
Nanoscale Phenomena in Ultrathin Catalyst Layers of PEM Fuel...
116
Nanoscale Phenomena in Ultrathin Catalyst Layers of PEM Fuel Cells: Insights from Molecular Dynamics by Amin Nouri Khorasani B.Sc., Sharif University of Technology, 2010 Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in the Department of Chemistry Faculty of Science Amin Nouri Khorasani 2013 SIMON FRASER UNIVERSITY Summer 2013
Transcript of Nanoscale Phenomena in Ultrathin Catalyst Layers of PEM Fuel...

Nanoscale Phenomena in Ultrathin Catalyst Layers of
PEM Fuel Cells: Insights from Molecular Dynamics
by
Amin Nouri Khorasani
B.Sc., Sharif University of Technology, 2010
Thesis Submitted in Partial Fulfillment of the
Requirements for the Degree of
Master of Science
in the
Department of Chemistry
Faculty of Science
Amin Nouri Khorasani 2013
SIMON FRASER UNIVERSITY
Summer 2013

ii
Approval
Name: Amin Nouri Khorasani
Degree: Master of Science
Title of Thesis: Nanoscale Phenomena in Ultrathin Catalyst Layer of
PEM Fuel Cells: Insights from Molecular Dynamics
Examining Committee: Chair: Krzystof Starosta
Associate Professor
Michael H. Eikerling
Senior Supervisor
Professor
Kourosh Malek
Co-supervisor
Adjunct Professor
Gary W. Leach
Supervisor
Associate Professor
Byron D. Gates
Supervisor
Associate Professor
Noham Weinberg
Internal Examiner
Adjunct Professor
Department of Chemistry
Date Defended: May 09, 2013

iii
Partial Copyright Licence

iv
Abstract
Ionomer-free ultrathin catalyst layers have shown promise to enhance the
performance and reduce the platinum loading of catalyst layers in polymer
electrolyte fuel cell. The nanostructure of a catalyst layer affects the distribution
and diffusion of reactants, and consequently its effectiveness factor. We
employed classical molecular dynamics to simulate a catalyst layer pore as a
water-filled channel with faceted walls, and investigated the effect of channel
geometry and charging on hydronium ion and water distribution and diffusion
in the channel.
Equilibrium hydronium ion distribution profiles on the catalyst channel were
obtained to calculate the effect of channel structure on the electrostatic
effectiveness factor of the channel. Furthermore, we calculated the self-diffusion
coefficient and interfacial water structure in the model channel. Results on
proton concentration, diffusion and kinetics are discussed in view of catalyst
layer performance.
Keywords: polymer electrolyte fuel cells; molecular modeling; electrostatic
effects; local reaction conditions; catalyst layer effectiveness

v
Dedication
Dedicated to my dear parents,
Dr. Fahimeh Fatemi, and Dr. Saied Nouri;
all I have and will accomplish
is only due to their love and sacrifice.

vi
Acknowledgements
Firstly, I would like to sincerely thank my senior supervisor, Professor Michael
Eikerling, for giving me the chance to conduct research under his supervision.
His extensive knowledge and expertise in electrochemistry always assured me of
stepping in the right path. His continuous support, encouragement, and patience
helped me obtain a solid knowledge in electrochemistry. He taught me how to
think logically, read critically, research productively and analyze the results
diligently.
I would like to thank my co-supervisor, Dr. Kourosh Malek, for his great help
and support. He patiently taught me high level molecular dynamics technique
from scratch, machine-level programming language and Linux. He kindly
monitored my progress on a day-to-day basis, discussed my results with me and
supported me with precise advice.
I extend my thanks to my supervisory committee members, Dr. Gary Leach and
Dr. Byron Gates, for the high level discussions and recommendations during my
committee meetings. Those discussions helped me understand the larger picture
of my research project, and helped me recognize the significance of my model
beyond the fuel cell context.
I must thank all the past and present members of Dr. Eikerling research group,
for the heated discussions in the group meeting. Not only the scientific
discussion during our group meeting nurtured my mind, but also helped me
improve my technical and public presentation skills. I must specially thank Dr.

vii
Liya Wang for teaching Density Functional Theory, Karen Chan for teaching
COMSOL to me, as well as answering my endless questions in electrochemistry,
Dr. Ehsan Sadeghi for great discussions about fuel cells science and engineering,
and Dr. Anatoly Golovnev for patiently explaining and discussing the relevant
physical concepts and suggesting creative ideas about my work.
I must acknowledge the support from NRC-EME staff for facilitating the logistics
of this research, providing resources to run the computationally extensive
simulations and maintaining the computer network.
Finally, I must thank my family for their emotional as well as academic support
during my studies.

viii
Table of Contents
Approval ........................................................................................................................... ii
Partial Copyright Licence .............................................................................................. iii
Abstract............................................................................................................................. iv
Dedication ......................................................................................................................... v
Acknowledgements ........................................................................................................ vi
Table of Contents .......................................................................................................... viii
List of Tables ..................................................................................................................... x
List of Figures .................................................................................................................. xi
List of Acronyms ........................................................................................................... xvi
1. Introduction .............................................................................................................1
1.1. The global energy infrastructure ...........................................................................1
1.2. Overview of fuel cells technologies .......................................................................2
1.3. Advances and challenges in design and operation of cathode catalyst
layer............................................................................................................................5
1.4. Ionomer-free ultrathin catalyst layers ...................................................................9
1.5. Local reaction conditions in catalyst layer .........................................................17
1.6. Water in nanoconfinement ...................................................................................18
1.7. Scope of the thesis ..................................................................................................20
2. Model Description ................................................................................................22
2.1. UTCL structural parameters ................................................................................22
2.2. Model system ..........................................................................................................23
2.3. Effectiveness of the catalyst layer channel .........................................................25
2.4. Limitations for using continuum modeling .......................................................28
2.5. Metal surface charge distribution ........................................................................29
3. Simulation Methodology ....................................................................................32
3.1. Overview .................................................................................................................32
3.2. Pt structure construction .......................................................................................35
3.3. Prediction of charge distribution on Pt nanochannel surface .........................38
3.4. Classical molecular dynamics ..............................................................................40
3.4.1. Description of the forcefields used ..........................................................40
3.4.2. Computational details ...............................................................................44
3.5. Simulation data analysis .......................................................................................45

ix
3.5.1. Hydronium ion density map and electrostatic effectiveness ..............46
3.5.2. Surface and bulk water diffusion ............................................................46
3.5.3. Interfacial water structure ........................................................................47
4. Results and Discussion ........................................................................................48
4.1. Overview .................................................................................................................48
4.2. Surface charge distribution in the nanostructured channel ............................48
4.3. Hydronium ion distribution in catalyst layer channels ...................................51
4.3.1. Hydronium ion density distribution in smooth Pt channels ...............51
4.3.2. Hydronium ion distribution in nanostructured channels ...................54
4.4. Electrostatic effectiveness factor as a function of channel structure ..............59
4.4.1. Effect of channel opening and structuring angle ..................................59
4.4.2. Effect of groove width ...............................................................................63
4.4.3. Effect of metal surface charge density ....................................................66
4.5. Structure and dynamics of water in the nanochannel ......................................67
4.5.1. Two phase water structure in the channel .............................................68
4.5.2. Effect of the channel opening and structuring angle on water
self-diffusion coefficient ............................................................................69
4.5.3. Effect of groove width on water self-diffusion coefficient ...................72
4.5.4. Effect of metal charging on water self-diffusion coefficient ................73
4.6. Sensitivity analysis to MD run parameters ........................................................75
4.6.1. Total simulation run time .........................................................................76
4.6.2. Choice of forcefield and intermolecular interactions ...........................76
4.6.3. Long range electrostatic interaction treatment ......................................77
4.6.4. Temperature................................................................................................78
4.6.5. Nanochannel material ...............................................................................79
4.6.6. Pt-Pt Lennard-Jones interaction parameters ..........................................81
4.7. Insight to the effect of nanostructuring on ORR kinetics .................................83
5. Conclusions ............................................................................................................84
5.1. Results Summary ...................................................................................................84
5.2. Suggestions for future work .................................................................................86
References ........................................................................................................................88

x
List of Tables
Table 1.1. Characteristic differences of Pt/C and UTCL ......................................10
Table 2.1 Range of parameters for investigation of channel structuring
effects ........................................................................................................25
Table 2.2. Typical values of surface charge density and metal potential,
system with θ=45°, dc=60 Å, dpp=30 Å, direct relation
between metal potential and surface charge density.........................31
Table 3.1. Van der Waals interaction parameters for the system species .........44
Table 4.1. Range of parameters for investigation of channel opening
effect ..........................................................................................................59
Table 4.2. Range of parameters for investigating the effect of groove
width .........................................................................................................63
Table 4.3. Range of parameters for investigating the effect of surface
charge ........................................................................................................66
Table 4.4. Physical properties of Pt, Cu, and Au for MD set-up ........................79
Table 4.5. Pt-Pt Lennard-Jones parameters for sensitivity analysis ..................81

xi
List of Figures
Figure 1.1. Basic design of PEM fuel cell; hydrogen oxidation reaction
occurs in the anode and oxygen reduction reaction in the
cathode. Protons transport through the PEM, and electrons
through the outer electric circuit. ...........................................................4
Figure 1.2. Schematic cross section of a single fuel cell, adapted from
Ref [2] (not to scale). Current conduction between the cells is
through the bipolar plates; flow fields feed fuel/air to the
cell, and GDL influences the transport of gases and liquid
to/from the cell. ..........................................................................................5
Figure 1.3. TEM image of a)Pt/C catalyst, b)PtCo/C alloy catalyst [16],
random dispersion of Pt particles on the substrate; Reprinted
with permission from Elsevier ................................................................7
Figure 1.4. SEM images of: a)Pt/C electrode, b) UTCL with 3M NSTF
design, c) Magnified view of NSTF [43], ionomer-free nature
of the UTCL makes the performance depend on the local
reaction conditions on the CL. Reproduced by the
permission of the Electrochemical Society ..........................................10
Figure 1.5. a) SEM image of NPG. b) High resolution TEM image of
NPG. c) TEM of Pt-coated NPG UTCL at a loading of 0.05
mg.cm-2. d) High resolution TEM of Pt-coated NPG, image
from Ref. [44], Reproduced by the permission of the
Electrochemical Society ..........................................................................11
Figure 1.6. SEM image of Ni75Pt25 nanoparticle for UTCL fabrication,
Ni(II) acetylaacetonate concentration in fabrication process:
A) 0.1, B)0.2, C)0.3, D)0.5 M, Reprinted with permission from
[45], Copyright (2013) American Chemical Society ...........................12
Figure 1.7. SEM image of 3M NSTF electrode a) Catalyst coated
membrane (CCM). b) Zoomed view of a microstructured
catalyst transfer substrate (MCTS) peak. c)Zoomed view of
catalyst electrodes. d) Lower catalyst loading, reprinted from
Ref. [47] with permission from the Electrochemical Society ............14

xii
Figure 1.8. SEM and STEM images of CL whiskerettes, nanostructuring
of whiskerettes shown in e and f. Reprinted from Ref. [47],
with permission from the Electrochemical Society ............................14
Figure 1.9. Schematic illustration of Chan and Eikerling continuum-type
pore-scale model system [51] ................................................................16
Figure 2.1. Electrostatic effectiveness vs potential for different pore
radii. Inset: corresponding surface charge density vs
potential, image from Ref. [51], reproduced by the
permission of the Electrochemical Society. .........................................23
Figure 2.2. Model system schematics, three geometric parameters (θ,dc,
dpp) and metal potential(φM) determine the system ...........................24
Figure 3.1. Overall scheme of the calculations .......................................................34
Figure 3.2. Primitive cell of Pt crystal with FCC structure ..................................35
Figure 3.3. Full atomistic Pt channel structure, incompletely constructed
to better illustrate build-up procedure (left), schematic
representation (right). Water and hydronium ions would be
added to the space in the middle of the structured channel. ...........36
Figure 3.4. 2-d Schematic picture of the model system with physical
boundary conditions ...............................................................................36
Figure 3.5. Force calculation scheme .......................................................................42
Figure 3.6. Radial distribution functions: a)SPC/E, b) SPC (modified), c)
TIP3P (modified)solid line, and the experimental data
dotted. Curves are shifted 2 units for clarity [86]. Reprinted
with permission from the American Chemical Society. ....................43
Figure 4.1. Typical distribution maps in the nanochannel predicted by
PB theory: a) potential distribution, b) proton concentration,
c) Electric field contour lines, d) metal surface charge density ........49
Figure 4.2. Effect of ɸM on the surface charge distribution...................................50

xiii
Figure 4.3. Effect of θ on surface charge distribution, constant dc=20 Å,
dpp=30 Å and ɸM=-50 mV for all cases ...................................................50
Figure 4.4. Pt structured channel with θ=15°, predicted charge
distribution close to homogeneity ........................................................51
Figure 4.5. Schematic of analytically solved electrostatic model system ...........52
Figure 4.6. Wall position definition for PB theory, the wall position is
not defined precisely in an MD simulation; however the
positions of all atoms nuclei are known. .............................................53
Figure 4.8. Hydronium density map (left) and nanostructured channel
(right). The structure is rotated to better represent the
stepped surface, θ=60°, dc=20 Å, dpp=40 Å, and φM=-50 mV. .............55
Figure 4.9. (a) Distribution map of hydronium ions in the nanochannel,
(b) surface hydronium ion density profile, (c) surface
potential, (d) local effectiveness. Dashed lines on the top left
figure indicate the region considered as surface. Channel
with θ=45°, dc=20 Å, dpp=40 Å, φM=-50 mV .......................................56
Figure 4.10. (a) Distribution map of hydronium ions in the nanochannel,
(b) surface hydronium ion density profile, (c) surface
potential, (d) local effectiveness. Dashed lines on the top left
figure indicate the region considered as surface. Channel
with θ=75°, dc=20 Å, dpp=30 Å, and φM=-50 mV ..................................57
Figure 4.11. Relative H3O+ density at channel peak region vs. dc, effect of
walls electrostatic repulsion on H3O+ depletion from peak
region, with constant θ=45°, dpp=30 Å, and φM=-50 mV ....................58
Figure 4.12. Effect of dc on electrostatic effectiveness, Γ; constant dpp=30 Å ........60
Figure 4.13. Direct effect of overall H3O+ concentration on channel
effectiveness, dispersion of data in the low concentration
region is the channel structure-specific effects. ..................................61

xiv
Figure 4.14. Effect of θ on channel effectiveness, Γ, enhanced
effectiveness at smooth surfaces, constant dpp=30 Å, φM=-50
mV. ............................................................................................................62
Figure 4.15. Inverse effect of groove width on electrostatic effectiveness
of nanochannel, constant dc=20 Å .........................................................64
Figure 4.16. Direct effect of overall H3O+ density on Γ, constant dc=20 Å ............65
Figure 4.17. Effect of θ on Γ, constant dc, inverse relation with surface
step fraction..............................................................................................66
Figure 4.18. Direct effect of σM on effectiveness, Γ, due to stronger
Coulomb interaction of H3O+ with walls .............................................67
Figure 4.19. Typical water density map, high contrast of colors indicate a
highly structured phase .........................................................................69
Figure 4.20. Effect of dc on water self-diffusion coefficient, Dw .............................70
Figure 4.21. Analyzing the effect of channel opening on Dw in terms of
ratio of bulk-like (nb) to surface (ns) water ratio .................................71
Figure 4.22. Dependence of Dw to channel structure, a) surface water, b)
bulk-like water .........................................................................................72
Figure 4.23. Effect of groove width on water self-diffusion coefficient, Dw,
dc=20 Å constant. .....................................................................................73
Figure 4.24. Effect of Pt surface charge density on water self-diffusion
coefficient, constant dc=20 Å dpp=30 Å .................................................74
Figure 4.25. Dependence of Dw on Pt surface charge, a) bulk-like water,
b) surface water .......................................................................................75
Figure 4.26. Sensitivity of calculated effectiveness factor to analyzed
trajectory length ......................................................................................76
Figure 4.27 Sensitivity of simulation result and calculation speed to PME
short range force calculation .................................................................78

xv
Figure 4.28. Temperature dependence of channel effectiveness for a
channel with θ=45°, dc=20 Å, dpp=30 Å, σ=-14 μC/cm2 .........................79
Figure 4.29 Sensitivity of Γ results to nanochannel material ................................80
Figure 4.30. Inverse relation of channel effectiveness with the metal
lattice constant .........................................................................................80
Figure 4.31. Sensitivity of nanochannel effectiveness to Pt-Pt interaction
parameters ................................................................................................82
Figure 4.32. Sensitivity of water self-diffusion coefficient to Pt-Pt
interaction parameters ............................................................................82

xvi
List of Acronyms
BV Butler-Volmer
CCL cathode catalyst layer
CCM catalyst coated membrane
CFD computational fluid dynamics
CL catalyst layer
DFT density functional theory
DME dropping mercury electrode
ECSA electrochemically active surface area
EVB empirical valence bond
FFT fast Fourier transform
GDL gas diffusion layer
LJ Lennard Jones
MCFC molten carbonate fuel cell
MCTS microstructured catalyst transfer substrate
MD molecular dynamics
MEA membrane electrode assembly
MEMS microelectromechanical systems
MPL microporous layer
NPG nano-porous gold
NPGM non-precious group metals
NSTF nanostructured thin film
PB Poisson Boltzmann
PBC periodic boundary condition
PEFC polymer electrolyte fuel cell
PEM polymer electrolyte membrane
PME particle mesh Ewald

xvii
PTFE polytetrafluoroethylene
PZC potential of zero charge
SAX small angle X-ray spectroscopy
SEM scanning electron microscopy
SOFC solid oxide fuel cell
SPC single point charge
SPC/E extended single point charge
STEM scanning transmission electron microscopy
TEM transmission electron microscopy
TIP3P transferable intermolecular potential, three- position
TIP4P transferable intermolecular potential, four- position
UTCL ultrathin catalyst layer

1
1. Introduction
1.1. The global energy infrastructure
Since the mid-19th century, fossil fuels have been the primary energy
source for human society, with applications ranging from fueling the steam
engines, to automobiles and industries, generating electricity, as well as heating
houses. In 2008, 87% of the world’s primary energy was supplied from non-
renewable sources [1]. Energy consumption of a society per capita is directly
related to its welfare standards. An ever-increasing energy consumption rate has
given human society unprecedented welfare and health standards in its history.
However, the global energy infrastructure is undergoing a dramatic
transition: The oil production cannot grow with the same pace as energy demand
due to reduction of reservoirs pressure and ultimate depletion, leading to the oil
peak phenomenon. Fossil fuels are finite resources and the lavish use of energy
from their burning has adverse environmental effects. The main product of the
combustion process, CO2, is a greenhouse gas and contributes significantly to the
global warming phenomenon. The importance of the use of environmentally-
friendly energy sources is most notable in urban areas: The exhaust gas from
cars’ internal combustion engines contains toxic species of CO and NOx, which
represent human health hazards.

