and Y. Furuyama. New and better protocols for a short-term Caco-2 cell culture system. J Pharm Sci 91: 669-79 (2002).
98. E. Liang, K. Chessic, and M. Yazdanian. Evaluation of an accelerated Caco-2 cell permeability model. J Pharm Sci 89: 336-45 (2000).
99. D. Sun, H. Lennernäs, L. S. Welage, J. L. Barnett, C. P. Landowski, D. Foster, D. Fleisher, K. D. Lee, and G. L. Amidon. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm Res 19: 1400-16 (2002).
100. S. Chong, S. A. Dando, K. M. Soucek, and R. A. Morrison. In vitro permeability through caco-2 cells is not quantitatively predictive of in vivo absorption for peptide-like drugs absorbed via the dipeptide transporter system. Pharm Res 13: 120-3 (1996).
101. T. Litman, T. E. Druley, W. D. Stein, and S. E. Bates. From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol Life Sci 58: 931-59 (2001).
102. R. Evers, M. Kool, L. van Deemter, H. Janssen, J. Calafat, L. C. Oomen, C. C. Paulusma, R. P. Oude Elferink, F. Baas, A. H. Schinkel, and P. Borst. Drug export activity of the human canalicular multispecific organic anion transporter in polarized kidney MDCK cells expressing cMOAT (MRP2) cDNA. J Clin Invest 101: 1310-9 (1998).
103. F. Tang, K. Horie, and R. T. Borchardt. Are MDCK cells transfected with the human MRP2 gene a good model of the human intestinal mucosa? Pharm Res19: 773-9 (2002).
104. F. Tang, K. Horie, and R. T. Borchardt. Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa? Pharm Res19: 765-72 (2002).
105. W. S. Putnam, L. Pan, K. Tsutsui, L. Takahashi, and L. Z. Benet. Comparison of bidirectional cephalexin transport across MDCK and caco-2 cell monolayers: interactions with peptide transporters. Pharm Res 19: 27-33 (2002).
59

106. K. Balon, B. U. Riebesehl, and B. W. Muller. Drug liposome partitioning as a tool for the prediction of human passive intestinal absorption. Pharm Res 16:882-8 (1999).
107. A. Avdeef, K. J. Box, J. E. Comer, C. Hibbert, and K. Y. Tam. pH-metric logP 10. Determination of liposomal membrane-water partition coefficients of ionizable drugs. Pharm Res 15: 209-15 (1998).
108. F. Beigi, Q. Yang, and P. Lundahl. Immobilized-liposome chromatographic analysis of drug partitioning into lipid bilayers. J Chromatogr A 704: 315-21 (1995).
109. P. Mueller, D. Rudin, H. Tien, and W. Westcott. Reconstitution of cell membrane structure in vitro and its transformation into an excitable system. Nature 194: 979-980 (1962).
110. M. Thompson, L. Lennox, and R. McClelland. Structure and electrochemical properties of microfiltration filter lipid membrane systems. Anal Chem 54: 76-81 (1982).
111. K. Sugano, H. Hamada, M. Machida, and H. Ushio. High throughput prediction of oral absorption: improvement of the composition of the lipid solution used in parallel artificial membrane permeation assay. J Biomol Screen 6: 189-96 (2001).
112. F. Wohnsland and B. Faller. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J Med Chem44: 923-30 (2001).
113. A. Avdeef. Physicochemical profiling (solubility, permeability and charge state). Curr Top Med Chem 1: 277-351 (2001).
114. M. T. DeMeo, E. A. Mutlu, A. Keshavarzian, and M. C. Tobin. Intestinal permeation and gastrointestinal disease. J Clin Gastroenterol 34: 385-96 (2002).
115. M. L. Marin, A. J. Greenstein, S. A. Geller, R. E. Gordon, and A. H. Aufses, Jr. A freeze fracture study of Crohn's disease of the terminal ileum: changes in epithelial tight junction organization. Am J Gastroenterol 78: 537-47 (1983).
116. A. Fasano, B. Baudry, D. W. Pumplin, S. S. Wasserman, B. D. Tall, J. M. Ketley, and J. B. Kaper. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc Natl Acad Sci U S A 88: 5242-6 (1991).
117. I. Sasaki, H. Tozaki, K. Matsumoto, Y. Ito, T. Fujita, M. Murakami, S. Muranishi, and A. Yamamoto. Development of an oral formulation of azetirelin, a new thyrotropin-releasing hormone (TRH) analogue, using n-lauryl-beta-D-maltopyranoside as an absorption enhancer. Biol Pharm Bull 22:611-5 (1999).
118. A. Fasano and S. Uzzau. Modulation of intestinal tight junctions by Zonula occludens toxin permits enteral administration of insulin and other macromolecules in an animal model. J Clin Invest 99: 1158-64 (1997).
60

