
Nanocatalysis Synthesis and Applications (Polshettiwar/Nanocatalysis) || Nanocatalysis: Activation...
33
19 NANOCATALYSIS: ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK Suresh Babu Kalidindi and Balaji R. Jagirdar INTRODUCTION Small molecules such as H 2 ,N 2 ,O 2 , CO, CO 2 , and CH 4 are sources of a variety of useful feedstock in many important industrial processes. 1 These molecules are thermo- dynamically stable, and their successful conversion into useful feedstock often depends on surmounting quite significant kinetic barriers. It is well established that catalysts pro- vide low-barrier reaction pathways through adsorption or binding and activation events. Issues such as activity, selectivity, and durability of catalysts remained as core issues of research in the field of heterogeneous catalysis (including processes involving small molecules) for scientists. The approaches for addressing such issues took a new shape during the last two decades due to the collaboration of heterogeneous catalysis with nanoscience. 2–6 Due to the various advantages offered by nanoscience, such as fine- tuning of size, shape, composition, and morphology of catalytic surfaces, heterogeneous catalysis is slowly changing from an empirical art to an understandable science. This transition could also be attributed partly to modern analytical techniques. Nanoscience has been a part of catalysis long before this term was introduced, and nanoparticles were used as active catalysts in activation of various small molecules. An excellent example is the Haber–Bosch process (discovered in 1913), which produces ammonia directly from N 2 and H 2 using an iron catalyst (BASF S6-10, composition: Nanocatalysis: Synthesis and Applications, First Edition. Edited by Vivek Polshettiwar and Tewodros Asefa. © 2013 John Wiley & Sons, Inc. Published 2013 by John Wiley & Sons, Inc. 679
Transcript of Nanocatalysis Synthesis and Applications (Polshettiwar/Nanocatalysis) || Nanocatalysis: Activation...

19NANOCATALYSIS: ACTIVATION
OF SMALL MOLECULESAND CONVERSION INTO
USEFUL FEEDSTOCKSuresh Babu Kalidindi and Balaji R. Jagirdar
INTRODUCTION
Small molecules such as H2, N2, O2, CO, CO2, and CH4 are sources of a variety ofuseful feedstock in many important industrial processes.1 These molecules are thermo-dynamically stable, and their successful conversion into useful feedstock often dependson surmounting quite significant kinetic barriers. It is well established that catalysts pro-vide low-barrier reaction pathways through adsorption or binding and activation events.Issues such as activity, selectivity, and durability of catalysts remained as core issuesof research in the field of heterogeneous catalysis (including processes involving smallmolecules) for scientists. The approaches for addressing such issues took a new shapeduring the last two decades due to the collaboration of heterogeneous catalysis withnanoscience.2–6 Due to the various advantages offered by nanoscience, such as fine-tuning of size, shape, composition, and morphology of catalytic surfaces, heterogeneouscatalysis is slowly changing from an empirical art to an understandable science. Thistransition could also be attributed partly to modern analytical techniques.
Nanoscience has been a part of catalysis long before this term was introduced, andnanoparticles were used as active catalysts in activation of various small molecules. Anexcellent example is the Haber–Bosch process (discovered in 1913), which producesammonia directly from N2 and H2 using an iron catalyst (BASF S6-10, composition:
Nanocatalysis: Synthesis and Applications, First Edition. Edited by Vivek Polshettiwar and Tewodros Asefa.© 2013 John Wiley & Sons, Inc. Published 2013 by John Wiley & Sons, Inc.
679

680 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
(a) (b)
Figure 19.1. (a) High-resolution scanning electron microscopy image of the BASF S6-10 cat-
alyst after reduction used in ammonia synthesis in the Haber–Bosch process; (b) transmission
electron microscopy (TEM) image of the Au/α-Fe2O3 catalyst used in the CO oxidation reaction.
(Reproduced with permission from Refs 7 (Figure 19.1a) and 9 (Figure 19.1b), Copyright 1983,
1989, Elsevier.)
Fe(40.5%), K(0.35%), Al(2.0%), Ca(1.7%), and O(53.2%)) that contains nanometer-sized particles with surface area of 20 m2/g.7,8 During the reaction, iron oxides undergoreduction to metallic iron. The iron particles formed are protected against agglomerationby a framework of Al2O3 and CaO under drastic reaction conditions. The iron particlesare also surrounded by the K+O adsorbed layer, which acts as an “electronic” promoter(Figure 19.1).
The recent example for activation of small molecules on nanoparticles (1989) is alow-temperature oxidation of carbon monoxide (CO) on nano-Au/�-Fe2O3 catalysts.9
Gold was generally considered a noble and relatively inactive metal for catalysis. Thediscovery that gold nanoparticles (Au-NPs) smaller than 5 nm supported on �-Fe2O3were very active for CO oxidation by Haruta and coworkers brought about a newparadigm in gold chemistry. This example brought out the concept of “nano-sized effecton catalytic properties” into limelight.
The following examples are chosen for the discussion due to their industrial andenvironmental importance andmore discussion is devoted to recent advancements, ratherthan reviewing the entire literature:
1. CO oxidation on Au/metal oxide catalysts
2. Direct synthesis of H2O2 from H2 and O23. Methanol synthesis from CO and CO2

CO OXIDATION ON AU/METAL OXIDE CATALYSTS 681
CO OXIDATION ON AU/METAL OXIDE CATALYSTS
In an era of increasing greenhouse gas emission levels, one of the important applicationsof heterogeneous catalysis is removal of toxic substances such as CO from car exhausts.The car exhaust gas flows through the catalytic converter and interacts with an activecatalyst. Thereafter, dissociatively chemisorbed (Oad) oxygen molecules interact withchemisorbed CO molecules to form CO2, which is immediately released into the gasphase (Figure 19.2). Currently used Pt/Pd-based catalysts work only above 200 ◦C, somost of the CO release into the environment takes place in the initial minutes afterstarting the car engine. The nano-Au/metal oxide catalysts present a potential solutionto this cold startup problem.10 Since the last two decades, an unprecedented level of
1 2
43
Figure 19.2. Illustration of a car exhaust catalyst and major steps (1–4) involved in CO oxi-
dation. However, there is still an ambiguity regarding the type of active oxygen intermediate
(atomic vs. molecular) involved in the CO oxidation. (Reproduced with permission from Ref. 8,
Copyright 2008, Wiley-VCH.) (See color insert.)

682 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
00 100 200
Catalyst temperature (°C)
300 400
50
Oxid
ati
on
eff
icie
ncy o
f H
2 (
%) 100
(a)(b) (c) (d)
(e)
(f)
(h)
(g)
Figure 19.3. CO oxidation efficiencies in presence of various catalysts: (a) Au/α-Fe2O3 (Au/Fe
= 1/19, coprecipitation, 400 ◦C); (b) 0.5 wt% Pd/γ-Al2O3 (impregnation, 300 ◦C); (c) Au fine
powder; (d) Co3O4 (carbonate, 400 ◦C); (e) NiO (hydrate, 200 ◦C); (f) α-Fe2O3 (hydrate, 400 ◦C);
(g) 5 wt% Au/α-Fe2O3 (impregnation, 200 ◦C); (h) 5 wt% Au/α-Al2O3 (impregnation, 200 ◦C).
(Reproduced with permission from Ref. 9, Copyright 1989, Elsevier.)
research activity has been carried out on gold nanocatalysis, which has been termed as“gold rush,” most of which has been devoted to the understanding of the unexpectedactivity of the so-called noble metal (Au).
Haruta and coworkers prepared Au-NPs supported on metal oxide catalysts (�-Fe2O3, � -Al2O3, NiO, and Co3O4) by coprecipitation from aqueous solution of HAuCl4and nitrates of transition metal, followed by calcination at high temperatures.9 Surpris-ingly, the Au/�-Fe2O3 catalyst (calcination temperature= 400 ◦C,�10 nm) was able tooxidize CO even at –70 ◦C. However, the Au-NPs synthesized using the impregnationmethod were active only at temperatures above 100 ◦C. The comparison of activitiesbetween various impregnated, coprecipitated gold catalysts and a supported Pd catalystis shown in Figure 19.3. The data shown in Figure 19.3 indicate that the catalyst activityis sensitive toward the synthesis method and structure of the catalyst. It is noteworthythat simple powders of Au consisting of larger particles are also active for CO oxidationat high temperatures.
The origin of the nano-sized effect in the catalytic properties of Au/metal oxidecatalysts has been widely debated, but no clear consensus has been reached yet. Thecomplexity of the Au/metal oxide catalyst introduces a variety of possible factors thatcan contribute to the activity of the catalysts. For simplicity, factors related to Au-NPsand metal oxide support are discussed separately.
Gold Nanoparticles
It is widely agreed that particle size has definite effect on the activity of the gold catalystseven though the physical explanation remains unclear. Amarked increase in the turnoverfrequency of Au/TiO2 catalysts is observed once the cluster size reaches below approx-imately 3.5 nm (Figure 19.4), but further decrease in cluster size below approximately3 nm results in a decrease of the activity. Similar trends were observed for the Au-NPs

