
nat's paper!!!! 2016
11
Lab on a Chip PAPER Cite this: DOI: 10.1039/c5lc01392e Received 13th November 2015, Accepted 7th January 2016 DOI: 10.1039/c5lc01392e www.rsc.org/loc A fully integrated paperfluidic molecular diagnostic chip for the extraction, amplification, and detection of nucleic acids from clinical samples† Natalia M. Rodriguez, a Winnie S. Wong, a Lena Liu, a Rajan Dewar bc and Catherine M. Klapperich * a Paper diagnostics have successfully been employed to detect the presence of antigens or small molecules in clinical samples through immunoassays; however, the detection of many disease targets relies on the much higher sensitivity and specificity achieved via nucleic acid amplification tests (NAAT). The steps in- volved in NAAT have recently begun to be explored in paper matrices, and our group, among others, has reported on paper-based extraction, amplification, and detection of DNA and RNA targets. Here, we inte- grate these paper-based NAAT steps into a single paperfluidic chip in a modular, foldable system that al- lows for fully integrated fluidic handling from sample to result. We showcase the functionality of the chip by combining nucleic acid isolation, isothermal amplification, and lateral flow detection of human papillo- mavirus (HPV) 16 DNA directly from crude cervical specimens in less than 1 hour for rapid, early detection of cervical cancer. The chip is made entirely of paper and adhesive sheets, making it low-cost, portable, and disposable, and offering the potential for a point-of-care molecular diagnostic platform even in remote and resource-limited settings. Introduction Integrated molecular diagnostics to enable sample-to-answer nucleic acid amplification testing (NAAT) have traditionally required sophisticated instrumentation to provide pressure driven fluid handling, cyclic thermal control, and optical as- say detection. These requirements result in expensive equip- ment and costly disposables unsuitable for use in resource- limited settings. Since the start of lab-on-a-chip microfluidic technologies in the 90s, their use in remote settings has been perceived as potentially one of their most powerful applica- tions, by taking advantage of their small size, portability, low volume requirement for samples, and rapid analysis without the need for an expert operator. 1 However, because these systems require pumps or pressure in order to drive fluid flow, they aren't yet equipment-free and rely on instrumenta- tion and electricity. An alternative that has garnered much at- tention recently is “paperfluidics”, where paper is used as a substrate to construct microfluidic devices for use in rapid di- agnostic tests. 2–6 Paper's ability to passively transport fluids through capillary action, or wicking, eliminates the need for pumps or other fluid handling equipment. In regard to mo- lecular diagnostics, a number of studies have reported on the success of individual NAAT steps (extraction, amplification, and detection) within paper matrices. 7–12 Our group has pre- viously shown paper-based nucleic acid extraction, isothermal amplification, and lateral flow detection for DNA 13 and RNA, 14 in separate paper-based modules. To date, only one other device has been reported that combines paper-based ex- traction, amplification and detection steps, 15 which consists of a magnetic sliding strip to run each step serially. While this device does encompass a fully integrated NAAT system, it relies on a UV light source and imaging for endpoint detec- tion, requiring equipment that may not be readily available in remote low-resource settings. Furthermore, this device has only been shown to detect E. coli DNA that was spiked into plasma, rather than extracting DNA from clinical specimens. In this study, we sought to integrate all three modules into a Lab Chip This journal is © The Royal Society of Chemistry 2016 a Department of Biomedical Engineering, Boston University, Boston, MA, USA. E-mail: [email protected] b Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA c Department of Pathology, University of Michigan Medical School, Ann Arbor, MI, USA † Electronic supplementary information (ESI) available: Additional figures containing primer sequences, a chip side-view schematic, HPV 16 DNA extrac- tions and in situ LAMP in PES results, and on-chip extraction images are in- cluded. See DOI: 10.1039/c5lc01392e Published on 20 January 2016. Downloaded by Boston University on 20/01/2016 18:42:39. View Article Online View Journal
-
Upload
winnie-wong -
Category
Documents
-
view
218 -
download
0
Transcript of nat's paper!!!! 2016

Lab on a Chip
PAPER
Cite this: DOI: 10.1039/c5lc01392e
Received 13th November 2015,Accepted 7th January 2016
DOI: 10.1039/c5lc01392e
www.rsc.org/loc
A fully integrated paperfluidic moleculardiagnostic chip for the extraction, amplification,and detection of nucleic acids from clinicalsamples†
Natalia M. Rodriguez,a Winnie S. Wong,a Lena Liu,a Rajan Dewarbc
and Catherine M. Klapperich*a
Paper diagnostics have successfully been employed to detect the presence of antigens or small molecules
in clinical samples through immunoassays; however, the detection of many disease targets relies on the
much higher sensitivity and specificity achieved via nucleic acid amplification tests (NAAT). The steps in-
volved in NAAT have recently begun to be explored in paper matrices, and our group, among others, has
reported on paper-based extraction, amplification, and detection of DNA and RNA targets. Here, we inte-
grate these paper-based NAAT steps into a single paperfluidic chip in a modular, foldable system that al-
lows for fully integrated fluidic handling from sample to result. We showcase the functionality of the chip
by combining nucleic acid isolation, isothermal amplification, and lateral flow detection of human papillo-
mavirus (HPV) 16 DNA directly from crude cervical specimens in less than 1 hour for rapid, early detection
of cervical cancer. The chip is made entirely of paper and adhesive sheets, making it low-cost, portable,
and disposable, and offering the potential for a point-of-care molecular diagnostic platform even in remote
and resource-limited settings.
Introduction
Integrated molecular diagnostics to enable sample-to-answernucleic acid amplification testing (NAAT) have traditionallyrequired sophisticated instrumentation to provide pressuredriven fluid handling, cyclic thermal control, and optical as-say detection. These requirements result in expensive equip-ment and costly disposables unsuitable for use in resource-limited settings. Since the start of lab-on-a-chip microfluidictechnologies in the 90s, their use in remote settings has beenperceived as potentially one of their most powerful applica-tions, by taking advantage of their small size, portability, lowvolume requirement for samples, and rapid analysis withoutthe need for an expert operator.1 However, because these
systems require pumps or pressure in order to drive fluidflow, they aren't yet equipment-free and rely on instrumenta-tion and electricity. An alternative that has garnered much at-tention recently is “paperfluidics”, where paper is used as asubstrate to construct microfluidic devices for use in rapid di-agnostic tests.2–6 Paper's ability to passively transport fluidsthrough capillary action, or wicking, eliminates the need forpumps or other fluid handling equipment. In regard to mo-lecular diagnostics, a number of studies have reported on thesuccess of individual NAAT steps (extraction, amplification,and detection) within paper matrices.7–12 Our group has pre-viously shown paper-based nucleic acid extraction, isothermalamplification, and lateral flow detection for DNA13 andRNA,14 in separate paper-based modules. To date, only oneother device has been reported that combines paper-based ex-traction, amplification and detection steps,15 which consistsof a magnetic sliding strip to run each step serially. Whilethis device does encompass a fully integrated NAAT system, itrelies on a UV light source and imaging for endpoint detec-tion, requiring equipment that may not be readily available inremote low-resource settings. Furthermore, this device hasonly been shown to detect E. coli DNA that was spiked intoplasma, rather than extracting DNA from clinical specimens.In this study, we sought to integrate all three modules into a
Lab ChipThis journal is © The Royal Society of Chemistry 2016
aDepartment of Biomedical Engineering, Boston University, Boston, MA, USA.