2
These concerns drive international initiatives to developing renewable
energy technologies, relying both on primary energy sources (bioenergy, direct
solar, hydropower, wind, and geothermal), as well as secondary sources (fuel
cells, batteries, supercapacitors).
The electrochemical reaction of hydrogen with oxygen produces water
and generates electricity. Utilizing hydrogen as fuel for the propulsion of
vehicles is a highly efficient and potentially sustainable option for the future of
transportation, which could drastically reduce the health and environmental
concerns. A hydrogen fuel cell is the device in which this reaction occurs.
Prototypes of fuel cell powered vehicles were produced by major automobile
companies such as Toyota, Nissan, Mercedes Benz, GMC, Chevrolet, and Honda;
commercial models are planned to be produced by 2017.
So far, fuel cell vehicles have met the required power density, durability,
and stability levels for use in commercial vehicles, and the current challenge for
their commercialization is their cost. Recent cost models of fuel cell vehicles [2],
[3] show strong dependence of the cost on the loading of platinum which is used
as the catalyst. High platinum price and its low utilization in current catalyst
layer (CL) designs motivate extensive research toward increasing the Pt
utilization and lowering the Pt loading in fuel cells.
1.2. Overview of fuel cells technologies
Fuel cells are electrochemical cells that convert the energy of a chemical
reaction to electrical energy with high thermodynamic efficiency. The reaction
happens between a fuel and an oxidant at anode and cathode electrodes,

3
respectively. Depending on the range of operating conditions and the intended
application, different types of fuel cells are distinguished which use different
fuels: solid oxide fuel cells (SOFC) and molten carbonate fuel cells (MCFC)
operate at high temperature. They can be fed with a variety of chemicals as fuel,
such as hydrogen, natural gas or carbon monoxide from gasified coal [4], and can
as well operate on a mix of all those. The operating temperature for SOFC is
generally between 500 °C to 1000 °C, and above 650 °C for MCFCs. Recent
advances in fabrication techniques have enabled lowering the temperature range
of SOFCs to 650-850 °C [5] without compromising cell efficiency. The high
temperature is essential for the performance of electrolyte, which is usually a
ceramic material conducting oxygen ions. The ionic conductivity of the
electrolyte decreases at low temperatures, which leads to an increase in the
Ohmic resistance of the cell. The main applications of SOFC and MCFC are
distributed power and backup power generation, and combined heat and power
generation [6]. Few efforts have explored application of SOFCs in mobile
applications [7].
Polymer electrolyte fuel cells (PEFC) operate at temperatures below 100
°C. PEFC have mainly been developed for vehicular devices, most importantly
fuel cell powered automobiles. PEFCs use hydrogen as the fuel in the anode, and
oxygen from air in the cathode, producing only water as a chemical product and
generating electricity. The reactions occurring in PEFC are hydrogen oxidation
reaction (HOR) at the anode, ( ) ( ) , with an equilibrium
electrode potential of , and the oxygen reduction reaction (ORR) at the
cathode, ( ) ( ) ( ) with . The overall cell

4
reaction is ( ) ( ) ( ) with an equilibrium cell potential of
.
The simplified layout of a PEFC is shown in Figure 1.1, and Figure 1.2
shows the typical design of a PEFC single cell essential components.
Figure 1.1. Basic design of PEM fuel cell; hydrogen oxidation reaction occurs
in the anode and oxygen reduction reaction in the cathode. Protons
transport through the PEM, and electrons through the outer electric
circuit.

5
Figure 1.2. Schematic cross section of a single fuel cell, adapted from Ref [2]
(not to scale). Current conduction between the cells is through the
bipolar plates; flow fields feed fuel/air to the cell, and GDL
influences the transport of gases and liquid to/from the cell.
1.3. Advances and challenges in design and operation of
cathode catalyst layer
Platinum is the most widely used monometallic catalyst for fuel cells to
date, mainly due to its proper oxygen adsorption energy [8]. The Pt loading,
defined as the mass of Pt per interfacial area of fuel cell electrode, is the most

6
significant parameter affecting the fuel cell cost, according to recent PEFC cost
models [2], [3], [9]. Therefore, a drastic reduction of Pt loading is a central goal to
meet the stringent cost targets for PEFC commercialization in automotive
applications. The current cost target for automotive fuel cell systems is $30/kWh
for the year 2015. The overall cost of a PEFC stack was reduced from $73/kWh in
2008 [10] to $48/kWh in 2012, achieved with a Pt loading of 0.196 mg/cm2 [3].
The ORR at the cathode has sluggish kinetics and it incurs much higher
irreversible potential losses than the HOR; hence it is currently the main barrier
to cheaper and more efficient PEM fuel cells [11]. Major efforts in materials
science and electrode design strive to reduce the voltage losses. Until the mid-
1980s, PEFC catalyst layer was composed of platinum nanoparticles dispersed on
high surface area carbon support, known as Pt-black. An example of carbon
support is Vulcan carbon with 200 m2/g specific surface area. Pt-black was used
at the typical Pt loading rate of 4 mg/cm2 in the cathode. Pt-black showed great
performance and durability features, though at a very high cost that made it
unfeasible for most consumer applications. A milestone in the cathode catalyst
layer (CCL) design was at 1989, when adding Nafion® to the CCL improved
proton transport in the CCL, and enabled reducing Pt loading to below 0.5
mg/cm2 [12] under laboratory setup with excessive gas supply. Wilson et al. [13]
showed that using thermoplastic ionomers enables melt-processing of electrodes,
which enhances the long term performance. Ralph et al. [14] developed a method
to produce the CL at lower cost and high volume with Pt loading of 0.6 mg/cm2.
It consists of mixing Pt nanoparticles with a size distribution in the range of 20-50
Å, a thermoplastic ionomer and carbon support with given ratio to make an ink,
and use a printing apparatus to coat the ink on a carbon fiber paper. This design

7
enabled removing polytetrafluoroethylene (PTFE) from the membrane electrode
assembly (MEA) design. The design concept is being used to date to produce the
CL. Due to the nature of the mixing process during MEA preparation, the local
distribution of Pt, carbon and ionomer cannot be controlled. The typical
thickness of the CL produced this way is 5-10 µm. Fuel cell systems using this
type of catalyst have shown sufficient energy density for use in automotive
applications. However, further cost reduction is required for fuel cell
commercialization [15]. Figure 1.3 shows the typical Pt/C catalyst material
morphology, and a rival PtCo/C CL exhibiting improved durability features [16].
Figure 1.3. TEM image of a)Pt/C catalyst, b)PtCo/C alloy catalyst [16],
random dispersion of Pt particles on the substrate; Reprinted with
permission from Elsevier
Reducing the Pt loading has been pursued through three major strategies:
- Reducing the voltage losses induced by mass transport by modifying the
MEA structure [17],

8
- Enhancing the intrinsic activity of Pt catalyst by using Pt-alloys, which
increases the exchange current density (j0) for the ORR [16], [18–22], or using
non-precious group metals [17], [23] which allows catalyst cost reduction,
- Development of the ultrathin catalyst layer (UTCL) design, relying on
low-resistance mass transport of species through water-filled pores, and
enhancing Pt utilization by increasing the Pt/water interface. We will explain this
strategy in section 1.4.
A major shortcoming of the Pt/C catalyst is low utilization of Pt due to the
random distribution of Pt inside CCL. Lee et al. [24] found that the effectiveness
factor of Pt utilization, UPt, in conventional Pt/C CL is around 5%. UPt was defind
as the ratio of the electrochemically active surface area to the total area of Pt in
the CL, estimated from TEM images [25]. The value of 5% UPt, despite the
imprecision, nurtures expectations that a tremendous Pt loading reduction
should be achievable without compromising performance. Another motivation
to develop more stable and durable CLs and catalyst support layers rises from
pronounced electrochemically active surface area (ECSA) loss due to Pt
degradation [26–29].
The CCL is a porous structure, and includes hydrophobic and hydrophilic
pores. Hydrophobic pores allow major fraction of the oxygen transport through
the CL, while hydrophilic pores allow transport of water. The balance between
hydrophobic and hydrophilic pores helps transport of both water and oxygen.
The CCL thickness relates directly with the transport resistance of oxygen
through the CCL. The thickness relates inversely with the effectiveness of Pt in
the catalyst layer [30]. Furthermore, Pt and ionomer distribution in the CL can

9
affect the performance of the cell via tuning current density generation across the
CL. An analytical model was developed to predict and optimize the cell current
density [31] by obtaining uniform reaction rates across the CL.
1.4. Ionomer-free ultrathin catalyst layers
In the conventional CCL, oxygen diffuses mainly through gas-filled pores,
proton concentration is controlled by the ionomer loading, and the water product
diffuses out of CCL through the hydrophilic pores, as well as through
vaporization out of the cell. Therefore, the utilization of Pt depends strongly on
the distribution of these phases in the catalyst layer channel. Since the
distribution of Pt, C and ionomer in the CL is essentially random because of the
fabrication method, relatively low amount of Pt would be effectively utilized
[32]. Furthermore, due to the thickness of catalyst layer, there will be non-
homogeneous distribution of reactants [33] in the catalyst layer, resulting in the
non-homogeneous reaction rate along the catalyst layer.
One of the approaches to overcome the low utilization of Pt is using
ionomer-free ultrathin catalyst layers [34], where oxygen and protons would
diffuse through water to react on the Pt catalyst. Continuity of Pt film catalyst
ensures electrical conductivity of catalyst to the current collector, thus removing
the need for carbon substrate. Thinness of the catalyst layer reduces transport
resistance for oxygen diffusion and proton migration; in fact, the reaction
penetration depth [35] is typically longer than the UTCL thickness.
Different methods for producing the UTCLs include magnetron
sputtering of either several discrete patches or a continuous film of Pt or Pt alloy

10
nanoparticles on a support material [36–38], deposition on nafion membrane[39],
or deposition on gas diffusion layer [40]. UTCLs are 20-30 times thinner than
conventional CL [41], and have drastically lower Pt loading than conventional
CL [42]. Table 1.1 summarizes the major characteristic differences of UTCL with
conventional Pt/C CLs. Figure 1.4 shows a schematic view of a UTCL design,
fabricated in 3M Company.
Table 1.1. Characteristic differences of Pt/C and UTCL
Property Pt/C CL UTCL
Thickness 5-10 μm 0.1-0.5 μm
Main structural characteristic Ionomer-impregnated
Ionomer-free
Proton concentration Fixed by ionomer Determined by electrostatic interaction with charged walls
Pt loading in fuel cell 0.4 mg.cm-2 0.1 mg.cm-2
Figure 1.4. SEM images of: a)Pt/C electrode, b) UTCL with 3M NSTF design,
c) Magnified view of NSTF [43], ionomer-free nature of the UTCL
makes the performance depend on the local reaction conditions on
the CL. Reproduced by the permission of the Electrochemical
Society

11
Depositing Pt nanoparticles on nanoporous gold leaf is another method
for UTCL fabrication. Figure 1.5 shows images of the substrate and Pt coating.
Unlike the 3M design, there is no necessity for continuity of the Pt film, since the
porous substrate is conductive.
Figure 1.5. a) SEM image of NPG. b) High resolution TEM image of NPG. c)
TEM of Pt-coated NPG UTCL at a loading of 0.05 mg.cm-2. d) High
resolution TEM of Pt-coated NPG, image from Ref. [44],
Reproduced by the permission of the Electrochemical Society
Pt deposition on dealloyed nickel through a solvothermal reduction
process is another option for UTCL fabrication. In this method, Ni(II)
acetylaacetonate Ni(acac)2 and Pt(II) acetylaacetonate Pt(acac)2 are boiled
together and then cooled down. Then the organic capping material is removed
and the alloy will then nucleate and form nanoparticles [45]. Figure 1.6 shows an
example of such UTCL.

12
Figure 1.6. SEM image of Ni75Pt25 nanoparticle for UTCL fabrication, Ni(II)
acetylaacetonate concentration in fabrication process: A) 0.1, B)0.2,
C)0.3, D)0.5 M, Reprinted with permission from [45], Copyright
(2013) American Chemical Society
Experimental studies on UTCLs have shown enhancements in
performance, durability, and stability in these layers [36], [41–43]. However, the
lack of fundamental knowledge in the principles of UTCL operation, as well as
practical issues such as water management [43] and metal cation contaminations
diffused to the catalyst pores from the membrane or GDL, originated from the
fabrication process, leave challenge for research on UTCLs.
The UTCL entails different challenges from Pt/C CL for modeling studies.
Thinness of the catalyst layer (100-500 nm) reduces the resistance of oxygen
transport in the catalyst layer to small values. Water, which can cause flooding in
the conventional catalyst layer, is the carrier of both protons and oxygen to the
reaction sites; therefore, the catalyst layer is utilized best under flooded
condition.

13
An important example of industrially commercialized UTCL is the
nanostructured thin film (NSTF) CLs fabricated by 3M Company. The design
specification that differentiates 3M NSTF from other types of UTCL is the organic
molecular solid substrate, which forms a crystalline whisker, and acts as support
for Pt layer. Since 3M NSTF is the only commercially available type of UTCL to
date, several studies have been carried out on characterizing these layers, and
recent fuel cell models consider this type of CL as a benchmark for fuel cell mass-
production economic calculations [3].
Figure 1.7 shows that NSTF electrode has 0.2-0.6 µm thickness, and the
channel size (areas between substrate whiskers) exhibit distribution between 2 to
200 nm. On the substrate whiskers of NSTF, Pt is deposited to form a continuous
layer. Continuity of the Pt layer is important to assure electric connection of all Pt
site to the electrode. The deposited Pt film forms a corrugated surface, as shown
in Figure 1.8, that consist of so-called whiskerettes. Pt exhibits different surface
facets, depending on the size of the formed whiskerettes [46].
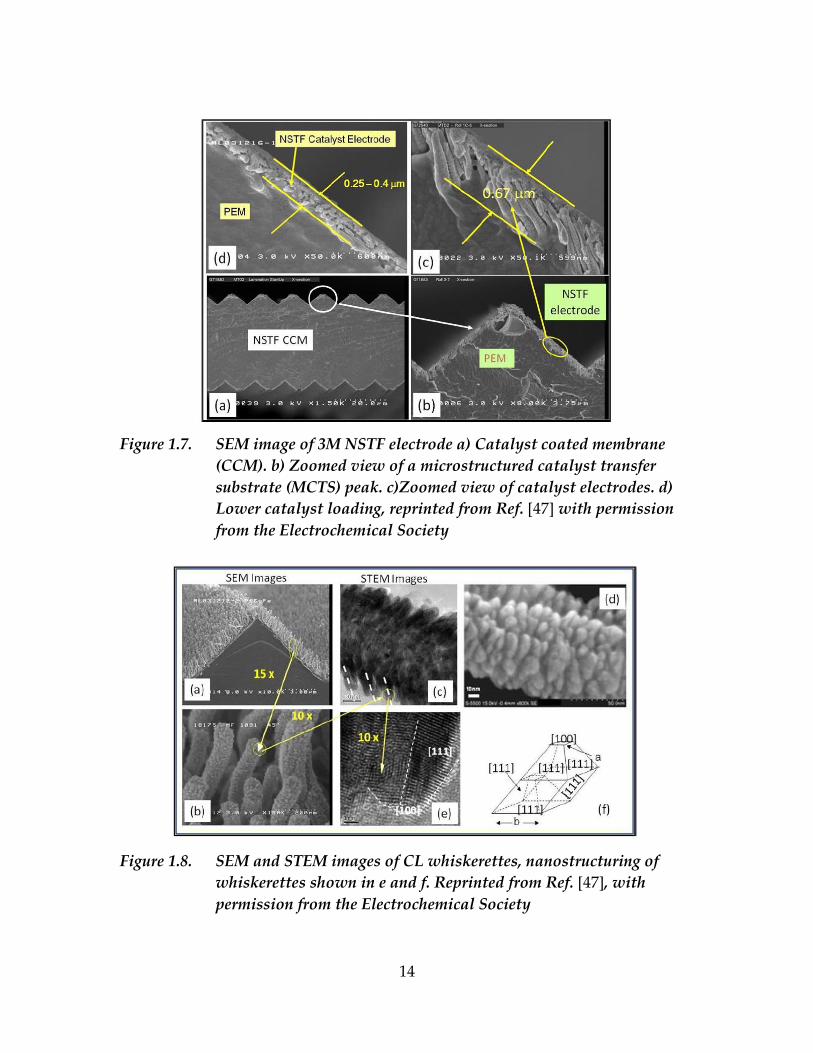
14
Figure 1.7. SEM image of 3M NSTF electrode a) Catalyst coated membrane
(CCM). b) Zoomed view of a microstructured catalyst transfer
substrate (MCTS) peak. c)Zoomed view of catalyst electrodes. d)
Lower catalyst loading, reprinted from Ref. [47] with permission
from the Electrochemical Society
Figure 1.8. SEM and STEM images of CL whiskerettes, nanostructuring of
whiskerettes shown in e and f. Reprinted from Ref. [47], with
permission from the Electrochemical Society

15
Unlike conventional Pt/C catalyst layers, all NSTF pores are hydrophilic,
and produce significantly more water per unit volume than their conventional
counterparts. Thus, the catalyst layer is flooded more easily due to the thinness
of NSTFs. There are strategies to prevent flooding in catalyst layer, such as
removing the water produced in cathode from anode side GDL [48].
NSTF CCLs have shown significant improvements of fuel cell
performance. The tests under well controlled conditions that suppress the
flooding of porous gas transport layers show that MEA with NSTF technology
on the cathode side exhibits improved performance compared to the MEA with
conventional CCL. The NSTF layers obliterate the need for ionomer
impregnation, while still providing a large ECSA due to the improved water
absorption in the hydrophilic pore network. Furthermore, experimental studies
on UTCLs with NSTF technology have shown significant enhancements in
catalyst durability and stability [36], [41], [49].
There have been efforts to develop analytical models of the
electrochemical phenomena in UTCLs [32], [50–53]. In a recent pore-scale model
by Chan and Eikerling [51], catalyst layer pores were modeled as water-filled
cylindrical channels with smooth walls at given radius, length and metal
potential. A schematic of the investigated model system is shown in Figure 1.9.
In this model, the proton and potential distribution along the channel was
calculated using Poisson-Nernst-Planck theory, and the oxygen distribution is
governed by Fickian diffusion of oxygen in water. The channel property
controlling the potential distribution is the surface charge density at the channel
walls. The Stern double layer model was used to relate the surface charge density

16
with the Pt potential of zero charge (PZC). Solution of the model provided the
distributions of reactants, electrolyte phase potential, reaction rates along the
pore, as well as the overall pore effectiveness of Pt utilization. Channel
performance was characterized via the effectiveness factor of Pt. The
effectiveness factor was defined as the actual (predicted) current density
normalized by the reference current density that would be obtained, if reactants
and potential were distributed uniformly in the channel.
Smooth Pt walls
Constant metal potential
PEM boundary
conditions:
Constant proton
concentration
No oxygen flux
GDL boundary
conditions:
Constant oxygen
concentration
No proton flux
Smooth cylindrical Pt channel
O2 diffusion H+ migration
r
L
Reaction plane (OHP)
Reaction plane (OHP)
Figure 1.9. Schematic illustration of Chan and Eikerling continuum-type pore-
scale model system [51]
An important aspect in modeling cathode CL is sluggish ORR kinetics,
which enables decoupling electrostatic and kinetic contributions to the
effectiveness factor. This concept was employed in Ref. [51], and it is used in the
current work as well, to assess the effect of electrostatic effectiveness of the
catalyst without explicitly accounting for kinetic effects.
The impedance model of such system revealed the effect of metal surface
charge density on the resistive and capacitive elements of the equivalent circuit
of a water-filled pore [52]. In the limiting cases of fast diffusion of either of the
species, and the case of blocked electrode, exact analytical expressions were

17
derived. Schmuck and Berg [53] scaled up the results of a the single pore model,
to obtain the macroscopic transport equations based on the microscopic pore
level phenomena.
1.5. Local reaction conditions in catalyst layer
The ionomer-free nature of UTCL entails different local reaction
conditions than conventional Pt/C CLs. Distributions of protons and electrolyte
phase potential in UTCL are not determined by the distribution of ionomer but
by electrostatic effects in water-filled pores, as rationalized in [51].
Unlike the random porous structure of conventional Pt/C catalyst layers,
the geometry of UTCL is controlled during the fabrication process. Therefore, the
reactant concentrations can be predicted using established physical theories:
metal surface charge distribution at any given point inside catalyst could be
predicted by an electric double layer model, proton distribution by Poisson-
Nernst-Planck theory, oxygen concentration by Fickian diffusion, and water
transport by Navier-Stokes transport equations [51], [54]. Therefore, local current
generation and reactivity could be predicted, and the local effectiveness could be
determined. This insight introduces an advantage for modeling: we could
perform pore scale or sub-pore scale modeling, and obtain useful insight into
electrocatalytic phenomena in the CL.