CO OXIDATION ON AU/METAL OXIDE CATALYSTS 683
(a) (b)
1.5 2.5 3.5
Cluster diameter (nm)
4.5 5.5
Tu
rno
ve
r fr
eq
uen
cy (
1/s
ite s
)
0.00
0.05
0.10
0.15
0.20
0.25
Figure 19.4. (a) TEM image of the Au (3.3 wt%)/TiO2 catalyst prepared by the deposition–
precipitation method, and then calcinated at 400 ◦C; (b) CO oxidation turnover frequencies
at 27 ◦C of the Au/TiO2 catalyst as a function of the average size of Au-NPs. (Reproduced
with permission from Ref. 12 (Figure 19.4a), Copyright 1993, Elsevier; adapted from Ref. 11
(Figure 19.4b).)
supported on extended TiO2(110) support. It has been suggested that the catalytic activityof nano-Au/metal oxide catalysts is due to different electronic properties associated withquantum size effects such as layer thickness. Scanning tunneling spectroscopy measure-ments on Au clusters supported on TiO2(110) suggest metal-to-nonmetal transition for achange in the thickness of metal clusters from two to three. The band gaps (in the rangeof 0.2–0.6 V) of two-atom-thick gold clusters (2.5–3.0 nm sized) are larger than the gapmeasured for three-atom-thick gold clusters.11, 12 Also, the cluster size corresponding tothe highest isosteric heats of CO adsorption (−� Hads) is in very strong correlation tothat exhibiting maximum catalytic activity.13
However, raising questions about the explanation based on quantum size effects,CO adsorption on nano-Au/FeO(111) catalyst was found to be independent of particlesize.14 Furthermore, particle size was found to have barely an effect on the activity inpresence of oxygen atoms, that is, when the nano-Au/TiO2 catalyst was oxidized witha supersonic beam of oxygen atoms.15 All these observations suggest that the particlesize plays a major role in supplying atomic oxygen or activated O2 to CO, rather than indetermining the binding site for CO molecules.
Oxygen does not dissociate to anymeasurable extent on extended gold surfaces evenat high temperatures and pressures. However, the role of undercoordinated Au atomsas possible sites for O2 activation is actively investigated. The theoretical calculationspredict that step density (i.e., the fraction of atoms in the particle having seven or lessneighbors) increases with decrease in size of Au particles.16 The relation between theadsorption energies for both CO and O atoms and the Au coordination number is shownin Figure 19.5.17 The adsorption energies are lowered by up to 1 eV as the coordinationnumber is decreased from 9 (extended Au(111)) to 4 (Au10 cluster). Therefore, greater

684 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
–0.6
4 5 6 7
Coordination number
CO
OAu10
Kink
Au(211)
Au(111)B
ind
ing
en
erg
y (
eV
)
8 9 10
–0.4
–0.2
0
0.2
0.4
0.6
Figure 19.5. Correlation between the binding energies, for CO molecules and O atoms, with
respect to the coordination number of Au atoms in a series of environments. Binding energies,
in eV, reported are referred to as gas-phase CO and O2, for O2 the energies are given per O
atom. (Reproduced with permission from Ref. 17, Copyright 2004, Elsevier.)
activity observed for small particles could be attributed to the greater number of undercoordinated atoms.
Corroborating these observations, undercoordinated gold atoms present on theextended surfaces also induce O2 activation. These undercoordinated Au atoms werecreated on Au(111)-extended surface by deposition of oxygen atoms using electronbombardment of condensed NO2 at 100 K.18 Once atomic oxygen is present on Au,nano as well as extended surfaces the CO oxidation takes place readily, even at temper-atures as low as 35 K.19 This suggests that the barrier for the reaction between CO andatomic oxygen is very low.
The charge on Au also plays a vital role in dictating the activity of the catalyst. Forexample, Au(III) supported on MgO was reduced using CO at various partial pressures,to obtain Au+1 and Au(0) at different concentrations.20 Lower partial pressures of CO(higher concentration of Au+1) lead to higher activity. The correlation between thecatalytic activity and the percentage surface concentration of cationic and zerovalentgold is shown in Figure 19.6.
The following factors related to Au-NPs could help in increasing their catalyticactivities for CO oxidation:
1. Different electronic properties arising from quantum size effects
2. Increasing the undercoordinated metal atoms
3. Positive charge on the gold
Metal Oxide Supports
Even though it has been shown that simple Au powders can act as catalysts for COoxidation at higher temperatures, the metal oxide support definitely plays a role. The
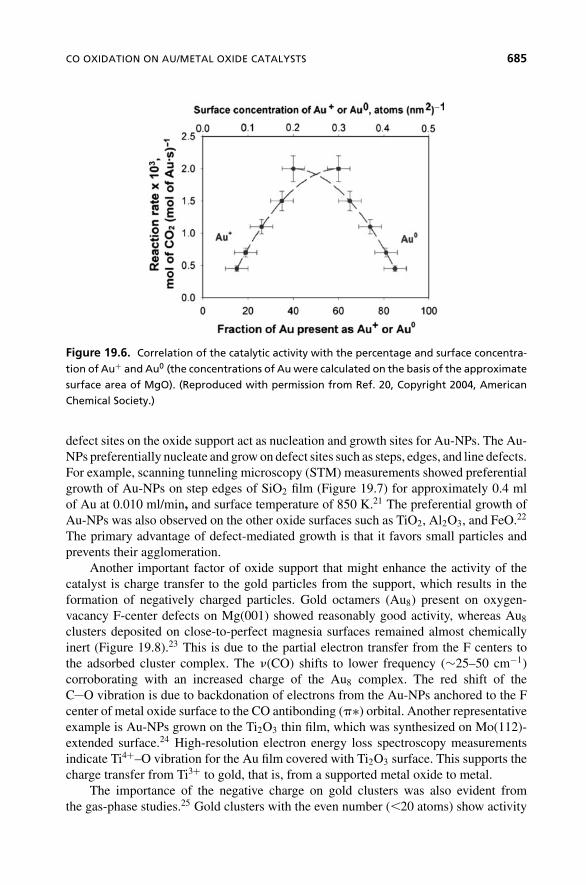
CO OXIDATION ON AU/METAL OXIDE CATALYSTS 685
Figure 19.6. Correlation of the catalytic activity with the percentage and surface concentra-
tion of Au+ and Au0 (the concentrations of Au were calculated on the basis of the approximate
surface area of MgO). (Reproduced with permission from Ref. 20, Copyright 2004, American
Chemical Society.)
defect sites on the oxide support act as nucleation and growth sites for Au-NPs. The Au-NPs preferentially nucleate and growon defect sites such as steps, edges, and line defects.For example, scanning tunneling microscopy (STM) measurements showed preferentialgrowth of Au-NPs on step edges of SiO2 film (Figure 19.7) for approximately 0.4 mlof Au at 0.010 ml/min, and surface temperature of 850 K.21 The preferential growth ofAu-NPs was also observed on the other oxide surfaces such as TiO2, Al2O3, and FeO.22
The primary advantage of defect-mediated growth is that it favors small particles andprevents their agglomeration.
Another important factor of oxide support that might enhance the activity of thecatalyst is charge transfer to the gold particles from the support, which results in theformation of negatively charged particles. Gold octamers (Au8) present on oxygen-vacancy F-center defects on Mg(001) showed reasonably good activity, whereas Au8clusters deposited on close-to-perfect magnesia surfaces remained almost chemicallyinert (Figure 19.8).23 This is due to the partial electron transfer from the F centers tothe adsorbed cluster complex. The �(CO) shifts to lower frequency (∼25–50 cm−1)corroborating with an increased charge of the Au8 complex. The red shift of theC O vibration is due to backdonation of electrons from the Au-NPs anchored to the Fcenter of metal oxide surface to the CO antibonding (�∗) orbital. Another representativeexample is Au-NPs grown on the Ti2O3 thin film, which was synthesized on Mo(112)-extended surface.24 High-resolution electron energy loss spectroscopy measurementsindicate Ti4+–O vibration for the Au film covered with Ti2O3 surface. This supports thecharge transfer from Ti3+ to gold, that is, from a supported metal oxide to metal.
The importance of the negative charge on gold clusters was also evident fromthe gas-phase studies.25 Gold clusters with the even number (�20 atoms) show activity

686 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
Figure 19.7. STM images (200 nm × 200 nm, UT = 1.8 V, and I = 0.08 nA) of Au clusters on
a 1200 K annealed SiO2 film after deposition of approximately 0.4 ml at 0.010 ml/min, surface
temperature of 850 K. (Reproduced with permission from Ref. 21, Copyright 2004, American
Chemical Society.)
Figure 19.8. Mass spectrometric data pertaining to the formation of CO2 on Au8 complex
supported on (a) F-center-rich MgO(001) thin film and (b) F-center-free MgO(001) thin film.
(Adapted from Ref. 23.)

DIRECT SYNTHESIS OF H2O2 FROM H2 AND O2 687
toward O2 adsorption, suggesting their strong activity for CO oxidation, whereas clusterswith the odd number are inactive toward O2 adsorption. The even-numbered negativelycharged clusters have an odd number of electrons, which will be donated to adsorbedO2 molecules to achieve closed shell configuration.26
All these observations demonstrate that negative charges on gold clusters increasethe activity of Au/metal oxide catalysts. However, this assertion is opposite to previousdiscussions (Figure 19.6) that positively charged Au clusters are most active.20 Thiscontradiction could be due to the different synthetic procedures used for Au-NPs sup-ported on extended surfaces and micron-sized metal oxide particles. Overall, althoughtheir specific roles are not well defined, both positively and negatively charged particlescan play vital roles in CO oxidation.
In addition, oxide support plays a vital role in O2 adsorption. The CO–O2 titrationexperiments have shown that a significant amount of O2 can adsorb on the Fe2O3 supportand this oxygen readily migrates to the interface followed by the subsequent reactionwith CO.27 Water adsorbed on oxide surface can then facilitate the transfer of O2 tometal oxides. Water present in the reaction mixtures increases the coverage of OHgroups. According to theoretical studies, the O2 adsorption on metal oxide takes placevia charge transfer from TiO2 to O2 in the presence of hydroxyl groups.28 Briefly, inaddition to providing stability to nanoparticles, oxide supports activate O2 moleculesvia direct (through charge transfer to Au) or indirect processes (through transfer of O2via surface hydroxyl groups).
Therefore, themechanism and factors that affect the activity of the catalyst are highlydependent on the nature of the catalyst (both Au and oxide) and reaction conditions(pressure and temperature) used. Although the mechanistic aspects of nano-Au/metaloxide-catalyzed CO oxidation reported in the literature are controversial, these studiesdefinitely give an outline of the factors that need to be taken care of for given conditions.
DIRECT SYNTHESIS OF H2O2 FROM H2 AND O2
Hydrogen peroxide is a widely used oxidizing agent for industrial as well as house-hold applications and can be synthesized via the activation of small molecules, H2 andO2. Since the only by-product of reactions involving H2O2 as an oxidizing agent iswater, it is widely accepted as a green oxidizing agent.29, 30 Especially in industry, it isenvironmentally benign alternative to chlorine-based bleaches. At present, H2O2 is man-ufactured industrially by the oxidation of a substituted anthrahydroquinone to the corre-sponding anthraquinone, in the so-called indirect anthraquinone method (Scheme 19.1),developed by Riedl and Pfleiderer in 1939.31,32 The high cost of production, difficul-ties in the recycling of the anthraquinone derivatives, and requirement of large quan-tities of extraction solvents are the problematic issues associated with this indirectprocess.
In this context, the direct synthesis of H2O2 from molecular H2 and O2 (Eq. 19.1) isan economical and environmentally benign alternative approach to the industrial indirectanthraquinone method.33 However, the key for the development of economically viableprocess for large-scale production of H2O2 lies in maximum utilization of H2 for the

