
Multilevel approach to the prediction of properties of organic compounds in the framework of the...
4
ISSN 0012-5008, Doklady Chemistry, 2009, Vol. 427, Part 1, pp. 172–175. © Pleiades Publishing, Ltd., 2009. Original Russian Text © I.I. Baskin, N.I. Zhokhova, V.A. Palyulin, A.N. Zefirov, N.S. Zefirov, 2009, published in Doklady Akademii Nauk, 2009, Vol. 427, No. 3, pp. 335–339. 172 Nowadays, the development of methodology of constructing quantitative structure–activity and struc- ture–property relationship (QSAR/QSPR) models aimed at improving the descriptor representation of chemical compounds and at applying increasingly sophisticated methods of analysis has achieved the sat- uration level when the available methods make it possi- ble to extract from databases almost all information useful for prediction. As stated in [1], in most cases, the predictive power of models constructed with the use of “fairly good” sets of descriptors and fairly good meth- ods of data processing depends only slightly on both the descriptor set and the method used and is nearly completely determined by the database used for con- structing a model. Thus, further improvement of the descriptor representation of chemical compounds and the introduction of new machine-learning methods will lead only to little progress, whereas radically new ideas are required for the actual breakthrough in this direc- tion to overcome the limitations caused by a lack of useful information in chemical databases. Meanwhile, there is a fundamental difference between machine-learning and human-learning meth- ods [1]. Machine data analysis needs a very large vol- ume of data to construct a reliable statistical model of some complexity, whereas humans require a surpris- ingly small number of examples to learn much more complicated concepts. One reason for this is that every new statistical model is constructed almost from the beginning and the models thus obtained are isolated from each other. When solving a problem, humans are always guided by the experience acquired while solv- ing other problems. Even when humans digest novel information, they always use analogies taken from pre- vious knowledge. Finally, the components of acquired knowledge are tightly interconnected in the human brain, which accelerates and facilitates many times over the process of obtaining new knowledge. The under- standing of this fact in recent years has lead to the development of the machine learning theory in a new direction, conventionally referred to as inductive knowledge transfer, which studies how interrelation of different tasks of data processing improves the quality of the resulting models [1]. Thus, one way of overcoming the limitations caused by insufficient information contained in separate chem- ical databases is to consider various properties of chem- ical compounds in interrelation to one another and, with allowance for this, to construct interrelated rather than isolated structure–property models. It is expected that such a strategy will lead to data integration where the useful information volume for each property will be considerably increased owing to the efficient use of information concerning other properties tightly related to a given one. We can also assume that the less the amount of available experimental data on a target prop- erty and the larger the amount of experimental data on other properties related to the given one, the more effi- cient will be the transfer of necessary information in constructing a model for prediction of this property. The possibility of constructing interrelated struc- ture–property models was demonstrated by us back in 1993 for an artificial neural network with seven outputs which could simultaneously predict seven physical properties of alkanes [2]. Inasmuch as this study was undertaken long before the first works concerning inductive knowledge transfer in machine-learning methods were initiated, no systematic studies of the effect of the simultaneous prediction of several proper- ties as compared with separate prediction of these prop- erties were at that moment performed. Such a system- atic study was recently carried in [1] dealing with the prediction of 11 tissue–air partition coefficients. It was demonstrated that inductive knowledge transfer approaches, such as multitask learning (i.e., parallel construction of interrelated models performed using a neural network and PLS method with several outputs) and feature net (i.e., sequential construction of related Multilevel Approach to the Prediction of Properties of Organic Compounds in the Framework of the QSAR/QSPR Methodology I. I. Baskin, N. I. Zhokhova, V. A. Palyulin, A. N. Zefirov, and Academician N. S. Zefirov Received January 16, 2009 DOI: 10.1134/S0012500809070076 Moscow State University, Moscow, 119991 Russia CHEMISTRY
Transcript of Multilevel approach to the prediction of properties of organic compounds in the framework of the...

ISSN 0012-5008, Doklady Chemistry, 2009, Vol. 427, Part 1, pp. 172–175. © Pleiades Publishing, Ltd., 2009.Original Russian Text © I.I. Baskin, N.I. Zhokhova, V.A. Palyulin, A.N. Zefirov, N.S. Zefirov, 2009, published in Doklady Akademii Nauk, 2009, Vol. 427, No. 3, pp. 335–339.