119. T. Uchiyama, T. Sugiyama, Y. S. Quan, A. Kotani, N. Okada, T. Fujita, S. Muranishi, and A. Yamamoto. Enhanced permeability of insulin across the rat intestinal membrane by various absorption enhancers: their intestinal mucosal toxicity and absorption-enhancing mechanism of n-lauryl-beta-D-maltopyranoside. J Pharm Pharmacol 51: 1241-50 (1999).
120. E. J. van Hoogdalem, C. D. Heijligers-Feijen, R. A. Mathot, A. T. Wackwitz, J. B. van Bree, J. C. Verhoef, A. G. de Boer, and D. D. Breimer. Rectal absorption enhancement of cefoxitin and desglycinamide arginine vasopressin by sodium tauro-24,25-dihydrofusidate in conscious rats. J Pharmacol Exp Ther 251: 741-4 (1989).
121. T. Lindmark, J. D. Söderholm, G. Olaison, G. Alvan, G. Ocklind, and P. Artursson. Mechanism of absorption enhancement in humans after rectal administration of ampicillin in suppositories containing sodium caprate. Pharm Res 14: 930-5 (1997).
122. W. Wang, S. Uzzau, S. E. Goldblum, and A. Fasano. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci 113: 4435-40 (2000).
123. J. A. McRoberts, R. Aranda, N. Riley, and H. Kang. Insulin regulates the paracellular permeability of cultured intestinal epithelial cell monolayers. JClin Invest 85: 1127-34 (1990).
124. B. Hoffmann, I. Nagel, and W. Clauss. Aldosterone regulates paracellular pathway resistance in rabbit distal colon. J Comp Physiol 160: 381-8 (1990).
125. M. C. Chen, J. Goliger, N. Bunnett, and A. H. Soll. Apical and basolateral EGF receptors regulate gastric mucosal paracellular permeability. Am J Physiol Gastrointest Liver Physiol 280: G264-72 (2001).
126. J. H. Hochman, J. A. Fix, and E. L. LeCluyse. In vitro and in vivo analysis of the mechanism of absorption enhancement by palmitoylcarnitine. J Pharmacol Exp Ther 269: 813-22 (1994).
127. T. Lindmark, Y. Kimura, and P. Artursson. Absorption enhancement through intracellular regulation of tight junction permeability by medium chain fatty acids in Caco-2 cells. J Pharmacol Exp Ther 284: 362-9 (1998).
128. B. Gumbiner. Structure, biochemistry, and assembly of epithelial tight junctions. Am J Physiol 253: C749-58 (1987).
129. E. Sinaga, S. D. Jois, M. Avery, I. T. Makagiansar, U. S. Tambunan, K. L. Audus, and T. J. Siahaan. Increasing paracellular porosity by E-cadherin peptides: discovery of bulge and groove regions in the EC1-domain of E-cadherin. Pharm Res 19: 1170-9 (2002).
130. M. Furuse, M. Itoh, T. Hirase, A. Nagafuchi, S. Yonemura, and S. Tsukita. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol 127: 1617-26 (1994).
61