688 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
OH
OH
R = alky group, – ethyl (in general)
R
O
O
R
H2/Pd catalyst
Oxygen-rich air
Scheme 19.1. Indirect anthraquinone process for the synthesis of H2O2.
production of H2O2. This can be achieved by minimizing the nonselective processes(Eqs 19.2, 19.3, and 19.4) such as formation of water via combustion or hydrogenationreactions. This is possible only through the development of highly selective catalysts. Inthe following sections, how systematic catalytic designing led to the higher selectivityof Equation (19.1) is discussed.
H2 + O2 → H2O2 (19.1)
H2 + O2 → H2O+ 12O2 (19.2)
H2O2 + H2 → 2H2O (19.3)
H2O2 → H2O+ 12O2 (19.4)
For the direct synthesis of H2O2 from H2 and O2, the first patent was filed in1914 using Pd catalysts.34 Since then, methods for making H2O2 directly from H2 andO2 have been investigated over the past several decades. In most of the earlier studies,H2/O2 mixtures were used in the explosive region over Pd catalysts at high pressures andelevated temperatures. However, for industrial applications, such conditions are highlydangerous; therefore, rightly, recent studies are focused in carrying out the studies wellbelow the explosive limits using diluted H2/O2 mixtures.
Until recently, Pd (supported on metal oxide) is the highly active among varioustransition metals for the direct production of H2O2 from H2 and O2. In case of Pd, theselectivity is very low in the absence of any external additives such as protons (H+)or suitable anions (Cl− or Br−) and water is obtained as the major product. However,in the presence of 2 × 10−5 M Br− and 0.12 M H2SO4 with O2/H2 ratio of 15, theselectivity is close to 80%. The effect of acid addition on the H2O2 formation is shownin Figure 19.9.35
At higher concentrations of acid, Pd leaches into the reaction medium (PdCl42−)from the Pd/SiO2 catalyst and under the reaction conditions undergoes reduction to givePd-NPs.36 The in situ formed colloidal Pd-NPs act as highly active catalysts. At loweracid concentrations, the presence of Br− ions decreases the dissolution of Pd from the

DIRECT SYNTHESIS OF H2O2 FROM H2 AND O2 689
Figure 19.9. The net formation rate of H2O2 at H2/O2 = 4 as a function of H2SO4 concentra-
tion at (�) 4 × 10−4 M HCl and (�) 4 × 10−4 M NaCl without H2SO4. The data were obtained
after 2 h of reaction. (Reproduced with permission from Ref. 35, Copyright 2006, Elsevier.)
support, resulting in catalysts that are highly selective for the direct synthesis of H2O2.The effect of addition of Br− is shown in Figure 19.10. Selectivities close to 90% canbe obtained in presence of Br−.37 The major disadvantage of this approach is removaland cleanup of the additive ions, which pose serious nongreen issues.
Recent studies have shown that Au-NPs are also active for the direct synthesis ofH2O2. In fact, Au/Al2O3 (1530mmol (g of catalyst)−1 h−1) is more active than Pd/Al2O3
00.00
(a) (b)
0.050.100.150.200.25
H2O
2 w
t (%
)
Co
nvers
ion
/sele
cti
vit
y (
%)
H2O
2 w
t (%
)
0.300.350.400.450.50
0.00.20.40.60.81.01.21.41.61.82.0
1 2 3Time (h)
4 5 6 7 0Time (h)
5 10 15 20 250
20
40
60
80
100
Co
nvers
ion
/sele
cti
vit
y (
%)
0
20
40
60
80
100
Figure 19.10. Formation of H2O2 (a) in the absence of Br− and (b) in the presence of Br−
(0.01 M): (•) wt% H2O2, (�) H2 conversion, and (�) selectivity for H2O2. The solution was
0.1 N in HCl and the O2/H2 = 4 : 1. (Reproduced with permission from Ref. 37, Copyright 2004,
Elsevier.)

690 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
(370 mmol (g of catalyst)−1 h−1) at 2 ◦C in methanol. In these reactions, the use of lowtemperatures favors high selectivity.38 Alloying of Pd with Au has drastic effect on theactivity and selectivity of the reaction. Interestingly, using Au–Pd-NPs high selectivitiescan be achieved in the absence of external additives such as acids and halides. Therate of H2O2 formation under the same reaction conditions, for Au–Pd(1 : 1)/Al2O3catalysts, is 4460 mmol (g of catalyst)−1 h−1. The exact role of Au was not outlinedin the literature; however, the enhanced catalytic activity could be attributed to “thesynergistic effect between two kinds of metals”—a commonly given explanation inthe literature for greater activity of bimetallic systems compared to individual systems.Among various precious metals, alloying of Au or Pt with Pd has positive effect onthe catalytic properties, whereas Rh and Ru are detrimental for H2O2 yields.39 TheAu–Pd/metal oxide or carbon is the most studied system for the direct synthesis ofH2O2.
The Au–Pd/Al2O3 catalysts were prepared using the coimpregnation method via anincipient wetness technique using aqueous solutions of PdCl2 and HAuCl4·3H2O, fol-lowed by calcination at high temperatures. The calcination temperature has pronouncedeffect on the activity and stability of the Au–Pd catalyst. The effect of calcination temper-ature on H2O2 yields is shown Figure 19.11.40,41 The activity of Au–Pd/Al2O3 catalystsdecreases with increase in temperature from 200 to 400 ◦C. The sample calcined at120 ◦C loses 75–80% of metal content from the support oxide surface into solutionafter the first cycle. On the other hand, the sample calcined at 400 ◦C is quite stablefor reuse. Therefore, calcination at higher temperature is necessary to prevent leaching
Figure 19.11. H2O2 productivity using Au–Pd/Al2O3 catalysts as a function of calcination
temperature to make the catalysts. (Reproduced with permission from Ref. 41, Copyright
2008, American Chemical Society.)

DIRECT SYNTHESIS OF H2O2 FROM H2 AND O2 691
(i)
(ii)
(iii)
(a)
Au-Mαα Pd-Lαα RGB
(b) (c)
(a) (b) (c)
(a) (b) (c)
Figure 19.12. Montage of STEM-XEDS data from Au–Pd/Al2O3 catalysts at various calcination
temperatures: (i) uncalcined, (ii) calcined at 200 ◦C, and (iii) calcined at 400 ◦C. (a) Au-Mα, (b)
Pd-Lα, and (c) RGB reconstruction, where red, Al; green, Pd; and blue, Au. (Reproduced with
permission from Ref. 41, Copyright 2008, American Chemical Society.) (See color insert.)
of the metal from the metal oxide surface into solution, even though it results in lossof some activity. The loss of activity is mainly due to the structural changes (includingchanges in oxidation state) with a change in calcination temperature. The change fromhomogeneous alloy to Pd-rich shell/Au-rich core structure is shown in Figure 19.12.
In contrast to metal oxides, carbon-supported Au–Pd alloy NPs retain their alloystructure even after calcination at 400 ◦C and thereby their activity. The activities

692 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
TABLE 19.1. Formation of hydrogen peroxide using Au–Pd-NP-supported catalystsa
CatalystHydrogen peroxide formation/
mol H2O2 h−1 kgcat−1 Hydrogen selectivity (%)
Au–Pd/carbon 110 80Au–Pd/Al2O3 15 14Au–Pd/TiO2 64 70
aReaction conditions: 0.01 g of catalyst, H2/O2 = 1 : 2 at a total pressure of 3.7 MPa in methanol and watersolvent mixture; Au and Pd are 2.5 wt%.Adapted from Ref. 42.
of Au–Pd catalysts on different supports are listed in Table 19.1. The activity order isAu–Pd/carbon�Au–Pd/TiO2 �Au–Pd/Al2O3.42 The carbon-supported Au–Pd catalystshows selectivity up to 80%. Treatment of a carbon ormetal oxide support with acid priorto the addition of metal salt has pronounced effect on the selectivity of the reaction.43
For the carbon support treated with 2% HNO3, the 2.5 wt% Au–2.5 wt% Pd/carboncatalyst showed more than 98% selectivity at 2 ◦C for the direct synthesis of H2O2from H2 and O2. This is due to the complete inhibition of one of the possible sidereactions, that is, H2O2 hydrogenation/decomposition (Eqs 19.3 and 19.4). Pretreatmentof carbon support with acids leads to the formation of smaller sized Au–Pd alloyparticles (Figure 19.13a); these small particles presumably block the reaction centersfor hydrogenation/decomposition of H2O2.
Pretreatment of metal oxide supports with acids has similar effect on the selectiv-ity of direct H2O2 synthesis. For acid-pretreated Au–Pd/TiO2 catalysts, the selectivity
Acid pretreatedUntreated
10
5
10
15
20
25
2 3 4 5 6 7 8 9 10
Particle size (nm)(a) (b)
Fre
qu
en
cy (
%)
0
5
10
15
20
25
30
35
40
Nu
mb
er
of
part
icle
s
11 12 13 14 15 16 17 18 19 >20 2 3 4 5 6 7 8 9 14
Particle size (nm)
15 17 55 87 115
128
132
142
162
185
Acid pretreatedUntreated
Figure 19.13. Particle size distribution of alloy particles for the untreated and acid-pretreated
(a) Au–Pd/carbon catalysts and (b) Au–Pd/Al2O3. (Adapted from Ref. 43 (Figure 19.13a;
reprinted with permission from Ref. 44, Copyright 2009, Wiley-VCH.)