E-mail: [email protected] of Pathology, Beth Israel Deaconess Medical Center, Harvard
Medical School, Boston, MA, USAcDepartment of Pathology, University of Michigan Medical School, Ann Arbor, MI,
USA
† Electronic supplementary information (ESI) available: Additional figurescontaining primer sequences, a chip side-view schematic, HPV 16 DNA extrac-tions and in situ LAMP in PES results, and on-chip extraction images are in-cluded. See DOI: 10.1039/c5lc01392e
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article OnlineView Journal

Lab Chip This journal is © The Royal Society of Chemistry 2016
single paperfluidic chip, and demonstrate detection of a tar-get nucleic acid analyte directly from crude clinical samples.
To showcase the full sample-to-answer functionality of thechip, we chose to design a test for cervical cancer, a diseasethat disproportionately affects the developing world whereearly detection is made difficult by a lack of screeningmethods suitable for these low-resource settings, and forwhich a rapid, low-cost, point-of-care (POC) molecular test ofthis nature is greatly needed. Each year, over half a millionnew cases of cervical cancer and over a quarter of a milliondeaths caused by cervical cancer occur worldwide.16,17 De-spite its alarming global mortality, cervical cancer is highlypreventable and easily treated through early detection and re-moval of precancerous lesions.16,18 Unfortunately, cervicalcancer is most prevalent in areas where no effective screeningprograms have been established. From a total of 528 000 newcases worldwide in 2012, 445 000 cases (84%) occurred in thedeveloping world,17 where, according to the World Health Or-ganization (WHO), less than 5% of women have access toscreening even once in their lifetime.19,20
The traditional method to detect cervical cancer is the Papsmear, where trained pathologists analyze cervical smears un-der a microscope and determine if there is evidence of abnor-mal cells. Although cytology-based screening has dramaticallyreduced the incidence and mortality of cervical cancer inmany developed countries, it is a qualitative process that suf-fers from very low sensitivity (as low as 53%),21 and success-ful cytological screening programs have been difficult to im-plement and sustain in remote and low-income settingsbecause they are resource-intensive and require highlytrained personnel.22 Moreover, many developing regions lackthe necessary infrastructure to transport Pap smears to andfrom a laboratory for processing and interpretation in atimely manner, thus requiring up to three patient visits tothe clinic for screening, communication of results, and treat-ment. The delay in getting results back to patients is knownto be a particularly significant barrier to screening, because alarge number of women won't return for results.18 Efforts toreduce global cervical cancer incidence and mortality need tomore effectively target developing regions via alternativemethods that are less costly, less dependent on existing labo-ratory infrastructure and trained professionals, and capableof rapid, accurate diagnosis at POC.
Visual inspection with acetic acid (VIA) is a rapid and inex-pensive detection method that has been promoted globally asa reasonable alternative to the Pap smear, but studies haverevealed unacceptably low sensitivity and specificity, resultingin very high rates of false negative and false positive re-sults.23,24 Moreover, like the Pap smear, VIA does not providemolecular information on the presence of the etiologicalagent of cervical cancer, human papillomavirus (HPV).
HPV is a common sexually transmitted infection, which inthe majority of cases is transient, asymptomatic, and clini-cally insignificant. In some women, however, the infectionbecomes persistent and may lead to the development of cervi-cal cancer. Over 99% of cervical cancer cases are caused by
HPV,25 more than half of which are caused by the HPV 16subtype.26 Given the limitations of cytology, much work hasbeen focused on molecular diagnostics for cervical cancerthrough HPV DNA testing. These methods typically have veryhigh sensitivity (>96–100%) and specificity (>90–100%).21 Alandmark study in rural India showed that a single round ofHPV testing was associated with a significant reduction inthe number of advanced cervical cancers and deaths fromcervical cancer over time compared to cytology or VIA.27
While these results validate the use of HPV DNA testing, asignificant drawback is the high cost and the need for sophis-ticated laboratory equipment. Furthermore, current HPVDNA tests still require highly trained laboratory personneland incur turnaround times of hours to days, depending onhow far the sample has to travel to a central laboratory.28–30
Translating the molecular testing process to the POC canminimize these limitations by providing results faster, on theorder of minutes, allowing doctors to diagnose, advise andpotentially treat patients in the same visit. Asymptomatic pa-tients positive for high-risk HPV strains like 16 could be scre-ened more closely, thus allocating precious resources tothose most at risk. A POC diagnostic device could be taken toremote settings beyond a standard clinic or laboratory, elimi-nating transport turnaround time. Additionally, a user-friendly, self-contained diagnostic device, with a simple vi-sual readout similar to an at-home pregnancy test, couldeliminate the need for highly trained specialists and would re-quire only minimal training of a community health worker.
Here, we present a paperfluidic chip made entirely of pa-per and adhesive sheets that combines nucleic acid extrac-tion, isothermal loop-mediated amplification, and lateralflow detection via immunochromatographic strips that en-able an immediate visual readout. This low-cost, portable,and disposable chip provides a simple, rapid molecular diag-nostic platform for POC detection of nucleic acids. We devel-oped a fully integrated, on-chip, sample-to-answer NAAT as-say for the detection of HPV 16 DNA directly from patientcervical specimens in less than an hour. This novel diagnos-tic platform could overcome many barriers currently faced inlimited-resource settings and increase access to cervical can-cer screening to those most in need.
Materials and methodsHPV 16 cloned DNA standards
HPV 16 DNA standards were generated by cloning the E7gene for HPV 16 into the pGEM-T Easy Vector (Promega,Madison, WI). The E7 gene was PCR amplified from HPV-16transformed cell DNA (Advanced Biotechnologies, Inc,Eldersburg, MD) with gene-specific forward and reverse clon-ing primers (Table S1†) containing restriction endonucleasesequences SpeI and AatII, respectively, using the standardTaq Polymerase protocol (New England Biolabs, Ipswich,MA). The PCR product was purified via phenol chloroform ex-traction and ethanol precipitation. The cleaned PCR productwas digested overnight with SpeI and AatII restriction
Lab on a ChipPaper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online

Lab ChipThis journal is © The Royal Society of Chemistry 2016
endonucleases (New England Biolabs, Ipswich, MA). The rele-vant band was gel extracted and ligated to the pGEM vectorand transformed into Top 10 cells from Life Technologies(Grand Island, NY). Plasmid DNA was extracted using a MiniPrep Kit (Qiagen, Valencia, CA) and sequenced (GeneWiz,Inc, Cambridge, MA) to confirm proper E7 insertion. A MidiPrep Kit (Qiagen, Valencia, CA) was used to generate largescale plasmid stocks of the correctly sequenced DNA. Theplasmid stocks were linearized with ZraI restriction endonu-clease (New England Biolabs, Ipswich, MA). The correct sizefragment was gel extracted using the QIAquick Extraction Kit(Qiagen, Valencia, CA), treated with phenol chloroform, andprecipitated in ethanol. The concentration of the purifiedDNA was determined by measuring the OD260 with a Nano-Drop ND-2000c apparatus (Thermo Scientific, Waltham, MA).The DNA copy number was calculated and 1 mL aliquotswere made and stored at −20 °C.
