
Nanoporous gold as an active low temperature catalyst ... · PDF fileNanoporous gold as an...
7
Nanoporous gold as an active low temperature catalyst toward CO oxidation in hydrogen-rich stream Dongwei Li 1 , Ye Zhu 1 , Hui Wang 1 & Yi Ding 1,2 1 Center for Advanced Energy Materials & Technology Research (AEMT), and School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China, 2 Shandong Applied Research Center for Gold Technology (Au-SDARC), Yantai 264005, China. Preferential CO oxidation (PROX) was investigated by using dealloyed nanoporous gold (NPG) catalyst under ambient conditions. Systematic investigations were carried out to characterize its catalytic performance by varying reaction parameters such as temperature and co-existence of CO 2 and H 2 O, which revealed that NPG was a highly active and selective catalyst for PROX, especially at low temperature. At 206C, the exit CO concentration could be reduced to less than 2 ppm with a turnover frequency of 4.1 3 10 22 s 21 at a space velocity of 120,000 mL h 21 g 21 cat . and its high activity could retain for more than 24 hours. The presence of residual Ag species in the structure did not seem to improve the intrinsic activity of NPG for PROX; however, they contributed to the stabilization of the NPG structure and apparent catalytic activity. These results indicated that NPG might be readily applicable for hydrogen purification in fuel cell applications. P roton exchange membrane fuel cells (PEMFCs) have been attracting much attention because of its high energy efficiency and environmental compatibility. Hydrogen as the fuel of PEMFC is traditionally pro- duced by steam reforming, but the resulted gases always contain a small quantity of CO (0.5–2%), which poisons platinum on anode of PEMFCs. Among the popular methods for CO removal from H 2 steam, preferential CO oxidation (PROX) appears to be the most promising one 1–4 . For an ideal PROX catalyst, it should meet primarily the following key requirements: (1) high CO oxidation conversion, (2) low H 2 oxidation activity, (3) resistance to deactivation caused by CO 2 and H 2 O. Considering system integration and miniaturization, it is also highly desired that the catalyst is durable at the start-up (room temperature) and operation (,80uC) temperature of PEMFCs. Among various catalysts studied for PROX, supported gold catalysts 5–7 are found to be more active than Pt- based catalysts 8,9 and metal oxides (Cu-based catalysts) 10,11 , especially at low temperature. However, it has been shown that supported gold catalysts tended to deactivate under realistic conditions because of the accumulation of carbonate-like species at the support interface 12,13 . In comparison, although no carbonate-like species were detected for unsupported gold powder (mean particle size 20 nm) by diffuse reflectance infrared Fourier trans- form spectroscopy 14 , it was rarely active for CO oxidation. In the past few years, dealloyed nanoporous gold (NPG) fabricated by selectively leaching less noble species (Ag, Cu or Al) from the corresponding gold alloys, has received increasing attention in catalysis 15–17 , biological detection 18 and electrochemistry 19 , due to its unique structural properties, such as high surface-to-volume ratio, excellent electrical and thermal conductivity, and superior chemical stability. While it has been reported that unsupported NPG can display exceptional catalytic performance for CO oxidation at low temperature 20,21 , it is quite surprising that this new catalyst does not receive enough attention for gas-phase catalysis, although more recent studies have revealed very intriguing catalytic activities for NPG, such as methanol oxidative coupling 22,23 , and selective benzyl alcohol oxidation 24 . To date, the catalytic properties of NPG for PROX in hydrogen stream, which is of practical significance for PEMFC development, has not been reported. Because NPG is made from a chloride-ion free dealloying process, it possesses an interesting unsupported structural feature, which is radically different from the traditional supported Au nanocatalysts. In this work we present an in-depth study on the catalytic performance of NPG for PROX. OPEN SUBJECT AREAS: HETEROGENEOUS CATALYSIS POROUS MATERIALS FUEL CELLS STRUCTURAL PROPERTIES Received 20 August 2013 Accepted 3 October 2013 Published 22 October 2013 Correspondence and requests for materials should be addressed to Y.D. ([email protected]. cn) SCIENTIFIC REPORTS | 3 : 3015 | DOI: 10.1038/srep03015 1
Transcript of Nanoporous gold as an active low temperature catalyst ... · PDF fileNanoporous gold as an...

Nanoporous gold as an active lowtemperature catalyst toward COoxidation in hydrogen-rich streamDongwei Li1, Ye Zhu1, Hui Wang1 & Yi Ding1,2
1Center for Advanced Energy Materials & Technology Research (AEMT), and School of Chemistry and Chemical Engineering,Shandong University, Jinan 250100, China, 2Shandong Applied Research Center for Gold Technology (Au-SDARC), Yantai264005, China.
Preferential CO oxidation (PROX) was investigated by using dealloyed nanoporous gold (NPG) catalystunder ambient conditions. Systematic investigations were carried out to characterize its catalyticperformance by varying reaction parameters such as temperature and co-existence of CO2 and H2O, whichrevealed that NPG was a highly active and selective catalyst for PROX, especially at low temperature. At206C, the exit CO concentration could be reduced to less than 2 ppm with a turnover frequency of 4.1 31022 s21 at a space velocity of 120,000 mL h21 g21
cat. and its high activity could retain for more than 24hours. The presence of residual Ag species in the structure did not seem to improve the intrinsic activity ofNPG for PROX; however, they contributed to the stabilization of the NPG structure and apparent catalyticactivity. These results indicated that NPG might be readily applicable for hydrogen purification in fuel cellapplications.