METHANOL SYNTHESIS FROM CO AND CO2 693
increased to 95% from approximately 70% for untreated catalysts.44 However, in thiscase both hydrogenation and decomposition reactions do not get affected by acid treat-ment as in the case of carbon-supported catalysts. But similar to the carbon-supportedcatalysts, the Au–Pd-NPs are more dispersed and larger sized particles decreased sig-nificantly for acid-pretreated samples (Figure 19.13b). In this case, the enhanced selec-tivity for acid-pretreated samples is due to more dispersed small-sized particles. Also,acid-pretreated catalysts can be successfully used even at ambient temperatures. Theacid-pre-treated Au–Pd/TiO2 catalyst at 40 ◦C retains greater activity than the untreatedcatalyst at 2 ◦C.
Now,with selectivities as high as 95%at ambient temperatures, these stable catalystsdefinitely find their applications in industrial processes. The major advantage with thedirect H2O2 synthesis process includes the possibility to easily produce H2O2 withconcentrations of 3–8% levels required in most medical and chemical applications. Thismethod also allows for local production of H2O2 whenever required, which thereforeeliminates the need for transport and storage.
METHANOL SYNTHESIS FROM CO AND CO2
With a capacity of about 30 million tons per year worldwide, methanol is one of themost important feedstock for many organic chemicals. Hydrogenation of CO or CO2(derived from fossil fuels) over a heterogeneous catalyst is the widely used methodologyfor large-scale production of methanol. In the early 1920s, large-scale industrial produc-tion of methanol was based on the sulfur-resistant zinc oxide–chromium oxide catalyst.However, this catalyst requires high pressure (250–350 bar) and high temperature (320–450 ◦C). In the 1960s, ICI developed a highly selective Cu/ZnO/Al2O3 catalyst, puttingan end to the high-pressure methanol synthesis.45, 46 In general, industrial catalysts con-tain more than 50% of Cu. The copper-based catalysts could be operated at fairly milderconditions (i.e., 50–100 bar and ∼250 ◦C). Today’s methanol is produced industriallyfrom syn-gas (CO + H2) using this catalyst (Eqs 19.5 and 19.6). In addition to hydro-genation of CO and CO2 reactions, the water–gas shift reaction plays an important rolein determining the overall efficiency of the reaction (Eq. 19.7).
CO+ 2H2 � CH3OH (19.5)
CO2 + 3H2 � CH3OH+ H2O (19.6)
CO+ H2O � CO2 + H2 (19.7)
Industrial Cu/ZnO/Al2O3 catalysts for methanol synthesis are commonly synthe-sized by coprecipitating copper zinc hydroxycarbonates from Cu(NO3)2 and Zn(NO3)2solutions. Subsequent treatments of the precipitate (i.e., aging, washing, drying, calci-nation, and reduction) have an effect on the microstructure of the resulting structureand thereby the activity.47 Therefore, this is a subject of intensive study and severalsuggestions have been made as to what factors affect the activity of the catalyst.48, 49
However, due to the complex nanostructure of the industrial catalyst, the structure–activity relationship is rather poorly understood. The best way to understand such a

694 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
complex relationship is to deduce the information from simpler model systems first andthen extend the understanding to complex catalysts. Extensive research has been carriedout in this direction during the last few decades and several well-detailed reviews areavailable in the literature on this topic.47,50–52 Therefore, in the following discussion, wedo not attempt a systematic review of the literature, but rather we emphasize more on theemerging trends since the last decade. For simplicity, the established structure-activityrelationship for Cu/ZnO, Cu/ZnO/Al2O3 catalysts is discussed separately.
Cu/ZnO Binary Catalysts
The individual counterparts of Cu/ZnO binary catalysts show very less activity towardmethanol synthesis from syn-gas. The copper particles synthesized using the coprecipi-tation method showed an activity less than 10−8 kgCH3OH m−2 h−1 from CO/CO2/H2 =24/6/70 mixture at 75 atm and 250 ◦C. The ZnO catalyst, with a surface area of 40 m2/g,showed an activity of 10−9 kgCH3OH m−2 h−1 at the same standard reaction conditions.Under similar conditions, Cu/ZnO binary catalysts showed several orders of activitygreater than those of individual components (3.63 × 10−5 kgCH3OH m−2h−1).53 In someof the earlier studies, an assumption was made that the activity of the Cu/ZnO catalystincreased linearly with increase in the Cu surface area.54 According to these studies, therole of metal oxides is limited to only providing stabilization to Cu-NPs. In these studies,the Cu surface areas were measured by chemisorption of N2O. However, promotion ofbulk oxidation of copper in Cu/ZnO/Al2O3 catalysts led to an overestimation of thesurface copper atoms.55 Therefore, later studies have ruled out this linear relation, androle of metal oxides was extended beyond stabilization of Cu-NPs. These results sug-gest strong synergetic effects between the metal and metal oxide and undoubtedly playcrucial roles in deciding the activity and selectivity of the binary catalyst for methanolsynthesis from syn-gas. Many models and explanations are available in the literature todetermine the active site and to understand the structure–activity relationship. Most ofthese results are controversial due to the complex nature of Cu/ZnO, similar to the caseof the heterogeneous catalysts. Some of the proposals include
1. copper oxidation state (active site);a) insertion of Cu+1 onto ZnO47,56, 57
b) metallic copper (Cu0) supported on ZnO46,58, 59
c) Schottky–Mott junction at Cu–ZnO interface—electron transfer from ZnO toCu to leave a partial negative charge on copper60
2. formation of dynamic surface and bulk alloy, depending on the reduction poten-tial of the synthesis gas61,62;
3. segregation of ZnO on copper surface.63–65
Recent advances in characterization techniques, especially electron microscopytechniques, helped establish some of these aspects with more certainty. For instance,“change in the morphology of the Cu-NPs in Cu/ZnO binary catalysts with respectto potential of synthesis gas mixtures” is one of the well-studied aspects. Reversible

METHANOL SYNTHESIS FROM CO AND CO2 695
Figure 19.14. In situ HRTEM images of a Cu/ZnO catalyst in various gas environments: (a) at
a pressure of 1.5 mbar of H2 at 220 ◦C; (b) in a gas mixture of H2 and H2O, H2/H2O = 3 : 1 at a
total pressure of 1.5 mbar at 220 ◦C; (c) in a gas mixture of H2 (95%) and CO (5%) at a total
pressure of 5 mbar and 220 ◦C. (Adapted from Ref. 68.)
structural changes of Cu-NPs in the binary catalyst were predicted from in situ X-raydiffraction (XRD) and X-ray absorption fine structure spectroscopy data.66,67 Visualiza-tion of these changes was demonstrated by in situ high-resolution microscopy.
In situ characterization techniques provide very useful information about the state ofcatalytic nanomaterials during catalysis. In situ high-resolution TEM studies performedon Cu/ZnO model catalysts showed reversible shape changes in response to changes ingaseous environment.68 The Cu/ZnO model catalyst was synthesized by impregnationof copper acetate solution on ZnO support, and the Cu-NPs were produced in situby reduction of CuO with hydrogen. The high-resolution TEM (HRTEM) images inFigure 19.14 a,b, and c show equilibrium shapes of Cu/ZnO in various gas environments.The Cu nanocrystals obtained after reduction with H2 were exposed to more oxidizingatmosphere (i.e., H2O/H2 = 3 : 1 mixture for 1 h). This resulted in the shape change ofparticles to the more spherical morphology. Also, Cu crystals are terminated by morefraction of (110) and (111) facets compared to the case wherein pure hydrogen is used.However, at the same time theCu/ZnO interface is notmuch affected byH2/H2Omixture.This suggests that adsorption of water on different exposed Cu facets is the main reasonfor the observed shape change. Notably, such shape changes are reversible. Additionof CO (more reducing gas) to H2 gas has more remarkable effect on the shape of Cuparticles. Disc-like Cu particles are formed due to an increase in the interfacial area byabout 50%. As shown in Figure 19.14c, now Cu-NPs are predominately terminated by(111) and (100) facets with the (111) facet parallel to the ZnO surface. This is probablydue to the decrease in the interfacial energy by the reduction of ZnO surface layer in themore reducible environment. The observed restructuring of the catalyst in presence ofH2, CO, and H2O demonstrates that generation of the relevant catalytic sites is possibleduring methanol synthesis.
Tailoring of microstrain in Cu-NPs is the crucial step in a rational catalyst design.69
Although each step in the preparation of the catalyst has an effect on the final microstruc-ture of the catalyst, aging of the precursor has strong influence on the activity. The