Clinical cervical specimens
The cervical specimens were accrued from the BIDMC cytol-ogy laboratory, on already tested and to be discarded speci-mens. The IRB approval and patient consent for research useof these de-identified and discarded specimens was waivedby the BIDMC Institutional Review Board. The specimenswere obtained in PreservCyt® solution. Testing was done onan FDA approved platform (Cervista; hrHPV), which evaluates14 of the most common high-risk HPV genotypes (16, 18, 31,33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68) using InvaderChemistry. Any patient health identifying information wascompletely removed, and the specimens were labeled with asample number and as HPV positive or negative only beforethey were transferred to the Klapperich Laboratory.
Samples were then transferred to 50 mL conical tubes andcentrifuged for 10 min at 4000 RPM. The supernatant was re-moved and the cell pellet was washed with 3 mL of PBS,vortexed and centrifuged for 10 min at 4000 RPM. This wasrepeated twice, leaving a cell pellet that was resuspended in 3mL of PBS and divided into (3) 1 mL aliquots. Each 1 mL ali-quot was centrifuged for 5 min at 13 000 RPM. The superna-tant was removed and pellets were frozen at −80 °C for long-term storage. Prior to use, pellets were resuspended in 1 mLof PBS, subdivided into 200 μl aliquots, centrifuged for 10min at 13 000 RPM, and the supernatant was removed,resulting in single-use pellets for experiments. It is importantto note that these centrifugation steps were required here be-cause the patient samples we received as part of our IRB-approved study were cervical tissue specimens fixed in largevolumes of ethanol-containing PreservCyt® solution, which isunsuitable for direct use in a POC device. In a real-world set-ting, a fresh or dried cervical swab would be placed directlyinto our lysis buffer, thereby eliminating these extra centrifu-gation steps.
For gold standard extraction experiments, DNA wasextracted from a single-use pellet of each specimen using the
DNeasy Blood & Tissue Kit (QIAGEN) and eluted into a finalvolume of 200 μL.
qPCR
To ascertain the DNA extraction yields, 5 μl of extracted DNAwas amplified via quantitative PCR (qPCR). Using theSurestart Taq DNA polymerase (Agilent, Santa Clara, CA),real-time PCR was performed on an Applied Biosystems 7500thermocycler under the following conditions: 10 min at 95 °Cfor polymerase activation, followed by 30 cycles of 30 s at 95°C, 15 s at 55 °C for primer annealing, and 90 s at 60 °C foramplification. The 25 μL reaction mixture contained 1×TaqMan buffer, 3.5 mM MgCl2, 8% DMSO, 200 μM dNTPs,200 nM primers and TaqMan probes (Table S1†), 0.1× RoxReference Dye, 0.625 U Taq DNA polymerase, and 5 μL ofsample or standard DNA. For clinical specimen gold standardextraction experiments, a multiplexed HPV 16 and RNasePqPCR assay was run following the same reaction conditionswhere RNaseP served as a DNA control to confirm that eachclinical specimen did in fact contain cells and that theQiagen extractions were performed properly. If a clinical sam-ple was negative for RNaseP (cycle threshold value >30), thesample was deemed invalid and was not used for furtherexperiments.
In each qPCR run, a cycle threshold value versus DNA con-centration standard curve was generated from a serial dilu-tion of our cloned HPV 16 DNA standards. For each patientsample, the effective viral DNA concentration was quantifiedvia standard curve interpolations.
Isothermal loop-mediated amplification assay
An isothermal loop-mediated amplification (LAMP) assay wasdeveloped for rapid amplification and detection of the HPV16 E7 gene using primer sequences previously designed byLuo et al.31 listed in Table S1.† The assay was first optimizedin a tube, and then translated to a chip format. The assaytakes place in situ, in a PES membrane in the sample inletport, as previously described by our group.14,32 The in-tubereaction was carried out in a final volume of 25 μL with 1 μLof the DNA sample, 1× Isothermal Amplification Buffer (NewEngland Biolabs), 8 U large fragment Bst 2.0 DNA polymer-ase, 0.8 M Betaine, 1 mM dNTPs, 5 pmol each of forward andreverse outer primers (F3 and B3), 20 pmol each of forwardand reverse loop primers (LF and LB), and 40 pmol each offorward and reverse inner primers (FIP and BIP). The on-chipLAMP reaction recipe was identical but was carried out in afinal volume of 12.5 μL. Fresh, single-use aliquots of each re-agent were used each time. The reaction was run for 30 mi-nutes at 63 °C. Forward and reverse loop primers (LF and LB)were tagged with fluorescein isothiocyanate (FITC) and bio-tin, respectively, to enable immediate downstream detectionof the amplified products on immunochromatographic, lat-eral flow detection (LFD) test strips (Ustar Biotechnologies,Hangzhou, China) consisting of streptavidin-conjugated gold
Lab on a Chip Paper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online

Lab Chip This journal is © The Royal Society of Chemistry 2016
nanoparticles, an anti-FITC test line, and a biotin (anti-streptavidin) flow control line.
For in-tube LAMP assay experiments, the amplified prod-ucts were analyzed by 2% agarose gel electrophoresis. Thespecificity of the products was confirmed by restriction en-zyme digestion with the PvuII restriction endonuclease (NewEngland Biolabs) with a single cutting site within the FIP re-gion. Following digestion at 37 °C overnight, the digestedproducts were analyzed by 2% agarose gel electrophoresisand by 10% acrylamide gel electrophoresis for higher resolu-tion analysis.
LFD strips were imaged using an iPhone 5 camera (Apple).LFD test line and control line intensities were quantifiedusing the Gel Analysis feature in ImageJ (National Institutesof Health) and analyzed by dividing the intensity of the testline by the intensity of the control line to obtain the percent-age of control intensity for each sample. The resulting inten-sities were plotted as median with range. Unpaired, two-tailed Student's T-tests were used to determine the signifi-cance of each sample readout compared to the experimentalnegative control sample readout.
Paperfluidic chip fabrication
Standard letter size self-adhesive laminating sheets (Fellowesproduct # 5221502) served as the base material for buildingour paperfluidic chip, providing a hydrophobic (tape) barriersurrounding the paper components that is low-cost and opti-cally transparent to enable our visual readout. We createdblueprint drawings for the adhesive base of the chip (Fig. 1b)using computer-aided design software (AutoCAD), and cutthe adhesive sheets accordingly using an electronic craft cut-ting tool (Graphtec Craft Robo Pro S with Graphtec Studiosoftware) using a standard blade (CB09U) and the followingsettings – cut force: 27, speed: 7 cm s−1, acceleration: 1. Theblueprint drawings include perforations in the adhesivesheets that were specifically designed to enable easy foldingand ripping as needed during chip fabrication and operation,such that the integrity of the chip would remainuncompromised. The cut adhesive sheets were peeled fromthe protective backing and placed adhesive side-up on thebenchtop as shown schematically in Fig. 1c, step i. Poly-ethersulfone (PES) filter paper (Millipore, cat# GPWP04700)was cut into 0.375 inch diameter discs using a 3/8″ craft holepunch (EK Tools, 54-10061). A single 0.375 inch diameter PESdisc is manually placed directly over the 0.3 inch diameterhole in the adhesive sheet, and the top tab is folded downalong the perforation over the PES (Fig. 1c, step ii) to createthe sample port (Fig. 1c, step iii). Next, the bottom tab isfolded up along the perforation to create a toehold for whatwill become the sample port cover film to prevent evapora-tion during the LAMP heat step (Fig. 1c, step iv). The 0.3 inchdiameter circle of tape that had been cut out of the adhesivesheet to make the sample port is peeled off the protectivebacking where it stayed behind and is manually placed adhe-sive side-down onto the adhesive sheet 2 inches down from
the center of the sample port (Fig. 1c, step v). This will alignwith the sample port when the cover film is placed duringLAMP and prevent the DNA and/or PES membrane fromsticking to the adhesive cover film.
