
MutationSpectrumofthep53GeneinBoneandSoftTissueSarcomas1cancerres.aacrjournals.org/content/52/22/6194.full.pdf ·...
7
ICANCER RESEARCH 52.6194-6199, November 15, 1992] Mutation Spectrum of the p53 Gene in Bone and Soft Tissue Sarcomas1 Junya Toguchida, Toshikazu Yamaguchi, Bruce Ritchie, Roberta L. Beauchamp, Siri H. Dayton, Guillermo E. Herrera, Takao Yamamuro, Yoshihiko Kotoura, Musan S. Sasaki, John B. Little, Ralph R. Weichselbaum, Kanji Ishizaki, and David W. Yandell2 Howe Laboratory of Ophthalmology, Massachusetts Eye and Ear Infirmary, Boston, Massachusetts 02114 [J. T., R. L. B., S. H. D., G. E. H., D. W. Y.]; Department of Orthopaedic Surgery fJ. T., To. Y., Ta. Y., Y. K.J and Radiation Biology Center fTo. Y., M. S. S., K. /./, Kyoto University, Kyoto 606, Japan; Department of Biochemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2H7 [B. R.f; Department of Cancer Biology, Harvard School of Public Health, Boston, Massachusetts 02115 [ J. B. L., D. W. Y.J; and Department of Radiation and Cellular Oncology, University of Chicago Medical Center, Chicago, Illinois 60637 /R. R. W.] ABSTRACT We present here an analysis of the spectrum of mutations of the p53 gene seen in 127 bone and soft tissue sarcomas of various histológica! classifications. Gross rearrangements were analyzed by Southern blot ting using a complementary DNA probe from the p53 gene, and subtle alterations in the entire coding sequence (exons 2 through 11) were identified by a combination of single-strand conformation polymor phism analysis and direct genomic sequencing. A total of 42 somatic alterations of the p53 gene were found, of which 21 were gross rear rangements and 21 were subtle alterations. These included 17 cases of a single base substitution, 3 small deletions, and one single base inser tion. In contrast to reported findings for other types of cancer, we found that mutations of the p53 gene in sarcomas are quite heterogeneous both in their distribution throughout the gene and in the type of genetic alterations that result. All 13 missense mutations we found occurred at highly conserved residues, whereas 8 nonsense mutations occurred at sites that spanned the gene from codons 46 to 316. Surprisingly, approximately one-half of the osteosarcomas with allelic deletions on 17p did not have detectable alterations in the coding sequence of the p53 gene. INTRODUCTION The p53 gene, located at chromosome 17pl3, encodes a M, 53,000 nuclear phosphoprotein that has been characterized ex tensively. Several studies have shown that reintroduction of a wild-type p53 gene into p53-deficient cells leads to suppression of the neoplastic phenotype; hence this gene has been catego rized as a tumor suppressor (1, 2). This suppressor activity may be exerted through the transcriptional control of proliferation- related genes, and several experiments have identified a tran scription-activating sequence in the p53 protein (3, 4). Another possible function of this protein is the regulation of DNA rep lication through its association with DNA replication com plexes. In simian virus 40-transformed cells, the p53 protein has been shown to bind large T-antigen, inhibiting T-antigen from binding a viral replication origin (5) or DNA polyrnerase «(6). Recently, it was shown that DNA fragments that bind to the p53 protein contain sequences similar to potential replica tion origins (7). Although the exact function of this gene is still unknown, alterations of this gene have been found to be closely associated with the formation of various human cancers (8, 9). Intensive analysis of p53 gene mutations has been carried out on a variety of human cancers, and tumor-specific mutation spectra have been found in several types of malignant tumors. Received 2/27/92; accepted 9/11/92. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accord ance with 18 U.S.C. Section 1734 solely to indicate this fact. 1Supported by grants from the NIH (D. W. Y., R. R. W.), the Center for Radiation Therapy (D. W. Y., R. R. W.), and the Japanese Ministry of Education, Science and Culture (M. S. S., T. Y.). D. W. Y. is a Research to Prevent Blindness Dolly Green Scholar. 2To whom requests for reprints should be addressed, at Howe Laboratory. Molecular Genetics, Room 575, Massachusetts Eye and Ear Infirmary, 243 Charles Street. Boston. MA 02114. These include a high mutation frequency at CpG sites in col- orectal cancer (10) and lymphoid malignancies (11), a predom inance of G to T transversions in non-small cell lung cancer (12), and a predominance of G to T transversions at codon 249 in hepatocellular carcinomas (13, 14). Osteosarcoma, a repre sentative of malignant tumors derived from bone and soft tis sue, is one of the first human tumors in which p53 gene muta tions were found (15). We describe here a comprehensive study of the spectrum ofp53 gene mutations found in a large number of bone and soft tissue sarcomas. Our goal was to compare the spectrum ofp53 mutations in sarcomas with that seen in other tumors in the hope that useful information about the role of p53 in sarcoma progression could be found. MATERIALS AND METHODS DNA Samples. Tumor tissues were obtained at surgical resection, dissected, rinsed to remove adjacent normal tissue and blood, flash frozen, and stored at -70°C until DNA extraction. Nonmalignant in filtrate or stromal cell content was not quantitated, but histopatholog- ical examination was carried out, and only those samples that contained predominantly neoplastic cells were used for molecular studies. High- molecular-weight DNA from tumor tissues and peripheral leukocytes were prepared as described elsewhere according to standard protocols (16). Southern Blot Analysis. Gross rearrangements of the p53 gene were analyzed using a cDNA3 probe for the p53 gene (pR4-2) (17). This cDNA probe hybridizes with three £coRI-digested fragments (15, 15, and 3.5 kilobases) (18). The 3.5-kilobase fragment, corresponding to exon 1 sequences, is too faint to be seen on normal autoradiographic exposure. The presence of these