18
1.6. Water in nanoconfinement
During the past few decades, tremendous progress has been made in
synthesis of carbon nanotubes, artificial zeolites, porous electrodes and
miniaturization of channels with diameter in the order of a few water molecules.
These developments led to remarkable advances in many fields of application,
including but not limited to electrocatalysis, separation, filtering,
microelectromechanical systems (MEMS), textile engineering and biophysics.
Carbon nanotube has shown diverse applications, ranging from support
material for fuel cell catalyst [21], to nanopipettes [55] and molecular detection
[56]. The ability to transport fluids with precise rate is central to many of the
applications. Zeolite has been applied in separation of either gas [57] or liquid
[58] from gas mixtures. Its separation depends on the pore size distribution in the
zeolite, and affects the adsorption isotherm of the adsorbant. Both carbon
nanotube and zeolites are examples of nanostructured porous media, in which
knowledge of the transport of fluids has led to technological advancements.
The developments in design and application of the nanostructured porous
media have aroused the interest of researchers in the fundamental theories
governing the behavior of fluids under confinement at the nanometer scale. One
of the most important liquids considered is water. Water is the most abundant
and important liquid in nature, however it is by no means a simple fluid: Polarity
and hydrogen bond network of water have caused anomaly in both its bulk and
confined properties.

19
Navier-Stokes equations are the general approach to describe the
transport phenomena of fluids; they form the basis of computational fluid
dynamic (CFD) methods. However, these equations are based on the continuity
of fluids, and break down when the critical dimension of flow becomes
comparable to the size of molecules. The critical size of pores at which continuity
equations break down is a topic of controversy in the scientific community [59],
[60].
Molecular modeling offers an alternative for continuum modeling of
confined water, by explicitly considering the effects of molecular size of water,
bond configurations, charge distributions, and dipole effects. Theories for
describing water potential date back to 1933, when the work of Bernal and
Fowler [61] presented consistent properties of water with those observed in X-
ray spectroscopy. Within the realm of classical molecular simulation, the work of
Rahman and Stillinger [62] was one of the first. Later models improved the
description of water in computer simulations. TIP3P [63] and SPC [64] models
could predict structural properties of water close to experimentally observed
values from neutron diffraction experiment [65]; however, self-diffusion
coefficients are significantly higher than the experimentally observed value [66].
Modifying the SPC model resulted in development of SPC/E model [67],
which improved the prediction of self-diffusion coefficient. SPC/E remains the
most popular 3-site water model to date.
In TIP4P [68] and TIP5P models [69], fictitious particles were added,
which represent the lone electron pair of oxygen. This modification increases the

20
accuracy of dipole moment prediction. However, these models are more
computationally expensive to implement in an MD simulation.
Describing the independent H+ species is not practical in classical MD
simulation; however, the forcefield could be parameterized to account for proton
in the form of hydronium ion (H3O+). In this case, bonded interaction is assumed
between oxygen with three indistinguishable hydrogen atoms. However, it
cannot account for the exchange of protons between two neighboring water
molecules. A more realistic view of proton transport in water was presented in
the empirical valence bond (EVB) model [70], which links ab-initio calculations to
classical MD. The model accounts for exchange of protons by considering a
superposition of two states such as (H3O+…H2O) or (H2O…H3O+) [71]. Using
coupling between different states, it could be used to predict formation of Eigen
and Zundel forms of proton [72]. Expansions of the two-state to multistate EVB
model has been used by the group of Schmickler to describe proton transport on
Pt catalyst [73].
1.7. Scope of the thesis
Chapter 2 of the thesis explains the model of catalyst layer nanochannel
with controlled corrugation, along with its limitations and assumptions. Chapter
3 explains the methodology used in this work: It couples a combination of
continuum-type modeling and classical molecular dynamics to predict the
proton and potential distributions in a catalyst layer nanochannel of Pt with
corrugated walls. Chapter 4 presents and discusses the findings of this work,
how nanostructuring affected proton and water distribution in the catalyst layer

21
channels, and how it is related to the effectiveness factor of CL. We conclude and
highlight the major findings of this work in chapter 5, and make suggestions for
future work.

22
2. Model Description
2.1. UTCL structural parameters
The main microscopic characteristics of a generic UTCL are the ionomer-
free nature, nano-porosity with high surface area, thinness (compared to
conventional CLs), and excess electric charge on the metal surface. Therefore, a
model of UTCL performance must accommodate these aspects.
The nano-porous structure of the catalyst corresponds to high ECSA,
which is a major advantage for fuel cell catalysts. Typical pore sizes in UTCL are
in range of 5-50 nm. Pt deposited on the nanoporous gold leaf has an ECSA of 60
m2/g for a Pt loading of 0.03 mg/cm2 [44]. NiPt nanoparticles have an ECSA of 48
m2/g for 8 nm-diameter NiPt nanoparticle supported on glassy carbon [45], and
3M NSTF shows an ECSA of 10-25 m2/g [47]. The effectiveness of Pt utilization
has reverse dependence on the pore size, as shown for the model of a smooth
cylindrical pore [51]. Figure 2.1 shows an inverse relation of Γ with pore radius,
and an inverse relationship of Γ with , due to an increase in the negative
excess charge accumulated on the pore walls upon a decrease in potential. The
inset plot shows the charge-potential relation, derived from the Stern electric
double layer model.

23
Figure 2.1. Electrostatic effectiveness vs potential for different pore radii.
Inset: corresponding surface charge density vs potential, image
from Ref. [51], reproduced by the permission of the Electrochemical
Society.
The thickness of UTCLs is in the range of 0.2-0.6 μm, which is 20-30 times
smaller than conventional Pt/C CL. Comparing this thickness with the typical
oxygen penetration depth inside the pore [74] (20 μm for ηc=-0.5 V [52]) shows
small oxygen diffusion resistance in the channel; therefore the concentration of
oxygen can be assumed as uniform along the channel.
The relation between metal surface charge density and metal potential is
not clearly known; however classical double layer models and an estimation of
the potential of zero charge could be used to establish such a relation. This
approach will be discussed in section 2.5.
2.2. Model system
In this study, we consider a nanostructured water-filled Pt channel that
represents the interior part of cathode catalyst layer. We consider saw-tooth-

24
shaped modulations on two Pt surfaces, which are mirror images of each other.
The structure is determined by three independent geometrical parameters:
structuring angle θ, channel opening dc and groove width dpp. A schematic
illustration of the channel geometry is shown in Figure 2.2. The values of these
parameters that were considered in this work are shown in Table 2.1. In each
simulation set, combinations of θ and one other structural parameter was
investigated. The present model can be considered an extension to the model of
Chan and Eikerling [51], with the effect of geometrical surface roughness at the
atomistic level being added.
In addition to the geometric effect, saw-tooth-shaped structuring of the
metal walls gives rise to an inhomogeneous charge distribution on the metal
surface. The surface charge density distributes in a way that makes an
equipotential surface with the potential of φM. Distribution of charge on the CL
surface will be explained in the next chapter.
Figure 2.2. Model system schematics, three geometric parameters (θ,dc, dpp)
and metal potential(φM) determine the system

25
The channel walls are made of Pt, and the channel is filled with water and
hydronium ions, H3O+. Periodic boundary conditions (PBC) are applied to the
model system in all directions. The simulated system consists of two identical
grooves. The reason for this choice is to minimize the effect of periodic boundary
conditions on the proton distribution maps.
Table 2.1 Range of parameters for investigation of channel structuring
effects
Parameter Value Unit
Structuring angle (θ) 0, 15, 30, 45, 60, 75 Degrees
Channel opening (dc) 10, 20, 30, 40, 50, 60, 80, 100 Å
Roughness Opening (dpp) 20, 30, 40, 50, 60, 70, 80 Å
Metal Potential (φM) -10 -50 -70 -100 -110 mV
2.3. Effectiveness of the catalyst layer channel
In electrocatalysis, the effectiveness factor quantifies the comparison of
different catalysts: It is defined as the ratio of actual reaction rate to an reference
rate that would be obtained if the whole catalyst surface was exposed to equal
concentrations of reactants and equal potentials. Using the cathodic branch of
Butler-Volmer (BV) equation, the local current density, j, generated at the catalyst
surface is calculated by
(
)
(
)
(
( )) 2.1

26
where j0 is the exchange current density, the oxygen concentration on the
Helmholtz plane, the proton concentration on the Helmholtz plane, the
oxygen reaction order of ORR, the proton reaction order of ORR, and αc the
cathodic transfer coefficient. The superscript “0” indicates the reference
condition, under which the exchange current density j0 was calculated. We
defined Helmholtz plane to be a plane parallel to the channel walls, at 4 Å
distance from the channel walls.
Local and total effectiveness can be calculated from equations 2.3 and 2.4,
comparing the actual current generation with the reference case (i), where the
proton and water distributions are equal to the well-defined values at the
boundary,
(
)
(
)
(
( )) 2.2
∫
2.3
The integration in equation 2.3 is done on the surface of Helmholtz plane,
and ds represents a surface element of the Helmholtz plane. Thinness of UTCL
implies small resistance of oxygen transport in the channel, and sluggish kinetics
of ORR allows for assumption of separation of electrostatic and kinetic
contribution to effectiveness, and assuming electrochemical equilibrium inside
the channel, the electrostatic effectiveness depends solely on the proton and
potential distribution, and it is defined as in equation 2.4.

27
(
)
(
( )) 2.4
Assumption of electrochemical equilibrium allows relating the potential
and concentration of protons inside the channel, thus expressing the
effectiveness relation only in terms of proton concentration,
(
) 2.5
where ̃ is the species’ electrochemical potential, and is the chemical potential.
We chose the reference potential at a point in solution, where proton
density is 1.25 M, corresponding to a typical effective proton density in the PEM.
However, PEM proton concentration is the maximum for the CL channel,
occurring on the PEM/CL boundary; typical proton density value in the CL
would be significantly smaller. However, choosing a high value of proton
concentration corresponds to large cathode overpotential values, assuring that at
the given potential, the Pt is not oxidized. Furthermore, having larger number of
protons in the channel offers a better statistical ensemble for protons. The system
consisted of 10-100 protons, 8000-20000 water molecules, and 6000-9000 Pt atoms
in the channel structure.

28
2.4. Limitations for using continuum modeling
In our model, each facet of the structured Pt channel is only a few
nanometers wide. At this scale, surface roughness at molecular scale is
significant, and has notable impact on the surface structure. Therefore, the
approximation of a “smooth” surface fails. Classical MD accounts for all Pt atoms
explicitly, and assigns mass, charge and Lennard-Jones interaction parameters to
each atom.
At the continuum level, Poisson-Boltzmann model describes the relation
between proton concentration and potential. It neglects the finite size of protons,
and proton-proton interactions. The role of solvent is treated in continuum
approximation. A full atomistic MD simulation, on the other hand, accounts for
molecular correlation and interactions, leading to a more accurate description of
the proton interactions at the interface.
The structure of water is not accounted for in continuum modeling.
Physical properties of water at interfaces are different from those in the bulk.
Hydrophobic or hydrophilic nature of the surface and its electric charge affect
the structure of adjacent water. In classical double layer models, the role of
interfacial water has either been neglected, or water is considered as a medium
with known dielectric constant [75]. In the case of nanochannels, important
phenomena occur at the interface, where effects of interfacial charging and the
structure of interfacial water layers must be accounted for.

29
2.5. Metal surface charge distribution
In a classical MD simulation, we can control the metal surface charge
distribution. However, translating the surface charge distribution to an
equivalent metal potential value requires a few assumptions and simplification.
Gauss’ law and classical double layer models relate the surface charge
distribution to the total potential drop in the solution phase across the double
layer for a smooth electrode [76]:
( ) (
) 2.6
For a dilute solution of protons at room temperature, evaluation of
constants in the above equation reveals the monotonic relationship of potential
drop at the interface with the electrode state of charge. The famous Gouy-
Chapman model gives
( ), where C is the bulk proton
concentration (mol/L), and σ the surface charge density (μC/cm2). The
fundamental assumption of the Gouy-Chapman model for electric double layer
is formation of a diffuse layer of point-like ions in electrochemical equilibrium.
The assumption of point-like ions causes inaccuracy to the prediction of Gouy-
Chapman model at high surface charge densities.
In order to find an approximate relation between surface charge density
and metal potential, we followed the procedure proposed in Ref. [51] to integrate
the double layer capacitance from PZC to metal potential ɸM. The double layer

30
capacitance in the Stern model [76] is given by
, where CH is the
Helmholtz layer capacitance, and Cdiff the diffuse layer capacitance, given by
( )
( ) 2.7
where HP indicates the position of Helmholtz plane. A typical value for CH is
~0.2 F.m-1, and a typical value for Cdiff is ~3 F.m-1 [76].
Since CH is usually significantly smaller than Cdiff, the capacitance can be
assumed dominated by CH; furthermore we assume CH to be constant. Therefore
we can integrate CH to obtain surface charge density:
∫ ( ) ( )
2.8
For ( ) constant it gives ( ), and thus
( ) .
Therefore, in order to calculate ɸM, we need to estimate CH and have prior
knowledge of ɸPZC. Measuring ɸPZC is straightforward for liquid electrodes such
as dropping mercury electrode (DME), where varying the potential affects the
surface tension of mercury, and enables measuring the differential capacitance of
mercury. Consequently, the minimal capacitance is measured at ɸPZC. However,
studying solid electrodes is experimentally difficult. There exist experimental
difficulties for studying solid electrodes such as Pt, including reproducibility of
electrode surfaces, solution contamination, specific adsorption of anions, oxidie

31
formation on the surface, as well as maintaining a clean surface during the
experiment. Moreover, the value of ɸPZC is not unique for a polycrystalline
surface. Mimicking the double layer phenomena has been attempted by
ultrahigh vacuum systems [77] as well as DFT calculations on well-defined
surfaces [78], [79]. The values of ɸPZC for Pt(111) range from 0.1 to 1.0 V in the
literature. However, for an estimate of metal potential, we assume ɸPZC=1.0 V,
and CH=0.24 F.m-1, a typical value measured from differential capacitance
measurement experiments. Table 2.2 shows typical corresponding values of
metal potential, potential drop across the double layer and metal surface charge
density.
Table 2.2. Typical values of surface charge density and metal potential,
system with θ=45°, dc=60 Å, dpp=30 Å, direct relation between metal
potential and surface charge density
ɸHP-ɸ0(mV)
(M)
σ(μC.cm-2) ɸM(V)
-1 0.20 -5.0 0.78
-10 0.25 -6.1 0.69
-50 0.57 -13.9 0.35
-70 0.84 -20.4 0.05

32
3. Simulation Methodology
3.1. Overview
In this chapter, we explain how model systems are generated, MD
simulations are run, and the trajectory results are analyzed. The general
procedure is explained, and the detailed discussions follow in the subsequent
sections.
As mentioned in the previous chapter, each model system is identified
with four parameters (θ, dc, dpp, φM). With these parameters we construct specific
Pt channel geometries (section 3.2) and calculate partial charges on Pt surface
atoms (section 3.3). Then we calculate the total charges on the Pt surface, and add
an equal number of protons to the channel interior to counterbalance the
negative charge on the walls. The concentration of water in the channel is
maintained at a value close to 55 mol/L.
In the next step, we performed energy minimization on the system.
Thereafter, the system was thermally annealed between 800 K and 300 K.
Production runs were performed for 4 ns on the system (section 3.4).
Production runs were analyzed to explore equilibrium properties of the
system (section 3.5). Our data analysis focused on three major aspects: (i) density
maps of hydronium ion distributions were analyzed to determine potential
distribution and the channel electrostatic effectiveness factor (section 3.5.1), (ii)

33
temporal water displacement data was analyzed to calculate water self-diffusion
coefficient in the channel (section 3.5.2), (iii) water distribution maps were
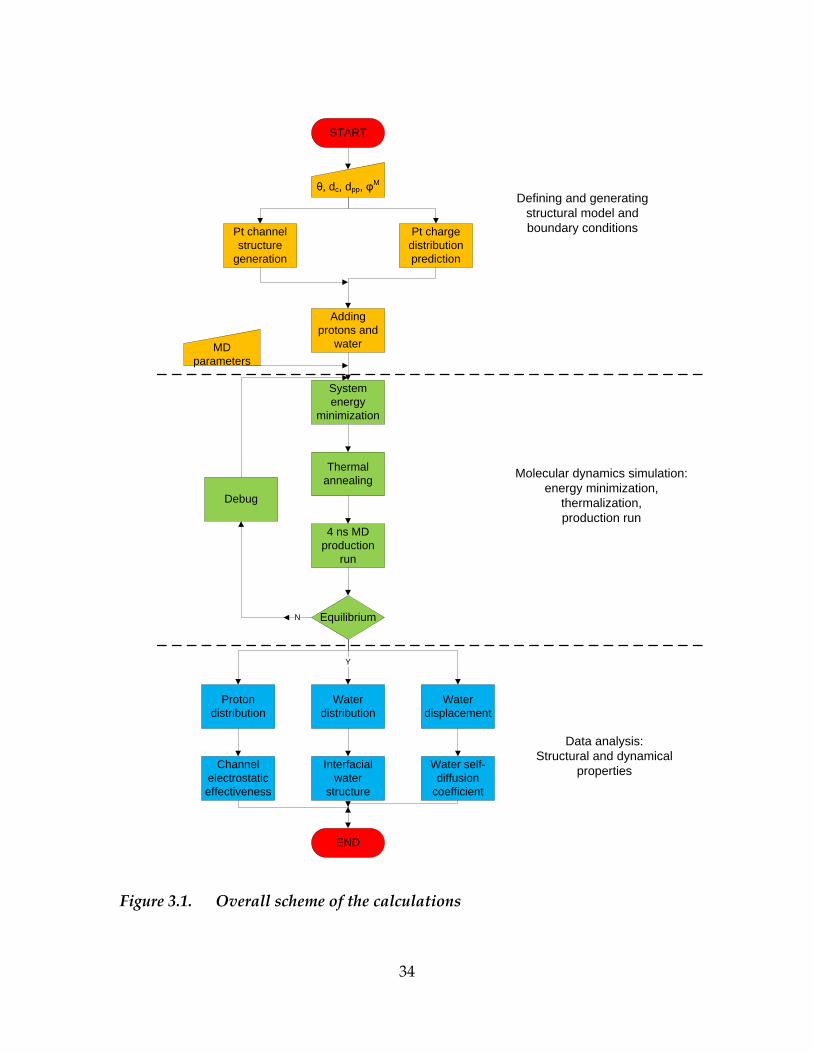
analyzed to identify interfacial water layers (section 3.5.3). Figure 3.1 shows the
overall scheme of calculations. In the following sections, we will explain the
detailed approaches.
34
Pt channel
structure
generation
θ, dc, dpp, φM
Pt charge
distribution
prediction
Adding
protons and
water
System
energy
minimization
Thermal
annealing
4 ns MD
production
run
EquilibriumN
MD
parameters
Water
distribution
Y
Proton
distribution
Water
displacement
Water self-
diffusion
coefficient
Interfacial
water
structure
Channel
electrostatic
effectiveness
END
START
Defining and generating
structural model and
boundary conditions
Molecular dynamics simulation:
energy minimization,
thermalization,
production run
Data analysis:
Structural and dynamical
properties
Debug
Figure 3.1. Overall scheme of the calculations

35
3.2. Pt structure construction
Platinum is a metal with face centred cubic (FCC) crystalline structure,
with lattice constant of a=3.92 Å. The unit cell of the Pt crystal lattice is shown in
Figure 3.2.
Figure 3.2. Primitive cell of Pt crystal with FCC structure
In order to construct a Pt nanochannel, we first created a cube of Pt by
replicating the unit cell in x,y,z directions. Then, the structural parameters were
used to calculate the plane equations of the channel walls, and the Pt atoms
located between two walls were removed. Figure 3.3 shows an incomplete Pt
channel, for the purpose of better illustration. The depth of the channel was fixed
for all cases, w=5.88 nm, corresponding to 15 times the Pt lattice constant. A 7 Å-
thick Pt layer was considered to both sides of the channel as a clearance layer, in
order to minimize the image charge effects on H3O+ distribution.