696 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
30
Irel
40 50 60 70
120 min
0 min
2θ / deg
80 90
Figure 19.15. In situ X-ray powder diffraction patterns of two Cu/ZnO catalysts (�, Cu phase;
◦, Zn phase) obtained from precursors, aged for 0 and 120 min, measured at 250 ◦C under
methanol steam-reforming conditions (Irel = intensity). The Pawley method was used to sim-
ulate the experimental patterns by a sum of profile functions and a background polynomial.
(Reprinted with permission from Ref. 69, Copyright 2004, Wiley-VCH.)
samples aged for more than 30 min in the mother liquor showed significant activitycompared to the nonaged samples. The corresponding structure–activity relationshipwas studied using XRD, nuclear magnetic resonance (NMR), and HRTEM techniques.A range of samples were prepared from differently aged (0, 15, 30, and 120 min) precur-sor samples for Cu–Zn = 70 : 30. Figure 19.15 shows the powder XRD patterns of thesamples aged for 0 min and 120 min. The Cu(111) reflection analysis showed that Cuparticle size decreased from 11 nm (0 min) to 7 nm (120 min) and the microstrain in theCu particles increased from 0.09% (0 min) to 0.26% (120 min). The63Cu NMR spectralstudies (Figure 19.16) further confirmed these observations. The line shape changesrevealed the development of microstrain with increase in aging time. An increase in themicrostrain leads to asymmetric profiles or even additional signals. On the other hand,unsupported Cu-NPs prepared using similar procedure showed almost negligible strainof 0.06%. This clearly indicates the strong role played by ZnO support in inducing strainin Cu-NPs. The origin of the microstrain could be from the epitaxial orientation of Cuand ZnO or lattice imperfections or incomplete reduction of Cu.
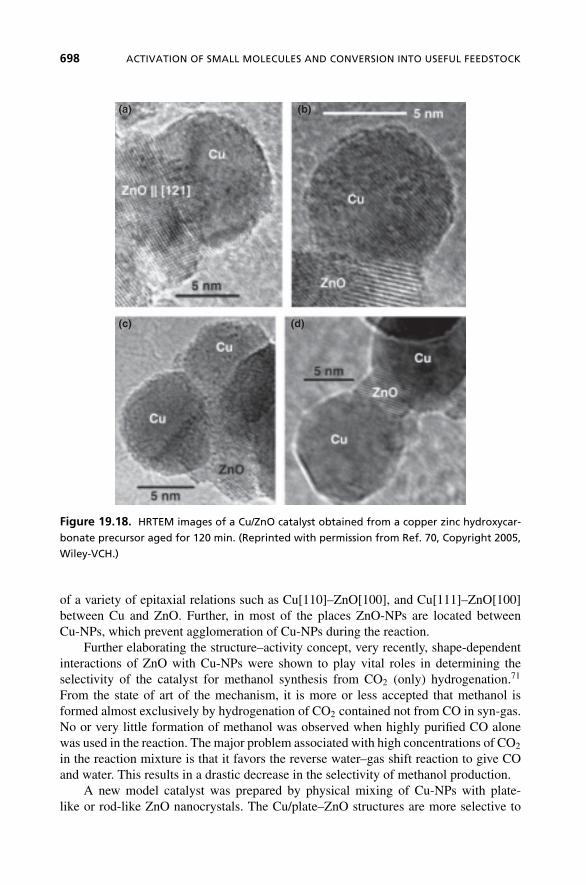
Further evidence for the presence of microstrain in Cu-NPs of Cu/ZnO catalystwas deduced using HRTEM analysis.70 The Cu/ZnO catalyst showed drastic changes inthe structure for differently aged samples. The bright field TEM images of the unagedsample showed the presence of a heterogeneous mixture of large and small particlesand isolated Cu and ZnO particles. On the other hand, the sample aged for more than30 min revealed to be more homogeneous with intimate contact between Cu and ZnO.This structural change exactly correlates with the high catalytic activity (Figure 19.17).The HRTEM images (Figure 19.18) of more aged Cu/ZnO samples reveal the presence

METHANOL SYNTHESIS FROM CO AND CO2 697
63Cu NMR
0 min
15 min
30 min
120 min
0 1000 2000
Irel
3000
δ / ppm
4000
Figure 19.16.63Cu NMR spectra of Cu/ZnO catalysts prepared from precursors, aged for 0, 15,
30, and 120 min, measured at 4.2 K (Irel = intensity). (Reprinted with permission from Ref. 69,
Copyright 2004, Wiley-VCH.)
0
200 nm
20 nm
120 min
600
700RH2
800
900
0 min
20 40 60 80 100 120
Precipitate aging time (min)
Figure 19.17. Bright field TEM images of Cu/ZnO catalysts obtained from hydroxycarbon-
ate precipitates aged for 0 min and 120 min together with the H2 production rate RH2
[mmol g−1 s−1] of four catalysts as a function of precipitate aging time (methanol–synthesis
activity and aging time exhibited a similar correlation). (Reprinted with permission from Ref.
70, Copyright 2005, Wiley-VCH.)
698 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
(a) (b)
(c) (d)
Figure 19.18. HRTEM images of a Cu/ZnO catalyst obtained from a copper zinc hydroxycar-
bonate precursor aged for 120 min. (Reprinted with permission from Ref. 70, Copyright 2005,
Wiley-VCH.)
of a variety of epitaxial relations such as Cu[110]–ZnO[100], and Cu[111]–ZnO[100]between Cu and ZnO. Further, in most of the places ZnO-NPs are located betweenCu-NPs, which prevent agglomeration of Cu-NPs during the reaction.
Further elaborating the structure–activity concept, very recently, shape-dependentinteractions of ZnO with Cu-NPs were shown to play vital roles in determining theselectivity of the catalyst for methanol synthesis from CO2 (only) hydrogenation.71
From the state of art of the mechanism, it is more or less accepted that methanol isformed almost exclusively by hydrogenation of CO2 contained not from CO in syn-gas.No or very little formation of methanol was observed when highly purified CO alonewas used in the reaction. The major problem associated with high concentrations of CO2in the reaction mixture is that it favors the reverse water–gas shift reaction to give COand water. This results in a drastic decrease in the selectivity of methanol production.
A new model catalyst was prepared by physical mixing of Cu-NPs with plate-like or rod-like ZnO nanocrystals. The Cu/plate–ZnO structures are more selective to

METHANOL SYNTHESIS FROM CO AND CO2 699
0.26 nm
[0001]
200 nm 500 nm
(b)(a)
(001)
(100)[0001]
(00-1)
(c) (d)
Figure 19.19. Bright field TEM images of (a) ZnO nanoplates and (b) ZnO nanorods. (c) HRTEM
image of ZnO nanorod and (d) crystallographic planes of ZnO nanorods along the [0001] rod
axis. (Reprinted with permission from Ref. 71, Copyright 2011, Wiley-VCH.)
methanol formation from CO2 and H2 than the Cu/rod–ZnO structure. For example,the selectivities for methanol formation from CO2 and H2 (1:2.2) are 39.1 and 71.6for Cu/rod–ZnO/Al2O3 and Cu/rod–ZnO/Al2O3 catalysts, respectively, at 543 K. Theobserved selectivity difference can be explained on the basis of the shape-dependentinteractions at the interfaces.
The plate-like ZnO nanocrystals predominately possess (002) polar facets, whereasrod-like structures have relatively higher nonpolar (100) and (101) planes along the[0001] rod axis (Figure 19.19). The interactions between Cu and ZnO are strongerin case of plate-like particles in comparison to those in rod-like particles due to thepolar facets covering the structure. When mixed with Cu, the temperature programmedreduction (TPR) peak (at 750 ◦C) corresponding to the reduction of the (002) facet ofZnO plates is strongly affected, and a peak at 750 ◦C is replaced by a reduction peakwith an onset from 200 ◦C and peaking at 500 ◦C. On the other hand, in case of rodsthe TPR peak (at 600 ◦C) corresponding to the reduction at the nonpolar facets changedmarginally. The strong interactions between the polar facets of ZnO plates and Cuwere further confirmed by X-ray photoelectron spectroscopy and electron paramagnetic

700 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
EV
Cu ZnO
ECEf
EVAC
4.2 eV
0.45 eV = φ(b) φ(t)
Cu
ZnO
(a) (b)
Figure 19.20. Band diagrams for Cu/ZnO junction: (a) unrelaxed Cu/ZnO junction, (b) Cu/ZnO
junction after vacancy creation and electron transfer. Here, Evac is vacuum level, Ev is valency
band edge, Ef is Fermi level, Ec is conduction band edge, φ(b) is Schottky barrier height, and
�(t) is band-bending height. (Adapted from Ref. 60.)
resonance studies. These data show transfer of electrons and oxygen atoms from ZnOto Cu through the Schottky–Mott junction formation at the interface. This results in theformation of CuO and oxygen vacancies in ZnO phase at the interface. Formation ofthe Schottky–Mott junction at the Cu–ZnO interface was proposed by Frost in 1988(Figure 19.20).60 The transfer of a pair of electrons to a lower energy metal Fermi levelfrom the oxide conduction band effectively reduces the enthalpy of formation of doublyionized oxygen vacancy by 2(Ec − Ef) = 2(b), where Ec is conduction band edge, Efis Fermi level, and (b) is Schottky barrier height.
Cu/ZnO/Al2O3 Ternary Catalysts
As stated earlier, the structure of Cu/ZnO/Al2O3 ternary catalysts is much more complexthan the Cu/ZnO binary catalyst. However, it should be possible to extend some of therecently developed concepts of the binary system to the ternary catalyst, with the aid ofadvanced characterization techniques.
A Cu/ZnO/Al2O3 ternary catalyst was prepared by coprecipitation of metal nitratesolution with sodium carbonate followed by calcination at 330 ◦C and reduction inhydrogen gas at 250 ◦C.72 This catalyst exhibits a molar ratio of Cu/Zn/Al= 60 : 30 : 10,which is characteristic of the commercial catalyst. Complex nanostructure features ofternary catalysts were investigated by HRTEM studies (Figure 19.21). Similar to thebinary system, the ZnO-NPs serve as spacers between particles. Cu-NPs are sphericaland covered partially or completelywith ZnO. TheAl2O3 is presentmostly in amorphousor poorly crystallized forms in the catalyst. Also, particles containing traces of Cu andZn were noted. The HRTEM images of such particles showed the presence of spinel-likecubic structure with lattice distances closer to those of ZnAl2O4. The EDX data fromsuch clusters further confirm the presence of ZnAl2O4 spinel in the ternary catalyst.Interestingly, most of the Cu-NPs contained in the ternary catalyst contained defects