fragments was confirmed by additional, longer exposure of all blots (data not shown). Allelic deletions on 17p were analyzed using six polymorphic probes (pi0.5, pA 10-41, pHF12-l, pmCT35.1, pmCT35.2, and pYNZ22) as previously de scribed (19, 20). Restriction endonuclease digestion of DNA samples, agarose gel electrophoresis, Southern hybridization, labeling of probes, and autoradiography were performed by standard procedures ( 16). SSCP Analysis. Each primer sequence was obtained from published data or by sequencing genomic clones of the human p53 gene provided by Dr. L. Crawford. PCR fragments were generated from 50 ng of genomic DNA in a 50-^1 mixture containing: 20 UMdATP, dTTP, dGTP; 2 UMdCTP; 1.0 to 2.5 min MgCl2; 20 pmol of each primer; 20 mivi Tris (pH 8.4 or 8.6); 50 mivi KCl; 50 Mg/ml bovine serum albumin; 0.5 units Taq polyrnerase (Perkin-Elmer Cetus); and 0.1 ^1 (7 nm) [«-12P]dCTP(3000 Ci/mmol). PCR was carried out using 30 cycles (30 s at 94°C,90 s at 52°to 60°C,and 120 s at 71°C)on a programmable thermal controller (MJ Research, Inc.). One-tenth of the amplified product was diluted with 15 to 40 u\ of a 0.1% sodium dodecyl sulfate-10 miviEDTA mixture, followed by 1:1 dilution with a 95% formamide, 89 mM Tris, 2 min EDTA, 89 miviboric acid, 0.05% bromophenol blue, 0.05% xylene cyanol loading solution. Dilute sam ples were heat denatured at 95°Cfor 2 min and then were loaded on 6% nondenaturing polyacrylamide gels: once on a gel containing 10% glycerol, and a second time on a gel that did not contain glycerol. 1The abbreviations used are: cDNA, complementar)1 DNA; SSCP, single-strand conformation polymorphism: PCR, polyrnerase chain reaction. 6194 on July 14, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Transcript of MutationSpectrumofthep53GeneinBoneandSoftTissueSarcomas1cancerres.aacrjournals.org/content/52/22/6194.full.pdf ·...

ICANCER RESEARCH 52.6194-6199, November 15, 1992]
Mutation Spectrum of the p53 Gene in Bone and Soft Tissue Sarcomas1
Junya Toguchida, Toshikazu Yamaguchi, Bruce Ritchie, Roberta L. Beauchamp, Siri H. Dayton,Guillermo E. Herrera, Takao Yamamuro, Yoshihiko Kotoura, Musan S. Sasaki, John B. Little,Ralph R. Weichselbaum, Kanji Ishizaki, and David W. Yandell2
Howe Laboratory of Ophthalmology, Massachusetts Eye and Ear Infirmary, Boston, Massachusetts 02114 [J. T., R. L. B., S. H. D., G. E. H., D. W. Y.]; Departmentof Orthopaedic Surgery fJ. T., To. Y., Ta. Y., Y. K.J and Radiation Biology Center fTo. Y., M. S. S., K. /./, Kyoto University, Kyoto 606, Japan; Department ofBiochemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2H7 [B. R.f; Department of Cancer Biology, Harvard School of Public Health, Boston,Massachusetts 02115 [ J. B. L., D. W. Y.J; and Department of Radiation and Cellular Oncology, University of Chicago Medical Center, Chicago, Illinois 60637/R. R. W.]
ABSTRACT
We present here an analysis of the spectrum of mutations of the p53gene seen in 127 bone and soft tissue sarcomas of various histológica!classifications. Gross rearrangements were analyzed by Southern blotting using a complementary DNA probe from the p53 gene, and subtlealterations in the entire coding sequence (exons 2 through 11) wereidentified by a combination of single-strand conformation polymorphism analysis and direct genomic sequencing. A total of 42 somaticalterations of the p53 gene were found, of which 21 were gross rearrangements and 21 were subtle alterations. These included 17 cases ofa single base substitution, 3 small deletions, and one single base insertion. In contrast to reported findings for other types of cancer, we foundthat mutations of the p53 gene in sarcomas are quite heterogeneous bothin their distribution throughout the gene and in the type of geneticalterations that result. All 13 missense mutations we found occurred athighly conserved residues, whereas 8 nonsense mutations occurred atsites that spanned the gene from codons 46 to 316. Surprisingly,approximately one-half of the osteosarcomas with allelic deletions on17p did not have detectable alterations in the coding sequence of thep53 gene.
INTRODUCTION
The p53 gene, located at chromosome 17pl3, encodes a M,53,000 nuclear phosphoprotein that has been characterized extensively. Several studies have shown that reintroduction of awild-type p53 gene into p53-deficient cells leads to suppression
of the neoplastic phenotype; hence this gene has been categorized as a tumor suppressor (1, 2). This suppressor activity maybe exerted through the transcriptional control of proliferation-related genes, and several experiments have identified a transcription-activating sequence in the p53 protein (3, 4). Anotherpossible function of this protein is the regulation of DNA replication through its association with DNA replication complexes. In simian virus 40-transformed cells, the p53 proteinhas been shown to bind large T-antigen, inhibiting T-antigenfrom binding a viral replication origin (5) or DNA polyrnerase«(6). Recently, it was shown that DNA fragments that bind tothe p53 protein contain sequences similar to potential replication origins (7). Although the exact function of this gene is stillunknown, alterations of this gene have been found to be closelyassociated with the formation of various human cancers (8, 9).
Intensive analysis of p53 gene mutations has been carried outon a variety of human cancers, and tumor-specific mutationspectra have been found in several types of malignant tumors.
Received 2/27/92; accepted 9/11/92.The costs of publication of this article were defrayed in part by the payment of
page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1Supported by grants from the NIH (D. W. Y., R. R. W.), the Center forRadiation Therapy (D. W. Y., R. R. W.), and the Japanese Ministry of Education,Science and Culture (M. S. S., T. Y.). D. W. Y. is a Research to Prevent BlindnessDolly Green Scholar.
2To whom requests for reprints should be addressed, at Howe Laboratory.Molecular Genetics, Room 575, Massachusetts Eye and Ear Infirmary, 243 CharlesStreet. Boston. MA 02114.
