mx - prr.hec.gov.pk
237
i mx o C 4. C d. c C cT. >v ‘Proclaim! Sind thy Lord is most ‘Bountiful. Me who taught (the use cf) the Ta&06 ‘Taught man that which he {new noth207t %
Transcript of mx - prr.hec.gov.pk

i
mxo
C4. C
d. cC
cT.>v
‘Proclaim!Sind thy Lord is most ‘Bountiful. Me whotaught (the use cf) theTa&06
‘Taught man that which he{new noth207t
%

Isolation and StructuralElucidation of Chemical
Constituents from Fumariaindica, Ferula oopoda and
Withania somnifera
Thesis submitted
for
The fulfilment of the Degree of
DOCTOR OF PHILOSOPHYBY
SHAKIL AHMED
%'ieraInternational Centre for Chemical Sciences
H.E.J. Research Institute of ChemistryUniversity of Karachi
Karachi
1998

:
‘Dedicatedto
My Dearest‘Parents

ACKNOWLEDGEMENTS
I take pride in acknowledging my assiduous supervisor Prof. Atta-ur-
Rahmart, H.I., S.I., T.I., Director of H.E.J. Research Institute of
Chemistry for his valuable guidance and constructive comments
during the course of this research work as well as for providing an
environment conducive for research. He has been always a source of
inspiration for me.
There has been a significant contribution of Dr. M. Iqbal
Choudhary, to the evolution of this thesis. I wish to express my
gratitude to him for his kind interest and helpful suggestions.
The task of this work would have been more difficult, without the
help of my colleagues. I sincerely gratitude Dr. Shaiq Alit Mr.
Shahid Malik, Mr. M. Nur-e-Alam, Miss Aniqa Naz, and Mr.
Mohammad Aslam for their co-operation and encouraging
comments. I would also like to thank Mr. Riaz Ali Khan, Dr. Jtfgan
Farooq, Dr. Shahid Badar Usmani, Syed Muddasser Kazmi,
Salma Shehnaz, Humera Naz, Mr. and Mrs. Abdul Jabbar and Dr.
Bakht Baidar Ali for their constant support.
I am deeply indebted to my dear parents, a debt which is impossible
to repay. A special thanks are due to my brothers and sisters for
their continuous encouragement and appreciation.
i
I am most grateful to my better-half Iffat for her patience,
encourangements and understanding.

My little angels Gul and Aeliya deserve a big thanks as they were
sometimes neglected by me during my research work.
Our librarian Mr. Aijaz Ahmed Soofi deserves a special bow in
providing library informations in time. I extend my special thanks to
Mr. Mahmood Alam who took a keen interest in solving my problems
whenever they arose.
A great deal of appreciation is due to the technical and non-technical
staff of the institute especially to Mr. Sarjrazullah for his kind help
and to Mr. Naveed Aslam for his photocopying services.
SHAKEEL AHMED

CONTENTS
I. SUMMARY .1
1.0 GENERAL INTRODUCTION l
1.1 ALKALOIDS
1.1.1 Heterocyclic Alkaloids
1.1.2 Isoquinoline Alkaloids
1.1.3 Pharmacology of Isoquinoline Alkaloids
1.1.4 Biosynthesis of Isoquinoline Alkaloids..
1.1.4.1 Isoquinolines
1.1.4.2 Simple Tetrahydrolsoquinoline Alkaloids .. 27
3
5
10
19
25
25
1.1.4.3 Benzylisoqulnoline Alkaloids
1.1.4.4 Pavine and Isopavine
1.1.4.5 Aporphine Alkaloids
1.1.4.6 Protoberberine Alkaloids
1.1.4.7 Protopine Alkaloids
1.1.4.8 Phthalideisoquinoline Alkaloids
1.1.4.9 Secophthalideisoquinoline Alkaloids
1.1.4.10 Spirobenzylisoquinoline Alkaloids
30
32
35
35
38
39
39
40
2.0 INTRODUCTION OF FUMARIA MDICA. 44
2.1 Literature Survey
2.2 Results and Discussion
44
64

3.2.4 Isolation of 2,3-Dihydrowithaferine-A (297)
3.3 Plant Material
3.3.1 Extraction and Purification
3.3.2 Isolation of Withanone (295)
3.3.3 Isolation of Quresimine (296)
3.3.4 Isolation of Withaferine-A (306)
3.3.5 Isolation of 2,3-Dihydrowithaferine-A (297)
163
170
170
170
172
173
174
4.0 INTRODUCTION OF FERULA OOPODA 176.
4.1 Biosynthesis of Sesquiterpenes
4.2 Results and Discussion
4.2.1 Isolation of Feralleate (303)
4.2.2 Isolation of Guaianolide (304)..
4.2.3 Isolation of Grilactone (305)....
4.3 P’ant Material
4.3.1 Extraction and Purification
4.3.2 Isolation of Feralleate (303)
4.3.3 Isolation of Guaianolide (304)..
4.3.4 Isolation of Grilactone (305) ....
177
182
182
190
194
198
198
200
201
202
5.0 REFERENCES 204
LIST OF PUBLICATIONS 221

2.2.1 Fumaileal (144)..
2.2.2 Fumaileate (149)
2.2.3 Papraline (143) ...
2.2.4 9,10-Methylenedloxy karachine (255)
2.2.5 Vanillin (142)
2.2.6 Cryptopine (61)
2.2.7 (+)-p-Hydrastlne (232)
2.2.8 Corydaldine (147)
2.3 General Experimental
2.3.1 Plant Material
2.3.2 Extration and Purification
2.3.3 Isolation of Fumaileal (144)
2.3.4 Isolation of Fumaileate (149)
2.3.5 Papraline (143)
2.3.6 9,10-Methylenedloxy karachine (255)
2.3.7 Vanillin (142)
2.3.8 Cryptopine (61)
2.3.9 (+)-p-Hydrastine (232)
2.3.10 Coiydaldine (147)
64
67:
73
77
87
89
94
99
101
104
104
109
no111
112
113
114
116
117
3.0 INTRODUCTION OF WITHANIA SOMNIFERA . 119
3.1 Biosynthesis of Withanolides ....
3.2 Results and Discussion
3.2.1 Isolation of Withanone (295) ....
3.2,2 Isolation of Quresimine (296)....
3.2.3 Isolation of Withaferine-A (306)
123
133
133
147
159
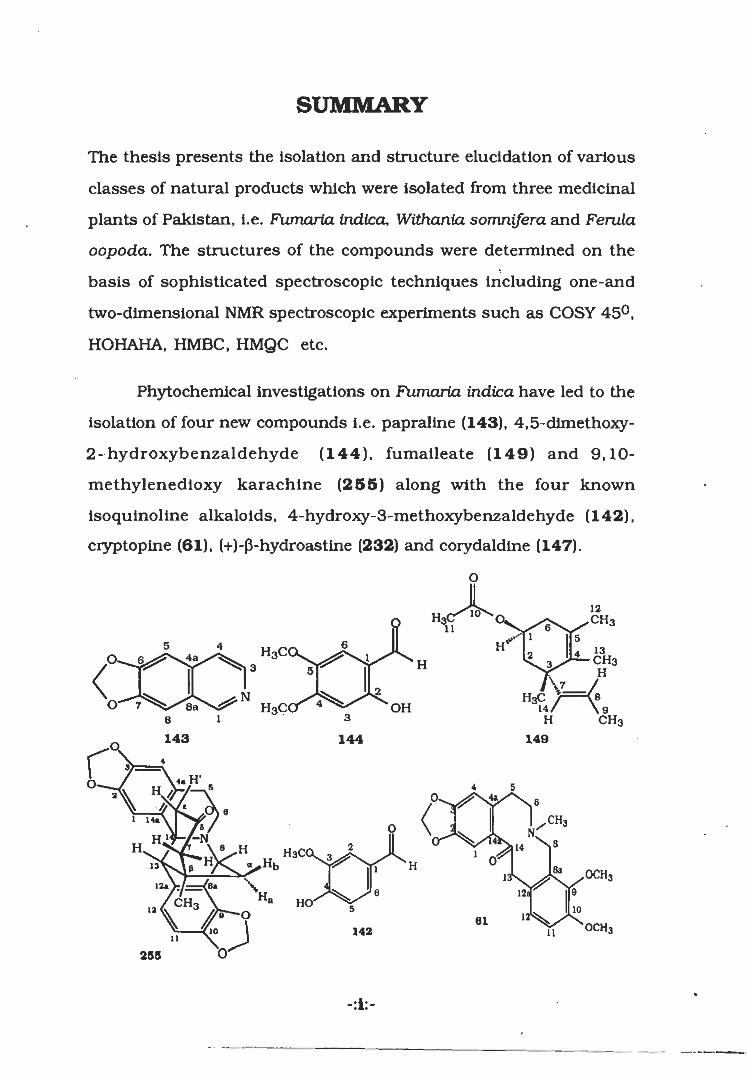
SUMMARY
The thesis presents the isolation and structure elucidation of various
classes of natural products which were isolated from three medicinal
plants of Pakistan, i.e. Fumaria indica, Withanta somnifera and Ferula
oopoda. The structures of the compounds were determined on the
basis of sophisticated spectroscopic techniques including one-and
two-dimensional NMR spectroscopic experiments such as COSY 45°,
HOHAHA, HMBC, HMQC etc.
Phytochemical investigations on Fumaria indica have led to the
isolation of four new compounds i.e. papraline (143), 4,5-dimethoxy-
2- hydroxybenzaldehyde (144), fumaileate (149) and 9,10-
methylenedioxy karachine (255) along with the four known
isoquinoline alkaloids, 4-hydroxy-3-methoxybenzaldehyde (142),
cryptopine (61), (+)-(3-hydroastine (232) and corydaldine (147).
o
H3C''ÿ 12CH3
L *Jk-CH36
H'T 56
3
ON
H3c<
H5] JL AH
H C
4 OHCH3318
143 144 149
4. H14 5
P>ÿ\4a 6
vH-N B2 14
r&b HaC< 3 O'HI31 1 83 .0CH3m P\i2» w—ra»
or
Ha HO5 10
1!61OCH3142 11
11
255
-:i:-
5
<O'8a N.
H' CH382* 5
H3C<H- IT 3'
4'I .N.9'
8aOCH3 H3C H5' 88’o
OCH3 o232 147
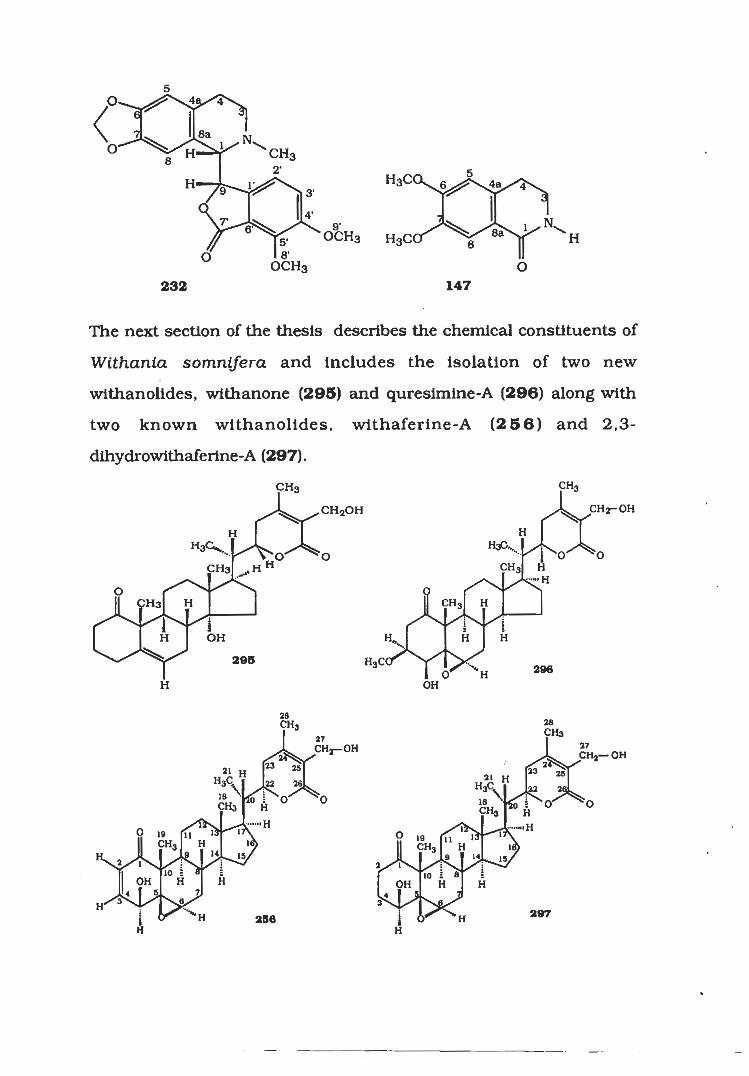
The next section of the thesis describes the chemical constituents of
Withania somnifera and Includes the isolation of two new
wlthanolides, withanone (295) and quresimine-A (296) along with
two known wlthanolides, withaferine-A (256) and 2,3-
dlhydrowithaferine-A (297).
CH3CH3
CH2-OHCH2OH %HH
H3Cv..UVCH3 ,HH T° ‘oo o
CHH
O:H3 H CH3 H
I IOHH H H
29S H3C< 2980* HH OH
28
CH3 28
CH327
27CHj— OHCHj— OH
H&"o o
H ~—H0 19 fn i!CH3 j H \\ CH3flH 1:1:u
M I5y 149 13.a/T 2V?fio s'
OH H OH[4 3ÿ-Hÿ3 0,
297286 O' HH H

The last section deals with the studies on the chemical constituents
from Ferula oopoda which have led to the isolation of ferulaileate
(303) along with two known sesquiterpene lactones guaianolide
(304) and grilactone (305) for the first time from this species.
H-bVH14
H2<H O
rCH3 i 'z
i
k r 5
6oj— lkÿi.3 6
\ 13L CH34. 15ch3hHF H Il5CH3CH3 z k /xl!0ÿ2/ÿH
* O
lOCH3ÿÿ3>CH3H3
30BH O304303

SECTION A
GENERAL INTRODUCTION

GENERAL INTRODUCTION
1.0 THE HISTORY OF MEDICINE
The history of modem medicine and pharmacy is considered to
begin from Hippocrates, the father of modern Medicine. In his
writings nearly 400 substances were named as medicinal substances.
Theophrastus wrote a book on "The History of Plants", in which he
mentioned 500 herbal drugs. His famous book "Materia Medica" was
first published in Greece, which served as the standard
phytopharmacological handbook for a long time. Following this
period the role of herbal medicine was greatly extended in the Islamic
age.
The most famous physician and philosopher of the early
Muslim era, Ibne-Sina, described 760 herbal drugs in his famous
book "Kanun fl al-Tibb" which was known as "the Canon" in Europe
till the 17th century A.D., and formed the basis of the Greco-Arab
system of medicine known as ’Tibbe-Unani". Indeed, no history of
medical science can be complete without reference to the scholarly
pioneering contributions of Alhazen (Ibn-al-Haitham), Rhazes (Al-
Razi) Jildaki, Al-Khawarazmi, Al-Kindi, Al-Biruni, Al-Farabi, Abual-
Kasim and Al-Zahrwi.
Herbs have been widely used since time immemorial as crude
vegetable drugs or extracts for the treatment of many diseases and to
maintain health [1]. For example an important herb "cinchona" has
been used as an antimalarial drug for thousands of years [2]. Over
-:1:-

the years plants have been shown to be rich In anti-cancer agents
(vinblastine and vincristine), narcotic analgesics (morphine, codeine),
antlmalarlal drugs (quinine, artemisinin), anticholinergic drugs
(atropine, hyoscyamine), adrenolytic agents (reserplne),
antiasthmatic drugs (ephedrine, khellin), liver protective agents
(catechin, silymarin), cardiac drugs (digitalis) etc.
Polysaccharides, proteins, fats and nucleic acids are the
fundamental building blocks of living organisms and are considered
as "primary metabolites". These primary metabolites are used by the
living cells in vivo for various purposes and they also give rise to
secondary metabolites such as alkaloids, terpenes, pigments etc. The
secondary metabolites are synthesized in the plants or living
organisms through enzyme catalysis by using the primary metabolites
as building blocks.
Most of these secondary metabolites play some important but
unknown roles in the life of a plant. Many of these secondary
metabolites have also been found to possess interesting
pharmacological activities and some have served as cures for human
and livestock diseases. Although man’s search to-date to Isolate
therapeutic agents from medicinal plants has yielded only modest
success, we should not forget that with the advent of new high
through-put screening techniques, much better ways of looking for
new plant drugs have become available. Knowledge about the basis of
plant metabolism, plant analysis, biosynthesis in plants and even
plant production has also considerably expanded. It is reasonable to
-:2:-

predict that the most exciting and productive period of medicinal
plant research lies ahead of us.
1.1 THE ALKALOIDS
Alkaloids are secondary metabolites which form one of the
most important classes of plant constituents. The term "alkaloid" or
"alkali-like" was first proposed by the pharmacist, W. Meissner in
1818 [31. Meyer presented the first comprehensive definition of
alkaloids in 1896. According to him "Alkaloids" (plant bases) occur
characteristically in plants, and are frequently distinguished by their
remarkable physiological activity. They contain carbon, hydrogen and
nitrogen, and in most cases oxygen as well. However in 1931 Trier
wrote "As a result of scientific progress, a collective term of this kind
(i.e., alkaloids) will have to be abandoned" [4]
Alkaloidal substances can be defined in a broad sense as
basic nitrogen-containing compounds of either plant or animal
origin, which have complex molecular structures and which can
manifest significant pharmacological activities.
A modem definition of alkaloids was given by S.W. Pelletier
[5]. He defined alkaloids "as cyclic organic compounds containing
nitrogen in a negative oxidation state which are of limited
distribution among living organisms." In the light of these definitions
many nitrogen containing natural products are excluded from the
class of alkaloids. For instance, cholchicine (1) is an N-acetyl
derivative and has a neutral nitrogen atom, while aristolochic acid
(2) is not basic and has no heterocyclic ring.
-:3:-

There is another class of related compounds which are called
"protoalkaloids". This class consists of simple amines in which the
amino acid nitrogen is not in the ring. These are the so-called
"biological amines” e.g., ephedrine (3), mescaline (4) etc.
COOH
<“O-
H3C< o.....NH— c— CH3 NO2H3C< X
OCH3
oOCH3
OCH3
Aristolochlc acid (2)(-)-Colchicine (1)
H3C<
\ H/
HO“ÿC— Cÿ-'CH3H5C6
NH2H3C<NH— CH3
OCH3Mescaline (4)
(-)-Ephedrine (3)
Another group of nitrogen containing compounds are called
"pseudoalkaloids". They are not derived from amino acid precursors.
e.g., steroidal alkaloids such as terminaline (5), solasodine (6) and
purine bases such as caffeine (7).
-A.-

CH3H3Cÿ /
\ CH3CH3
CH3 H
IH
HO*i HOH
Terminaline (5)
HOH3
\ NJlMe
ICH3P CH3 H3ÿN!
oCH3 H
\Ii H
H CH3
HO*
Caffeine (7)(-)-Solasodlne (6)
Alkaloids may be further sub-classified on the basis of their
skeleton and biogenesis.
l.i.i Heterocyclic alkaloids
Alkaloids containing atleast one heterocyclic ring are
classified under this category. Examples are:
**
-:5:-
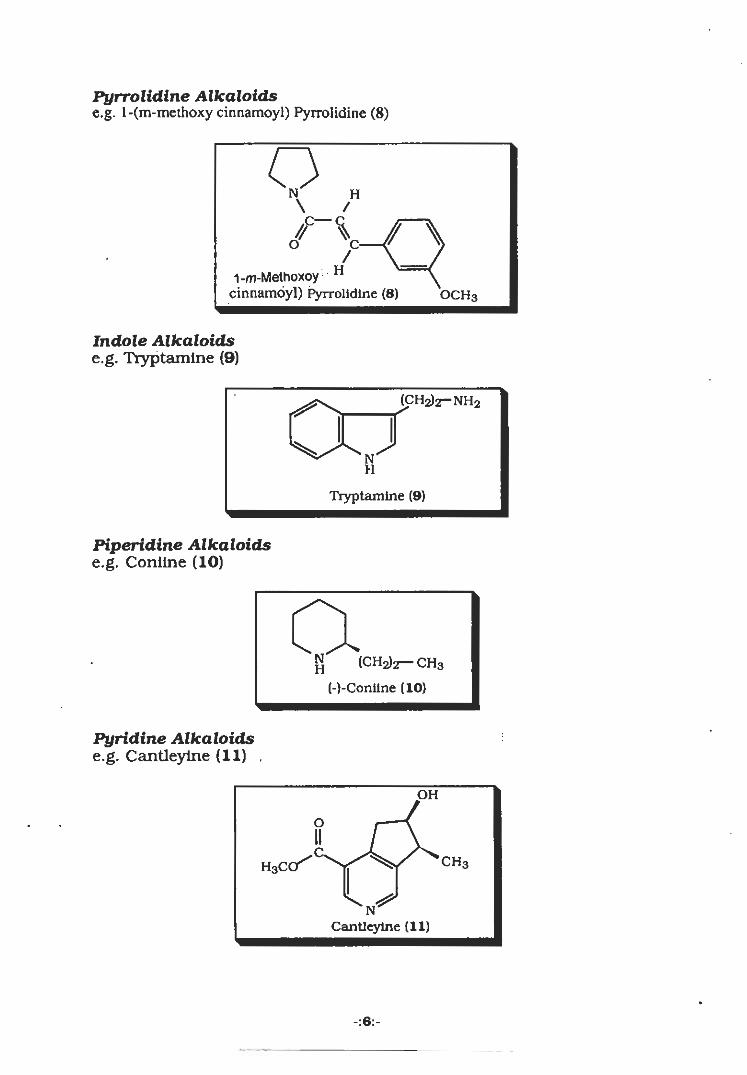
Pyrrolidine Alkaloidse.g. l-(m-methoxy cinnamoyl) Pyrrolidine (8)
QH
/~SÿTS/
1-m-Methoxoy H
cinnamoyl) Pyrrolidine (8) OCH3
Indole Alkaloidse.g. Tryptamine (9)
(CH2)2r-NH2
Tryptamine (9)
Piperidine Alkaloidse.g. Coniine (10)
aH
(CH2)2— CH3(-)-Coniine (10)
Pyridine Alkaloidse.g. Cantleylne (11) .
•:
OH
O
H3corÿ CH3
NCantleylne (11)
-:6:-

Tropane and Related Basese.g. Tropinone (12)
H3(ÿN,
OTropinone (12)
Histamine Alkaloidse.g. (±)-Glochldine (13)
/Ti>cy°n-H13C6
(±)-Glochldlne (13)
Isoquinoline Alkaloidse.g. Hydrohydrastinlne(14)
N\o- CH3
Hydrohydrastlnlne (14)
Quinoline Alkaloidse.g. Quinoline (15)
ooQuinoline (15)
-:7:-

Izidine Alkaloidse.g. PyrroHzidine (16)
H
CDPyrrolizidine (16)
Alkaloids with an Exocyclic Nitrogene.g. (-)-Cassaine (17)
CH3/CH-CO— O— CH2— CH2— N,
\CH3
CH3"VCH3
H
OHO*
(-)-Cassalne (17)H3C CH3
Putrescine Alkaloidse.g. Paucine (18)
OH
HO.
.O
NH
Paucine (18)
-:8:-

Peptide Alkaloidse.g. Integerrine (19)
HN
NH.
\O
N HN
rkH3<\
H3</
CeH5CH- CH
IN(CH3)2
Integerrine (19)
Diterpene Alkaloidse.g. Veatchine (20)
,CH2\
I
,N" / }
\
HII
I
OH
CH3Veatchine (20)
Steroidal Alkaloidse.g. Funtumine (21)
O. CH3CH3
CH3
iH
Hal
Funtumine (21)
-:9:-

ISOQUINOLINE ALKALOIDS1.1.2
The isoquinoline alkaloids form an important class of
secondary metabolites. The first alkaloid isolated i.e. morphine has
an isoquinoline skeleton [6,7]. The isoquinoline class has many
pharmaceutically important compounds which occur mainly in the
plant families Papaueraceae, Magnoliaceae, Annonaceae, Lauraceae,
Alangiacease, Berberidaceae, Ancistrocladaceae and Menispermaceae
[8].
Phytochemical studies have been mostly carried out on plants
of family Menispermaceae. Many of these are used in folk medicine, or
as a local food such as plants of the genera Tinospora, Stephania,
Cycled, Archangelisia, Fibrauria and Tiliacora.
Isoquinoline alkaloids can be further divided into the
following sub-groups on the basis of their structures and biogenesis
[9].
Simple Isoquinoline Alkaloids
e.g. Noroxyhydrastinine (132) [10)
<°'O',N.
H
O
Noroxyhydrastinine (132)

1-Phenylisoquinoline Alkaloidse.g. (+)-Ciystostyline I (22) [11]
H3C<
NSH3C< CH3
H’
H3C< OCH3
OCH3(+)-Crystostyline I (22)
N-Benzylisoquinoline Alkaloidse.g. N-Methylviguine (23) [12]
CXX)<"
N-Methylvlgulne (23)OCH3
4-Phenylisoquinoline Alkaloidse.g. (+)-Latlflne (24) [13]
OH
OHH
H3C<
N.CH3
(+)-LaUflne (24)

Isoquinolinequinon.ese.g. Renierol (25) [14]
O
H3<
NH3C<
OOH
Renierol (26)
Benzylisoquinoline Alkaloidse.g. (±)-Fumarizine (26) 115]
<°'o- NN
CH3
OCH3o'
•o(±)-Fumarizine (26)
Bisbenzylisoquinoline alkaloids with Aryl Links Onlye.g. Nor-2'-pisopowlaridine (27) [16]
,OH HO,
HSCÿN N.OCH3 H3C< H
VJ H
OCH3 HO'
Nor-2’-pisopowiaridine (27)
12:-

Bisbenzylisquinoline Alkaloids with One Ether Linke.g. Northallbroline (28) [17]
.OCH3 H3C<
RH3C<N HO'OH N HH
OH
Northallbroline (28)
Bisbenzylisoquinoline Alkaloids with One Ether/One Aryl Linke.g. (-)-Cordobimine (29) [18]
,OCH3 H3C<
H3C<N OHH'*" .0
OCH3 HO'
(-)-Cordoblmine (29)
Bisbenzylisoquinoline Alkaloids with Two Ether Linkse.g. Pangkorimine (30) [19]
.OCH3 H3C<
NHRHO'
H-1 O,
OH
Pangkorimine (30)

i u vaiaiiiuiÿ
OCH3
,0HN, NH
NH* O
OCH3 HO'
Pachyovatamlne (31)
Bisbenzylisoquinoline Alkaloids with Three Ether Linkse.g. Kurramine (32) [21]
HO„
ON;
O
oOH
Kurramine (32)
Seco Bisbenzylisoquinoline Alkaloidse.g. (-)-PunJabine (33) [22]
OCH3
,0HN,
CH3H1 O
O
CHOO
HO'(-)-PunJabine (33)

Cularine Alkaloidse.g. (+)-Norcularidine (34) [23]
NHHO' C"" H
WOCH3
(+)-Norcularldlne (34)H3C1
Seco-cularine Alkaloidse.g. Norsecocularidlne (35) [24]
<CHsH
HO‘\
wOCH3
Norsecocularidlne (35)
H3C1
Cancentrine Alkaloidse.g. Dehydrocancentrtne-A (36) [25]
H3C(
NCH3
\H3C< o
HO' \ NO
\J Dehydrocancentrine-A (36)
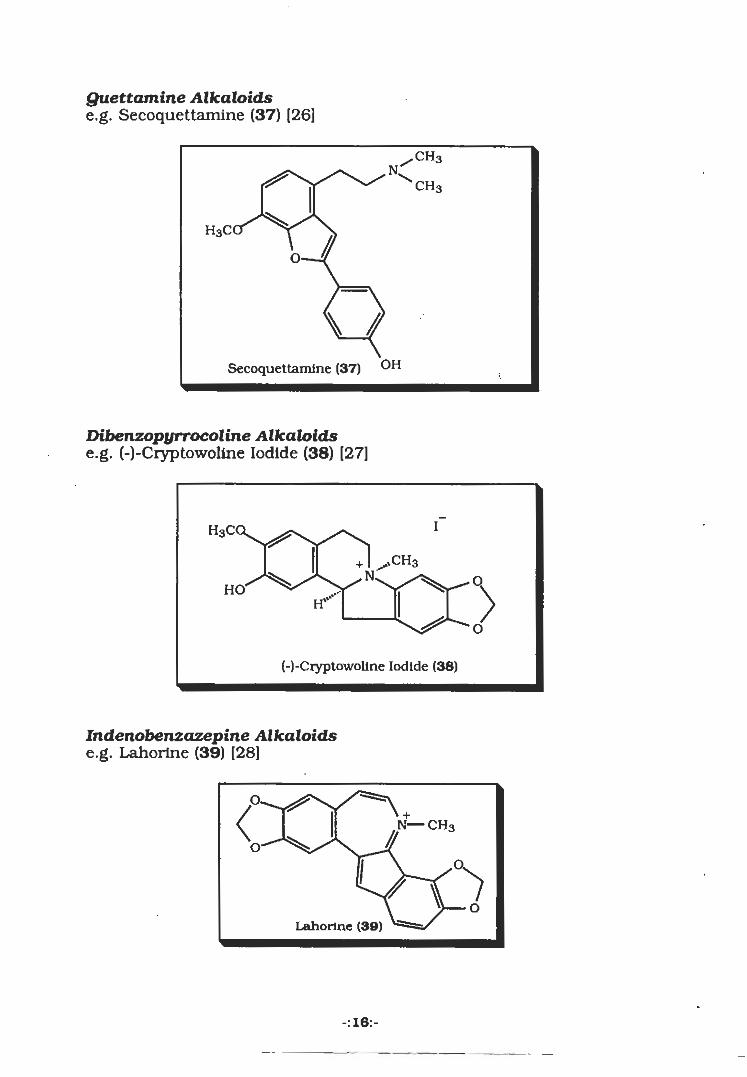
Quettamine Alkaloidse.g. Secoquettamine (37) (26]
/CH3N\
CH3
H3C( uW
Secoquettamine (37) OH
Dibenzopyrrocoline Alkaloidse.g. (-)-Cryptowoline Iodide (38) [27]
H3C<
+ IÿCH3
HO' H''lO
(-)-Cryptowollne Iodide (38)
Indenobenzazepine Alkaloidse.g. Lahorine (39) (28]
<fo-N— CH3
h
Lahorine (30)
-.16:-
i
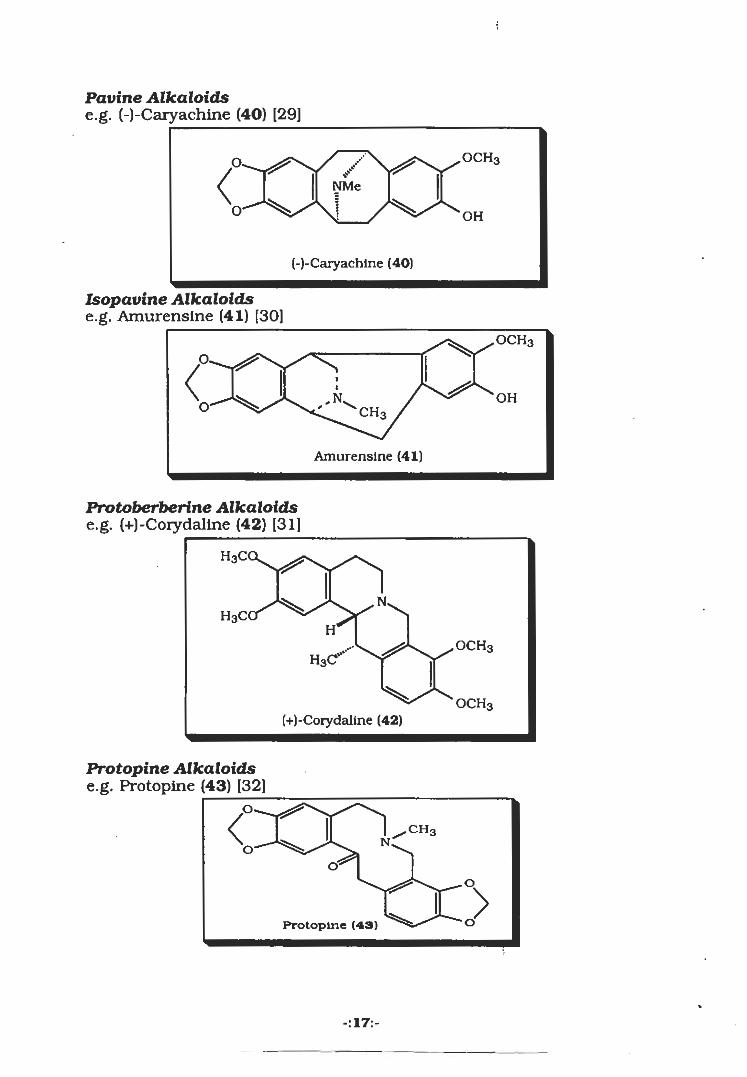
Pavine Alkaloidse.g. (-)-Caryachine (40) [29]
.OCH3a.NMe
O' OHj
(-)-Caryachlne (40)
Isopavine Alkaloidse.g. Amurensine (41) [30]
OCH3
<“O'N OH
CH3
Amurensine (41)
Protoberberine Alkaloidse.g. (+)-Corydaline (42) [31]
H3C<
N.H3C<
H1,OCH3
HaC*'"
OCH3(+)-Corydaline (42)
Protopine Alkaloidse.g. Protopine (43) [32]
<: 1 /CH3NCO'
DProtopine (43)
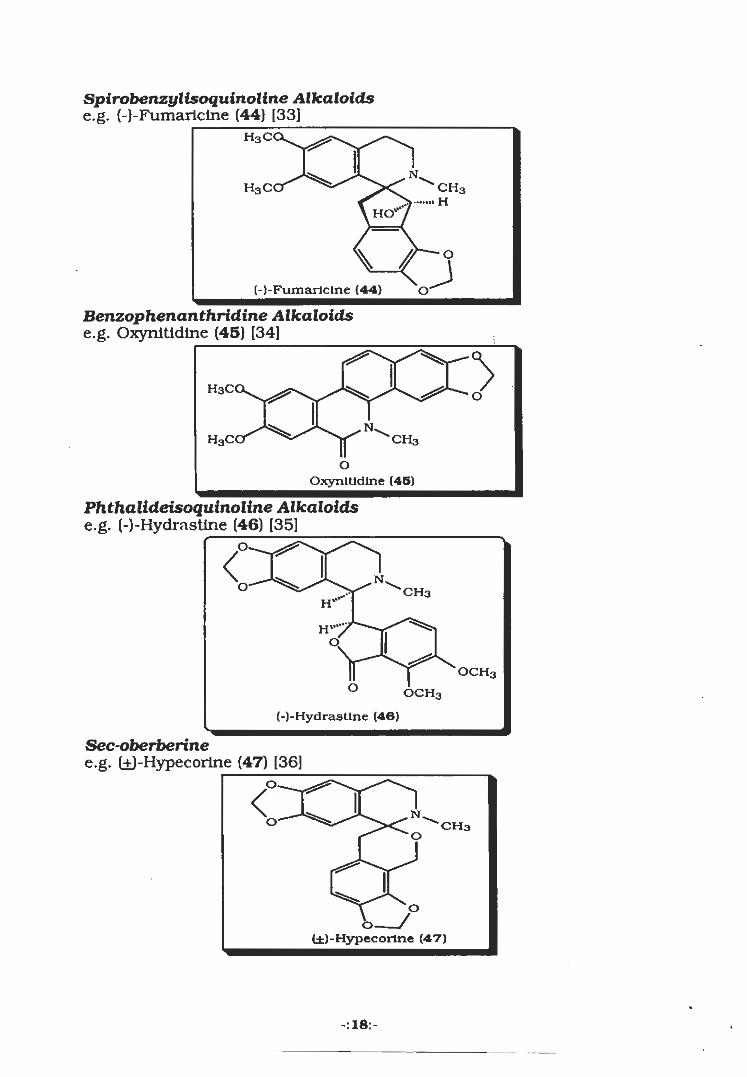
Spirobenzylisoquinoline Alkaloidse.g. (-)-Fumarlciiie (44) [33]_
H3C<
N.CH3H3C< . H
HO"
(-)-FumarlcIne (44)
Benzophenanthridine Alkaloidse.g. Oxynitidine (45) [34]
>H3C<
N.H3C< CH3
oOxynitidine (45)
Phthalideisoquinoline Alkaloidse.g. (-)-Hydrastine (46) [35]
<°'o- N.CH3
OCH3o OCH3
(-)-Hydrastlne (40)
Sec-oberberinee.g. (±)-Hypecorine (47) [36]
<: oCHa
O
1o
(±)-Hypecorlne (47)

Aporphine Alkaloidse.g. Cassyfillne (48) [37]
OCH3
<fO'
NH
N
H3C<
OHCassyfillne (48)
Morphine Alkaloidse.g. Carococcullne (49) [38]
H3C<
HO'
N.H CH3
O' OCH3
OH
Carococcullne (49)
1.1.3 PHARMACOLOGY OF ISOgUINOLINES ALKALOIDS
Pharmacology Is the study of the interactions between drugs
and the biological system. Pharmacology has a long history. The
primitive man believed that disease was caused by evil spirits
inhabiting the body. This belief persisted throughout the early
civilization till the Egyption period of medicine until it was
challenged by Hippocrates in 460 BC [39]. After Hippocrates the
Arabs adopted a scientific approach to the study of medicine.