172
Nowadays, the development of methodology ofconstructing quantitative structure–activity and struc-ture–property relationship (QSAR/QSPR) modelsaimed at improving the descriptor representation ofchemical compounds and at applying increasinglysophisticated methods of analysis has achieved the sat-uration level when the available methods make it possi-ble to extract from databases almost all informationuseful for prediction. As stated in [1], in most cases, thepredictive power of models constructed with the use of“fairly good” sets of descriptors and fairly good meth-ods of data processing depends only slightly on boththe descriptor set and the method used and is nearlycompletely determined by the database used for con-structing a model. Thus, further improvement of thedescriptor representation of chemical compounds andthe introduction of new machine-learning methods willlead only to little progress, whereas radically new ideasare required for the actual breakthrough in this direc-tion to overcome the limitations caused by a lack ofuseful information in chemical databases.
Meanwhile, there is a fundamental differencebetween machine-learning and human-learning meth-ods [1]. Machine data analysis needs a very large vol-ume of data to construct a reliable statistical model ofsome complexity, whereas humans require a surpris-ingly small number of examples to learn much morecomplicated concepts. One reason for this is that everynew statistical model is constructed almost from thebeginning and the models thus obtained are isolatedfrom each other. When solving a problem, humans arealways guided by the experience acquired while solv-ing other problems. Even when humans digest novelinformation, they always use analogies taken from pre-vious knowledge. Finally, the components of acquiredknowledge are tightly interconnected in the humanbrain, which accelerates and facilitates many times over
the process of obtaining new knowledge. The under-standing of this fact in recent years has lead to thedevelopment of the machine learning theory in a newdirection, conventionally referred to as inductiveknowledge transfer, which studies how interrelation ofdifferent tasks of data processing improves the qualityof the resulting models [1].
Thus, one way of overcoming the limitations causedby insufficient information contained in separate chem-ical databases is to consider various properties of chem-ical compounds in interrelation to one another and,with allowance for this, to construct interrelated ratherthan isolated structure–property models. It is expectedthat such a strategy will lead to data integration wherethe useful information volume for each property will beconsiderably increased owing to the efficient use ofinformation concerning other properties tightly relatedto a given one. We can also assume that the less theamount of available experimental data on a target prop-erty and the larger the amount of experimental data onother properties related to the given one, the more effi-cient will be the transfer of necessary information inconstructing a model for prediction of this property.
The possibility of constructing interrelated struc-ture–property models was demonstrated by us back in1993 for an artificial neural network with seven outputswhich could simultaneously predict seven physicalproperties of alkanes [2]. Inasmuch as this study wasundertaken long before the first works concerninginductive knowledge transfer in machine-learningmethods were initiated, no systematic studies of theeffect of the simultaneous prediction of several proper-ties as compared with separate prediction of these prop-erties were at that moment performed. Such a system-atic study was recently carried in [1] dealing with theprediction of 11 tissue–air partition coefficients. It wasdemonstrated that inductive knowledge transferapproaches, such as multitask learning (i.e., parallelconstruction of interrelated models performed using aneural network and PLS method with several outputs)and feature net (i.e., sequential construction of related
Multilevel Approach to the Prediction of Properties of Organic Compounds in the Framework
of the QSAR/QSPR Methodology
I. I. Baskin, N. I. Zhokhova, V. A. Palyulin, A. N. Zefirov, and
Academician
N. S. Zefirov
Received January 16, 2009
DOI:
10.1134/S0012500809070076
Moscow State University, Moscow, 119991 Russia
CHEMISTRY
DOKLADY CHEMISTRY
Vol. 427
Part 1
2009
MULTILEVEL APPROACH TO THE PREDICTION OF PROPERTIES 173
models), significantly improve the predictive power ofstructure–property models as compared with the rou-tine method of constructing isolated models.
The essence of the suggested multilevel approach tothe prediction of properties of organic compounds inthe framework of the QSAR/QSPR methodology is asfollows. Properties of organic compounds are predictedin the framework of the fragmental approach [3, 4].This makes it possible to use the advantages of the frag-mental approach, such as the high speed and unique-ness of calculations, as well as the natural interpretation
of the models in terms of elements of structural formu-las of organic compounds. In addition, due to the basischaracter of fragment descriptors [5], the latter shouldprovide the opportunity to approximate any structure–property relationships, including very complicatedones. At the same time, it is suggested to use a multi-layer network of models, in which the outputs of themodels of previous layers are the inputs for the modelsof the next layers, rather than the single-level model,which uses the values of fragment descriptors as inputsand yields the values of predicted properties as outputs.It is worth noting that such an organization of the mod-els resembles stepwise information processing thatoccurs in the multilayer structures of the cerebral cor-tex. It is required that each intermediate model gives atthe output either experimentally measureable quantitiesor clearly interpreted calculated quantities. This allowseach model to use its structure–property database thatshould be used for constructing this model. In this case,the multilevel organization of models makes it possibleto efficiently perform inductive knowledge transferfrom the models of a previous layer to the models of a
Predicted property
Model
Fragmentdescriptors
Predictedproperty
Model
2
Model
1–1
Model
1–
N
Fragment descriptors
(‡)
(b)
2
1
Prediction
3
2
1
0 1
Experiment
Prediction
2
7
1
–1–2–3–1
–2
–3
–4
Experiment
65432
3
4
5
6
7(‡)
(b)
Fig. 1.