These include a high mutation frequency at CpG sites in col-orectal cancer (10) and lymphoid malignancies (11), a predominance of G to T transversions in non-small cell lung cancer(12), and a predominance of G to T transversions at codon 249in hepatocellular carcinomas (13, 14). Osteosarcoma, a representative of malignant tumors derived from bone and soft tissue, is one of the first human tumors in which p53 gene mutations were found (15). We describe here a comprehensive studyof the spectrum ofp53 gene mutations found in a large numberof bone and soft tissue sarcomas. Our goal was to compare thespectrum ofp53 mutations in sarcomas with that seen in othertumors in the hope that useful information about the role of p53in sarcoma progression could be found.
MATERIALS AND METHODS
DNA Samples. Tumor tissues were obtained at surgical resection,dissected, rinsed to remove adjacent normal tissue and blood, flashfrozen, and stored at -70°C until DNA extraction. Nonmalignant in
filtrate or stromal cell content was not quantitated, but histopatholog-ical examination was carried out, and only those samples that containedpredominantly neoplastic cells were used for molecular studies. High-molecular-weight DNA from tumor tissues and peripheral leukocyteswere prepared as described elsewhere according to standard protocols(16).
Southern Blot Analysis. Gross rearrangements of the p53 gene wereanalyzed using a cDNA3 probe for the p53 gene (pR4-2) (17). ThiscDNA probe hybridizes with three £coRI-digested fragments (15, 15,and 3.5 kilobases) (18). The 3.5-kilobase fragment, correspondingto exon 1 sequences, is too faint to be seen on normal autoradiographicexposure. The presence of these fragments was confirmed by additional,longer exposure of all blots (data not shown). Allelic deletions on17p were analyzed using six polymorphic probes (pi0.5, pA 10-41,pHF12-l, pmCT35.1, pmCT35.2, and pYNZ22) as previously described (19, 20). Restriction endonuclease digestion of DNA samples,agarose gel electrophoresis, Southern hybridization, labeling of probes,and autoradiography were performed by standard procedures (16).
SSCP Analysis. Each primer sequence was obtained from publisheddata or by sequencing genomic clones of the human p53 gene providedby Dr. L. Crawford. PCR fragments were generated from 50 ng ofgenomic DNA in a 50-^1 mixture containing: 20 UMdATP, dTTP,
dGTP; 2 UMdCTP; 1.0 to 2.5 min MgCl2; 20 pmol of each primer;20 mivi Tris (pH 8.4 or 8.6); 50 mivi KCl; 50 Mg/ml bovine serumalbumin; 0.5 units Taq polyrnerase (Perkin-Elmer Cetus); and 0.1 ^1(7 nm) [«-12P]dCTP(3000 Ci/mmol). PCR was carried out using 30cycles (30 s at 94°C,90 s at 52°to 60°C,and 120 s at 71°C)on a
programmable thermal controller (MJ Research, Inc.). One-tenth ofthe amplified product was diluted with 15 to 40 u\ of a 0.1% sodiumdodecyl sulfate-10 miviEDTA mixture, followed by 1:1 dilution with a95% formamide, 89 mM Tris, 2 min EDTA, 89 miviboric acid, 0.05%bromophenol blue, 0.05% xylene cyanol loading solution. Dilute samples were heat denatured at 95°Cfor 2 min and then were loaded on
6% nondenaturing polyacrylamide gels: once on a gel containing 10%glycerol, and a second time on a gel that did not contain glycerol.
1The abbreviations used are: cDNA, complementar)1 DNA; SSCP, single-strand
conformation polymorphism: PCR, polyrnerase chain reaction.
6194
on July 14, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

p53 GENE MUTATIONS IN SARCOMAS
Electrophoresis was performed at room temperature with a constantpower of 8 W for 8 to 16 h. Autoradiography was carried out for 12-24
h without an intensifying screen.Direct Genomic Sequencing. Direct sequencing of double-stranded
PCR fragments was performed as previously described (21). AmplifiedPCR products were purified through Sepharose CL-6B (Pharmacia)and combined with 7 pmol of 32P-end-labeled primer. Primer-templatemixtures were heat-denatured, and sequencing'reactions were carried
out with Sequenase (U.S. Biochemical). Electrophoresis was performedon 6% denaturing polyacrylamide gels. Autoradiography was carriedout for 12-16 h without the use of an intensifying screen.
RESULTS
Rearrangements of the p53 Gene. DN A from 127 bone andsoft tissue sarcomas was examined by Southern blot analysisusing a cDNA probe from the p53 gene (pR4-2) (17). Thepathological diagnosis for these samples was: osteosarcoma in76 cases; malignant fibrous histiocytoma in 13 cases; chondro-sarcoma in 9 cases; synovial sarcoma and Ewing's sarcoma
(5 cases each); liposarcoma and rhabdomyosarcoma (4 caseseach); chordoma in 3 cases; fibrosarcoma in 2 cases; malignantschwannoma; alveolar soft part sarcoma; leiomyosarcoma; malignant hemangiopericytoma; epithelioid sarcoma; and ter-atoma (one case each). Gross rearrangements of the p53 locuswere detected in 21 tumors (Table 1). These gross rearrangements were identified by an abnormal restriction pattern, usually involving the presence of additional fragments of abnormalsize (Fig. 1). Abnormally migrating bands in Fig. 1 have arisenfrom structural rearrangements within a 15-kilobase EcoRlfragment containing exons 2-10 of thep53 gene; the remainingfaint 15-kilobase band is due to a second EcoRl fragment containing the unrearranged 3' end of the gene (18). In all cases the
rearrangements were not observed in DNA of leukocytes takenfrom the same patient, excluding the possibility of neutral polymorphisms at the enzyme recognition sites (data not shown).Of the 21 rearrangements we found, 18 occurred in osteosarcoma tumors, 2 in malignant fibrous histiocytomas, and 1 in afibrosarcoma.