Plant extracts have been used to cure diseases from the very
beginning of civilization. Over the last century plant extracts have
been shown to be responsible for providing relief against many
diseases due to the presence of some biologically active primary and
secondary metabolites which are produced in them. These
metabolites can be isolated and identified by using different
chromatographic and spectroscopic techniques.
Out of the many diverse classes of secondary metabolites the
isoquinoline alkaloids represent one of the largest groups of
alkaloids. Many alkaloids belonging to this group have shown
interesting pharmacological activities. A number of simple
isoquinolines alkaloids have also shown pharmacological effects on
the nervous system [40].
Many benzylisoquinoline alkaloids have also exhibited
interesting pharmacological activities. For example dioxyline (50) is
used in the form of its phosphate salts as a coronary and peripheral
vasodilator[4l]. Its methylenedioxy analogue, 3-methyl-6,7-
H3C< CH3.CH3
<H3C(
H3C< Vo7OCH33-Methyl-6,7-methylenedloxy-1-plperonytsoqulnollne (81)Dioxyline (80)
-:20:-

methylenedioxy-1-piperoxylisoquineline (51) is effective as a smooth
muscle relaxant [42]. Corydaldine (52) another isoquinolone
alkaloid, showed antirheumatic properties [43].
H3C(
N-HH3C<
oCorydaldine (52)
During pharmacological investigations on bisbenzylisoquino-
line alkaloids (+)-tubocurarine (53) was found to have potent
poisonous properties and was used as a poison by the South
American Indians. They used this alkaloid on the tip of arrows and
when this poison was injected into the blood stream of the victim, it
quickly blocked neuromuscular action, causing immediate death.
H3C< 0ci
?<:o' CH3H' CH2
OH
CH2H O
Hÿ;H3CT |
.OH
Cl® OCH3(+)-Tubocurarine chloride (53)

This alkaloid is also used in abdominal surgery in very small doses as
a complement to a local anesthetic since it causes paralysis of the
abdominal muscles without stopping the natural movement of the
intestines [44], The alkaloid is also an effective musqle relaxant.
Glaziovine (54), a proaporphine alkaloid, showed
antidepressive activity [45].
H3C<
N.H3C< CH3
o(±)-Glaziovine (B4)
The aporphine alkaloids are also important pharmacologically.
Many of them exhibit prominent pharmacological activity. For
example apocodeine (56) has antiemetic activity [46].
Boldine (55), an aporphine alkaloid, was found to have mild
sedative, diuretic and antiparasitic actions and it also increases the
secretions of the liver and salivary glands [47]. Another aprophine
alkaloid, xylopine (57), exhibits sedative and analgesic activity.
-:22

<fO- N-HN.
H3C< CH3HH
H3C<
OMeOH
CH3 (±)-Xyloplne (57)(+)-Boldlne (BB)
H<
HHO,
H3C<(±)-Apocodelne (BB)
Protoberberlne alkaloids have also shown pharmacological
activity. For instance the tetrahydroprotoberberlne alkaloids, which
belong to this class, have shown promising transquilizing properties
[481.
Berberine (58) has found use in the treatment of
gastrointestinal disorders. It is reported to depress intestinal
<: NX
,OCH3
OCH3Berberine (68)
-:23:-

peristalsis and to remove inflammatory congestion of the mucosal
surface of the intestine. Therefore, it is used in the treatment of
diarrhea of infancy and childhood [49].
)H3C<
Nv.CH3H3C<
OCH3(+)-8-Methoxydihydronltidine (59)
The benzophenanthridines alkaloid, 8-methyoxydihydro-
nitidlne (59), has shown anticancer activity. Protopine (43) and
cryptopine (61) are known to stimulate the uterus [50].
H3C<
<°'o-
•CH3,CH3 N.N;
H3C<o'-o-
ODoSs
Cryptopine (61)Protopine (43)
Some pathalideisoquinoline alkaloids also exhibit
pharmacological activity. For example narcotine (62) has antitussive
activity and it can be used to suppress cough [51].
-:24:-

CH3
OCH3,o
OCH3w(-)-a-Narcotlne (62)
OCH3
1.1.4 BIOSYNTHESIS OF ISOgUINOLINES ALKALOIDS
1.1.4.1 Isoquinolines
Biosynthesis is the experimental study of the formation of
secondary metabolites. Biosynthetically most alkaloids are derived
from simple amino acids such as ornithine (63), lysine (64),
phenylalanine (65), tyrosine (66), tryptophan (67), histidine (68) and
anthranilic acid (69).
CO2H
<H2NH2fr * CO2H H NH2l C02HH;
HH
Lysine (64) Phenylalanine (65)Ornithine (63)
CO2H CO2H
<<NH2 NH2HO' N
H
Tryptophan (67)Tyrosine (66)
CO2Hrpc CO2H
H NH2Histidine (68) Anthranilic acid (69)
4-:25:-
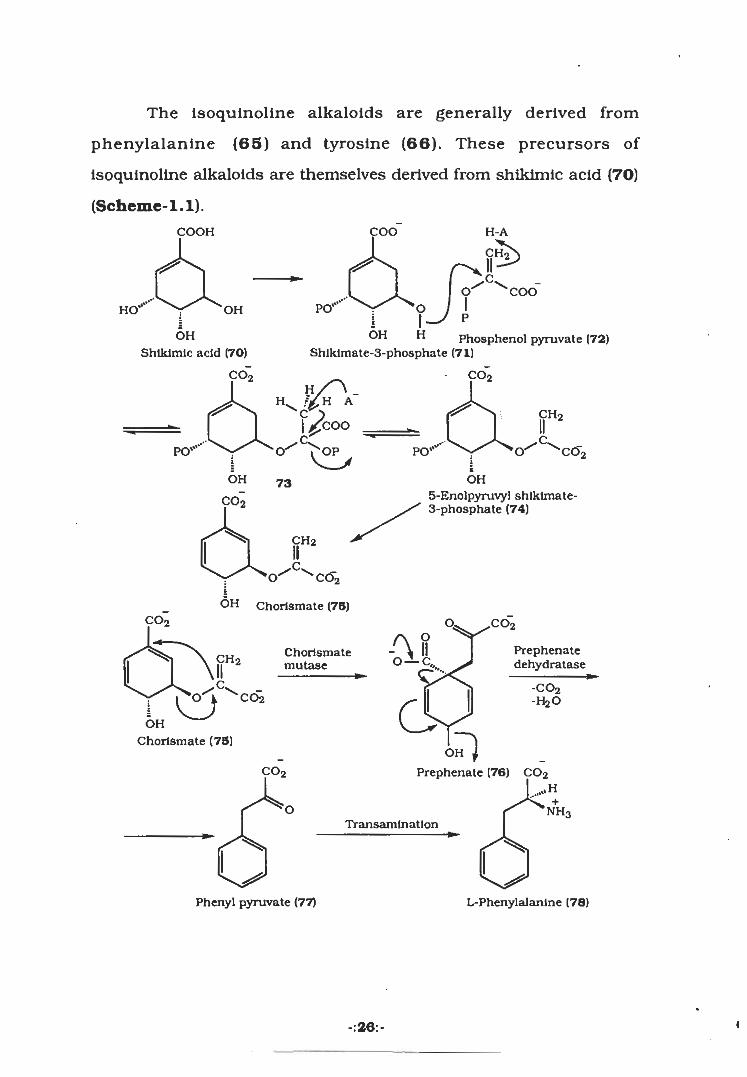
The isoquinoline alkaloids are generally derived from
phenylalanine (65) and tyrosine (66). These precursors of
isoquinoline alkaloids are themselves derived from shikimic acid (70)
(Scheme-1.1).
COOH COO H-A
&C
O COOIPO'" oOHHO
i ptOH H
Shlklmate-3-phosphate (71)
OH Phosphenol pyruvate (72)
Shikimic acid (70)
co2 co2
CH2coo
.'OPvly
Oÿ OÿÿCCÿPO"‘‘‘ PO"‘”I IOH 73 OH
5-EnoIpyruvyl shlklmate-3-phosphate (74)C02
CH2
.C.co2O'
iOH Chorlsmate (75)
,co2co2 O,
AffO-I-C,,,Prephenatedehydratase
ChorlsmatemutaseCH2
C -co2-HaOro cdi
OH
Chorlsmate (75)<3
oAco2 Prephenate (76) C02
UH+
O NH3Transamination
Phenyl pyruvate (77) L-Phenylalanlne (78)
1-:26.-

(Scheme-1.1) contd....
,C02co2
Prephenatedehydratase
ChorlsmalemutaseCH2 o2c.vI C
-co2-H2Or 1 ° y co2
Chorlsmate (75)
OH
iOH
Prephenate (76) QQ2
L.Hco2
o NH3Transamination
X
OHOH
p-Hydroxyphenyl pyruvic acid (79) L-Tyroslne (66)
Scheme-1.1
1.1.4.2 Simple Tetrahydroisoquinoline Alkaloids
The biosynthesis of the simple tetrahydroisoquinoline
alkaloids starts from the conversion of tyrosine to dopamine. This is
O-methylated to 4-hydroxy-3-methoxyphenylethylamine (60) which
undergoes oxidation to give 4,5-dihydroxy-3-methoxyphenethylamine
(82) and 5-hydroxy-3,4-dimethoxyphenethylamine (87) [52.53]. This
is the key intermediate in the formation of various
tetrahydroisoquinoline alkaloids such as anhalonidine (84), pellotine
(85), etc. (Scheme-1.2). It was originally assumed that the two-
carbon unit i.e., C-l and C-9, which is incorporated in cactaceae
alkaloids such as anhalonidine (84) and pellotine (85) was derived
from acetic acid. However after feeding acetic acid labelled at C-l to
the Peyote cactus, the pellotine isolated had the activity equally
divided between the C-1 and C-9 carbons.
-:27:-

Leete has shown that tyrosine (66) acts as a precursor of
anhalonidine (84) [53].
COOH HO, ,COOH
NH2H NH2
HO'TVroslne (06) DOPA (80) nH3C(
HO. H3ONH2
HO' NH2 NH2HO' HO'OH Dopamine (81)604.5-Dlhydroxy-3-methyoxy
phenylethylamlne (82)
IH3CI H3C( H30
NH2 NH2NH2H3C< H30HO'
OH 0CH3Mescaline (88)
OCH387 83
IH3OH3c<
N"HH3OH3C< H”iOH CH3
Anhalonidine (84)Anhalamine (88)
IH3OH3C(
N.,N.H3C( CH3H3CI CH3 H'"1
OH CH3Pellotlne (85)Anhalldlne (89)
Scheme-1.2
The biosynthesis of anhalonidine (84) in Laphophora williamsii
(Lemaire) was followed by feeding labelled sodium pyruvate. The C-3
of pyruvate was found to be incorporated into C-9 of anhalonidine
<-:28:~

(84). It was suggested that tetrahydrolsoquinoline alkaloids having a
methyl group at C-l are formed through the intermediacy of acetyl
coenzyme A and N-acetylphenethyl amine (90) (Scheme-1.3) [53].
H3C<CC&Nac=o
.1CHa
Labeled sodium pyruvate
O. II
H3Q-C— S— CoAL. willtamsU
NH2H3C<
OH
75
1H3C*
H3C<N— HIH3CI
H3C<OH
OH "CH3Anhalonldlne (84)
BO
Scheme-1.3
The biosynthesis of lophocerine (96) In the cactus Lophocercus
schottii was investigated by using labelled mevalonic acid (91) as a
precursor and resulted in the formation of tyrosine (66) which can
,OP .OH
T*ik. COOH
93Pyrophosphate (92)Labeled mevalonic acid (91)
Hal OP
V -xxCOOH
NH2
Labelled tyrosine (66)95Leucine (94)
H3C<
L. schottti N,HO' CH3
•CH3H*
CH3Labelled lophocerine (96)
Scheme-1.4
-:29:-

act as a precursor to the phenylelthylamine portion of the
tetrahydroisoquinoline nucleus (Scheme 1.4) (55].
1.1.4.3 Benzylisoquinoline Alkaloids
Winterstein and Trier suggested in 1910 that
benzylisoquinoline alkaloids might be derived in nature from two
molecules of 3,4-dihydroxyphenylalanine DOPA (80) via N-
norlaudanosoline (100) (Scheme-1.5) (54).
.COOH H< , ,COOH
H" NH2NH2
H< H<
(+)-Tyros1ne (66) DOPA (80)
ICOOHHOOCL. .NH2
OHOH
OHOH3,4-Dihydroxy phenyl pyruvic acid (98)
3,4-Dlhydroxy phenethylalanine (97)
IHOH<
OH+
NH2H<
Dopamine (81) OH99
H3ClH<
NH NHH3C<HO' H-fH'-t
.OH .OCH3
OH OCH3N-Norlaudanosoline (100) Papaverine (101)
Scheme-1.5
-:30:-
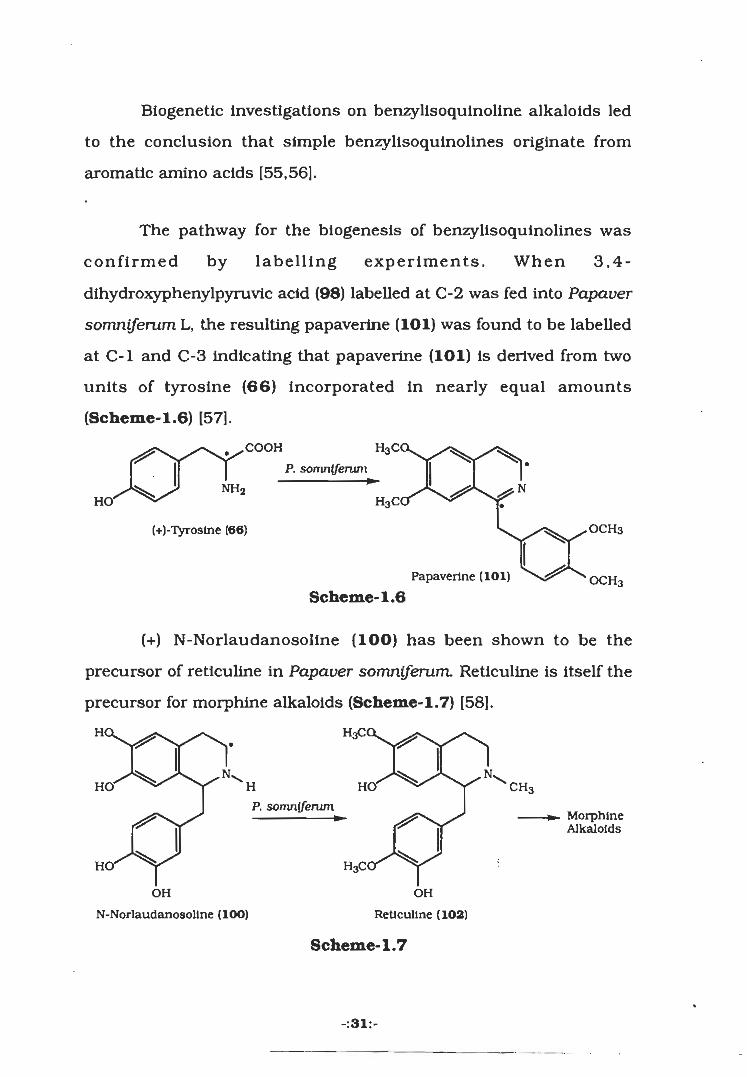
Biogenetic investigations on benzylisoquinoline alkaloids led
to the conclusion that simple benzylisoquinolines originate from
aromatic amino acids [55,56].
The pathway for the biogenesis of benzylisoquinolines was
confirmed by labelling experiments. When 3,4-
dihydroxyphenylpyruvic acid (98) labelled at C-2 was fed into Papaver
somniferum L, the resulting papaverine (101) was found to be labelled
at C-l and C-3 indicating that papaverine (101) is derived from two
units of tyrosine (66) incorporated in nearly equal amounts
(Scheme-1.6) [57].
, .COOH H3C<P. somniferum
NH2HO' H3C(
OCH3(+)-Tyroslne (66)
Papaverine (101) OCH3Scheme-1.6
(+) N-Norlaudanosoline (100) has been shown to be the
precursor of reticuline in Papaver somniferum. Reticuline is Itself the
precursor for morphine alkaloids (Scheme-1.7) [58].
H< HaC<
HO' H HO' CH3P. somniferum
MorphineAlkaloids
H< H3C(
OH OH
N-Norlaudanosollne (lOO) Reticuline (102)
Scheme-1.7

1.1.4.4 Pavine and Isopavine
A probable biosynthetic sequence of pavine and isopavine
starts from laudanosine (N-methyl tetrahydropapaverine).
Argemonine (108), the initial product, is converted to norargemonine
(110) and bisnorargemonine (109) on successive demethylation [59].
OH )H
M< M<,0Me ,0MeX V
NMe NMeV V/HO' MeO'
(-l-Platyccrtne (106)(-)-Munltaglne (1M)
IA O-coupUng0X
Met M<. Route A Route BNMe Ns. .NMe
HO' CHa CH<HO'
.OH
y y'OMe ‘OMe 'OMe
Imlnlum salt (103) (+)-ReLlculIne (102) M-Laudanldine (106)
p-coupling
Met.OMe .OMe
lNMe .NMe
(-)Blsnorargemonlne (109)
HO' Mt'OH
H-lsonorargemonlne (107)
M« MeO,.OMe ,OMe
,NMe ,NMeHO' MeO'
OMe 'OMe
I)-Argemonine (10©(-)- Norargemonine (110)
Scheme-1.8
The biogenetic connection between (+)-reticuline (102) and (-)-
argemonine (108) was investigated by feeding experiments on mixed
plants of Argemone mexicana and Argemone hispida [60].
-:32:-

A possible biosynthetic route based on the intermediacy of (+)-
reticuline (89) is presented In (Scheme-1.8). (+)-Reticuline (102) is
converted to the corresponding iminium salt by the in vivo oxidation.
This salt can then undergo Intramolecular para and ortho phenolic
coupling to afford (-)-bisnorargemonine (109) and (-) argemonine
(108) (Scheme-1.8).
(+)-Reticuline (102) can be converted, as shown in route B
(Scheme-1.8) to (+)-laudanidine (106) and then, by para-coupling, to
(-)-norargemonine (110).
Since pavine and isopavine alkaloids have the same absolute
configuration, it is therefore supposed that both skeletons are derived
biogenetically from the same precursor (4-hydroxybenzyltetrahydro-
isoquinoline (Scheme-1.9) [61,62].
OH>H
L-HHO,H<O
HaC< CH3CH3H3C< H-H-1 HaC<H3C(
H3C<H3C<Roemecarine-2a-N-oxlde (112)Roemecarine (111)
Scheme-1.9
This was later supported by the isolation of the 4-
hydroxybenzyltetrahydroisoquinoline base roemecarine (111) along
with it corresponding N-oxide (112).
-:33:-

H3CIH3C<
NNHO‘
HO'HH
H3C<H3CiOHOH
Retlculine (102)(+)-Norretlcullne (113)
H3CH3O
CH3+N.N.
CH3CH3 HO'HO'
HIH1
H3OH3CIMagnlflorlne (116)
Isoboldlne (114)
IHad
N.
IS. CH3HO'
H3C
OH
Boldine (116)
H3Ci H3O
N, N.CH3HO' CH3HO'
HH i
//
H3OO
OH(+)-Crotonoslne (118)N-Methylcoclaurlne (117)
Scheme-1.10
-:34:-

1.1.4.5 Aporphines Alkaloids
Aporphine alkaloids are derived from the corresponding
phenolic tetrahydrobenzylisoquinolines by direct oxidative coupling
of dienone derivatives (proaporphines), which then rearrange into
aporphines through dienone-phenol rearrangement (Scheme-1.10).
Feeding experiments using labelled reticuline in Papaver
somnifenim showed that isoboldine (114) was derived from reticuline
(102) by direct ortho-para oxidative coupling [63). Moreover, it has
been demonstrated that (±)-4'-0-methylnorlaudanosine (249), (±)-
reticuline (102) and (±)-norreticuline (113) were effective precursors
for boldine (116) in Litsea glutinosa [64).
H<
N.CH3HO*
HO'
OH 249
Since (+)-reticuline (102) but not (-)-reticuline is a possible
precursor for boldine (116) and since (+)-isoboldine (114) was
specifically incorporated into boldine (116), the postulated
biosynthesis of boldine (116) is believed to proceed as follows:
(+)-Norreticuline (113) -> (+)-reticuline (102) -> (+)-isoboldine
(114) -> (+)-Boldine (116) (Scheme-.1.10).
-:35:-

In investigations on the mode of biosynthesis of isocorydine
(120), labelled norreticuline (113),
norlaudanidine and reticuline (102) were fed to Annona sequimosa,
and It was shown that only reticuline was incorporated (Scheme-
1.11) (65).
norprotosinominine,
H3C< H30
N, N.CH3H< HO' CH3H'1 N,
HH< HO,
HaC. H3OIsocorydine (120)Protoslnomlnlne (119)
Scheme-l.il
Another aporphine base, isothebaine (124), may arise by the
dienolbenzene rearrangement from (+)-orientaline (121) via the
intermediate orientalinone (122) and orientalinol (123) (Scheme-
1.12) (66,67).
H3C< H3C
N, N,HO' CH3 HO'
h„ CHa
H3CI
/H3C(
/O Orientalinone (122)Orientallne (121)
IH3O H3C<
N. N.CH3Hi CH3HO'
"H•"HH3C1
/H3C< /Hi
HO Orientalinol (125)Isothebaine (124)
Scheme-1.12
-:30:-

1.1.4.6 Protoberberine Alkaloids
The protoberberine alkaloids are formed from a
benzyltetrahydroisoqulnoline base by condensation with
formaldehyde [68-70].
Spenser and co-workers have shown that tyrosine is a very
efficient precursor for berberine and is incorporated both into the
"top" and "bottom" parts of the alkaloid (Scheme-1.13) [71].
a N!,C00H
I H'" .OCH3NH3
H(
Tyrosine (66) Berberine (58)OCH3
Scheme-1.13
Barton [72] and Battersby [73] proposed that N-
methyltetrahydroisoquinollne could be converted to a protoberberine
H3C< o
<3 £ÿCH2 HN-CHH< 3H'
.OH O
OCH3 OCH3(+)-ReticulIne (102)
12B
,N*
,OCH3
Berberine (B8) OCH3
Scheme-1.14
-.37-

by oxidative cyclization 125 of the N-methyl function rather than by
a Mannich-type condensation with formaldehyde (Scheme-1.14).
1.1.4.7 Protopine Alkaloids
Reticuline (102) has been shown to be an efficient precursor
in the biosynthesis of protopine (43). Thus when labelled (+)-
reticuline (102) hydrochloride was fed to Dicentra spectabilis it gave
rise to labelled protopine (43) (Scheme-1.15) 174].
H3C( H3C<
N.HO' CH3 HO'
H'.OH ,OH
Reticuline (102) OCH3 OCH3Scoulerlne (126)o CH3
krO-
OoProtopine (43)
Scheme-1.15
(+)-Reticuline (102) labelled at the methoxy carbon was also
significantly Incorporated into protopine (60) and the protoberbertine
alkaloids (Scheme-1.16) [75).
H3C< oo— CH3krN-CH3HO' H-i o''ÿ
.OH ooOCH3 Protoplne (43)Reticuline (102)
Scheme-1.16
-:38:

1.1.4.8 Pthalideisoquinoline Alkaloids
The pthalideisoquinoline alkaloids are formed in nature by
oxidative rearrangement of tetrahydroprotoberberine isoquinoline
alkaloids [70]. Tyrosine (66) and methionine serve as the basic
building units in the biosynthesis of tetrahydroprotoberberine
isoquinoline alkaloids [77,78]. The protoberberine alkaloid scoulerine
(126) is itself derived from reticuline (102) in Papaver somniferum[79].
Experiments with scoulerine (126) stereospeclfically labelled
at C-13 have shown that the 13-pro-S-hydrogen of scoulerine (126)
is removed in its conversion to narcotine (62) in Papaver somniferum,
but its H-14 retains its stereochemistry during the conversion of
scoulerine (126) to narcotine (62). The intermediates between
scoulerine (126) and narcotine (62) have not yet been identified
although it has been suggested that ophiocarpine (127) and (+)-
enganine (128) could be the intermediates [80].
In this connection, it is interesting to note that the alkaloid
(+)-enganine (128), containing a hemiacetal group, has been isolated
from P. somniferum (Scheme-1.17) [81].
1.1.4.9 Secophthalideisoquinoline Alkaloids
The secophthalideisoquinoline alkaloids can be subdivided
into enol lactone keto acids, diketo acids, and ene lactams. A
blogenetic scheme is proposed for the secophthalideisoqulnolines
which includes the following sequence:
-:39:-

H3C< H3C<
N\; ,N,
HO'HO' CH3H'i H',OH .OH
ReUcuIlne (102) Scoulerine (126)OCH3“OCH3 IoO'
% aCH3 N.
H‘ H-1H--J .OCH3HÿlCHsOCO
“O OHOphlocorplne (127)Enganlne (128) £H O-
oJ<J
O— NVCH3
H"H1 j
OCH3NarcoUne (62) O OCH3
Scheme-1.17
Classical phthalideisoquinoline (60) phthalideisoquinoline
N-metho salt (129) -» secophthalide enol lactone (130) ->
secophthalide keto acid (131) -» secophthalide diketo acid (132)
fumariflorine (133) - type alkaloid (Scheme-1.18) [83-85],
1.1.4.10 Spirobenzylisoquinoline Alkaloids
Shamma proposed that phenolic protoberberine N-metho salt
could be the possible biogenetic precursor for spirobenzyliso¬
quinoline alkaloids [85,86],
-:40:-

o<J :!<CH3CH3 CH3
H* H'H"7
O ‘oo o o-
(-)-BIcucullln N-metho salt (129)(-)-Blcucullln (60)
ICH3/
3 /CHa,N.
CH3o CH3oo)OOH
O
O T O
U/Adlumldlcelne (131) 0
__J
I /Ha
o
Aobamedlne (130)
Nv. CH3CH3 /uO"
NsoCOOH CH3
.oO'ooOH
Fumariflorlne (133)Blcuculllnlne (132)
Scheme-1.18
Further biosynthetic divisions could be explained by
considering the substituents present on the aromatic ring of the
skeleton. The protoberberine N-metho salt can undergo cleavage to
the corresponding quinoid intermediates which could then form the
spiro system by an electrocyclic Michael condensation process. A
tautomeric shift would yield phenolic spirobenzylisoquinolines which
could then lead to the corresponding alkaloids via modification of the
oxygenated substituents.

H3C<HaC<
:Ar-CHs .N-CH3
fÿrV H3C(H3C<
c °N
NH; W H H3«
kd OH Vf135Dihydroprotobcrberlne (134)H
\H3C<H3C<
X XX c»3H3c< H3C< CH3
H3< VA-OH \-OHV
137OH O
Spirane (136)
*H3C<
XCH3H3C< XH2I
/\--O
AOchotenslmlne (138)
Scheme-1.19
In a proposed mechanism it was suggested that the N-metho
salt can undergo cleavage to the intermediate in basic medium,
which after electrocycllc ring closure (135) can form the spirane
(136). A tautomeric shift can afford (124) which can be easily
converted Into ochotenslmlne (138) (Scheme-1.19). If an exocyclic
methylene is present on C-13, the precursor of the compound is likely
to be the N-metho salt of the C-13 methylated dihydroprotoberberine
[87].
-:42:-

Those splrobenzylisoquinoline (141) having one or two oxygen
functions in ring "C" are probably derived from oxygenated 13-
oxotetrahydroberberine metho salt (139) via intermediate (140). This
photolytic rearrangement has been proposed by Manske and co¬
workers (Scheme-1.20) [88-90].
o aJ/CH3 *N-CH3CHJ
.OCH3 .OCH3O'
OCH313-Oxotetrahydroberberine metho salt (139)
OCH3140
o___
oCH3%
/> OCH3
0CH3Splrobenzylisoquinoline alkaloid (141)
Scheme-1.20
-:43:-

2.0 INTRODUCTION OF FUMARIA INDICA
F\imaria indica (family Fumariaceae) locally known as "Papra"
Is a small herb which is widely distributed in northern Pakistan
(91,92]. Fumaria indica has been used in the indigenous system of
medicine for the treatment of various diseases [93].
The plant extract is regarded as a laxative, diuretic and
alterative, and is said to be beneficial in dyspepsia and scrofulous
skin infections. The seeds of the plant are also used as a mild
analgesic [94],
The taxonomic delineation of families Papaveraceae and
fumariaceae is not uniform. Fedde [95], Tutin [96] and Melchior [97]
classified the Fumariaceaous plants in Papveraceae, as subfamily
Fiunarioideae, whereas Hutchinson [98] considered the Fumariaceae
to be an independent family.
2.1 LITERATURE SURVEY
The distribution of the alkaloids isolated from Fumaria species
is presented in Table-2.1.
-:44:-

Table-2.1 List of compounds Isolated from various plants of Fumariaceae
Exact8er. M. Formula Name Source Str. Ref.No. No.
[99]4-Hydroxy-3-methoxybenzaldehyde
Fumcuia indlca 142152.047 C8H8O31
Fumaria indica [94]C10H7NO2 Papraline 1432 173.054
C9H10O4 4,5- Fumaria indlca [99]3 182.057 144Dlmethoxy-1-hydroxybenzaldehyde
C10H9NO3 Noroxyhydras-tinlne
Fumaria indica,Fumariaparviflora
145 [100]191.1864
FUmaria indica.FumariaJlabeUata,Fumariaschleicheri
[101,102]205.213 CnHnNOs Oxyhydrasti-nlne
1465
[102]207.093 C11H13NO3 Corydaldine Fumaria
JlabeUata1476
1102,103)N-Methylcorydaldine
148221.255 C12H15NO3 Fumariavailantii,FumariaJlabeUata,Fumaria indica
7
Fumaria indica [99]222.161 C14H22O2 Fumaileate 1408
Fumariflorine [104.97]237.255 C12H15NO4 FumariaparvifloraFumaria indica
1509
Fumariflorineethyl ester
151 [106,107,265.308 C14H19NO4 Fumariaparviflora
10108|
Fumaria indica 152 [104]11 283.128 C17H17NO3 Paprazlne
153 [106,107]285.137 C17H19NO3 Coclaurine Fumariaparviflora
12
154 [107,109]285.137 C17H19NO3 Norjuziphlne Fumariavailantii
13
[110]Juziphlne Fumariavailantii
155299,369 C18H21NO314
156 [104,111]C18H19NO4 N-Feruloyltyra-mlne
Fumaria indica15 313.129
-:4S:-

(Table 2.1) contd.
mm-Ser. M. Formula Name Source Str. Ref.MassNo. No.
16 317.300 CigHi 1NO4 Norsanguina-rine
Fumaria indica.Fumariavailantii
1S7 [112]
320.324 Coptisine17 158Fumaria indica,Fumariadensijlom
[113.114]CI9H14N+04
18 321.100 CigHi5N04 Dlhydrocoptl-sine
Fumaria indica 159 [104,115.116]
19 322.108 Dehydroche-llanthefoline
Fumaria IndicaCI9HI6N+04 160 1113]
20 323.348 (+)-StyloplneCigHi7N04 Fumaria indica,Fumariaparviflora
[107,116]161
323.348 (±)-StylopineC19H i7N04 [102,117,21 Fumariadensijlora,Fumaria
Jlabellata
161118]
323.348 Ci9Hi7N04 (-)-Stylopine22 Fumariaofficinalis,Fumariashcleicheri
[119,1201161
23 325.363 CigHi9N04 Cheilanthifo- FumariaJudaica,Fumariavailantii
[107,115,162line 117)
24 327.379 Ci9H2iN04 Isoboldlne Fumariaparviflora,Fumariavailantii
[106,107|163
25 327.379 CigH2iN04 Lastourvilline Fumaria indica 1109)164
26 327.379 Ci9H2lN04 (+)-Scoulerine [112|165Fumariaparviflora,Fumariavailantii
327.379 Ci9H2iN04 (±)-Scoulerine27 Fumariaofficinalis
[120,121]165
327.379 (-)-Scoulerine28 CigH2iN04 165 [122]Fumariaofficinalis
CigH23N0429 329.395 Reticullne [121,123,Fumariavailantii
166124]
-:46:-

(Tibia 2.1) coDCd.
ExactMass
M. Formula Ref.Name Source Str.Ha. No.
332.335 LahorineC20Hi4N+O4 Fumariaparvljlora
(123,125,30 167126]
Sangulnarlne31 332.335 Fumaria indica,Fumariacapreolata,Fumariaofficinalis
168 [115,1181C20Hl4N+O4
C20H15NO4 dihydrosan-guinarine
32 333.343 Fumariaofficinalis,Fumariauailantii
1106,127)169
8-Oxocoptlsine Fumaria indica.Fumariaparvljlora,Fumariauailantii
33 335.079 C19H13NO5 U28]102
(115,126,129.130]
Berberlne Fumaria kraiikil34 336.367 170C20Hi8N+O4
C2qH2iN04339.390 Canadlne 1115,130.35 Fumariakralikii,Fumariaofficinalis
172131]
339.390 H-Sinactlne36 C2qH2iN04 |117|Fumaria
officinalis171
C2QH2IN04339.390 (±)-Slnactine37 Fumariaojjicinalis
1117,120]171
38 341.406 C2qH23N04 Isocorydine Fumariauailantii
[124,132.173133]
C19H19NO539 341.363 Ledecorine Fumariauailantii
I134|174
C19H19NO5 Norfumaiitlne FUmaria kralikii40 341.363 175 [1351
347.326 C2qH13NO5 Oxysangutna-rlne
Fumaria indica 1127.136]17641
42 348.378 Chelerynthe-rine
(1271C2)Hi8N+04 Fumariaschlicheri
177
1125)43 348.396 LahoramlneC2iHi8N+04 Fumariaparvijlora
178
351.358 C2qH17NO5 Fumarillne (125,137)Fumaria indica.Fumariaofficinalis
44 179
-:47:-
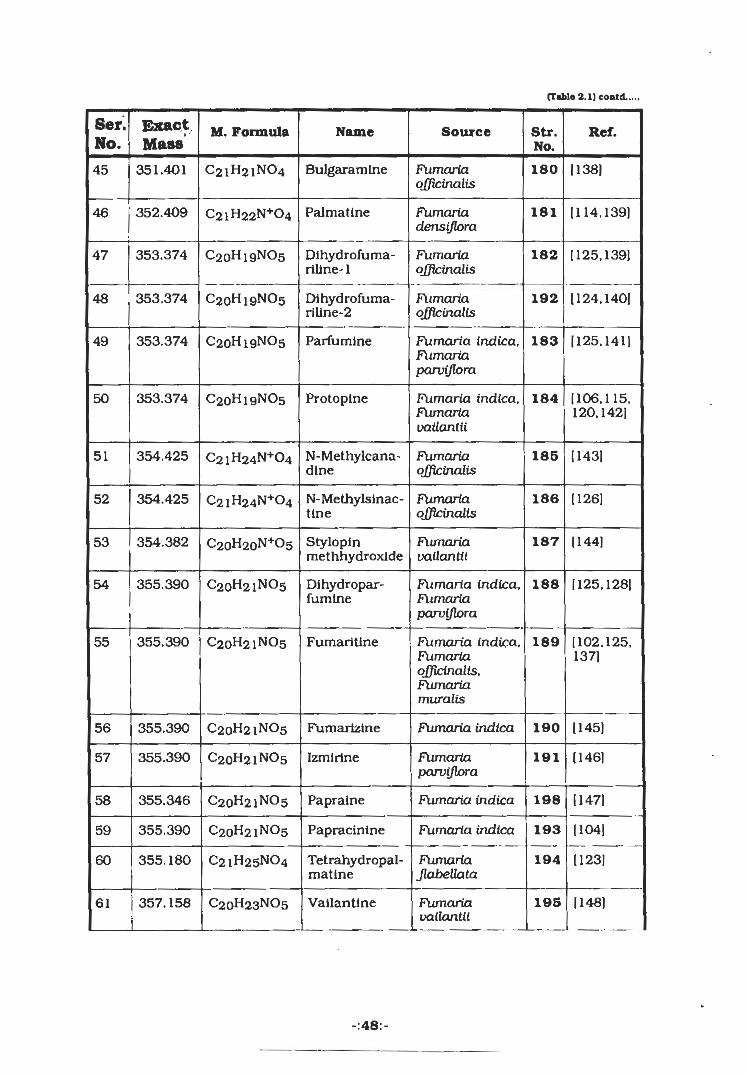
(Tmblo 2.1) cootd....,
Ser. ExactMass
M. Formula Name Source Str. Ref.No. No.
C21H21NO4 Bulgaramlne Fumariaofficinalis
1138)351.401 18045
46 352.409 Palmatine Fumariadensiflora
181 [114.139]C2lH22N+04
353.374 C20H19NO5 Dihydrofuma-rUlne-1
Fumariaojfficinalis
(125,139)47 182
353.374 C20h19no5 Dihydrofuma-rillne-2
(124,140148 Fumariaojfficinalis
192
Parfumlne353.374 C20H19NO5 Fumaria indica,Fumariaparvijlora
(125,141)49 183
50 353.374 C20H19NO5 Protoptne Fumaria indica.Fumariavailantii
[106.115.120,142]
184
51 354.425 N-Methylcana- 185 (1431Fumariaojfficinalis
C2lH24N+04dine
N-Methylslnac- Fumariaojfficinalis
186 (126)52 354.425 C2lH24N+04tine
53 354.382 Stylopinmethhydroxlde
Fumariavailantii
187 (144)c2oH2oN+o5
355.390 C2oH2lN°5 Dlhydropar-fumlne
Fumaria indica.Fumariaparvijlora
[125.128154 188
355.390 C20H21NO5 FUmaria indica.Fumariaojficinalis,Fumariamuralis
[102.125,55 Fumaritine 189137]
Fumaria indica56 355.390 C20H21NO5 Fumarlzine [145]190
355.390 C20H21NO5 Fumariaparvijlora
[146]57 Izmlrlne 191
[147]355.346 C20H21NO5 Papraine Fumaria indica 19858
Fumaria indica [104]355.390 C20H21NO5 Papracinlne 19359
[123]60 355.180 C2lH25N04 Tetrahydropal-matine
FumariaJlabellata
194
1148)357.158 C20H23NO5 Vallantlne Fumariavailantii
19561
-:48:-

(Table 2.1) contd.
Ser. ExactNo. Mass
Ref.M. Formula Name Source Str.No.
8-methoxydl-hydro-sanguinarlne
1149]C21H17NO5 Fumaria indica 196363.36962
[150]C20H15NO6 Densiflorine Fumariadensijlom
197365.34263
Fumaria indica.Fumariaparvijlora
1151]C20H17NO6 (+)-Adlumldlne 20464 367,106
(±)-AIdumldtne [152,153]367.106 C20H17NO6 Fumariavailantii
20565
[154.155]C20H17NO6 t-)-Blcuculline Fumaria indica.Fumariaparvijlora,Fumariavailantii
206367.10666
[1021(+)-Bicuculline F. vailantii367.106 C20H17NO6 20767
[102,120.C20H17NO6 (+)-Blcuculllne FumariavailantiiFumaria
JlabeUata
20868 367,106155]
Parfumldine [125,156]C21H21NO5 Fumariaparvijlora,Fumariaofficinalis,Fumariavailantii
203367.16569
(-)-Capnoldine 1154,155,C20H17NO6 Fumariavailantii
70 367.106 210157)
[158|Fumaria indica,Fumariavailantii
205368.150 N-Methylprotopine
71 C2IH22N+05
[159]C20H19NO6 Fumariavailantii
212369.373 H-Corledine72
Corlumldine [1551369.373 C20H19NO6 Fumariaparviflora
21373
[100.146]369.416 C21H23NO5 Cryptopine Fumariadens{flora,Fumaria kralvii,
Fumariaojfficinalis
22074
[128]C20H19NO6 209369.373 Fumariavailantii
75 Enganlne
-:49:-
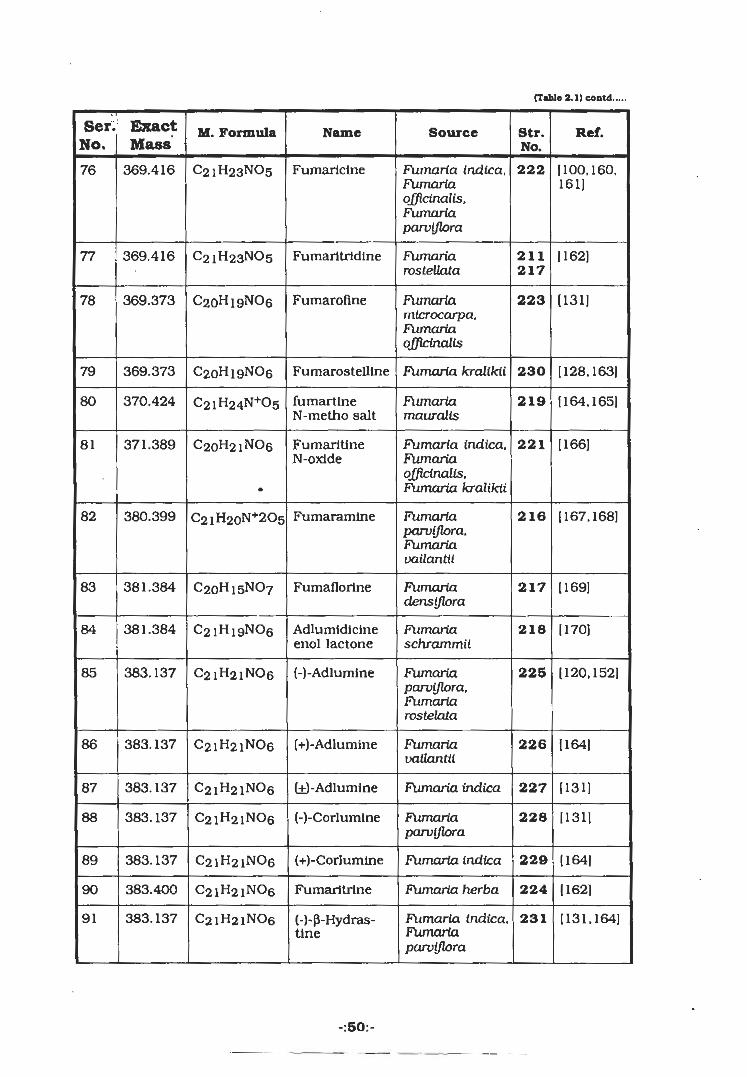
(Table 2.1) contd.
Ser. ExactNo. Mass
Ref.M. Formula Name Source Str.No.
(100.160.369.416 C21H23NO5 Fumarlclne Fumaria indlca,Fumariaofficinalis,Fumariaparviflora
22276161]
1162]369.416 c2lH23N°5 Fumaritrldine Fumariarostellata
77 211217
1131]Fumaroflne369.373 C20H19NO6 Fumariamicrocarpa,Fumariaofficinalis
22378
369.373 Fumaria kralikii [128,1631C20H19NO6 Fumarostelltne 23079
fumartlneN-metho salt
370.424 Fli/nariamauralis
219 [164.165]80 C2iH24N+05
[166]C20H21NO6 Fumaria indica,Fumariaofficinalis,Fumaria kralikii
221FumaritineN-oxlde
371.38981
(167.168]82 380.399 Fumaramine Fumariaparviflora.Fumariavailantil
216C2lH20N+2O5
1169]83 381.384 C2qH15NO7 Fumaflorine Fumariadensiflora
217
Adlumidicineenol lactone
11701381.384 C2iHi9N06 21884 Ftimariaschrammii
383.137 (-)-Adlumlne 225 (120,152]85 C21H21NO6 Fumariaparviflora,Fumariarostelala
(+)-Adlumine (1641383.13786 C21H21NO6 Fumariavailantil
226
(131](±)-Adlumine Fumaria indica383.137 C21H21NO6 22787
(1311383.137 C21H21NO6 (-)-Corlumlne 228Fumariaparviflora
88
383.137 (+)-Corlumlne FUmarta indica 229 1164]C21H21NO689
Fumaria herba (162]383.400 C21H21NO6 Fumaritrine 22490
FUmarta indica,Fumariaparviflora
(131.164]383.13791 C21H21NO6 231(-)-p-Hydras-tlne
-:50:-