131. J. Haskins, L. Gu, E. S. Wittchen, J. Hibbard, and B. R. Stevenson. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol 141: 199-208 (1998).
132. M. Itoh, K. Morita, and S. Tsukita. Characterization of ZO-2 as a MAGUK family member associated with tight as well as adherens junctions with a binding affinity to occludin and alpha catenin. J Biol Chem 274: 5981-6 (1999).
133. E. S. Wittchen, J. Haskins, and B. R. Stevenson. Protein interactions at the tight junction. Actin has multiple binding partners, and ZO-1 forms independent complexes with ZO-2 and ZO-3. J Biol Chem 274: 35179-85 (1999).
134. C. M. Van Itallie and J. M. Anderson. Occludin confers adhesiveness when expressed in fibroblasts. J Cell Sci 110: 1113-21 (1997).
135. M. Saitou, M. Furuse, H. Sasaki, J. D. Schulzke, M. Fromm, H. Takano, T. Noda, and S. Tsukita. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 11: 4131-42 (2000).
136. T. Tsukamoto and S. K. Nigam. Role of tyrosine phosphorylation in the reassembly of occludin and other tight junction proteins. Am J Physiol 276:F737-50 (1999).
137. L. Gonzalez-Mariscal, A. Betanzos, P. Nava, and B. E. Jaramillo. Tight junction proteins. Prog Biophys Mol Biol 81: 1-44 (2003).
138. M. Itoh, M. Furuse, K. Morita, K. Kubota, M. Saitou, and S. Tsukita. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 147: 1351-63 (1999).
139. M. Furuse, H. Sasaki, K. Fujimoto, and S. Tsukita. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol 143: 391-401 (1998).
140. S. Tsukita, M. Furuse, and M. Itoh. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2: 285-93 (2001).
141. M. Furuse, H. Sasaki, and S. Tsukita. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol 147:891-903 (1999).
142. M. Furuse, K. Furuse, H. Sasaki, and S. Tsukita. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol 153: 263-72 (2001).
143. C. Rahner, L. L. Mitic, and J. M. Anderson. Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology 120: 411-22 (2001).
144. I. Martin-Padura, S. Lostaglio, M. Schneemann, L. Williams, M. Romano, P. Fruscella, C. Panzeri, A. Stoppacciaro, L. Ruco, A. Villa, D. Simmons, and E. Dejana. Junctional adhesion molecule, a novel member of the immunoglobulin
62

superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142: 117-27 (1998).
145. Y. Liu, A. Nusrat, F. J. Schnell, T. A. Reaves, S. Walsh, M. Pochet, and C. A. Parkos. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci 113 ( Pt 13): 2363-74 (2000).
146. T. W. Liang, H. H. Chiu, A. Gurney, A. Sidle, D. B. Tumas, P. Schow, J. Foster, T. Klassen, K. Dennis, R. A. DeMarco, T. Pham, G. Frantz, and S. Fong. Vascular endothelial-junctional adhesion molecule (VE-JAM)/JAM 2 interacts with T, NK, and dendritic cells through JAM 3. J Immunol 168:1618-26 (2002).
147. S. C. Sutton, E. L. LeCluyse, L. Cammack, and J. A. Fix. Enhanced bioavailability of cefoxitin using palmitoyl L-carnitine. I. Enhancer activity in different intestinal regions. Pharm Res 9: 191-4 (1992).
148. P. D. Ward, T. K. Tippin, and D. R. Thakker. Enhancing paracellular permeability by modulating epithelial tight junctions. 3: 346-358 (2000).
149. J. Behrens, L. Vakaet, R. Friis, E. Winterhager, F. Van Roy, M. M. Mareel, and W. Birchmeier. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol 120: 757-66 (1993).
150. B. Rothen-Rutishauser, F. K. Riesen, A. Braun, M. Gunthert, and H. Wunderli-Allenspach. Dynamics of tight and adherens junctions under EGTA treatment. J Membr Biol 188: 151-62 (2002).
151. M. Sakai, T. Imai, H. Ohtake, and M. Otagiri. Cytotoxicity of absorption enhancers in Caco-2 cell monolayers. J Pharm Pharmacol 50: 1101-8 (1998).
152. N. G. Schipper, S. Olsson, J. A. Hoogstraate, A. G. deBoer, K. M. Vårum, and P. Artursson. Chitosans as absorption enhancers for poorly absorbable drugs 2: mechanism of absorption enhancement. Pharm Res 14: 923-9 (1997).
153. N. G. Schipper, K. M. Vårum, P. Stenberg, G. Ocklind, H. Lennernäs, and P. Artursson. Chitosans as absorption enhancers of poorly absorbable drugs. 3: Influence of mucus on absorption enhancement. Eur J Pharm Sci 8: 335-43 (1999).
154. A. Fasano, C. Fiorentini, G. Donelli, S. Uzzau, J. B. Kaper, K. Margaretten, X. Ding, S. Guandalini, L. Comstock, and S. E. Goldblum. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J Clin Invest 96: 710-20 (1995).
155. R. El Asmar, P. Panigrahi, P. Bamford, I. Berti, T. Not, G. V. Coppa, C. Catassi, and A. Fasano. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology 123: 1607-15 (2002).
63