Screening for Point Mutations of the pS3 Gene. Mutationstoo small to be resolved by Southern blotting were identified intwo steps. In the first step, SSCP analysis was used to identifysamples with DNA sequence variations. SSCP was performedwith only minor modifications of the original published proce-
Table 1 Gross alterations of the p53 gene in sarcoma
123456789 10 11
IDKS-41KS-61KS-73KS-102KS-162KS-72KS-115KS-121KS-149KS-150KS-74K.S-82KS-103KS-1
11KS-127KS-I63KS-1
68KS-173KS-4SKS-71KS-80Tumor
typeOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaOsteosarcomaMFH"MFHFibrosarcomaProposedalterationHomozygous
partialdeletionHomozygouspartialdeletionHomozygouspartialdeletionHomozygouspartialdeletionHomozygouspartialdeletionHomozygous
rearrangementHomozygousrearrangementHomozygousrearrangementHomozygousrearrangementHomozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygousrearrangementHeterozygous
rearrangement" Malignant fibrous histiocytoma
4.4
Fig. 1. Rearrangements of the p53 gene in osteosarcomas. High-molecular-weight DNA was digested with fcoRI, and fragments were separated on a 0.7%agarose gel, blotted onto a nylon membrane, and hybridized with a cDNA probefrom lhep53 gene (pR4-2). Lane 1, leukocyte DNA from a normal control; Lanes2-11, DNAs from osteosarcoma tumors (see "Materials and Methods").
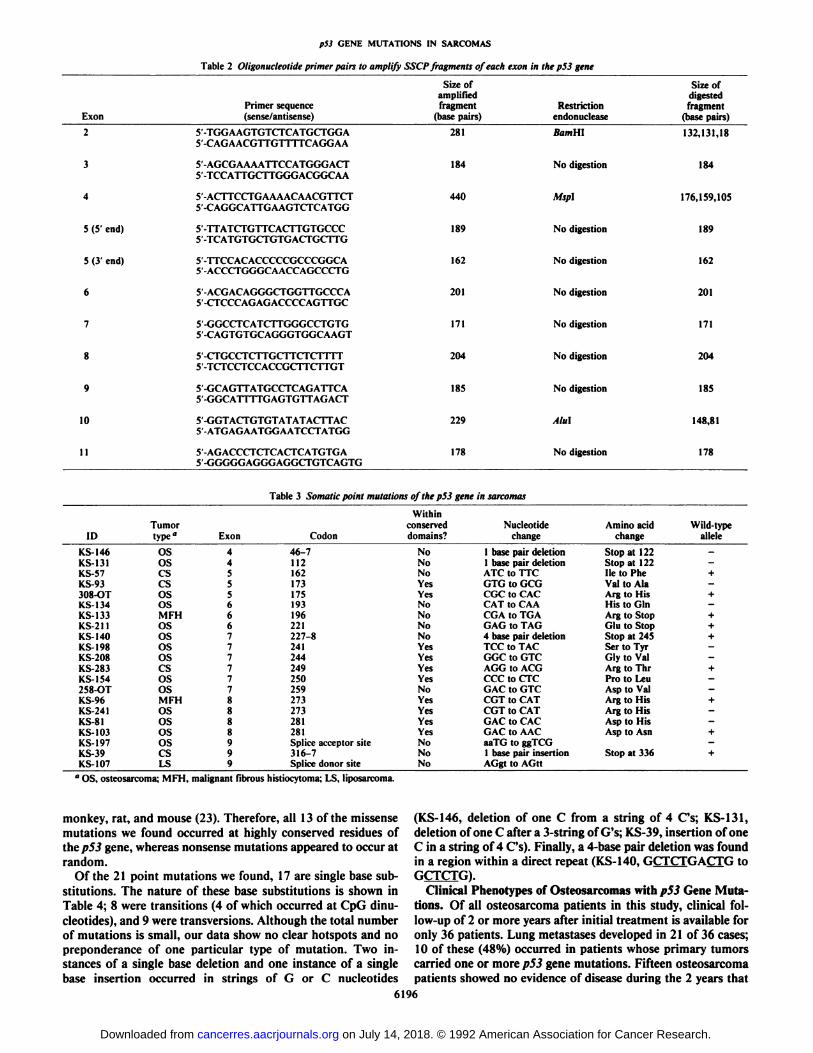
dure (22). Separate pairs of oligonucleotide primers were usedto amplify each of the/753 exons 2-11 except exon 5, for whichtwo overlapping PCR fragments were analyzed (Table 2). Theexon-containing PCR fragments included at least 10 base pairsof 5' and 3' flanking sequences surrounding each exon. To
increase the sensitivity of the SSCP technique for some regions,PCR-amplified DNA fragments from some exons were digestedby an appropriate restriction endonuclease to reduce the size ofthe PCR fragments to approximately 200 base pairs (Table 2).To further characterize variants, samples that showed a variantalÃeleby SSCP analysis were analyzed by direct genomic sequencing (21). To confirm that the majority of mutations in thep53 gene were detected by this screening process, we carried outDNA sequence analysis on 36 "SSCP-negative" samples in
exon 5 and 48 samples in exon 7. This analysis revealed noadditional point mutations (data not shown).
A total of 21 subtle alterations including point mutations,small deletions, and an insertion were identified and characterized by this screening method (Table 3). Examples of SSCPanalysis and sequences showing mutations are presented inFigs. 2 and 3, respectively. Of these 21 mutations, 14 werefound in osteosarcomas, 4 occurred in chondrosarcomas, 2 inmalignant fibrous histiocytoma, and one mutation was found ina liposarcoma. None of these tumors had a gross alteration ofthep53 gene except one case (KS-103), in which a heterozygous
rearrangement was found. The point mutation found in thistumor was also heterozygous. These results suggest that twodifferent mutations occurred in each of two copies of the p53gene in this case. Although we cannot exclude the possibilitythat two mutations occurred on the same alÃele,we believe thatthis is unlikely.