Cellulose blotting paper (Whatman GB003, cat# 09-301-400) was cut using a 30 W Epilog Zing laser cutter (speed =70%, power = 28%, frequency = 200) to make absorbent padsshaped as 2.5 inch long sectors that extend radially from0.375 inch at the base of the sample port to an ultimatewidth of 0.75 inch (drawn in SolidWorks, Waltham, MA). Theabsorbent pad is manually aligned and placed over the sam-ple port extending towards the left side of the chip as shownin Fig. 1c, step vi. The lower middle section of the chip isthen folded over the centerline perforation over the absor-bent pad as shown in Fig. 1c, steps vii–viii to create a hydro-phobic (tape) barrier between the absorbent pad and the LFDstrip. The LFD strip is then manually aligned with the sampleport center and placed down extending over the right side ofthe chip as shown in Fig. 1c, step ix. Next, the bottom tworemaining sections of the adhesive sheet are folded up overthe perforations to seal the chip from the bottom (Fig. 1c, steps x–xi). The fabrication is now complete, and the chip is then flippedover so that the PES membrane sample port is right side-up andready for use (Fig. 1c, step xii).
Integrated on-chip assay
DNA extraction and purification. A single-step cell lysisand DNA extraction recipe was developed based on thechaotropic lysis and alcohol precipitation methods of Boomet al.33 and Linnes et al.13 A single-use pellet of each clinicalcervical specimen (or 6 μL of cloned HPV16 DNA during pre-liminary experiments) was resuspended in a lysis buffercontaining 3 M guanidinium thiocyanate, 300 mM sodiumchloride, 35% v/v 1-butanol (Sigma Aldrich, St. Louis, MO),and 3 μL of 15 mg mL−1 Glycoblue coprecipitant (Life Tech-nologies, Grand Island, NY) in a total volume of 100 μL. Thismixture was pipetted onto the sample port of the paperfluidicchip. The liquid phase wicks through the absorbent pad di-rectly underneath the PES membrane by capillary forces, leav-ing the precipitated DNA-Glycoblue solid phase. A series ofethanol washes (200 μL of 70% ethanol, followed by 100 μLof 100% ethanol) were then pipetted through the sampleport, removing impurities while leaving the purified DNA-glycogen precipitate on the PES membrane. The left side ofthe chip containing the soiled absorbent pad was then rippedalong the perforation and discarded. The rest of the chip isleft on the benchtop for at least 2 minutes to allow for com-plete drying of ethanol prior to addition of the LAMP reactionmix.
Isothermal amplification. A 12.5 μL LAMP reaction mixwas pipetted directly onto the sample port and was fullyabsorbed by the PES, presumably dissociating the DNA-Glycoblue complexes. The bottom tab of the chip is thenfolded up along the perforation and pressed down to sealover the absorbent pad and serves as a cover film to prevent
Lab on a ChipPaper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online

Lab ChipThis journal is © The Royal Society of Chemistry 2016
Fig. 1 Paperfluidic molecular diagnostic chip. a) Image of paperfluidic chip. Scale bar = 1 inch. b) Blueprint drawings and dimensions for theadhesive base of the chip. c) Schematic of chip fabrication steps: i. the cut adhesive sheets are peeled from the protective backing and placedadhesive side-up on the benchtop (the white area is adhesive sheet, the dark grey areas are holes that have been cut out of the adhesive sheet); ii.a 0.375 inch diameter PES disc is manually placed directly over the 0.3 inch diameter hole in the adhesive sheet, and the top tab is folded downalong the perforation over the PES; iii. the sample port is now created (the light grey color indicates areas where the adhesive sheet has beenfolded over onto itself, rendering the area non-adhesive); iv. the bottom tab is folded up along the perforation to create a toehold for what will be-come the sample port cover film to prevent evaporation during the LAMP heat step; v. the 0.3 inch diameter circle of tape that had been cut outof the adhesive sheet to make the sample port is peeled off the protective backing where it stayed behind and is manually placed adhesive side-down onto the adhesive sheet 2 inches down from the center of the sample port. This will align with the sample port when the cover film is placedduring LAMP and prevent the DNA and/or PES membrane from sticking to the adhesive cover film; vi. the absorbent pad is manually aligned andplaced over the sample port extending towards the left side of the chip; vii–viii. the lower middle section of the chip is then folded over the center-line perforation over the absorbent pad to create a hydrophobic (tape) barrier between the absorbent pad and the LFD strip; ix. the LFD strip isthen manually aligned with the sample port center and placed down extending over the right side of the chip; x–xi. the bottom two remaining sec-tions of the adhesive sheet are folded up over the perforations to seal the chip from the bottom; xii. the fabrication is now complete, and the chipis then flipped over so that the PES membrane sample port is right side-up and ready for use.
Lab on a Chip Paper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online

Lab Chip This journal is © The Royal Society of Chemistry 2016
evaporation during the incubation period for LAMP. The chipis then placed face-down on a 63 °C heat block or hot platefor 30 min.
Lateral flow detection. Following the LAMP incubation,the cover film was peeled back using the toehold tab to
expose the sample port on top, and peeled under the chip toexpose the sample port outlet on the bottom, thereby remov-ing the hydrophobic (tape) barrier between the sample portand the LFD strip (see side view schematic in Fig. S1†). 50 μLof nuclease free water was then pipetted onto the sample
Fig. 2 Fluidic demonstration of chip operation. i) A lysed sample, demonstrated here using 100 μL of blue dye, is placed onto the sample port ofthe paperfluidic chip using a pipette or dropper. ii) The prevailing capillary forces generated by the absorbent pad directly underneath the sampleport quickly wick the liquid waste through the PES membrane and away from the sample port leaving the solid phase behind. iii) A first wash of70% ethanol, demonstrated here using 200 μL of yellow dye, is filtered through the sample port. iv) The wash buffer will wick through to theabsorbent pad, removing impurities and leaving behind the purified precipitated DNA. v–vi) A final wash of 100% ethanol, demonstrated here using100 μL of water, is filtered through the sample port, leaving just the purified DNA on the PES membrane. vii) The waste absorbent pad is discardedby ripping off the left side of the chip at the designated perforation. viii) The LAMP reaction mix is placed directly onto the sample port where thepurified DNA remains, and the bottom tab of the chip is folded up over the designated perforation to act as a cover film for the sample port andprevent evaporation during the heat step. ix) After the heat incubation for LAMP, the cover film is peeled back using the toehold to expose thesample port on top, and peeled under the chip to expose the sample port outlet on the bottom, thereby removing the hydrophobic (tape) barrierbetween the sample port and the LFD strip. The PES membrane is now in direct contact with the LFD strip and the amplified products are theneluted onto the strip by adding 50 μL of water to the sample port. x) The eluted products wick through the LFD strip towards the right.