(Table 2.1) contd.
Ser. ExactMass
M. Formula Ref.Name Source Str.No. No.
1152)C21H21NO6 232FumariavaUantii
383.13792 (+)-P-Hydroas-tlne
1152]C21H21NO6 Fumaria indica 23393 383.137 (+)-P-Hydras-tlne
1154]C21H21NO6 23494 383.137 Fumariaparvijlora.Fumariaschelicheri
(+)-a-Hydras-tlne
FumariavaUantii
[164]C21H21NO6 23595 383.137 (-)-a-Hydras-tine
Fumariaparvijlora,Fumariaschelicheri
[1661C21H21NO6 23696 383.137 (±}'<x-Hydras-tine
1100]C21H23NO6 Fumaria indica 24997 385.152 Raddeantne
Acetodihydro-sangulnarine
[165]C23H19NO5 FumariavaUantii
19998 389.407
[168]Fumaramidine396.442 C22H24N2O5 Fumariaparvijlora
20099
[168]Fumaridine Fumariaparvijlora,FumariavaUantii
396.442 C22H24N2O5 201100
[139,170]397.427 Fumariaschrammii
202C22H23NC)6 Adlumiceineenol lactone
101
[171]397.427 C22H23NO6 Fumarophy-cine
Fumariakralikii,Fumaria
officinalis.Fumariamuralis
214102
1172]397.384 C22H19NO7 Narlumidtne Fumaria indica 237103
[120.173]397.427 C21H23NO6 N-Methyl-hydrastlne
Fumariaparvijlora,FumariavaUantii
238104
1100]Fumaria indicaC21H19NO7 Paprarine 239105 397.113
[174]Fumschlel-cherlne
398.415 Fumariaschleicheri,Fumariaschrammii
240106 C2iH24N+06
-:S1:-

(Table 3.1) conld.,
UaMfs*So.Ref.M. Formula Source Str.Name
No.
(130)N-Methyladlu-mlne
215Fumartavailantii
107 398.415 C2lH24N+06
Fumarta tndica [1751C2iH2iNC>7 Narlumlclne 242399.399108
[120,1701Fumartaschrammli
243C2iH2iNC>7 Adlumldlcelne109 399.399
O-Methylfuma-rophyclne
(153)C23H25N06411.454 Fumartakraukii,Fumariaofficinalis
244110
(119,1661Fumaria tndica,Fumariaschrammit
C21H19NO8 Narceimine 245413.383111
1120,140,246C22H25N07 Adlumlceine FumariaschrammU
415.163112170]
[176.177JNaraceimlclne Fumaria tndica 247C21H21NO8415.399113
[168.1731C22H25n07 N-Methyl-hydrastelne
248415.163 Fumariaparvijlora,Fumariaschleicheri,Fumariavailantii
114
Fumaria indica 255 [1821C25H23NC>5115 417.157 9,10-Methylenedloxykarachlne
Fumaria indica 250 [168,178]116 426.468 C23H26N2C>6 Nareclneimlde
C22h23NOs Bicuculllnldlne 251 [102,174]FumariaschrammiLFumarialabellata
429.426117
paprafumlne 253 [100]C22H23N08 Fumaria inidca119 429.142
254 [1021Fumaria indica457.177 C24H27NO8 Papraclne120
255 [1791515.516 C26H29NOl0 Parvlflorine Fumariaparviflora
121
[1801Fumariadensijlora
C30H34N2O6 Fumadenslne 241122 518.608
-:52:-

NO'
143
R3<
N.RiR2<
o146 Rj = H, R2 + R3 = CH3146 Rj = CH3> R2 + R3 = CH3147 R1 = H, R2 = R3 = CH3148 Rj = R2 = R3 = CH3
CH3N.O
CH3< ORO
O
160 R = H161 R=C2H5
R2.
N.R3 RI
54R:
Re Re
R7163 Ri = R4 = R5 = R7 = Ra = H, R2 = OCH3. R3 = Re = OH164 Rj = R2 = R5 = R7 = Ra = H, R3 = OCH3, R4 = Re = OH166 R! = CH3, R2 = R5 = R7 = Ra = H. Ra = OCH3, R4 = Re = OH166 Rx = CH3. R2 = Re = OCH3> R4 = R7 = Ra = H. R3 = R5 = OH174 Ri = CH3> R2 + R3 = Re + R7 = 0-CH2, R4 = R5 = H, Re = OH190 Ri = CH3, R2 + R3 = Re + R7 = 0-CH2-0. R4 = R5 = H, Ra = OC
-:63:-

R2<
CH3RI«H
R;
R4<
RS
163 Ri = R3 = R4 = H. Ffe = CH3, R5 = OC1104 Ri = R2 = R3 = H, R4 = CH3> R5 = OCl173 Rj = R2 = R4 = H. R3 = CH3, R5 = H
< N RioRi
O
>O159 Ri = R2 = H102 Ri + R2 = O
Ri
+AR2<
OR3
;
OR,158 Rj + R2 — R3 + R4 — CH2160 RL = CH3( R2 = H. Rs + R4 = CH2170 Rj + R2 = CH2l R3 = R4 = CH3181 Ri = R2 = R3 = R4 = CH3
-:54:-

Rl<
N,
R2<H
OR3
OR4
161 Ri + R2 = R3 + R4 = CH2162 Ri = CH3. R2 = H, R3 + R4 = CH2166 R1 = R4 = CH3> R2=R3 = H171 Ri + R2 = CH3, R3 + R4 = CH2172 Ri + R2 = CH2, R3 = R4 = CH3194 Rl = R2 = R3 = R4 = CH3
Ri'
CH3+N:
R2«
OR3R5
OR4187 Ri + R2 = R3 + «4 = CH2, R5 = OH186 Rx + R2 = CH2. R3 = R4 = CH3, R5 = H186 R1 = R2 = CH3>R3+R4 = CH2, R5=H
°>o
N
O
\-i 167
°>O
JKR2< CH3
ORI 168 RI + R2- CH2177 R1 = R2 = CH3
-:55:-

o
>o
N— CH3O\ / H2 Rl\— o
169 Rj = R2 = H176 R] + R2 = 0196 Ri= H. R2 = OCH3199 R, = H. R2 = CH2-CO-CH3
R2'
IÿCH3NC
Hro'
.ORs
OR4184 Rj + R2 = R3 + R4 — CH2191 Rj = CHs, R2 = H. Ra + R4 = CH2196 R] = R2 = H, R3 = R4 = CH3220 Rj = R2 = CH3, Ra + R4 = CH2
O
< + I/ch3+Nÿ-CH3O'
O'
o
Q206
Rl+ N— CH3/)
Hi
167 Rj + Rz = CH2178 R1 = R2 = CH3
-:66:-
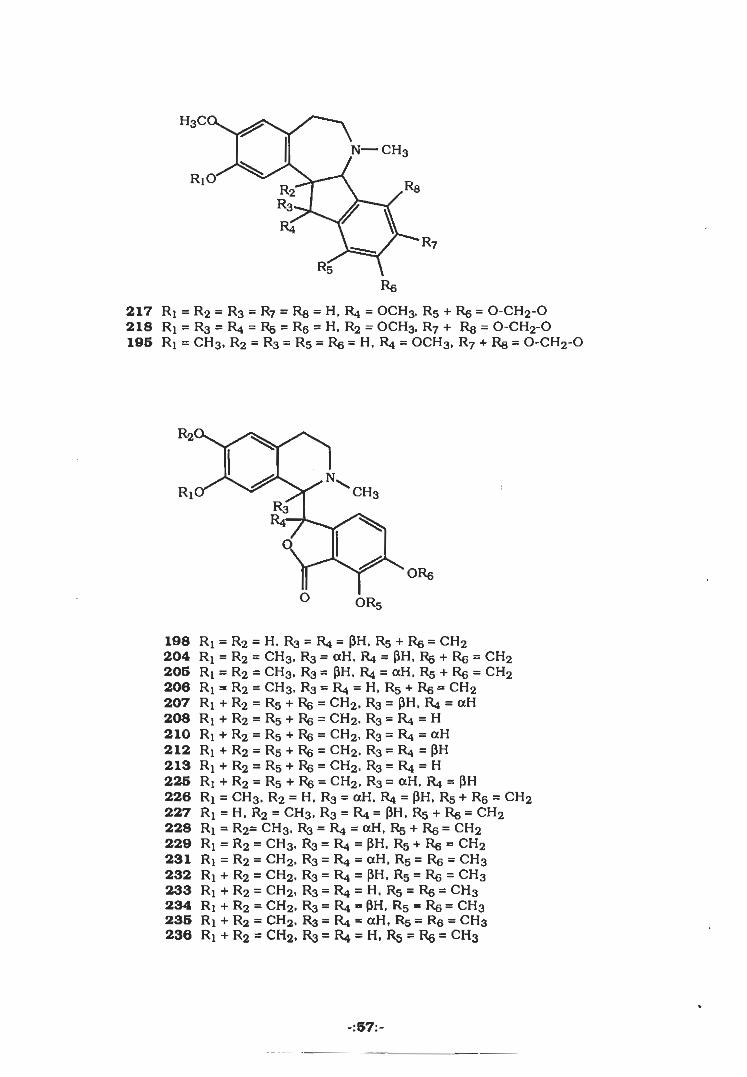
H3C(
N— CH3
Ri' ReR2R3 nR4
R7
R5Re
217 Rj = R2 — R3 = R7 = Rg = H, R4 = OCH3, R5 + Re = 0-CH2-0218 Rj = R3 = R4 = Rg = Rg = H, R2 — OCH3, R7 + Rg — 0-CH2-0196 Rx = CH3, R2 = R3 = r5 = Re = H. R4 = OCH3, R7 + Rg = 0-CH2-0
R2«
CH3RIR3
ORg
O OR5
198 RI = R2 = H,R3 = R4 = (JH. R5 + Rg = CH2204 Rj = R2 = CH3, Rs = aH. R4 = pH, Rg + Rg = CH2205 Ri = R2 = CH3> R3 = PH. R4 = aH. Rg + Rg = CH2206 Ri = R2 = CH3, R3=R4 = H>R5 + Rg = CH2207 Ri + R2 = R5 + Rg = CH2, RS = PH, R4 = aH208 R; + R2 = R5 + Rg = CH2, R3 — R4 = H210 Rj + R2 = R5 + Rg = CH2, R3 = R4 = aH212 Rj + R2 = Rg + Rg = CH2> R3 = R4 = PH213 Ri + R2 = R5 + Rg = CH2, R3 = R4 = H225 R! + R2 = R5 + Rg = CH2. R3 = aH, R4 = pH226 Rx = CH3, R2 = H, Rs = aH. R4 = pH, Rg + Rg = CH2227 Rx = H, R2 = CH3, R3 = R4 = PH. Rg + Rg = CH2228 Ri = R2= CH3, R3 = R4 = aH, R5 + Rg = CH2229 Rx = R2 = CH3, R3 = R4 = PH. Rg + Rg » CH2231 Ri = R2 = CH2, Ra = R4 = aH. R5 = Rg = CH3232 Ri + R2 = CH2, R3 = R4 = PH, R5 = Rg = CH3233 Rx + R2 = CH2, R3 = R4 = H, R5 = Rg = CH3234 Ri + R2 = CH2, R3 = R4 = pH, R5 = Rg = CH3235 Rx + R2 = CH2. R3 = R4 = aH, R5 = Rg = CH3236 Ri + R2 - CH2. R3 = R4 = H. R5 = Rg = CH3
-:57:-

<1 N.O CH3
HH.
Q
OH wOH215
H30
CH3
CH3H3C«H
H-
Q
O
Wo241
CH3N:H3C<
CH3
HH3C(
O
o\—o o 218
CH3N,R2<
CH3
R5RI
OR3
O OR4222 Ri + R2 = R3 + R4 = CH2. R5 = H202 Rj = R-j = CH3, R3 + R4 = CH2, Re = H238 Rt + Rz - CH2. R3 - R4 - CH3. Re - H239 Ri + R2 = R3 + R4 = CH2, R5 = OH
-:68:

Ri
NR21 COOR3
O
o
217 R1 = R2 = CH3> R3 = H
R, ,N.
Re
OR7R2(
.O
OR3
IIV0R4
242 R! = R2 = R3 = R4 = CH3, R5 = R6 = CH3. R7 = H
-:59:
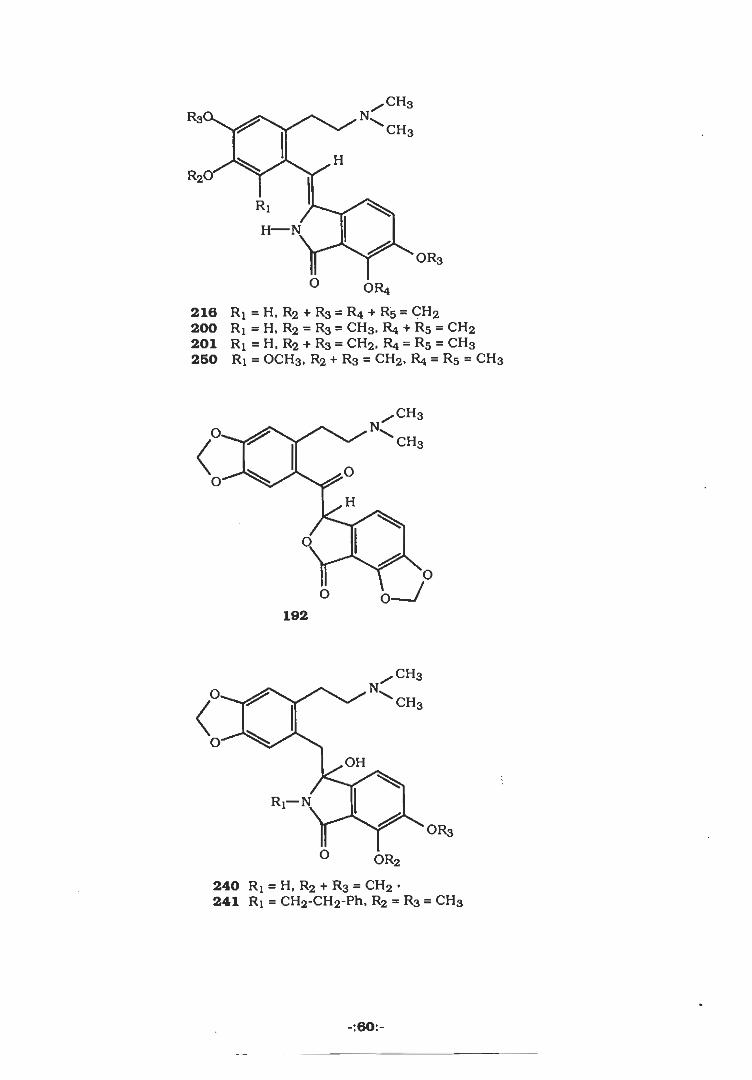
CH3NRa<
CH3
HR2<
RIH— N
OR3
O OR4216 Rj = H, R2 + R3 = R4 + R5 = CH2200 R! = H, R2 = R3 = CH3. R4 + R5 = CH2201 Ri = H, R2 + R3 = CH2> R4 = R5 = CH3250 R! = 0CH3, R2 + R3 = CH2, R4 = R5 = CH3
CH3N;o
CH3< .0oH
Q
Ouo o192
CH3N;o
CH3<oOH
Rl— N
ORg
O OR2
240 R! = H, R2 + R3 = CH2 •241 Ri = CH2-CH2-Ph, R2 = R3 = CH3
-.60:-

CH3NR3< s
CH3o
R5 OReR2< R4
RIO-
OR7
ORe
248 Rj = R4 = R5 = Re = H, R2 + R3 = CH2. Re = R7 = CH3243 R2 + R3 = Re + R7 = CH2, RI = R4 = R5 = Re = H245 Ri = Re = H, R2 + R3 = Re + R7 - CH2, R4 + R5 = O246 Rj = R4 = R5 = Re = H, R2 = R3 = CH3. Re + R7 = CH2251 Rj = Re - H, R2 - R3 = CH3, R4 + R5 = O, Re + R7 = CH2252 Ri = Re = H, R2 + R3 = CH2, R4 + R5 — O, Re = R7 = CH3254 Rj = H, R2 + R3 = CH2, R4 + R5 = O, Re = R7 = CH3, R3 = C2Hs
CH3NS
CH3< oOH\\ .ORo
HO'
O
247 R = H253 R = CH3
.61:-

Re R7R3 V/
N
•' Oc'
-7R4 R5 o
R2O OR,
175 R, = R3 = R4 = Re = R7 = H, R2 = CH3, R5 = OH179 R, + R2 = CH2, R3 = CH3, R4 + R5 = O, Re = R7 = H182 R, + R2 = CH2, R3 = CH3> R4 = Re = R7 = H. R5 = OH237 R, + R2 = CH2, R3 = CH3, R4 = R6 = R7 = H183 R, = Re = R7 = H, R2 = R3 = CH3, R4 + R5 = O189 R, = R4 = R7 = H, R2 = = R3 = CH3, R5 = OH188 R, = R5 = R6 = R7 = H. R2 = R3 = CH3. R4 = OH203 R, = R2 = R3 = CH3, R4 + R5 = O. Re = R7 = H222 R, = R2 = R3 = CH3, R4 = R6 = R7 = H, R5 = OH211 R, = R5 = Re = R7 = H, R2 = R2 = R3 = CH3, R4 = OCH3223 R, = R7 = H, R2 = R3 = CH3, R4 + R5 = O. Re = OH214 Ri = R4 = R6 = R7 = H, R2 = R3 = CH3> R5 = OAC249 R, = R2 = R4 = Re = H, R3- CH3> R5 = R7= OH230 R, = R4 = H, R2 = R3 = CH3. R5 = OH. Re + R7 = O224 R, =R2 = R3 = CH3> R4 = OCH3, R5 = Rg = R7 = H244 R, = R2 = R3 = CH3. R4 = Rg = R7 = H, R5 = OAC255 R, = D-glucose, R2 = R3 = CH3, Rj + R5 = O, Rg = R7 = H
CH3/
N
O_JK i> 0 O
O O
197
-:62:-

Ri ,Ra\
CH3/
0--N
O
R3R4 -Jo
H3C0 OH
193 R! = R2 = R3 = R4 = H221 Rj a R2 = R3 = H, R4 = OH
H3C ru
'V3+ N
J 0_yH OH '
H3CO OH
219
OHO
N
HHO'
152 R = H156 R = OCH3
Ri'
N—Rs
R2< ,OR3
r\OR4
180 Rj = R2 = R5 = CH3. R3 + R4 = CH2
-:63:-

SECTION B
ISOLATION AND STRUCTUREELUCIDATION OF FUMARIA INDICA

2.2 RESULTS AND DISCUSSION
Four new and four known compounds have been isolated from
Fumarta indica. Various spectroscopic studies and other experimental
techniques were carried out for the structural determination of these
compounds. The results of these studies are discussed in this
chapter. The extraction and isolation procedures of these compounds
are discussed in detail in Chapter-3 of the "Experimental section".
New Compounds from Fumaria indica
2.2.1 Fumaileal (144)
The chloroform extract obtained by extraction (scheme-2.8,
Experimental) was chromatographed over a silica gel column using
pet.ether : acetone (8:2) as eluent to obtain fraction FN-2 (Scheme-
1.2, Experimental). This fraction was further purified by preparative
TLC on silica gel (GF-254, 0.2 mm) using pet.ether : acetone (8.1:1.5
+ two drops of NH4OH) as the developing solvent. This afforded
Fumaileal (4,5-dimethoxy-2-hydroxy-benzaldehyde,144, 7.2 mg)
which gave a dark blue colour test with ceric sulphate spray.
o6
H3C( 1H5
,24H3C< OH
3
Fumaileal (144)
The HREIMS showed molecular ion at m/z 182.0594
corresponding to the molecular formula C9H10O4 (calcd. 182.0579)
-:64:-

showing five degree of unsaturations in the molecule. The 1R
spectrum of 144 showed an absorption at 1718 cm*1 corresponding
to the carbonyl functionality.
The UV spectrum showed strong absorption at Xmax (MeOH)
250 (log e) (2.873) nm. The spectrum (CDCI3. 400 MHz)
displayed three sharp singlets in the downfield region one of which at
8 9.8 was assigned to the aldehydic proton. Only two aromatic
protons resonated at 8 7.13 and 7.23, from this it was apparent that
the benzene ring was tetra-substituted. Two methoxy groups
resonated as a sharp 6H "singlet" at 83.69.
The 13C-NMR spectrum (broadband) of 144 showed nine
carbon signals. The DEPT spectrum established that the molecule
contained two methyl, two methine and hence five quaternary
carbons atoms (105). The 13C-NMR assignments are shown in Table-
2.2.
Table-2.2. 13C-NMR assignments of 144
8 (DEPT)13C-NMR ChemicalShift (8)
Carbon No
C-l 132.2 -C-02 147.4 -C-03 105.8 CH04 142.2 -C-05 142.1 -C-06 106.7 CH07 56.5 -OCH308 56.6 -OCH3
o09 190.7 II-C— H
-:65:-

The IR spectrum (CHCI3) of 144 showed an absorption at
1718 cm-1 due to the conjugated carbonyl group present in the
molecule. The HREIMS (m/z 182.0579} revealed the molecular
composition, CgHioC>4, which was consistent with five degree of
unsaturations. Four of these were accounted for a benzene ring and
the fifth for an aldehydic carbonyl group. Two oxygen atoms were
present in the two methoxy groups, the third as a carbonyl and
fourth was due to a hydroxy function. The spectrum
showed a signal at 8 190.7 due to the aldehydic carbonyl group. The
*H-NMR spectrum showed a 1H singlet at 5 9.8 for the aldehydic
proton.
The presence of a hydroxyl group at C-2 was inferred from the
IR, *H and 13C-NMR spectroscopic studies. The IR spectrum showed
a strong absorption band at 3340 cm-1 due to the hydroxyl group.
The 13C-NMR spectrum contained a signal at 8 147.0 for the
aromatic carbon bearing the OH group.
The presence of methoxy groups at C-4 and C-5 positions was
established by *H-NMR and 13C-NMR spectra. The *H-NMR spectrum
showed the two methoxy groups as an overlapping 6H signal at 8
3.96. However in the i3C-NMR spectrum, the two methyl signals of
the methoxy groups resonated separately at 8 56.5 and 8 56.6,
establishing the presence of two methoxy groups in molecule.
The structure of (4,5-dimethoxy-2-hydroxy benzaldehyde) 144
was supported by the mass spectrum. The M+ ion in the HREI MS of
our compound 144 was at m/z 182.0513 corresponding to the
-:66:-

molecular formula C9H10O4. Another important fragment showing a
peak at m/z153.0549(C8HgO3) was due to the loss of the aldehydic
group from the molecular ion. The mass fragmentation pattern is
shown in Scheme-2.1.
t
H3C<H
OHH3C( H3C< ‘OH
m/z 153m/z 182
Scheme-2.1
2.2.2 Acetyl-3,4,5-trimethyl-3-propane-4-cyclohexane
(Fumaileate,149)
The chloroform extract obtained by extraction (Scheme-2.8,
Experimental) was loaded on a silica gel column (70-230 mesh.
ASTM), which was eluted with increasing polarities of pet.ether:
acetone. The fraction "FN-3" obtained on elution with pet.ether:
acetone (7:3, 48 mg) contained four minor compounds and one major
compound. The mixture was subjected to preparative TLC on silica
gel precoated plates which were developed in pet.ether : acetone
(7.5:2.5), This afforded a pure compound, fumaileate (149) as a
colourless amorphous solid (7.0 mg).
O
H3CÿO 12
CH3
CH3
6
H"T 5
H C
H3CH3140
-:67:-

The high resolution mass spectrum afforded the molecular ion
peak at m/z 222.1610 corresponding to the molecular formula
C14H22O2 (calcd. 222.1619) leading to the presence of four double
bonds equivalents in the molecule.
The UV spectrum showed absorptions at A,max (MeOH) (log e)
201 (3.169) and 226 (3.365) nm. The IR spectrum showed bands at
2912, 1718, 1254 cnr1 which were due to (C-H), (C=0), (C-O-C)
groups respectively.
The 1H-NMR spectrum (CDCI3, 500 MHz) of 149 showed
resonances for three downfield protons at 8 5.10 m(lH, H-la), 6.13 m
(1H, H-7) and 6.70 dq (1H, H-8). Five signals for the methyl groups
appeared at 8 0.9 s (3H, H-10). 8 1.15 s (3H, H-12), 1.51 s (3H. H-ll),
1.90 dd (3H, H-9) and 2.03 s (3H, COCH3). Four methylene protons
resonated at 8 1.58 dd (1H, H-2a)1.74 d (1H, H-2p), 2.08 dd (1H, H-
6a) and 2.40 d (1H, H-6P).
The 13C-NMR spectrum (CDCI3, 100 MHz), showed resonances
for fourteen carbon atoms in the molecule. The 13C-NMR chemical
shift assignments are presented in Table-2.3. A signal at 8170.59
corresponded to the presence of an ester functionality.
This ester group was further confirmed from the IR spectrum
which showed an absorption at 1718 cm1. The downfield proton at
(8 5.10 m) showed the attachment of the carbon bearing this proton
to an oxygen atom. Two methyl groups resonated as singlets at 8 0.90
and 1.15 showing that they were attached to a quaternary carbon.
-:68:-

Table-2.3. 13C-NMR assignment of 149
Carbon No. iT* *T>y'i
c-i 68.1 -CH-
43.6C-2 -CH2
C-3 35.7 -C-
C-4 140.0 -C-
C-5 127.5 -C-
C-6 37.0 -CH2
C-7 145.8 -CH
C-8 134.4 -CH
C-9 29.0 -CH3
OC-10 170.6
C-ll 21.0 -CH3
C-12 20.6 -CH3:
C-13 29.4 -CH3
C-14 18.2 -CH3
The protons of one methyl group resonated as a double
doublet at 5 1.90 dd (Jit2 = 2.6 Hz, Ji,3 = 6.9 Hz), Two downfield
oleflnic protons 5 6.13 and 6.70 were assigned in a trans relationship
to each other on the basis of "J" value {J = 15.7 Hz).
The HREIMS of 149 showed the M+ at m/z 222.1610
(C i4H2202, calcd. 222.1620). The peak at m/z 161.1329 (C12H17
icalcd. 161.1330) was due to loss of a acetate group from the
-:69:

molecular Ion. A sizable peak at m/z 121.1041 (C9H13 calcd.
121.1017) showed the loss of propene group from m/z 161.1329.
Another peak at m/z 81.0814 (C6H9 calcd. 81.0704) was due to loss
of ethene group and methyl radical from the m/z 121.1041 fragment.
The mass fragments are presented in (Scheme-2.2).
r ;
CH3.CH3- CHiCOOH
H" CH3CH3 H3<CH3CHa
m/z 222loss of H"
.CH3
CH3
m/z 16l'
Scheme-2.2: Mass fragmentation pattern of fumalleate.
The 13C NMR spectra (Broadband decoupled and DEPT ) [181.
182] showed all signals representing fourteen carbon atoms (Table-
2.3). The DEPT and broadband decoupled spectra exhibited the
presence of five methyls, two methylene, three methine and four
quaternary carbons. The most downfield signal at 8170.5 could be
assigned to the C-10 carbonyl functionality. The signals at 8 21.0,
20.6, 29.5 and 18.2 were ascribed to the C-ll, C-12, C-13 and C-14
methyls respectively. The two methylene signals at 8 43.6 and 37.0 were
attributed to the C-2 and C-6 respectively. Three signals observed in
-:70:-

the sp2 chemical shift range were ascribed to the C-l (8 68.07), C-7 (
8145.8) and C-8 (8 134.5) methines. The remaining three signals were
tantatively assigned to the C-3, C-4 and C-5 quaternary carbon
resonating at 8 35.8, 140.0 and 127.5 respectively. It needs further
support of heteronuclear experiment for an unambiguous
assignment.
The HMBC (Heteronuclear Multiple Bond Connectivity) [182]
was performed to unambiguously assign the 13C NMR chemical shift
values. The methyl protons resonating at 8 2.03 showed 2J
heteronuclear interaction with the C-10 (8 170.5) carbonyl carbon.
Another proton at 8 5.10 (H-l) exhibited interaction with the C-
10 (8 170.5) thereby confirming the position of carbonyl in this region
unambiguously, H-l also showed connectivity with the C-3
quaternary carbon resonating at 8 35.8. Methyl protons (H3-I2)
exhibited HMBC interactions with the C-4 (8 140.0). Another methyl
resonating at 8 1.15 (H3-I3) also displayed heteronuclear correlation
with the C-4. The chemical shift assignments in the isolated propene
unit were also confirmed by HMBC experiment. The H-7 (8 6.7)
displayed 2J and heteronuclear correlation with the C-8 (8 134.5)
and C-9 (8 29.0) respectively. These heteronuclear correlations are
presented around the structure, Fig. 2.3.
The COSY-450 spectrum showed homonuclear correlation of
H-l geminal to ester functionality at 8 5.10 with the H-2 a and -p
methylene protons resonating at 8 2.08 and 2.40 respectively. It also
exhibited vicinal interaction with H-6 methylene protons (8 1.74 and
1.70). H-2 a and -P methylene protons also displayed geminal
-:71:-

interactions. Antother proton at 5 6.70 (H-7) showed COSY 45°
interations with H-8 (5 6.13). H-7 and H-8 displayed allylic
interactions with H-9 methyl (8 1.91).
The nOe difference experiment was performed to establish the
stereochemistry of protons and functional groups at various
asymmetric centers. For example the irradiation of H3.I4 methyl at 8
0.99 exhibited enhancement of the H-2(5 at 8 2.40 which in turn
showed 1.8 % nOe with the H-6|3 resonating at 8 1.74. Irradiation of
another proton at 8 2.08 (H-2oc) displayed the 3.2 % nOe interaction
with the H-la thereby confirming the /)-stereochemistry of ester
functionality at this position. The H-la (8 5.10) also showed
interaction with the H-6a (8 1.58). The nOe interactions are shown
around the structure Fig. 2.1.
3.1%
O3.4%
H HH3C o< .CH3
HH 2.1%
o*'' CH3H HH3C'3.2%
CH3H1.8%
Fig. 2.1: nOe difference mearurements on fumaileate (149).
-:72:-

o J<xi 6|j=14.3H2Jio.6a = 4.5. 1.5Hz, dd
1.74 d
H H 12 1.51(s)
CH3H3C 10 o5.10(m) H'
2.40 dH
62.03(s)51
2 1.15(s)4133
CH32.08 dd H 14 H 6.1pdq j3 )= ! ?Hzi J3.j=15.7 Hz
J2o.2P”ÿ*ÿz*-ÿ2a. io=ÿ.8,Hz H3CT
0.99(B)pj
6.70(m)CH3
1.91 dd JI,3=1.7HZJI,J= 6.9 Hz
Fig. 2.2: 1H-NMR assignments of fumaileate (149).
O
1.58di15.1
H170.521.03 11 12 20 ICH3ÿo''10 C37.0O.
6Ha 5ll127.
140/1.152]2.40d
46.63
134.48ÿÿ)2.08d
’•"zZ145.8
8H3C r99(s)8.25
gCH3HI j6.70m
Fig. 2.3: HMBC assignments of fumaileate (149).
2.2.3 Papraline (143)
The ethanolic extract of aerial parts of Fumaria indica was
diluted with distilled water. Then it was deffated with pet.ether and
remaining aqueous portion was shaken with chloroform. The
chloroform layer was concentrated under reduced pressure which
-:73:-

resulted the fraction "FN" . The remaining aqueous layer was treated
with dilute acetic acid (pH~2.5) and was shaken with chloroform and
separated from the aqueous layer. This chloroform extract was
evaporated to a gum and named as fraction "FA". The aqueous layer
was than basified with ammonium hydroxide (pH~9.0) and extracted
with chloroform and the concentrated chloroform extract was coded
"FB".
The fraction "FB" was subjected to silica gel column
chromatography using chloroform-methanol as the eluent system.
The fraction FB-1 was obtained from this column at increasing
polarity of 19:1 (chloroform-methanol) which showed two compounds
(Scheme-2.9, Experimental). This fraction was subjected to
preparative thin layer chromatography (TLC) using pet. ether :
acetone (82:18) which yeilded a colorless compound (143) along with
a yellowish red colored substance (unidentified). The compound 143
has never been isolated so far from the natural source .
5 4
4a.3
I18
143
The LREIMS of 143 displayed the molecular ion peak at m/z
173 Which was further confirmed by HREIMS at m/z 173.0469
(calcd. 173.0474) indicating the eight degrees of unsaturation. The
mass fragmentation pattern of 143 was distinctly similar to the
simple isoquinoline alkaloids [100]. The peak at m/z 144, is
characteristic for isoquinoline alkaloids and was due to the fragment
-:74:-

Ion CgHeNO. Other major fragment observed In the mass spectrum
was at m/z 115 which arose due to the loss of CsHsN. The mass
fragmentation pattern is presented in Scheme-2.3.
The UV spectrum of 143 displayed absorption maximum at
325,300 and 200 nm characteristic of isoquinoline chromophor.
These absorption suggested that the molecule is fully aromatic.
The IR spectrum showed absorptions at 937, 1480, 2738, 2950
indicating the presence of methylenedioxy, aromatic C=C,
conjugated C=N and C-H respectively.
-1cm
The *H-NMR spectrum (CDCI3, 500 MHz) showed six signals
in the downfield region. A two-proton singlet resonating at 6 6.31
could be assigned to the methylenedioxy protons. A set of two
protons resonating as two singlets at 5 7.31 and 7.45 assigned to the
H-5 and H-8 respectively. The lack of any interaction between these
two protons suggested their para disposition. The most downfield
proton resonating at 6 9.61 exhibited close proximaitly to nitrogen
atom and could be assigned to H-l. Two protons resonated as
doublets at 6 8.20 and 7.91 (J=5.7Hz) were assigned to the H-3 and H-
4 based on coupling constant and COSY-450 experiment-
The DEPT and broadband decoupled 13C-NMR spectra
(CDCI3,125 MHz) showed ten carbon atoms (Table-2.4) indicating
the presence of one methylene, five methine carbon and four
quaternary carbons in the molecule. The methylenedioxy carbon
showed a characteristic signal at 6103.5. The two downfield signals at
-:75:-

8 104.7 and 103.3 could be attributed to the C-5 and C-8 respectively.
Another signal integrated for two carbons at 8123.2 was assigned to
the C-l and C-3 adjacent to nitrogen functionality. The remaining
methine carbon at 8 103.3 was ascribed to the C-4. The assignment
of quatemaiy carbons 8135.1 (C-4a), 142.3 (C-6), 146.0 (C-7) and
136.3 (C-8a) were based on HMBC spectrum.
Table-2.4.
Carbon No. Multiplicity13C-NMR Chemical Shift (8)
C-l 123.2 -CH
C-2
123.2C-3 -CH
104.65C-4 -CH
I
TC-4a 135.1
C-5 104.7 -CH
C-6 142.3 -C-
146.0C-7 -C-
C-8 103.3 -CH
C-8a 136.3 -C-
0-CH2-0 103.5 -CH2
Knowing the Basic skeleton of the compound 143, the
assignment of quaternary carbon were confirmed by HMBC
experiment. The most downfield proton at 8 9.16 (H-l) exhibited 2j
and 3jinteraction with the C-8a (8 136.3), C-8 (8 103.3 ) and C-3 (8
123.2). Another proton at 8 7.92 (H-4) showed hetronuclear
correlation with the C-4a quaternary carbon resonating at 8135.1
and also exhibited connectivity with the C-5 methine at 8104.7. The
-:76:-

H-8 (d 7.45) showed heteronulear connectivity with the C-l, C-8a, C-
7 and C-6 resonating at 8 123.2, 136.3, 146.0 and 142.3 respectively.
The HMBC connectivities are shown around the structure 143 ( Fig.
2.4}
H 7.3] H 7.91
J-J 8.20o.104.6142.3 104.7
123.3103.!
146.0JL. 103.3 ii M
/\,H H 9
O
Fig. 2.4: HMBC assignments of papraline (143).
+•
Nr -H0 4j
P
0-s i
m/z 173 m/z 1726Xo
-HCON
b*m/z 144
m/z 115
Scheme-2.3
9,10-Methylenedioxy Karachine (255)2.2.4
The ethanolic extract of the aerial parts of Fumaria indica was
evaporated and the resulting gum trituraled with chloroform (pH 2.5)
-:77:-

to afford a gummy material (15 gm), which was then to column
chromatography. On elution with chloroform : methanol (99.7:0.3) a
semi-pure substance was obtained as fraction FA-3 (Scheme-2.10,
experimental). This was subjected to preparative thin layer
chromatography on precoated silica gel plates using chloroform :
methanol (99.5:0.5) as the developing solvent system (experimental
section) to afford a pure alkaloid 9,10-methylenedioxy karachine
(255) as colourless amorphous solid (7.5 mg).
r°w4a H'O 5HJ
// e ,P\61 14a
h
-NH
Hb13
\12a V/-(8a
Ha,2<c»3 V,v_/ÿ°>2-*10 J11
o255
The HREIMS of (255) showed the molecular ion at m/z
417.1549 corresponding to the molecular formula C25H23NO5 (calcd.
417.1576) indicating the presence of fifteen double bond equivalents
in the molecule. The exact mass of the molecule (255) obtained by
FAB positive mass spectrometry [248,249] was 418.158 (calcd.
418.1654) indicating the fifteen degree of unsaturations.
-:78:-