Characteristics of Point Mutations Found in Sarcomas. Thelocations of the 21 point mutations we found are shown in Fig.4. Of these, 13 mutations were single-base changes that causeamino acid substitutions in the p53 protein, and 6 are nonsensemutations that result in premature termination codons due todeletion, insertion, or the direct creation of a stop codon due toa single base substitution. The remaining two mutations wereintronic mutations at canonical splice junctions. The majorityofp53 gene mutations in various types of cancers were found infive evolutionarily conserved domains (8, 9, 23). Of the 21 pointmutations described in this study, 10 (48%) occurred in theconserved domains; all 10 of these were missense mutations.Three more missense mutations (162"c to 162phc, 193His to193Gln, 259Asp to 259Val) were found outside the conserved
domains but occurred at codons that are identical in human,6195
on July 14, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
p53 GENE MUTATIONS IN SARCOMAS
Table 2 Oligonucleotide primer pairs to amplify SSCP fragments of each exon in the p53 gene
Exon2345
(5'end)5
(3'end)678910IIPrimer
sequence(sense/antisense)5
-TGGAAGTGTCTCATGCTGGA5-CAGAACGTTGTTTTCAGGAA5'-AGCGAAAATTCCATGGGACT5
-TCCATTGCTTGGGACGGCAA5'-ACTTCCTGAAAACAACGTTCT5'-CAGGCATTGAAGTCTCATGG5
-TTATCTGTTCACTTGTGCCC5-TCATGTGCTGTGACTGCTTGS'-TTCCACACCCCCGCCCGGCA5
-ACCCTGGGCAACCAGCCCTG5
-ACGACAGGGCTGGTTGCCCA5'-CTCCCAGAGACCCCAGTTGC5
-GGCCTCATCTTGGGCCTGTG5-CAGTGTGCAGGGTGGCAAGT5
-CTGCCTCTTGCTTCTCTTTT5-TCTCCTCCACCGCTTCTTGT5'-GCAGTTATGCCTCAGATTCA5
-GGCATTTTGAGTGTrAGACT5
GGTACTGTGTATATACTTAC5-ATGAGAATGGAATCCTATGG5
-AGACCCTCTCACTCATGTGAS'-GGGGGAGGGAGGCTGTCAGTGSize
ofamplifiedfragment(base
pairs)281184440189162201171204185229178Restrictionendonucleaseft/millNo
digestionMsplNo
digestionNo
digestionNo
digestionNo
digestionNo
digestionNo
digestionAlulNo
digestionSize
ofdigestedfragment(base
pairs)132,131,18184176,159,105189162201171204185148,81178Table
3 Somatic point mutations of the p53 gene insarcomasIDKS-146KS-131KS-57KS-93308-OTKS-134KS-133KS-211KS-140KS-198KS-208KS-283KS-1S4258-OTKS-96KS-241KS-81KS-103KS-197KS-39KS-107Tumortype"OSOScscsOSOSMFHOSOSOSOScsOSOSMFHOSOSOSOScsLSExon
Codon4
46-741125162517351756193619662217227-87241724472497250725982738273828182819
Splice acceptorsite9316-79
Splice donor siteWithinconserveddomains?NoNoNoYesYesNoNoNoNoYesYesYesYesNoYesYesYesYesNoNoNoNucleotide
Aminoacidchangechange1
base pair deletion Stop at 1221base pair deletion Stop at 122ATC
to TTC Ile toPheGTGto GCG Val toAlaCGCto CAC Arg toHisCATto CAA His toGinCGAto TGA Arg toSlopGAGto TAG Glu toSlop4
base pair deletion Stop at245TCCto TAC Ser toTyrGGCto GTC Gly toValAGGto ACG Arg toThrCCCto CTC Pro toLeuGACto GTC Asp toValCGTto CAT Arg toHisCGTto CAT Arg toHisGACto CAC Asp toHisGACto AAC Asp toAsnaaTGtoggTCG1
base pair insertion Stop at336AGgtto AGttWild-typealÃele-—+—+—+++——+——+——+—+
'OS, osteosarcoma; MFH. malignant fibrous histiocytoma; LS. liposarcoma.
monkey, rat, and mouse (23). Therefore, all 13 of the missensemutations we found occurred at highly conserved residues oflhep53 gene, whereas nonsense mutations appeared to occur atrandom.
Of the 21 point mutations we found, 17 are single base substitutions. The nature of these base substitutions is shown inTable 4; 8 were transitions (4 of which occurred at CpG dinu-
cleotides), and 9 were transversions. Although the total numberof mutations is small, our data show no clear hotspots and nopreponderance of one particular type of mutation. Two instances of a single base deletion and one instance of a singlebase insertion occurred in strings of G or C nucleotides
(KS-146, deletion of one C from a string of 4 Cs; KS-131,deletion of one C after a 3-string of G's; KS-39, insertion of oneC in a string of 4 Cs). Finally, a 4-base pair deletion was foundin a region within a direct repeat (KS-140, GCTCTGACTG toGCTCTG).
Clinical Phenotypes of Osteosarcomas with pS3 Gene Mutations. Of all osteosarcoma patients in this study, clinical follow-up of 2 or more years after initial treatment is available foronly 36 patients. Lung métastasesdeveloped in 21 of 36 cases;10 of these (48%) occurred in patients whose primary tumorscarried one or more p53 gene mutations. Fifteen osteosarcomapatients showed no evidence of disease during the 2 years that
6196
on July 14, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

p53 GENE MUTATIONS IN SARCOMAS
a b
Fig. 2. a, SSCP analysis of exon 7 of the p53gene in a patient with osteosarcoma (KS-154).Control and KS-154 are DNAs from leukocytes,and KS-154-T is DNA from tumor tissues, b,SSCP analysis of exon 9 of the p53 gene in apatient with malignant fibrous histiocytoma(KS-39). c, SSCP analysis of exon 9 of the p53gene in a patient with liposarcoma (KS-107).Arrows, variant bands.
a
248Ara< G KS-154-T 319Lys/A
252L
c\T \250LeuC7
CTAG CTAG
ro
KS-107-T
CTAG C I A G
- g/t
Fig. 3. Direct genomic sequence analysis of the p53 gene in patients shown in Fig. 2. a, homozygous C to T transition at codon 250 in exon 7. h, homozygousinsertion of one C in a string of 4 Cs spanning codons 316-317 in exon 9. c, heterozygous G to T transversion at the splice donor site of intron 9.