156. V. Wong and B. M. Gumbiner. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol 136: 399-409 (1997).
157. F. Lacaz-Vieira, M. M. Jaeger, P. Farshori, and B. Kachar. Small synthetic peptides homologous to segments of the first external loop of occludin impair tight junction resealing. J Membr Biol 168: 289-97 (1999).
158. I. Toth, J. P. Malkinson, N. S. Flinn, B. Drouillat, A. Horvath, J. Erchegyi, M. Idei, A. Venetianer, P. Artursson, L. Lazorova, B. Szende, and G. Keri. Novel lipoamino acid- and liposaccharide-based system for peptide delivery: application for oral administration of tumor-selective somatostatin analogues. J Med Chem 42: 4010-3. (1999).
159. M. Seto, U. Jaeger, R. D. Hockett, W. Graninger, S. Bennett, P. Goldman, and S. J. Korsmeyer. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. Embo J 7: 123-31. (1988).
160. D. Aharoni, A. Dantes, M. Oren, and A. Amsterdam. cAMP-mediated signals as determinants for apoptosis in primary granulosa cells. Exp Cell Res 218:271-82 (1995).
161. T. Mosman. Rapid colorometric assay for cellular growth and survival:application to prolifiration and cytotoxicity assays. J Immunol Methods 65: 55-63 (1983).
162. E. K. Anderberg, C. Nyström, and P. Artursson. Epithelial transport of drugs in cell culture. VII: Effects of pharmaceutical surfactant excipients and bile acids on transepithelial permeability in monolayers of human intestinal epithelial (Caco-2) cells. J Pharm Sci 81: 879-87 (1992).
163. S. Tavelin, J. Gråsjö, J. Taipalensuu, G. Ocklind, and P. Artursson. Applications of epithelial cell culture in studies of drug transport. MethodsMol Biol 188: 233-72 (2002).
164. F. Curry. Mechanics and thermodynamics of transcapillary exchange. In Handbook of Physiology, Vol. 4, Am Physiol Soc, Bethesda, 1984, pp. 309-374.
165. A. Quaroni and J. F. Beaulieu. Cell dynamics and differentiation of conditionally immortalized human intestinal epithelial cells. Gastroenterology113: 1198-213 (1997).
166. E. C. Paul, J. Hochman, and A. Quaroni. Conditionally immortalized intestinal epithelial cells: novel approach for study of differentiated enterocytes. Am J Physiol 265: C266-78 (1993).
167. P. S. Jat and P. A. Sharp. Cell lines established by a temperature-sensitive simian virus 40 large- T-antigen gene are growth restricted at the nonpermissive temperature. Mol Cell Biol 9: 1672-81 (1989).
168. S. M. Frisch and H. Francis. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 124: 619-26 (1994).
64