Proton exchange membrane fuel cells (PEMFCs) have been attracting much attention because of its highenergy efficiency and environmental compatibility. Hydrogen as the fuel of PEMFC is traditionally pro-duced by steam reforming, but the resulted gases always contain a small quantity of CO (0.5–2%), which
poisons platinum on anode of PEMFCs. Among the popular methods for CO removal from H2 steam, preferentialCO oxidation (PROX) appears to be the most promising one1–4. For an ideal PROX catalyst, it should meetprimarily the following key requirements: (1) high CO oxidation conversion, (2) low H2 oxidation activity, (3)resistance to deactivation caused by CO2 and H2O. Considering system integration and miniaturization, it is alsohighly desired that the catalyst is durable at the start-up (room temperature) and operation (,80uC) temperatureof PEMFCs.
Among various catalysts studied for PROX, supported gold catalysts5–7 are found to be more active than Pt-based catalysts8,9 and metal oxides (Cu-based catalysts)10,11, especially at low temperature. However, it has beenshown that supported gold catalysts tended to deactivate under realistic conditions because of the accumulationof carbonate-like species at the support interface12,13. In comparison, although no carbonate-like species weredetected for unsupported gold powder (mean particle size 20 nm) by diffuse reflectance infrared Fourier trans-form spectroscopy14, it was rarely active for CO oxidation.
In the past few years, dealloyed nanoporous gold (NPG) fabricated by selectively leaching less noble species(Ag, Cu or Al) from the corresponding gold alloys, has received increasing attention in catalysis15–17, biologicaldetection18 and electrochemistry19, due to its unique structural properties, such as high surface-to-volume ratio,excellent electrical and thermal conductivity, and superior chemical stability. While it has been reported thatunsupported NPG can display exceptional catalytic performance for CO oxidation at low temperature20,21, it isquite surprising that this new catalyst does not receive enough attention for gas-phase catalysis, although morerecent studies have revealed very intriguing catalytic activities for NPG, such as methanol oxidative coupling22,23,and selective benzyl alcohol oxidation24. To date, the catalytic properties of NPG for PROX in hydrogen stream,which is of practical significance for PEMFC development, has not been reported. Because NPG is made from achloride-ion free dealloying process, it possesses an interesting unsupported structural feature, which is radicallydifferent from the traditional supported Au nanocatalysts. In this work we present an in-depth study on thecatalytic performance of NPG for PROX.
OPEN
SUBJECT AREAS:HETEROGENEOUS
CATALYSIS
POROUS MATERIALS
FUEL CELLS
STRUCTURAL PROPERTIES
Received20 August 2013
Accepted3 October 2013
Published22 October 2013
Correspondence andrequests for materials
should be addressed toY.D. ([email protected].
cn)
SCIENTIFIC REPORTS | 3 : 3015 | DOI: 10.1038/srep03015 1
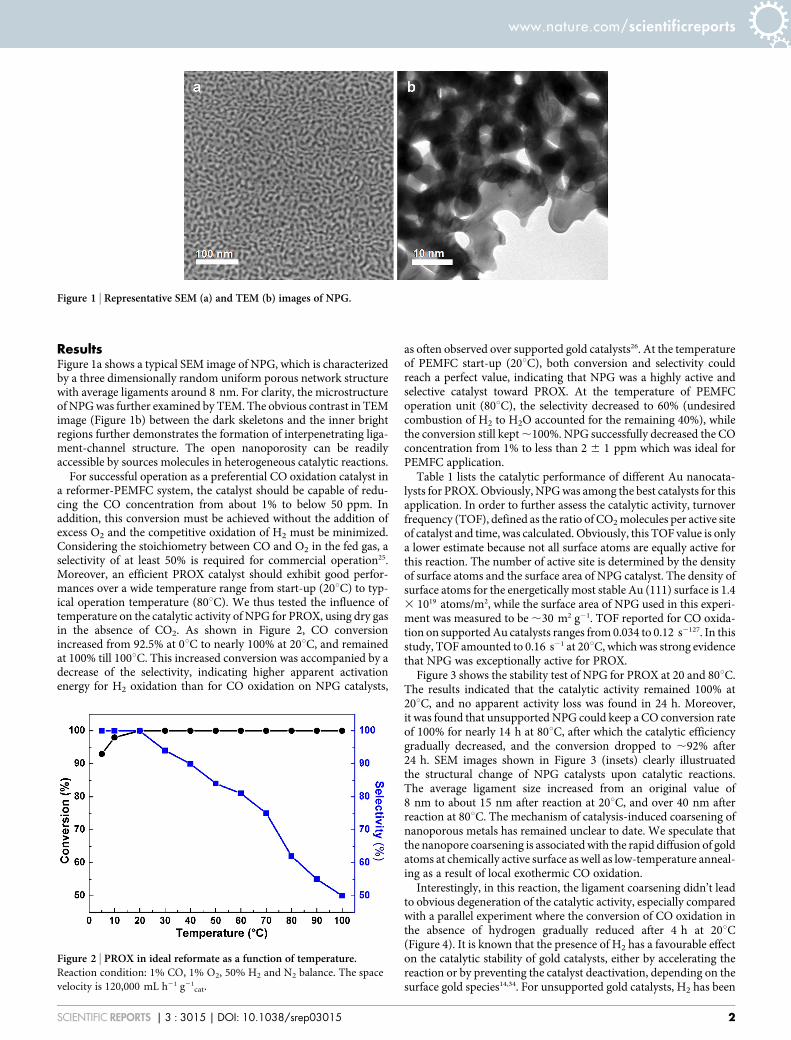
ResultsFigure 1a shows a typical SEM image of NPG, which is characterizedby a three dimensionally random uniform porous network structurewith average ligaments around 8 nm. For clarity, the microstructureof NPG was further examined by TEM. The obvious contrast in TEMimage (Figure 1b) between the dark skeletons and the inner brightregions further demonstrates the formation of interpenetrating liga-ment-channel structure. The open nanoporosity can be readilyaccessible by sources molecules in heterogeneous catalytic reactions.
