
The Hartree-Fock-Bogoliubov Theory Bose-Einstein Condensates
Upload
shyue-ping-ongCategory
view
337download
6
The Hartree-Fock Approximation
Shyue Ping Ong

Stationary Schrödinger Equation for a System of Atoms
where
NANO266 2
Eψ = Hψ
H = −h 2
2me
∇i2
i∑ −
h 2
2mk
∇k2 −
e2Zk
rikk∑
i∑ +
e2
rijj∑
i∑
k∑ +
ZkZle2
rkll∑
k∑
KE of electrons
KE of nuclei
Coulumbic attraction between nuclei and electrons
Coulombic repulsion between electrons
Coulombic repulsion between nuclei

Stationary Schrödinger Equation in Atomic Units
To simplify the equations a little, let us from henceforth work with atomic units
NANO266 3
Dimension Unit Name Unit Symbol Mass Electron rest mass me
Charge Elementary Charge e Action Reduced Planck’s constant ħ Electric constant Coulomb force constant ke
H = −12∇i2
i∑ −
12mk
∇k2 −
Zk
rikk∑
i∑ +
1rijj
∑i∑
k∑ +
ZkZlrkll
∑k∑

The Variational Principle
We can judge the quality of the wave functions by the energy – the lower the energy, the better. We may also use any arbitrary basis set to expand the guess wave function.
How do we actually use this?
NANO266 4
φHφ dr∫φ 2 dr∫
≥ E0
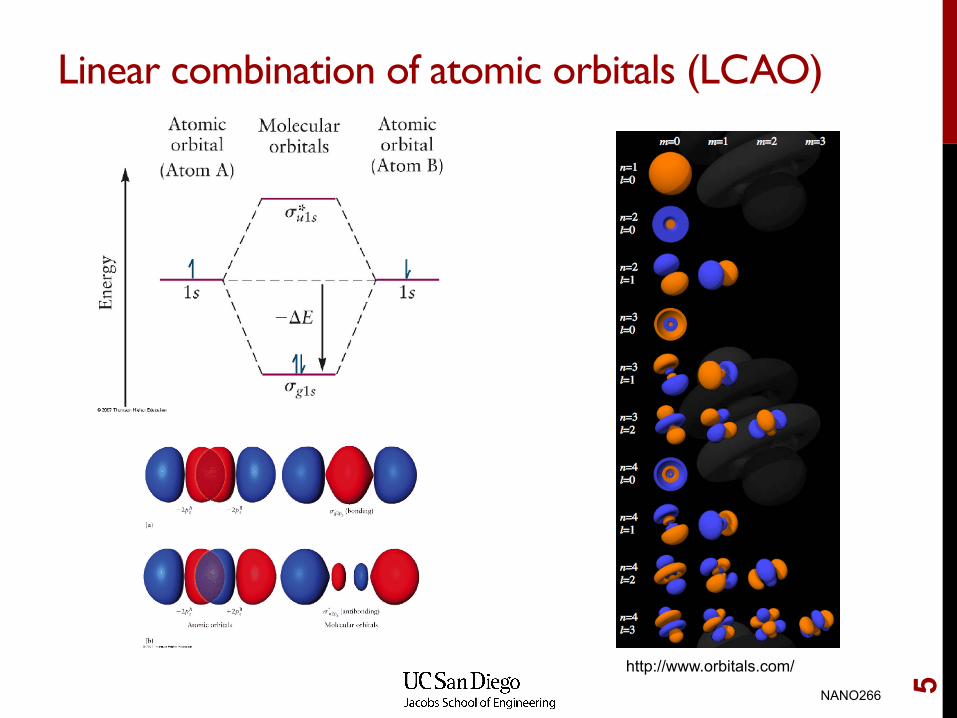
Linear combination of atomic orbitals (LCAO)
NANO266 5
http://www.orbitals.com/