Lab on a ChipPaper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online

Lab ChipThis journal is © The Royal Society of Chemistry 2016
port, which filtered through the PES and wicked directly ontothe LFD strip for immediate detection of amplified products.
Results and discussionFluidic demonstration of chip operation
A lysed sample, demonstrated here using 100 μL of blue dye,is placed onto the sample port of the paperfluidic chip usinga pipette or dropper (Fig. 2i). The prevailing capillary forcesgenerated by the absorbent pad directly underneath the sam-ple port quickly wick the liquid waste through the PES mem-brane and away from the sample port (Fig. 2ii). A hydropho-bic (tape) barrier between the absorbent pad and the LFDstrip prevents the liquid waste from wicking through to theLFD strip (see side view schematic in Fig. S1†). Any solidphase within the sample, most importantly the precipitatedDNA, will remain on the sample port surface. Next, a firstwash of 70% ethanol, demonstrated here using 200 μL of yel-low dye, is filtered through the sample port (Fig. 2iii–iv). Thewash buffer will wick through to the absorbent pad, remov-ing most impurities like cell debris, proteins, and salts andleaving behind the purified precipitated DNA. Because etha-nol can inhibit the subsequent LAMP reaction, it is importantto completely dry the sample port. To that end, a final washof 100% ethanol, demonstrated here using 100 μL water, isfiltered through the sample port (Fig. 2v), leaving just the pu-rified DNA precipitates on the PES membrane (Fig. 2vi).
The waste absorbent pad is no longer needed at this pointand can be discarded by ripping off the left side of the chipat the designated perforation (Fig. 2vii). Next, 12.5 μL of theLAMP reaction mix is placed directly onto the sample portwhere the purified DNA remains, and the bottom tab of thechip is folded up over the designated perforation to act as acover film for the sample port and prevent evaporation dur-ing the heat step (Fig. 2viii). The chip is then placed face-down onto a heat block or hot plate set to 63 °C for 30 min(not shown). After the heat incubation, the cover film ispeeled back using the toehold to expose the sample port ontop, and peeled under the chip to expose the sample port out-let on the bottom, thereby removing the hydrophobic (tape)barrier between the sample port and the LFD strip. The PESmembrane is now in direct contact with the LFD strip andthe amplified products are then eluted onto the strip byadding 50 μL of water to the sample port (Fig. 2ix). Theeluted products wick through the LFD strip towards the ab-sorbent pad on the right. As the liquid wicks through theconjugate pad, the streptavidin-conjugated gold nanoparticlesbind the biotin probes on the LB primers within theamplicons. As the liquid continues to wick over the detectionzone, amplicons that also contain the FITC probe on the LFprimers will aggregate at the anti-FITC test line. Any excessstreptavidin-conjugated gold nanoparticles will continue towick through the LFD strip and bind the biotin control line,which confirms that the strip functioned properly. Here, asthis was just a demonstration using water, the test result is
obviously negative, thus only the control line appears on thestrip (Fig. 2x).
HPV 16 E7 LAMP assay
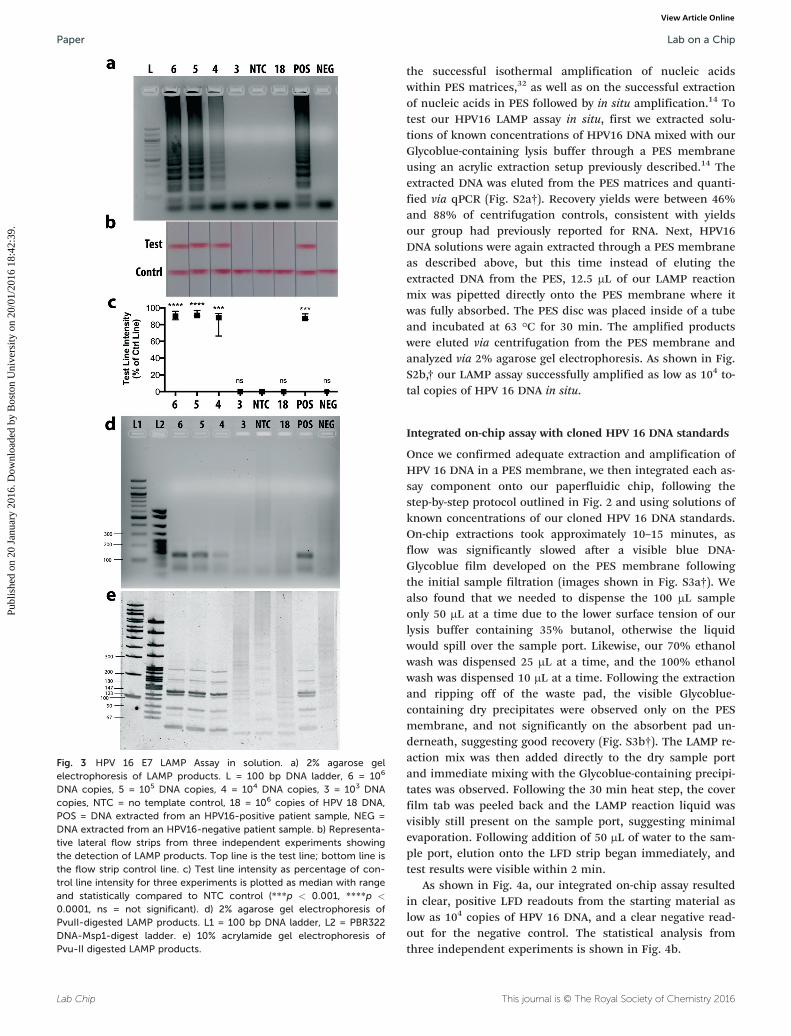
The HPV 16 E7 LAMP assay was first optimized in-tube usingour cloned HPV16 DNA standards. We ran the optimizedLAMP assay using serial dilutions of our DNA standards andfound that our lower limit of detection was 104 total copiesas confirmed by agarose gel electrophoresis (Fig. 3a) and LFDstrips (Fig. 3b). The LFD strips enable immediate detection ofamplified products with the naked eye. Test line intensitieswere quantified as a percentage of control line intensitiesand results from three independent experiments are plottedin Fig. 3c. DNA quantities down to 104 total copies show aclear visible test line that is statistically different from thenegative control. While 103 DNA copies were not amplified todetectable levels, the HPV literature has shown that a viralload below 104 copies is not indicative of cervical cancerprogression.34,35
We included a no template control (NTC) and anonspecific DNA control (106 total copies of HPV 18 DNA),both of which were negative on both the gel and LFD strips,demonstrating primer specificity. Additionally, we ran ourLAMP assay on Qiagen kit-extracted DNA from a patient sam-ple that tested positive for HPV 16 and from a clinical samplethat tested negative for HPV 16. It is important to note thatthese samples contain large amounts of human DNA and po-tentially other viral genomes. Our LAMP assay correctly iden-tified these patient samples as positive and negative, respec-tively, thus further confirming the specificity of our LAMPassay.