For successful operation as a preferential CO oxidation catalyst ina reformer-PEMFC system, the catalyst should be capable of redu-cing the CO concentration from about 1% to below 50 ppm. Inaddition, this conversion must be achieved without the addition ofexcess O2 and the competitive oxidation of H2 must be minimized.Considering the stoichiometry between CO and O2 in the fed gas, aselectivity of at least 50% is required for commercial operation25.Moreover, an efficient PROX catalyst should exhibit good perfor-mances over a wide temperature range from start-up (20uC) to typ-ical operation temperature (80uC). We thus tested the influence oftemperature on the catalytic activity of NPG for PROX, using dry gasin the absence of CO2. As shown in Figure 2, CO conversionincreased from 92.5% at 0uC to nearly 100% at 20uC, and remainedat 100% till 100uC. This increased conversion was accompanied by adecrease of the selectivity, indicating higher apparent activationenergy for H2 oxidation than for CO oxidation on NPG catalysts,
as often observed over supported gold catalysts26. At the temperatureof PEMFC start-up (20uC), both conversion and selectivity couldreach a perfect value, indicating that NPG was a highly active andselective catalyst toward PROX. At the temperature of PEMFCoperation unit (80uC), the selectivity decreased to 60% (undesiredcombustion of H2 to H2O accounted for the remaining 40%), whilethe conversion still kept ,100%. NPG successfully decreased the COconcentration from 1% to less than 2 6 1 ppm which was ideal forPEMFC application.
Table 1 lists the catalytic performance of different Au nanocata-lysts for PROX. Obviously, NPG was among the best catalysts for thisapplication. In order to further assess the catalytic activity, turnoverfrequency (TOF), defined as the ratio of CO2 molecules per active siteof catalyst and time, was calculated. Obviously, this TOF value is onlya lower estimate because not all surface atoms are equally active forthis reaction. The number of active site is determined by the densityof surface atoms and the surface area of NPG catalyst. The density ofsurface atoms for the energetically most stable Au (111) surface is 1.43 1019 atoms/m2, while the surface area of NPG used in this experi-ment was measured to be ,30 m2 g21. TOF reported for CO oxida-tion on supported Au catalysts ranges from 0.034 to 0.12 s2127. In thisstudy, TOF amounted to 0.16 s21 at 20uC, which was strong evidencethat NPG was exceptionally active for PROX.
Figure 3 shows the stability test of NPG for PROX at 20 and 80uC.The results indicated that the catalytic activity remained 100% at20uC, and no apparent activity loss was found in 24 h. Moreover,it was found that unsupported NPG could keep a CO conversion rateof 100% for nearly 14 h at 80uC, after which the catalytic efficiencygradually decreased, and the conversion dropped to ,92% after24 h. SEM images shown in Figure 3 (insets) clearly illustruatedthe structural change of NPG catalysts upon catalytic reactions.The average ligament size increased from an original value of8 nm to about 15 nm after reaction at 20uC, and over 40 nm afterreaction at 80uC. The mechanism of catalysis-induced coarsening ofnanoporous metals has remained unclear to date. We speculate thatthe nanopore coarsening is associated with the rapid diffusion of goldatoms at chemically active surface as well as low-temperature anneal-ing as a result of local exothermic CO oxidation.
Interestingly, in this reaction, the ligament coarsening didn’t leadto obvious degeneration of the catalytic activity, especially comparedwith a parallel experiment where the conversion of CO oxidation inthe absence of hydrogen gradually reduced after 4 h at 20uC(Figure 4). It is known that the presence of H2 has a favourable effecton the catalytic stability of gold catalysts, either by accelerating thereaction or by preventing the catalyst deactivation, depending on thesurface gold species14,34. For unsupported gold catalysts, H2 has been
Figure 1 | Representative SEM (a) and TEM (b) images of NPG.
Figure 2 | PROX in ideal reformate as a function of temperature.Reaction condition: 1% CO, 1% O2, 50% H2 and N2 balance. The space
velocity is 120,000 mL h21 g21cat.
www.nature.com/scientificreports
SCIENTIFIC REPORTS | 3 : 3015 | DOI: 10.1038/srep03015 2
reported to activate O2 apparently via highly oxidizing intermediatespecies, thus weakening the deactivation tendency14,34.
According to the mechanism of PROX over unsupported goldpowder14, a possible mechanism with regard to this reaction overNPG is shown in Figure 5. It is believed that molecularly adsorbedO2 is first activated on NPG by reaction with H2 to form highlyoxidative intermediates such as OOH35,36. Then superficial CO andintermediates react to form CO2 and OH. The adsorbed OH wouldfurther react with superficial CO to produce CO2 and H. The cycle isclosed when two H atoms recombine into H2 or react with O2 mole-cules to form new OOH species.