36
Figure 3.3. Full atomistic Pt channel structure, incompletely constructed to
better illustrate build-up procedure (left), schematic representation
(right). Water and hydronium ions would be added to the space in
the middle of the structured channel.
Figure 3.4. 2-d Schematic picture of the model system with physical boundary
conditions
The plane equations indicating channel walls are as follows:

37
( )
( )
3.1
( )
( )
3.2
( )
( )
3.3
( )
( )
3.4
( )
( )
3.5
( )
( )
3.6
( )
( )
3.7
( )
( )
3.8
The value θ determines the surface Miller indices. The Miller indices will
be multiples of (1, tan(θ), 0). Therefore, in each case we simulate a stepped Pt
surfaces.
For each case, step fraction is defined as 1/n, where n is the number of
atoms on the terrace of each step.

38
( ) 3.9
( ) 3.10
The two cases of θ=0° and θ=45° generate known smooth surfaces of
Pt(100) and Pt(110), respectively. Cases with 0< θ<45° have terraces parallel to the
xz plane, and cases with 45°< θ<90° have terraces parallel to the yz plane. This
change in the plane direction is the same reason for having two definitions for
step fraction.
3.3. Prediction of charge distribution on Pt nanochannel
surface
According to Gauss’s law, the surface of a conductor is equipotential, and
all free charge on the metal is accumulated on the surface. For a smooth surface,
the metal surface charge density is homogeneous. However, for a “rough”
surface, the charge distribution is inhomogeneous. In this section, we explain
how we assigned partial electric charge to each Pt surface atom in the
nanochannel.
Potential distribution inside the channel was predicted using the
numerical solution of Poisson-Boltzmann (PB) equation,
( )
(
( )
) 3.11

39
where ( ) is the number density of protons, dielectric constant of water,
the reference proton concentration, kB Boltzmann constant, and T the absolute
temperature, set to 300 K.
We assumed dielectric constant of , where is the vacuum
permittivity, and
, as the reference concentration. Boundary
condition for this problem is constant metal potential on the walls ( ) for
boundaries 1-4 and 7-10 in Figure 3.4, and symmetry (
) on boundaries
number 5 and 6.
The PB equation has an analytical solution only for simple geometries. For
geometries like the present case, the PDE could only be solved numerically. We
used triangular meshing and finite element method in COMSOL Multiphysics ®
package to obtain the numerical solution to this partial differential equation.
We calculated the surface charge density by evaluating the potential
gradient at the metal boundary and using
. The “surface Pt atoms” are
located within a distance of 3Å away from ideal wall positions. The total surface
area is
( ), and the interfacial area per surface atom is
,
where nsurf is the number of surface Pt atoms.
Therefore, the partial charge of each atom, q’, is calculated as:
( ) 3.12

40
where
is the elementary charge. The total charge on the
surface is calculated by adding up all charges on surface atoms ∑
.
The ensemble is made electroneutral by adding a corresponding correct
number of hydronium ions to the system. Thus, the total charge on the surface
must be a correct number. Since Q’ is not essentially an integer, we cut it to the
closest integer value, and normalized all q’ values: [ ] ⇒
, where
is the partial charge assigned to each surface Pt atom. Q is the total charge on
the surface, and |Q| equals the number of hydronium ions that must be added to
the system to counterbalance the wall charges.
3.4. Classical molecular dynamics
We have used classical MD to find equilibrium distribution of protons and
water in the nanochannel. In this chapter, we first introduce the basic concept of
MD, and explain the procedures and parameters we used to simulate the system.
The theory is discussed in detail in Ref. [80]. The calculations were carried out
using the GROMACS [81] open source package.
3.4.1. Description of the forcefields used
Classical MD calculation is based on integrating Newton’s equation of
motion for each species in the system,
. The force exerted on each atom is
calculated as in equation 3.13. (Bold-faced variables indicate vectors.)
3.13

41
where FB is the bonded (intramolecular) interactions of an atom, described
by harmonic oscillator approximation as stated in equations 3.14 and 3.15.
( )
( )
3.14
( ) ( )
| | 3.15
FB was calculated only on hydronium ions and water molecules, since Pt channel
atoms were assumed frozen at their positions.
Forces due to non-bonded interactions, FNB, include Van der Waals
interactions, described by Lennard Jones interactions (FLJ), and charge-charge
interactions described by Coulomb’s law (FCoulomb),
( )
( )
, (
( )
( )
)
| |, 3.16
| | 3.17
Having calculated the force, new position and velocity of each atom can
be calculated by discretizing the equation of motion using leapfrog finite
differencing scheme [82] for velocity calculation, (
) (
)
( ) , and position calculation, ( ) ( ) (
) . The time step
chosen was 2 fs, commonly used in the literature. It was tested, though, that the

42
simulation systems would crash with time steps larger than 2.4 fs, due to
interaction of species beyond the cut-off range, leading to blowing up of the
molecules within the system.
The overall scheme of force calculation parameters is shown in Figure 3.5.
Figure 3.5. Force calculation scheme
In our model, Pt atoms were fixed to their position throughout the
simulation. Calculation of partial charges of Pt atoms was explained in the
previous section. The interaction of Pt with other atoms was calculated by fitting
an n-body Sutton-Chen potential [83] to Lennard Jones form, the same procedure
as followed in Ref. [84]. Forcefield parameters of H3O+ were taken from Ref. [85].
We chose the SPC/E water model [67] for this study. If used with the
GROMOS43 forcefield, this water model gives the self-diffusion coefficient of
( )
[86], which is close to the experimental
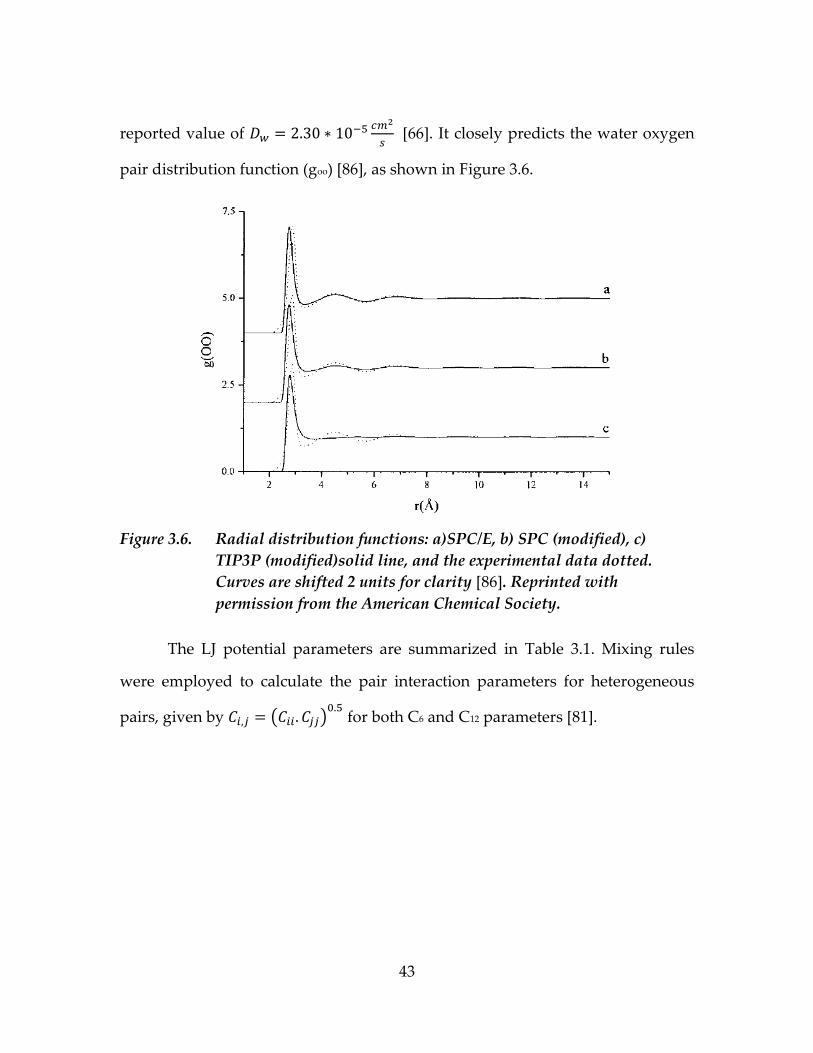
43
reported value of
[66]. It closely predicts the water oxygen
pair distribution function (goo) [86], as shown in Figure 3.6.
Figure 3.6. Radial distribution functions: a)SPC/E, b) SPC (modified), c)
TIP3P (modified)solid line, and the experimental data dotted.
Curves are shifted 2 units for clarity [86]. Reprinted with
permission from the American Chemical Society.
The LJ potential parameters are summarized in Table 3.1. Mixing rules
were employed to calculate the pair interaction parameters for heterogeneous
pairs, given by ( )
for both C6 and C12 parameters [81].

44
Table 3.1. Van der Waals interaction parameters for the system species
Pair C6(kJ.nm6) C12(kJ.nm12) q (e)
Pt-Pt 0.015 2.98*10-6 See text
O-O (water) 0.003 2.63*10-6 -0.848
O-O (hydronium ion) 0.003 2.63*10-6 -0.248
H-H (water) 0.00 0.00 0.424
H-H (hydronium ion) 0.00 0.00 0.416
3.4.2. Computational details
We simulated an infinite nanochannel, using periodic boundary condition.
Therefore, we had to make sure that the finite cell size did not introduce
significant artifacts on the proton distribution diagrams of the system, which
could stem from insufficient number of exchange of species through the periodic
boundaries. The effect of finite simulation cell size is noticeable especially when
the system consists of ions with long range electrostatic interactions, such as the
system of this study. The artifacts caused due to long range electrostatic
treatment are due to artificial stabilization of the system, which inhibit
conformational fluctuations. Such artifacts have been reported in references [46],
[87], [88]. The Particle Mesh Ewald (PME) is a method [89] employed for systems
with long range electrostatic interactions. In order to capture effects of dipole
(O(1/r3)) and Coulomb (O(1/r)) potentials efficiently, the PME method calculates
the short range forces directly, and calculates the long range forces in Fourier
space[88]. With this method, the computational time scales with the size of the
system,N, as N.log(N); compared to the scaling as N2 in the Ewald summation
method [88].

45
Van der Waals forces have short effective range. Therefore, the forces on
each atom could be estimated with the sum of forces exerted from atoms closer
than a specific “neighboring radius” (chosen 0.9 nm in this work). This choice
drastically increases the force calculation speed. After each 10 MD steps (20 fs)
the list of neighboring atoms must be updated. However, the neighbor searching
algorithm mandates using a small time step of 2 fs. The small time step is
essential for inhibiting dissociation of molecules.
We used the canonical ensemble for this model. Therefore, the system was
coupled to a Berendsen thermostat [90] with a time constant of 1 ps, to keep the
ensemble temperature at 300 K.
All the calculations were done on the scientific computation clusters of
NRC-Institute for Energy, Mining and Environment of Canada in Vancouver,
BC. We used MATLAB v2006a and COMSOL 3.5a for pre-processing and
analysing the simulation results. Gromacs v4.0.3 [81] was used to run MD
calculations. The simulation was run on one node with eight processors. The
typical simulation run time was ~2 ns/day. VMD [91] and RASMOL [92] software
were used for visualization of the results.
3.5. Simulation data analysis
The last 2 ns of the MD trajectory (from a total of 4 ns) represent
equilibrated configurations. After the initial 2 ns of the trajectory, the potential
and kinetic energy of the system do not deviate from the average value by more
than 2%. Proton and water distribution maps are generated by averaging over

46
statistically independent configurations, and water self-diffusion coefficients
were calculated from analyzing water molecule displacements over time.
The analyses lead to the calculation of the electrostatic effectiveness factor
of the channel, water structure and water self-diffusion coefficient.
3.5.1. Hydronium ion density map and electrostatic effectiveness
In order for the electrochemical reaction to occur, hydronium ions need to
approach the surface. We considered the hydronium ions within a distance of 4
Å from the ideal location of walls as “surface protons”. This definition enabled
attributing a single value of proton concentration to each point on the Helmholtz
plane, and mapping the 2-dimensional proton distribution map onto a 1-
dimensional distribution profile. Therefore, calculation of channel effectiveness
reduces to a 1-d integration problem. This task was achieved using MATLAB
image processing toolbox.
The density profile was used to back-calculate the potential profile
adjacent to the surface, assuming a Boltzmann distribution of proton
concentration,
(
). Electrostatic effectiveness factor was
calculated as explained in equation 2.4.
3.5.2. Surface and bulk water diffusion
The self-diffusion coefficient of water can be calculated from
| ( ) ( ) |
. We analyzed Dw by distinguishing surface water
and bulk-like water. The water molecules within a distance of 4 Å from channel
ideal wall position in the final trajectory frame were considered surface water,

47
and the rest were considered bulk-like water. Dw for these two groups was
calculated over a period of 200 ps. The trajectory length is chosen short enough,
so that the exchange between the two phases of water is not significant.
3.5.3. Interfacial water structure
In order to investigate the effect of nanoconfinement on water structure,
we calculated the pair correlation function g(r) defined as
( )
∑ ∑
( )
. In statistical mechanics, gi,j(r) shows the
probability of finding species i at radial distance r from species j. g(r) is an
indication of the structure and density of a certain phase. We calculated goo(r) for
both surface water and bulk-like water, and compared the distributions.

48
4. Results and Discussion
4.1. Overview
In this chapter, the results of surface charge, proton and water distribution
in the nanochannel are presented and discussed. Section 4.2 presents the results
of surface inhomogeneity on the electric charge distribution on the metal surface.
Section 4.3 presents a qualitative analysis of the proton distribution in the
nanochannel. In order to quantify the impact of varying degrees of surface
structuring on electrocatalytic properties of the nanochannel, we calculated the
channel electrostatic effectiveness factor. The results are presented in section 4.4.
In section 4.5, we present the impact of channel nanostructure on water diffusion
and structure, and section 4.6 explains the sensitivity of obtained results to the
parameters employed for simulation.
4.2. Surface charge distribution in the nanostructured channel
On a smooth surface, all the metal atoms have equal surface charge
density. In a structured channel, we noticed that peak positions have the highest
surface charge, and deep positions have the lowest surface charge. Typical
potential, surface charge distribution and proton concentration maps are shown
in Figure 4.1.