The peak at m/z 320.091 C19H14NO4 showed the loss of
C6H9O from the molecule. The loss of 97 a.m.u. corresponds to
C6H9O, or more specifically to 2 molecules of acetone minus the
elements of water. The loss of 97 mass units, from the molecular ion
can occur via cleavage a to the nitrogen atom (C14 to Ce bond)
followed by a retro-Diels-Alder process. The remaining
fragmentation pattern of 255 was distinely similar to the
protoberberine isoquinoline alkaloids. The mass fragmentation
pattern is presented in Scheme-2.4.
mass
The UV spectrum showed strong absorptions at Xmax (MeOH)
(loge) 285 nm (3.942) characteristic for tetrahydro coptlsine (158)
[250].
The 1R spectrum (CHCI3) showed an absorption band at 1710
cm'1 due to the presence of a nonconjugated carbonyl group.
The *H-NMR spectrum (CDCI3, 500 MHz) of (255) showed two
singlets for the C-l and C-4 aromatic protons at 8 6.92, and 8 6.19
respectively, along with two doublets at 8 6.38 (J = 7.3 Hz) and 8 6.47
(7.3 Hz) for C-l1 and C-12 respectively. The methylenedioxy protons
were centered at 8 5.87 dd (2H, Jgem = 5.93 Hz) and 5.80 dd (2H, Jgem
= 3.5 Hz). A 1H double doublet at 8 1.12 (Ja-x = 2.5 Hz, Ja-b = 16.6
Hz) was assigned to Ha while another 1H double doublet centered at
8 2.1 dd (Jb-x = 5.0 Hz, Jb-a = 16.6 Hz) corresponded to Hb, the
upheld singlet at 8 0.82 was assigned to the bridgehead methyl group
attached to C-p. Two sets of 2H doublets at 8 2.38/8 2.41 (Jgem =14.0 Hz) and 8 2.62/2.66 were assigned to two sets of gemenal
-:79-

coupled methylene protons and Joining the carbonyl groups. The
methyline (H-8) proton adjacent to the nitrogen resulted at 5 4.01 q
(J8a = 1.8 Hz, Js.ap = 4.01 Hz). The iH-NMR of 255 is shown in Fig.
2.5.
The 13C-NMR spectrum (CDCI3, 125MHz) indicated that there
were 25 carbon signals. The status of various carbons in the
molecules were made by DEPT experiments. From these experiments
it was clearly that there were seven CH2, two of which were at 5
101.03 and 100.79. Due to its characteristic chemical shifts of
methylenedioxy groups,(substituted at C-2/C-3. C-9/C-10). the three
upheld methylenes at 8 52.97, 55.14 and 53.68 were assigned to C-6,
C-e and C-y carbons respectively, while the other two methylenes (C-5
and C-a) resonating at 8 29.36 and 29.71.
DEPT experiments also indicated that there were four
aromatic methine carbon signals and they all have nearly the same
chemical shift i.e., 8 106.49, 105.04, 108.49 and 110.0 due to the C-
1, C-4, C-ll and C-12 respectively. There were nine quaternary
carbon signals appeared in broadband decoupled 13C-NMR spectrum.
The non-oxygenated quaternary carbons C-4a, C-8a, C-12a and C-
14a resonated at 8 126.44, 129.52, 129.80 and 119.87 respectively.
Two positions in each of the benzene rings were oxygenated and these
quaternary carbons appeared at 8 144.42, 143.64, 146.20 and 146.49
which were designated to the C-2, C-3, C-9 and C-10 carbons.
To establish direct !H/13C connectivites, the HMQC
(Heteronuclear Multiple Quantum Coherence) experiment was
-:80:-

performed. The methyl group resonating at 8 31.38 (C-fi CH3)
showed cross-peak with the proton signal at 8 0.84. The protons at 8
5.87 and 5.80 were found to be cross-linked with carbons resonating
at 8 101.03 and 100.79 assigned to the two methylenedioxys. The
methylene protons of C-5 and C-6 at 8 2.40 and 3.09 exhibited
peaks with carbon signals at 8 29.36 and 52.97 respectively. The C-13
proton (8 3.10) showed heteronuclear interaction with the carbon at 8
35.77 (C-13). The iH/13C connectives of all 25 carbons with their
respective protons. The various carbons resolved with the help of
DEPT experiments are given in Table-2.5.
Table-2.5: 13C-NMR Assignment of 255
cross-
MultiplicityCarbon No. 13C-NMR Chemical Shift (8)
106.49 -CHC-l
C-2 144.42 -C-
143.64 -C-C-3
105.4C-4 -CH
C-4a 126.44 -C-
C-5 29.36 -CH2
C-6 52.97 -CH2
C-8 58.80 -CH
C-9 146.20 -C-
-c-c-10 146.49
-CHC-ll 108.49
C-12 110.0 -CH
129.80 -C-C-12a
35.77 -CHC-13
68.45C14 -C-
119.87 -C-C-14a
O-CH2-O* 101.03 -CH2
81:-

(Table 2.0.) contd.
MultiplicityCarbon No. 13C-NMR Chemical Shift (5)
O-CH2-O* 100.79 -CH2
O
weakC
-CH229.71a - CH2
weakP -c31.38 -CH3P-CH353.68 -CH27 - CH2
55.14 -CH2e - CH2
•Interchangeable values
The presence of incorporated ’’acetone units" in 9,10-
methylenedioxy karachine (255) (like its analogue karachine reported
previously) was established mass, !H- and 13C-NMR spectroscopy
studies.
The HERIMS (m/z 417.154) revealed the molecular
composition C25H23NOs, indicating presence of fifteen double bond
equivalents in the molecule. Four oxygens atoms were accounted for
by the two methylenedioxy groups. Twelve double equivalents are
accounted for by the basic protoberberine skeleton having two
methylenedioxy groups. This indicated that the fifth oxygen might be
present as a ketonic carbonyl and there may be two additional rings
in the molecule. The HRE1MS also showed a peak at m/z 320.091
corresponding to the molecular composition C19H14NO4 due to loss
of (C6H9O) unit from the molecule.
-:82:-

The *H-NMR spectrum of (255) showed a 1H double doublet
at 6 1.12 which was assigned to Ha. Another 1H signal double
doublet 8 2.1 corresponded to Hb. An upheld 3H singlet at 8 0.82 was
assigned to the methyl protons of the bridgehead methyl group
attached to C-(3. The compound 255 was compared with LH-NMR
data of 256 which was already isolated from Befberis aristata [251J.
The comparative proton NMR data of both the compounds
(255 and 256) which are arranged in tabular form Table-2.6 showed
the big difference at ring D of 256. In case of 256 the ring D
contains two methoxyl groups which appeared in *H-NMR at 8 3.82
and 3.77, whereas in compound 255 there were no methoxyl signal
found in !H-NMR spectrum. Instead of disappearance of two methoxy
signals, an additional methylene signal at 8 5.80 was appeared as a
doublet having coupling constant 3.5 Hz.
The 13C-NMR spectrum showed the resonance of the a, P, y
and e carbon at 8 29.71, 8 31.38, 8 53.68, 8 55.14 respectively. The p
quaternary carbon and carbonyl carbons were too weak to be detected
In the 13C-NMR spectrum. While the P-CH3 carbon appeared at 8
31.38. Similarly, the same extra methylene signal was appeared at 8
100.79 in carbon spectrum. The status of this signal was determined
by DEPT experiment.
With the help of above discussion it has been concluded that
an additional ring is formed as methylenedioxy. The formation of this
ring was further confirmed with the help of HRMS showing an extra
-:83:-

degree of unsaturation. The remaining signals are closely matched
with the reported data of 256.
The above evidence confirmed that 255 is a derivative of 256
and named as 9,10-methylenedioxy karachine 255.
Q
O 4
4a H'5
// E6
1 14a5
H<7Y N>L WHr»><G
H,
Hb13
\12a )r/=<8aHa/CH3 \
12\_r OCH3
li
OCH3Karachine (256)
These spectral studies and the close correspondence of the
data with that previously reported by us for karachine (256), let to
the assignments of its structures 9,16-methylenedioxy karachine
(255).
-:84:-

Table-2.6. 1H-NMR chemical shift (5) value of compounds 255and 256
;':iv •-?/-
H. No. 256
6.29 sH-l 6.62 s
6.19 sH-4 6.17, s
2.40 mH-5 2.22 - 2.30 m
3.09 mH-6 3.10 m
4.10 q. J = 4.0, 1.8 Hz)H-8 4.12 q, J = 4.0, 1.8 Hz
6.38 d, J = 7.3 Hz 6.52 d. J = 8.2 HzH-l 1
6.47 d. J = 7.3 HzH-l2 6.55 d, J = 8.2 Hz
3.10 sH-13 3.07 s
2.10 d., J = 16.6, 5.0 Hz 2.08 q, J = 12.6, 1.8 HzH-ab
1.12 dd, J = 16.6, 2.5 Hz 1.11 q. J = 12.6, 1.8 HzH-aa
0.84 s 0.82 s(}-Me
2.38 d, J= 14.6 Hz 2.46 d. J= 14.0 HzH-Y
2.41 d, J = 14.6 Hz 2.46 d, J= 14.0 HzH-Y
2.66 d, J= 14.16 Hz 2.70 d, J = 14.3 HzH-e
2.62 d, J = 14.2 Hz 2.72 d, J = 14.3 HzH-e’
5.80 d. J = 3.5 Hz (OCH2O) 3.82 s. (OMe)
3.77 s. (OMe)
5.87 d. J = 5.9 Hz (OCH2O) 5.82 d, J = 1.5 Hz (OCH2O)
-:86:-

Qr v=?5.87 d. Jgem=5.9Hz 6.19 s2.62
H'2.66do H4ÿ1 14avÿ
2.40 m
\ ;o5II 6> 3.09 m
6.92 s S3.28 s-/-N 4.01 q. Jÿl.SHz. J„b=4.0Hz
2. ldd, Jb)(=5.0Hz. Jha=l 6.6Hz
HH JTHV;3.1 s Hb13
##Ha1.12 dd. Jlut=2.5H2. Jab=16.6Hz
0.84& O
8a12a>—1—
6.47 d. J=7.3Hz /12CH3
Vll
\ JQ>
6.38 d. J=7.3Hz5.8d, Jgem =3.5Hz
Fig. 2.5. of 9,10-methylenedioxy karachine (255).
<„< VNt
H3C
m/z 320 OOm/z 417
I
Om/z 174
m/z 148
Scheme 2.4 Mass fragmentation pattern of 9,10-methylenedioxykarachine (255).
-:86:-

Known Compounds Isolated for the First Time from the
Aerial Parts of Fumaria indica
4-Hydroxy-3-Methoxy Benzaldehyde (Vanillin) (142)2.2.5
The CHCI3 extracts (fraction FN-1, (Scheme-2.8,
Experimental), was chromatographed on a silica gel column, elution
being with pet.ether : acetone (9:1) to afford a fraction FN-1 (Scheme-
2, Experimental). Fraction FN-1 was then subjected to preparative
TLC employing pet.ether : acetone (8.5:1.5) as eluent to give a
colourless powder (6.4 mg).
O
H3CO\3H1
4j6
HO'5
142
The UV (MeOH) spectrum of the compound showing
absorptions at Xmax (MeOH) (log e) 195 (2.962), 204 (3.337), 230
(3.291), 278 (3.114), 308 (3.093) [1051 characteristic for vanillin type
compounds.
The IR (CHCI3) spectrum showed absorptions at 3408 cm'1
(OH), 1670 cm'1 ((conjugated C=0) and 1584 cm1 (C=C).
The 1H-NMR spectrum (CDCI3, 500 MHz) indicated the
presence of four downfleld protons, due to the aldehydic proton at 6
-:87:-

9.87 (s), the two aromatic protons resonating at 8 7.40 and 7.01 (C-
2H, C-5H) and the O-H proton which resonated as a broad singlet at
5 6.17. One 3H singlet at 5 3.94 was due to the methoxy protons
substituted at the C-3 position.
The high resolution mass spectrum afforded the molecular ion
peak at m/z 152.0481 leading to the molecular formula CsHsOs and
indicating five double bond equivalents in the molecule. The
molecular ion peaks was further confirmed by FAB positive mass
spectrometry. The ion at m/z 123.0452 (C7H7O2. calcd. 123.0446)
was due to the loss of CHO group from the molecular ion indicating
the presence of an aldehydic functionality.
The DEPT and broadband decoupled 13C-NMR Spectra
(CDCI3, 100MHz) showed eight carbon atoms (Table 2.7) indicating
the presence of one methoxy, three methine and four quaternary
carbons in the molecule. The methoxy carbon showed a characteristic
signal at 8 56.10. The three downfield methine signals at 8 127.5,
114.4 and 108.8 could be attributed to the C-6, C-5 and C-2 aromatic
methine carbons. A quaternary carbon signal at 8 190.8 was
characteristic for carbonyls carbon. The remaining quaternary carbon
signals at 8 129.11, 140.01 and 51.7 were ascribed to C-l, C-3 and C-
4 respectively, the assignment of quaternary carbons were based on
HMBC spectrum.
The 13C-NMR spectrum (CDC13, 125 MHz) of the compound
showed the presence of only eight carbon atoms in the molecule. The
13C-NMR spectrum is presented in Table-2.7.
-:88:-

Table-2.7. 13C-NMR Assignments of 142
Multiplicity (DEPT )Carbon No. 13C-NMR Chemical Shift (5)
129.11 -C-C-l
-CHC-2 108.8
147.01C-3 -C-
151.7C-4 -C-
C-5 -CH114.4
127.50C-6 -CH
OII 190.8 -C-c
56.10 -OCH3-OCH3
By comparing the spectral data ( UV, IR, MS, 13C-NMR) of
142 with those reported earlier compound 142 was identified as 4-
hydroxy-3-methoxy benzaldehyde (Vanillin).
Cryptopine (61)2.2.6
The CHC13 extract (Fraction FA-4, Scheme-2.10,
Experimental) obtained at pH 2.5 was chromatographed on a silica
gel column which was eluted with increasing polarities of chloroform
: methanol. Elution with chloroform : methanol (9.5:0.5) afforded a
fraction FA-4 (52 mg) (experimental, Scheme-2.10) which was
subjected to preparative thin layer chromatography (TLC) by using
pet.ether : acetone (78:28) as a developing solvent system to afford an
alkaloid as a white crystalline solid (18 mg, 9 x 10’5% yield), mp =156-158 °C named "cryptopine (61)”. The substance give positive test
with DragondrofFs reagent.
-:89:-

54
o. 4a6
3
.CH32 N.
O' 14a 814
1 O'8a .OCH313
912a
1012
OCH3n61
The UV (MeOH) spectrum was characteristic of cryptopine and
protopine [94, 106, 146], showing absorptions at Xmax (MeOH) (log E)
286 (2.801), 206 (3.750) and 193 (3.769) nm. The IR spectrum showed
C-H streching vibration at 2898 cm-1, while the band at 1718 cm'1
indicated the presence of a 0=0 group in the molecule.
The *H-NMR spectrum (CDCI3, 500 MHz) afforded signals for
four downfield aromatic protons. The 1H singlets at 6 7.02 and 6.71
were assigned to the protons C-1H and C-4H respectively. Two others
downfield protons resonated as overlapping double doublets at 8 6.71
and 6.82 was assigned to the C-l1 and C-12 respectively ( Ji1,12 = 7.9
Hz). The assignments of 01 are given in Fig. 2.6.
The C-5 and C-6 methylene protons appeared as two methyl
groug of 2H multiplets, centred at 8 2.11 and 2.83 respectively.
Another 2H multiplet centred at 8 3.75 was assigned to C-13H in the
ten membered flexible ring of cryptopine.
-:90:-
The HREIMS of cryptopine exhibited the molecular ion at m/z
369.1560.(C21H23NO5, calcd. 369.1576)suggesting the presence of ten
degree of unsaturations. The M+ was confirmed by fast atom
bombardment (FAB) mass spectrometiy [240].
Other major peaks appeared at m/z 288, 267, 221, 170, 178
and 148. The fragmentation pattern was similar to that reported for
cryptopine (Scheme-2.5).
CH2H3C<H2(CH3
DN;
H3C<O'
D m/z 148
m/z 369
H3C< H3C<
N\
H3C( CH3 H3C< co o
m/z 221 m/z 178
Scheme-2.5
The 13C-NMR spectrum (CDCI3, 100 MHz) of cryptopine (61)
indicated that there were 21 carbon signals. The multiplicity
assignments were made by DEPT experiments which are presented in
Table-2.7. From these experiments it was clear that there were five
CH2, one of which was at 8 101.27 due to its characteristic chemical
shift, it was apparent that this methylene is that of a methylenedloxy
-:91:

group (substituted at C-2/C-3). The two upfield methylenes at 8
31.76 and 46.48 were assigned to C-5 and C-13 carbons respectively.
while the other two methylenes (C-6 and C-8, present a to nitrogen)
have a downfleld chemical shift, resonating at 8 57.85 and 50.85.
DEPT experiment also indicated that there were four aromatic
methlne carbon signals and they all have nearly the same chemical
shifts i.e.. 8 106.75, 110.54, 108.20 and 110.58 due to the C-l, C-4,
C-ll and 012 respectively. There were nine quaternary carbon
signals including one carbonyl carbon 014, which appeared at 8
194.43. The non-oxygenated quaternary carbons 04a, 08a, 012a
and C-14a resonated at 8 136.20, 129.04, 117.95 and 132.82
respectively. The N-CH3 and two OCH3 carbons resonated at 8 41.51,
56.02 and 56.35 respectively. Two positions in each of the benzene
rings were oxygenated and these quatemaiy carbons appeared at 8
146.41, 148.10, 148.90 which were designated to the 02, 03, 09
and OlO carbons respectively.
On the basis of these spectral studies the compound (61) was
identified as cryptopine.
The one-bond iH/ÿC correlation for 61 were determined on
the basis of HMQC experiments (Tablc-2.8).
-.92:-

Table-2.8. 13C-NMR Assignments of 61
Carbon No. 13C-NMR Chemical Shift (8) :
C-l 106.75 -CH
C-2 146.41 -C-
C-3 148.10 -C-
C-4 110.54 -CH
C-4a 136.2 -C-
C-5 31.76 -CH2
C-6 57.85 -CH2
-CH250.85C-8
C-8a 129.04 -C-
C-9 148.90 -C-
146.09C-10 -C-
c-u 108.20 -CH
C-12 110.58 -CH
C-l2a 117.95 -C-
C-13 46.48 -CH2
C-l4 194.43 -C-
C-14a 132.82 -C-
O-CH2-O 101.27 -CH2
-OCH3 56.02 -OCH3
-OCH3 56.35 -OCH3
N-CH3 41.51 -CH3
-:93:-
6.71 s 2.11 m
O 2.83 m5
< 6l4a 2.21 s
.CH35.95 s2 14a N.1o 3.09 m14
7.02 s a 3.92 s
.OCH313 8<3.75 m
9|12a
3.93s
OCH36.71 t. J=7.9Hz
Fig 2.6. LH-NMR spectral data of cryptopine (61).
(+)-p-Hydxastine (232)2.2.7
The chloroform extract (obtained as shown Scheme-2.10) by
extraction at pH ~2.5 was subjected to column chromatography on a
silica gel column using chloroform : methanol (9 :1) as eluent to
afford fraction FA-5 (Scheme-2.10 Experimental). This fraction was
subjected to preparative TLC using chloroform : methanol (9.7:0.3)
saturated with vapours of ammonia as the developing system to
afford (+) P-hydrastine as a colourless amorphous solid 6.9 mg,
[<X)D25 = +48°. the alkaloid gave a red coloured reaction with
dragendroffs reagent.
The UV (MeOH) spectrum was characteristic of
secophthalideisoquinollnes chromophores (173] showing absorptions
at Xmax (MeOH) (log e) 290 (3.889), 264 (3.655), 218 (4.150) nm. The
IR (CHCI3) spectrum showed an absorption at 1740 cm1 which
indicated the presence of a lactone carbonyl group. Other Intense IR
-:94:-

absorptions were at 1610 (C=C aromatic) and 1130 cm'1 (C-O-C)
[170],
5
O. 43/436
7} 8a N.O H CH38
2'
H9 3‘
Q4'
7'9'6'
OCH35’
o 8‘
OCH3(+)-(}-Hydrastine (232)
The •H-NMR spectrum (CDC13, 500 MHz) Indicated the
presence of four downlleld protons resonating at 5 7.11 d (J = 7.96), 8
6.83 (J = 7.96), 6 6.64 s and 8 6.35 s due to the C-3’H, C-2H, C-5H
and C-8H respectively. Protons of methylenedioxy group, C-1H and
C-9H appeared at 8 6.03, 8 4.07 and 8 5.67 respectively.
The presence of 2-methoxy groups was indicated by the two
3H resonances at 8 3.72 and 8 3.76 which must be located at C-4‘
and C-5’, since the other two aromatic protons (C-2H and C-3H) were
in an ortho relationship. The !H-NMR of 232 are shown in Fig 2.7.
The high resolution electron-impact mass spectrum of (+)-(5-
hydrastine (232) showed the molecular ion at m/z 383.1245. in
agreement with the molecular formula C21H21NO6 (calcd. 383.1368.
-.95:-
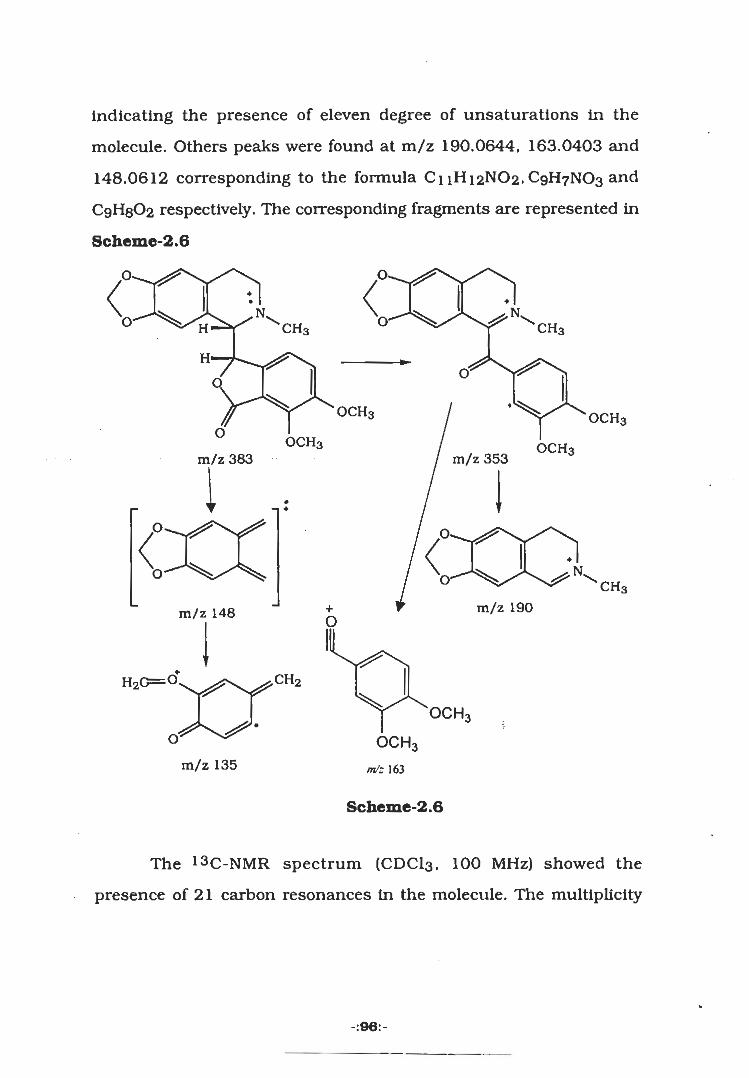
indicating the presence of eleven degree of unsaturations in the
molecule. Others peaks were found at m/z 190.0644, 163.0403 and
148.0612 corresponding to the formula CiiHi2N02,CgH7N03 and
C9H8O2 respectively. The corresponding fragments are represented in
Scheme-2.6
<: NS
CH3 CH3H
H-O'
OCH3 OCH3O
OCH3 OCH3m/z 353m/z 383
lO a
o O'CH3
m/z 190+m/z 148O
H2G=°*> CH2
OCH3i
o OCH3m/z 135 m/z 163
Scheme-2.6
The 13C-NMR spectrum (CDCI3, 100 MHz) showed the
presence of 21 carbon resonances in the molecule. The multiplicity
-:96:-

assignments were made by carrying out the DEPT pulse sequence
which are presented in Table-2.9.
Table-2.9. 13C-NMR Assignments of 62
MultiplicityCarbon No. 13C-NMR Chemical Shift (5)
66.30 -CHC-l
50.36 -CHC-3
C-4 41.14 -CH2
126.1 -C-C-4a
107.5 -CHC-5
149.6 -C-C-6
145.45 -C-C-7
106.8 -CHC-8
128.4 -C-C-8a
C-9 80.1 -CH
116.2 -C-C-l’
C-2' 117.6 -CH
C-3' 118.1 -CH
C-4' 150.1 -C-
C-5’ 151.2 -C-
-C-C-6' 140.2
COc=o 167.8
101.5 -CH20-CH2-0
-OCH3 54.8 -OCH3
55.6 -OCH3-OCH3
-N-CH3 44.5 -N-CH3
-:97:-

The C-1 methine carbon and C-3 methylene carbon appeared
at 8 66.30 and 50.36. The downfield chemical shifts of these carbons
were due to the adjacent to nitrogen function. The C-9 methine
carbon appeared at 880.1 due to the acetyl function. The signal at 6
101.5 was assigned to the methylenedioxy group at C-6 and C-7
position. The chemical shifts of the aromatic methine carbons C-5,
C-8, C-2' and C-3' were assignes as 6 107.5, 106.8, 117.6 and 118.1
respectively. DEPT spectrum established that there were eight
quaternary carbons in (+)-|5-hydrastin (232). Out of these the four
oxygenated carbons were C-6, C-7, C-4’ and C-5' which resonated at
8 149.6, 145.45, 150.1 and 151.2 respectively, while the rest of the
carbon atoms appeared at 6 125.12, 128.4, 116.2 and 140.2 due to C-
4a, C-8a, C-l' and C-6’ carbons respectively resonating in broadband
13C-NMR spectrum. The lactonic carbonyl carbon appeared at 8
167.8. The N-CH3 carbon appeared at 6 44.5 while the methoxy
carbons resonated at 5 54.8 and 55.6.
2.40 m6.64 s
P 2.74 m
6.03
2.63 s
CH3o H6.35 s 4.07
6.83 d. J=7.96Hz
5.67*1 7.1) d. J=7.96Hz
Q
3.72
OCH3o 3.76
OCH3
Fig 2.7. iH-NMR spectral data of (+)-P-hydrastine (232).
-:98:-

Corydaldine (147)2.2.8
The CHCI3 extract (pH -8.5) (Scheme-2.9) was
chromatographed on a silica gel (70-230 mesh) column with
increasing polarities of chloroform : methanol (8:2) to afford a
fraction FB-3 (Scheme-2.9, Experimental). Fraction FB-3 was seen by
(6.0 mg) and corydaldine (147) (10 mg) was obtained as a colourless
powder.
H3Ca'"*ÿ 4a 4
3
8aH3CO' H8
oCorydaldine (147)
The UV (MeOH) spectrum was typical of isoquinolone bases
[100,186) showing absorption maxima at \max 295 (2.610), 260
(2.730), 220 (3.240) and 208 (3.182) nm.
The IR spectrum showed an absorption at 3672 cm1 which
indicated the presence of an N-H function. Other intense IR
absorptions were at 1710 (C=0) and 1595 (C=C) cm-1.
-:99:-

EXPERIMENTAL
*

2.3 General Experimental
All chemical and instrumental analyses were performed at the
International Centre for Chemical Sciences, H.E.J. Research
Institute of Chemistry, University of Karachi. All solvents used for
thin layer chromatography and for final elution thereafter were
purchased from E. Merck. For other chromatographic techniques
commercially available solvents were used after distillation at their
respective boiling points except were mentioned. Distilled pet. ether
was collected between 64°C and 68°C. Hydrochloric acid, acetic acid
and ammonium hydroxide were purchased from E. Merck.
Instrumental Details
Physical Constants
All melting points were recorded in glass capillary tubes using
Buchi melting point apparatus. Optical rotations were measured on
JASCO DIP-360 digital polarimeter in chloroform.
Spectroscopy
The UV spectra were recorded in methanol on Shimadzu UV-
240 (Shimadzu Corporation, Kyoto, Japan) spectrophotometers.
The IR spectra were recorded in chloroform on Shimadzu IR-
240 (Shimadzu Corporation, Koyoto, Japan) or (Japan Spectroscopic
Co. Ltd.) instruments.
101:-

The Nuclear Magnetic Resonances (NMR) spectra were
recorded in CDCI3, CD3OD and C5D5N using TMS as on internal
standard on Bruker AM-300 FT NMR, AM-400 FT NMR and AM-500
FT NMR spectrometers.
The 13C-NMR spectra were recorded at 75, 100 and 125 MHz
on Bruker AM-300, Am-400 and Am-500 FT NMR respectively.
Mass spectra were recorded on Finnigan MAT-112 and 312
double focusing mass spectrometers connected to IBM-AT Compatible
PC based system. Peak matching, linked scan, field desorption (FD)
and fast atom bombardment (both +ve and -ve FAB) experiments were
performed on MAT-312 or Jeol HX-110 mass spectrometers. FABMS
were recorded in a glycerol-water (1:1) matrix in the presence of KI.
Accurate mass measurements high resolution electron-impact mass
spectra were recorded on a Jeol-JMS H x 110 mass spectrometer.
Chromatography
Column chromatography was performed with silica gel (E.
Merck, type 60, 70-230 mesh). Precoated silica gel GF-254 preparative
plates (20 x 20 cm 0.2 mm thick) (E. Merck) were used for preparative
thin layer chromatography. The purity of the samples was checked on
the precoated plates.
X-ray Diffraction Studies
All the data were gathered using nicolet R3m/v (now Siemens) four
circle diffractometer with graphite monochromated Cu Ka radiation
(\ = 1.54184 A)
-:102:-

Spray Reagents Used During Detections
Ceric Sulphate
Saturated solution of ceric sulphate in 65% sulfuric acid was
used for detection of terpenes which give a pink/blue colouration
after spraying the spots on TLC plates and upon heating for 10-15
min at 120°C.
Dragendorffs Reagent
A mixture of 25 ml acetic acid, 2.6 gm basic bismuth
carbonate and 7 gm sodium iodide was boiled for a few minutes.
The copious precipitates of sodium acetate were filtered
through a sintered glass filter after about 12 h. 20 ml of the clear
red-brown filtrate was mixed with 80 ml ethyl acetate and 0.5 ml
water was added and the material stored in an amber glass bottle.
This was the stock solution. The spray reagent was prepared by
mixing 10 ml stock solution, 100 ml acetic acid and 240 ml ethyl
acetate. Alkaloids and occasionally a number compounds containing
no nitrogen, appear as orange-coloured spots after spraying.
Iodine
A few iodine crystals were placed in a TLC tank and warmed
for a couple of minutes on a water bath (40-50°C). Spots appeared on
the TLC plate when kept inside the tank for a minute.
103:-

Plant Material2.3.1
The plant material (Fresh aerial parts of Fumaria indica) (40
kg) was collected from Dera Ghazi Khan, Punjab (Pakistan). The
plant was identified by taxonomist, Department of Botany at the
Karachi University, Karachi, where the sample specimen was also
deposited. The plant material was air-dried under shade.
Extraction and Purification2.3.2
The air-dried aerial parts (18 kg) of Fumaria indica were
crushed and soaked in ethanol (40 liters) for seven days and then
filtered. The filtrates were concentrated to give a crude gum (815 g)
which was then fractionated into different fractions by solvent-
solvent extraction (Scheme-2.7). The ethanolic extract was
suspended in distilled water and defeated with pet.ether. The defatted
aqueous layer than extract with chloroform. The chloroform soluble
layer (78 gm) was named "FN". The aqueous layer then acidified with
acetic acid upto pH=2.5, was again extracted with CHCI3. The
chloroform soluble portion (36 gm) was named as "FA". The
remaining aqueous layer was basified with NH4OH at pH=8.5 and
extracted with CHCI3. The chloroform soluble portion (54 gm) was
named "FB". The detailed fractionation procedure is summarized in
Scheme-2.7. These extracts were subjected to column
chromatography and preparative thin-layer chromatography to afford
pure compounds (Scheme-2.8, 2.9, 2.10).

Aerial part of air-dried Fumaria indica(18kg)
Powdered and extracted withethanol (40 litre) and concentratedunder vaccuum
Ethanolic gum(815 gm)
Suspended in distilled water 5.0 litre
Aqueous extract
defatted with pet. ether (40-65°C)
(70 litre)
1IAqueous extract (515 gm) Pet. ether extract
(221 g)Extracted with chloroform(10 litre)
1(Chloroform extract
(78 g)Aqueous extract
Extracted withCHC13 at pH -2.5
( tFN
Aqueous extractChloroform extract(36 g)
Extracted withCHCI3 at pH -8.5
( ]FA
Aqueous layerChloroform extract(54 g)
FB
Scheme-2.7
-:105:-
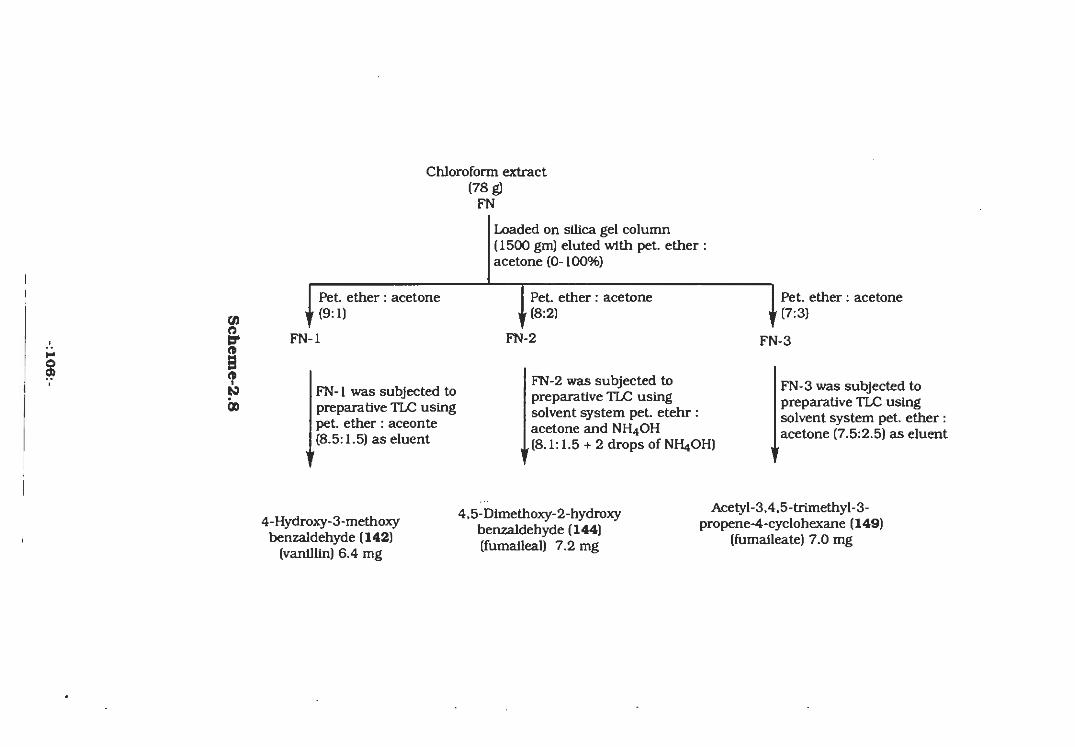
Chloroform extract(78 g)
FN
Loaded on silica gel column(1500 gm) eluted with pet. ether :acetone (0- 100%)
I 1( Pet ether : acetone(7:3)
Pet. ether : acetone(9:1)
Pet ether : acetone(8:2)
</ia
FN-2FN-1er FN-3Ms 3
FN-2 was subjected topreparative TLC usingsolvent system pet etehr :acetone and NH4OH(8.1:1.5 + 2 drops of NH4OH)
FN-3 was subjected topreparative TLC usingsolvent system pet. ether :acetone (7.5:2.5) as eluent
FN-1 was subjected topreparative TLC usingpet ether : aceonte(8.5:1.5) as eluent
to00
' ftt
Acetyl-3,4,5-trimethyl-3-propene-4-cyclohexane (149)
(fumaileate) 7.0 mg
4,5-Dimethoxy-2-hydroxybenzaldehyde (144)(fumaileal) 7.2 mg
4-Hydroxy-3-methoxybenzaldehyde (142)
(vanillin) 6.4 mg1

Chloroform extract(pH ~8.5) (54 g)
FB
Loaded on silica gel column(1200 gm) eluted with chloroform :methanol (0-100%)
I
I
( IW CHCI3 : MeOHCHCI3 : MeOH(19:1)
CHCI3 : MeOH
U (8:2)
FB-3
oP* (9:1)aao FB-1 FB-2>4 ft
bOFB-2 was subjected topreparative TLC usingsolvent system peL ether :acetone (8:2) as eluent
FB-3 was subjected topreparative TLC usingsolvent system pet ether :acetone (7.5:2.5) as eluent
CO FB-1 was subjected topreparative TLC usingpet ether : aceonte(82:18) as eluent
t1 'Fumaramine (216) (6.0 mg)Corydaldine (147) (10 mg)
Protopine (184) (10.0 mg)jParfumihe (183) (7.0 mg)
Oxysanguinarine (176) (7.5 mg)
Papraline (147)i
I
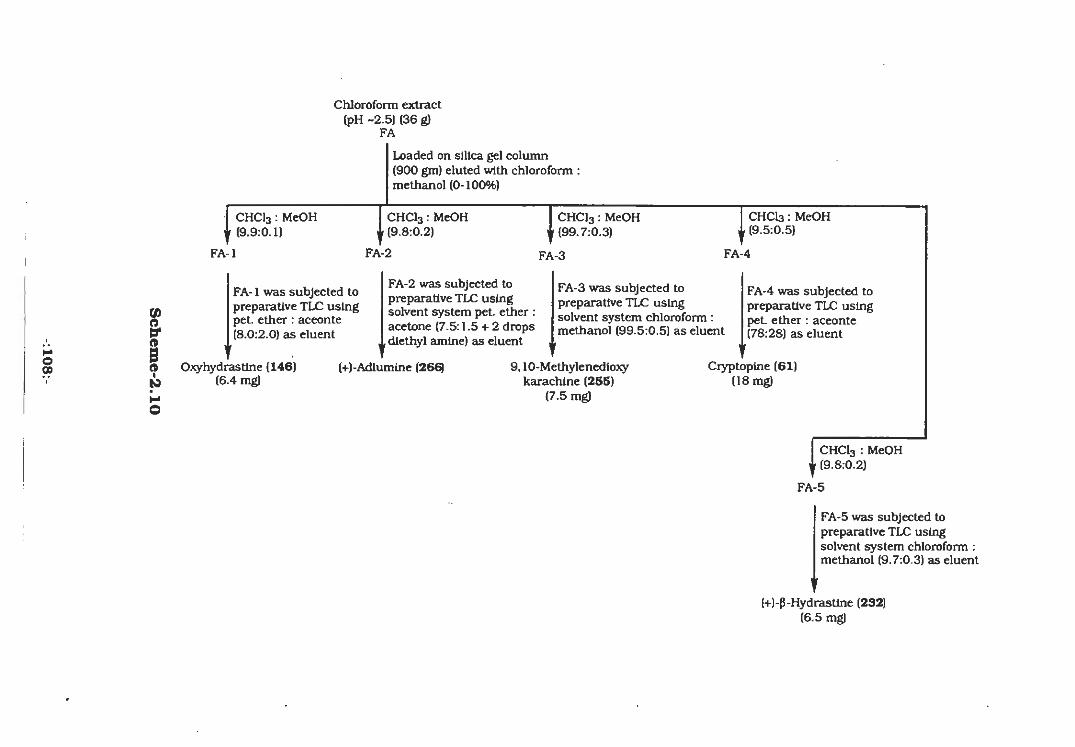
Chloroform extract(pH -2.5) (36 g)
FA
Loaded on silica gel column(900 gm) eluted with chloroform :methanol (0-100%)
\\ ICHCla : MeOH(9.5:0.5)
CHC13: MeOH(9.8:0.2)
CHCI3: MeOH(99.7:0.3)
CHC)3: MeOH1 f (9.9:0.1)
FA-2 FA-4FA-1 FA-31
FA-2 was subjected topreparative TLC usingsolvent system pet. ether :acetone (7.5:1.5 + 2 dropsdiethyl amine) as eluent
(+)-Adlumine (266)
FA-3 was subjected to
preparative TLC usingsolvent system chloroform :methanol (99.5:0.5) as eluent
FA-4 was subjected topreparative TLC usingpeL ether : aceonte(78:28) as eluent
FA-1 was subjected to
preparative TLC usingpet. ether : aceonte(8.0:2.0) as eluent
0)Otf
Bo Cryptopine (61)
(18 mg)9.10-Methylenedtoxy
karachine (265)(7.5 mg)
Oxyhydrastlne (146)(6 4 mg)
00
10Mo
[CHCI3 : MeOH(9.8:0.2)
FA-5
FA-5 was subjected topreparative TLC usingsolvent system chloroform :methanol (9.7:0.3) as eluent
' '(+)-(!-Hydrastine (232)
(6.5 mg)