DiscussionThe above results indicated that NPG was a highly active and select-ive catalyst toward CO oxidation in ideal stream. Considering thatrealistic reformate in the fuel processing section contains consid-erable amount of H2O (10–15%) and CO2 (10–20%), it is thereforeof practical importance to investigate whether the exceptional per-formance of NPG observed in ideal reformate can be maintainedunder more realistic conditions.
We thus evaluated the influence of CO2 on PROX by comparingthe conversion and selectivity in feed gas with and without 10% CO2
at the temperature of PEMFC operation unit (80uC). To obtain morerealistic information, here the loading of NPG catalyst was reducedto 10 mg, corresponding to a space velocity of 240,000 mL h21 g21
cat.Figure 6a shows the effect of CO2 on the NPG catalytic activity. It canbe seen that the presence of CO2 had slightly negative effect on COconversion of the catalyst from about 95% in the absence of CO2 to
around 92% in the presence of CO2, but the selectivity increased from75 to 90%. For the catalysts of gold supported on oxides, it wasreported the presence of CO2 often inhibited the CO conversionmuch more seriously. In the case of Au/MnOx for PROX, Hoflundand co-workers reported that the presence of CO2 had a large det-rimental effect on the catalytic performance, and would decrease itsreactivity from 70 to 30%37. This inhibiting effect was often assoc-iated with the oxide support, including formation of carbonate spe-cies on the support, and chemical-state change of both active goldsurface species and the oxides38,39. In comparison with supported Aucatalysts, NPG is truly support-free and thus such deactivation can beruled out. Moreover, the reverse water gas shift reaction has neg-ligible effect in our system, especially at low temperature. This is whyNPG is more tolerable to CO2 as compared with supported goldcatalysts. As far as the enhanced selectivity, CO2 molecules adsorbedon the active sites might inhibit the intimate contact between hydro-gen and oxygen molecules.
The effect of H2O in the feed stream on PROX is shown inFigure 6b. H2O was fed continuously through a syringe pump andwas vaporized prior to the reaction bed, yielding ,10% water vaporin the reactant gas. Under the humidified condition, CO was stillremoved absolutely and the selectivity increased to 100% at 80uC.With respect to the origin of the H2O effect, the possibility of watergas shift reaction (WGSR) can be ruled out, because a separateWGSR (1% CO 1 10% H2O) on NPG showed no CO2 formationat the corresponding temperature. The increased selectivity could beattributed to the reaction equilibrium for H2 oxidation, which shiftedto the left in the presence of H2O. The second reason might be relatedto the adsorption of H2O molecules which led to the formation of
Table 1 | Comparison of the catalytic performance of different Au nanocatalysts for PROX
Catalyst Preparation method T (uC) Conversion (%) Selectivity (%) Exit CO Concentration (ppm) Ref.
Au/Al2O3 Impregnation 70 85 52 3000 28Au/CeO2 Deposition-Precipitation 40 100 51 - 29Au/Fe2O3 Co-precipitation 80 99.8 51 - 30Au/CeO2-Al2O3 Impregnation 50 99 63 200 28Au/MnOx-CeO2 Deposition-Precipitation 120 90 48 - 31Au/ZnO-TiO2 Impregnation 80 85 43 - 32Au/Co3O4-TiO2 Impregnation 25 100 80 33Au powder Commercial 200 - 30 - 14NPG Dealloying 20 100 100 2 6 1 (negligible) This workNPG Dealloying 80 100 60 2 6 1 (negligible) This work
Figure 3 | The stability of NPG for PROX at 206C (&) and 806C (.).Insets are SEM images of NPG samples before and after catalytic reactions.
Scale bars: 100 nm.
Figure 4 | Catalytic performance of NPG for CO oxidation at 206C. The
reaction gas composition: 1% CO, 10% O2 and N2 balance. The space
velocity is 120,000 mL h21 g21cat.
www.nature.com/scientificreports
SCIENTIFIC REPORTS | 3 : 3015 | DOI: 10.1038/srep03015 3
carboxyl group that accelerated the adsorption of oxygen to oxidizeCO into CO2
40,41. Figure 6c depicts the influence of co-presence of10% CO2 and 10% H2O in the feed stream on PROX over NPGcatalyst. Very apparently, feeding both CO2 and H2O in the streamhas remarkably positive effect on CO removal. The CO conversionwas nearly perfect and the selectivity increased from 75 to 100% at
80uC. The variation of CO conversion and selectivity with reactiontime in different feed gases can be more clearly appreciated in adesigned experiment as illustrated in Figure 6d. During the first30 min, NPG catalysts behaved quite normal as observed inFigure 6a–c, with a conversion slightly over 95% and selectivityaround 75%. During the second stage, 10% CO2 was added intothe stream, which slightly reduced the conversion to about 92%.After another 30 min (stage 3), 10% H2O was introduced into thestream, which immediately increased the conversion from 92 tonearly 100%. And over the entire reaction period, continuousincrease of selectivity was observed, indicating a favorable effect fromthe presence of H2O and/or CO2.
Regarding the possible contribution of silver to gold catalysis, Mouand co-workers reported that AuAg alloy particles on mesoporoussupports possessed much higher activity for low temperature COoxidation as compared to monometallic Au catalyst42. Consideringthat some silver atoms might be trapped within the ligaments of NPGduring dealloying, a question often arises as whether the apparentcatalytic activity is solely from NPG itself or the residual Ag atomsplay an important role. In deed, several groups have noted the effectof the residual elements on CO oxidation and methanol oxida-tion43,44. For example, Baumer and co-workers considered NPG cata-lyst as a bimetallic catalyst rather than a pure Au catalyst, and thesuperior catalytic activity for low temperature CO oxidation wasattributable to the residual Ag45. To investigate the possible effectof Ag in the present system, control experiments were performed
Figure 5 | Reaction mechanism of CO oxidation in the presence of H2
over NPG.