METHANOL SYNTHESIS FROM CO AND CO2 701
(a) (b)
(c) (d)
Figure 19.21. Microstructural features of the Cu/ZnO/Al2O3 catalyst revealed with TEM and
HRTEM. (Reprinted with permission from Ref. 72, Copyright 2007, Wiley-VCH.)
such as twins and stacking faults. Most of the observed twin boundaries were onlypartially coherent; that is, regular packing of atoms was interrupted by missing line inthe direction of twin boundary. In case of small Cu-NPs, most of the lattice defectsterminate at the surface, which creates useful sites for catalysis. It has been shown thatthe activity is proportional to the overall faulting probability 1.5� + , where � isthe stacking fault probability and is the twinning probability. Also, another closelyrelated parameter, the strain that is strongly interlinked with defects, is proportional tothe activity. These results suggest that the presence of stable nonequilibrium structures(twin and stacking faults) in Cu creates unique geometric and electronic situations,which accounts for the high activity of the Cu/ZnO/Al2O3 catalyst.
By twomajor modifications to the regular protocol for the synthesis of the industrialcatalyst, Behrens et al. synthesized a novelCu/ZnO/Al2O3 catalystwith excellent activityfor methanol synthesis.73 The twomodifications included (1) the precipitation of Cu/Zn–nitrate solution using sodium aluminate and carbonate solution, instead of precipitating

702 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
Figure 19.22. HRTEM image of the Cu/ZnO/Al2O3 catalyst obtained from modified procedure
(see the text). (Reprinted with permission from Ref. 73, Copyright 2010, Wiley-VCH.)
Cu/Zn/Al–nitrate solution using sodium carbonate solution; (2) the conventional agingstep, which was replaced by a continuous process with direct spray-drying of the freshprecipitate suspension. The microstructure of the resultant catalyst was analyzed byHRTEM technique (Figure 19.22). The Cu-NPs are embedded partly in disorderedZnO/Al2O3 matrix. Unlike the industrial catalysts, separate metal oxide nanoparticlesare not observed, which means that the typical Cu/ZnO arrangement is absent in thiscatalyst. On the other hand, similar to the industrial catalyst, Cu-NPs are spherical withsizes in the range of 4–9 nm. As expected, the Cu surface area of this novel catalyst is64% of the industrial catalyst due to its partly embedded nature. Surprisingly, in caseof the novel catalyst, intrinsic activity of Cu is greater by 64%. Embedding of Cu-NPsin oxide matrix leads to unavoidable loss of Cu surface area; at the same time, it helpsto achieve higher degree of metal–metal oxide interface area. The average coverage ofCu-NPs with oxide (ZnO/Al2O3) is 35% for the industrial catalyst and 50% for this novelcatalyst. This makes the available Cu surface highly active for methanol formation fromthe syn-gas reaction due to the presence of strong metal support interactions betweenCu and ZnO/Al2O3. This example stresses the need for giving more focus on attainingoptimal Cu metal oxide interface/surface area of the Cu ratio rather than just increasedCu surface areas in Cu/ZnO/Al2O3 catalysts.
Quasihomogeneous Catalyst
It is clear from the above discussion that model catalysts are highly useful in under-standing the structure–activity relationship. In this regard, use of colloidal nanoparticlesas catalysts (quasihomogeneous catalysts) for methanol synthesis is a new concept that

METHANOL SYNTHESIS FROM CO AND CO2 703
CuO
O
N
N
HDA
Squalane, 200 οC
Cu +
Me2N
O
Me2N
OH
ZnO/Cu + ......
Squalane, 200 οC
ZnEt2
Scheme 19.2. Reaction pathways involved in synthesis of HDA/Cu and ZnO/Cu colloids.
(Adapted from Ref. 77.)
has evolved recently. This allowed further elaboration of understanding of the structure–activity relationship.
Copper is the key component that plays a major role in deciding the catalytic activ-ity of the Cu/ZnO/Al2O3 catalyst. Single crystalline surfaces of Cu were also reportedto be active for methanol synthesis.74, 75 However, the extent of activity is much lesscompared to the binary system. Copper colloidal nanoparticles were studied as quasi-homogeneous catalysts for methanol synthesis from syn-gas. Deep red copper colloidswere obtained by reduction of copper acetylacetonate in tetrahydrofuran (THF) withAl(n-octyl)3 or Al(n-butyl)3. The organoaluminum compounds stabilize Cu-NPs byforming an organometallic protecting shell. Using this procedure, Cu-NPs with a sizerange of 3–5 nm were obtained. Freshly prepared Cu colloids in THF showed excep-tional activities toward methanol synthesis in a quasihomogeneous phase. At 150 ◦C, aproductivity of 4.0 molmethanol/(kgCu h) was obtained. Under similar reaction conditions,the commercial catalyst gave a productivity of 5.5 molmethanol/(kgCu h). However, thesestudies have shown that the presence of Zn is not essential to induce higher activity formethanol synthesis from syn-gas.
The quasihomogeneous catalysis for methanol synthesis was later extended to theZnO/Cu binary system. Synthesis of such binary systems in colloidal state is a syn-thetic challenge, and well-designed synthetic protocols are necessary to synthesizesuch systems, especially in nonaqueous medium.76 The ZnO surface-decorated Cu-NPs(ZnO/Cu) were synthesized by sequential copyrolysis of [Cu(OCHMeCH2NMe2)2] andZnEt2 in squalane at 200 ◦C (Scheme 19.2).77 Surprisingly, the colloids obtained usingthis procedure were stable to precipitation even in the absence of capping agents andthe particles obtained were very small (1–3 nm). The extended X-ray absorption finestructure (EXAFS) data confirmed the presence of small Cu-NPs together with a nonag-gregated oxygen-deficient ZnO phase. The catalytic activity tests were carried out insqualane due to its high boiling point, low viscosity, and higher solubility of gases usedin the reaction. The comparison of catalytic activities with reference to industrial cata-lysts is given in Table 19.2. The ZnO/Cu (50 : 50) catalyst showed a remarkable activityof 84.1% of the ternary reference catalyst. These results again reinforce the outcomeof experiments on single crystalline surfaces that the presence of ZnO enhances thecatalytic activity of Cu for methanol synthesis from the syn-gas reaction.

704 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
TABLE 19.2. Catalytic activity of HDA/Cu and ZnO/Cu colloids referenced to an industrialcatalyst (slurry) under the same conditions
Sample Productivity/�mol gcat−1 h−1 Activity (%)
HDA/Cu 9 0.4ZnO/Cu (25 : 75) 1371 58.5ZnO/Cu (50 : 50) 1971 84.150 CuO/35 ZnO/15 Al2O3 2342 100
Adapted from Ref. 77.
In another such approach, zinc stearate and copper stearate in squalane were reducedwith diluted H2 to yield dark red Cu colloids (5–10 nm) with Zn stearate shell.78 Here,zinc stearate acts as a stabilizer and is not reduced by H2. These colloids showed excep-tional activities toward methanol synthesis. It has been found that Zn stearate undergoeshydrolysis under reaction conditions to yield ZnO. The active state of catalysts wasfound to have ZnO prism nanoparticles with truncated edges connected to Cu nanopar-ticles (Figure 19.23). The sequence of reactions involved in the synthesis methodology
Figure 19.23. HRTEM and corresponding fast Fourier transform images of the active state of
the Cu/ZnO catalyst after heat pretreatment, severe reduction, and 53 h on mixture of 72 vol%
H2, 10 vol% CO, 4 vol% CO2, and balance N2 stream. (Reprinted with permission from Ref. 78,
Copyright 2010, Wiley-VCH.)

METHANOL SYNTHESIS FROM CO AND CO2 705
Figure 19.24. Rate of methanol formation from syn-gas (72 vol% H2, 10 vol% CO, 4 vol%
CO2, and balance N2) over Cu–Zn stearate colloids with different Cu/Zn ratios and a reference
catalyst (�, Cu/ZnO/Al2O3; �, 25 : 75; ◦, 50 : 50; �, 75 : 25). Reaction conditions: 493 K, 2.6 MPa.
The dashed line in the figure indicates the mixing time. (Reprinted with permission from Ref.
78, Copyright 2010, Wiley-VCH.)
is shown in Equations (19.8) and (19.10).
Cu{CH3 (CH2)16 COO}2 + H2 → Cu+ 2CH3 (CH2)16 COOH (19.8)
CH3 (CH2)16 COOH+ 2H2 → CH3 (CH2)16 CH2OH+ H2O (19.9)
Zn {CH3 (CH2)16 COO}2 + H2O → ZnO+ 2CH3 (CH2)16 COOH (19.10)
The Cu–Zn colloids obtained using these methodologies are more active than theindustrial catalyst for methanol synthesis. The rate of reaction was found to be twofoldfaster than the industrial catalyst (Figure 19.24). The productivity of methanol is 6408,6222 �molCH3OH g−1
Cu h−1 at 493 K for Cu–Zn stearate (50 : 50) and Cu/ZnO/Al2O3(slurry) catalysts, respectively.
The observed enhanced activity of Cu–Zn colloids could be ascribed to themigrationof ZnOx species to the Cu surface. Here, ZnO acts as a reservoir for ZnOx species. TheCu–ZnOx- interface was confirmed by attenuated total reflection-infrared spectroscopy(ATR-IR) results. The IR bands observed at 1970 cm−1 and 1930 cm−1 were assignedto CO adsorbed on Cu sites with Zn adatoms. These results are in accordance with theobservation made in case of the single crystalline model catalysts that migration of ZnOadatoms to Cu increases the activity of the catalyst.63, 64