Isolation of Fumaileal (144)2.3.3
The fraction FN-2 obtained from the silica gel column was
eluted with pet.ether : acetone (8:2). It was concentrated In a round
bottomed flask to give a gum (70 mg). This material was triturated
with chloroform to give chloroform soluble portion (45.0 mg)
containing one compound along with some impurities. Fraction FN-2
was further purified by preparative TLC on silica gel plates (GF-254,
0.2 mm) when were developed with pet.ether : acetone (8.1:1.5) + two
drops of NH4OH, to give a pure UV active band, fumaileal (144) as an
amorphous solid (7.2 mg, yield 4.00 x 10*5%), (Rf = 0.65).
Spectral Data
la]20 D = 0o (c = 0.258, MeOH)
UV (MeOH), nm (log e): Xmax 250 (2.873).
IR (CHCI3), vmax cm 1: 3340 (OH),1718 (C=0), 1598 (C=C).
EIMS m/z: (rel.int.%): 182 (M+. 100), 167 (22). 53 (14), 139 (12), 111
(12), 93 (15), 65 (18) and 53 (11).
HREIMS m/z: (rel. int. %): 182.0594 (C9H10O4. calcd. 182.0579)
(100), 167.0312 (18), (C8H704, calcd. 167.0344, 153.0549 (10),
(C8H903, calcd. 153.05516), 93.0692 (C7H9. calcd. 93.0704), 53.0412
(8), (C4H5, calcd. 53.0391).
-:I09.-

1H-NMR (CDC13, 400 MHz) 5: 9.8 (1H. s, aldehydic proton), 7.23 (1H.
s, H-3), 7.13 (1H, s,H-6) and 3.69 (6H, s, 2OCH3).
13C-NMR (CDCI3, 100 MHz) 8: See Table-2.3.
Fumaileate (149)2.3.4
The fraction FN-3 was obtained from the silica gel column
(containing CHCI3 extract of Fumaria indica) on elution with pet.
ether : acetone (7:3). This fraction was subjected to preparative TLC
using pet.ether : acetone (7.5:2.5) to afford fumaileate (149) as an
amorphous solid (7.0 mg, yield 3.8 x 10_5%), (Rf = 0.62). This spot of
fumaileate give a positive test on TLC with ceric sulphate spray
reagent after heating.
Spectral Data
[a]25D; +51 (c = 0.0322 CHCI3).
UV (MeOH), nm (log e): Xma* 226 (3.365), 201 (3.169).
IR (CHCI3), vmax cm1: 2912 (C-H), 1718 (C=Q) and 1254 (C-O-C).
HREIMS m/z: (rel. int. %): 222.1610 IC14H22O2. calcd. 222.1619,
(10)), 161.1329 IC12H17, calcd. 161.1330, (5)1, 121.1009 [C9H13,
calcd. 121.1017. (70)1 and 69.0701 [C5H9. calcd. 69.07041.
1H-NMR (CDCI3, 500 MHz): 8 2.03 (3H. s, acetyl methyl), 5.10 (1H,
m, H-l), 1.58 (1H, dd, J2a,2p = 11-75 Hz, J2o,la = 4.0 Hz, H-2a). 1.74

(d. J2a.2p = 11.75, H-20), 2.08 (1H, dd, J6a,6p = 10.5, Jea.ia = 3.5 Hz,
H-6a), 2.40 (1H, d, J6a,63 = 10.5 Hz, H-6p), 6.70 (1H, dq, J7.8 = 13.8
Hz. J8,g = 6.9 Hz, H-8), 1.90 (3H. d, Jg,8 = 6.9 Hz, H-9), 0.99 (3H. s,
H-10), 1.51 (3H, s, H-l1) and 1.15 (3H, s, H-12).
1SC-NMR (CDC13, 125 MHz): See Table-2.4.
Isolation of Papraline ( 143)2.3.5
Papraline (143 ) was isolated from fraction FB-1 (Scheme-
2.9). It was obtained by chromatography on a silica gel column (100
g) on elution with chloroform : methanol (19:1). This fraction when
subjected to TLC using pet.ether : acetone (82:18) afforded a
compound which was identified as papraline (143) after spectroscopic
studies, papraline was obtained as an amorphous solid (5.0 mg) (yield
2.7 x 10'5%) (Rf = 0.54).
Spectral Data
[a]25D = 0° (c = 0.04 MeOH).
,
UV (MeOH), nm (log e): Xmax 200 (2.431, 300 (2.221) and 325 (2.143).
IR (CHC13): vmax cm 1: 2915, 2840 (C-H), 2738 (C=N-) and 1518
(C=C).
EIMS m/z: (rel.int.%): 173 (M+, 100), 172 (82), 149 (10), 134 (14), 126
(5), 115 (25), 104 (5), 88 (11), 87 (12), 62 (32), 58 (16) and 57 (13).
-till:-

HREIMS m/z (rel.int.%): 173.0512 (M+, C10H7NO2. calcd. 173.0714,
100), 172.0368 (CIOH6NC>2, calcd. 172.0398, 82), 148.0471 (C9H802,
calcd. 148.0477, 10), 134.0418 (C8H802, calcd. 134.0376, 14) and
104.0197 (C7H4O, calcd. 104.0252, 5).
1H-NMR (CDCI3, 500 MHz) 8: 9.16 (1H, s, H-l), 8.20 (1H, br.s, H-3),
7.91 (1H, br.s, H-4), 7.31 (1H, s, H-5), 7.45 (1H, s, H-8) and 6.31 (2H,
s, O-CH2-O).
13C-NMR (CDCI3, 100 MHz) 8: See Table-2.5.
Isolation of 9,10-Methylenedioxy Karachine (255)2.3.6
Fraction FA-3 was obtained by column chromatography of the
CHCI3 extract of Fumaria indica obtained at pH 2.5 on a silica gel
column on elution with chloroform : methanol (99.7:0.3). This
fraction when purified by preparative TLC using chloroform :
methanol (99.5:0.5) as the solvent system to afford a pure alkaloid
which was identified after spectroscopic studies as 9,10-
methylenedioxy karachi (7.5 mg, 3.5 x 10-5% yield). This compound
give positive test with dragendroffs reagent.
Spectral Data
[a]25D = +250° (c = 0.204, MeOH)
UV (MeOH), nm (log e): Xmax 220 (3.052), 285 (3.942).

i
IR (CHC13): vmax cm-l; 1710 (C=0). 1605 (C=C) and 1450 (CH2
band).
HREIMS m/z (rel. int. %): 417.1549 (C25H23NO5, calcd. 417.1576,
60), 320.0919 (C19H14NO4, calcd. 320.0922, 100). 174.05481
(CIOH8N02, calcd. 174.0554) and 148.0512 (C9H8O2, calcd. 148.0524.
15).
1H-NMR (CDCI3, 500 MHz) 5: 6.92 (1H, s, H-l), 6.19 (1H, s, H-4),
6.38 (1H, d, J = 7.3 Hz. H-ll), 6.47 (1H, d, J = 7.3 Hz, H-12), 5.87
(2H, dd, Jgem = 5.93 Hz, 0-CH2*-0-), 5.80 (2H, dd, Jgem = 3.57 Hz)
1.12 (1H, dd Ja-x = 2.5 Hz, Hb), 0.82 (3H, s, C-P-CH3), 2.38 (1H, d,
Jgem = 14.0 Hz, H-7*), 2.41 (1H, d, Jgem = 14.0, H-y), 2.62 (1H, d, J =14.16 Hz, He*), 2.66 (1H, d, J = 14.16 Hz. H-e*), 4.0 (1H, q, Jxa = 1.8
Hz, Jxb = 4.0 Hz, H-8), 3.05 (1H. s, H-13), 2.62 - 2.29 (2H, m H-5)
and 3.0 - 3.1 (2H, m, H-6).
(CDCI3, 100 MHz) 5: See Table-2.6.
Isolation of Vanillin (142)2.3.7
Fraction FN-1 ( Scheme-2.5, Experimental) was obtained by
column chromatography of the CHCI3 extract of Fumaria indica on a
silica gel column on elution with pet.ether : acetone (9:1). This
fraction then purified by preparative TLC using pet.ether : acetone
(8.5:1.5) as the solvent system to afford a pure compound which was
identified after spectroscopic studies as 4-Hydroxy-3-methyoxy
benzaldeyhde (142, 6.4 mg, 3.3 x 10*5% yield).
;

Spectral Data
[a]25D = 0° (c = 0.305, MeOH)
UV (MeOH), nm (log e): Xmax 195 (2.962), 204(3.337), 230 (3.291), 278
(3.114), 308 (3.093)
m (CHC13) vmax cm'1: 3408 (OH),1670 (0=0) and1584 (C=C).
HREIMS m/z (rel. int. %): 152.0481 (C8H803), calcd. 152.0451 (45),
123.0452 (C7H7O2, calcd. 123.0467, 20),
1H-NMR (CDCI3, 500 MHz) 6: 9.87 (1H, s, aldlhydlc-H), 7.40(2H, m,
H-2 and H-5), 7.01 (1H, dd, J = 1.8Hz, 8.47Hz, H-6), 6.17 (1H, br.s,
OH) and 3.94 (3H. s, OCH3).
13C-NMR (CDCI3, 125 MHz) 5: See Table-2.7.
Isolation of Cryptopine (61)2.3.8
The chloroform extract obtained at pH ~2.5 was loaded on a
silica gel column (900 gm). This column was eluted with chloroform :
methanol on elution with chloroform : methanol mixtures (9.5:0.5)
fraction FA-4 was obtained. It was concentrated to give a gum (52
mg), which was subjected to preparative thin layer chromatography
using pet.ether : acetone (78:28) as the developing solvent system to
afford an alkaloid which gave red colouration with Dragendorffs
reagent and was identified as cryptopine (61) on the basis of the
spectral data.

Spectral Data
M.P.: 156- 158°C
[a]25D : 0° (c = 0.032. MeOH)
UV (MeOH). nm (log e): Xmax 193. (2.063). 206 (3.750) and 286
(2.801).
IR (CHCI3) vmax cm-*: 2898 (C-H).1718 (OO) and1585 (C=C).
EIMS m/z: (rel.int.%): 369 (M+. 12). 288 (5). 267 (7). 221(22). 190 (9).
178 (30). 170 (15).148 (100), 100 (6). 83 (16), 71 (4). 58 (39) and 55
(8).
HREIMS m/z (rel. int. %): 369.166 (M+, C21H23NO5, calcd. 369.1576,
12). 190.0579 (C10H8NO3, calcd. 190.0658. 9). 179.0658 (C10H11O3.
calcd. 179.0708, 22), 148.0609 (C9H8O2, calcd. 148.024, 100) and
58.0642 (C3H8N, calcd. 58.0418, 39). i
iH-NMR (CDCI3, 400 MHz) 5: 7.02 (1H, s, H-l), 6.71 (1H. s, H-4),
2.11 (2H, m, H-5), 2.83 (2H, m, H-6), 3.09 (2H, m, H-8), 6.71 (2H. t,
H-l1, 12), 3.75 (2H, m. H-13). 5.95 (2H. s, 0-CH2-0), 3.93 (3H, s,
OCH3) and 2.21 (3H, s, N-CH3).
13C-NMR (CDCI3, 100 MHz) 5: See Table-2.9.

(+)-p-Hydrastine (232)2.3.9
The fraction FA-5 obtained from the silica gel column was
eluted with chloroform : methanol (9:1). It was concentrated In a
round bottomed flask on rotary evaporator to give a gum (48 mg).
This material was triturated with chloroform to give chloroform
soluble portion (30 mg) containing one compound along with some
minor compounds. Fraction FA-5 was further purified by preparative
TLC on silica gel plates (GF-254, 0.2 mm) and developed with
chloroform : methanol (9.7:0.3) to give a pure UV active band (+)-p-
hydrastlne (232) as a gum (6.5 mg, yield 3.6 x 10-5%). The spot of
(+)-p-hydrastine (232) give red colour with Dragendorffs reagent.
Spectral Data
[a]25D = +48 (c=.0367 CHC13).
UV (MeOH), nm (log e): Xmax 290 (3.889), 264 (3.655) and 218 (4.150).
IR (CHCI3) vmax cm-1: 1740 (lactone C=0), 1610 (C=C aromatic) and
1130 (C-O-C).
EIMS m/z: (rel.int.%): 383 (M+, 5), 353 (19), 281 (10). 267 (10), 237
(5), 163 (45), 148 (100), 91 (12) and 58 (10).
HREIMS (rel. int. %): 383.1245 (C21H21NO6. calcd. 383 .1368,5)
190.0644 (C11H12NO2. calcd. 190.0868, 20), 163.0403 (C9H7NO3,
calcd. 163.0633. 45) and 148.0612 (C9H8O2, calcd. 148.0524, 100).

1H-NMR (CDC13, 500 MHz) 5: 7.11 (1H, d, J = 7.96 Hz. H-3'), 683
(1H, d, J = 7.96. H-2'), 6.64 (1H. s, H-5), 6.35 (1H. s, H-8), 6.03 (2H.
s. O-CH2-O). 4.07 (1H. d, Ji,9 = 2.9 Hz. H-l), 5.67 (1H. d, J9,l = 2.9
Hz, H-9), 3.72 (3H, s, OCH3), 3.76 (3H, s. OCH3), 2.63 (3H. s, N-
CH3). 2.40 (2H. m. H-4) and 2.74 (2H. m. H-3).
13C-NMR (CDCI3, 100 MHz) 5: See Table-2.9.
2.3.10 Corydaldine (147)
The fraction FB-3 (Scheme-2.9) was concentrated to a gum
(400 mg) and subjected to precoated silica gel plates using pet. ether :
acetone (7.5:2.5) to afford (10 mg) of a pure alkaloid, corydaldine
(147) (5.5 x 10-7% yield).
Spectral Data
[aj25D 0° ( c=.867 MeOH)
UV (MeOH). nm (log e): Xmax 295 (2.610), 260 (2.730), 220 (3.240) and
208 (3.182).
m (CHCI3) vmax cm 1: 3672 (N-H), 1710 (C=0), 1595 (C=C).
HREIMS m/z (rel. int. %): 207.089, (C11H13NO3 calcd. 207.081, 40).
178.068 (C10H10O3. calcd. 178.063), 150.069 (C9H10O2. calcd.
150.064) and 104.026 (C7H4O, calcd. 104.026).

1H-NMR (CDCI3, 500 MHz) 8: 7.55 (1H. s, H-8), 6.67 (1H, s, H-5),
6.30 (1H, br. s, N-H), 3.91 (3H. s, OCH3), 3.t>2 (3H, s, OCH3), 3.55
{2H, t, J3>4 = 4.8 Hz, H-3) and 2.93{2H, t, J4,3 = 4.8 Hz, H-4).

SECTION C
ISOLATION AND STRUCTUREELUCIDATION OF
STEROIDAL LACTONES FROM WITHANIA
SOMNIFERA

3.0 INTRODUCTION
Withania somnifera, also called as the "nightingale", belongs to
the family solanaceae. Withania somnifera L. Dunal occurs over a large
area of tropical and sub-tropical region of the world including
Pakistan, India, Sri-Lanka, Mediterranean regions, Canaries, S.
Africa, Iran, Iraq, Syria and Turkey. Economically this family is veiy
important because it affords important foods and drugs. Plants
belonging to this family include potato, tomato, tobacco, red-pepper
etc. [187].
somnifera (Dunal) is locally known as
"Ashwagandha” or "Asgandh" and it is an ancient Ayurvedic drug
used to treat various body disorders [188, 189].
Withania
The family Solanaceae is divided into 84 genera and 3000
species. Out of these 14 genera and 52 species of this family are
found in Pakistan as herbs and shrubs. Withania somnifera is a
shrub, 30-150 cm long with ascending branches and stellate-
tomentosic shoots. The roots are stout and fleshy, whitish brown in
colour. The leaves are simple, ovate and glabrous, flowers are
greenish yellow, the berries are small, globose, orange-red when
amateur, enclosed in a persistent calyx, seeds are yellow and
reniform [189]
The plant finds extensive use in the indigenous medicinal
systems (Ayurvedic and Unani systems). Its leaves are bitter in taste
and used as an antihelmantic. The infusion is given in fever.
-:119:-

Bruised leaves and fruits are locally applied to tumors and
tubercular glands, carbuncles and ulcers [191, 192].
The roots are used as nutrient and health restorative In
pregnant women and old people. The decoction of the root boiled with
milk and "ghee" Is recommended for curing sterility In women. The
roots are also used in doses of 30 grains in constipation, senile
debility, rheumatism, in cases of general debility, nervous
exhaustion, loss of memory, loss of muscular energy and
spermatorrhoea.
The extract of Withania somnifera was found to have
significant activities. For example the methanolic and chloroform
extracts of the aerial parts of this plant showed significant
antimicrobial activity against gram (+}-bacteria [193]. Withania
somnifera is a plant which showed antitumor activity against
urethane-induced lung adenomas in adult male albino mice by
inducing a state of nonspecific increase in resistance and
immunostimulant properties [193, 194].The aqueous extract of the
Withania somnifera .when administered to a dog, showed a slight
soporific action followed by a complete return to normality [195].
The plant also showed biphasic activity in membrane
stabilization when fresh sheep erythrocytes (SRBC) were subjected to
hypotoxic and heat strains [197]. The fruit is diuretic and its
poisonous seeds have shown mild coagulating effects on milk. The
plant has alkaloids with sedative properties [198].The root extract of
Withania somnifera contains an ingredient which has a GABA-:

mimetic activity [199J.The antinflammatory activity and protective
effect against CCLj-induced hepatotoxicity of the alcoholic extract of
leaves of W. sorrmifera have also been assessed. The extract (at lg/kg
dose) was found to be as active as 50 mg/kg of phenylbutazone and
10 mg/kg of hydrocortisone [200].
The fruits of this plant contain a high proportion of free
amino acids as evident by a positive ninhydrin color reaction. The
presence of these amino acids may be explained by the fact that the
proteolytic enzyme chymase is present in the berries of this plant
[201]. The tubers of Withania somnifera are used in psoriasis
bronchitis, ulcers and scabies [198].
Phytochemical studies on this plant have led to the isolation
of a number of steroidal lactones (withanolldes). Withaferin A (306}
is the most important withanolide isolated so far. It has been
receiving a good deal of attention because of its antibiotic and
antitumor activities. Withaferin-A (306), in concentrations of 10
pg/ml, inhibited the growth of various gram-positive bacteria, acid
fast and aerobic bacilli and pathogenic fungi, but was inactive
against gram-negative organisms and anaerobes. It partially inhibited
the activity of glucose-6-phosphatedehydrogenase-A [202].

,CHaOH
H
V.O' o
ST" HO
iH
O’Withaferin-A (250)OH
The reaction between the antibiotic and glutathione in
Bacillus subtiUls results in the inactivation of the antibiotic activity,
indicating that its carbonyl group is largely responsible for its
antibacterial activity of withaferin-A (306) and this enhancement is
reversed by EDTA [247J.
Withanolide-D (267) was found to active against mouse
leukaemia L5178Y cells [2031.
OH
\ o'H
O
SV-HO
IH
O* Wlthanollde-D (257)OH
-:122:-

Several withanolides are Insect antifeedants e.g. withanolide-
E (258) is a potent antifeedant for larvae of Spodoptera lithorates
[204].
iOH
V I/O oHOH
OH
IH OH
O'WlthanoUdes-E (258)
Withanolide-E (258) also showed immunosuppressive activity
on human p and T-lymphocytes, as well as on mice thymocytes [205].
4p-Hydroxywithanolide E showed a considerable long lasting and
enhanced activity against L-1210 leukemia [206]
3.1 Biosynthesis of Withanolides
Withanolides are naturally occurring C-28 steroidal lactones
having an ergostane skeleton in which C-22 and C-26 have been
appropriately oxidized in order to from a 8 lactone ring. The C28
sterols such as compesterol, 24-methyl-cholesta-5,24-diene-3P-ol and
24-methylene cholesterol are regarded as possible steroidal biogenetic
precursor of withanolides. assuming that withanolide biosynthesis
does not diverge from the major sterol biosynthetic pathway before
the A-desmethyl sterol stage. The possible sterol 24-methylene
-:123 -

cholesterol (284) and compesterol were regarded as precursors of
withanolides. This was confirmed by labelling experiments [207].
oo o
H3G— c— SCoA
H3G— C— CH2— C— SCoA
Acetoacetyl CoA (260)cfI oH2C=C— SCoA
/ H3C— C— SCoA259
\ CoA — SH
fHoG— C— SEnz
IHgO— C OH
H2G— C— SCoAEnzSH
H30~C OHI I
H20— COOH H2O— COOH
NADPHr2623-Hydroxy-3-methyl-glutaryl-SCoA (261)
V NADP+OH
I NADPH NADP+H2G— CH— SEnz H2G— CH2OH
H3O— C OHV
__/I
H3»— C OHI I
H20— COOH H2G— COOH
Mevalonic acid (264)263
Scheme-3.1
The biosynthesis of withanolides was first investigated by
Goodwin and co-workers (208] by administration of [28-3H]-24-
methylenecholesterol to young leaves of Withania somnifera, either
directly or via the stem. The second procedure gave a significantly
higher degree of incorporation.
Squalene (275), a linear C-30 compound which was
discovered in 1916, is now known to be the key precursor of
124:-

cholesterol (284). The biosynthesis of squalene (275) starts with
acetyl CoA (259). Two units of acetyl CoA (259) combine to form
acetoacetyl CoA (260) which then produces 3-hydroxy-3-methyl
glutaryl-SCoA [HMG-SCoA] (261). The (HMG-SCoA] (261) on
reduction with NADPH gives rise to the hemithioacetal (263) which
is converted into mevalonic acid (264) after reduction with NADPH
[209] Scheme-3.1.
Only the JR-form of mevalonic acid is utilized by the living
system for producing terpenes, while the S-form is metabolically
inert.
H2O— CHJJOH H2G— CH2OPPATP ADPI IHgO— C OH
H20— COOH
H30—C OH
H20— COOH
265264
2ATP
V\ ADP
H2rOPPHaO-CHÿOPP C02+ P
I IfH3O—1? H3O—
by— OH
CH2OH3-Isopentenyl pyrophosphate UPP) (267)
H20— jj:— Oÿ-H
OCH— CH2— OPPHgO—C
3-Phospho-5-pyrophosphatemevalonic acid (266)
IIsomeraseCH3
3.3-Dlmethylallyl pyrophosphate (268)
Scheme-3.2

The mevalonic acid (264) is phosphorylated in the presence of
ATP to yield mevalonic -5 - phosphate (265) which is then
decarboxylated and dehydrated to yield isopentenyl pyrophosphate
(IPP, 267) by the route shown in Scheme-3.2, IPP (267) is then
isomerized in the presence of an enzyme "isomerase" to produce
dimethyl allyl pyrophosphate (DMAPP, 268, Scheme-3.2).
IPP (267) can condense in a head-to-tail manner with DMAPP
(267) by formally a combined SN2-E2 process to yield geranyl
pyrophosphate (GPP, 270, Scheme-3.3).
OPP
OPPH
1NPP (269)
266
OPP
GPP (270)
Scheme-3.3
Condensation of GPP (270) with IPP (267) gives famesyl
pyrosphosphate (FPP, 271). FPP is the biosynthetic precursor
sesquiterpenes and diterpenes [210].
-:126:-

The coupling of two FPP (271) units in a head-to-head
manner, produces presqualene (272) which on rearrangement gives
rise to squalene (275, Scheme-3.4).
.OPP
CHaOPP CH3<>CH3oOPP H
CH3
CH3CH3
GPP (270)FPP (271)
+
OPPCH3PPO, head-to-head
IWl]
WÿCH3CH3%ÿCH3
CH3CH3 FPP (271)CH3 Enz CH3
CH3 FPP
Presqualene (272)
CH3PPO.
<n,CH3
CH3CH3273
CH31.2 shift
CH3 CH3
,CH3 ,CH3
nCH3 CH3CH3 CH31©©CH3 CH3NADPH NADPCH3 CH3
CH3H3< H3« CH3274Squalene (270)
Scheme-3.4

The squalene thus formed gets oxidized by atmospheric oxygen
catalyzed by NADPH-linked oxidize to form squalene 2,3-epoxide
(276), which then undergoes cyclization to yield lanosterol (278,
Scheme-3.5).
oSqualene (275)
(O)CH3
CH3
CH3CH3
O* SquaIene-2.3-epoxide (270)
IH3< CH3
f CH3I
VH CH3
HO* 277
|H3<CH3H3<
CH3
CHalCH3
CH3
CH3
HO*’CH3 Lanosterol (278)
Schcmc-3.5H3<
-.128:-

H3< CHJ H3< CÿJ
CH3.H 1 :H3LHCH3 CH3
cn3cn3 14 14
10}
X,CH3 CHO
HO4 HO*H 279’CH3H3< ’CH3
Lanoaterol (278)
j101H3< CHJ H3<
CH3L*HCH3 CH3
CH3 OHb
iM H COOH
HO’ HO'281H 280
H3C CH3H3C CH3 IHI
H3( CHI H3< CH>
.H :H3L-HIsCH3 CH3
CHa CHa
I DemeLhylaUonalC-4H H 283
HO’ HO'
1H3C CHÿ 282
H3( CK,
OH3I.HCH3
CHa
H
HO'Cholesterol (284)
Scheme-3.6
The bio-converslon of lanosterol (278) to cholesterol (284)
involves a number of metabolic changes which occur on a cellular
level and which are not interdependent. The sequence of reactions
and intermediates may also differ slightly among different organisms
(Scheme-3.6). Addition of the methylene group at C-24 may occur
:
-:129:-

with S-adenosyl methionine to form 24-methylene cholesterol, (287)
which Is a possible precursor of the withanolides (Scheme-3.7) (211).
H
H
HO*Cholesterol (284)
/H3C—S\
S-Adenosyl methionine
©/
HaT/NUH
IH H 285
H(
CHa— H
CH3 H
CH3
H H 286
HO*CH2
CUa
CH3 H
CH3
CH3
H
HO*24-Methylene cholesterol (287)
Scheme-3.7
130:-

CH2OH
HH3CH3C,
IO]CH3 CH3
CH3CH3
IH
288HO'HO*
24-Methylene cholesterol (287)[O] CH3loi0°H CH3
Hlr CH2OHH3(H3GV OHOHCH3LHHCH3
CH3CH3
1I HH292289
HO1HO'
HCH3
HCH3
COHH3H3Cÿ
CH3OHO'
CH3L*HH
CH3CH3
IH
HO1 293Hi 290
CH3CH3(O)
CH3CH3
H3C,H3C. V.V oO' OHCH3OHs
10)CH3 CH3
iH
HO' 294Scheme-3.8
131:-

24-Methylene cholesterol (287) Is actually one of the
intermediates leading to the 6-lactone (Scheme-3.7). In each case
(288) should undergo C-22 hydroxylation to (289), a well known
reaction in the steroidal biosynthetic pathway (212).
Two alternative hypotheses for withanolide biosynthesis were
mentioned in the literature (Scheme-3.8). The first involves
cyclization of (289) to (290) and then oxidation to (294) while the
second involves oxidation of the allylic isomer followed by cyclization
to form the lactol (191), present in a number of withanolides.
Further oxidation can then lead to the 6-lactone (194).
-:132:-

RESULTS AND DISCUSSION3.2
3.2.1 Isolation of Withanone (295)
The methanolic extract of aerial parts of Withania somniferawas evaporated under vacuum to a gum. The material was partitioned
between distilled water and pet.ether followed by removal of pet. ether
and extraction with chloroform. The chloroform soluble extract
(Scheme-3.12) was loaded on a silica gel column (mesh 70-230
ASTM) and eluted with pet.ether : chloroform. The fraction (WF-1)
was thus obtained on elution with pet.ether : chloroform (2:8). This
fraction (WF-1) was then subjected to column chromatography on
silica gel (mesh 70-230 ASTM) and the column was eluted with
pet.ether : chloroform (2:8) to afford fractions WSF-1 and WSF-2.
The fraction WSF-2 was subjected to preprative TLC elution having
with pet.ether : chloroform (2:8) , to afford a pure compound,
withanone (295) as a colourless amorphous solid (6.5 mg, yield 1.8 x
lO'7)
28
CH3
!4 CH2OH23 25
21 1,22 126
20 i O O18 20 s
CH3 HH
17n X12N
\\ iU'-H13II I "Is I 14
16
15
2
3
Withanone (205)H
-:133:-

The IR spectrum of withanone (295) exhibited bands at 1660
cm-1 due to a, (i-unsaturated lactone, 1690 cm'1 (non conjugated
ketone) and 3340 cm-1 for the hydroxyl group in the molecule [214].
The UV spectrum of withanone (295) showed absorption
maximum at 215 nm characteristic of a six membered a, (3-
unsaturated lactone chromophore [214].
The iH-NMR spectrum (400 MHz, CDCI3) of withanone (295)
showed three singlets each integrating for three protons at 8 0.81,
1.25 and 2.03 which were assigned to the protons of the tertiary
methyl groups. A doublet at 8 1.03 (J21.20 = 7.0 Hz) was due to the
protons of the C-21 secondary methyl group. Two AB doublets at 8
4.31 and 4.40 (J27a,27(l = 10.4 Hz) were due to the gleminally coupled
C-27 hydroxy methylenic protons. A downfield doublet of double
doublets centered at 8 4.65 (J22.20 = 13.3 Hz, J22,23(3 = 3.5 Hz, J22.23a
= 3.5 Hz) was assigned to the C-22 methine proton geminal to the
lactone oxygen. The !H-NMR assignments of (295) are given in
Fig.3.1
The high resolution electron impact mass spectrum afforded
the molecular ion at m/z 456.2801 establishing the molecular
formula as C28H40O5 (calcd. 456.2875 ) indicating nine degrees of
unsaturation in the molecule. The molecular ion was further
confirmed by +ve fabms.
TheiH-iH long range shift correlations were determined by
recording a series of HOHAHA (TOCSY) spectra with variable delays
134:-

(20, 60, 100 ms). The C-21 methine proton displayed interactions
with the C-22 methine and with the C-23a and C-23P protons. C-
22H also exhibited long-range couplings with the C-21 methyl
protons. The C-28 methyl protons showed homoallylic coupling with
the C-27 methylene protons in the HOHAHA spectra.The HOHAHA
interactions of (295) are presented in Flg.3.2
The COSY-450 spectrum of withanone (295) revealed the
presence of several spin systems in the molecule. One of the spin
systems "A" starts with the C-2 methylene protons at 5 1.5 (m) and 5
2.1 (m) which showed connectivity with the C-3 methylene protons (5
1.62 and 1.9). The C-2 and C-3 methylene protons also showed cross-
peaks with the C-4 methylene protons (5 1.15 and 51.31). This set of
3 adjacent methylenes constitutes spin system "A" Fig. No. I.
1.5
,,if L1.62 m H
1.9 m
V.2
4
H / '''H H
t1.31 m 1.15
Spin system "A"
Fig. No. I
Another larger spin system ”B" starts with the vinylic proton
at 5 5.45. This proton showed interactions in the HOHAHA spectra
with the C-7 methylene protons (5 1.6 and 1.9 ). The vinylic proton
also displayed a long-rang coupling with H-8 methine proton at 5

2.21 and H-4 methylene protons at (8 1.15 and 1.31). On the other
hand the H-9 methine proton displayed long-rang interaction with H-
8 methine proton and H-ll methylene protons. The H-7 methylene
proton resonating at (8 1.60, 1.91) also showed interaction with H-9
and H-6 methine protons respectively, The C-12 protons (81.51, 1.40)
did not show any Interaction in the COSY-450 spectrum except with
the C-ll protons, indicating that the H-12 methylene carbon Is
attached to a quaternary carbon (Fig. No. II).
H.x|
Hr>.H
K3Spin system "B"
Fig. No. n
The broad band decoupled 13C-NMR spectrum (CDCI3, 125
MHz) showed resonances for all twenty eight carbon atoms in the
molecule, The DEPT spectrum showing signals for the C-2, C-3 and
C-4 methylene carbons at 8 32.87, 26.25 and 31.06 respectively.
The HMQC spectrum showed that these carbon atoms were
directly attached to the C-2 protons (8 1.5 and 2.1), C-3 protons (8
1.62 and 1.9) and C-4 protons (8 1.15 and 1.31) respectively which
-:136:-

are presented in Table 3.1.The HMQC spectrum also showed the
following respective carbon - proton- one-bond connectives 122.8 (8
5.45 m). 36.52 (5 1.6 and 1.9 m), 42.0 (8 2.21 m), 32.2 8 1.9 m), 22.26
(8 1.41 and 1.45 m) and 23.67 (8 1.4 and 1.5 m) for the C-6, C-7, C-8,
C-9, C-l1 and C-12 carbons and the corresponding attached protons.
The DEPT spectrum indicated that there were four methyls,
ten methylene, six methine and hence seven quaternary carbons in
the molecule [215], This further confirmed that there were 28 carbons
in the molecule. Downfield signals at 8 167.1 and 8 214.7 were
assigned to the a, p-unsaturated lactonic (C-26) and ketonic (C-3)
carbonyls respectively. The remaining four downfield carbons which
resonated at 8 125.1 (C), 8 154.3 (C), 8 122.8 (CH) and 8 146.5 (C)
were assigned to the C-25, C-24, C-6 and C-5 carbons respectively.
The 3H signals at 8 9.39, 14.9, 9.28 and 19.98 were assigned to the
C-l9, C-18, C-21 and C-28 methyls respectively. The chemical shift
assignments of various carbons of compound (295) are presented in
Fig.3.5
The third spin system "C" (Fig. No. in) starts from the C-17
methine proton centered at 8 1.56 which showed interactions in the
HOHAHA spectra with the C-l6 methylene protons (8 1.45 and 1.51
m) as well as with the C-15 methylene protons (8 1.67 and 1.95 m).
The C-17 methine proton also showed connectivity with the C-20
methine proton (8 2.0 m). This methine proton showed coupling with
the C-21 methyl protons centered at 8 1.01 and with the C-22
methine proton at 8 4.5. These protons were further coupled with the
C-23 methylene protons at 8 2.5 and 2.3 m.
-:137:-

[22
Spin system "C"
Fig. No. Ill
The long-range !H/13C interactions in withanone (29B) were
determined by the HMBC experiment. Some of the couplings were
between C-6 (8 5.45) with the C-4 methylene protons(8 31.0) as well
as between C-4 (8 31.0) with the C-6 proton (8 5.45) hereby
interconnecting the spin system "A" and "B". Long-range coupling of
the C-9 methine proton (8 1.9) was also seen with the C-19 methyl
carbon (8 9.39) and with the C-l carbonyl carbon (8 214.7). The C-2
methylene carbon (8 32.87) also showed long-rang connectivity with
the C-l carbonyl carbon (8 214.7). The C-19 methyl protons (8 1.25)
showed connectivity with the C-10 quaternary carbon . On the basis
of these interactions it was possible to combine fragments "A" and
"B" as shown in Fig. No. IV
-:138:-

H Hn/.<rs o
.. Ill!Jr i
+2 9
H"43 H10 f
I 4 JL 7tvH'
3a H123b
n ft!*'CH3
S'H.9
2 10 8H‘
,3 75
6
[A+B]
Fig. No. IV
The structure of another part "C" of the molecule was deduced
as follows: Long-range iH/ÿC correlations of the 015 methylene
protons (8 1.67, 1.95m) with the C-14 oxygen-bearing quatemaiy
carbon (8 85.09) and with the C-8 methine carbon (8 42.0) were seen
in the HMBC spectrums. This region also showed long-range coupling
of the C-17 methine proton (8 1.56) with the 013 quaternary carbon
(8 48.04) as well as with the C-18 methyl carbon (8 14.90). The C-12
methylene protons in fragment "B" also showed connectivity with the
013 quaternary carbon (8 48.04), C-18 methyl and (8 14.90) the C-17
methine carbon (8 50.6). Similarly, the C-18 methyl protons showed
coupling with the C-13 quaternary carbon (8 48.04). The combination
of fragments (A +B) with ”C" led to a larger structural fragment "D"
shown in Fig. V.
-:139:-
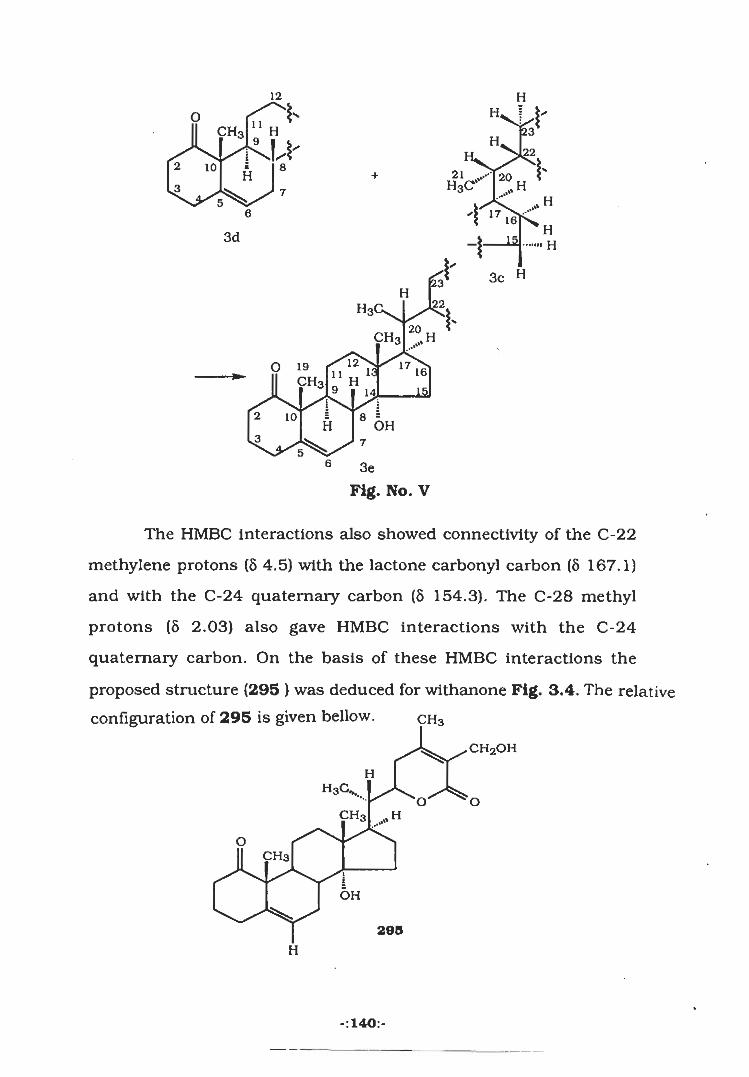
12
HJ1- Idri CHJ” hr H,12
*82 10 ft
fÿi<+
.3 75
6
H3d
i3c H
4ÿCH3J>H
H3
12O 19
151
2 10H
8OH
3 75
6 3e
Fig. No. V
The HMBC interactions also showed connectivity of the C-22
methylene protons (8 4.5) with the lactone carbonyl carbon (8 167.1)
and with the C-24 quaternary carbon (8 154.3). The C-28 methyl
protons (8 2.03) also gave HMBC interactions with the C-24
quaternary carbon. On the basis of these HMBC interactions the
proposed structure (295 ) was deduced for withanone Fig. 3.4. The relative
configuration of 295 is given bellow. CH3
,CH2OH
H3Gÿ.
fH3UHo- o
oCHa
OH
205
H
-:140:-