Figure 6 | (a) Effect of CO2 on PROX over NPG at 80uC. Reaction condition: 1% CO, 1% O2, 50% H2, 10% CO2 and N2 balance. (b) Effect of H2O on
PROX over NPG at 80uC. Reaction condition: 1% CO, 1% O2, 50% H2, 10% H2O and N2 balance. (c) Effect of co-adding CO2 and H2O in the feed gas on
PROX. Reaction condition: 1% CO, 1% O2, 50% H2, 10% CO2, 10% H2O and N2 balance. (d) Variation of PROX with reaction time in different
feed gases. Conversion (&%); Selectivity (.#). The space velocity is 240,000 mL h21 g21cat.
www.nature.com/scientificreports
SCIENTIFIC REPORTS | 3 : 3015 | DOI: 10.1038/srep03015 4
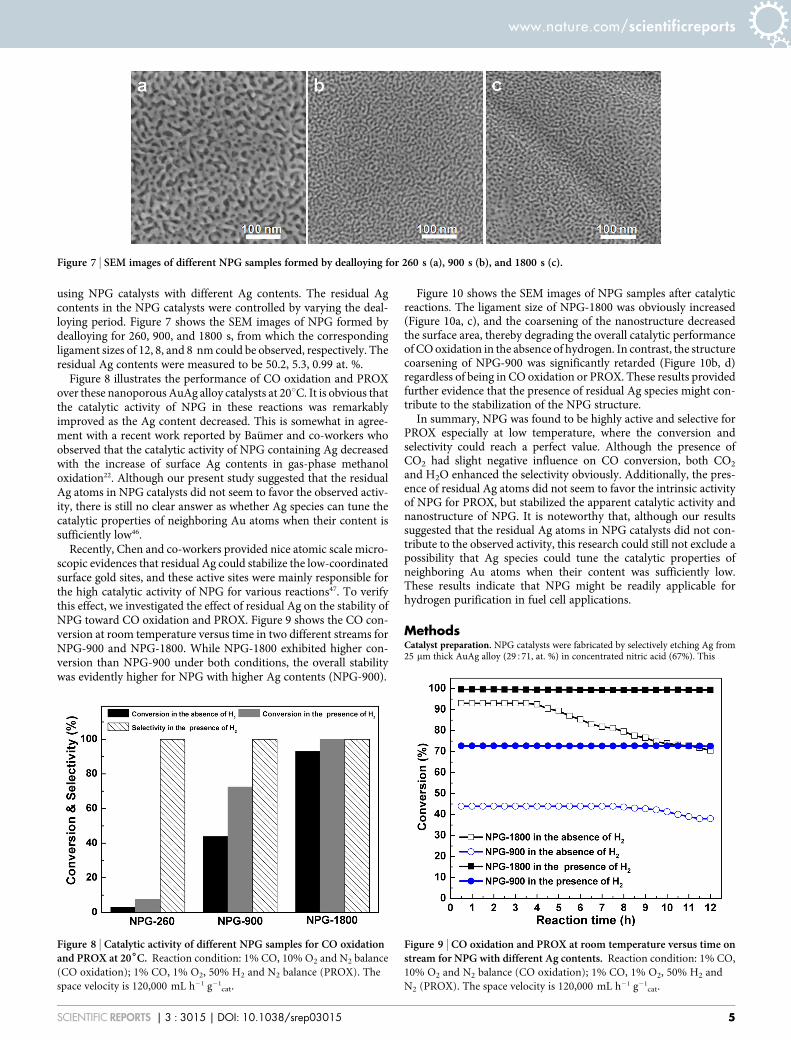
using NPG catalysts with different Ag contents. The residual Agcontents in the NPG catalysts were controlled by varying the deal-loying period. Figure 7 shows the SEM images of NPG formed bydealloying for 260, 900, and 1800 s, from which the correspondingligament sizes of 12, 8, and 8 nm could be observed, respectively. Theresidual Ag contents were measured to be 50.2, 5.3, 0.99 at. %.
Figure 8 illustrates the performance of CO oxidation and PROXover these nanoporous AuAg alloy catalysts at 20uC. It is obvious thatthe catalytic activity of NPG in these reactions was remarkablyimproved as the Ag content decreased. This is somewhat in agree-ment with a recent work reported by Baumer and co-workers whoobserved that the catalytic activity of NPG containing Ag decreasedwith the increase of surface Ag contents in gas-phase methanoloxidation22. Although our present study suggested that the residualAg atoms in NPG catalysts did not seem to favor the observed activ-ity, there is still no clear answer as whether Ag species can tune thecatalytic properties of neighboring Au atoms when their content issufficiently low46.
Recently, Chen and co-workers provided nice atomic scale micro-scopic evidences that residual Ag could stabilize the low-coordinatedsurface gold sites, and these active sites were mainly responsible forthe high catalytic activity of NPG for various reactions47. To verifythis effect, we investigated the effect of residual Ag on the stability ofNPG toward CO oxidation and PROX. Figure 9 shows the CO con-version at room temperature versus time in two different streams forNPG-900 and NPG-1800. While NPG-1800 exhibited higher con-version than NPG-900 under both conditions, the overall stabilitywas evidently higher for NPG with higher Ag contents (NPG-900).