169. M. Heiskala, P. A. Peterson, and Y. Yang. The roles of claudin superfamily proteins in paracellular transport. Traffic 2: 93-8 (2001).
170. C. U. Nielsen, J. Amstrup, B. Steffansen, S. Frokjaer, and B. Brodin. Epidermal growth factor inhibits glycylsarcosine transport and hPepT1 expression in a human intestinal cell line. Am J Physiol Gastrointest Liver Physiol 281: G191-9. (2001).
171. N. Yanai and M. Obinata. Apoptosis is induced at nonpermissive temperature by a transient increase in p53 in cell lines immortalized with temperature-sensitive SV40 large T-antigen gene. Exp Cell Res 211: 296-300 (1994).
172. C. Conejero, R. Wright, and W. Freed. Glutamate and antimitotic agents induce differentiation, p53 activation, and apoptosis in rodent neostriatal cell lines immortalized with the tsA58 allele of SV40 large T antigen. Exp Neurol158: 109-20. (1999).
173. P. H. Gallimore, P. S. Lecane, B. Hann, and R. J. Grand. Differentiation is inhibited and a senescence pathway is activated when simian virus 40 tsA 58-transformed human retinoblasts are grown at the restrictive temperature. CellGrowth Differ 8: 763-71. (1997).
174. J. L. Vayssiere, P. X. Petit, Y. Risler, and B. Mignotte. Commitment to apoptosis is associated with changes in mitochondrial biogenesis and activity in cell lines conditionally immortalized with simian virus 40. Proc Natl Acad Sci U S A 91: 11752-6. (1994).
175. A. Taher, N. Yanai, and M. Obinata. Properties of incompletely immortalized cell lines generated from a line established from temperature-sensitive SV40 T-antigen gene transgenic mice. Exp Cell Res 219: 332-8. (1995).
176. E. M. Eves, L. H. Boise, C. B. Thompson, A. J. Wagner, N. Hay, and M. R. Rosner. Apoptosis induced by differentiation or serum deprivation in an immortalized central nervous system neuronal cell line. J Neurochem 67:1908-20. (1996).
177. V. Chadwick, S. Phillips, and A. Hofmann. Measurements of intestinal permeability using low molecular weight polyethylene glycols (PEG 400). II. Application to normal and abnormal permeability states in man and animals. Gastroenterology 73: 247-251 (1977).
178. R. Lobenberg and G. L. Amidon. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur J Pharm Biopharm 50: 3-12 (2000).
179. H. Lennernäs, K. Palm, U. Fagerholm, and P. Artursson. Comparison between active and passive drug transport in human intestinal epithelial (Caco-2) cells in vitro and human jejunum in vivo. Int J Pharm 127: 103-107 (1996).
180. H. Lennernäs, S. Nylander, and A. L. Ungell. Jejunal permeability: a comparison between the ussing chamber technique and the single-pass perfusion in humans. Pharm Res 14: 667-71 (1997).
65

181. S. Winiwarter, N. M. Bonham, F. Ax, A. Hallberg, H. Lennernäs, and A. Karlen. Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach. J Med Chem 41: 4939-49 (1998).
182. A. H. Dantzig and L. Bergin. Uptake of the cephalosporin, cephalexin, by a dipeptide transport carrier in the human intestinal cell line, Caco-2. BiochimBiophys Acta 1027: 211-7. (1990).
183. D. T. Thwaites, C. D. Brown, B. H. Hirst, and N. L. Simmons. Transepithelial glycylsarcosine transport in intestinal Caco-2 cells mediated by expression of H(+)-coupled carriers at both apical and basal membranes. J Biol Chem 268:7640-2. (1993).
184. G. M. Pauletti, S. Gangwar, B. Wang, and R. T. Borchardt. Esterase-sensitive cyclic prodrugs of peptides: evaluation of a phenylpropionic acid promoiety in a model hexapeptide. Pharm Res 14: 11-7 (1997).
185. A. Yamamoto. Improvement of intestinal absorption of peptide and protein drugs by chemical modification with fatty acids. Nippon Rinsho 56: 601-7 (1998).
186. I. Toth. A novel chemical approach to drug delivery: lipid amino acid conjugates. J Drug Target 2: 217-39 (1994).
66


![Review Paper Writing Workshop Prof Azman Slides...675$7(*,(6 72 :5,7( 5(9,(: 3$3(56 2UJDQLVHGE\ )DFXOW\ RI 6FLHQFH 8QLYHUVLWL7HNQRORJL0DOD\VLD WK 'HFHPEHU $]PDQ+DVVDQ 'HSXW\ 'HDQ 5HVHDUFK](https://static.fdocuments.in/doc/165x107/5f24e4c3ebdfb1370f1e8189/review-paper-writing-workshop-prof-azman-slides-67576-72-57-59-3356.jpg)














![OYDQLD /DZ 6FKRRO · 2019-06-04 · $zdugv )ulho 6fdqodq $zdug 0dufk sul]h iru ehvw 7hpsoh 8qlyhuvlw\ 6fkrro ri /dz vfkroduvkls 7hpsoh /dz 5hylhz )dfxow\ krqruhh dw 7hpsoh /dz 5hylhz](https://static.fdocuments.in/doc/165x107/5f44fe819a2d3b6295504c4b/oydqld-dz-6fkrro-2019-06-04-zdugv-ulho-6fdqodq-zdug-0dufk-sulh-iru-ehvw-7hpsoh.jpg)