6197
on July 14, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

p53 GENE MUTATIONS IN SARCOMAS
NH 1iExons
21Amino
AcidsV
V13
41!100T
1TTWWTi11ii567l
ll200Ì
WVll8
910l!300113<
Fig. 4. The distribution of somatic point mutations of the p53 gene in sarcomas. Top. schematic diagram of the p53 coding sequences with five conserveddomains (3). Bottom, regions spanned by exons 2 through 11. T. missense mutation: ". other types of mutations (nonsense or splicing mutations).
Table 4 Nature of base substitutions in the p53 gene mutations in sarcomas
SubstitutionsTransitionG:C
toA:TA:TtoG:CTransversionG:C
toT:AG:CtoC:GA:TtoC:GA:Tto T:ANumber
(%)8(47)6(35)2(12)9(53)4(23)2(12)0
(0)3(18)
followed their initial treatment. p53 gene mutations were identified in 7 patients from this group (47%). Therefore, no definite association was observed between p53 gene mutation andthe incidence of lung metastasis in the 36 patients for whom a24-month clinical follow-up was available.
Association of pS3 Gene Mutations with AlÃeleLoss on 17p.Allelic deletions on 17p were previously found in approximately 70% of osteosaroma tumors, and the common regionof alÃeleloss was assigned to 17pl3, where the p53 gene islocated (19, 24). These data suggest that allelic deletions on 17poccur as a second event following an initial mutation on thehomologous copy of p53 gene. Of 76 cases of osteosarcomaanalyzed in this study, 57 cases were informative (constitutionally heterozygous) for the analysis of alÃeleloss on 17p (Table5). In 39 of 57 cases (68%), tumor cells lost one alÃeleat one ormore polymorphic loci on 17p. Alterations in the p53 gene,including either gross rearrangements or point mutations, wereidentified in 51% (20 of 39) of tumors with alÃeleloss on 17p.No detectable mutations were found in the remaining 49% oftumors with alÃeleloss on 17p. Genetic alterations of the p53gene were found in 50% (9 of 18) of tumors that were informative for 17p markers and which retained both alÃelesat the p53locus.
DISCUSSION
A number of studies have been reported concerning the incidence of p53 gene mutations in human cancers, allowing acomparison of the mutation spectrum we found in sarcomaswith that seen in other types of malignant tumors. Our studyrevealed a relatively high incidence (21 of 126) of gross rearrangements of thep5J gene in sarcomas. This is consistent withprevious reports describing p53 gene mutations in sarcomas(15, 25-27). Point mutations of the p53 gene have also beenfound with a high frequency in various other types of cancers.However, only chronic myelogenous leukemia at blast crisisshowed an equivalent frequency of gross rearrangements of thep53 gene (28, 29). In these previous studies, the majority ofrearrangements in both sarcomas and chronic myelogenous leu
kemia were found to occur in intron 1, and tumors with rearrangements were shown to have no detectable mRNA of the/753gene (27-29), indicating that rearrangements of the p53 gene
COOH usually cause loss-of-function mutations. A similar pattern ofrearrangements was observed in our study, although a preciseanalysis of the rearrangement could not be performed in manycases because DNA from the primary tumor was limited.
The fraction of sarcomas with loss-of-function mutations inthe p53 gene increases significantly if point mutations leadingto nonsense errors are included. Of the 21 point mutations wefound, 6 mutations create a premature stop codon downstream,leading to the truncation of important regions known to complex with viral antigens or wild-type p53 protein (30, 31). Although it is likely that the two mutations found at splicing sitesof exon 9 are also loss-of-function mutations, neither RNA norprotein studies could be carried out on these tumors. Therefore,at least 27 (21 rearrangements and 6 nonsense mutations) of 42(64%) of thep53 gene mutations we identified in 127 sarcomascan be regarded as loss-of-function mutations.
Interestingly, we found a similar mutation spectrum amonggerm-line p53 gene mutations in sarcoma patients. Althoughthe number of cases was much smaller, 4 of 8 patients withgerm-line p53 gene mutations carried loss-of-function mutations (32). This predominance of loss-of-function mutations insarcomas is considerably different from the mutation spectrumreported for other types of cancer such as colorectal carcinomasor non-small cell lung cancer, where missense errors accountedfor 94% (30 of 32 cases) or 91% (21 of 23) of all mutations,respectively (10, 12). Since the number of potential targets fornonsense mutations is theoretically much greater than that forfunctionally significant missense mutations, it is reasonable toexpect that the spectrum of single base substitutions would bebroad in tumors with a tendency toward loss-of-function mutations. Indeed, the mutations we observed in 127 sarcomasspanned a wide region of the p53 gene, from codons 46 to 316.Our findings emphasize the importance of screening, at a minimum, the entire p53 coding region if an accurate evaluation ofthe spectrum of p53 gene mutations is sought.