49
Figure 4.1. Typical distribution maps in the nanochannel predicted by PB
theory: a) potential distribution, b) proton concentration, c)
Electric field contour lines, d) metal surface charge density
Within the investigated range of 1 nm< dc< 10 nm, we observed that dc did
not affect the prediction of surface charge density distribution. Therefore, the
-0.08
-0.07
-0.06
-0.05
-0.04
-0.03
-0.02
-0.01
0
0.E+0
5.E+7
1.E+8
2.E+8
2.E+8
3.E+8
3.E+8
0 20 40 60 80
x (Å)
E q
( 𝑚)
𝑜
𝑚
a b
c d
𝜙( 𝑉
) 𝐸
(𝑉 𝑚)
𝑐 𝐻 𝑂
( 𝑀
)
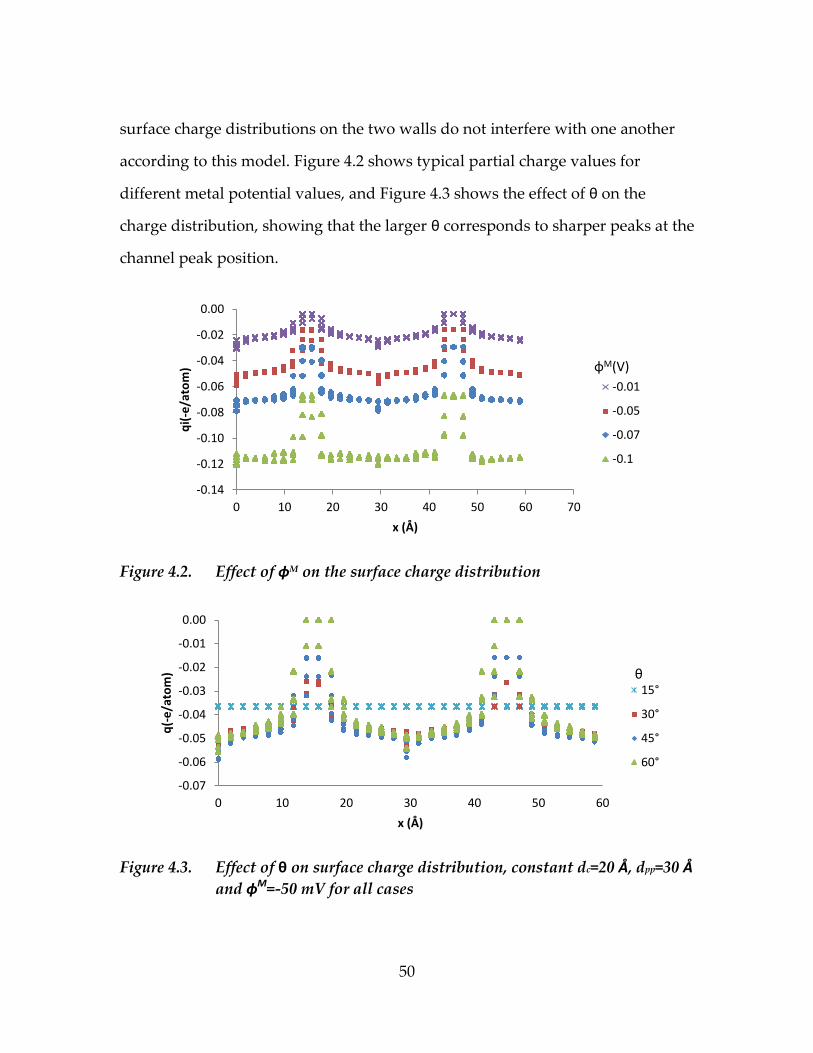
50
surface charge distributions on the two walls do not interfere with one another
according to this model. Figure 4.2 shows typical partial charge values for
different metal potential values, and Figure 4.3 shows the effect of θ on the
charge distribution, showing that the larger θ corresponds to sharper peaks at the
channel peak position.
Figure 4.2. Effect of ɸM on the surface charge distribution
Figure 4.3. Effect of θ on surface charge distribution, constant dc=20 Å, dpp=30 Å
and ɸM=-50 mV for all cases
-0.14
-0.12
-0.10
-0.08
-0.06
-0.04
-0.02
0.00
0 10 20 30 40 50 60 70
qi(
-e/a
tom
)
x (Å)
-0.01
-0.05
-0.07
-0.1
ɸM(V)
-0.07
-0.06
-0.05
-0.04
-0.03
-0.02
-0.01
0.00
0 10 20 30 40 50 60
q(-
e/a
tom
)
x (Å)
15°
30°
45°
60°
θ

51
Figure 4.3 shows that the surface charge density distribution for θ=15° is
almost homogeneous. This observation is expected from such channel geometry,
since the structure is similar to a Pt (100) smooth surface with weak roughening,
as shown in Figure 4.4. The observation of almost homogeneous σ distribution
holds for all θ<15° values.
Figure 4.4. Pt structured channel with θ=15°, predicted charge distribution
close to homogeneity
4.3. Hydronium ion distribution in catalyst layer channels
4.3.1. Hydronium ion density distribution in smooth Pt channels
Equilibrium distribution of hydronium ions on a smooth catalyst surface
is uniform along the surface. Variation of ion density perpendicular to the
surface is determined according to the electrochemical equilibrium in the
channel. Continuum-type models have been able to predict distribution
prediction for simple geometries of the channel. Engstroem and Wennerstroem
have analytically solved the Poisson-Boltzmann (PB) equation for geometry of
two parallel slabs [93], with the boundary condition of constant surface charge
density on two parallel charged slabs. Andelman has solved the PB equation for

52
other cases with simple geometry [94]. In the absence of counter-ions in the
solution, potential and hydronium ion distribution were calculated as below.
Figure 4.5. Schematic of analytically solved electrostatic model system
( )
( ( )), with
, 4.1
( )
( )
,
4.2
where nm denotes the hydronium ion density at the center of the channel (z=0),
and 1/K is the characteristic length scale of the system.
In order to confirm the consistency of molecular dynamics calculations
with this theory, we performed 10 cases of MD simulations of hydronium ions
and water distribution on slab-like channels with smooth walls of Pt(111) at
various openings and surface charge densities. We investigated the hydronium
ion density distribution perpendicular to the Pt walls. The results indicated
overall agreement between the two approaches in the channel interior, although
the effect of finite size of atoms is not captured in the PB model prediction. A

53
simple schematic of this difference is shown in Figure 4.6. Due to these
differences between the two models, the maximum proton concentration was
observed 1 Å further from the surface in an MD calculation. We also observed a
typical layering behavior, which is not predicted in the PB theory. The
discrepancy between the two models was reflected more clearly in systems with
high surface charge density and with smaller groove width. The two predictions
could be deemed matching for systems with channel spacing over 7 nm and
surface charge density smaller than
. Sample results from the calculations
are shown in Figure 4.7.
Figure 4.6. Wall position definition for PB theory, the wall position is not
defined precisely in an MD simulation; however the positions of all
atoms nuclei are known.

54
a
b
c
Figure 4.7. Hydronium ion number density at σ=-16.8 μC.cm-2 and channel
openings of a)2 nm, b) 3 nm, c) 4 nm, better agreement between PB
and MD results at larger channel opening
4.3.2. Hydronium ion distribution in nanostructured channels
We investigated hydronium ion density distributions on CL channels, to
observe how metal nanostructuring causes deviation from uniform distribution
0
1
2
3
4
5
-4 -2 0 2 4
c x(1
/ n
m3)
x(nm)
MD
PB
0
1
2
3
4
-4 -2 0 2 4
c x(1
/ n
m3)
x (nm)
MD
PB
0
0.5
1
1.5
2
2.5
-4 -2 0 2 4
c x(1
/ n
m3)
x(nm)
MD
PB
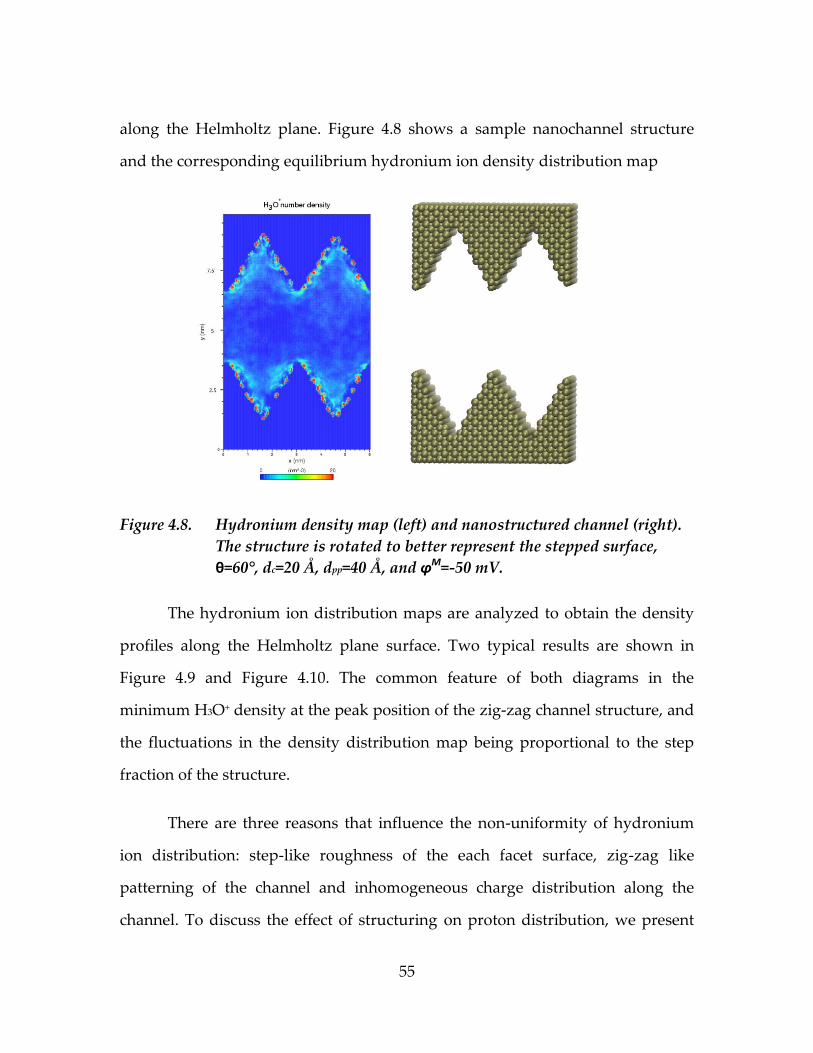
55
along the Helmholtz plane. Figure 4.8 shows a sample nanochannel structure
and the corresponding equilibrium hydronium ion density distribution map
Figure 4.8. Hydronium density map (left) and nanostructured channel (right).
The structure is rotated to better represent the stepped surface,
θ=60°, dc=20 Å, dpp=40 Å, and φM=-50 mV.
The hydronium ion distribution maps are analyzed to obtain the density
profiles along the Helmholtz plane surface. Two typical results are shown in
Figure 4.9 and Figure 4.10. The common feature of both diagrams in the
minimum H3O+ density at the peak position of the zig-zag channel structure, and
the fluctuations in the density distribution map being proportional to the step
fraction of the structure.
There are three reasons that influence the non-uniformity of hydronium
ion distribution: step-like roughness of the each facet surface, zig-zag like
patterning of the channel and inhomogeneous charge distribution along the
channel. To discuss the effect of structuring on proton distribution, we present
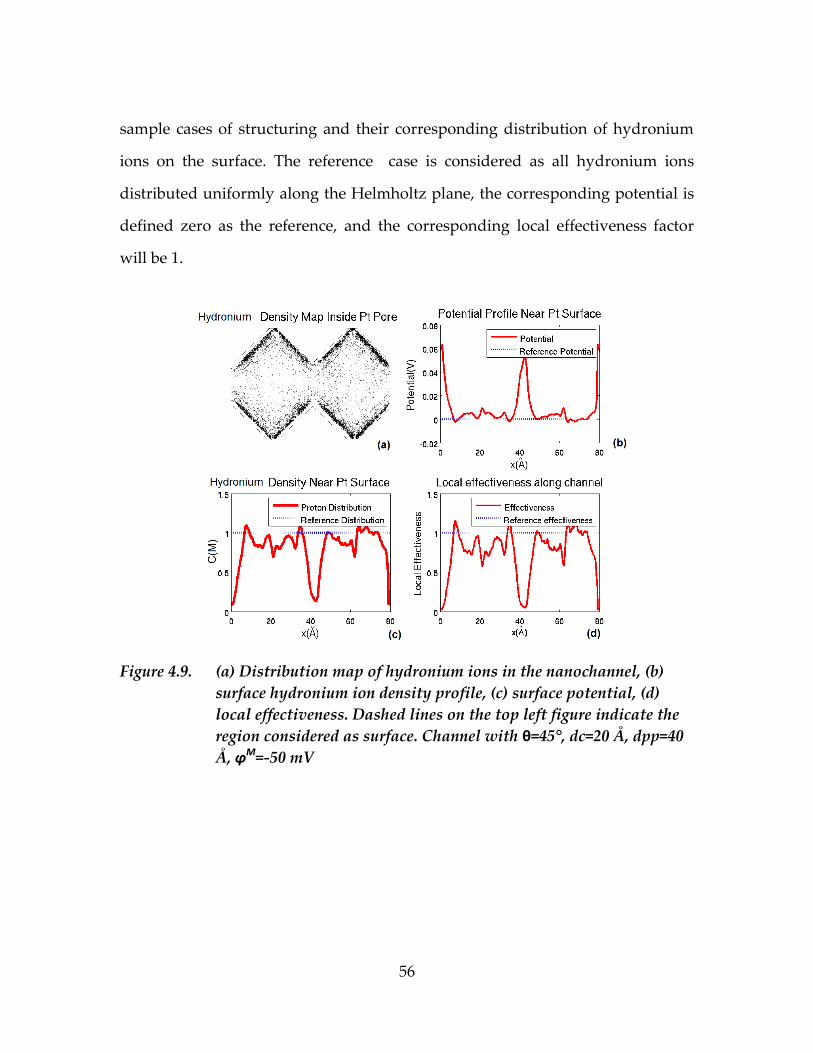
56
sample cases of structuring and their corresponding distribution of hydronium
ions on the surface. The reference case is considered as all hydronium ions
distributed uniformly along the Helmholtz plane, the corresponding potential is
defined zero as the reference, and the corresponding local effectiveness factor
will be 1.
Figure 4.9. (a) Distribution map of hydronium ions in the nanochannel, (b)
surface hydronium ion density profile, (c) surface potential, (d)
local effectiveness. Dashed lines on the top left figure indicate the
region considered as surface. Channel with θ=45°, dc=20 Å, dpp=40
Å, φM=-50 mV

57
Figure 4.10. (a) Distribution map of hydronium ions in the nanochannel, (b)
surface hydronium ion density profile, (c) surface potential, (d)
local effectiveness. Dashed lines on the top left figure indicate the
region considered as surface. Channel with θ=75°, dc=20 Å, dpp=30 Å,
and φM=-50 mV
Although the two chambers in the simulation cell are identical, the proton
density profile along the surface does not show complete symmetry in all cases.
The observation suggests that either the equilibrium condition has not been
obtained, or the sampling has been insufficient. We changed the Van der Waals
and electrostatic force calculation cut-off values; however, no significant change
was observed neither in proton distribution maps nor water mean square
displacement plots. Some results are shown in sections 4.6.2, 4.6.3, and 4.6.6.
Appearance of a local minimum hydronium ion density at the peaks of
the zig-zag pattern was unexpected, since these points carry the highest negative

58
surface charge density. A possible reason for this behavior is the walls maximum
repulsion at the peaks.
The electrostatic repulsion between two nanochannel walls reaches
maximum at the peak position. The peak positions have the shortest distance
from each other, and carry the highest amount of similar charge. We suggested
that these effects lead to depletion of H3O+ in the peak region. In order to test this
hypothesis, we compared H3O+ density at the peak position for different channel
openings. Increasing channel opening decreases overlapping, and is expected to
increase H3O+ density at the peak. For this comparison, we set θ=45° and dpp=30
Å and
constant, and varied dc between 10 Å and 100 Å. We observed
that H3O+ density at the peak increased with increase of the channel opening, but
the minimum at the peak position remains. Reducing double layer overlap by
increasing dc does not remove the unexpected minimum at the peak. The results
are shown in Figure 4.11.
Figure 4.11. Relative H3O+ density at channel peak region vs. dc, effect of walls
electrostatic repulsion on H3O+ depletion from peak region, with
constant θ=45°, dpp=30 Å, and φM=-50 mV
0.15
0.2
0.25
0.3
0.35
0.4
0 20 40 60 80 100 120
C/C
0
dc(Å)

59
4.4. Electrostatic effectiveness factor as a function of channel
structure
We showed how the channel structure affects proton distribution in the
catalyst layer channel. However, so far we did not comment on whether this
structuring led to enhancement or deterioration of channel performance. To
address this question, electrostatic effectiveness factor was introduced in chapter
2. Since the activity is normalized to the total Helmholtz plane area, the
electrostatic effectiveness factor shows the effect of structuring on channel
performance beyond changing the active surface area.
4.4.1. Effect of channel opening and structuring angle
In order to investigate the effect of channel opening on electrostatic
effectiveness, we varied the structural parameters as per table below.
Table 4.1. Range of parameters for investigation of channel opening effect
Parameter Value Unit
Structuring angle (θ) 0, 15, 30, 45, 60, 75 Degrees
Channel opening (dc) 10, 20, 30, 40, 50, 60, 80, 100 Å
Groove width (dpp) 30 Å
Metal potential (φM) and
average surface charge density (σ)
-10 (-5), -50 (-14) mV (
)
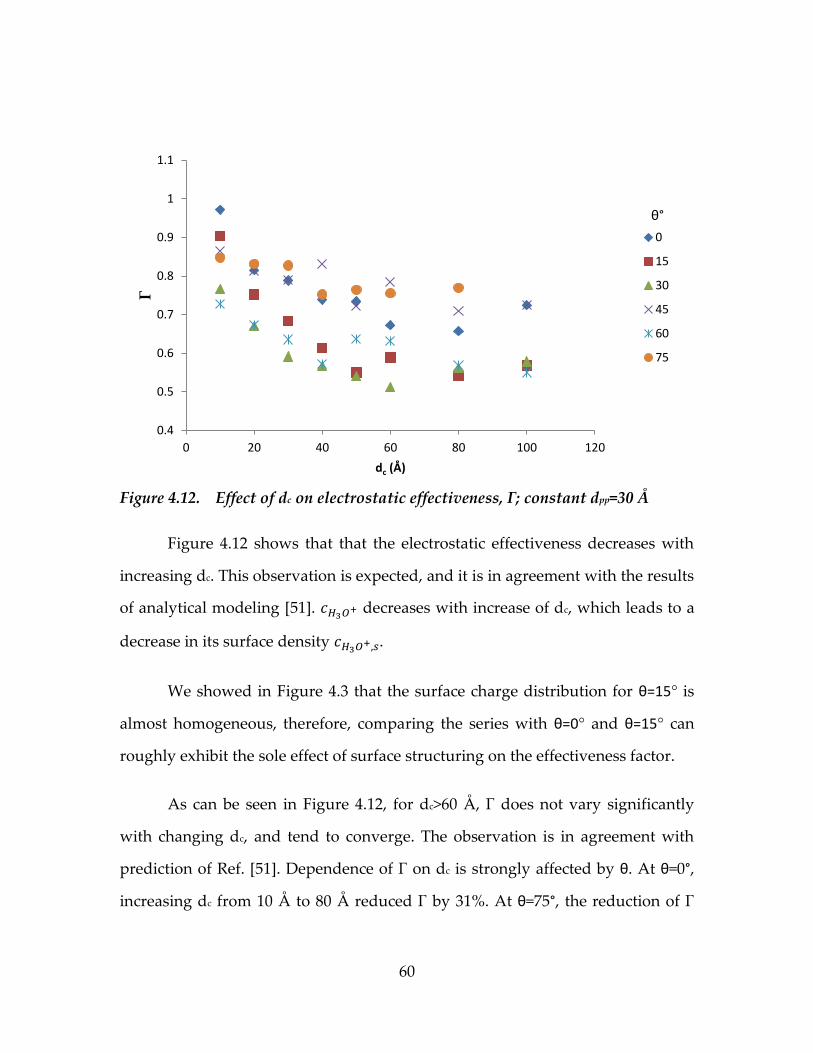
60
Figure 4.12. Effect of dc on electrostatic effectiveness, Γ; constant dpp=30 Å
Figure 4.12 shows that that the electrostatic effectiveness decreases with
increasing dc. This observation is expected, and it is in agreement with the results
of analytical modeling [51]. decreases with increase of dc, which leads to a
decrease in its surface density .
We showed in Figure 4.3 that the surface charge distribution for θ=15° is
almost homogeneous, therefore, comparing the series with θ=0° and θ=15° can
roughly exhibit the sole effect of surface structuring on the effectiveness factor.
As can be seen in Figure 4.12, for dc>60 Å, Γ does not vary significantly
with changing dc, and tend to converge. The observation is in agreement with
prediction of Ref. [51]. Dependence of Γ on dc is strongly affected by θ. At θ=0°,
increasing dc from 10 Å to 80 Å reduced Γ by 31%. At θ=75°, the reduction of Γ
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
0 20 40 60 80 100 120
Γ
dc (Å)
0
15
30
45
60
75
θ°