706 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
Overall, the quasihomogeneous catalysts were found to be very useful model cata-lysts for methanol synthesis from syn-gas. Using these catalysts, it is possible to attainactivities higher than that of the industrial catalyst by proper designing of the Cu–ZnOinterface as discussed previously.
SUMMARY AND OUTLOOK
Traditionally, it is considered very difficult to gain an understanding of heterogeneouscatalysts, especially the interface of multicomponent industrial catalysts. With theintroduction of nanoscience to this field, it is possible to separate the complex fac-tors (e.g., size, shape, and support effects) involved in a multicomponent catalyst andevaluate the effect of each factor on the activity and selectivity of the catalyst in asystematic manner.
This is possible through an apt designing of the model catalysts that mimic thereal industrial catalyst in certain aspects. For example, the particle size effect on theAu/TiO2 catalytic system is very well established from Au particles deposited on singlecrystalline TiO2 surfaces. In the three reactions discussed here—CO oxidation, directsynthesis of H2O2, and methanol synthesis from syn-gas—this type of strategy hasunveiled various factors that affect the catalytic properties of a catalytic system undera set of conditions. This type of understanding is very important for rational designingof heterogeneous catalysts, especially for reactions involving small molecule activation,which require highly active catalytic surfaces with greater selectivity. At the same time,care should be taken while generalizing concepts derived from certain model catalysts.This is why some of the concepts are controversial because comparisons were madebetween different kinds of model catalysts.
As discussed in this chapter, the collaboration of nanoscience with heterogeneouscatalysis will help in the development of new catalysts with novel properties for vari-ous reactions, involving energy-intensive processes such as CO oxidation, H2O2 syn-thesis from H2 and O2. Also, this collaboration led to a better understanding of thestructure–activity relationship of well-used industrial catalysts such as Cu/ZnO/Al2O3.Such understanding is essential for the development of next-generation heterogeneouscatalysts, which can activate very stable small molecules such as methane at relativelymilder conditions. Selective oxidation of methane to syn-gas in presence of metal dis-persed metal oxide catalysts is a well-established industrial process, which requires highenergy input.79, 80 The catalysts that activate small hydrocarbons at relatively milder con-ditions will pave a way for the emergence of new technologies for the use of alternateenergy sources.
REFERENCES
1. TolmanW. B. Activation of Small Molecules: Organometallic and Bioinorganic Perspectives.New York: John Wiley & Sons, Inc.; 2006.
2. Somorjai G. A. Introduction to Surface Chemistry and Catalysis. New York: John Wiley &Sons, Inc.; 1994.

REFERENCES 707
3. Thomas J. M., Thomas W. J. Principles and Practice of Heterogeneous Catalysis. Weinheim:Wiley-VCH Verlag GmbH; 1997.
4. Finke R. G. In: Feldheim D. L., Foss C. A., Jr., editors. Metal Nanoparticles: Synthesis,Characterization and Applications. New York: Marcel Dekker; 2002, p. 17–54.
5. Fendler J. H. Nanoparticles and Nanostructured Films. Preparation, Characterization andApplications. Weinheim: Wiley-VCH Verlag GmbH; 1998.
6. Schmid G. In: Klabunde K. J., editor. Nanoscale Materials in Chemistry. New York: Wiley-Interscience; 2001, p. 15–60.
7. Ertl G., Prigge D., Schloegl R., Weiss M. Surface characterization of ammonia-synthesiscatalysts. J. Catal. 1983;79:359–377.
8. Ertl G. Reactions at surfaces: from atoms to complexity (Nobel Lecture). Angew. Chem. Int.Ed. 2008;47:3524–3535.
9. Haruta M., Yamada N., Kobayashi T., Lijima S. Gold catalysts prepared by coprecipitationfor low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989;115:301.
10. Bell A. T. The impact of nanoscience on heterogeneous catalysis. Science 2003;299:1688–1691.
11. Valden M., Lai X., Goodman D. W. Onset of catalytic activity of gold clusters on titania withthe appearance of nonmetallic properties. Science 1998;281:1647–1650.
12. HarutaM., Tsubota S., Kobayashi T., KageyamaH., GenetM. J., DelmonB. Low-temperatureoxidation of CO over gold supported on TiO2, �-Fe2O3, and CO3O4. J. Catal. 1993;144:175–192.
13. Meier D. C., Goodman D. W. The influence of metal cluster size on adsorption energies: COadsorbed on Au clusters supported on TiO2. J. Am. Chem. Soc. 2004;126:1892–1899.
14. Lemire C., Meyer R., Shaikhutdinov S., Freund H. J. Do quantum size effects control COadsorption on gold nanoparticles? Angew. Chem. Int. Ed. 2004;43:118–121.
15. Kim T. S., Stiehl J. D., Reeves C. T., Meyer R. J., Mullins C. B. Cryogenic CO oxidationon TiO2-supported gold nanoclusters precovered with atomic oxygen. J. Am. Chem. Soc.2003;125:2018–2019.
16. Mavrikakis M., Stoltze P., Norskøv J. K. Making gold less noble. Catal. Lett. 2000;64:101–106.
17. Lopez N., Janssens T. V. W., Clausen B. S., Xu Y., Mavrikakis M., Bligaard T., Norskøv J. K.On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation.J. Catal. 2004;223:232–235.
18. Deng X., Min B. K., Guloy A., Friend C. M. Enhancement of O2 dissociation on Au(111)by adsorbed oxygen: implications for oxidation catalysis. J. Am. Chem. Soc. 2005;127:9267–9270.
19. Kim J., Dohnalek Z., Kay B. D. Cryogenic CO2 formation on oxidized gold clusters synthe-sized via reactive layer assisted deposition. J. Am. Chem. Soc. 2005;127:14592–14593.
20. Guzman J., Gates B. C. Catalysis by supported gold: correlation between catalytic activityfor CO oxidation and oxidation states of gold. J. Am. Chem. Soc. 2004;126:2672–2673.
21. Min B. K., Wallace W. T., Santra A. K., Goodman D. W. Role of defects in the nucleationand growth of Au nanoclusters on SiO2 thin films. J. Phys. Chem. B 2004;108:16339–16343.
22. Shaikhutdinov S. K., Meyer R., Naschitzki M., Baumer M., Freund H. J. Size and supporteffects for CO adsorption on gold model catalysts. Catal. Lett. 2003;86:211–219.

708 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
23. Yoon B., Hakkinen H., Landman U., Worz A. S., Antonietti J.-M., Abbet S., Judai K., HeizU. Charging effects on bonding and catalyzed oxidation of CO on Au8 clusters on MgO.Science 2005;307:403–407.
24. Chen M. S., Goodman D. W. The structure of catalytically active gold on titania. Science2004;306:252–255.
25. Wallace W. T., Whetten R. L. Coadsorption of CO and O2 on selected gold clusters: evidencefor efficient room-temperature CO2 generation. J. Am. Chem. Soc. 2002;124:7499–7505.
26. Stolcic D., Fischer M., Gantefor G., Kim Y. D., Sun Q., Jena P. Direct observation of keyreaction intermediates on gold clusters. J. Am. Chem. Soc. 2003;125:2848–2849.
27. Schubert M. M., Hackenberg S., van Veen A. C., Muhler M., Plzak V., Behm R. J. COoxidation over supported gold catalysts – “Inert” and “Active” support materials and theirrole for the oxygen supply during reaction. J. Catal. 2001;197:113–122.
28. Liu L. M., McAllister B., Ye H. Q., Hu P. Identifying an O2 supply pathway in CO oxidationon Au/TiO2(110): a density functional theory study on the intrinsic role of water. J. Am.Chem. Soc. 2006;128:4017–4022.
29. Hage R., Lienke A. Applications of transition-metal catalysts to textile and wood-pulp bleach-ing. Angew. Chem. Int. Ed. 2006;45:206–222.
30. Esmelindro M. C., Oestreicher E. G., Marquez-Alvarez H., Dariva C., Egues S. M. S.,Fernandes C., Bortoluzzi A. J., Drago V., Antunes O. A. C. Catalytic oxidation of cyclohexaneby a binuclear Fe(III) complex biomimetic to methane monooxygenase. J. Inorg. Biochem.2005;99:2054–2061.
31. Riedl H.-J., G. Pfleiderer (I.G. Farbenindustrie AG), Hydrogen peroxide. DE 671318, 1939.
32. Riedl H.-J., G. Pfleiderer (I.G. Farbenindustrie AG), Production of hydrogen peroxide. US2158525, 1939.
33. Edwards J. K., Hutchings G. J. Palladium and gold-palladium catalysts for the direct synthesisof hydrogen peroxide. Angew. Chem. Int. Ed. 2008;47:9192–9198.
34. Henkel H., Weber W. Manufacture of hydrogen peroxide. US 1108752, 1914.
35. Liu Q., Lunsford J. H. Controlling factors in the direct formation of H2O2 from H2 and O2over a Pd/SiO2 catalyst in ethanol. Appl. Catal. A 2006;314:94–100.
36. Dissanayake D. P., Lunsford J. H. Evidence for the role of colloidal palladium in the catalyticformation of H2O2 from H2 and O2. J. Catal. 2002;206:173–176.
37. Chinta S., Lunsford J. H. A mechanistic study of H2O2 and H2O formation from H2 and O2catalyzed by palladium in an aqueous medium. J. Catal. 2004;225:249–255.
38. Landon P., Collier P. J., Papworth A. J., Kiely C. J., Hutchings G. J. Direct formation ofhydrogen peroxide from H2/O2 using a gold catalyst. Chem. Commun. 2002;2058–2059.
39. Choudhary V. R., Samanta C., Choudhary T. V. Direct oxidation of H2 to H2O2 over Pd-based catalysts: Influence of oxidation state, support and metal additives. Appl. Catal. A2006;308:128–133.
40. Edwards J. K., Solsona B. E., Landon P., Carley A. F., Herzing A., Kiely C. J., HutchingsG. J. Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au-Pdcatalysts. J. Catal. 2005;236:69–79.
41. Herzing A. A., Carley A. F., Edwards J. K., Hutchings G. J., Kiely C. J. Microstructuraldevelopment and catalytic performance of Au−Pd nanoparticles on Al2O3 supports: theeffect of heat treatment temperature and atmosphere. Chem. Mater. 2008;20:1492–1501.