Our detection of a disproportionately low frequency of p53gene mutations among tumors with alÃeleloss on 17p is quiteinteresting with respect to the "two-hit hypothesis" of tumor
suppressor gene action. By comparison, a study ofp53 mutationand allelic loss on 17p in colorectal carcinoma has shown that86% of tumors with 17p allelic loss carried a mutation in thep53 gene (10). Several explanations could account for the disparate findings in these two studies. First, studies that do notinclude a detailed analysis of the promoter and exon 1 mightunderestimate the overall frequency ofp53 mutations by failingto detect mutations in regulatory regions, as have been described for another tumor suppressor, the retinoblastoma gene(33). Since the promoter region of the p53 gene was not analyzed in our study, the possibility exists that we missed suchmutations; hence, it is possible that most of the tumors withalÃeleloss in our study do carry p53 mutations, but they wentundetected. A second possibility is that the p53 gene was not
Table 5 The association of allelic deletions on 17p with the p53 gene mutations
LOHon17p+Detectable
mutationRearrange
ments107Point mutations102Total20
9No
detectablemutation
Total19
9
6198
on July 14, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

p53 GENE MUTATIONS IN SARCOMAS
mutated in some tumors with allelic losses on 17p. The presence of another tumor suppressor gene on 17p telomeric to thep53 locus has been postulated in studies of breast cancer (34)and hepatocellular carcinoma (35) and could account for ourdata. Finally, a third possibility is that allelic loss at the p53locus might itself have oncogenic potential in some target cellsand would thus precede detectable mutations at the remaininglocus in these cases. In colorectal carcinomas, missense mutations are supposed to precede allelic deletions; the mutant proteins are assumed to create a growth advantage through a "dominant negative" effect. Therefore, cells with one mutant alÃele
may lose the remaining normal alÃeleduring a phase of rapidproliferation (10). Thus, a simple quantitative decrease of thep53 gene product might be enough to endow cells with a growthadvantage in a tissue-specific manner. The fact that approximately one-half of tumors without allelic deletions on 17p inthis study have an alteration of the p53 gene may support thismodel. This hypothesis would be more compelling if expressionof the two alÃelesof the p53 gene is not equivalent in all cells, ashas been shown for other genes (36, 37). A precise analysis ofthe expression of the p53 gene in tumors without detectablemutations would further our understanding of this issue.
ACKNOWLEDGMENTS
We thank Dr. T. Dryja for his cooperation; Drs. N. Takada, T.Umeda, N. Kawaguchi, Y. Kaneko, I. Spiro, A. Rosenberg, and H.Halprin for providing materials and clinical information; Dr. Y. Naka-mura for polymorphic probes on 17p; Drs. H. Koeffler and C. Miller fora cDNA clone of the p53 gene; Drs. L. Crawford and S. Tuck for agenomic clone containing the p53 gene; and Drs. B. Ludeke, R. Chung,J. Whaley, G. Cowley, and B. Seizinger for technical advice.
REFERENCES1. Finlay. C., Hinds, P., and Levine, A. The p53 proto-oncogene can act as a
suppressor of transformation. Cell, 57: 1083-1093, 1989.2. Baker, S., Markowitz, S., Fearon, E., Willson, J., and Vogelstein, B. Sup
pression of human colorectal carcinoma cell growth by wild-type p53. Science(Washington DC). 249: 912-915, 1990.
3. Fields. S., and Jang, S. Presence of a potent transcription activating sequencein the p53 protein. Science (Washington DC), 249: 1046-1049, 1990.
4. Raycroft, L., Wu, H., and Lozano, G. Transcriptional activation by wild-typebut not transforming mutants of thep53 anti-oncogene. Science (WashingtonDC), 249: 1049-1051, 1990.
5. Braithwaite, A., Sturzbecher, H-W., Addison, C., Palmer, C.. Rudge. K., andJenkins, J. Mouse p53 inhibits SV 40 origin-dependent DNA replication.Nature (Lond.), 329: 458-460, 1987.
6. Gannon, J., and Lane, D. pS3 and DNA polymerase a compete for binding toSV 40 antigen. Nature (Lond.), 329: 456-458, 1987.
7. Kern, S.. Kinzler, K., Bruskin, A.. Jarosz, D., Friedman, P., Prives, C., andVogelstein, B. Identification o(p53 as a sequence-specific DNA-binding protein. Science (Washington DC), 252: 1708-1711, 1991.
8. Levine, A., Momand, J., and Finlay. C. The p53 tumor suppressor gene.Nature (Lond.), 351: 453-456, 1991.
9. Hollstein, M., Sidransky, D., Vogelstein, B., and Harris, C. p53 mutations inhuman cancer. Science (Washington DC), 25J: 49-53, 1991.
10. Baker. S.. Presinger. A., Jessup, J., Paraskeva, C., Markowitz. S.. Willson. J.,Hamilton, S., and Vogelstein. B. p53 gene mutations occur in combinationwith 17p allelic deletions as late events in colorectal tumorigenesis. CancerRes., 50:7717-7722, 1990.
11. Gaidano. G., Ballerini, P., Gong, J., Inghirami. G., Neri, A., Newcomb, E.,Magrath, I., Knowles, D., and Dalia-Pavera. R. pS3 mutations in humanlymphoid malignancies: association with Burkitt lymphoma and chronic lym-phocytic leukemia. Proc. Nati. Acad. Sci. USA, 88: 5413-5417, 1991.
12. Chiba, I., Takahashi, T., Ñau, M., D'Amico, D., Curie!, D., Mitsudomi, T.,Buchhagen, D., Carbone. D., Piantadosi, S., Koga, H., Reissman, P., Sla-mon. D., Holmes, E., and Minna. J. Mutations in the p53 gene are frequentin primary, resected non-small cell lung cancer. Oncogene. 5: 1603-1610,1990.
13. Hsu, I., Metcalf, R.. Sun, T., Welsh, J., Wang, N., and Harris, C. Mutationalhotspot in the p53 gene in human hepatocellular carcinomas. Nature (Lond.).550:427-428, 1991.
14. Bressac, B., Kew. M.. Wands, J., and Ozturk, M. Selective G to T mutationsof p53 in hepatocellular carcinoma from southern Africa. Nature (Lond.),550:429-431. 1991.
15. Masuda, H., Miller. C.. Koeffler, H., Battifora, H., and Cline, M. Rearrangement of the p53 gene in human osteogenic sarcomas. Proc. Nati. Acad. Sci.USA, «4:7716-7719, 1987.
16. Maniatis, T., Fritsch, J., and Sambrook, J. (eds.). Molecular Cloning: aLaboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Press,1982.