One disadvantage of the LAMP method is the possibilityfor interaction and self-priming of the oligonucleotides dur-ing the reaction. This phenomenon has been widely reportedin the literature,36–39 and is usually circumvented by optimi-zation of assay conditions and setting an assay cutoff timefar before these events are likely to occur. Nonetheless, be-cause our ultimate assay detection method is based onprimer-tagged probes, it was important to ensure that a posi-tive result on the LFD strip correlated with a LAMP productspecific to our target sequence. To this end, the amplifiedproducts were digested with the PvuII restriction endonucle-ase and analyzed by 2% agarose gel electrophoresis (Fig. 3d)and at higher resolution by 10% acrylamide gel electrophore-sis (Fig. 3e). The HPV 16 E7 gene sequence contains a singlePvuII cutting site within the FIP region, and positive productdigests were in agreement with the expected product bandsizes,40 while the negative product digests showed nothingon a low-resolution agarose gel (Fig. 3d) and showed irregularband patterns inconsistent with expected product band sizeson a high-resolution acrylamide gel (Fig. 3e).
Having confirmed the lower detection limit and specificityof our HPV16 LAMP assay in-tube, we then tested our LAMPassay in situ, directly within a paper matrix containing freshlyextracted HPV16 DNA. Our group has previously reported on
Lab on a Chip Paper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online
Lab Chip This journal is © The Royal Society of Chemistry 2016
the successful isothermal amplification of nucleic acidswithin PES matrices,32 as well as on the successful extractionof nucleic acids in PES followed by in situ amplification.14 Totest our HPV16 LAMP assay in situ, first we extracted solu-tions of known concentrations of HPV16 DNA mixed with ourGlycoblue-containing lysis buffer through a PES membraneusing an acrylic extraction setup previously described.14 Theextracted DNA was eluted from the PES matrices and quanti-fied via qPCR (Fig. S2a†). Recovery yields were between 46%and 88% of centrifugation controls, consistent with yieldsour group had previously reported for RNA. Next, HPV16DNA solutions were again extracted through a PES membraneas described above, but this time instead of eluting theextracted DNA from the PES, 12.5 μL of our LAMP reactionmix was pipetted directly onto the PES membrane where itwas fully absorbed. The PES disc was placed inside of a tubeand incubated at 63 °C for 30 min. The amplified productswere eluted via centrifugation from the PES membrane andanalyzed via 2% agarose gel electrophoresis. As shown in Fig.S2b,† our LAMP assay successfully amplified as low as 104 to-tal copies of HPV 16 DNA in situ.
Integrated on-chip assay with cloned HPV 16 DNA standards
Once we confirmed adequate extraction and amplification ofHPV 16 DNA in a PES membrane, we then integrated each as-say component onto our paperfluidic chip, following thestep-by-step protocol outlined in Fig. 2 and using solutions ofknown concentrations of our cloned HPV 16 DNA standards.On-chip extractions took approximately 10–15 minutes, asflow was significantly slowed after a visible blue DNA-Glycoblue film developed on the PES membrane followingthe initial sample filtration (images shown in Fig. S3a†). Wealso found that we needed to dispense the 100 μL sampleonly 50 μL at a time due to the lower surface tension of ourlysis buffer containing 35% butanol, otherwise the liquidwould spill over the sample port. Likewise, our 70% ethanolwash was dispensed 25 μL at a time, and the 100% ethanolwash was dispensed 10 μL at a time. Following the extractionand ripping off of the waste pad, the visible Glycoblue-containing dry precipitates were observed only on the PESmembrane, and not significantly on the absorbent pad un-derneath, suggesting good recovery (Fig. S3b†). The LAMP re-action mix was then added directly to the dry sample portand immediate mixing with the Glycoblue-containing precipi-tates was observed. Following the 30 min heat step, the coverfilm tab was peeled back and the LAMP reaction liquid wasvisibly still present on the sample port, suggesting minimalevaporation. Following addition of 50 μL of water to the sam-ple port, elution onto the LFD strip began immediately, andtest results were visible within 2 min.
As shown in Fig. 4a, our integrated on-chip assay resultedin clear, positive LFD readouts from the starting material aslow as 104 copies of HPV 16 DNA, and a clear negative read-out for the negative control. The statistical analysis fromthree independent experiments is shown in Fig. 4b.
Fig. 3 HPV 16 E7 LAMP Assay in solution. a) 2% agarose gelelectrophoresis of LAMP products. L = 100 bp DNA ladder, 6 = 106
DNA copies, 5 = 105 DNA copies, 4 = 104 DNA copies, 3 = 103 DNAcopies, NTC = no template control, 18 = 106 copies of HPV 18 DNA,POS = DNA extracted from an HPV16-positive patient sample, NEG =DNA extracted from an HPV16-negative patient sample. b) Representa-tive lateral flow strips from three independent experiments showingthe detection of LAMP products. Top line is the test line; bottom line isthe flow strip control line. c) Test line intensity as percentage of con-trol line intensity for three experiments is plotted as median with rangeand statistically compared to NTC control (***p < 0.001, ****p <
0.0001, ns = not significant). d) 2% agarose gel electrophoresis ofPvuII-digested LAMP products. L1 = 100 bp DNA ladder, L2 = PBR322DNA-Msp1-digest ladder. e) 10% acrylamide gel electrophoresis ofPvu-II digested LAMP products.
Lab on a ChipPaper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online

Lab ChipThis journal is © The Royal Society of Chemistry 2016
Integrated on-chip assay with clinical cervical specimens
DNA from cervical tissue sample pellets was extracted via thegold standard Qiagen DNeasy Tissue Kit. Each sample extrac-tion was analyzed by qPCR for HPV16 E7 and RNaseP DNA.RNaseP serves as a human gene internal control to ensurethat the cervical swab sample contained cervical cells andthat DNA was properly extracted. Any samples that testednegative for RNaseP by qPCR were considered “invalid” and
were not used in further experiments. Five HPV 16 positiveand five HPV 16 negative samples (Fig. 5 table) were selectedfor on-chip testing to demonstrate proof-of-concept clinicalutility of our paperfluidic chip.
A single-use pellet from each sample A–J was resuspendedin 100 μL of lysis buffer, vortexed thoroughly, and pipettedonto the sample port of the chip. During preliminary experi-ments, significant accumulation of debris and salts from thelysed samples left a visible grainy film on the PES membrane,which greatly inhibited the subsequent LAMP reaction. Thisprompted an increase from 100 to 200 μL of our 70% ethanol
Fig. 4 Integrated on-chip assay with cloned HPV 16 DNA standards. a)Representative lateral flow strips from three independent on-chip ex-periments showing the detection of LAMP products from 1E4 to 1E6DNA copies, NTC = no template control. Left line is the test line; rightline is the flow strip control line. b) Test line intensity as percentage ofcontrol line intensity for three experiments is plotted as median withrange and statistically compared to NTC control (**p < 0.01, ***p <
0.001, ****p < 0.0001).