Figure 10 shows the SEM images of NPG samples after catalyticreactions. The ligament size of NPG-1800 was obviously increased(Figure 10a, c), and the coarsening of the nanostructure decreasedthe surface area, thereby degrading the overall catalytic performanceof CO oxidation in the absence of hydrogen. In contrast, the structurecoarsening of NPG-900 was significantly retarded (Figure 10b, d)regardless of being in CO oxidation or PROX. These results providedfurther evidence that the presence of residual Ag species might con-tribute to the stabilization of the NPG structure.
In summary, NPG was found to be highly active and selective forPROX especially at low temperature, where the conversion andselectivity could reach a perfect value. Although the presence ofCO2 had slight negative influence on CO conversion, both CO2
and H2O enhanced the selectivity obviously. Additionally, the pres-ence of residual Ag atoms did not seem to favor the intrinsic activityof NPG for PROX, but stabilized the apparent catalytic activity andnanostructure of NPG. It is noteworthy that, although our resultssuggested that the residual Ag atoms in NPG catalysts did not con-tribute to the observed activity, this research could still not exclude apossibility that Ag species could tune the catalytic properties ofneighboring Au atoms when their content was sufficiently low.These results indicate that NPG might be readily applicable forhydrogen purification in fuel cell applications.
MethodsCatalyst preparation. NPG catalysts were fabricated by selectively etching Ag from25 mm thick AuAg alloy (29571, at. %) in concentrated nitric acid (67%). This
Figure 7 | SEM images of different NPG samples formed by dealloying for 260 s (a), 900 s (b), and 1800 s (c).
Figure 8 | Catalytic activity of different NPG samples for CO oxidationand PROX at 206C. Reaction condition: 1% CO, 10% O2 and N2 balance
(CO oxidation); 1% CO, 1% O2, 50% H2 and N2 balance (PROX). The
space velocity is 120,000 mL h21 g21cat.
Figure 9 | CO oxidation and PROX at room temperature versus time onstream for NPG with different Ag contents. Reaction condition: 1% CO,
10% O2 and N2 balance (CO oxidation); 1% CO, 1% O2, 50% H2 and
N2 (PROX). The space velocity is 120,000 mL h21 g21cat.
www.nature.com/scientificreports
SCIENTIFIC REPORTS | 3 : 3015 | DOI: 10.1038/srep03015 5

dealloying process was carried out in a standard three-electrode cell with a CHI 760Cpotentiostat under an applied voltage (0.4 V) at room temperature, using platinumelectrode as the counter electrode and saturated calomel electrode (RHE) as thereference electrode. Before dealloying, AuAg alloy foils were annealed at 850uC for16 h and subsequently degreased by sonicating in ethanol and rinsed with distilledwater. The residual Ag contents in NPG catalysts were controlled by varyingdealloying period. All NPG samples were filtered out and thoroughly washed withdistilled water until no acid was detected in the filtrate, and then dried in a vacuumdesiccator at room temperature.
Catalyst characterization. The microstructure of samples was characterized with aJEOL JSM-6700F field emission scanning electron microscope (SEM), equipped withan Oxford INCA x-sight energy-dispersive spectrometer (EDS) for compositionalanalysis. The surface area was measured with Quadrasorb SI-MP (QuantachromeInstruments) using the BET method.
Catalytic test. In all experiments, NPG catalysts were (20 mg) crushed into powderand then diluted with 0.5 g quartz sand (1-mm) to ensure adequate contact betweengas reactants and the catalysts. The mixed catalysts were placed into a 4-mm-i.d.quartz tube and tested in a continuous flow fixed bed micro-reactor at atmosphericpressure. With regard to the temperature control during testing, the reactor wassettled in a cold trap at 0uC and in a heating furnace at higher temperatures to keepisothermal. The reactant gas containing 1% CO, 1% O2, 50% H2, and N2 for balancewas guided into the reactor with a space velocity of 120,000 mL h21 g21
cat (the typicalcomposition in reforming stream is 45–75% H2, 10–20% CO2, 0.5–2% CO, and H2O).The effect of CO2 and H2O was investigated, at separate runs, with addition of 10% inthe respective feed gas. The amounts of CO and CO2 were measured by an on-lineinfrared gas analyzer (Gasboard-3121, China Wuhan Cubic Co.). The resolution ofthis analyzer is 1 ppm. O2 was analyzed by an online Shimadzu Type gaschromatograph (GC-14C) equipped with a thermal conductivity detector (TCD),using 5 A Molecular Sieve.
CO conversion was calculated based on CO2 formation as follows:
conversion%~½CO2�out
½CO�in|100%
When 10% CO2 was added in the reactant gases, the CO conversion was calculatedbased on CO consumption, considering otherwise possible errors in the quantifica-tion of small changes in CO2 concentration. The selectivity was calculated from theoxygen mass balance as follows:
selectivity%~0:5(½CO�in{½CO�out)
½O2�in{½O2�out
|100%
1. Potemkin, D. I. et al. Preferential CO oxidation over bimetallic Pt–Co catalystsprepared via double complex salt decomposition. Chem. Eng. J. 207–208, 683–689(2012).
2. Alayoglu, S., Nilekar, A. U., Mavrikakis, M. & Eichhorn, B. Ru–Pt core–shellnanoparticles for preferential oxidation of carbon monoxide in hydrogen. Nat.Mater. 7, 333–338 (2008).