The El mass spectrum of withanone (295) showed the
molecular ion at m/z 456. The exact mass m/z 456.2801 agreed with
the molecular formula (C28H40O5, calcd. 456.2875). The ion at m/z
438.2761 (C28H3804, calcd. 438.2769) was due to the loss of a water
molecule from the molecular ion. The base peak at m/z 141.0561
arose by cleavage of lactone ring. The mass fragmentation patteren of
withanone (295) is shown in Scheme-3.2.
Stereochemical Assignments
(3-Stereochemistry of the 18 and 19 methyls were deduced due
to the fact that withanolides are biosynthesised from
cholesterol with (3-oriented 18 and 19 methyls.
1.
2 The a-stereochemistry of the 14-OH was assigned based on
the reason that ring C/D is tran-fused in cholesterol (a
biogenetic precursor of the withanolides).
The (3-orientation of C-17 side chain was assigned due to the
biogenetic consideration.
3
Hence, we can draw the complete structure of wihanone
(295) with stereochemistry. The COSY-450 spectrum exhibited
coupling interactions which were found to be in full agreement with
the assigned structure (295) are shown in Fig. 3.5.
141:-

5 2.03
CH35 2.30 m
8 2.51 m 9CH2-OH5 4.316 4.40
HJ2i.22=70Hz 6 2.0 m
8 1.01 d HH3c,
H
8 1.42 m \
/W5 1.51 m
5 1.41 m H5 1.45 m 9
V
' oH 5 4.5 ddd J22.20=13-3H2
•ÿ’’6 1.56 mH J22 23(J='ÿ*5HzJ21 23a=ÿ*ÿÿzP 5 1.25 s
CH3H5 2.21 m5 1.5m
6 1,45 mHr
H8 2.1mH 5 1.51 mH
::= 1 HOH H
5 1.62 m zHH 6 1.95 m
8 1.9 m 8 1.67 m
H5 1.9 m
H 6 1.6 mHH
H8 1.15 m 8 1,9 rn5 1.31 m H8 5.45 m
Fig. 3.1. iH-NMR assignments of withanone (295).
8 2.03
CH3y/ 8 2.30 m
5 2.51 m9:! CH2-OH
'*8 4.318 4.40
H> N8 2.0 m
H"-. y- 6 1.51 m g 1.42 my 5 1.41 m H
(h 1.45 m HA?0ÿÿ0\
H! 8 4.5 ddd
pj 5 1.56 m'
H .-•*
Q 8 1.25
CH35 2.21 myr 5 1.5 m
5 2.1m 9 H,
H H 6 1.5 m .I I I"'/’5 1.62 m
HH OH 5 1.67 m
8 1.95 m /C8 1.9 mlV
••18 1.9 m "HH 5 1.6 mH'"
8 1.15 mH
H 5 1.9 m5 1.31 m H. * 5 5,45 m
Fig. 3.2. HOHAHA interactions of withanone (295).
-:142:-
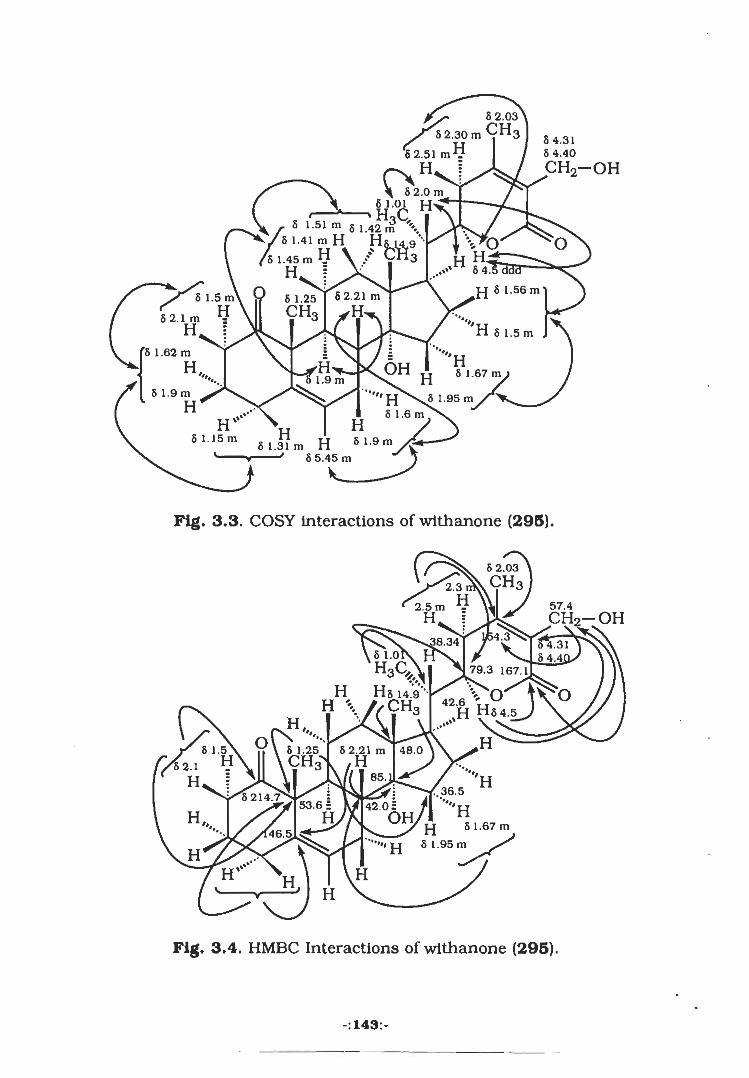
8 2.03
CH3y/ 8 2.30 m
6 2.51 m8 4.318 4.40
CH2-OH\ 8 2.0 m
r
__H>
\ /ÿ8 151 m 8 1.42 m \
<V 8 1.41 m H/s 1.45 m H \ /MS
yH 8 L56
"'H 8 1.5 m .
H
"D8 2.21 myS 8 1.5 m
8 2.1m H8 1.25
CH3| /H-iH
’6 1.62 m
OH HH, H 8 1.67 m
s8 1.95 m
1.9 m8 1.9 m
HH 8 1.6 mH H
H8 1.15 m 8 1.9 m8 1.31 m H-' 8 5.45 m
Fig. 3.3. COSY interactions of wlthanone (295).
5r2H2.3
2.5 m y 57.4
CH2-OHH
18.34 4.318 1.0 8 4.4'
HaS., 79.3 167.1
JVH VO
H HS4.542.6H % CH3£H,
9 \ 8 1.25
\CH3>H8 1.5 82.21 m 48.0
H8 2.1:\
85.H HI 36.58214.7 42ÿ0:53.6 HH,. H OH8 1.67 mH46.3
•*»# 5 1.95 m'"HHo--'H
iL HV
Fig. 3.4. HMBC Interactions of wlthanone (295).

6 19.98
CH36 57.4
CH2-OH6 125.16 38.34
6 9.28 X_T
H3C...6 14.9
CH3
6 79.3 6 167.1
6 42.6' U UH6 23.67
6 5.06
Q 6 9.39 6 22.26CHgL H
5 48.046 23.67
6 85.096 214.7
6 42.0\5 32.87 6 36.526 53.67
H OH6 26.25xTvÿ 6 36.52
631.06 6 122.8
Fig. 3.5. 13C-NMR assignments of withanone (295).
The perspective view of withanone(295) is presented in Fig.
3.6 [217].
21
,.CH3 CH3H 26'
18
CH3 a*HOOH£H3 Ji 13
.1712 H &2HO A®5
8 1610 [92 14NOH HH I H O743 5
6
H
H
Fig. 3.6. The perspective view of withanone (295).
-:144>

CH3
CH2-OH
H3G..V r°- oCH3 CH3
o CH2-OHCH3 H
TH O' o
m/z 438 m/z 141& n\°1
ACH3
CH2-OH
HHac...•v
r°- oCH3
sr»HO
CH3
1 OH
m/z 456<C.
CH3OH
n CH2-OHCH3 to§O
H;.CH3 3 O' o
m/z 138 m/z 167mIV
CH3
oCH3 H
TH OH
m/z 288
VScheme-3.9. Mass fragmentation pattern of withanone (295).
145:-

Table-3.1. HMQC Assignments of Withanone (295)
No 6 13C8 H
C-l 214.7
C-2 1.5, 2.1 M 32.87
C-3 1.62, 1.90 M 26.5
C-4 1.15, 1.31 M 31.06
C-5 146.5
122.8C-6 5.45 m
C-7 1.60, 1.91 m 36.52
2.21 m 42.0C-8
1.9 m 32.2C-9
C-10 53.67
22.26C-ll 1.41, 1.45 m
C-12 1.40, 1.51 m 23.67
C-13 48.04
85.09C-14
C-15 1.67, 1.95 m 36.52
C-l6 1.45, 1.51 m 23.67
1.56 mC-l7 50.6
C-18 0.81 s 14.90
C-19 1.25s 9.39
2.00 mC-20 42.6
C-21 1.01 d 9.28
4.5 dddC-22 79.3
C-23 2.3, 2.5 m 38.34
154.3C-24
C-25 125.1
C-26 167.1
4.31, 4.40 d 57.4CT27
C-28 2.03 s 19.98
146'.'

Quresimine-A (296)3.2.2
The fraction WF-4 (Scheme-3.12, Experimental) was obtained
on eluting the silica gel column loaded with defatted CHCI3 extract
of Withania somnifera with chloroform : methanol (9.5:0.5). This
fraction was subjected to preparative TLC and elution was carried out
with pet. ether : chloroform (2:8) + 3 drops of diethyl amine. This
yielded quresimine-A as a colourless amorphous solid (7.5 mg) yield
2.08 x 10-5%.
28
CH327
24l CH2-OH23 25
21 H
H3CV18 20 =
26
‘ooCH3 H
17
12o 1611 1319
CH3 H159 14
I1102 8
H H H5 7
6Hgcor
o' HOH
Quresimine-A (296)
The UV absorption maximum at 214 nm was characteristic
and an a,P-unsaturated lactone chromophore [218).
147:-

The IR spectrum displayed absorptions at 1683 cm-1 for a, P-unsaturated lactone, at 3455 cm1 for (O-H) and 1590 cm1 (C=C)
[219].
The high resolution mass spectrum of 296 showed the
molecular ion peak at m/z 502.2910 corresponding to the molecular
formula C29H42O7 which indicated the presence of nine double bond
equivalents in the molecule. The peaks appearing at m/z 484.2804
and 470.2667 showed the loss of water and methylene group
respectively. The peak which appeared at m/z 452.2530 again
exhibited the loss of water molecule. The peak at m/z 387.2522
(C24H35O4) and 86 (C4H6O2) resulted due to the cleavage of C-l/C-4
bond which further indicated that four oxygens were present in major
portion. The mass fragment at m/z 334.1979(C2oH3oO<i) formed by
the cleavage of ring C was indicative of the presence of four oxygen
functions and the remaining fragment appeared at m/z 169.0810
(C9H13O3) and indicated the presence of a six-membered lactone
substituent at the C-20 side chain, the prominent peak at m/z
141.571 (C7H9O3) was originated by cleavage of the C-20/C-22
bonds. The complete mass fragmentation is presented in Scheme-
3.10.
The 1H-NMR spectrum (300 MHz, CDCI3) of 296 Fig. 3.7,
showed three 3H singlets for the quaternary methyls at 6 0.65, 5 1.29
and 5 2.02 assigned to C-18, C-19 and C-28 protons respectively,
while a 3H doublet at 5 0.98 (J = 6.6 Hz) was due to the C-21
secondary methyl protons. A doublet of double doublets appeared at 8
4.41 (J1 J2 = 4.46. J3 = 15.2 Hz) and was assigned to the C-22 proton
-:148:

of the lactone moiety [220]. Two AB doublets at 8 4.36 and 8 4.38
(J27a,27(}=13.5 Hz.) centred due to the C-27 methylenic protons. A
broad 1H signal at 8 3.20 was assigned to the C-6a proton of oxirane
ring. A 1H doublet at 8 3.48 (J = 3.18 Hz) was due to the C-4a
proton. The multlplet for the C-3a proton resonating at 8 3.69 was
found to be coupled with the C-4a methine proton while the C-2
methylene protons appeared as as multiplets at 8 2.58 and 8 3.00. A
three proton singlet resonating at 8 3.33 was due to the C-3 methoxy
group.
The COSY-450 spectrum of 296 served to establish the
proton-proton connectivities which were found to be very helpful in
deducing the structure (296). The signal at 8 3.69 (C-3a proton)
showed strong connectivities with the protons at 8 2.58 and 8 3.00
(C-2 methylene protons) as well as with the protons at 8 3.48 (C-4a
methine proton). The proton at 8 3.20 (C-6a) protons showed
connectivities with those at 8 1.30 and 8 2.15 (C-7 methylene
protons).
O
fsoodd II]2.58 dd1 -TH
H
H3CO<
OH
Spin System "A”
Fig. No. I

The COSY-450 spectrum was used to explore the spin systems
in the molecule. The first spin system "A" starts with the C-4
methine proton (8 3.48) which showed connectivities with another
downfield C-3 methine proton (8 3.09). This latter methine was
further coupled with the C-2 methylene protons (8 2.58 and 8 3.00)
shown in Spin System "A" Fig. No. I.
Another spins systems "B" starts with the C-6 proton of the
oxirane ring which resonated at 8 3.20 and was coupled with the C-7
methylene protons (8 1.30 and 8 2.15). These methylene (C-7) protons
showed further connectivities with the C-8 methine proton at 8 1.39.
The C-8 methine proton in turn showed connectivities with the C-9
methine proton(8 2.10) and with C-14 (82.10). The C-9 methine
proton (8 1.20) and with the C-14 methine proton (d 2.10). the C-9
methine proton also showed coupling with the C-ll methylene
protons (8 1.21). The C-ll proton was in turn connected with the C-
12 proton at (8 1.65). The C-14 methine afforded a cross-peak with
the C-15 methylene proton (8 1.28, 1.25) which in turn showed
connectivities with C-16 methylene protons (8 1.58 and 1.63). The C-
16 proton exhibited cross-peaks with the C-17 methine proton at 8
1.09. This proton was further coupled with the C-20 methine proton
(8 1.90) which was in turn coupled to the C-22 methine proton (8
4.40). The latter showed cross-peaks with the C-23 methylene proton 1
(8 1.90 and 2.45). These Interactions let to the deduction of the spin
system shown in Spin System "B" Fig. No. n.
-:150:-

H 2.451.90 IH.o1.90
n 0.97
5 H3C,,,o-H1.21
H H
PH H
1.39 H
5i.2o , y \
C'*'Ho1
3.20
Spin System "B"
Fig. No. H
The 13C-NMR spectra (CDC13, 125 MHz, DEPT and B.B)
indicated that there were five methyls, eight methylenes, nine
methines and seven quaternary carbons in the molecule, which was
further confirmed that there were 29 carbons associated with 42
hydrogens. Downfield signals at d 209.8 and 167.0 were assigned to
the ketonic (C-l) and a,(i-unsaturated lactonic (C-26) carbonyls
respectively. The remaining two downfield carbons which resonated at
8 152.8 and 125.7 were assigned to the C-25 and C-24 quaternary
carbons respectively. The methyl signals at 8 11.6, 15.7, 13.3, 20.0
and 56.8 were ascribed to C-18, C-l9. C-21, C-28 and -OCH3 carbons
151:-

respectively. The chemical shift assignments of various carbons of
compound 296 are presented in Fig. 3.9.
and the structures of theThe spin systems A and B
remaining fragments were investigated with the help of HOHAHA
spectra recorded with the times of 20, 60 and 100 ms.
To establish direct iH/ÿC connectivites, the HMQC
(Heteronuclear Multiple Quantum Coherence) experiment [182, 221]
was performed. The live methyls groups resonating at 8 11.6 (C-18).
15.7 (C-19), 13.3 (C-21), 20.0 (C-28) and 56.8 (-OCH3) showed cross-
peaks with the proton signals at 8 0.65, 1.29, 0.98, 2.02 and 3.33
respectively. The proton at d 4.41, 3.69, 3.48 and 3.20 were showed
cross-linked with the carbons resonating at 8 78.7, 77.7, 75.1 and
60.3 assigned to the C-22, C-3, C-4 and C-6 methine carbons
respectively. The C-27 methylene protons at 8 4.36 and 4.38 exhibited
cross-peaks with the carbon signal at 8 57.4 in the spectrum.The
1H/13C connectivities of all 29 carbons with their respective protons
are given in Table-3.2.
The assembly of structure 296 on the basis of long-range
heteronuclear correlation observed between carbons and protons of
various spin system, in the HMBC spectrum Fig. 3.8. The HMBC
spectrum of quresimine-A showed the correlations of H-4a (8 3.48),
C-13 (8 209.8), C-2 (8 39.13), C-3 (8 77.50), C-5 (8 64.93) and C-8 (8
29.36) which confirmed the assignments of ring A and B. Another
important signal at 8 4.41 (H-22a) showed interactions with C-20 (8
38.7), C-23 (8 29.36), C-26 (8 167.0), C-25 (8 152.8) and C-24 (8
-:162:-

125.7) and confirmed the assignments of the lactone moiety. So the
proposed structure of 296 establish from various sub-structures
obtained from COSY-450 Fig. 3.10 and HOHAHA spectrum Fig.3.11.
Table-3.2. HMQC Assignments of Quresimine-A (296)
No. 5 1H 8 13C
C-l 209.8
2.58m. 3.00 m 39.13C-2
3.69 m 77.50C-3
3.48 d(J = 3.18 Hz) 75.17C-4
64.93C-53.20 br.s 60.37C-6
1.30 m, 2.15 m 31.17C-7
29.36C-8 1.39 m
C-9 1.20 m 42.78
50.43C-101.21 m 21.62C-ll
24.27C-l2 1.65 m
42.68C-l3
2.10m 56.06C-14
C-l5 1.22 m 27.28
39.56C-16 1.61 m
1.09 mC-l7 51.96
0.65 s 11.56C-18
C-19 1.29 s 15.72
C-20 1.90 m 38.77
0.98 d J = 6.6 Hz 13.34C-21
4.41 dd (Ji=J2 = 4.46 Hz, J3 = 15.2 Hz) 78.34C-22
1.90 m, 2.45 m 29.36C-23
C-24 125.7
152.8C-25
167.0C-26
4.36 dd, 4.38 dd J27a,27|J = 13.5 Hz 57.44C-27
2.02 s 20.0C-28
3.33 56.83-OCH3
163:-

CH3
CH2-OH
H3Cÿi O' o
CH3 H(-•ÿÿIII j~J
oCH3 H
IHHH
V.H3CCy
Ho4OH
Proposed structure of 296.
The steroechemical assignments at C-4 and C-6 are based on
the observed coupling constants. Assuming cts-decline type chair
conformations of ring A and B, the C-4 methine proton with p-
oriented C-4 hydroxyl group (i.e. a-orientation of proton) should
show one diaxial couplings with the C-3 methine proton. The
coupling constants (Ja>e = 3.18 Hz) of the H-4 signal, therefore,
convincingly indicates a p-orientation (equatorial) of the hydroxyl
substituent.
The stereochemistry assignment of the oxirane ring is based
on the chemical shift comparison with earlier reported withanolides.
This was further confirmed by recording the nOe spectrum
where no enhancement of the methyl signal at 8 1.29 (CH3-19) was
164:-

observed by irradiating the H-6 signal at 8 3.20 (H-6) which should
otherwise be observed if the oxirane was a-oriented Fig. 3.12.
2.02 s
28
CH31.90 m
2.45 m H 2724z
CH2-OHH4.36 dd.4.38 dd.J27a.27b=13.5Hz
1.90 m 123
0.98 d, J=6.6Hz 211.65 m H3C
1.21 m 8 20 T=/S' H \ f IÿCH3 H 4.41 ddd.J1.2=4.46Hz. J3=15.2Hz
H j/l2 Jj7
o‘Sfta H19 J
9 14
25
Nx>N>1.61 m
13 i1.39 m 1
2.58 m HH,
H H15 1J'H
1.09 m1
7I I*•-. 3.48 d.ÿ
J=3.18Hz
3 H 5
2 H3.69 m 10 1.22 m85 :J-J 2.10 mHH1.20 m
: 1.30 m\H3ca
6""H 2.15 m4
3.33 s O'OH 3.20 br.s
Fig. 3.7. iH-NMR assignments of 296.
'Z.V’2 a
CH3v* 1.90 m/ TT
2.45 m f? 57.4CH2-OHH
KJ4.361.90 m4 38
H13.3 167.078.4H3C,„;
0.65 s 38.7 r=:CH3S H
O o
:O 1298
CH3 42ii2.58 mH H
S.Cÿmi209.8
3.48 a3.69 m 39 i 50.4 i 29.3iH H HH
771.5 =175.7 64.9= 60.37
H3C3.33 a O’ H
OH
Fig. 3.8. HMBC interactions of 296.
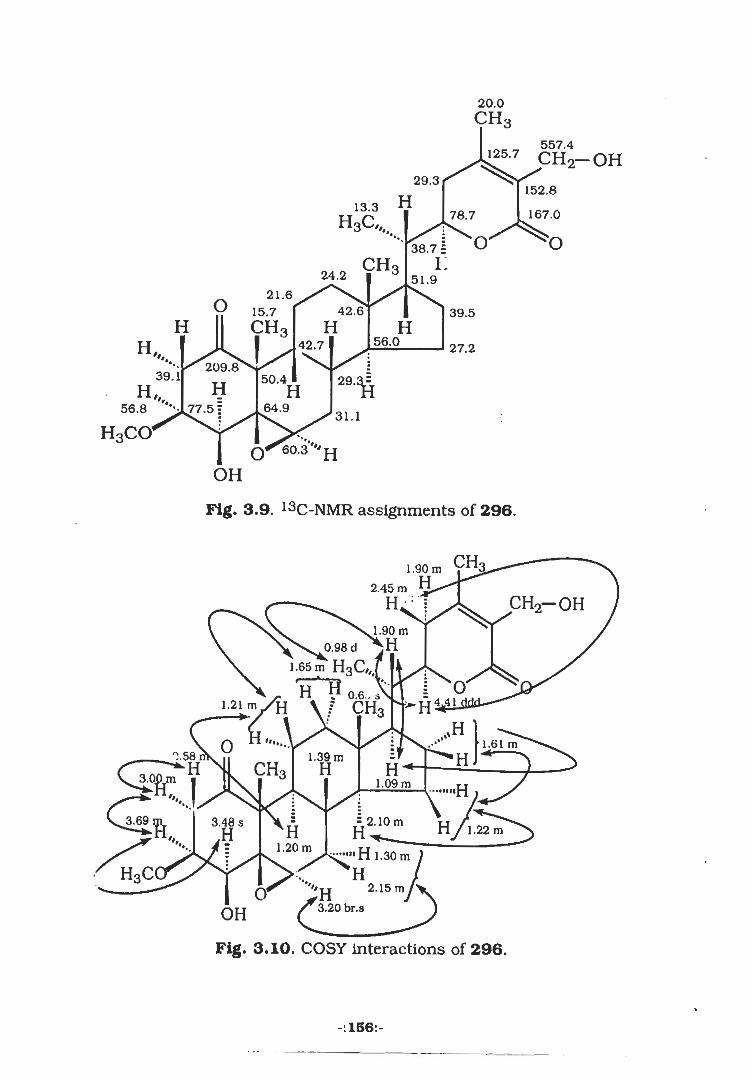
20.0
CH3557.4
CH2— OH
152.8
125.7
29.3
13.3 HH3C„. 167.078.7
38.7 = U oHCH324.2 51.9
21.6
9 157
CH342.6 39.5
H H H56.042.7 27.2
:209.839.1 50.4 29.ftHH... H
64.956.8 77.5: 31.1:
H3CO0ÿ603'"H
OH
Fig. 3.9. 13C-NMR assignments of 296.
CH31.90 m
2.45 m
CH2-OH%1.90 m
H0.98 d
1.65 m H3C/,j
TTR 0.6/4.41i.2i m yy[ ! CH3
:H
3.58ÿ9 1.61 m:if1.39 m HCH3 H H "*
1.09 m H•
(72 :
iz v2.10 m3.48 sK H /1.22 mH HH1.20 m**.. z
H 1.30 m:
' H3C HX 2.15 mO' H3.20 br.sOH
Fig, 3.10. COSY interactions of 296.
-:156:-

CHsH
I CH2-OH
H3CVH H / ‘
v(H0
H \:ÿ
f"'2.58 m H
CH33.(ÿm
HH
V.3.69 m 3.48 s
:ÿHH
Fig. 3.11. HOHAHA interactions of 296.
2i
-C«3Hÿ2o 2819
H3C18 PH*P«3 23n [2
1311Hlo
\\\25UHO. O
HO158 H9o !,267l\ 5 I166H CH227 2H2 4
ott3Co, OH3
HFig. 3.12. Perspective view of 296.
Xu
nx—o
XK>
<?*A Ix°''W°n b 1
I
oV)o Clcer i—oA (O XoB X a
05A Xi
CO V'NjII
I Iom/z 502
(C29H4207)2 O m/z 141
(C7H9O3)P n oI
Om uM IX bOi cCD
P
1 tA
XX +uK>o2P n xx —orr X10
o oD X x x
05 n(0 x—oo .o :i>-h ac10o ooto ac acacxo X XCD o%X03 o05 1o £ O 'O
X m/z 170(C9H14O3)
X o oX X m/z 334(C20H30O4)
5*5 m/z 234(CHH18O3 )
m/z 264(C15H2o04)
O05
XV nIV m

Reported Withanolides from Withania somnifera
Withaferine-A (306)3.2.3
The fraction WF-1 (500 mg) (Scheme 3.12, experimental
section), was subjected to chromatography on a small column packed
with silica gel (150 g, 60-120 mesh size) using pet. ether : chloroform
as the packing solvent. Elution was with chloroform and then with
increasing polarities of chloroform : methanol.
The fraction WSF-1 obtained on elution with pet. ether :
chloroform (1:9) (45 mg) was further purified by preparative TLC,
elution being with pet. ether : chloroform (1:9) to give one major UV
active band. This gave an orange colour with Dragendorffs spray
reagent (30 mg, 8.3x 10-5% yield).
The high resolution mass spectrum afforded the molecular ion
peak at m/z 470.2701, leading to the molecular formula C28H38O6
(calcd. 470.2668) indicating ten double bond equivalents in the
molecule.
The UV spectrum showed an absorption at 225 nm indicative
of an a, p-unsaturated lactone [218].
The IR spectrum the absorption bands for the hydroxyl, and a,
p-unsaturated ketone and lactone groups appeared at 3420, 1725,
1685 cm_1 respectively [214].
159:-

28
CH327
CH2-OH
21 HHsQ 122 26L
tV'Ki'o-ÿoH
t H12o 171319 11
CH3 16,149H 15,
I2ÿÿ110 = 8
OH H H*4
A3H
\a HH
Withaferin-A (306).
The iH-NMR spectrum (CDCI3, 500 MHz) of 306, showed a
double doublet at 6 5.90 (J2,3 = 10.4, J2 4 = 2.3 Hz) for the C-2
proton. Another double doublet resonated at 5 6.48 (J2.3 = 10.4 Hz,
J3,4 = 2.2 Hz) and was assigned to the C-3 methlne proton. The LH-
NMR spectrum also showed three 3H singlets at 8 0.64, 1.30 and 2.12
which were assigned to the C-18, C-19 and the C-28 tertiary methyl
protons respectively. A 3H doublet at 8 1.00 was due to the C-21
secondary methyl protons. A multiplet at 8 4.42 was assigned to the
C-22 methine proton. Other proton chemical shifts are assigned on
Fig. 12.
The COSY 45° spectrum showed that the H-22 at 8 4.42 was
coupled with H-20 methine (8 1.79) and H-23 methylene protons (8
-:160:-

1.69, 2.25). The H-4a (8 3.49) was coupled with H-3 olefinic methine
proton (8 6.48).
&ras27
CH2-OH1.79 dJ21,20=5.0HZ
J2i.20-5.OHz 21 l-I
H3C,0.64 m
CH3
J ABd1.0 d 4.3023 25 4.39
, 1 22 26. J27a.27p=10.4Hz\ ><xÿÿs»J27P,27a=10.4Hz
20 * O O3 H1.35 m .„
18U
11 1.60 m ]j3 *
H jA 1.90 mm /
4.42 m1.43 m
M 1.30 s
CH3
5.90 d
J2.3=10.4Hz
J2.4=2.3Hz 1.601419 9H.
I2ÿ11.29 mJO 5 8
OH H H 2.15 m1.95 m
H\63 2.10mH H4
O6.48 dJ3,2=10.4HzJ3,4ÿ2.2Hz
H 1.40m
H 4.09 d
J0O.7H=5OHZ5.10 br.s
Fig. 3.12. iH-NMR assignments of 306.
The 13C-NMR spectrum showed resonances for all twenty
eight carbons. The lH/13C connectivities of compound 306 were
established on the basis of HMQC experiments. The long-range
Interaction in wlthaferin-A (306) were determined by the
HMBC (Heteronuclear Multiple Bond Connectivity) experiment. In
HMBC spectrum the H-2 proton at (8 5.90) showed interaction with
C-l, C-10, C-3 and C-4 resonating at 8 200.0, 49.5, 143.8 and 69.0
respectively. The H-4 proton at 8 5.10 showed coupling with C-5
quaternary carbon at (8 65.0) as well as C-3, C-6 and C-10 resonating
at 5 143.8, 59.2 and 49.5 respectively.

mr57.0
1155.4 CH2-OH30.1
4.301.79£5H 4.39
%c: 168.2
L 4.421.35
1.43Sÿ26- yo\, .60/ 44ÿ'CH3
21.539.2) 1.90
5.90. 1.6015.0H 56.1 7.5128.< 7
Fig. 3.13. HMBC interactions of Withaferin-A (306).
cif5327 57.0
[5ÿ CH2-OH25ÿ90
30A
13.5 21 JJH3Cÿ
Cfta”22176.0 26U68.2
\ i$9.5= O oH
26.0 18 52.5: H1221.5.
Ts 17\H 16/ 39.2
19 11 44.0
CH3j-f 200.a\l
128xm2 49.5
OH
43.0 56.015.0.|9
27.510 = 29.118H H
i (T 6 'H
Fig. 3.14 13C-NMR assignments of 306.
30.2
H

Another downfteld proton resonating at 8 4.42 assigned for H-
22 showed interaction with C-20, 026 and 023 resonating at 8
39.5, 168.2 and 30.6 respectively. The complete HMBC interaction are
shown in Fig. 3.13.
The long-range connectivities were determined from
the HMBC experiment. The 13C-NMR assignments are shown in Fig.
3.14
21 28
CH3 CH323
22,
O HO19 nH o 9H3 fir
13l t 27 IH. CH2262 fa 16To [9 14
4
OH H o3 5 6
HHH
Fig. 3.IB. Perspective view of withaferine-A (306).
By comparing the spectral data (UV, IR, MS, 13C-
NMR) of (256), the compound was identified as withaferin-A [223].
3.2.4 2,3-Dihydrowithaferin-A (297)
The fraction WF-3 (450 mg, Scheme-3.12) was subjected to
chromatography over silica gel (100 g, 60-120 mesh size). Elution was
carried out with increasing polarities of pet. ether : chloroform. The
fraction "WF-5" obtained on elution with pet. ether : chloroform (2:8)
on evaporation afforded a pure compound as white crystals (10 mg).
-:163:
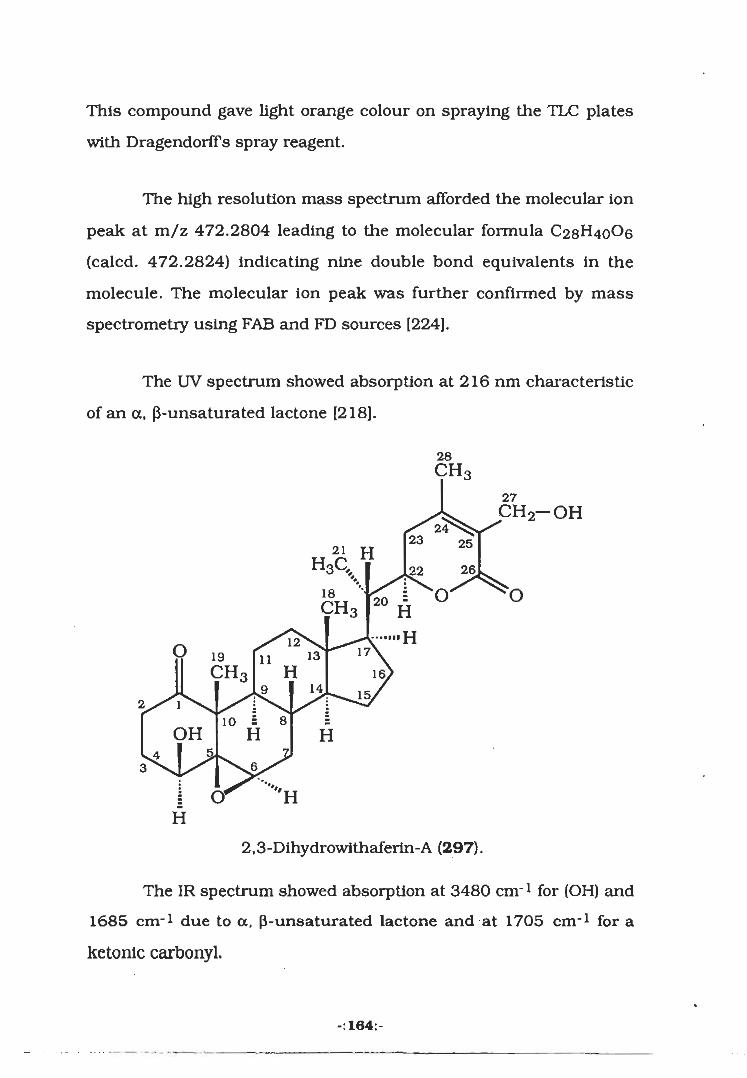
This compound gave light orange colour on spraying the TLC plates
with Dragendorffs spray reagent.
The high resolution mass spectrum afforded the molecular ion
peak at m/z 472.2804 leading to the molecular formula C28H40O6
(calcd. 472.2824) indicating nine double bond equivalents in the
molecule. The molecular ion peak was further confirmed by mass
spectrometry using FAB and FD sources [224].
The UV spectrum showed absorption at 216 nm characteristic
of an a, p-unsaturated lactone [218].
28
CH327
CH2-OH23
21 HH3<\
18
26l22
I20 = O O
CH3 H
IT H12Q 19 171311
CH3 16,149 !5,
2 1
810 =OH H H74
A3
'''/i O' HH
2,3-Dihydrowithaferin-A (297).
The IR spectrum showed absorption at 3480 cm-1 for (OH) and
1685 cm-1 due to a, (i-unsaturated lactone and at 1705 cm-1 for a
ketonlc carbonyl.
-:184:-

28 2.16 s
CH32.35 m
27l.|ÿ m
1.82 d
J2°220=6.7HZ J2i2°H’7HZH3C
CH2-OH4.34 d4.39 d
2?23 25
00 nfi J22a,20p=12.6Hz
\ 12.6Hz
20 - O O1.53 m 1.00 m
H H1.10 m
f CH3• 18 .
HH1.23 m
H 4.42 ddd J22a-2op=3.5Hz
J-J d22a,23P=7- 1
d22a.23o=7•1Hz
112"
0.95 m
o11 1.29 m
1.80CH3 H1.90 m Hf
%1419 9 H15.0.901
o.8oinNH2 810OH H
H 1.53 m0.85 m 75 H2.45 m 1.20m\62.10 mH1 H
H \ O H 1.20 m1.90 m 3.09 br.s
3.41 lJ=3.1Hz
Fig. 3.16 !H-NMR assignments of 297.
The 1H-NMR spectrum (CDCI3, 500 MHz) exhibited three 3H
singlets at 5 0.67, 1.32 and 2.16 which were assigned to the C-18, C-
19 and the C-28 tertiary methyl protons respectively. A doublet at 8
1.02 was due to the C-21 secondary methyl proton. Two AB doublets
at 8 4.34 and 4.39 were due to the C-27 hydroxymethylene protons.
Two multiplets, one at 8 1.85 and the other at 8 2.35, were assigned
to the C-23 methylene protons. A multiplet at 8 4.42 was assigned to
the C-22 methine proton. A triplet at 8 3.41 was due to the C-4
methine proton geminal to a hydroxyl group in ring A. A broad singlet
at 8 3.09 was due to the C-6 methine proton of the oxirane ring.
-:165:-

crir27 55.9
25P5-229.1
2312.9 21 H
H3c„H
22178.5 26LI66.8\ i oO'u.i
cHr 385HM-Hm 17\
20
24.0
20.8ÿ/I2SNNVf 19 fn 42.9
II CHÿ H 16) 38.7212.m} 42.9 553
29.3=i26.925.8 \2 50.5 10 = 8
OH H H31.231.1
O HH
Fig. 3.17 13C-NMR assignments of 297.
The iHÿH connectivities (COSY 45°) showed that H-22 at 8
4.42 was coupled with H-20 methine proton (8 1.82) and with H-23
methylene protons (8 1.85, 2.35). The H-20 methine showed further
couplings with H-21 (8 1.02) and H-22 (8 4.42). Similarly the H-4a (8
3.41) was coupled with H-3a (8 1.90) and H-3p (8 2.45). The iH-NMR
assignments of 297 are shown in Fig. 3.16.
The 13C-NMR spectrum (CDCI3, 125 MHz) exhibited signals
for twenty eight carbons, in agreement with the molecular formula
C28H40O6. There were four methyl, nine methylene, eight methine
and seven quaternary carbons in the molecule. The 13C chemical
shift assignments of 2,3-dihydrowithaferin-A shown on Fig.3.17 were
made with the help of HMQC and HMBC spectra.
-:166:-

21 28CHg CH3
23
<ÿ3 A24\ HOV 27 I
Q,7HO fH3 *
H
13]
H* CH22 26r8 16|9Ho.H/ H O
H[3 5 6
HHH
Fig. 3.18. Perspective view of 2,3-dihydroxywithaferine-A (297).
By comparing the spectral data (UV, 1R, MS, iH-NMR and
13C-NMR) of (297) with those reported in the literature [223] was
identified as 2,3-dihydrowithaferin-A.

Dried aerial parts ofWithania somnifera
(36 kg)
Powdered and extractedwith methanol (70 litres)
Methanolic extract (1.5 kg)
Suspended in distilledwater (4,5 litres)
defatted with pet. ether (10 litres)
Defatted aqueous layer Pet. ether extract(325 g)
Extracted with chloroform
Chloroform extract(154 g)
Aqueous extract
Scheme 3.11.