17. Harlow. E., Williamson, N.. Ralston, R., Helfman. D.. and Adams, T. Molecular cloning and in vitro expression of a cDNA clone for human cellulartumor antigen p53. Mol. Cell. Biol., 5: 1601-1610, 1985.
18. Lamb. P., and Crawford, L. Characterization of the human p53 gene. Mol.Cell. Biol.. 6: 1379-1385, 1986.
19. Toguchida, J., Ishizaki, K., Nakamura. Y., Sasaki, M., Ikenaga, M., Kalo,M., Sugimoto, M.. Kotoura, Y., and Yamamuro. T. Assignment of commonalÃeleloss in osteosarcoma to the subregion 17pl3. Cancer Res., 49: 6247-6251, 1989.
20. Nakamura, Y., Lathrop. M., O'Connell, P., Leppert, M.. Barker, D., Wright.
E.. Skolonick, M.. Kondoleon, S., Litt, S., Lalouel, J., and White, R. Amapped set of DNA markers for human chromosome 17. Genomics, 2:302-309, 1988.
21. Yandell, D., and Dryja, T. Detection of DNA sequence polymorphisms byenzymatic amplification and direct genomic sequencing. Am. J. Hum. Genet., 45: 547-555, 1989.
22. Orila. M., Suzuki. Y., Sekiya, T., and Hayashi, K. Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerasechain reaction. Genomics, 5: 874-879, 1989.
23. Soussi, T., de Fromentel, C., and May. P. Structural aspects of the p53protein in relation to gene evolution. Oncogene, 5: 945-952, 1990.
24. Toguchida. J., Ishizaki, K., Sasaki, M., Ikenaga, M., Sugimoto, M., Kotoura,Y.. and Yamamuro, T. Chromosomal reorganization for the expression ofrecessive mutation of rctinoblastoma susceptibility gene in the developmentof osteosarcoma. Cancer Res., 48: 3939-3943, 1988.
25. Mulligan, L., Matlashewski, G., Scrable, H., and Cavenee, W. Mechanismsof p53 loss in human sarcomas. Proc. Nati. Acad. Sci. USA, 87: 5863-5867,1990.
26. Stratton, M., Moss. S., Warren, W., Patterson, H., Clark. J., Fisher, C.,Fletcher, C., Ball, A., Thomas, M., Gusterson, B.. and Cooper, C. Mutationof the p53 gene in human soft tissue sarcomas: association with abnormalitiesof the RBI gene. Oncogene, 5: 1297-1301. 1990.
27. Miller, C, Aslo, A., Tsay, C., Slamon, D.. Ishizaki, K., Toguchida. J., Yamamuro. T., Lampkin. B., and Koeffler, H. Frequency and structure of p53rearrangements in human osteosarcoma. Cancer Res., 50: 7950-7954. 1990.
28. Ahuja, H., Bar-Eli. M., Advani, S., Benchimol, S., and Cline, M. Alterationsin the p53 gene and the clonal evolution of the balst crisis of chronic myelo-cytic leukemia. Proc. Nati. Acad. Sci. USA. «6:6783-6787, 1989.
29. Kelman, Z., Prokocimer, M., Peller, S., Kahn, Y.. Rechavi. G., Manor, Y.,Cohen. A., and Rotter, V. Rearrangements in the p53 gene in Philadelphiachromosome positive chronic myelogenous leukemia. Blood, 74: 2318-2324,1989.
30. Soussi, T., de Fromental, C, Sturzbecher, H., Ullrich, S., Jenkins. J., andMay, P. Evolutionary conservation of the biochemical properties of p53:specific interactions of Xenopus laevis p53 with simian virus large T antigenand mammalian heat shock proteins 70. J. Virol., 63: 3894-3901, 1989.
31. Milner, J., Medcalf. E.. and Cook, A. Tumor suppressor p53: analysis ofwild-type and mutant p53 complexes. Mol. Cell. Biol., //: 12-19, 1991.
32. Toguchida, J.. Yamaguchi, T., Dayton. S., Beauchamp, R.. Herrera, G.,Ishizaki. K., Yamamuro.T., Meyers. P., Little, J., Sasaki, M., Weichselbaum,R., and Yandell. D. Prevalence and spectrum of germ-line p53 gene mutations among patients with sarcoma. N. Engl. J. Med., 326:1301-1308. 1992.
33. Sakai, T.. Ohtani, N., McGee, T., Robins, P., and Dryja, T. Oncogenicgerm-line mutations in Spl and AFT sites in the human retinoblastoma gene.Nature (Lond.), 353: 83-86, 1991.
34. Sato, T., Tanigami, A., Yamakawa, K., Akiyama, F., Kasumi, F., Sakamoto,G., and Nakamura. Y. Allelotypc of breast cancer: accumulative alÃelelossespromote tumor progression in primary breast cancer. Cancer Res.. 50: 7184-7189, 1990.
35. Fujimori, M., Tokino, T., Hiño,O., Kitagawa. T.. Imamura, T.. Okamolo,E., Mitsunobu, M., Ishikawa, T., Nakagama, H., Harada, H., Yagura. M.,Matsubara, K.. and Nakamura. Y. Allelotype study of primary hepatocellularcarcinoma. Cancer Res., 51: 89-93, 1991.
36. Barlow. D., Sloger, R., Herrmann, B., Saito, K., and Schweifer, N. Themouse insulin-like growth factor type-2 receptor is imprinted and closelylinked to the 7m? locus. Nature (Lond.), 349: 84-87. 1991.
37. DeChiara, T., Robertson, E., and Efstratiadis. A. Parental imprinting of themouse insulin-like growth factor II gene. Cell, 64: 849-859, 1991.
6199
on July 14, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

1992;52:6194-6199. Cancer Res Junya Toguchida, Toshikazu Yamaguchi, Bruce Ritchie, et al. Sarcomas
Gene in Bone and Soft Tissuep53Mutation Spectrum of the
Updated version
http://cancerres.aacrjournals.org/content/52/22/6194
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/52/22/6194To request permission to re-use all or part of this article, use this link
on July 14, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from


