61
was only 8%. This sensitivity is attributed to the change of channel volume at
different θ, as given by
( ( )) ⇒
( ) 4.3
Therefore, at large θ, the volume change is weakly affected by dc. Consequently Γ
is weakly related to dc. Γ is a monotonic function of surface proton ratio, as
expected from equation 2.4. We investigated the correlation between Γ and the
overall in the channel. Figure 4.13 shows this relationship.
Figure 4.13. Direct effect of overall H3O+ concentration on channel effectiveness,
dispersion of data in the low concentration region is the channel
structure-specific effects.
0.4
0.5
0.6
0.7
0.8
0.9
1
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5
Γ
0
15
30
45
60
75
( )
θ°
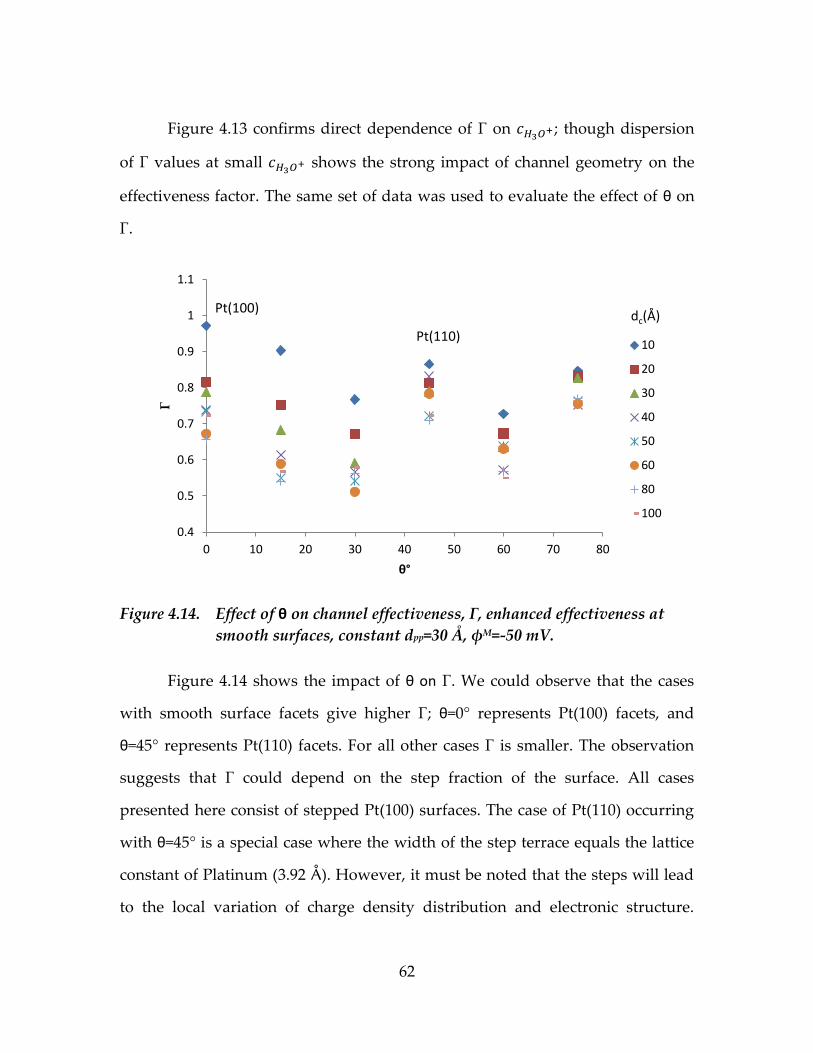
62
Figure 4.13 confirms direct dependence of Γ on ; though dispersion
of Γ values at small shows the strong impact of channel geometry on the
effectiveness factor. The same set of data was used to evaluate the effect of θ on
Γ.
Figure 4.14. Effect of θ on channel effectiveness, Γ, enhanced effectiveness at
smooth surfaces, constant dpp=30 Å, φM=-50 mV.
Figure 4.14 shows the impact of θ on Γ. We could observe that the cases
with smooth surface facets give higher Γ; θ=0° represents Pt(100) facets, and
θ=45° represents Pt(110) facets. For all other cases Γ is smaller. The observation
suggests that Γ could depend on the step fraction of the surface. All cases
presented here consist of stepped Pt(100) surfaces. The case of Pt(110) occurring
with θ=45° is a special case where the width of the step terrace equals the lattice
constant of Platinum (3.92 Å). However, it must be noted that the steps will lead
to the local variation of charge density distribution and electronic structure.
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
0 10 20 30 40 50 60 70 80
Γ
θ°
10
20
30
40
50
60
80
100
dc(Å) Pt(100)
Pt(110)

63
These effects, which have not been considered in our PB model to determine the
surface charge density distribution, could overshadow the bare geometry effect.
To broaden the conclusion about the effect of structuring angle on
electrocatalysis, the coupling between electrostatic and kinetic effects of stepped
surfaces must be considered. Increasing the step fraction is known to lower the
potential of zero charge of the metal [78], [95]. The work function of Pt is lower
on the step sites due to the change in spatial distribution of electrons on the step
sites [96–98]. Thus, although the stepped geometry was found to have a negative
impacts on Γ, steps have a positive impact on the kinetics of ORR on the surface,
which are not accounted for in this work. This consideration inhibits drawing a
definitive conclusion about the overall effect of θ on electrocatalytic phenomena
in nanochannels with corrugated walls.
4.4.2. Effect of groove width
We investigated the effect of dpp for the set of parameters specified in
Table 4.2. The main observed trend was a decrease of Γ with increasing dpp, as
illustrated in Figure 4.15.
Table 4.2. Range of parameters for investigating the effect of groove width
Parameter Value Unit
Structuring angle (θ) 0, 15, 30, 45, 60, 75 Degrees
Channel opening (dc) 20 Å
Groove width (dpp) 20, 30, 40, 50, 60, 70, 80 Å
Metal potential (ɸM) and
average surface charge density (σ)
-50 (-14) mV(
)

64
Figure 4.15. Inverse effect of groove width on electrostatic effectiveness of
nanochannel, constant dc=20 Å
Increasing dpp while keeping dc and σ fixed reduces in the
nanochannel. Lower causes decrease of the ratio of hydronium ions on the
surface. The relation between Γ and is shown in Figure 4.16.
0.4
0.5
0.6
0.7
0.8
0.9
1
0 20 40 60 80 100
Γ
dpp (Å)
0
15
30
45
60
75
θ

65
Figure 4.16. Direct effect of overall H3O+ density on Γ, constant dc=20 Å
We used the data from this set of simulations to evaluate the effect of θ.
The results attest to our former hypothesis of the effect of step fraction on
channel effectiveness. Step fraction does not vary with changing dc, nor with dpp.
Therefore, similar behavior is expected in the case of varying dpp. However, some
discrepancy is observed, which arises from the variation of inside the
channel. Increasing dpp reduces the overall inside the channel, consequently
reducing . Reduction of overall hydronium ion concentration on the
surface implies smaller Γ in the nanochannel.
0.4
0.5
0.6
0.7
0.8
0.9
1
0 0.5 1 1.5 2
Γ
0
15
30
45
60
75
θ
( )

66
Figure 4.17. Effect of θ on Γ, constant dc, inverse relation with surface step
fraction
4.4.3. Effect of metal surface charge density
A higher value of |σM| increases the Coulomb attraction between protons
and the negatively charged channel walls, and it increases in the channel.
The values chosen for this study are reported in Table 4.3. The results are
presented in Figure 4.18.
Table 4.3. Range of parameters for investigating the effect of surface charge
Parameter Value Unit
Structuring angle (θ) 0, 15, 30, 45, 60, 75 Degrees
Channel opening (dc) 20 Å
Groove width (dpp) 30 Å
Metal potential (ɸM) and
average surface charge density (σM)
-5, -10, -50, -70, -100, -110 (-5 .. -50) mV(
)
0.4
0.5
0.6
0.7
0.8
0.9
1
0 10 20 30 40 50 60 70 80
Γ
θ°
20
30
40
50
60
70
80
dpp(Å)
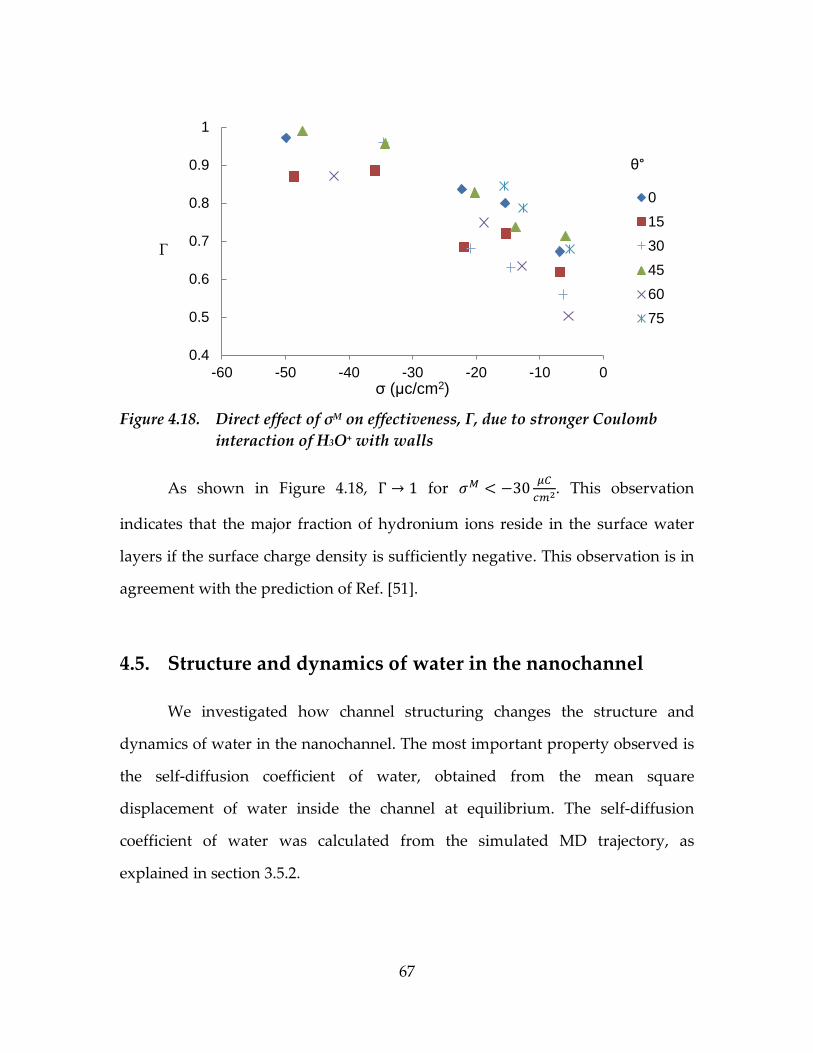
67
Figure 4.18. Direct effect of σM on effectiveness, Γ, due to stronger Coulomb
interaction of H3O+ with walls
As shown in Figure 4.18, for
. This observation
indicates that the major fraction of hydronium ions reside in the surface water
layers if the surface charge density is sufficiently negative. This observation is in
agreement with the prediction of Ref. [51].
4.5. Structure and dynamics of water in the nanochannel
We investigated how channel structuring changes the structure and
dynamics of water in the nanochannel. The most important property observed is
the self-diffusion coefficient of water, obtained from the mean square
displacement of water inside the channel at equilibrium. The self-diffusion
coefficient of water was calculated from the simulated MD trajectory, as
explained in section 3.5.2.
0.4
0.5
0.6
0.7
0.8
0.9
1
-60 -50 -40 -30 -20 -10 0
Γ
σ (μc/cm2)
0
15
30
45
60
75
θ°

68
For all cases, we evaluated whether a plot of log(MSD) vs log(t) is linear.
We employed this relation as a test whether equilibrium of water had been
obtained. We also investigated the structuring of water at the surface by
analyzing the auto-correlation function of water molecules, as explained in
section 3.5.3. In a refined analysis, we evaluated the structure and dynamics of
surface and bulk-like water. Several experimental and theoretical works have
confirmed the existence of the two distinct phases of water. In this work, we
considered water molecules in a slab of 4 Å distance from the metal wall position
as surface water, and the rest as bulk-like water. We assumed the subsets did not
exchange water molecules for 200 ps. The error bars on the self-diffusion
coefficient diagrams indicate the statistical RMS deviation of MSD vs. t from a
straight line.
The value of 4 Å is estimated to be the maximum distance up to which a
charge transfer reaction could occur. Increasing this value to higher numbers
diminishes the existing distinctions between different nanochannels to a great
extent.
4.5.1. Two phase water structure in the channel
Density maps of water, like the one shown in Figure 4.19, reveal that a
highly structured layer of water exists at the metal interface. Figure 4.19 shows
formation of two phases of water in the nanochannel, due to the confinement
effect. We refer to the phase adjacent to the channel wall “surface water” and the
phase with homogeneous density distribution “bulk-like water”.

69
Figure 4.19. Typical water density map, high contrast of colors indicate a
highly structured phase
4.5.2. Effect of the channel opening and structuring angle on water self-
diffusion coefficient
The self-diffusion coefficient of water, Dw, increases with dc. The value
approaches a plateau at the limit of dc>10 nm. Within the range of our
investigation, the maximum Dw value is 10% below the Dw predicted by the bulk
SPC/E water model [86].
The structuring angle θ has a weak yet discernible effect on Dw. The
variation of Dw with θ is due to the direct dependence of channel volume on θ.
Therefore, the effect of θ on Dw is more pronounced at small dc values.
Figure 4.20 shows that at dc=10 Å, the difference in Dw between θ=0° and θ=75°

70
cases is
, but the difference drops to
when dc=80 Å. The
competing effect of dc and θ is on volume change, due to the following relation,
( ( )) ( ) [ ( )
] 4.4
It shows that the effect of dc on volume is more pronounced at small θ values.
The result can be expressed in terms of the ratio of the number of bulk-like water
molecules to the number of surface water molecules.
Figure 4.20. Effect of dc on water self-diffusion coefficient, Dw
0
0.5
1
1.5
2
2.5
3
0 20 40 60 80 100 120dc(Å)
0°
15°
30°
45°
60°
75°
θ Dw,bulk= 2.
𝐷𝑤 ( 𝑐𝑚
𝑠)

71
Figure 4.21. Analyzing the effect of channel opening on Dw in terms of ratio of
bulk-like (nb) to surface (ns) water ratio
Figure 4.21 shows that the effect of dc and θ on Dw can be attributed to
tuning the ratio of number of bulk-like to number of surface water molecules. It
could equally be analyzed as the volume to area ratio of the channel, too. Self-
diffusion coefficient of each phase of water (bulk-like/surface) shows weak
dependence to dc. Figure 4.20 shows the existence of two distinct phases of water
with their specific Dw. However, the dependence of Dw,b to the bulk to surface
water ratio indicates the high exchange rate during the analysis time (200 ps)
between bulk-like and surface water at small bulk to surface ratios.
0
0.5
1
1.5
2
2.5
3
0 2 4 6 8 10 12
0°
15°
30°
45°
60°
75°
θ
(
𝑚
)

72
Figure 4.22. Dependence of Dw to channel structure, a) surface water, b) bulk-
like water
4.5.3. Effect of groove width on water self-diffusion coefficient
The effect of dpp is similar to dc. Increasing dpp increases the channel
volume to wall area ratio, as shown below,
( ( ) ( )) ⇒
( )
( ) 4.5
Consequently, the
ratio increases, which leads to the increase of Dw with dpp.
The results are shown in Figure 4.23.
0
0.5
1
1.5
2
2.5
3
0 5 10
(
𝑚
)
0
0.5
1
1.5
2
2.5
3
0 5 10
15°
30°
45°
60°
75°
θ
(
𝑚
)
a b

73
Figure 4.23. Effect of groove width on water self-diffusion coefficient, Dw, dc=20
Å constant.
4.5.4. Effect of metal charging on water self-diffusion coefficient
The interaction of water with the metal surface is a charge-dipole
interaction. Variation of σM influences the water dynamics in the channel. Over
the investigated range of parameters, we could observe a slight decrease of Dw
with |σM|. The reason is stronger immobilization and preferential ordering of
water at the interface due to increasing strength of interaction with the
negatively charged walls. The results are shown in Figure 4.24.
0
0.5
1
1.5
2
2.5
3
0 10 20 30 40 50 60 70 80 90
dpp(Å)
0
15
30
45
60
θ°
𝐷𝑤(
𝑐𝑚
𝑠)
𝑚

74
Figure 4.24. Effect of Pt surface charge density on water self-diffusion
coefficient, constant dc=20 Å dpp=30 Å
This effect can be evaluated separately for surface and bulk-like water. As
we would expect, the effect of σM on Dw is more pronounced on surface water
through charge-dipole interactions. The results are shown in Figure 4.25.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
-60 -50 -40 -30 -20 -10 0
σ(μC/cm2)
0°
15°
30°
45°
60°
𝑊(
𝑚
) θ

75
Figure 4.25. Dependence of Dw on Pt surface charge, a) bulk-like water, b)
surface water
4.6. Sensitivity analysis to MD run parameters
It is essential to make sure that the obtained results are not highly affected
by the choice of computational parameters. In chapter 2 we mentioned how the
range of structural parameters was chosen for this study. In chapter 3,
parameters used in the MD run were introduced, and the reasons for choosing
them were explained. In this section, we show how the results for different
channel geometries would vary with changing computational parameters, MD
run time, forcefield and long-range electrostatic treatment method, as well as the
nature of catalyst metal.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
-60 -40 -20 0
σ(µC/cm2)
(
𝑚
)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
-60 -40 -20 0
σ(µC/cm2)
15°
30°
45°
60°
75°
(
𝑚
)
θ θ
a b

76
4.6.1. Total simulation run time
All simulations were run for a total of 4 ns. The final 2 ns were sampled as
equilibrium configurations. The configuration ensemble was sampled each 10 ps,
resulting in 200 frames for analysis. For a nanochannel with θ=45°, dc=20 Å,
dpp=30 Å and average surface charge density of
we extended the
simulation to 10 ns, and extracted the coordinates every 10 ps. The results
showed negligible fluctuation of Γ values. This assured us that the chosen 4 ns
run time has been sufficient.
Figure 4.26. Sensitivity of calculated effectiveness factor to analyzed trajectory
length
4.6.2. Choice of forcefield and intermolecular interactions
Hydronium ion distribution in the nanochannel is determined by the
charge-charge interaction with surface Pt atoms. It does not change significantly
with variations to Lennard-Jones parameters of water and Pt. Though, a few
models were proposed for partial charges of a hydronium ion. In the current
0102030405060708090
[0-1] [0-2] [2-4] [4-6] [6-8] [8-10] [6-10]
Effectiveness% 84.6 83 81.3 80.7 80.5 81.6 80.6
84.6 83 81.3 80.7 80.5 81.6 80.6
Γ%
Trajectory analysis time (ns)
[0-1]
[0-2]
[2-4]
[4-6]
[6-8]
[8-10]
[6-10]

77
work we used the values reported by Goddard et. al. [85] consistently, and no
further investigations was done in this regard.
Water is described by SPC/E model, and the model is calibrated to be used
with the GROMOS43a forcefield. Test simulations were run with SPC and TIP3P
water models, too, and the results were compared to those of the SPC/E model.
We obtained similar Γ values. The predicted Dw was 10% smaller with TIP3P
model and 7% smaller for TIP3P model. This observation was expected, because
it follows the same trend as predictions of the bulk self-diffusion coefficient of
water, Dw.
4.6.3. Long range electrostatic interaction treatment
We used Particle Mesh Ewald (PME) method. This method is an
approximation to Ewald method, improving the scaling of calculation time with
system size, N, from N2 to N.log(N). The method is essential to implement,
because electrostatic forces are of long range nature, and the interaction of
species with those in neighboring cells must be accounted for.
The minimum recommended short range cut-off for calculation to be used
in the PME method is 0.9 nm [81]. For a sample case we increased the short range
cut-off up to 1.9 nm. The variation of Γ was small, and Dw was not affected at all.
Therefore, we did the calculation with the recommended 0.9 nm. It must be
noted that increasing the PME short range cut-off is accompanied with drastic
decrease in the simulation speed. This dependence was the motivation for
choosing the minimum acceptable PME short range cut-off.