Chloroform extract of Wilhania somnifera ( 154 (g)
Loaded on to a silica gel column (mesh 70-230 ASTM) (1400 g) andelution with pet. ether : chloroform (0-100%) than with chloroform :methanol (0-100%)
pet. ether : CHCI3 pet. ether : CHCI3 CHC13 : MeOH(9.5:0.5)
CHCI3: MeOHCHCI3(1:9)(2:8) (9:1)
11
WF-1(500 mg)
WF-2(200 mg)
WF-3(450 mg)
WF-5(50 mg)
WF-4(300 mg)
Subjected to preparativeTLC and eluted with
Loaded on smallcolumn and eluted withpet ether: CHCI3 ( l:9)
Loaded on smallcolumn and eluted withpet. ether : CHCI3 (2:8)
Subjected to preparativeTLC and eluted withCHC13: MeOH (9.5:0 5)
Subjected to preparativeTLC and eluted withpet. ether : CHC13 (2:8) +3 drops of diethyl amine
Opet ether : CHCI3+ 2 drops diethyl amineA O
BM(1:9)o>
CD1
CD 2,3-Dihydrowithaferin-A (297) Quresimine-A (296)(10 mg)
Withanolide-A(7.0 mg)
M* 470 (5.0 mg)(7.5 mg)
10
WSF-1(45 mg)
WSF-2(52 mg)
Subjected to preparative
TLC and eluted pet. ether :
CHCI3 (1:9)
Subjected to preparativeTLC and eluted pet. ether :CHCI3 (2:8)
Withaferine-A (306) (30 mg) Withanone (295) (6.5 mg)
1

3.3 Plant Material
The fresh plant (aerial parts) of Withania somnifera (70 kg) was
collected from Karachi (Pakistan), identified by the plant taxonomist
of Botany Department, University of Karachi and dried in the
absence of sun light. The aerial parts of the plant Withania somnifera(36 kg) were powdered and soaked in methanol (70 liter).
Extraction and Purification3.3.1
The methanolic extract was evaporated to afford a gum (1.5
kg). This gum was partitioned between distilled water and (4.5 liter)
pet. ether (10 liter). The aqueous extract was again extracted with
chloroform (7 liter). This chloroform extract was evaporated and then
loaded on a silica gel (mesh 70-230 ASTM) column, Scheme-3.11.
Isolation of Withanone (295)3.3.2
The chloroform extract (154 gm) was loaded on a silica gel
column and the column eluted with increasing polarities of pet. ether
: chloroform. On elution with pet. ether : chloroform (2:8) fraction
WF-1 (500 mg) was obtained. The WF-1 fraction exhibited several
spots on TLC. This fraction (500 mg) was then subjected to
chromatography on a small column (65 gm) elution being with pet.
ether : chloroform (1:9). Two major fractions obtained from this
column were WSF-1 and WSF-2. Fraction WSF-1 was demonstrated
to contain withaferin-A (30 mg, 8.0 x 10-5 yield) after spectroscopic
techniques.

The second fraction WSF-2 (52 mg) also showed one major
compound along with some minor compounds . This fraction
subjected to preparative TLC, elution being carried out with pet. ether
: chloroform (2:8), to afford withanone 295, 6.5 mg, yield 1.8 x 10-?%(Scheme-3.12).
was
Spectral Data
[oc]25D: +17° (c = 0.0892. CHC13)
UV (MeOH), nm (log e): Xmax 215 nm characteristic for cc.p-
unsaturated lactone.
(IR (CHCI3): vmax cm-1: 3440 (O-H). 1690 (a.(J- unsaturated lactone)
and.1615 (C=C).
EIMS m/z: (rel.int.%): 456 (82), 438 (15), 327 (26), 288 (35), 286 (10),
269 (36). 267 (30). 226 (100). 213 (47). 167 (10). 159 (30). 141 (31),
138 (15) and 95 (42).
HREIMS m/z: (rel. int. %): 456.2801 (C28H40O5. calcd. 456.2875)
(15). 438.2790 (100). (C28H3804. calcd. 438.276ÿ 288.2086 (20).
(C19H28O2, calcd. 288.2089 (28), 167.0705 (C9HUO3. calcd.
176.0708). 141.0546 (8)/ (C7H8O3, calcd. 141.0551).
FAB +ve: 457.2879.
iH-NMR (400 MHz. CDCI3) 8: See Fig. 3.1.
13C-NMR (CDCI3. 125 MHz) 8: See Fig. 3.5.
COSY-450 : See Fig. 3,3.
HOHAHA: See Fig. 3.2.
HMBC: See Fig. 3.4.
HMQC: See Table-3.1.

Isolation of Quxesimine-A (296)3.3.3
The chloroform soluble extract (154 gm) (Scheme-3.12) was
loaded on a silica gel (mesh 70-230 ASTM) column (1400 gm) which
was eluted first with chloroform and then with mixtures of methanol
: chloroform. The fraction WF-4 (300 mg) was thus obtained on
elution with chloroform : methanol (9.5:0.5). The fraction WF-4 was
then subjected to preparative TLC, the plates being eluted with pet.
ether : chloroform (2:8) + 3 drops of diethylamine to afford 296 (7.5
mg, yield 2.1 x 10_7%) as a colourless powder.
Spectral Data
[al25D = + 17° (c = 0.075, CHC13)
UV (MeOH), nm (log e) Xmax: 214 (3.45).
IR (CHCI3) vmax cm'1: 3455 (O-H), 1683 (a, p-unsaturated lactone)
and 1590 (C=C).
EIMS m/z (rel. int. %): 502.2910 (M+. 10), 484 (15), 452 (22), 434
(13), 347 (22). 311 (16). 281 (15), 241 (20), 213 (21), 197 (42) and 141
(100).
HREIMS m/z (rel. int. %): 502.2910 (M+,5),484.2804 (70), 470.2667
(20),452.2530 (12), 387.2522 (15), 334.1979 (25), 234.2795 (17),
264.3011(15),169.0810 (70),170.1932 (10) 141.571 (100) and 86.104
(18).
1H-NMR (CDCI3. 300 MHz) 5: See Fig. 3.7.

13C-NMR (CDC13I 125 MHz) 8: See Fig. 3.9.
COSY-45°: See Fig. 3.10.
HOHAHA: See Fig. 3.11.
HMBC: See Fig. 3.8.
HMQC: See Table-3.2.
Isolation of Withaferine-A (306)3.3.4
The chloroform extract (154 gm) was loaded on a silica gel
column and eluted with pet. ether : chloroform (2:8) on elution at
this polarity a fraction WF-1 was obtained (500 mg). This fraction
was subjected to chromatography on a small column packed with
silica gel (50 gm) and eluted with pet.ether : chloroform (1:9) to afford
another fraction WSF-1 (45 mg) which showed several minor
compounds along with a major UV active band on TLC. This fraction
was subjected to preparative TLC using pet.ether : chloroform (1:9) as
eluent to afford a pure compound (Withaferine-Ay30 mg) (8.3 x 10'5,
yield).
Spectral Data
[a]20D + 35o (c = 1.3 CHCI3)
UV (MeOH), nm (log e) Xmax‘ 225 (3.363).
-:173:-

IR (CHCI3) vmax cm'1: 3420, 1725 and 1685.
EIMS: m/z (rel. int. %): 470 [M+ (10)], 452 (10). 347 (15), 299 (8),
124.0 (95) and 95 (100).
HREIMS m/z (rel. int. %): 470.2701 (M+ 21), 452.2306 (15),95.1062(100).
1H-NMR (CDCI3, 500 MHz) 6: See Fig. 3.12.
13C-NMR (CDCI3, 100 MHz) 5: See Fig. 3.14.
Isolation of 2,3-Dihydrowithaferine-A 2973.3.5
The methanolic extract of Withania somnifera (1.5 kg)
suspended in water and defatted with pet. ether. This defatted
aqueous extract was then extract with chloroform after evaporation
of chloroform under vaccum condition afforded (154 g) dry chloroform
extract. This chloroform extract subjected to silica gel column (1400
g) using (mesh: 70-230 ASTM) and eluted with pet.ether : chloroform
and then chloroform : methanol with increasing polarity at pure
chloroform. The fraction WF-3 (450 mg) obtained UV active
compounds on TLC.
This fraction was again subjected to pencil column and eluted
with pet.ether : chloroform, which afforded a pure compound on
elution with pet.ether : chloroform (2:8) which give orange colour
with Dragendorff s spray.
Spectral Data
[a]25D +89° (c = 0.98 CHCI3)
-:174:-

TR (CHCI3) Vmax cm*1: 1705, 1685 and 3480.
EIMS m/z (rel. lnt. %): 472 [M+ (5)], 454 (54), 3474 (10). 283 (20), 197
(33). 141 (100) and 95 (65).
1H-NMR (CDC13, 400 MHz) 5: See Fig. 3.16.
13C-NMR (CDCI3, 125 MHz) 5: See Fig. 3,17.
-:176:-

SECTION D
ISOLATION AND STRUCTUREELUCIDATION OF
FROM FERULA OOPODA

INTRODUCTION4.0
Ferula oopoda belongs to the plant family Umbelliferae. It Is a
perennial herb distributed from the Mediterranean region to Central
Asia. This genus Ferula consists about 140 species [226J. Only 15
species have been identified in Pakistan [227]. These species are:
Ferula assafoetida, F. baluchistanica, F. communis, F. costata, F,
hindukushensis, F. kokonica, F. lehmanii, F. microlabe, F. narthex, F.
oopoda, Fovina, F. reppiea, F. rubicaulis and F. stewartiana. Some of
the species are important as sources of oleogum and are used in
indigenous medicine [228].
Some species of genus Ferula are extensively used in India for
flavouring the curries, sauces and pickles. They stimulate the
intestinal and respiratory tracts and the nervous system. They are
useful in asthma, whooping cough, chronic bronchitis and intestinal
flatulence. Some species are administered in hysteria, epileptic
affections, cholera and are often employed in veterinary medicine
[229-231].
The ethanolic extracts of Ferula sinaica roots inhibited the
spontaneous movements of rat and guinea pig uterine smooth
muscles and also the contractions induced by oxytocin stimulation.
These data suggest that the plant extract has some antioxytocic
potential [232]. The aqueous extract of Ferula ovina showed
anticholinergic and antihistaminic antispasmodic effects [233].
176:-

The essential oils of Ferula narthex, F. ovina and F. oopoda
when tested in liquid media against standard cultures of
Staphylococcus aureus, Escherichia coli, Salmonella typhi, Shigella
dysenteriae and vibrio cholerae showed good inhibitory activity. The
oils of Ferula narthex and F. ovina were more active against
Staphylococcus aureus, while the growth of the pathogens of dysentery
and cholera was inhibited by F. oopoda oil [234], Many sesquiterpene
lactones have been reported from F. oopoda. [235].
4.1 Biosynthesis of Sesquiterpenes
The biosynthesis of sesquiterpene can be divided into two
major parts. The first part is the biosynthesis of the acyclic precursor
(FPP) while second part is the cyclization of the acyclic precursor into
the sesquiterpenoidal skeleton [236,237].
Biosynthesis of Acyclic Precursor (FPP)
J.W. Comforth, in his work on the biosynthesis of steroids,
characterized two active forms of isoprene i.e., isopentenyl
pyrophosphate (IPP, 267) and dimethylallyl pyrophosphate (DMAPP,
268). These intermediates are obligatory for the synthesis of plant
terpenes.
The incorporation of these intermediates into terpenes is
catalyzed by various enzymes. The biosynthesis of the acyclic
precursor is presented in Scheme-4.1. Two molecule of acetyl co¬
enzyme A (259, derived from carbohydrate, fat or protein catabolism),
condense to yield acetoacetyl co-enzyme-A (260). This acetoacetyl co-
-:177:-

enzyme A further condenses with another molecule of 259 by aldol
type reaction to form 3-hydroxy-3-methyl gluteryl co-enzyme A (260).
H+o
oJ oAcetoacetyl-CoAthlolase
C— S.CoAM(
( o — Cr C— S.CoA
O 260©
©H2C— C— S.CoAH2G— C—S.CoA
Acetyl-coenzyme A (259)Acetyl-coenzyme A (259)
O
H2CS.CoA
3-Hydroxy-3-methyl glutaryl-S.CoAHMG-S.CoA (261)
NADPH
NADP+
HOO CH2OH
3R-Mevalonlc acid (264)
3 ATP
OOH
CH2OPPH— O
265
-co2H+
),CH2OPP- HR .CH2OPP
HMeHS
IsopentenylpyrophosphateaPP) (267)
8cheme-4.1. Biosynthesis of sesquiterpenes.
Dlmethylallylpyrophosphate(DMAPP) (268)
-:178:-

3-Hydroxy-3-methyl glutaryl co-enzyme A (261) Is then
irreversibly reduced through the intervention of NADPH to 3R-
mevalonic acid (264) . Only the R form of mevalonic acid is utilized
by organisms for producing terpenes, while the "S" form Is
metabollcally inert.
Phosphorylation of mevalonic acid (264) by ATP (adenosine
triphosphate) leads to mevalonic acid S-pyrophosphate (266). After
decarboxylation, it affords IPP (267).
Me ( OPP OPP
+
Me'HR(DMAPP) (268) HS(IPP) (267)
MeMe OPP Me OPP
HR V
+
Me'
HS(GPP) (270)(IPP) (267)
Me Me Me OPP
(FPP) (271)
OPPOPP
cis trans
272 273
Scheme-4.2.
-:179:-

IPP is then converted by an enzyme-catalyzed prototropy into
an equilibrium with dimethyl allyl pyrophosphate (DMAPP, 268) in
which the latter predominates.
These two intermediates IPP (267) and DMAPP (268) condense
in head-to-tail manner to produced geranyl pyrophosphate, GPP
(270), which then condenses with IPP (267) to afford famesyl
pyrophosphate (FPP, 271). This is the key acyclic precursor in the
biosynthesis of sesquiterpenes (Scheme-4.2).
The carbon skeleton of almost all the known sesquiterpenes
can be derived from trans-famesyl pyrophosphate (273) and the cis
famesyl pyrophosphate (FPP, 272), through appropriate cyclization
and rearrangements. The cfe-famesyl pyrophosphate (272) should be
derived from trans-famesyl pyrophosphate (273) via a reversible
mechanism.
y-Bisabolene (302) is considered to be the precursor of a
number of sesquiterpene systems. (Scheme-4.3).
-:180:-
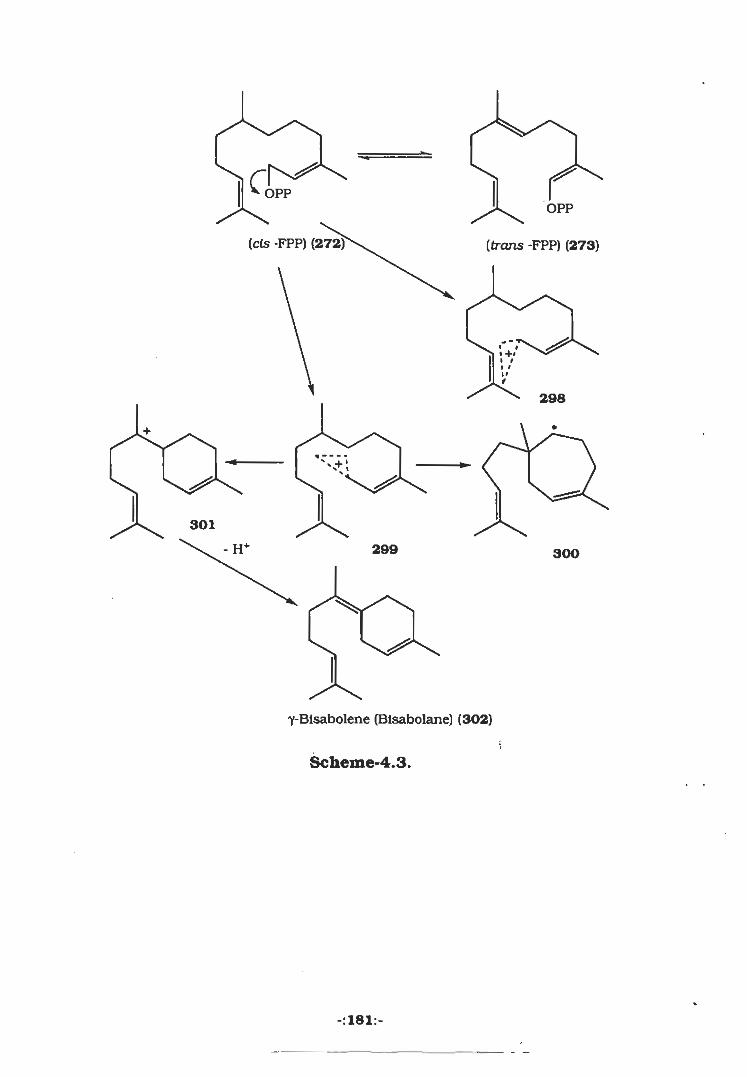
cOPPOPP
(c(s -FPP) (272) (irons -FPP) (273)
+<'
298
+
301
-H* 299 300
7-Blsabolene (Blsabolane) (302)
Scheme-4.3.

4.2 RESULTS AND DISCUSSION
New Compound from Aerial parts of Ferula oopoda
Feraileate (303)4.2.1
A new compound, feraileate (303), was isolated (fraction F0-
4, Scheme-4.6, experimental section) from the ethanolic extracts of
Ferula oopoda collected from the Quetta district, Pakistan. The
fraction FO-4 (40 mg) (experimental section, Scheme-4.6) was
further purified by preparative TLC on silica gel (GF-254, 0.2 mm)
using pet. ether : acetone (7:3) as the developing solvent. This
afforded a compound which gave a pink colour with Dragendorffs
reagent and dark brown colour with ceric sulphate reagent.
The UV (MeOH) spectrum of feraileate (303) was typical for
aromatic systems [242] showing absorption at 225 (log e = 3.05), 299
(log e = 2.19) and 303 (log e = 3.40) nm.
The IR spectrum showed an absorption at 1690 cm-1 which
indicated the presence of an a, p-unsaturated ester carbonyl group
(238]. Other intense IR absorptions were at 1120 cm*1 (C-O-C), 1600
(C=C) and 2900 (C-H) cm1.
The high resolution electron impact mass spectrum showed
the molecular ion peak at m/z 306.1103, corresponding to the
molecular formula, Ci6His06. indicating eight degrees of
unsaturation in the molecule. The mass fragmentation pattern of
feraileate (303) is presented in Scheme-4.4. The fragment "a" at m/z
i-:182:~

151.021 of composition C8H7O3 resulted from the cleavage of the
CO bond, indicating that the acyclic ketone moiety was probably
attached to C-l of the aromatic skeleton. The fragment "b" at m/z
179.029, C9H7O4 further suggested that the ketone group is directly
attached to the C-l aromatic carbon. The peak at m/z 55.054 (C4H7)
indicated that butene group is present in the molecule (Scheme-4.4).
The spectrum (CDCI3, 400 MHz) revealed the
presence of four methyl groups in the molecule i.e., a 3H doublet at 8
1.54 (J = 7.0 Hz) for the C-3' methyl protons and two methyl doublet
at 8 2.0 (J = 3.1 Hz) and 8 2.1 (J = 3.1 Hz) for C-4" and C-5"
respectively. A singlet at 8 3.9 was assigned to the methoxy protons.
Two downfleld protons resonating at 8 7.1 d (J = 1.5 Hz), and 8 7.3 (J
= 1.5 Hz) were due to C-6H and C-2H respectively. The
spectrum showed that the benzene ring is tetra substituted. The "J"
values of C-6H and C-2H suggested that these proton .were meta (or
possibly para) to each other. The C-3" methine proton resonating at 8
6.1 as a multiplet indicated the presence of a double bond. A 2H
singlets at 8 6.0 was assigned to the methylenedioxy protons. The C-
2' methine proton appeared as a quartet at 8 5.9 (7.0 Hz) establishing
that the methine proton has a vicinal methyl group. The LH-NMR
assignments of feraileate (303) are given in Fig. 4.1.
-:183:-

:
7.1 d. J=1.5Hz
H o1.54 d. J=7.0Hz
>CH3o. 1* 3'2 —•
"'H6.0 sA O 5.9 q. J=7.0Hz
O 4
1 7.3 d.H 1 1
I J=1.5Hz
OCH34"
CH3 2.0 d,J J=. -3-9SH3C 3.1Hz
2.0 d,J = 3.1Hz
H6.1 m
Fig. 4.1. iH-NMR chemical shifts of feraileate (303).
The COSY-450 spectrum of feraileate (303) exhibited coupling
interactions which were found to be in full agreement with the
assigned structure see Fig. 4.2.
H O
17.3102.9 195.0 CH3o 71.0.
1 r 3'
J!129.1 2i'%140.0
H102.4143.0 109.4
1o 4
149.0H 1670
HsC V
20.456.7
15.8
H
Fig. 4.2. 13C-NMR chemical shifts of feraileate (303).
The Me-3’ protons at 5 1.54 showed interactions with the H-2'
methine proton at 5 5.9. The Me-4” protons at 5 2.0 showed coupling
with H-3” the methine proton at 8 6.1. The H-3” methine proton was
;
-:184:-
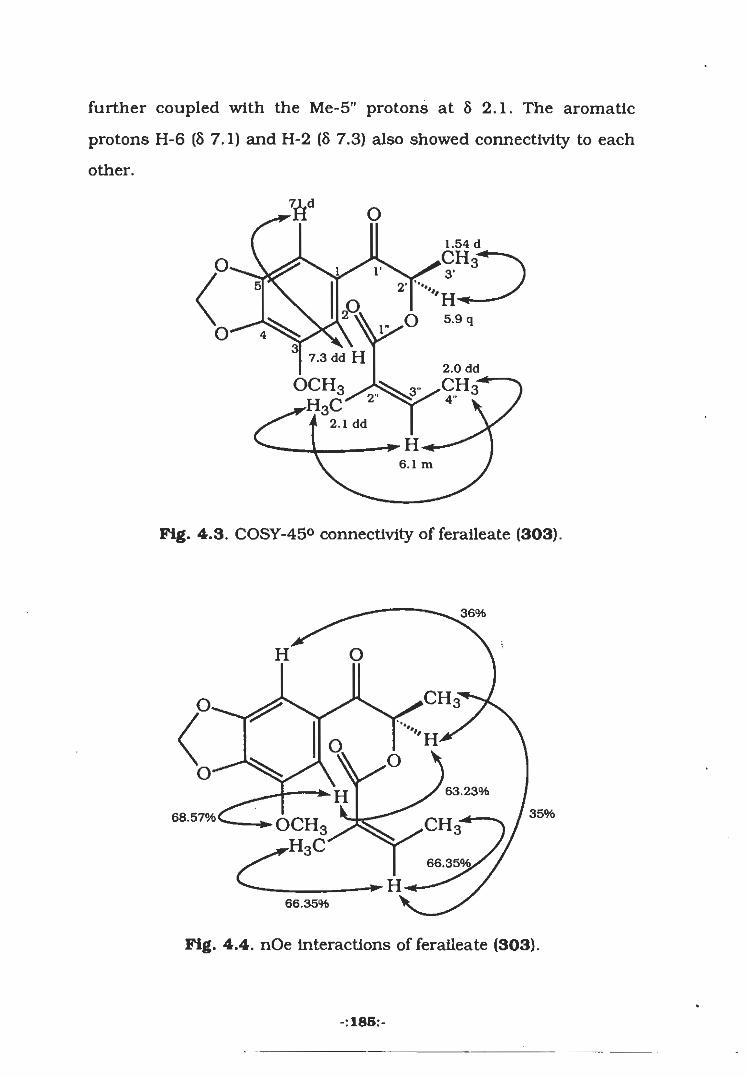
further coupled with the Me-5" protons at 8 2.1. The aromatic
protons H-6 (8 7.1) and H-2 (8 7.3) also showed connectivity to each
other.
%d o1.54 d
CH3o 1' 3'
< T'"-Hr O 5-9 q
5
o 4
7.3 dd H2.0 dd
OCH3H3C
<r 2.1 dd
6.1 m
Fig. 4.3. COSY-450 connectivity of feralleate (303).
36%
H O
O
< Ho4.O
O63.23%H
35%68.57%*OCH3 .CH3-
H3C66.3!
66.35%
Fig. 4.4. nOe Interactions of feralleate (303).
-:185:-

o;+o3 IIIo
< I oQ < Ioo
OCH30CH3
m/z 179m/z 306
+
<co
o.
o o< I (not observed)O
OCH3
m/z 151
Scheme-4.4. Mass fragmentation of feraileate (303).
!
*

oH
1.54102.91 195.0 71.0ÿCH
67.0,0 59
o6.0 02.4
129.1140.0
o.J43.0l 109.4
o149.0
20.456.7
OCH3ÿ
15.8
H 6.1
Fig. 4.5. HMBC interactions of feraileate (303).
The 13C-NMR spectra (CDC13, 100 MHz, DEPT) indicated that
there were four methyl, one methylene and four methine carbons in
the molecule. The 13C-NMR spectra (B.B) showed sixteen carbon
signals of these sixteen carbon atoms, nine appeared in DEPT spectra
so that there were seven quaternary carbons.
The downfleld signals at 8 167.0 and 195.0 were assigned to
the a, (5-unsaturated carbonyl carbon (C-l"), and the ketonic carbon
(C-T) respectively. Other downfield carbons resonated at 8 149 (C).
109.4 (CH), 129.1 (C), 102.9 (CH), 140 (C), 143 (C) and were assigned
to C-3, C-2, C-l, C-6, C-5 and C-4 respectively.
The methylene carbon at 8 102.4 was to the methylenedioxy
carbon. Other carbon atoms at 8 139 (CH), 127 (C), 71.0 (CH) were
assigned to C-3", C-2" and C-2' respectively.
-:187:-

The chemical shift assignments of various carbons of
feraileate (303) are presented in Fig. 4.2.
The one-bond !H/13C correlations for feraileate (303) were
determined on the basis of HMQC experiments (Table-4.1). The
vinylic C-3" (139.0) was directly correlated with the proton that
resonated at 6 6.1. The methine C-2' (8 71.0). showed a cross-peak
with H-2 at 8 5.9. The C-2 and C-6 carbons resonating at (8 109.4)
and (8 102.9) showed cross-peaks with the protons at 8 7.3 and 8 7.1
(H-2 and H-6 respectively).
The long-range *H/13C interactions in feraileate (303) were
determined by the HMBC experiment see Fig. 4.5. The signal at 8 6.1
(H-3”) showed couplings with C-2" quaternary carbon and C-4” (8 2.0)
methyl carbon. H-2’ (8 5.9) showed coupling with the C-1’ quaternary
carbon (8 195) and with the C-3' methyl carbon at (8 1.54). The H-6 (8
7.1) showed connectivity with C-1 (8 129.0), C-1’ (8 195.0) and C-5 (8
140).
The structure of feraileate (303) was further confirmed by nOe
experiment. nOe difference measurement established the relative
stereochemistry at C-2’ asymmetric centers and positions of the
functionalities in the molecule. Irradiation at 8 5.9 resulted in
63.25% nOe of the C-2 proton and 36% nOe of the C-6 proton.
Irradiation at C-2H caused a 68.57% reciprocal nOe on the C-3
methoxy (8 3.9) proton. This indicated the methoxy group is adjacent
to C-2 proton (i.e., C-3 position). Irradiation at C-3" proton showed
-:188:-

nOe 66.35% with C-5" and C-4” methyl groups. Detailed nOe
measurements are summarized around Fig. 4.4.
Table-4.1. HMQC Assignments of Feraileate (303)
Carbon No. 6lH 8 13C
129.1C-l
7.3 d (J= 1.5 Hz) 109.4C-2
C-3 149.0
C-4 143.0
C-5 140.0
7.1 d (J= 1.5 Hz) 102.9C-6
195.0C-l'
5.9 q (J = 7.0 Hz) 71.0C-2'
1.54 d (J = 7.0 Hz)C-3’ 17.3
C-l” 167.0
C-2" 127.0
6.1 mC-3" 139.0
C-4" 2.0 d, (J= 3.1Hz) 20.4
C-5" 2.1 d (J = 3.1 Hz) 15.8
O-CH2-O 6.0 s 102.4
3.9 sOCH3 56.7
-:189:-:

REPORTED SESQUITERPENE LACTONE ISOLATED
PROM FERULA OOPODA
Guaianolide (304)4.2.2
The pet. ether extract obtained by extraction of the ethanolic
extract of Ferula oopoda (Scheme - 4.6, experimental section), was
loaded on a silica gel column (70-230 mesh, ASTM), which was eluted
with increasing polarities of pet. ether : acetone. The fraction "FO-3"
obtained on elution with pet. ether : acetone (7:3) 48 mg contained
three compounds. The mixture was subjected to preparative TLC on
silica gel precoated plates which were developed in pet. ether :
acetone (9:1), this afforded one major compound which gave a dark
pink colour with ceric sulphate reagent after heating 120°C and was
identified as guaianolide (304), previously as isolated from Ferula
Arrtgonit bocchieri [239].
14
H2C
10 9'21
8]
n'•in," 13
CH3
oFig. 4.6. Structure of guaianolide (304).
-:10O:-

The UV absorption of gualanolide (304) at Xmax 254 (log e
2.516) indicated an a, p-unsaturated carbonyl chromophore [218].
The high resolution mass spectrum afforded the molecular ion
peak at m/z 230.1305 leading to the molecular formula C15H18O2
and indicating seven double bond equivalents in the molecule. The
molecular ion peak was further confirmed by mass spectrometery
using FAB and FD sources [240, 241].
The peak at m/z 215. 1072 (C14H15O2) corresponded to the
loss of "CH3" from the M+ ion. Other prominent peaks in HREIMS
were at m/z 185, 151 and 120. The mass fragmentation pattern of the
compound (304) was characteristic of sesquiterpene lactone type
compound [235].
The IR spectrum displayed an absorptions at 1740 cm'1
attributable to a conjugated y-lactone. The presence of a cabon -
carbon double bond was indicated by an absorption band at 1645
cm'1 [242].
The spectrum {CDCI3, 400 MHz) of compound (304)
showed only one terminal methylene group with the methylene
protons resonating at 8 4.81 and 4.89 as a br. singlets. Another
downfield proton resonated 8 4.57 as a multiplets was assigned to H-
6a. A three-proton singlet at 8 1.69 was assigned to the allylic methyl
group. The resonance at 8 5.50 is assigned to H-3 in trisubstituted
double bond, on the basis of its coupling with the methyl group at C-
15 (8 1.79 br. s) and with a complex group of signals attributable to
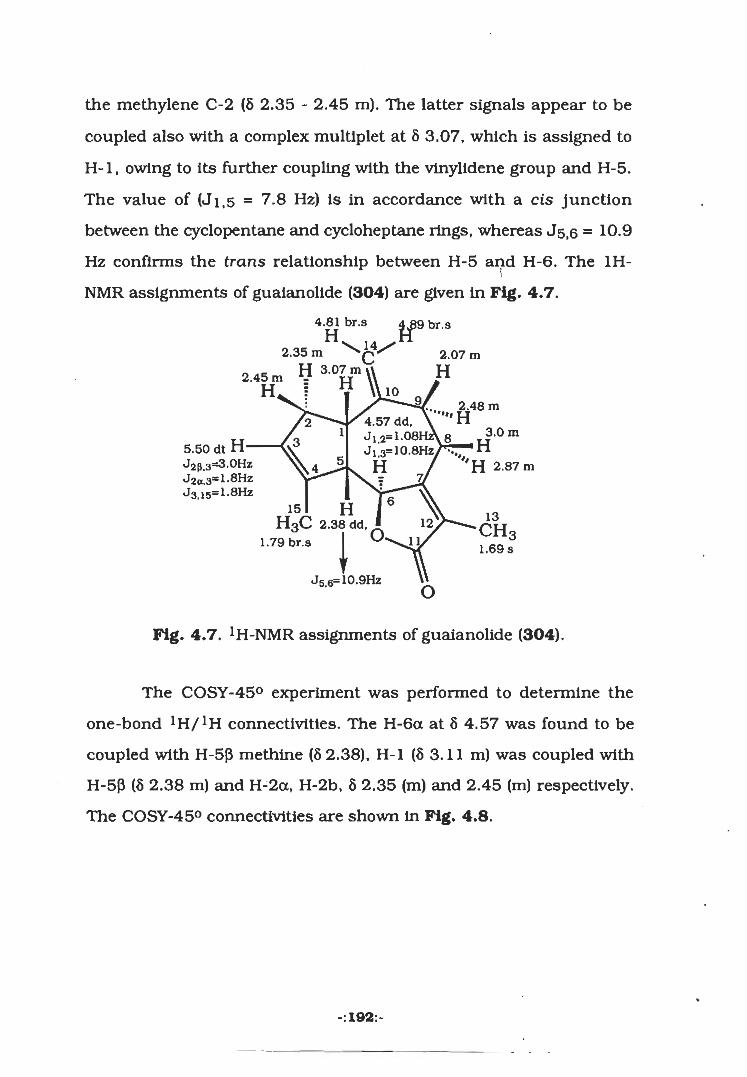
the methylene C-2 (8 2.35 - 2.45 m). The latter signals appear to be
coupled also with a complex multlplet at 5 3.07, which is assigned to
H-l, owing to its further coupling with the vinylidene group and H-5.
The value of (Ji,5 = 7.8 Hz) is in accordance with a cis Junctionbetween the cyclopentane and cycloheptane rings, whereas J5,6 = 10.9
Hz confirms the trans relationship between H-5 and H-6. The 1H-
NMR assignments of guaianolide (304) Eire given in Fig. 4.7.
4.81 br.s 9 br.sH
2.35 m
H 3.07 m « HH \\ 10 _f
K fl
2.07 m
2.45 m
?2.48 m:
4.57 dd,
J12=1.08HZ\8 TT
Jl>3=10.8Hzrÿ7H
'23.0 m1
5.50 dt H-
J3,15=1-8Hz
H H 2.87 m111
6 \!2>15
13H3C 2.38 dd*1.79 br.s |° CH3
1.69s1
J5.6=10.9Hz o
Fig. 4.7. assignments of guaianolide (304).
The COSY-450 experiment was performed to determine the
one-bond connectivities. The H-6a at 8 4.57 was found to be
coupled with H-5fJ methine (8 2.38). H-l (8 3.11 m) was coupled with
H-5P (8 2.38 m) and H-2a, H-2b, 8 2.35 (m) smd 2.45 (m) respectively.
The COSY-450 connectivities Eire shown in Fig. 4.8.
-:192:~

4.81 4.89H H
2.35 2.07C
if w\\ H2.45
H2.48
V H4.573.0
H HHV5.50 XH 2.87
\H3C 2.38CH3o.1.79 1.69
o
Fig. 4.8. COSY-450 interactions of guaianolide (304).
The 13c-NMR spectrum (CDC13, 100 MHz) exhibited all fifteen
carbons, which were in complete agreement with the molecular
formula C15H18O2. There were two methyl, four methylene and four
methine carbons (by difference from the DEPT spectrum) and five
quaternary carbons in the molecule. The C-l1 carbonyl appeared at 8
174.0. The two tertiary methyl carbons resonating at 8 8.4 and 18.36
were assigned to C-13 and C-l5 respectively. The downfield carbons
appeared at 8 122, 162, 142, 126.8 and 149.5 which were assigned to
C-12, C-7, C-4, C-3 and C-10 carbons respectively. The 13C-NMR
assignments of guaianolide are presented in Fig. 4.9.
By comparison of the spectra data (UV, IR, MS, !H-NMR, 13C-
NMR) with those reported earlier the compound was identified as
guaianolide (304) [239],
-:193-

H H
'“'112.6
\' 149.534.7 31.11° 9*r2
49.51
H 8) 30.3126.8' 53.7
83.07/162.0142.0
6 \12 >
15 122.013H3C CH3°ÿu
174.0V
18.08.4
o
rig. 4.9. 13C-NMR assignment of guaianolide (304).
Isolation of Grilactone (305)4.2.3
The pet. ether soluble fraction was subjected to silica gel
column using pet. ether : acetone as solvent. The fraction "FO-l" was
obtained in pet. ether : acetone (9:1, Scheme-4.6, Experimental) was
further chromatographed by preparative TLC in pet. ether acetone
(9.5:0.5). This afforded a sesquiterpene lactone. It gave a
characteristic red coloured reaction with ceric sulphate solution and
a yellow colour with Dragendorffs reagent. The sesquiterpene lactone
was identified grilactone after spectroscopic studies [243, 244].
The UV spectrum (MeOH) of the compound (305) showed only
terminal absorption. The IR spectrum indicated the presence of a
flve-membered y-lactone with absorption at 1730 cm1.
-:194:-

]HU 14
CwH10 9>
21
8H
V613
nVj......CH3A i15CH3 =0ÿ12 H
O
Fig. 4.10. Structure of grllactone (305).
The high resolution mass spectrum of compound showed the
molecular ion at m/z 232.1442 corresponding to the molecular
formula C15H20O2 (calcd. 232.1463) indicating six degrees of
unsaturation. A peak at m/z 217 was due to the loss of a methyl
group from the molecular ion. A large peak at m/z 204.1140 was
ascribed the loss of an ethylene fragment from the molecular ion.
Another peak at m/z 188 represented the loss of CO2 (m.u. 44) from
the M+ ion while the peak at m/z 158 resulted from the loss of
lactone ring along with further loss of two protons. The peak at m/z
105.0721 was due to the loss of a fragment C5H7O2 from the M+-
C2H4 ion. Other major peaks were at m/z 176, 119, 91 and 79.
The iH-NMR spectrum (CDCI3, 400 MHz) of compound (305)
showed two methyl signals 5 1.14 d (J = 6.85 Hz) and 8 1.83 d (J =0.6 Hz) for the Me-13 and M-15 protons respectively. The downfield
proton at 8 4.32 dd (J©p,5a = 11.5 Hz, Jep.7p =* 6.5 Hz) for the oxygen

bearing proton H-6 [245, 246]. Other downfteld protons resonated at
8 4.81 (br. s), 4.82 (br. s) and 5 5.45 which were assigned to H-14a,
H-14P and H-3 respectively. The *H-NMR assignments are shown in
Fig. 4.11.
Jla,20=®-3HzJla,5a=®-6Hz 4.81 br.s
4.82 br.s H14/HC2.44 m
H 3.06 dt.u
¥ VH 2.41 m
H,HI/10 ?
2.72 m \ T T
4ÿ2ddH sWH2 36p.5a=l 1-5 Hz
Jep.7p=6.5Hz1
%H 1.87 mN,
H5.45 br peak
7\\ 13 1.14 d. J=6.85Hz
i IV..-'"CH315 1 2.
CH3 £
12 H1.83 d. J=0.6Hz2.70 m
J5o.6p=ll-0Hz O35a.ia=8.8Hz
Fig. 4.11. iH-NMR assignments of grilactone (305).
The 13C-NMR spectrum (CDCI3, 100 MHz) showed a signals at
5 179.30 due to the carbonyl carbon. The two signals at 5 125.44 and
142.40 were due to the C-3 and C-4 carbons. The terminal methylenic
carbon atom appeared at 5 111.90. The 13C-NMR multiplicity
assignments were made by DEPT and GASPE experiment [181, 182].
The 13C-NMR assignments are shown in Fig. 4.12. The
stereochemistry at Cl, C-5, C-6 and C-7 was established by X-ray
crystallography (Fig. 4.13).
198:-i

AV'“H
147.634.5 33.710 9’r2
1 47.6125.4
H H% 7 19 5H-
1 38.01442.4] 6
'ÿH3H rÿcnr°ÿ12179.3 V
H17.3
O
Fig. 4.12. 13C-NMR assignments of grtiactone (305).
C(14)
C(3)
'CI4)
C(2)
,0(1)©cm C(\C(6! ,0(2)©t(12)
C(10) CI7)C(11)
:(15) C(l
30-0fC(9)
C(13)
Fig. 4.13. X-ray strcuture of grilactone (305).
-:197:-

i
4.3 Plant Material
The plant material (fresh aerial parts of Ferula oopoda) (15.0
kg) was collected from Quetta province, Pakistan in the month of
July 1994. The plant was identified by the plant taxonomist of
Botany Department of the Baluchistan University, Quetta, Pakistan.
Extraction and Purification4.3.1
The fresh plant material was dried in the absence of sunlight
of Ferula oopoda (9.5 kg). This dry plant material was crushed,
powdered, soaked in ethanol (25 litters) for one week, and filtered.
The filtrates were concentrated on a rotary evaporator under reduced
pressure at 45°C, which gave a crude gum (408 g).
The crude gum was suspended in water and extracted with
pet. ether, and the petroleum ether layer evaporated to dryness. The
resulting material (154 g) was found to contain terpenes and
designated as "FO” (Scheme-4.5). The resulting gummy material (154
g) was subjected to chromatography on a silica gel (70-230 mesh
ASTM) column (1.5 kg) subsequent TLC experiments afforded pure
compounds. The detailed fraction procedure is summarized in
Scheme-4.6.
The remaining aqueous layer was then extracted with
chloroform. The chloroform extract was evaporated to dryness on a
rotary evaporator to afford a gummy material (15.0 g) which was
found to contain some minor compounds on TLC.
-:108:-

Ferula oopoda<Vr(95kg)
Powdered and extracted withethanol (25 litres) and evaporatedunder vaccuum
Ethanollc extract (390 g)
Add distilled water
Aqueous extract
Extracted with pet. ether
Pet. ether extract (FO)(154 g)
Defatted aqueous extract
Extracted with chloroform
Chloroform extract(15.0 g)
Aqueous extract
Scheme-4.5.