Solving the one-electron molecular system with the LCAO basis set approach
In general, we may express our trial wave functions as a series of mathematical functions, known as a basis set. For a single nucleus, the eigenfunctions are effectively the hydrogenic atomic orbitals. We may use these atomic orbitals as a basis set for our molecular orbitals. This is known as the linear combination of atomic orbitals (LCAO) approach.
NANO266 6
φ = aiϕii=1
N
∑

The Secular Equation
NANO266 7
E =aiϕi
i=1
N
∑"
#$
%
&'H aiϕi
i=1
N
∑"
#$
%
&'dr∫
aiϕii=1
N
∑"
#$
%
&'
2
dr∫
=
aiaj ϕiHϕ j dr∫ij∑
aiaj ϕiϕ j dr∫ij∑
=
aiajHijij∑
aiajSijij∑
Resonance integral
Overlap integral

The Secular Equation, contd
To minimize the energy,
Which gives
Or in matrix form
NANO266 8
∂E∂ak
= 0, ∀k
ai (Hki −ESki )i=1
N
∑ = 0, ∀k
H11 −ES11 H12 −ES12 ! H1N −ES1NH21 −ES21 H22 −ES22 ! H2N −ES2N" " # "
HN1 −ESN1 HN 2 −ESN 2 ! HNN −ESNN
"
#
$$$$$
%
&
'''''
a1a2"aN
"
#
$$$$$
%
&
'''''
= 0

The Secular Equation, contd
Solutions exist only if Procedure:
i. Select a set of N basis functions. ii. Determine all N2 values of Hij and Sij. iii. Form the secular determinant and determine the N roots Ej. iv. For each Ej, solve for coefficients ai.
NANO266 9
H11 −ES11 H12 −ES12 ! H1N −ES1NH21 −ES21 H22 −ES22 ! H2N −ES2N" " # "
HN1 −ESN1 HN 2 −ESN 2 ! HNN −ESNN
= 0
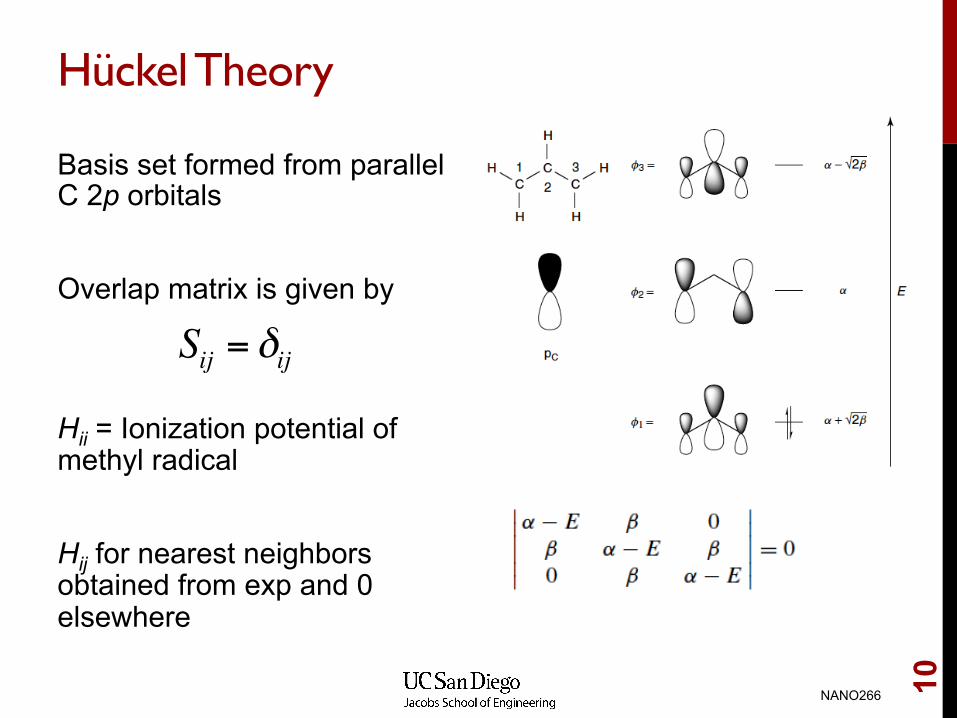
Hückel Theory
Basis set formed from parallel C 2p orbitals Overlap matrix is given by Hii = Ionization potential of methyl radical Hij for nearest neighbors obtained from exp and 0 elsewhere
NANO266 10
Sij = δij