78
Figure 4.27 Sensitivity of simulation result and calculation speed to PME
short range force calculation
4.6.4. Temperature
In this study we considered the constant temperature of 300 K. We
performed sensitivity analysis on temperature, and the effect on Γ was found to
be negligible. This observation is in line with the dominance of electrostatic over
thermal effects in the system. The results are shown in Figure 4.28.
Furthermore, we investigated the effect of changing the employed
thermostat algorithm from Berendsen to Nose-Hoover. For the same test system,
no significant deviation from the original Γ value was observed.
0
2
4
6
8
10
12
0
0.2
0.4
0.6
0.8
1
0 0.5 1 1.5 2
Sim
ula
tio
n s
pe
ed
(n
s/d
ay)
Γ
PME short range (nm)

79
Figure 4.28. Temperature dependence of channel effectiveness for a channel with
θ=45°, dc=20 Å, dpp=30 Å, σ=-14 μC/cm2
4.6.5. Nanochannel material
In the classical MD method, the metal is identified by four parameters:
crystalline structure, lattice constant and the two Lennard-Jones fitting
parameters. We simulated sample channels made of copper and gold, which
have FCC structure. The Lennard-Jones interaction parameters are taken from
Ref. [99], and are reported in Table 4.4. The form of Lennard-Jones potential used
is introduced in equation 3.14.
Table 4.4. Physical properties of Pt, Cu, and Au for MD set-up
Pair Lattice const. (Å) C6 (kJ.nm6.mol-1) C12 (kJ.nm12.mol-1)
Pt-Pt 3.92 1.51*10-2 2.98*10-6
Cu-Cu 3.61 2.23*10-2 3.11*10-6
Au-Au 4.08 5.07*10-2 1.46*10-5
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
280 290 300 325 350 365
Effectiveness 0.82 0.8 0.82 0.83 0.81 0.82
0.82 0.8 0.82 0.83 0.81 0.82
Γ
Temperature (K)

80
Sample calculations were done on channels with θ=45°, dpp=30 Å and ɸM=-
50 mV, and varying dc={20,50, 100} Å. The results are presented in Figure 4.29.
Figure 4.29 Sensitivity of Γ results to nanochannel material
The results presented in Figure 4.29 could be presented in terms of the
metal lattice constant, suggesting direct relation between the effectiveness factor
and the packing of metal atoms, as illustrated in Figure 4.30.
Figure 4.30. Inverse relation of channel effectiveness with the metal lattice
constant
0.5
0.6
0.7
0.8
0.9
1
0 20 40 60 80 100 120
Γ
dc(Å)
Pt
Au
Cu
0.5
0.6
0.7
0.8
0.9
1
3.5 3.6 3.7 3.8 3.9 4 4.1 4.2
Γ
Lattice constant (Å)
20
50
100
dc(Å)
Au Pt
Cu

81
Figure 4.29 and Figure 4.30 suggest that there is a slight dependence of Γ
on the nanochannel material. The observation indicates dominance of Coulomb
interaction over the Lennard-Jones interactions.
4.6.6. Pt-Pt Lennard-Jones interaction parameters
In this work, the Lennard-Jones interaction parameters for Pt-Pt pair were
adapted from a similar system [84]. However, to test the sensitivity of results to
the choice of Pt-Pt parameters, we hypothetically changed the parameters by an
order of magnitude and calculated the effectiveness factor. For this purpose,
sample calculations were done for a channel with θ=45°, dc=20 Å, dpp=30 Å and
ɸM=-50 mV with the Lennard-Jones parameters listed in Table 4.5.
Table 4.5. Pt-Pt Lennard-Jones parameters for sensitivity analysis
Pair C6 (kJ.nm6.mol-1) C12 (kJ.nm12.mol-1) Effectiveness
Pt-Pt (original) 1.51*10-2 2.98*10-6 0.813
Test 1 1.51*10-3 2.98*10-6 0.698
Test 2 1.51*10-1 2.98*10-6 0.890
Test 3 1.51*10-2 2.98*10-7 0.976
Test 4 1.51*10-2 2.98*10-5 0.624
The result confirms the direct relationship of Γ with the attractive branch
(c6) and inverse relationship with the repulsive branch (c12) of the Lennard-Jones
potential. Figure 4.31 shows that the effect of the repulsive branch is more
pronounced (~30% change) than the attractive branch (~20% change).

82
Figure 4.31. Sensitivity of nanochannel effectiveness to Pt-Pt interaction
parameters
The sensitivity of Dw to the Pt-Pt interaction parameters was also
investigated. We observed an inverse relation of Dw with the attractive coefficient
c6, implying that more hydrophilic confinement reduces water mobility. The
effect of the c12 repulsive parameter was not significant, due to its short range
nature.
Figure 4.32. Sensitivity of water self-diffusion coefficient to Pt-Pt interaction
parameters
0.698
0.813 0.89 0.976
0.813
0.624
0
0.2
0.4
0.6
0.8
1
1.E-7 1.E-6 1.E-5 1.E-4 1.E-3 1.E-2 1.E-1 1.E+0
Γ
c-value
c6
c12
1.51
1.224
0.797
1.045 1.224 1.148
00.20.40.60.8
11.21.41.61.8
1.0E-7 1.0E-5 1.0E-3 1.0E-1
c-value
c6
c12
(
𝑚
)

83
4.7. Insight to the effect of nanostructuring on ORR kinetics
The present research investigated the effect of metal nanostructure on the
local reaction conditions in a CL nanochannel. The implicit assumption for this
research is that the catalyst nanostructure did not affect the intrinsic
electrocatalytic activity of the catalyst. This assumption enabled comparison of
nanochannels based on the electrostatic effectiveness factor. The intrinsic reaction
rate is represented as the exchange current density j0 in the Butler-Volmer
equation. However, there is evidence showing that the metal basal plane
structure affects the intrinsic reaction rate. Therefore, it is essential to review the
impact of surface structure on the kinetics of ORR, to predict the effect of
nanostructuring on the overall catalyst effectiveness.
Smoluchowsky [100] showed that the local work function of metals is not
homogeneous on a rough metal surface. The local smoothing of electron density
gives rise to lowering of the work function at the step sites of the metal surface,
which enhances the electrocatalytic activity. The work function was later
correlated with the potential of zero (total) charge of the metal [101]: it was
shown that increasing the step density of a metal has an inverse effect on the
metal PZC for different surfaces as well as nanoparticles [87], [95], [102–105]. The
UTCL pore model of Chan and Eikerling [51] predicted a direct relation between
the value of Pt PZC and catalyst effectiveness factor at a given potential.

84
5. Conclusions
5.1. Results Summary
In this work, we simulated ionomer-free catalyst layer channels with zig-
zag patterning of platinum walls. We investigated the effects of channel
nanostructuring on the equilibrium hydronium ion and water distribution, and
the electrostatic effectiveness factor of Pt utilization in the nanochannel. The
major findings are as follows.
Different structuring angles change the step fraction on the
nanostructured channel. We observed that protons concentrate away from the
steps on each facet. Therefore, the electrostatic effectiveness factor is inversely
related to the step fraction on each facet. In terms of structuring angle, we
observed that smooth facets simulated with θ=0°, representing Pt(100) surface,
and θ=45°, representing Pt(110) surface, exhibited higher values of the
effectiveness factor.
The electrostatic effectiveness is inversely related to the channel opening.
The observation is linked to the reduction of the overall hydronium ion density
in the channel. At constant surface charge density, increasing the channel
opening causes dilution of hydronium ions in water, and consequently lowers
their concentration at the Helmholtz plane.

85
Increasing the groove width does not change the step fraction of facets,
but reduces overall proton concentration in the channel. Therefore, we obtained
an inverse relationship between groove width and effectiveness factor.
Compared to the channel opening effect, the effect of groove width is less
pronounced.
The effectiveness factor of the channel is directly related with the metal
surface charge density. Changing the surface charge density to more negative
values strengthens the attraction between hydronium ions and negatively
charged walls, hence increasing the effectiveness factor. We presented our results
based on surface charge density rather than metal potential, because finding the
exact relation between metal surface charge density and potential requires a
consistent model of metal-solution interface, including the effect of surface oxide
formation; at this time, the understanding of this relation is still sketchy.
However, the investigated potential should be smaller than the platinum
potential of zero charge.
We investigated the effect of nanochannel structuring on the structure and
self-diffusion coefficient of water inside the channel. The correlation was
described best by distinguishing the volume of water on the surface and in the
bulk-like phase. The nanochannel walls hinder the water dynamics strongly at
their surface, and have weaker effect on the bulk-like water phase. Surface water
has self-diffusion coefficient almost three times smaller than bulk-like water.
Water motion is hindered at the interface due to charge-dipole type interaction of
the charged channel walls with water. The bulk-like water phase exhibits a self-

86
diffusion coefficient smaller than that of bulk water, due to the confinement
effect.
The self-diffusion coefficient of water affects oxygen diffusivity in the
channel; however, the effect on the overall electrocatalysis in the cathode is
estimated to be less important than the electrostatic effect: oxygen diffusion
resistance is not the controlling step of ORR in ultrathin ionomer-free catalyst
layers [51].
5.2. Suggestions for future work
It is known that a variation in the step fraction on the catalyst surface
shifts the potential of zero charge [104]. Therefore, stepped surfaces would have
different charging behaviour than smooth surfaces at equal metal potential
values. This effect could be integrated to the prediction of metal surface charge
distribution (described in section 3.3). Considering this effect would lead to a
better understanding of the effect of structuring on the CCL performance.
In the current approach, metal surface charge distribution is fixed. It does
not change during the course of a simulation. However, in reality the
arrangement of hydronium ions leads to a redistribution of electric charges at the
interface. Since accounting for mobile surface charges is not already
implemented in the MD code, addressing this problem would be an
improvement of the classical MD code. This would mean considering fictitious
particles to represent electric charges on the metal surface, and allow those
particles to move on the metal surface.

87
We investigated a monoatomic catalyst, and the sensitivity analysis on the
metal material showed that the effect of material manifests itself by its lattice
constant. However, our model does not yet predict the interfacial hydronium ion
distribution for a metallic catalyst deposited on a conductive substrate. Such
systems are the prevailing fuel cell catalysts [38]. For such systems, we must
know the spatial distribution of the potential of zero charge at catalyst, to be able
to predict the surface charge distribution. The design space of the problem can be
then changed to a smooth substrate with different controlled configurations of
catalyst nanoparticles deposited on the surface. This model could be considered
as a piece of the multiscale modeling roadmap of fuel cell catalyst modeling, for
which DFT simulations at the nanoparticle level are conducted in separate works
in our group [22].
The other improvement to the model can come from a more accurate
description of proton and water at the interface. Especially, the two or multi-state
EVB model [70], [106] can be used to provide a more accurate insight on the
proton and water structure at the interface. It must be noted, however, that EVB
is a computationally expensive method, therefore the choice of simulated
systems must be carefully refined.
On the experimental side, the results of this work indicated that smaller
channel opening results in enhanced effectiveness factor of Pt utilization. In a
benchmark design such as 3M NSTF catalysts, this means recommending small
feature sizes of support whiskers.

88
References
[1] W. Moomaw, F. Yamba, M. Kamimoto, L. Maurice, J. Nyboer, K. Urama,
and T. Weir, “Introduction,” in IPCC Special Report on Renewable Energy
Sources and Climate Change Mitigation, O. Edenhofer, R. Pichs-Madruga, Y.
Sokona, K. Seyboth, P. Matschoss, S. Kadner, T. Zwickel, P. Eikemeier, G.
Hansen, S. Schloemer, and C. von Stechow, Eds. Cambridge, New York: ,
2011.
[2] B. D. James, J. A. Kalinoski, and K. N. Baum, Mass Production Cost
Estimation for Direct H 2 PEM Fuel Cell Systems for Automotive Applications :
2009 Update, US department of energy report. 2010.
[3] B. D. James, K. Baum, A. B. Spisak, and W. G. Colella, “Mass-Production
Cost Estimation for Automotive Fuel Cell Systems, US department of
energy report,” 2013.
[4] S. P. S. Badwal and K. Foger, “Solid Oxide Electrolyte Fuel Cell Review,”
Ceramics International, vol. 22, pp. 257–265, 1996.
[5] N. Shaigan, W. Qu, D. G. Ivey, and W. Chen, “A review of recent progress
in coatings, surface modifications and alloy developments for solid oxide
fuel cell ferritic stainless steel interconnects,” Journal of Power Sources, vol.
195, no. 6, pp. 1529–1542, Mar. 2010.
[6] E. Antolini, “The stability of LiAlO2 powders and electrolyte matrices in
molten carbonate fuel cell environment,” Ceramics International, pp. 1–16,
Nov. 2012.
[7] E. Wachsman and K. T. Lee, “Lowering the Temperature of Solid Oxide
Fuel Cells,” Science, vol. 18, pp. 935–939, 2011.
[8] J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, and H.
Jonsson, “Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell
Cathode,” Journal of Physical Chemistry C, vol. 108, pp. 17886–17892, 2004.

89
[9] A. Veziroglu and R. Macario, “Fuel cell vehicles: State of the art with
economic and environmental concerns,” International Journal of Hydrogen
Energy, vol. 36, no. 1, pp. 25–43, Jan. 2011.
[10] “Hydrogen and Fuel Cell Activities, Progress, and Plans; Report to the
Congress, US department of energy,” 2009.
[11] L. Carrette, K. a. Friedrich, and U. Stimming, “Fuel Cells - Fundamentals
and Applications,” Fuel Cells, vol. 1, no. 1, pp. 5–39, May 2001.
[12] I. D. Raistrick, “Electrode assembly for use in a solid polymer electrolyte
fuel cell,” U.S. Patent 4876115,1989.
[13] M. S. Wilson, J. A. Valerio, and S. S. Gottesfeld, “Low Platinum Loading
Electrodes for Polymer Electrolyte Fuel Cells Fabricated using
Thermoplastic Ionomers,” Electrochimica Acta, vol. 40, no. 3, pp. 355–363,
1995.
[14] T. R. Ralph, J. E. Keating, S. A. Campbell, D. P. Wilkinson, M. Davis, J. St-
Pierre, and M. C. Johnson, “Low Cost Electrodes for Proton Exchange
Membrane Fuel Cells,” Journal of The Electrochemical Society, vol. 144, no. 11,
pp. 3845–3857, 1997.
[15] D. Papageorgopoulos, “Fuel cells sub-program overview, DOE hydrogen
and fuel cell program annual progress report,” 2011.
[16] P. Yu, M. Pemberton, and P. Plasse, “PtCo/C cathode catalyst for improved
durability in PEMFCs,” Journal of Power Sources, vol. 144, no. 1, pp. 11–20,
Jun. 2005.
[17] H. a. Gasteiger, S. S. Kocha, B. Sompalli, and F. T. Wagner, “Activity
benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen
reduction catalysts for PEMFCs,” Applied Catalysis B: Environmental, vol. 56,
no. 1–2, pp. 9–35, Mar. 2005.
[18] J. X. Wang, H. Inada, L. Wu, Y. Zhu, Y. Choi, P. Liu, W.-P. Zhou, and R. R.
Adzic, “Oxygen reduction on well-defined core-shell nanocatalysts:
particle size, facet, and Pt shell thickness effects.,” Journal of the American
Chemical Society, vol. 131, no. 47, pp. 17298–302, Dec. 2009.

90
[19] A. Bauer, D. P. Wilkinson, E. L. Gyenge, D. Bizzotto, and S. Ye, “Liquid
Crystalline Phase Templated Platinum Catalyst for Oxygen Reduction,”
Journal of The Electrochemical Society, vol. 156, no. 10, p. B1169, 2009.
[20] L. Zhang, L. Wang, C. M. B. Holt, T. Navessin, K. Malek, M. H. Eikerling,
and D. Mitlin, “Oxygen Reduction Reaction Activity and Electrochemical
Stability of Thin-Film Bilayer Systems of Platinum on Niobium Oxide,” The
Journal of Physical Chemistry C, vol. 114, no. 39, pp. 16463–16474, Oct. 2010.
[21] L. Zhang, L. Wang, C. M. B. Holt, B. Zahiri, Z. Li, K. Malek, T. Navessin, M.
H. Eikerling, and D. Mitlin, “Highly corrosion resistant platinum–niobium
oxide–carbon nanotube electrodes for the oxygen reduction in PEM fuel
cells,” Energy & Environmental Science, vol. 5, no. 3, p. 6156, 2012.
[22] L. Wang, “Computational modeling of supported catalyst systems:
Investigation of free catalyst nanoparticles and support effects,” Simon
Fraser University, 2011.
[23] B. Wang, “Recent development of non-platinum catalysts for oxygen
reduction reaction,” Journal of Power Sources, vol. 152, pp. 1–15, Dec. 2005.
[24] M. Lee, M. Uchida, D. a. Tryk, H. Uchida, and M. Watanabe, “The
effectiveness of platinum/carbon electrocatalysts: Dependence on catalyst
layer thickness and Pt alloy catalytic effects,” Electrochimica Acta, vol. 56,
no. 13, pp. 4783–4790, May 2011.
[25] M. Lee, M. Uchida, H. Yano, D. a. Tryk, H. Uchida, and M. Watanabe,
“New evaluation method for the effectiveness of platinum/carbon
electrocatalysts under operating conditions,” Electrochimica Acta, vol. 55,
no. 28, pp. 8504–8512, Dec. 2010.
[26] S. G. Rinaldo, J. Stumper, and M. Eikerling, “Physical Theory of Platinum
Nanoparticle Dissolution in Polymer Electrolyte Fuel Cells,” The Journal of
Physical Chemistry C, vol. 114, pp. 5773–5785, 2010.
[27] S. G. Rinaldo, W. Lee, J. Stumper, and M. Eikerling, “Model- and Theory-
Based Evaluation of Pt Dissolution for Supported Pt Nanoparticle
Distributions under Potential Cycling,” Electrochemical and Solid-State
Letters, vol. 14, no. 5, p. B47, 2011.