3. Park, E. D., Lee, D. & Lee, H. C. Recent progress in selective CO removal in a H2-rich stream. Catal. Today 139, 280–290 (2009).
4. Nilekar, A. U., Alayoglu, S., Eichhorn, B. & Mavrikakis, M. Preferential COoxidation in hydrogen: reactivity of core-shell nanoparticles. J. Am. Chem. Soc.132, 7418–7428 (2010).
5. Naknam, P., Luengnaruemitchai, A. & Wongkasemjit, S. Au/ZnO and Au/ZnO-Fe2O3 prepared by deposition-precipitation and their activity in the preferentialoxidation of CO. Energy Fuels 23, 5084–5091 (2009).
6. Tu, Y. B., Luo, J. Y., Meng, M., Wang, G. & He, J. J. Ultrasonic-assisted synthesis ofhighly active catalyst Au/MnOx–CeO2 used for the preferential oxidation of CO inH2-rich stream. Int. J. Hydrogen Energy 34, 3743–3754 (2009).
7. Shodiya, T., Schmidt, O., Peng, W. & Hotz, N. Novel nano-scale Au/a-Fe2O3
catalyst for the preferential oxidation of CO in biofuel reformate gas. J. Catal. 300,63–69 (2013).
8. Liu, K., Wang, A. Q. & Zhang, T. Recent advances in preferential oxidation of COreaction over platinum group metal catalysts. ACS Catal. 2, 1165–1178 (2012).
9. Xu, J. et al. Mechanistic study of preferential CO oxidation on a Pt/NaY zeolitecatalyst. J. Catal. 287, 114–123 (2012).
10. Varghese, S. et al. CO oxidation and preferential oxidation of CO in the presenceof hydrogen over SBA-15-templated CuO-Co3O4 catalysts. Appl. Catal. A: Gen.443–444, 161–170 (2012).
11. Gamarra, D. et al. Preferential oxidation of CO in excess H2 over CuO/CeO2
catalysts: Characterization and performance as a function of the exposed facepresent in the CeO2 support. Appl. Catal. B: Environ. 130–131, 224–238 (2013).
12. Wang, H. et al. Deactivation of a Au/CeO2–Co3O4 catalyst during CO preferentialoxidation in H2-rich stream. J. Catal. 264, 154–162 (2009).
13. Kim, C. H. & Thompson, L. T. Deactivation of Au/CeOx water gas shift catalysts. J.Catal. 230, 66–74 (2005).
14. Quinet, E. et al. H2-induced promotion of CO oxidation over unsupported gold.Catal. Today 138, 43–49 (2008).
15. Ding, Y. & Chen, M. W. Nanoporous metals for catalytic and optical applications.MRS Bull. 34, 569–576 (2009).
16. Zhang, X. M. & Ding, Y. Unsupported nanoporous gold for heterogeneouscatalysis. Catal. Sci. Technol. DOI: 10.1039/C3CY00241A (2013).
17. Asao, N. et al. Nanostructured materials as catalysts: nanoporous-gold-catalyzedoxidation of organosilanes with Water. Angew. Chem. Int. Ed. 49, 10093–10095(2010).
18. Yu, F. et al. Simultaneous Excitation of Propagating and Localized SurfacePlasmon Resonance in Nanoporous Gold Membranes. Anal. Chem. 78,7346–7350 (2006).
19. Zhang, J. T., Liu, P. P., Ma, H. Y. & Ding, Y. Nanostructured porous gold formethanol electro-oxidation. J. Phys. Chem. C 111, 10382–10388 (2007).
20. Zielasek, V. et al. Gold catalysts: nanoporous gold foams. Angew. Chem. Int. Ed.45, 8241–8244 (2006).
21. Xu, C. X. et al. Low temperature CO oxidation over unsupported nanoporousgold. J. Am. Chem. Soc. 129, 42–43 (2007).
22. Wittstock, A., Zielasek, V., Biener, J., Friend, C. M. & Baumer, M. Nanoporousgold catalysts for selective gas-phase oxidative coupling of methanol at lowtemperature. Science 327, 319–322 (2010).
23. Kosuda, K. M., Wittstock, A., Friend, C. M. & Baumer, M. Oxygen-mediatedcoupling of alcohols over nanoporous gold catalysts at ambient pressures. Angew.Chem. Int. Ed. 51, 1698–1701 (2012).
24. Han, D. Q., Xu, T. T., Su, J. X., Xu, X. H. & Ding, Y. Gas-phase selective oxidationof benzyl alcohol to benzaldehyde with molecular oxygen over unsupportednanoporous gold. ChemCatChem 2, 383–386 (2010).
25. Landon, P. et al. Selective oxidation of CO in the presence of H2, H2O and CO2 viagold for use in fuel cells. Chem. Comm. 3385–3387 (2005).
26. Bamwenda, G. R., Tsubota, S., Nakamura, T. & Haruta, M. The influence of thepreparation methods on the catalytic activity of platinum and gold supported onTiO2 for CO oxidation. Catal. Lett. 44, 83–87 (1997).
27. Kandoi, S., Gokhale, A. A., Grabow, L. C., Dumesic, J. A. & Mavrikakis, M. WhyAu and Cu are more selective than Pt for preferential oxidation of CO at lowtemperature. Catal. Lett. 93, 93–100 (2004).
28. Fonseca, J. et al. Preferential CO oxidation over nanosized gold catalystssupported on ceria and amorphous ceria–alumina. Appl. Catal. B: Environ. 128,10–20 (2012).