The Born-Oppenheimer Approximation
Heavier nuclei moves much more slowly than electrons => Electronic relaxation is “instantaneous” with respect to nuclear motion
Electronic Schrödinger Equation
NANO266 11
(Hel +VN )ψel (qi;qk ) = Eelψel (qi;qk )
Electronic energy Constant for a set of nuclear coordinates

Stationary Electronic Schrödinger Equation
where
NANO266 12
Eelψel = Helψel
Hel = −12∇i2
i∑ −
Zk
rikk∑
i∑ +
1rijj
∑i∑
KE and nuclear attraction terms are separable
H = hii∑ where hi = −
12∇i −
Zk
rikk∑

Hartree-Product Wave Functions
Eigen functions of the one-electron Hamiltonian is given by
Because the Hamiltonian is separable,
NANO266 13
hiψi = εiψi
ψHP = ψii∏
HψHP = hii∑ ψk
k∏
= εii∑#
$%
&
'(ψHP

The effective potential approach
To include electron-electron repulsion, we use a mean field approach, i.e., each electron sees an “effective” potential from the other electrons
NANO266 14
hi = −12∇i −
Zk
rikk∑ +Vi, j
where
Vi, j =ρ j
rij∫
j≠i∑ dr
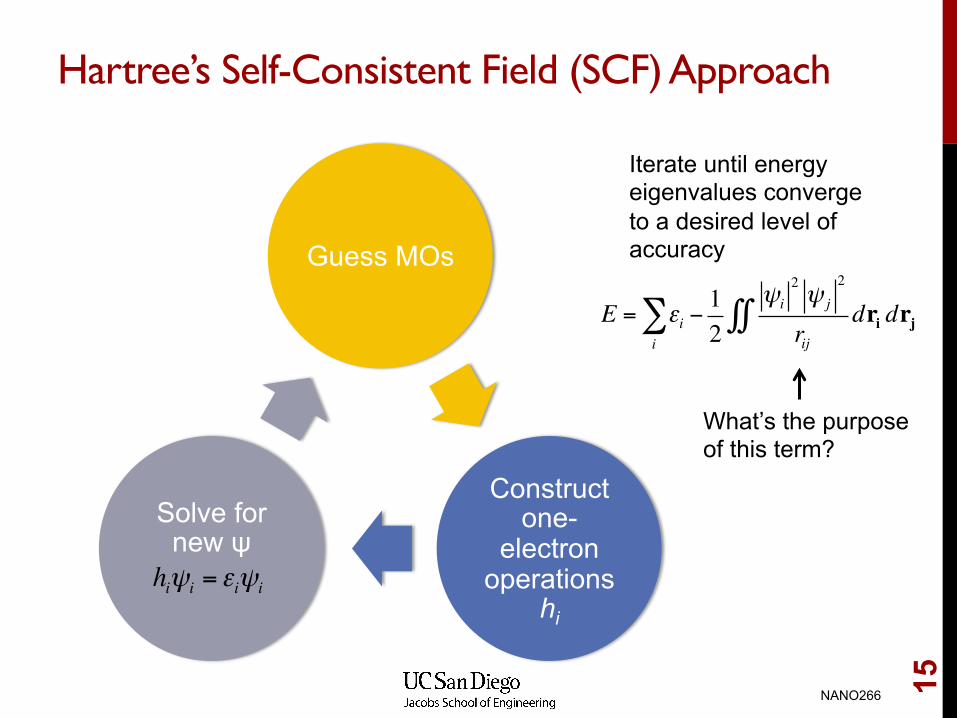
Hartree’s Self-Consistent Field (SCF) Approach
NANO266 15
Guess MOs
Construct one-
electron operations
hi
Solve for new ψ
hiψi = εiψi
Iterate until energy eigenvalues converge to a desired level of accuracy
E = εii∑ −
12
ψi2ψ j
2
rijdri drj∫∫
What’s the purpose of this term?