Fig. 5 Integrated on-chip assay with clinical cervical specimens. Tablelists gold standard assay (Qiagen-extraction quantified by qPCR) resultsfor HPV16 and RNaseP control DNA quantities for each of 10 patientsamples labeled A–J. NEG = negative result. Chip LFD strip images foreach sample A–J below.
Lab on a Chip Paper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online

Lab Chip This journal is © The Royal Society of Chemistry 2016
washes, which did not entirely remove the residue in allcases, but significantly improved the LAMP performancenonetheless.
All 5 positive samples resulted in clear, positive LFD re-sults as seen in Fig. 5 strips A–E. Of the 5 negative samples, 3resulted in a negative LFD result (strips G, I, J), but 2exhibited faint test lines (F, H) resulting in a false positive re-sult. As mentioned above, the interaction and self-priming oftwo or more of the six primers involved in LAMP is a widelyrecognized issue that may be the cause of our false positiveresults. Considering our LFD strip detection method, movingforward we suspect that a sequence-specific probe (notprimer-tagged), as reported by Curtis et al.,41 would be bettersuited for this type of assay.
It is also possible that cell debris and salts in the cervicalsamples left behind on the sample port are affecting ourprimer melting temperatures in ways we are unable to ac-count for. While further primer and/or probe optimizationmay be required, herein we have demonstrated that a fullyintegrated, sample-to-answer, molecular diagnostic assay on alow-cost, disposable paperfluidic chip platform is possible.
Furthermore, it is known that with HPV testing, clinicalfalse-positive results (positive screening tests without under-lying precancerous lesions) are common,42 largely due totransient levels of HPV infection in any given population thatmay never develop into cervical intraepithelial neoplasias orcancer. Consequently, American Cancer Society guidelines donot recommend screening by HPV testing alone for most clin-ical settings, but as co-testing with cytology every 5 years inwomen over 30.43 In resource-limited settings, where co-testing is likely not an option, our paperfluidic diagnosticplatform allows for primary HPV screening, where a positiveresult can be followed up by cytology, thereby allocating pre-cious resources to those most at risk. A recent systematic re-view concluded that HPV testing alone is very promising forthe primary screening of women aged 30 years and older,particularly when coupled with cytology testing for follow-upof HPV-positive results, which may reduce the number offalse-positive findings that would result from HPV test-ing.29,44 A study in Brazil indicated that HPV testing followedby cytology triage could be a very cost-effective strategy forlower-middle income countries, with results reflecting a syn-ergistic effect between a highly sensitive initial screening test(HPV DNA) in sequence with a highly specific test (cytol-ogy).45 In extremely resource-limited countries, the use of asimple, low-cost HPV DNA test like the one presented here,followed by immediate ‘screen and treat’ strategies based onlow-cost methods like VIA in those who test positive, is likelyto minimize the number of patient visits and make best useof limited resources.46
Future work will be required to address some of the cur-rent limitations of the device before implementation in thefield can be realized. Currently, the chip requires an externalsource of heat for the isothermal amplification step. Thework presented here was performed using a heat block butthis can also be achieved through several methods that do
not require electricity such as battery operated resistanceheaters47 or exothermic reactions like those in simple toewarmers.48 Nonetheless, incorporating an integrated heatingsystem within the paperfluidic platform would greatly in-crease its portability and usability. An additional limitationto the current chip design is that it requires a significantamount of pipetting and manipulation by the user. A numberof variables can be optimized to limit the number of steps,for example, increasing the surface area of the sample port,or optimizing buffers and assay conditions to reduce the vol-ume needed.
Conclusion
We present the first ever fully integrated rapid molecular di-agnostic made entirely of paper and tape. Our paperfluidicchip offers an equipment-free, modular NAAT platform thatis low-cost, easy to manufacture, and simple to use. We dem-onstrated full sample-to-answer functionality of our chip byimplementing an HPV 16 DNA extraction, amplification, anddetection assay directly from patient cervical samples. Ouron-chip HPV 16 assay addresses many of the limitations ofconventional cytology by providing highly sensitive molecularlevel information regarding the presence of high-risk HPV 16in cervical samples without the need for laboratory infrastruc-ture or highly trained pathologists. The device also addressesthe limitations of current molecular diagnostic techniques byallowing for rapid, point-of-care detection in less than anhour without the need for transportation to/from a labora-tory. Additionally, this paperfluidic chip can serve as a molec-ular diagnostic platform for any disease, requiring onlychanging of primer sequences and the corresponding optimi-zation of LAMP assay conditions. The device is made fromlow-cost paper and tape materials, making it inexpensive,portable, and disposable, allowing it to be applied in low-resource settings and reach a larger pool of patients who mayotherwise not get diagnosed due to insufficient resources andpersonnel.
Acknowledgements
The authors wish to thank Dr. Christopher Chen, Dr. MarioCabodi, Dr. Jacqueline Linnes, Dr. Sharon Wong, and Dr.Andy Fan for helpful discussions, and Angelina Jockovich forher assistance with AutoCAD. NMR acknowledges fundingfrom a National Science Foundation Graduate Research Fel-lowship and a Diversity Administrative Supplement underNIH grant U54-EB015403-S1.
References
1 P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tamand B. H. Weigl, Nature, 2006, 442, 412–418.
2 A. K. Yetisen, M. S. Akram and C. R. Lowe, Lab Chip,2013, 13, 2210.
3 A. W. Martinez, S. T. Phillips, G. M. Whitesides and E.Carrilho, Anal. Chem., 2010, 82, 3–10.
Lab on a ChipPaper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online

Lab ChipThis journal is © The Royal Society of Chemistry 2016
4 A. W. Martinez, S. T. Phillips, M. J. Butte and G. M.Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318–1320.
5 A. W. Martinez, S. T. Phillips and G. M. Whitesides, Proc.Natl. Acad. Sci. U. S. A., 2008, 105, 19606–19611.
6 J. Hu, S. Wang, L. Wang, F. Li, B. Pingguan-Murphy, T. J. Luand F. Xu, Biosens. Bioelectron., 2014, 54, 585–597.
7 S. R. Jangam, D. H. Yamada, S. M. McFall and D. M. Kelso,J. Clin. Microbiol., 2009, 47, 2363–2368.
8 A. V. Govindarajan, S. Ramachandran, G. D. Vigil, P. Yagerand K. F. Böhringer, Lab Chip, 2011, 12, 174.
9 M. M. Ali, S. D. Aguirre, Y. Xu, C. D. M. Filipe, R. Pelton andY. Li, Chem. Commun., 2009, 6640.
10 B. A. Rohrman and R. R. Richards-Kortum, Lab Chip,2012, 12, 3082.
11 W. Gan, B. Zhuang, P. Zhang, J. Han, C.-X. Li and P. Liu,Lab Chip, 2014, 1–11.
12 C. Liu, E. Geva, M. Mauk, X. Qiu, W. R. Abrams, D.Malamud, K. Curtis, S. M. Owen and H. H. Bau, Analyst(Cambridge, U. K.), 2011, 136, 2069.