29. Sakwarathorn, T., Luengnaruemitchai, A. & Pongstabodee, S. Preferential COoxidation in H2-rich stream over Au/CeO2 catalysts prepared via modifieddeposition–precipitation. J. Ind. Eng. Chem. 17, 747–754 (2011).
30. Landon, P. et al. Selective oxidation of CO in the presence of H2, H2O and CO2
utilising Au/a-Fe2O3 catalysts for use in fuel cells. J. Mater. Chem. 16, 199–208(2006).
31. Tu, Y. B. et al. CO preferential oxidation over Au/MnOx–CeO2 catalysts preparedwith ultrasonic assistance: Effect of calcination temperature. Fuel Process. Technol.93, 78–84 (2012).
32. Chen, Y. W., Lee, D. S. & Chen, H. J. Preferential oxidation of CO in H2 stream onAu/ZnO-TiO2 catalysts. Int. J. Hydrogen Energy 37, 15140–15155 (2012).
33. Chen, Y. W., Chen, H. J. & Lee, D. S. Au/Co3O4–TiO2 catalysts for preferentialoxidation of CO in H2 stream. J. Mol. Catal. A: Chem. 363–364, 470–480 (2012).
Figure 10 | SEM images of different NPG samples after catalytic reaction.(a). NPG-1800 (CO oxidation) (b). NPG-900 (CO oxidation) (c). NPG-
1800 (PROX) (d). NPG-900 (PROX).
www.nature.com/scientificreports
SCIENTIFIC REPORTS | 3 : 3015 | DOI: 10.1038/srep03015 6

34. Rossignol, C. et al. Selective oxidation of CO over model gold-based catalysts inthe presence of H2. J. Catal. 230, 476–483 (2005).
35. Quinet, E. et al. On the mechanism of hydrogen-promoted gold-catalyzed COoxidation. J. Catal. 268, 384–389 (2009).
36. Deronzier, T., Morfin, F., Massin, L., Lomello, M. & Rousset, J. L. Purenanoporous gold powder: synthesis and catalytic properties. Chem. Mater. 23,5287–5289 (2011).
37. Hoflund, G. B., Gardner, S. D., Schryer, D. R., Upchurch, B. T. & Kielin, E. J. Effectof COz on the performance of Au/lMnO, and Pt/SnO, low-temperature COoxidation catalysts. Langmuir 11, 3431–3434 (1995).
38. Avgouropoulos, G. et al. A comparative study of Pt/c-Al2O3, Au/a-Fe2O3 andCuO–CeO2 catalysts for the selective oxidation of carbon monoxide in excesshydrogen. Catal. Today 75, 157–167 (2002).
39. Schubert, M. M., Venugopal, A., Kahlich, M. J., Plzak, V. & Behm, R. J. Influence ofH2O and CO2 on the selective CO oxidation in H2-rich gases over Au/a-Fe2O3. J.Catal. 222, 32–40 (2004).
40. Date, M., Okumura, M., Tsubota, S. & Haruta, M. Vital role of moisture in thecatalytic activity of supported gold nanoparticles. Angew. Chem. Int. Ed. 43,2129–2132 (2004).
41. Bongiorno, A. & Landman, U. Water-enhanced catalysis of CO oxidation on freeand supported gold nanoclusters. Phys. Rev. Lett. 95, 106102 (2005).
42. Wang, A. Q., Liu, J. H., Lin, S. D., Lin, T. S. & Mou, C. Y. A novel efficient Au–Agalloy catalyst system: preparation, activity, and characterization. J. Catal. 233,186–197 (2005).
43. Wang, L. C., Zhong, Y., Widmann, D., Weissmuller, J. & Behm, R. J. On the natureof active sites on nanoporous Au catalysts for CO oxidation: a combined micro-reactor and TAP reactor study. ChemCatChem 4, 251–259 (2012).
44. Moskaleva, L. V. et al. Silver residues as a possible key to a remarkable oxidativecatalytic activity of nanoporous gold. Phys. Chem. Chem. Phys. 13, 4529–4539(2011).
45. Wittstock, A. et al. Nanoporous Au: An unsupported pure gold catalyst? J. Phys.Chem. C 113, 5593–5600 (2009).
46. Rohe, S. et al. CO oxidation on nanoporous gold: a combined TPD and XPS studyof active catalysts. Surf. Sci. 609, 106–112 (2013).
47. Fujita, T. et al. Atomic origins of the high catalytic activity of nanoporous gold.Nat. Mater. 11, 775–780 (2012).
AcknowledgmentsThis work was sponsored by the National 973 Program Project of China (2012CB932800),and the National Science Foundation of China (51171092) and the Research Fund for theDoctoral Program of Higher Education of China (20090131110019).
Author contributionsY.D. and D.L. designed this research; D.L. carried out the experiments and analyzed thedata; Y.Z. and H.W. contributed to the discussion; D.L. and Y.D. wrote the paper.
Additional informationCompeting financial interests: The authors declare no competing financial interests.
How to cite this article: Li, D.W., Zhu, Y., Wang, H. & Ding, Y. Nanoporous gold as anactive low temperature catalyst toward CO oxidation in hydrogen-rich stream. Sci. Rep. 3,3015; DOI:10.1038/srep03015 (2013).
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported license. To view a copy of this license,
visit http://creativecommons.org/licenses/by-nc-sa/3.0
www.nature.com/scientificreports
SCIENTIFIC REPORTS | 3 : 3015 | DOI: 10.1038/srep03015 7


