What about the Pauli Exclusion Principle?
Two identical fermions (spin ½ particles) cannot occupy the same quantum state simultaneously
è Wave function has to be anti-symmetric
For two electron system, we have
NANO266 16
ψSD =12ψa (1)α(1)ψb(2)α(2)−ψa (2)α(2)ψb(1)α(1)[ ]
=12
ψa (1)α(1) ψb(1)α(1)ψa (2)α(2) ψb(2)α(2)
where α is the electron spin eigenfunction
Slater determinant

For many electrons…
NANO266 17
ψSD =1N!
χ1(1) χ2 (1) ! χN (1)χ1(2) χ2 (2) ! χN (2)! ! " !
χ1(N ) χ2 (N ) ! χN (N )
where χ k are the spin orbitals

The Hartree-Fock (HF) Self-Consistent Field (SCF) Method
NANO266 18
fi = −12∇i2 −
Zk
rik+Vi
HF{ j}k
nuclei
∑
F11 −ES11 F12 −ES12 ! F1N −ES1NF21 −ES21 F22 −ES22 ! F21 −ES2N" " # "
FN1 −ESN1 FN 2 −ESN 2 ! FNN −ESNN
= 0
HF Secular Equation
Fµυ = µ |− 12∇i2 |υ − Zk µ | 1
rk|υ + Pλσ
λσ
∑ (µυ | λσ )− 12(µλ |υσ )
$
%&'
()k
nuclei
∑
Weighting of four-index integrals by density matrix, P
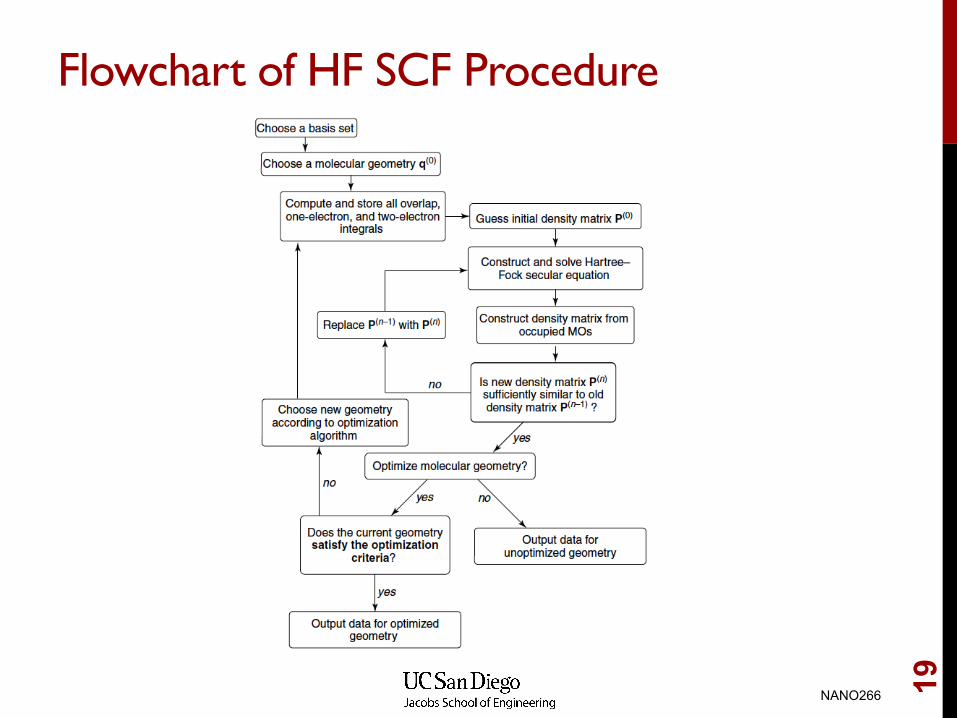
Flowchart of HF SCF Procedure
NANO266 19