91
[28] S. G. Rinaldo, W. Lee, J. Stumper, and M. Eikerling, “Nonmonotonic
dynamics in Lifshitz-Slyozov-Wagner theory: Ostwald ripening in
nanoparticle catalysts,” Physical Review E, vol. 86, no. 4, p. 041601, Oct.
2012.
[29] M. Wang, F. Xu, H. Sun, Q. Liu, K. Artyushkova, E. a. Stach, and J. Xie,
“Nanoscale graphite-supported Pt catalysts for oxygen reduction reactions
in fuel cells,” Electrochimica Acta, vol. 56, no. 5, pp. 2566–2573, Feb. 2011.
[30] D. Song, Q. Wang, Z. Liu, T. Navessin, M. Eikerling, and S. Holdcroft,
“Numerical optimization study of the catalyst layer of PEM fuel cell
cathode,” Journal of Power Sources, vol. 126, no. 1–2, pp. 104–111, Feb. 2004.
[31] D. Song, Q. Wang, Z. Liu, M. Eikerling, Z. Xie, T. Navessin, and S.
Holdcroft, “A method for optimizing distributions of Nafion and Pt in
cathode catalyst layers of PEM fuel cells,” Electrochimica Acta, vol. 50, no.
16–17, pp. 3347–3358, May 2005.
[32] Z. Xia, Q. Wang, M. Eikerling, and Z. Liu, “Effectiveness factor of Pt
utilization in cathode catalyst layer of polymer electrolyte fuel cells,” vol.
667, pp. 657–667, 2008.
[33] M. H. Eikerling, K. Malek, and Q. Wang, “Catalyst Layer Modeling :
Structure , Properties and Performance,” in PEM Fuel Cell Electrocatalysts
and Catalyst Layers: Fundamentals and Applications, no. i, J. Zhang, Ed.
Springer-Verlag, 2008, pp. 381–446.
[34] A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon, and W. van Schalkwijk,
“Nanostructured materials for advanced energy conversion and storage
devices.,” Nature materials, vol. 4, no. 5, pp. 366–77, May 2005.
[35] M. H. Eikerling and K. Malek, “Physical Modeling of Materials for PEFCs:
A Balancing Act of Water and Complex Morphologies,” in Proton Exchange
Membrane Fuel Cells: Materials Properties and Performance, D. P. Wilkinson, J.
Zhang, and R. Hui, Eds. Boca Raton, FL: CRC Press, 2009.
[36] M. K. Debe, A. K. Schmoeckel, G. D. Vernstrom, and R. Atanasoski, “High
voltage stability of nanostructured thin film catalysts for PEM fuel cells,”
Journal of Power Sources, vol. 161, no. 2, pp. 1002–1011, Oct. 2006.

92
[37] A. Bonakdarpour, K. Stevens, G. D. Vernstrom, R. Atanasoski, A. K.
Schmoeckel, M. K. Debe, and J. R. Dahn, “Oxygen reduction activity of Pt
and Pt–Mn–Co electrocatalysts sputtered on nano-structured thin film
support,” Electrochimica Acta, vol. 53, no. 2, pp. 688–694, Dec. 2007.
[38] D. van der Vliet, C. Wang, M. Debe, R. Atanasoski, N. M. Markovic, and V.
R. Stamenkovic, “Platinum-alloy nanostructured thin film catalysts for the
oxygen reduction reaction,” Electrochimica Acta, vol. 56, no. 24, pp. 8695–
8699, Oct. 2011.
[39] R. O’Hayre, S.-J. Lee, S.-W. Cha, and F. . Prinz, “A sharp peak in the
performance of sputtered platinum fuel cells at ultra-low platinum
loading,” Journal of Power Sources, vol. 109, no. 2, pp. 483–493, Jul. 2002.
[40] D. Gruber, N. Ponath, J. Müller, and F. Lindstaedt, “Sputter-deposited
ultra-low catalyst loadings for PEM fuel cells,” Journal of Power Sources, vol.
150, pp. 67–72, Oct. 2005.
[41] M. K. Debe, A. K. Schmoeckel, and R. Atanasoski, “Durability aspects of
nanostructured thin film catalysts for PEM fuel cells,” ECS Transactions,
vol. 1, no. 8, pp. 51–66, 2006.
[42] D. a. Stevens, J. M. Rouleau, R. E. Mar, a. Bonakdarpour, R. T. Atanasoski,
a. K. Schmoeckel, M. K. Debe, and J. R. Dahn, “Characterization and
PEMFC Testing of Pt[sub 1−x]M[sub x] (M=Ru,Mo,Co,Ta,Au,Sn) Anode
Electrocatalyst Composition Spreads,” Journal of The Electrochemical Society,
vol. 154, no. 6, p. B566, 2007.
[43] P. K. Sinha, W. Gu, A. Kongkanand, and E. Thompson, “Performance of
Nano Structured Thin Film (NSTF) Electrodes under Partially-Humidified
Conditions,” Journal of The Electrochemical Society, vol. 158, no. 7, p. B831,
2011.
[44] J. Erlebacher and J. Snyder, “Dealloyed Nanoporous Metals for PEM Fuel
Cell Catalysis,” ECS Transactions, vol. 25, no. 1, pp. 603–612, 2009.

93
[45] J. Snyder, I. McCue, K. Livi, and J. Erlebacher,
“Structure/processing/properties relationships in nanoporous
nanoparticles as applied to catalysis of the cathodic oxygen reduction
reaction.,” Journal of the American Chemical Society, vol. 134, no. 20, pp.
8633–45, May 2012.
[46] M. Arenz, K. J. J. Mayrhofer, V. Stamenkovic, B. B. Blizanac, T. Tomoyuki,
P. N. Ross, and N. M. Markovic, “The effect of the particle size on the
kinetics of CO electrooxidation on high surface area Pt catalysts.,” Journal of
the American Chemical Society, vol. 127, no. 18, pp. 6819–29, May 2005.
[47] M. K. Debe, “Nanostructured Thin Film Electrocatalysts for PEM Fuel Cells
- A Tutorial on the Fundamental Characteristics and Practical Properties of
NSTF Catalysts,” ECS Transactions, vol. 45, no. 2, pp. 47–68, Apr. 2012.
[48] A. J. Steinbach, M. K. Debe, M. J. Pejsa, D. M. Peppin, A. T. Haug, M. J.
Kurkowski, and S. M. Maier-Hendricks, “Influence of Anode GDL on
PEMFC Ultra-Thin Electrode Water Management at Low Temperatures,”
vol. 41, no. 1, pp. 449–457, 2011.
[49] M. K. Debe, “Electrocatalyst approaches and challenges for automotive
fuel cells.,” Nature, vol. 486, no. 7401, pp. 43–51, Jun. 2012.
[50] Q. Wang, M. Eikerling, D. Song, and Z.-S. Liu, “Modeling of Ultrathin
Two-Phase Catalyst Layers in PEFCs,” Journal of The Electrochemical Society,
vol. 154, no. 6, p. F95, 2007.
[51] K. Chan and M. Eikerling, “A Pore-Scale Model of Oxygen Reduction in
Ionomer-Free Catalyst Layers of PEFCs,” Journal of The Electrochemical
Society, vol. 158, no. 1, p. B18, 2011.
[52] K. Chan and M. Eikerling, “Impedance Model of Oxygen Reduction in
Water-Flooded Pores of Ionomer-Free PEFC Catalyst Layers,” Journal of The
Electrochemical Society, vol. 159, no. 2, p. B155, 2012.
[53] M. Schmuck and P. Berg, “Homogenization of a Catalyst Layer Model for
Periodically Distributed Pore Geometries in PEM Fuel Cells,” Applied
Mathematics Research eXpress, Jul. 2012.

94
[54] R. B. Bird, W. E. Stewart, and E. N. Lightfoot, Transport Phenomena, 2nd ed.
John Wiley and Sons, Inc., 2001.
[55] M. H. Hong, K. H. Kim, J. Bae, and W. Jhe, “Scanning nanolithography
using a material-filled nanopipette,” Applied Physics Letters, vol. 77, no. 16,
2000.
[56] J. Kong, N. Franklin, C. Zhou, M. Chapline, S. Peng, K. Cho, and H. Dai,
“Nanotube molecular wires as chemical sensors,” Science (New York, N.Y.),
vol. 287, no. 5453, pp. 622–5, Jan. 2000.
[57] S. Jakobtorweihen *, N. Hansen, and F. J. Keil, “Molecular simulation of
alkene adsorption in zeolites,” Molecular Physics, vol. 103, no. 4, pp. 471–
489, Feb. 2005.
[58] a. M. P. McDonnell, D. Beving, a. Wang, W. Chen, and Y. Yan,
“Hydrophilic and Antimicrobial Zeolite Coatings for Gravity-Independent
Water Separation,” Advanced Functional Materials, vol. 15, no. 2, pp. 336–
340, Feb. 2005.
[59] R. Paul and S. J. Paddison, “Structure and Dielectric Saturation of Water in
Hydrated Polymer Electrolyte Membranes: Inclusion of the Internal Field
Energy,” The Journal of Physical Chemistry B, vol. 108, no. 35, pp. 13231–
13241, Sep. 2004.
[60] P. Berg and J. Findlay, “Analytical solution of the Poisson-Nernst-Planck-
Stokes equations in a cylindrical channel,” Proceedings of the Royal Society A:
Mathematical, Physical and Engineering Sciences, vol. 467, no. 2135, pp. 3157–
3169, Jun. 2011.
[61] J. D. Bernal and R. H. Fowler, “A Theory of Water and Ionic Solution, with
Particular Reference to Hydrogen and Hydroxyl Ions,” The Journal of
Chemical Physics, vol. 1, no. 8, pp. 515–548, 1933.
[62] A. Rahman and F. H. Stillinger, “Molecular Dynamics Study of Liquid
Water,” The Journal of Chemical Physics, vol. 55, no. 7, p. 3336, 1971.
[63] W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L.
Klein, “Comparison of simple potential functions for simulating liquid
water,” The Journal of Chemical Physics, vol. 79, no. 2, p. 926, 1983.

95
[64] H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, and J. Hermans,
“Interaction models for water in relation to protein hydration,” pp. 331–
338, 1981.
[65] A. K. Soper and M. G. Phillips, “A new determination of the structure of
water at 25 C,” Chemical Physics, vol. 107, no. 1, pp. 47–60, 1986.
[66] R. Mills, “Self-diffusion in normal and heavy water in the range 1-45,” The
Journal of Physical Chemistry, vol. 77, no. 5, pp. 685–688, 1973.
[67] H. J. C. Berendsen, J. R. Grigera, and T. P. Straatsma, “The Missing Term in
Effective Pair Potentialst,” pp. 6269–6271, 1987.
[68] W. L. Jorgensen and J. D. Madura, “Temperature and size dependence for
Monte Carlo simulations of TIP4P water,” Molecular Physics, vol. 56, no. 6,
1985.
[69] M. W. Mahoney and W. L. Jorgensen, “A five-site model for liquid water
and the reproduction of the density anomaly by rigid, nonpolarizable
potential functions,” The Journal of Chemical Physics, vol. 112, no. 20, p. 8910,
2000.
[70] A. Warshel and R. M. Weiss, “An empirical valence bond approach for
comparing reactions in solutions and in enzymes,” Journal of the American
Chemical Society, vol. 102, no. 20, pp. 6218–6226, Sep. 1980.
[71] S. Walbran and a. a. Kornyshev, “Proton transport in polarizable water,”
The Journal of Chemical Physics, vol. 114, no. 22, p. 10039, 2001.
[72] M. Eikerling, A. Kornyshev, and E. Spohr, “Proton-Conducting Polymer
Electrolyte Membranes: Water and Structure in Charge,” in Fuel Cells I, no.
April, G. G. Scherer, Ed. Berlin Heidelberg: Springer-Verlag, 2008, pp. 15–
54.
[73] F. Wilhelm, W. Schmickler, and E. Spohr, “Proton transfer to charged
platinum electrodes. A molecular dynamics trajectory study.,” Journal of
physics: Condensed matter, vol. 22, no. 17, p. 175001, May 2010.
[74] M. Eikerling, “Water Management in Cathode Catalyst Layers of PEM Fuel
Cells,” Journal of The Electrochemical Society, vol. 153, no. 3, p. E58, 2006.

96
[75] E. Spohr, “Molecular dynamics simulation of a water/metal interface,” vol.
123, no. 3, pp. 218–221, 1986.
[76] A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and
Applications, 2nd ed. New York: John Wiley and Sons, Inc., 2000.
[77] M. J. Weaver, “Potentials of Zero Charge for Platinum ( 111 ) -Aqueous
Interfaces: A Combined Assessment from In-Situ and Ultrahigh-Vacuum
Measurements,” Langmuir, vol. 14, pp. 3932–3936, 1998.
[78] R. Gómez, V. Climent, J. M. Feliu, and M. J. Weaver, “Dependence of the
Potential of Zero Charge of Stepped Platinum (111) Electrodes on the
Oriented Step-Edge Density: Electrochemical Implications and
Comparison with Work Function Behavior,” The Journal of Physical
Chemistry B, vol. 104, no. 3, pp. 597–605, Jan. 2000.
[79] K. Letchworth-Weaver and T. a. Arias, “Joint density functional theory of
the electrode-electrolyte interface: Application to fixed electrode potentials,
interfacial capacitances, and potentials of zero charge,” Physical Review B,
vol. 86, no. 7, p. 075140, Aug. 2012.
[80] M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids. Oxford:
Oxford Science Publications, 1996.
[81] B. Hess, C. Kutzner, D. van der Spoel, and E. Lindahl, “GROMACS
4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular
Simulation,” Journal of Chemical Theory and Computation, vol. 4, no. 3, pp.
435–447, Mar. 2008.
[82] E. Cances, M. Defranceschi, W. Kutzelnigg, C. Le Bris, and Y. Maday,
“Computational Quantum Chemistry : A Primer,” in Handbook of numerical
analysis, vol. X, 2003, pp. 3–270.
[83] B. D. Todd and R. . Lynden-Bell, “Surface and bulk properties of metals
modelled with Sutton-Chen potentials,” Surface Science, vol. 281, pp. 191–
206, 1993.

97
[84] T. Mashio, K. Malek, M. Eikerling, A. Ohma, H. Kanesaka, and K.
Shinohara, “Molecular Dynamics Study of Ionomer and Water Adsorption
at Carbon Support Materials,” The Journal of Physical Chemistry C, vol. 114,
no. 32, pp. 13739–13745, Aug. 2010.
[85] S. S. Jang, V. Molinero, C. Tahir, and W. A. G. Iii, “Nanophase-Segregation
and Transport in Nafion 117 from Molecular Dynamics Simulations : Effect
of Monomeric Sequence,” no. i, pp. 3149–3157, 2004.
[86] P. Mark and L. Nilsson, “Structure and Dynamics of the TIP3P, SPC, and
SPC/E Water Models at 298 K,” The Journal of Physical Chemistry A, vol. 105,
no. 43, pp. 9954–9960, Nov. 2001.
[87] J. Solla-Gullom, F. J. Vidal-Iglesias, A. Lopez-Cudero, E. Garnier, J. M.
Feliu, and A. Aldaz, “Shape-dependent electrocatalysis: methanol and
formic acid electrooxidation on preferentially oriented Pt nanoparticles,”
Physical chemistry chemical physics : PCCP, vol. 10, no. 25, pp. 3689–3698, Jul.
2008.
[88] P. K. Jha, R. Sknepnek, G. I. Guerrero-Garcia, and M. O. de la Cruz, “A
Graphics Processing Unit Implementation of Coulomb Interaction in
Molecular Dynamics,” Journal of Chemical Theory and Computation, vol. 6,
pp. 3058–3065, 2010.
[89] T. Darden, D. York, and L. Pedersen, “Particle mesh Ewald: An N⋅log(N)
method for Ewald sums in large systems,” The Journal of Chemical Physics,
vol. 98, no. 12, p. 10089, 1993.
[90] H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, a. DiNola, and J.
R. Haak, “Molecular dynamics with coupling to an external bath,” The
Journal of Chemical Physics, vol. 81, no. 8, p. 3684, 1984.
[91] W. Humphrey, A. Dalke, and K. Schulten, “VMD - Visual Molecular
Dynamics,” J. Molec. Graphics, vol. 14, pp. 33–38, 1996.
[92] R. Sayle and E. Milner-White, “RasMol: Biomolecular graphics for all,”
Trends in biochemical sciences, 1995.

98
[93] S. Engstroem and H. Wennerstrom, “Ion condensation on planar surfaces.
A solution of the Poisson-Boltzmann equation for two parallel charged
plates,” The Journal of Physical Chemistry, vol. 82, no. 25, pp. 2711–2714,
1978.
[94] D. Andelman, “Introduction to electrostatics in soft and biological matter,”
in Soft Condensed Matter Physics in Molecular And Cell Biology, Taylor &
Francis, 2006.
[95] V. Climent, R. Gomez, and J. M. Feliu, “Effect of increasing amount of
steps on the potential of zero total charge of Pt (111) electrodes,” vol. 45,
pp. 629–637, 1999.
[96] K. Besocke, B. Krahl-urban, and H. Wagner, “Dipole moments Associated
with Edge Atoms; a Comparative Study on Stepped Pt, Au and W
Surfaces,” Surface Science, vol. 68, pp. 39–46, 1977.
[97] H. Ishida and A. Liebsch, “Calculation of the electronic structure of
stepped metal surfaces,” vol. 46, no. 11, pp. 7153–7156, 1992.
[98] N. P. Lebedeva, M. T. M. Koper, J. M. Feliu, and R. a. van Santen, “Role of
Crystalline Defects in Electrocatalysis: Mechanism and Kinetics of CO
Adlayer Oxidation on Stepped Platinum Electrodes,” The Journal of Physical
Chemistry B, vol. 106, no. 50, pp. 12938–12947, Dec. 2002.
[99] P. M. Agrawal, B. M. Rice, and D. L. Thompson, “Predicting trends in rate
parameters for self-diffusion on FCC metal surfaces,” Surface Science, vol.
515, no. 1, pp. 21–35, Aug. 2002.
[100] R. Smoluchowsky, “Anisotropy of the electronic work function of metals,”
Physical Review, vol. 60, pp. 661–674, 1941.
[101] W. Schmickler and E. Santos, Interfacial Electrochemistry, 2nd ed. New York:
Springer, 2010.
[102] J. Clavilier, J. M. Orts, R. Gomez, J. M. Feliu, and A. Aldaz, “Comparison of
electrosorption at activated polycrystalline and Pt (531) kinked platinum
electrodes: surface voltammetry and charge displacement on potentiostatic
CO adsorption,” vol. 404, pp. 281–289, 1996.

99
[103] K. Domke, E. Herrero, A. Rodes, and J. M. Feliu, “Determination of the
potentials of zero total charge of Pt(100) stepped surfaces in the [] zone.
Effect of the step density and anion adsorption,” Journal of Electroanalytical
Chemistry, vol. 552, pp. 115–128, Jul. 2003.
[104] Q.-S. Chen, J. Solla-Gullón, S.-G. Sun, and J. M. Feliu, “The potential of zero
total charge of Pt nanoparticles and polycrystalline electrodes with
different surface structure: The role of anion adsorption in fundamental
electrocatalysis,” Electrochimica Acta, vol. 55, no. 27, pp. 7982–7994, Nov.
2010.
[105] A. Bjoerling, E. Herrero, and J. M. Feliu, “Electrochemical Oxidation of Pt
(111) Vicinal Surfaces: Effects of Surface Structure and Specific Anion
Adsorption,” The Journal of Physical Chemistry C, vol. 115, pp. 15509–15515,
2011.
[106] H. Chen, Y. Wu, and G. a Voth, “Proton transport behavior through the
influenza A M2 channel: insights from molecular simulation.,” Biophysical
journal, vol. 93, no. 10, pp. 3470–9, Nov. 2007.














![Nanoscale Resistive Switching in ...strukov/papers/2014/AdvFuncMat2014.pdf · [ 5b , 8 ] Nanoscale electromechanical phenomena in functional oxide layers, ranging from fl exoelectricity](https://static.fdocuments.in/doc/165x107/5fc39ae3a436ea4b2b10ff1c/nanoscale-resistive-switching-in-strukovpapers2014advfuncmat2014pdf-.jpg)