Pet. ether extract of Ferula oopoda(154 g) "FO"
Loaded on to a silica gel column(mesh 70-230 ASTM) (1.5 g) andeluted with pet. ether : acetone(0-100%) then washed with methanol
Pet. ether :acetone(9:1)
Pet. ether :acetone(9:1)
Pet. ether :acetone(9:1)
Pet. ether :acetone(9:1)
FO-2FO-1(5.0 g)
FO-3(48 mg)
FO-4(40 mg)
Scheme-4.6.
4.3.2 Isolation of Feraileate (303)
The pet.ether extract (FO) 154 g was loaded on silica gel
column (1.5 kg) and was eluted with pet.ether : acetone with
increasing polarity. The fraction FO-4 (40 mg) obtained from this
column at pet. ether : acetone (7:3). The fraction FO-4 was than
subjected to preparative TLC using pet. ether : acetone (7:3) as the
developing solvent afford the pure compound feraileate (304) 18 mg,
1.8 x 10'6%). It gives blue colour with ceric sulfate reagent after
heating upto 120°C.
Spectra Data
[ot]25D = 40° (c = 0.002, CHC13)
UV (MeOH) nm (log e) Xmax: 225 (3.05), 299(2.19) and 303 (3.40).
m(CHCl3) Vmaxcm-l: 2900, 1690, 1600 and 1120.
-:200:-

EIMS m/z (rel. int. %): 306 (M+, 14), 179 (100), 151 (8). 83 (18) and
55 (14)HREIMS m/z (rel. int. %): 306.1103 (M+15), 179.029 (100), 151.021 (10)
and 55.054 (15).
1H-NMR (CDC13, 400 MHz) 8: See Fig. 4.1
13C-NMR (CDCI3, 125 MHz) 8: See Fig. 4.2.
COSY-450: See Fig. 4.3.
nOe: See Fig. 4.4.
HM9C: See Table-4.1.
HMBC: See Fig. 4.5.
Isolation of Guaianolide (304)4.3.3
The ethanolic extract (390 g) of Ferula oopoda extract with pet.
ether afforded a gummy material (154 g) named "FO". The fraction FO
loaded on silica gel column (1.5 kg) and was eluted with pet.ether :
acetone. The fraction "FO-3" (48 mg) was obtained at pet.ether :
acetone (7:3, Scheme-4.6). This fraction when subjected to
preparative TLC using Pet.ether: acetone (9:1) afforded a coumpound
identified as guaianolide (304) after spectroscopic studies
guaianolide (304) was obtained as a colourless amorphous solid (8.5
mg, yield 8.9 x 10'7%)
!Spectra Data
-:201:-

[cc]20D = -21° (c = 0.03, CHC13)
UV (MeOH) ran (log e) Xmax- 202 (2.98), 254 (2.51)
IR (CHCI3) vmax cm'1: 13030 (terminal methylene), 2990 (C-H),
1740 (C=0), 1645 (C=C) and 1120 (C-O).
ElMS m/z (rel. int. %): 230 (M+, 90). 215 (40). 185 (50), 121 (100),
105 (90) and 91 (97).
HREIMS m/z (rel. int. %): 230.1305 (M+ 100), 215.1072 (35) and 121.1017 (20).
1H-NMR (CDCI3, 400 MHz) 6: See Fig. 4.7
13C-NMR (CDCI3, 100 MHz) 8: See Fig. 4.8.
COSY-45®: See Fig. 4.9.
Isolation of Grilactone (305)4.3.4
The fraction "FO-1" was obtained by chromatography on the
silica gel column (loaded with extract of Ferula oopoda on elution
with pet. ether : acetone (9:1, Scheme-4.6). This fraction was
subjected to preparative TLC using pet.ether : acetone (9.5:0.5) as the
solvent system to afford grilactone (305, 2.5 g). This gave a dark
yellow colour with Dragendorffs reagent. The compound was
crystallized after evaporation of the solvent at room temperature,
m.p. 89-91°C.
Spectra Data
[al20D = 85° (c = 1.45, CHC13)
-:202:-

UV (MeOH) nm (log £) Xmax: terminal absorption.
IR (CHCI3) Vmax cm 1: 1730 (lactone C=0), 1600 (C=C) and 1090 (C-
O).
EIMS m/z (rel. Int. %): 232 (M+, 20), 217 (5), 204 (90), 188 (20), 176
(15), 159 (70), 158 (100), 119 (40). 91 (60) and 79 (58).
1H-NMR (CDCI3, 400 MHz) 8: See Fig. 4.11.
13C-NMR (CDCI3, 125 MHz) 8: See Fig. 4.12.
X-ray structure: See Fig. 4.13.i
HREIMS m/z (rel. int. %): 232.1442 (M+ 18), 204.1140(100),
105.0721(40)
-:203:-

5.0 REFERENCES
V
/

V.E. Tyler, PlantaMed.. 33(1), 1 (1987).1.
T. Mukheijee, Fitoterapia, LXD(3), 197 (1991).
M. Hesse "Alkaloid chemistry " John Wiley & Sons, New York,
p. 2 (1981).
2.
3.
G. Tirer, "Die Alkaloide", 2nd ed. Bomtrager-Verlag, Berlin,
(1931).4.
W. William Pelletier, "Alkaloids Chemical and BiologicalPerspectives", Vol. 1, p. 1-32, New York, (1934).
5.
6. T. Kametani 'The Chemistry of Isoquinoline Alkaloids",p. 1,
Hirokawa Publishing Company, Inc. Tokyo (1969).
7. Y. Kondo, Heterocycles, 4, 197 (1970).
V. Prelninger, "The Alkaloids", (ed. R.H.F. Manske), AcademicPress, New York, Vol. 15, p. 207 (1975).
8.
Atta-ur-Rahman "Hand Book of Natural Products Data,
Isoquinoline Alkaloids" Vol. 3, Elsevier Science Publishers;
Amsterdam, (1994).
9.
Atta-ur-Rahman, M.K. Bhatti, M.I. Choudhary and B. Sener,
Fitoterapia, LXm, 129 (1992).
10.
S. Agurell, I. Granelli, K. Leander, B. Luning and Rosenblom,
ActaChem. ScandL, 28B, 239 (1974).11.
I.A. Israilov, T. Irgashev, M.S. Yunusov and S.Y. Yunusov,
KhimPrir. Soedin, 13, 834 (1977).
12.
S. Kobayashi, T. Tokumoto, S. Iguchi, M. Kihara, Y. lmakuraand Z. Taira, J. Chem. Res., (5), 280 (1986).
13.
T.C. McKee and C.M. Ireland, J. Nat. Prod (Lloydia), 50, 754(1987).
14.
Atta-ur-Rahman, S.S. Ali, M.M. Qurehsi, S. Hassan and K.Bhatti, Fitoterapia, 60, 552 (1989).
15.
-:204:-

A. Jossang, M. Leboeuf, A. Cave and T. Sevenet, J. Nat. Prod.(Uoydia), 49, 1018 (1986).
16.
17. K.H.C. Baser and N. Kirlmer, PlantaMecL, 54, 513 (1988).
18. J. Saez, E. Fernandez, A. Jossang, A. Cave and A. Cave, Can.J. Chem., 67, 275 (1989).
19. M. Lavault, J. Bruneton, A. Cave, K.C. Chan, J.R. Deverre, T.Sevenet and H. Guinaudeau, Can. J. Chem., 65, 343 (1987).
20. M.U.S. Sultanabawa, S. Sotheeswraan, S. Balasubramanian,
M.Abd. El-Kawi, D.J. Slatkin and P.L. Schiff, Jr.,
Phytochemistry, 24, 589 (1985).
S.F. Hussain, L. Khan, H. Guinaudeau, J.E. Leet, A.J. Freyerand M. Shamma, Tetrahedron, 40, 2513 (1984).
21.
22. J.E. Leet, S.F. Hussain, R.D. Minard and M. Shamma,
Heterocycles, 19, 2355 (1982).
23. D.P. Allais and H. Guinaudeau, J. Nat. Prod (Lloydla), 53,
1280(1990).
L. Castedo, S. Lopez, A.R. de Lera and C. Villaverde.Phytochemistry, 28, 251 (1989).
24.
25. D.B. MacLean, L. Baczynsky), R. Rodrigo and R.H.F. Manske,
Can. J. Chem., 50, 862 (1972).
26. M.H.A. Zarga, G.A. Miana and M. Shamma, Tetrahedron Lett,
22, 541 (1981).
27. J. Ewing, G.K. Hughes, E. Ritchie and W.C. Taylor, Aust. J.Chem., 6, 78 (1953).
G. Blasko, S.F. Hussain, A.J. Freyer and M. Shamma,
Tetrahedron Lett, 22, 3127 (1981).
28.
29. S.S. Lee, Yi-Chuliu and C.-H. Chen, J. Nat. Prod. (Lloydla),
53. 1267 (1990).
30. F. Veznik, E. Taborska, H. Bochorakova, F. Tureck, V. Hanusand J. Slavik, Collect. Czech. Chem. Commun., 52, 1634(1987).
-:205:-

P.W. Jeffs, 'The Protoberberine Alkaloids", Vol. 9, 41 (1967),31.
R.H.F. Manske, "The Protopine Alkaloids", Vol. 4, 147 (1954).32.
S. McLean and J. Whelan, "SpirobenzylisoquinolineAlkaloids”, in Vol. 17 161.
33.
R.H.F. Manske, "a-Naphthaphenanthridine Alkaloids", Vol. 4,
253 (1954).
34.
35. J. Stanek, "Phthalideisoquinoline Alkaloids”, Vol. 4, 433(1954).
36. Yakhontova, L.D. et al., Khim Prir. Soedin, 8, 624 (1972);
Chem. Nat Compel, (Engl. Transl.), 592.
37. M.Tomita, Yakugaku Zasshi, 85, 827 (1965); Chem. Abst., 63,
18187e.
38. D.J. Slatkin, J. Ndt. Prod. (Lbydia). 37. 488 (1974).
39. A. B. Hahn "Pharmacology in Nursing" , C.V. Mosby Company,(1982).
40. A.M. Hjort, E.J. de Beer and D.W. Fassett, J. Pharmacol Exp.Ther., 62, 195 (1938).
"The Merck Index", 8th ed., p. 385 (1968).41.
:
42. "The Merck Index", 8th ed., p. 688 (1968).
43. A. Brossi, H. Besendrof, L.A. Pirk and A. Rheiner, Jr., Med.Chem., 5, 281 (1965).
J.D. Dutscher, J. Am. Chem. Soc., 74, 221 (1952).44.
45. S.A. Siphar, Belg. Pat, 688, 814 (1966).
46. M.V. Koch, J.G. Cannon and A.M. Burkman, J. Med Chem.,
11, 977 (1968).
47. H. Kreitmair, Pharmazie, 7, 507 (1952).
48. Roussel-UcLaf Patents, Chem. Abst 69, 59475m (1968).
D.C. Sharda, J. Indian. Med, A55, 54. 22 (1970).49.
-:206:-

E.L. McCauley and A.K. Reynolds, 'The Alkaloids", (ed. R.H.F.Manske), Vol, 5, p. 79,164, Academic Press, New York, (1955).
M.S. Segal, M.M. Goldstein and E.O. Attinger, Dis. Chest, 32,
305 (1957).
50.
51.
A.R. Batlersby, R. Binks and R. Huxtable, Tetrahedron Lett.
No.6 563 (1967).
52.
E. Leete, J. Am. Chem. Soc., 88, 4218 (1966).53.
A.R. Battersby, Tilden Lecture Proc. Chem. Soc., 189 (1963).
P. Winterstein and G. Trier, "Die Alkaloids", p. 307,
Bomtrager, Berlin, (1910).
54.
55.
R. Robinson, "The Structural Relation of Natural Products”,
p. 78, Oxford Univ. Press, London and New York, (1955).56.
A.R. Battersby and B.J.T. Harper, J. Chem. Soc., London3526(1962).
57.
A.R. Battersby, G.W. Evans, R.O. Martin, M.E. Warren andH. Rapoport, Tetrahedron Lett, 1275 (1965).
58.
F.R. Stermitz, S.Y. Low and G. Kallos, J. Am. Chem. Soc., 85,
1551 (1963).59.
D.H.R. Barton, R.H. Hesse and G.W. Kirby, J. Chem. Soc.,
6379 (1965).60.
S.F. Dyke and A.C. Ellis, Tetrahedron, 27, 3803 (1971).
D.W. Brown, S.F. Dyke, Hardy and M. Sainsbury, TetrahedronLett, 1515 (1969).
61.
62.
63. E. Brochmann-Hanssen, C.-C.Fu and L.Y. Misconi, J. Pharm.Set, 60, 1880 (1971).
D.S. Bhakuni, S. Tewari and R.S. Kapil, J. Chem. Soc., PerkinTrans, I, 706 (1977).
64.
O. Prakash, D.S. Bhakuni and R.S. Kapil, J. Chem. Soc.,
Perkin Trans, 1, 622 (1978).
65.
-:207:-

A.R. Battersby, R.T. Brown, J.H. Clements and G.G. Iverach.
Chem. Commun., 230 (1965).66.
S.M. Kupchan, V. Kameswaran, J.T. Lynn, O.K. Williams and
A.J. Liepa, J. Am. Chem. Soc., 97, 5622 (1975).67.
E. Spath and E. Kruta, Ber., 62, 1024 91929).68.
69. A. Pictet and T.Q. Chon, Ber.. 49, 370 (1916).
R. Robinson, 'The Structural Relation of Natural Products",
p. 86, Oxford University Press (clarendon), London and New
York. (1955).
70.
J.R. Gear and I.D. Spenser, Can. J. Chem., 41, 783 (1963).71.
D.H.R. Barton, Proc. Chem. Soc., London 293 (1963).72.
J.R. Gear and I.D. Spenser, Can. J. Chem., 41, 783 (1963).73.
M.-M. Janot and Pourat, Bull. Soc. Chim. Fr., 6, 823 (1955).74.
A.R. Battersby, R.J. Francis, E.A. Ruveda and J. Staunton,
Chem. Commun., 89 (1965).75.
76. E. Nasir and S.I. All "Flora of west Pakistan", p.628, Fkahriprinting press, (1972).
77. I.D. Spenser, Compr. Biochem., 20. 231 (1968).
78. E. Mcdonald "The Chemistry of Heterocyclic Compounds", (G.
Grethe, ed.). Vol. 38, part 1, p. 275, wiley (Interscience), NewYork, (1981).
A.R. Battersby, M. Hirst, D.J. McCaldin, R. Southgate and J.Staunton, J. Chem. Soc., C, 2163 (1968).
79.
A.R. Battersby, J. Staunton, H.R. Wiltshire, B.J. BIrcher andC. Fugantl, J. Chem. Soc., Perkin Trans. 1, 1162 (1975).
80.
B. Gdzler, T. Gftzler and M. Shamma, Tetrahedron, 39, 577(1983).
81.
F. Rorgacs, J. Provost, J.F. Descontois, A. Jehanno and M.Pesson, Acad. Set Ser, D, 279, 855 (1974).
82.
-:208>

S.F. Hussain and M. Shamma, Tetrahedron Lett, 21, 1693(1980).
83.
M. Shamma and C.D. Jones, J. Am. Chem. Soc., 91, 4009(1969); 92, 4943 (1970).
84.
M. Shamma and J.F. Nugent, J. Chem. Soc, Chem. Commun.,
1642 (1971).85.
M. Shamma and J.F. Nugent, Tetrahedron Lett, 2625 (1970).86.
T.T. Wu, J.L. Moniot and M. Shamma, Tetrahedron Lett., 3419(1978).
87.
H.L. Holland, M. Castillo, D.B. MacLean and I.D. Spenser,
Can. J. Chem., 52, 2818 (1974).88.
B. Nalliah, R.H.F. Manske, R. Rodrigo and D.B. MacLean,
Tetrahedron Lett, 2795 (1973).89.
M. Hanaoka and C. Mukai, Heterocycles, 6, 1981 (1977).90.
E. Nasir and S.I. Ali, "Flora of West Pakistan”, No. 73,
Ferozsons Karachi, p. 39 (1974).91.
K.M. Nadkami, "The Materia Medica", Popular Prakashan,
Bombay, p. 560 (1976).92.
K.R. Klrtlkar and B.D. Basu, "Indian Medicinal Plants",
Indian Press, Allahabad, p. 86 (1918).93.
Atta-ur-Rahman, S. Ahmad, M.K. Bhattl and M.I. Choudhaiy,Phytochemistry, 40, 593 (1995).
F. Fedde, “Das Pflanzenreich”, (A. Eng. ed.), Part 4, No. 40, p.288, Englemann Leipzig, (1909).
T.G. Tutin, V.H. Heywood, N.A. Burges, D.H. Valentine, S.M.
Walters and D.A. Webb, "Flora Europea", Vol. 1, CambridgeUniv. Press, London, (1964).
94.
95.
96.
H. Melchior, "Syllabus der Pflanzenfamilien", (A. Eng. ed.),
12th ed., Part II, p. 179, Bomtraeger, Berlin, (1964).97.
-:209>!

J. Hutchinson, "Families of Flowering Plants" 2nd ed. Vol. 2,
Oxford Univ. Press, London (1959).98.
Atta-ur-Rahman, S. Ahmed, F, Akhtar, M.K. Bhatti and M.l.
Choudhary, Phytochemistry, (in press).99.
R.W. Daskotch, P.L. Schiff, JR. and J.L. Beal, Tetrahedron,
25, 469 (1969).100.
S.F. Hussain, S. Nakkady, L. Khan and M. Shamma,
Phytochemistry, 22, 319 (1983).101.
102. Atta-ur-Rahman, M.K. Bhatti, M.l. Choudhary and B. Sener,
Fitoterapia, LXIQ, 129 (1992).
M. Shamma and Sr. M.A. Podczasy, Tetrahedron, 27, 727(1971).
103.
104. Atta-ur-Rahman, M.K. Bhatti, F. Akhter and M.l. Choudhary,Phytochemistry, 31, 2869 (1992).
105. J. Weijlard, E. Tashjian and M. Tishler, J. Am. Chem. Soc.,
69, 2070, (1947).
106. J. Fridrichsons and A. McL. Mathieson, Tetrahedron, 24, 5785(1968).
107. H. Hao and F. Qicheng, PlantaMecL, 52, 193 (1986).
S.F. Hussain and M. Shamma, Tetrahedron Lett., 21, 1693(1980).
108.
109. J. Eloumi-Ropivta, J. Beliveau and D.Z. Simon, J. Nat. Prod.,
48, 460 (1985).
110. R. Ziyaeu, T. lrgashev, l.A. Israilov, N.d. Abdullaev, M.S.Yunusov and S. Yu, Younusov, Khim-Prir. Soedin, 239 (1977);
Chem. Abst, 87, 114612e (1977).
111. N. Fukuda, M. Yonemitsu and T. Kimura, Chem. Pharm. Bull.31, 156 (1983).
112. A. Ikuta and H. Itokawa, Phytochemistry, 15, 577 (1976).
113. K. Jewers, J. Chem. Soc., Perkin Trans-2, 1393 (1972).

;
K. Iwasa and N. Takao, Phytochemistry, 21, 611 (1982).
Y.C. Tripathl, V.B. Pandey, Pharmazie, 42, 745 (1980); Chem.Abst 108, 109566p (1988).
P.W. Jeffs and J.D. Schaiver, J. Org. Chem., 40, 644 (1975).
114.
115.
116.
S.F. Cooper, J.A. Mockle and F. Stanavy, Planta Med., 21, 313(1972).
117.
G.V. Petri and P.T.M. Huang, Set Pharm., 48, 169 (1978).
J. Slavik, L. Slavlkova and L. Dolejs, Collect. Czech. Chem.Comm., 46, 2537 (1981).
118.
119.
120. H.G. Klryakov, Z.H. Mordirossian, D.B. MacLean and J.P.Ruder, Phytochemistry, 20, 1721 (1981).
W. Wan Chu Chan and P. Maitland, J. Chem. Soc. (C), 753(1966).
121.
E.B. Hanssen and B. Nielsen, Tetrahedron Lett., No.20, 2261(1966).
122.
T. Gozler, M.A. Onur, R.D. Minard and M. Shamma, J. Nat.Prod., 46, 414 (1983).
123.
R.R. Arndt and W.H. Baarschers, J. Chem. Soc., 2244 (1964).124.
G. Blasko, S.F., Hussain, A.J. Freyer and M. Shamma,
Tetrahedron Lett, 22, 3127, 3131, 3135, 3139, 3134 (1981).125.
A.R. Battersby, J. Staunton, H.R. Wiltshir, R.J. Francis andR. Southgate, J. Chem. Soc., Perkin Trans 1, 1140, 1147(1975).
126.
A. Ikuta, K. Syono and T. Furuya, Phytochemistry, 13, 2175(1974).
127.
V. Preininger, 'The Alkaloids", (ed. A. Brossi), vol, 29, p. 79(1985).
128.
G. Blasko, M. Shamma, A.A. Ansari and Atta-ur-Rahman,
Heterocycles, 9, 296 (1975).129.
:
-:211

T. Kametani, I.Noguchi, K. Saito and S. Kaneda, J. Chem.Soc. (C), 2036 (1969).
130.
D.W. Hughes, H.L. Holland and D.B. MacLean, Can. J.Chem., 64, 2252 (1976).
131.
J. Comin and V. Deulofeu, J. Org. Chem., 19, 1774 (1954).132.
133. A.J. Marsaioli, F. De, A.M. Reis, A.F. Magalhaes, E.A. Ruvedaand A.M. Kuck, Phytochemistry, 18, 165 (1979).
N. Murugesan and M. Shamma, Heterocycles, 14, 585 (1980).134.
M.D. Colton, H. Guinaudeau, M. Shamma, M.A. Onur and T.Gozler, J. Nat Prod., 48, 846 (1985).
135.
X.A. Dominguez, J.G. Delgado, A. Monroy, C.L.G. Armendariz,
A. Alcala, J. Quevedo and P. Rojas, Can. J. Chem., 43, 679(1965).
136.
137. D.B. MacLean, R.A. Bell., J.K. Saunders, C.Y. Chen andR.H.F. Manske, Can. J. Chem., 47, 3593 (1969).
138. G.I. Yakimov, J. Nat Prod. (Uoydia), 47, 1048 (1984).
G.A. Miana and M. Ikram, Pak. J. Ind. Res., 13, 49 (1970).139.
140. M.E. Ropova, A.N. Boeva, L. Dolejs, V. Preininger, V. Simanekand F. Santavy. PlantaMed., 40, 156 (1980).
I.A. Israilov, M.S. Yunusov and S.Yu. Yunusov, Khim Prir.Soedin, 6, 493 (1970).
141.
K. Iwasa M. Okada and N. Takao, Phytochemistry, 22, 627(1983).
142.
143. H.B. Dutschewska, B. Dimov, M. Mollov and L. Evstatieva,
PlantaMed., 39, 77 (1980).
J. Slavik and L.S. Slavikova, Collect. Czech. Chem. Commun.,
51, 2232 (1986).
144.
145. Atta-ur-Rahman, S.S. All, M.M. Qureshi, S. Hasan and M.K.Bhattl, Fitoterapia, 60, 552 (1989).
-:212:-

H. Guinaudeau, M. Shamma and T. Gozler, J. Nat. Prod., 46,
934 (1983).146.
Atta-ur-Rahman, M.K. Bhatti, H. Ahmed, Habib-ur-Rehmanand D.S. Rycroft, Heterocycles, 29, 1091 (1989).
147.
M.U. Ibragimova, I.A. Israilov, M.S. Yunusov, S.Yu. Yunusov,
Khim Prir. Soedin, 476 91974); J. Nat. Comp, 10, 481 (1974);
Chem Abst., 82, 28586n (1975).
148.
V.B. Pandey, A.B. Ray and B. Dasguputa, Phytochemistry, 18,
695 (1979).
149.
150. M.E. Popova, V. Simanek, J. Novak, L. Dalejs, P. Sedmera andV. Preringer, PlantaMed., 48, 272 (1983).
151. G. Blasko, S.F. Hussain and M. Shamma, J. Nat. Prod., 44,
475 (1981).
G. Blasko, D.J. Gula and M. Shamma, J. Nat. Prod., 46, 105(1982).
152.
153. N.N. Margvelashvili, O.N. Tolkachev, N.P. Prisyazhnyuk andA.T. Kiryanova, Khim. Prir. Soedin, 592 (1978); Chem. Nat.Comp., 509 (1978).
154. S. Teitel, J.O. Brien and S. Brossi, J. Org. Chem., 37, 1879(1972).
155. G. Snatzke, G. Wollenberg, J. Hrbek, Jr. and F. Santavy, K.Blaha, W. Klyne and R.J. Swan, Tetrahedron, 26, 5059 (1969).
156. I.A. Israilov, M.S. Yunusov and S.Yu. Yunusov, Khim Prir.Soedin, 6, 518 (1970).
157. N.N. Margvelashvili, A.T. Kiijanova and O.N. Tolkachev, KhimPrir. Soedin, 127 (1972); J. Nat Comp., 131 (1972).
N.M. Mollov and G.I. Yakimov, Dokl. Bulg. Akad. Nauk., 26,
59 (1972); Chem Abst, 77, 85674k (1972).
158.
159. I.A. Israilov, M.S. Yunusov, N.D. Abdullaev and S.Yu.Yunusov, Khim Prir. Soedin, 536 91975); Chem. Nat. Comp.,
. 572 91975).

J.K. Saunders, R.A, Bell, C.Y. Chen, D.B. MacLean andR.H.F. Manske, Can. J. Chem., 46, 2873 (1968).
160.
161. T. Kishimoto and S. Uyeo, J. Chem. Soc. C, 2600 (1969).
N.M. Mollov, H.G. Kiryakov and G.I. Yakimov, Phytochemistry.
11. 2331 (1972).
162.
C.K. Yu, J.K. Saunders and D.B. MacLean, Can. J. Chem.,
49. 3020(1971).
163.
K.L. Seitanidi, M.R. Yagaudaev, I.A. Israilov and M.S.Yunusov, Khim. Prir. Soedin, 465 (1978); Chem. Nat. Comp.,395 (1978).
164.
A. Loukis, J. Pharm. Pharmacol., 33, 16 (1981).165.
166. D.W. Hughes, B.C. Nalliah, D.B. MacLean and H.G. Kiryakov,Can. J. Chem., 57, 53 (1979).
167. R.G.A. Rodrigo, R.H.F. Manske, H.L. Holland and D.B.
MacLean. Can. J. Chem., 54, 471 (1970).
168. S.F. Hussain, R.D. Minard. A.J. Freyer and M. Shamma, J.Nat Prod., 44, 169 (1981).
169. Collect. Czech. Chem. Commun., 61 (7), 1064 (1996); Chem.Abst 125, 163290h.
170. I.V. Preininger, V. Simanek. O. Gasic, F. Santavy and L.Dolejs, Phytochemistry, 12, 2513 (1973).
N.M. Mollov, G.I. Yakimov and P.P. Panov, C.R. Acad. Bulg.Set, 20, 557 (1967).
K.K. Seth, V.B. Pandey, A.B. Ray, B. Dasgupta and S.A.H.Shah, Chem. Ind. (London), 744 (1979).
171.
172.
G. Blasko, V. Elango, B. Sener, A.J. Freyer and M. Shamma,
J. Org. Chem., 47, 880 (1982).
H.G. Kiryakov, Z.H. Mardirossian, D.W. Hughes and D.B.MacLean, Phytochemistry, 19, 2507 (1981).
173.
174.

V.K. Tripathi and V.B. Pandey, Phytochemistry, 31, 2188(1992).
175.
Y.C. Tripathi, V.B. Pandey, N.K.R. Pathak and M. Biswas,
Phytochemistry,
C.R. Addinall and R.T. Major, J. Am. Chem. Soc., 55, 1202,
2153 (1933).
176.
177.
J. Hodkova, Z. Vesely, Z. Koblicova, J. Holubek and J.
Trojanek, J. Nat. Prod.. 35, 61 (1972).178.
S.F. Hussain and M. Shamma, Tetrahedron Lett., 21, 1909(1980).
179.
M.H.A. Zarga, S.S. Sabri, S. Firdous and M. Shamma,
Phytochemistry. 26, 1233 (1987).180.
Atta-ur-Rahman, "Nuclear Magnetic Resonance
Spectroscopy", p.202, Springer-Verlag, New York,(1986).181.
Atta-ur-Rahman, "One and Two-Dimensional NMR
Spectroscopy" Elsevier Science Publishers, Amsterdam, (1989).182.
M. Shamma and C.D. Jones, J. Am. Chem. Soc., 91, 4009(1969).
183.
T.T. Wu, J.L. Monlot and M. Shamma, Tetrahedron Lett.,
3419. (1978).184.
Atta-ur-Rahman, S. Malik, S. Ahmed, M.I. Choudhaiy andHabib-ur-Rehman, Heterocycles, 23, 953 (1985).
T. Irgashev, I.A. Israllov, N.D. Abdullaev, M.S. Yunusov and
S.Yu. Yunusov, Khim Prir Soedin, 127 (1977) or Chem. J. Nat.
Compounds 13, 118 (1977).
185.
186.
S.M.H. Jafri, "The Flora of Karachi" p. 239. 298 (1966).187.
188. K.R. Klrtikar and B.D. Basu, "Indian Medicinal Plants" Vol. 3,
p.1783, Shiva Publishers, Dehradun, (1991).
M.L. Dhar, M.M. Dhar, B.N. Dhawn, B.N. Malhotra and C.Ray, Indian Journal of Experimental Biology, 8, 232 (1991).
189.
-:215:-

’The Wealth of India", Raw Materials, Vol. X, Publication and
Information Directorate CSIR, p. 581-585, New Delhi India(1976).
190.
W. Dymock, C.J.H. Warden and D. Hooper, "Pharmacographlaindica", The Institute of Health and Tibbi Research,
republished under the auspices of Hamdard NationalFoundation, Pakistan, Vol. 1, p.322 (1980).
R.N. Chopra. "Glossary of Indian Medicinal Plants", Council
of Scientific and Industrial Research, p. 259 New Delhi(1956).
191.
192.
H.J. Jaffer, A.L.M. Jawad, H.S. Saber and A.A. Naib,
Fitoterapia, 6, 497 (1988).193.
N. Singh, S.P. Singh, R. Nath, D.R. Gupta, R.P. Kohli and
K.P. Bhargava, Int. J. Crude Drug Res., 24 (2), 90 (1986).194.
W. Dymock, C.J.H. Warden and D. Hooper, "Pharmacographlaindica", The Institute of Health and Tibbi Research.republished under the auspices of Hamdard NationalFoundation, Pakistan, Vol. 1, p.566-72, (1980).
195.
196. K. Anbalagan and J. Sadique, J. Int. J. Crude Drug Res., 23(4). 177 (1985).
197. Peshney, N.L. Magha, P.G. Pku. Puhjairao Kirishi Vidya Peeth,
Res. J., 13, 119-32(1989).
198. E. Nasir, S.I.Ali and J. Yasin, "Flora of Pakistan" 168, 5,
30,31, Shamim Printing Press, Karachi (1985).
199. A.K. Mehta, P. Binkley, S.S. Gandhi and M.K. Ticku, Indian J.Med. Res. Sect B.. 94, 312 (1991).
200. S. Sudhir, R.D. Budhiraja, G.P. Arora , L.C.Gupta and K.N.Garg, Planta Medica , 61 (1986).
C.K. Atal and A.E. Schwarting, Current Science, (India), 29, 22(1960).
201.
K.R. Kirtikar and B.D. Basu, "Indian Medicinal Plants" Vol. 3,
p.1773, Shiva Publishers, Dehradun, (1918).202.
-:216:-

203. R.D. Budhiraja, S. Sudir and K.N. Gorg, Indian Journal ojPhysiol, 27, 129 (1983).
T. Elisha and Khalid, Int. J. Crude Drug Res., 27, (2) 109-112(1989).
204.
;
B. Shohat, I. Kirson and D. Lavie, Biomedicine, 28, 18 (1978).205.
K. Sakurai, H. Ishii, S. Kobayashi and T. LwaoChem. Pharm.Bull, 24, 1403 (1976).
206.
W.J.S. Lockley, H.H. Rees and T.W. Goodwin, Phytochemistry,22, 2253(1983).
207.
208. W.J.S. Lockley, H.H. Rees and T.W. Goodwin, Phytochemistry,15, 937 (1976).
I.Danishefsky, "Biochemistry for Medical Sciences" p.232,Little, Brown and Company, Boston, (1980).
209
210. P. Manitto, "Biosynthesis of Natural Products" Ellis HowardLimited, Chichester, p. 267,279 (1981).
211. V.V. Velde and D. Lavie, Phytochemistry, 20, 1359 (1981).
212. I. Kirson, H.E. Gottlieb and E. Glotter, J. Chem. Res., 125,
2134 (1980).
213. V.V. Velde, D. Lavie, R.D. Budhiraja, S. Sudhir and K.N. GargPhytochemistry, 22, 2253 (1983).
P. Clerc and S. Simon, "Strukturaufklarrung OrganischerVerbindungen", Springer-Verlag, Berlin, P(u) 20 (1981).
L. Muller, J. Am. Chem. Soc., 101, 4481 (1979).
214.
215.
216. Y. Oshima, A. Bagchi, H. Hiklno, S.C. Stnha, M. Shahi andA.B. Ray, Tetrahedron Letters, 20, 2025 (1987).
217. D. Lavie, I. Kirson, E. Glotter, D. Rabinovich and Z.J.Shakked, Chem. Soc. Commun., 877 (1972).
218. A.I. Scott "Introduction of Ultraviolet Spectra of NaturalProducts" Pergamon Press, Oxford, p. 64,82 (1964).

D, Lavie, I. Kirson and E. Glotter, Isr. J. Chem., 6, 671 (1968).219.
Atta-ur-Rahman, S.A. Jamal, E. Asif and M.I. Choudhary,Phytochemistry, 30, 3824 (1991).
220.
E. Derome, "Modern NMR Techniques for ChemistryResearch", Pergamon Press, Oxford, (1987).
221.
P. Clerc and S. Simon, "Strukturaufklarrung OrganischerVerbindungen", Springer-Verlag, Berlin, P.I 85, I 140, (1981).
D. Lavie, E. Glotter and Y. Shvo, J. Org. Chem., 36, 1774(1965).
222.
223.
224. I.D. Spenser, Compr. Biochem., 20, 231 (1968).
Information Directorate, New Delhi, p. 581 (1976).
E. Nasir and S.I. All, "Flora of West Pakistan" No. 20(Umbelliferae), Feroz Sons Ltd. Karachi, p. 519 (1972).
225.
226.
227. A. Waheed, R. Ahmad, A. Sattar and S.A. Khan, Pak. J. SetInd. Res. 32 (12), 807 (1989).
"Wealth of India", ICSIR, New Delhi. 4, 20 (1976).228.
Council of Scientific and Industrial Research, 'The Wealth ofIndia", (Raw Materials), Vol. 4, Publication and InformationDirectorate, p. 20-23, New Dehli (1956).
229.
230. L.M. Perry and J. Metzger, "Medicinal Plants of East andSouth East Asia", The MIT Press, p. 415-416 (1980).
231. K.R. Kirtikar, "Indian Medicinal Plants", BbhuwaneswariAsrama, Bahadur Gang, Allahbad, p. 628-629 (1981).
232. A.M.B.A. Khalil, S. Afifi, J. Ethnopharmacol, 31 (3), 291(1991).
233. A. Khalil, S. Aqel, S. Afifl, A. Eisawi, J. Ethnopharmacol, 30(1), 35 (1990).
S.M. Hanif, M. Choudhary, M.K. Bhatty, Pakistan Journal ofScientific and Industrial Research, 30 (1), 19 (1987).
234.

Atta-ur-Rahman, R. Zaidi, M.I. Choudhary, S. Firdous and
N.M. Hasan, Fitoterapia, LXV, 62, (1994).235.
J.D. Martin and Darlas, In “Marine Natural Products" (ed.
P.J. Scheuer), Vol. 1, Acadmic Press, New York, P. 125 (1978).236.
T.K. Devon and A.I. Scott, "Hand-Book of Naturally Occrring
Compounds", Vol. 2, Acadmic Press, New York, p. 55 (1972).237.
G. Blasko, D.J. Gula and M. Shamma, J.N. Prod., 45. 105(1982).
238.
239. C.G. Casinovi, L. Tomasslnl and M. Nlcolelti, Gazz. Chim.ItaL, 119, 563 (1989).
K.L.Rinehart, J. Science, 218, 254, (1982).240.
H.D. Beckey, H. Knoppel, G. Metzinger and P. Schalze,
"Advances In Mass Spectrometry" (ed. W.L. Lead), Vol. 3,
Institute of Petroleum, London, p. 35-67 (1966).
241.
D.L. Pavla, G.M. Lampman and G.S. Krinz, "Introduction ofSpectroscopy", Saunder s College Publishing Co. Philadelphia,p. 41 (1979).
N. P. Klr'yalov, Z. Obschei, Khim. Prir. Soedin., 34, 3831(1964).
242.
243.
244. N. P. Kir’yalov, T. V. Bukreeva and V. S. Gindin.Khim. Prir.
Soedin., 8, 446 (1972).
i245. W. Herz, H. Watanabe, M. Miyazak, Y. Kishida, J. Am Chem
Soc., 84, 2601 (1962).
246. T. J. Mahry, W. Renold, H. E. Miller. H. B. Kagan, J. Am.
Chem Soc., 31, 681 (1966).
Y.R. Chadha, "The Wealth of India", Publication and
Information Directorate, New Delhi, p.581 (1976).
247.
K.L. Rinehart J. Science, 218, 254 (1982).248.
249. H.D. Backey, H. Knoppel, G. Metzinger and P. Schalze,
“Advances in Mass Spectormetry" (ed., W.L. Lead), Vol.3
Institute of Petroleum, London, p.35-67 (1966).
-:219:-

M. Shamma "The Isoquinoline Alkaloids" Academic Press NewYork and London, 309 (1972).
250.
G. Blasko, N. Murgesan, A.J. Freyer and M. Shamma, J. Am.
Chem. Soc., Vol. 104, No.7, 2039 (1982).251.
i
-:220:-

;
LIST OF PUBLICATIONS
Alkaloidal constituents of Fumaria indica, Atta-ur-Rahman,
Shakil Ahmad, M. Khalid Bhattl and M. Iqbal Choudhary,
Phytochemistry. Vol. 40. p.593 (1995).
1.
New bioactive steroidal lactone from W. somnifera Atta-ur-
Rahman, S. Ahmad, F. Akhater and M. Iqbal Choudhary, J.
Nat Prod, (in press).
2.
3. Isoquinoline alkaloids from Fumaria sp. Atta-ur-Rahman, F.
Akhater S. Ahmad, and M. Iqbal Choudhary, Phytochemistry,
(in press).
Non alkaloidal contituents of Fumaria indica Atta-ur-Rahman,
Shakil Ahmad, M. Khalid Bhatti and M. Iqbal Choudhary.
Phytochemistry, (in press).
4.
5. Isolation and structural studies on the alkaloids of Rhazya
stricta, Atta-ur-Rahman, Habib-ur-Rehman, Shakil Ahmad
and M. Iqbal Choudhary, Heterocycles (in press).
-:221:-