Limitations of HF
Fock operators are one-electron => All electron correlation, other than exchange, is ignored
Four-index integrals leads to N4 scaling with respect to basis set size
NANO266 20
Ecorr = Eexact −EHF
Practical Aspects of HF Calculations
Basis Sets
Effective Core
Potentials
Open-shell vs Closed-
shell Accuracy
Performance
NANO266 21

Basis Set
Set of mathematical functions used to construct the wave function.
In theory, HF limit is achieved by an infinite basis set.
In practice, use finite basis sets that can approach HF limit as efficiently as possible
NANO266 22

Contracted Gaussian Functions
Slater-type orbitals (STO) with radial decay cannot be analytically integrated
-> Use linear combination of Gaussian-type orbitals (GTOs) with radial decay to approximate STOs
STO-3G • STO approximated by 3 GTOs • Known as single-ζ or minimal basis set.
NANO266 23
e−r2
e−r

Multiple-ζ and Split-Valence
Multiple-ζ • Adding more basis functions per atomic orbital • Examples: cc-pCVDZ, cc-pCVTZ (correlation-consistent polarized
Core and Valence (Double/Triple/etc.) Zeta)
Split-valence or Valence-Multiple-ζ • Still represent core orbitals with single, contracted basis functions • Valence orbitals are split into many functions (Why?) • Examples: 3-21G, 6-31G, 6-311G
NANO266 24
# of primitives in core
# of primitives in valence

Polarization and Diffuse Functions
Polarization functions • Description of MOs require more flexibility than provided by AOs,
e.g., NH3 is predicted to be planar if using just s and p functions • Additional basis functions of one quantum number of higher
angular momentum than valence, e.g., first row -> d orbitals • Notation: 6-31G* [old] or 6-31G(d) [new], 6-31(2d,p) [2d functions
for heavy atoms, additional p for H]
Diffuse functions
• Highest energy MOs of anions, highly excited states tend to be more diffuse
• Augment standard basis sets with diffuse functions • Notation: 6-31+G, 6-311++G(3df, 2pd), aug-cc-pCVDZ
NANO266 25

Effective Core Potentials
Heavy atoms have many electrons • Intractable to model all of them, even with a minimal basis set • However, most of the electrons are in the core
Solution: Replace core electrons with analytical functions (effective core potentials or ECPs) that represent combined nuclear-electronic core to the remaining electrons Key selection decision: How many electrons to include in the core?
NANO266 26

Open-shell vs closed-shell
Restricted HF (RHF) • Closed-shell systems, i.e., no unpaired electrons
Restricted open-shell HF (ROHF)
• Use RHF formalism, but with density matrix for singly occupied orbitals not multiplied by a factor of 2.
• Wave functions are eigenfunctions of S2
• But fails to account for spin polarization in doubly occupied orbitals
Unrestricted HF (UHF)
• Includes spin polarization • Wave functions are not eigenfunctions of S2, i.e., spin contamination
NANO266 27

Accuracy
Energetics • In general, extremely poor; correlation is extremely important in
chemical bonding! • Protonation energies are typically ok (no electrons in H+) • Koopman’s Theorem: First IE is equal to the negative of the
orbital energy of the HOMO
Geometry
• Typically relatively good ground state structures with basis sets of modest size
• But transition states (with partial bonding) can be problematic, as well as some pathological systems
NANO266 28

Performance
Formal N4 scaling But in reality, speedups can be achieved through:
• Symmetry • Estimating upper bounds to four-index integrals • Fast multipole and linear exchange integral computations
For practical geometry optimizations, frequently helps to first compute geometry with a smaller basis set to provide a better initial geometry and a guess for the Hessian matrix.
NANO266 29














![Hartree-Fock Theory Variational Principlehagino/lectures/notes3.pdfRemarks 1. Single-particle Hamiltonian: Direct (Hartree) term Exchange (Fock) term [non-local pot.] 2. Iteration](https://static.fdocuments.in/doc/165x107/60b10f0c822da453294ca21c/hartree-fock-theory-variational-haginolecturesnotes3pdf-remarks-1-single-particle.jpg)