REFERENCES 709
42. Edwards K., Carley A. F., Herzing A. A., Kiely C. J., Hutchings G. J. Direct synthesisof hydrogen peroxide from H2 and O2 using supported Au-Pd catalysts. Faraday Discuss.2008;138:225–239.
43. Edwards J. K., Solsona B. E., N E. N., Carley A. F., Herzing A. A., Kiely C. J., HutchingsG. J. Switching off hydrogen peroxide hydrogenation in the direct synthesis process. Science2009;323:1037–1041.
44. Edwards J. K., N E. N., Carley A. F., Herzing A. A., Kiely C. J., Hutchings G. J. Direct syn-thesis of H2O2 from H2 and O2 over gold, palladium, and gold-palladium catalysts supportedon acid-pretreated TiO2. Angew. Chem. Int. Ed. 2009;48:8512–8515.
45. Davies P., Snowdon F. F., Bridger G. W., Hughes D. O., Young P. W. U. K. Water-gasconversion and catalysts therefor. Patent No. 1010871.
46. Waugh K. C. Methanol synthesis. Catal. Today 1992;15:51–75.
47. Klier K. Methanol synthesis. Adv. Catal. 1982;31:243–313.
48. Waller D., Stirling D., Stone F. S., Spencer M. S. Copper-zinc oxide catalysts. Activityin relation to precursor structure and morphology. Faraday Discuss. Chem. Soc. 1989;87:107–120.
49. Bems B., Schur M., Dassenoy A., Junkes H., Herein D., Schlogl R. Relations betweensynthesis and microstructural properties of copper/zinc hydroxycarbonates. Chem. Eur. J.2003;9:2039–2052.
50. Catalysis Surveys from Asia 8, Development of high performance Cu/ZnO-based catalystsfor methanol synthesis and the water-gas shift reaction. December 2004, 285–294.
51. Liu X.-M., Lu G. Q., Yan Z.-F., Beltramini J. Recent advances in catalysts for methanolsynthesis via hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003;42:6518–6530.
52. Hansen J. B., In: Ertl G., Knozinger H., Weitkamp J., editors. Handbook of HeterogeneousCatalysis, vol. 4. Weinheim: Wiley-VCH Verlag GmbH; 1997, p. 1856.
53. Herman R. G., Klier K., Simmons G. W., Finn B. P., Bulko J. B., Kobylinski T. P. Catalyticsynthesis of methanol from CO/H2: I. Phase composition, electronic properties, and activitiesof the Cu/ZnO/M2O3 catalysts. J. Catal. 1979;56:407–429.
54. Chinchen G. C., Waugh K. C., Whan D. A. The activity and state of the copper surface inmethanol synthesis catalysts. Appl. Catal. 1986;25:101–107.
55. Evans J.W.,Wainwright M. S., Bridgewater A. J., Young D. J. On the determination of coppersurface area by reaction with nitrous oxide. Appl. Catal. 1983;7:75–83.
56. PonecV. Cu and Pd, two catalysts for CH3OH synthesis: the similarities and the differences.Surf. Sci. 1992;272:111–117.
57. Spencer M. S. The role of zinc oxide in Cu/ZnO catalysts for methanol synthesis and thewater-gas shift reaction. Top. Catal. 1999;8:259.
58. Clausen B.S., Topsøe H. In situ high pressure, high temperature XAFS studies of Cu-basedcatalysts during methanol synthesis. Catal. Today 1991;9:189.
59. Yoshihara J., Campbell C. T. Methanol synthesis and reverse water-gas shift kinetics overCu(110) model catalysts: structural sensitivity. J. Catal. 1996;161:776.
60. Frost J. Junction effect interactions in methanol synthesis catalysts. Nature 1988;334:577–580.
61. Topsøe N.-Y Topsøe H. On the nature of surface structural changes in Cu/ZnO methanolsynthesis catalysts. Top. Catal. 1999;8:267–270.

710 ACTIVATION OF SMALL MOLECULES AND CONVERSION INTO USEFUL FEEDSTOCK
62. Choi Y., Futagami K., Fujitani T., Nakamura J. The role of ZnO in Cu/ZnOmethanol synthesiscatalysts–morphology effect or active site model? Appl. Catal. A 2001;208:163–167.
63. Fujitani T., Nakamura I., Uchijima T., Nakamura J. The kinetics and mechanism ofmethanol synthesis by hydrogenation of CO2 over a Zn-deposited Cu(111) surface. Surf.Sci. 1997;383:285–298.
64. Nakamura I., Nakano H., Fujitani T., Uchijima T., Nakamura J. Evidence for a special formatespecies adsorbed on the Cu-Zn active site for methanol synthesis. Surf. Sci. 1998;402–404:92–95.
65. Jansen W. P. A., Beckers J., Heuvel J. C. v. d., Denier A.W., Gon v. d., Bliek A., BrongersmaH. H. Dynamic behavior of the surface structure of Cu/ZnO/SiO2 catalysts. J. Catal.2002;210:229–236.
66. Grunwaldt J. D., Molenbroek A. M., Topsøe N.-Y., Topsøe H., Clausen B. S. In situ investi-gations of structural changes in Cu/ZnO catalysts. J. Catal. 2000;194:452.
67. Clausen B. S., Schiotz J., Grabaek L., Ovesen C. V., Jacobsen K. W., Nørskov J. K., TøpseeH. Wetting/non-wetting phenomena during catalysis: evidence from in situ on-line EXAFSstudies of Cu-based catalysts. Top. Catal. 1994;1:367–376.
68. Hansen P. L., Wagner J. B., Helveg S., Rostrup-Nielsen J. R., Clausen B. S., Topsøe H.Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals. Science2002;295:2053–2055.
69. Kniep B. L., Ressler T., Rabis A., Girgsdies F., Baenitz M., Steglich F., Schlogl R. Rationaldesign of nanostructured copper-zinc oxide catalysts for the steam reforming of methanol.Angew. Chem. Int. Ed. 2004;43:112.
70. Ressler T., Kniep B. L., Kasatkin I., Schlogl R. The microstructure of copper zinc oxidecatalysts: bridging the materials gap. Angew. Chem. Int. Ed. 2005;44:4704–4707.
71. Liao F., Huang Y., Ge J., ZhengW., Tedsree K., Collier P., Hong X., Tsang S. C. Morphology-dependent interactions of ZnO with Cu nanoparticles at the materials’ interface in selectivehydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2011;50:2162–2165.
72. Kasatkin I., Kurr P., Kniep B., Trunschke A., Schlogl R. Role of lattice strain and defects incopper particles on the activity of Cu/ZnO/Al2O3 catalysts for methanol synthesis. Angew.Chem. Int. Ed. 2007;46:7324–7327.
73. Behrens M., Furche A., Kasatkin I., Trunschke A., Busser W., Muhler M., Kniep B., FischerR., Schlogl R. The potential of microstructural optimization in metal/oxide catalysts: higherintrinsic activity of copper by partial embedding of copper nanoparticles. Chem.Cat.Chem.2010;2:816–818.
74. Szanyi J., GoodmanD.W.Methanol synthesis on aCu(100) catalyst.Catal. Lett. 1991;10:383–390.
75. Rasmussen P. B., Holmblad P. M., Askgaard T. S., Ovesen C. V., Stoltze P., Nørskov J. K.,Chorkendorff I. Methanol synthesis on Cu(100) from a binary gas mixture of CO2 and H2.Catal. Lett. 1994;26:373–381.
76. Kalidindi S. B., Jagirdar B. R. Synthesis of Cu@ZnO core-shell nanocomposite throughdigestive ripening of Cu and Zn nanoparticles. J. Phys. Chem. C 2008;112:4042–4048.
77. Schroter M. K., Khodeir L., van den Berg M. W. E., Hikov T., Cokoja M., Miao S., GrunertW., MuhlerM., Fischer R. A. A colloidal ZnO/Cu nanocatalyst for methanol synthesis.Chem.Commun. 2006;2498–2500.

REFERENCES 711
78. Schimpf S., Rittermeier A., Zhang X., Li Z.-A., SpasovaM., van den BergM.W. E., Farle M.,Wang Y., Fischer R. A., Muhler M. Stearate-based Cu colloids in methanol synthesis: struc-tural changes driven by strongmetal-support interactions.Chem. Cat. Chem. 2010;2:214–222.
79. Ashcroft A. T., CheethamA. K., Foord J. S., GreenM. L. H., Grey C. P., Murrell A. J., VernonP. D. F. Selective oxidation ofmethane to synthesis gas using transitionmetal catalysts.Nature1990;344:319–321.
80. Ashcroft A. T., Cheetham A. K., Green M. L. H., Vernon P. D. F. Partial oxidation ofmethane to synthesis gas using carbon dioxide synthesis gas using carbon dioxide. Nature1991;352:225–226.


