13 J. C. Linnes, A. Fan, N. M. Rodriguez, B. Lemieux, H. Kongand C. M. Klapperich, RSC Adv., 2014, 4, 42245–42251.
14 N. M. Rodriguez, J. C. Linnes, A. Fan, C. K. Ellenson, N. R.Pollock and C. M. Klapperich, Anal. Chem., 2015, 87,7872–7879.
15 J. T. Connelly, J. P. Rolland and G. M. Whitesides, Anal.Chem., 2015, 87(15), 7595–7601.
16 J. Ferlay, H.-R. Shin, F. Bray, D. Forman, C. Mathers andD. M. Parkin, Int. J. Cancer, 2010, 127, 2893–2917.
17 J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers,M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Int. J.Cancer, 2014, 136, E359–E386.
18 S. J. Goldie, L. Gaffikin, J. D. Goldhaber-Fiebert, A. Gordillo-Tobar, C. Levin, C. Mahé, T. C. Wright and Alliance forCervical Cancer Prevention Cost Working Group, N. Engl. J.Med., 2005, 353, 2158–2168.
19 L. Denny, M. Quinn and R. Sankaranarayanan, Vaccine,2006, 24, S71–S77.
20 L. Denny, Reprod. Health Matters, 2008, 16, 18–31.21 H. Ying, F. Jing, Z. Fanghui, Q. Youlin and H. Yali, Sci. Rep.,
2014, 4.22 S. J. Goldie, Virus Res., 2002, 89, 301–309.23 A. Goel, G. Gandhi, S. Batra, S. Bhambhani, V. Zutshi and P.
Sachdeva, Int. J. Gynecol. Obstet., 2005, 88, 25–30.24 D. Hegde, H. Shetty, P. K. Shetty and S. Rai, J. Cancer Res.
Ther., 2011, 7, 454–458.25 M. Schiffman, P. E. Castle, J. Jeronimo, A. C. Rodriguez and
S. Wacholder, Lancet, 2007, 370, 890–907.26 G. Clifford, S. Franceschi, M. Diaz, N. Muñoz and L. L. Villa,
Vaccine, 2006, 24, S26–S34.27 R. Sankaranarayanan, B. M. Nene, S. S. Shastri, K. Jayant, R.
Muwonge, A. M. Budukh, S. Hingmire, S. G. Malvi, R.Thorat, A. Kothari, R. Chinoy, R. Kelkar, S. Kane, S. Desai,V. R. Keskar, R. Rajeshwarkar, N. Panse and K. A. Dinshaw,N. Engl. J. Med., 2009, 360, 1385–1394.
28 L. Kuhn, L. Denny, A. Pollack, A. Lorincz, R. M. Richart andT. C. Wright, J. Natl. Cancer Inst., 2000, 92, 818–825.
29 M. H. Mayrand, E. Duarte-Franco, I. Rodrigues, S. D. Walter,J. Hanley, A. Ferenczy, S. Ratnam and F. Coutlée, N. Engl. J.Med., 2007, 357, 1579–1588.
30 L. L. Villa and L. Denny, Int. J. Gynecol. Obstet., 2006, 94,S71–S80.
31 L. Luo, K. Nie, M. J. Yang, M. Wang, J. Li, C. Zhang, H. T.Liu and X. J. Ma, J. Clin. Microbiol., 2011, 49, 3545–3550.
32 J. C. Linnes, N. M. Rodriguez, L. Liu and C. M. Klapperich,Polyethersulfone improves the efficiency of nucleic acidamplification compared to current paper-based diagnosticmaterials, Submitted 2015.
33 R. Boom, C. J. Sol, M. M. Salimans, C. L. Jansen, P. M.Wertheim-van Dillen and J. van der Noordaa, J. Clin.Microbiol., 1990, 28, 495–503.
34 M. van Duin, P. J. F. Snijders, H. F. J. Schrijnemakers, F. J.Voorhorst, L. Rozendaal, M. A. E. Nobbenhuis, A. J. C. vanden Brule, R. H. M. Verheijen, T. J. Helmerhorst andC. J. L. M. Meijer, Int. J. Cancer, 2002, 98, 590–595.
35 D. C. Swan, R. A. Tucker, G. Tortolero-Luna, M. F. Mitchell,L. Wideroff, E. R. Unger, R. A. Nisenbaum, W. C. Reeves andJ. P. Icenogle, J. Clin. Microbiol., 1999, 37, 1030–1034.
36 N. Kuboki, N. Inoue, T. Sakurai, F. Di Cello, D. J. Grab, H.Suzuki, C. Sugimoto and I. Igarashi, J. Clin. Microbiol.,2003, 41, 5517–5524.
37 J. Inacio, O. Flores and I. Spencer-Martins, J. Clin. Microbiol.,2008, 46, 713–720.
38 H.-Y. Yeh, C. A. Shoemaker and P. H. Klesius, J. Microbiol.Methods, 2005, 63, 36–44.
39 P.-H. Teng, C.-L. Chen, P.-F. Sung, F.-C. Lee, B.-R. Ou andP.-Y. Lee, J. Virol. Methods, 2007, 146, 317–326.
40 T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K.Watanabe, N. Amino and T. Hase, Nucleic Acids Res.,2000, 28, E63.
41 K. A. Curtis, D. L. Rudolph and S. M. Owen, J. Med. Virol.,2009, 81, 966–972.
42 M. Rebolj, I. Pribac and E. Lynge, Eur. J. Cancer, 2011, 47,255–261.
43 D. Saslow, D. Solomon, H. W. Lawson, M. Killackey, S. L.Kulasingam, J. Cain, F. A. R. Garcia, A. T. Moriarty, A. G.Waxman, D. C. Wilbur, N. Wentzensen, L. S. Downs, M.Spitzer, A. B. Moscicki, E. L. Franco, M. H. Stoler, M.Schiffman, P. E. Castle and E. R. Myers, Am. J. Clin. Pathol.,2012, 137, 516–542.
44 Agency for Healthcare Research and Quality (AHRQ), 2011,AHRQ Publication No. 11-05156-EF-1.
45 T. Vanni, P. M. Luz, B. Grinsztejn, V. G. Veloso, A. Foss, M.Mesa-Frias and R. Legood, Int. J. Cancer, 2011, 131,E96–E104.
46 J. Cuzick, M. Arbyn, R. Sankaranarayanan, V. Tsu, G. Ronco,M.-H. Mayrand, J. Dillner and C. J. L. M. Meijer, Vaccine,2008, 26, K29–K41.
47 K. E. Herold, N. Sergeev, A. Matviyenko and A. Rasooly, inMethods in Molecular Biology, Humana Press, Totowa, NJ,2009, vol. 504, pp. 441–458.
48 S. Huang, J. Do, M. Mahalanabis, A. Fan, L. Zhao, L. Jepeal,S. K. Singh and C. M. Klapperich, PLoS One, 2013, 8, e60059.
Lab on a Chip Paper
Publ
ishe
d on
20
Janu
ary
2016
. Dow
nloa
ded
by B
osto
n U
nive
rsity
on
20/0
1/20
16 1
8:42
:39.
View Article Online


