(a) Single-level and (b) multilevel approaches toconstructing the QSAR/QSPR models
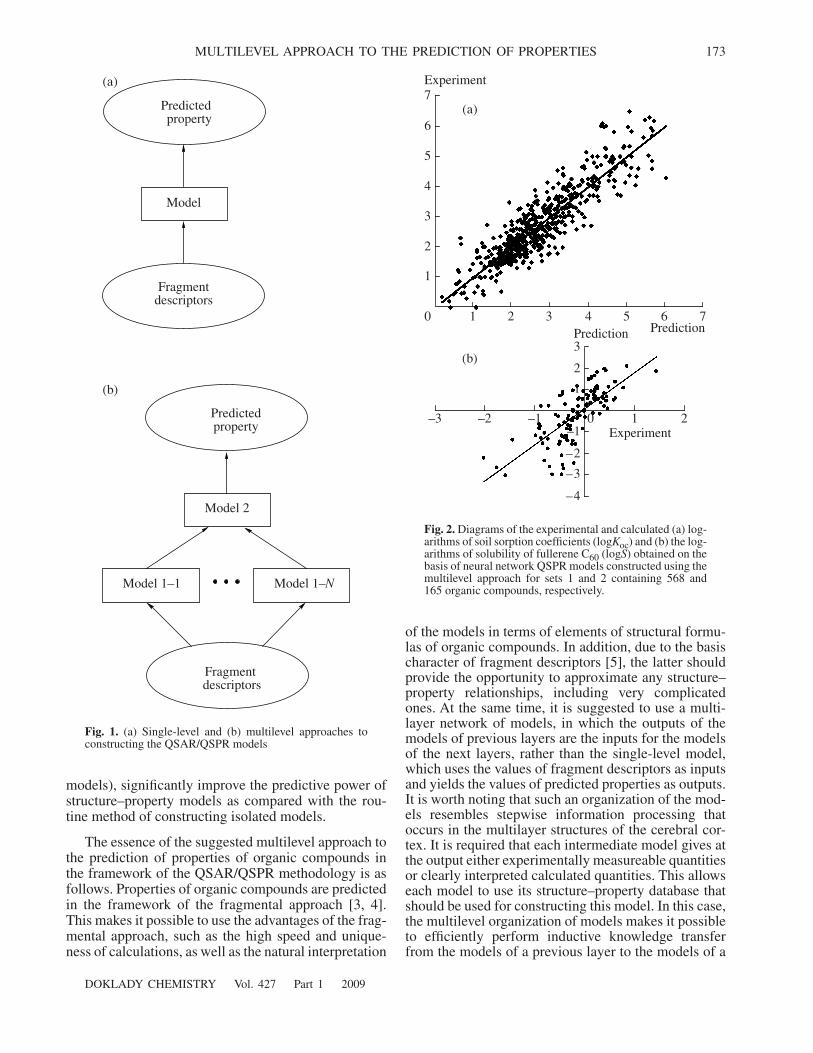
Fig. 2.
Diagrams of the experimental and calculated (a) log-arithms of soil sorption coefficients (log
K
oc
) and (b) the log-arithms of solubility of fullerene C
60
(log
S
) obtained on thebasis of neural network QSPR models constructed using themultilevel approach for sets 1 and 2 containing 568 and165 organic compounds, respectively.
0

174
DOKLADY CHEMISTRY
Vol. 427
Part 1
2009
BASKIN et al.
next layer, which should ensure the improvement of thelatter since they use additional information taken inex-plicitly from other databases. We believe that, for thisprocess to be efficient, the models of a previous layershould learn on significantly larger data sets than themodels of a next layer. The schemes of the single-leveland multilevel approaches to the construction ofQSAR/QSPR models are shown in Fig. 1.
Natural candidates for the role of output propertiesfor the intermediate models are physicochemical prop-erties related to fundamental types of interactions(hydrophobicity, polarizability, characteristics ofhydrogen bond strength, etc.), various constants of sub-stituents, and quantum-chemical characteristics(HOMO, LUMO, atomic charges). It is worth notingthat most of these characteristics have descriptors thathave long been used for constructing quantitative struc-ture–property relationships. A fundamental distinctionand advantage of the multilevel approach over thedirect use of physicochemical and quantum-chemicaldescriptors for constructing models is that there persiststhe aforementioned interpretation through fragmentdescriptors in terms of structural formulas. In addition,the universality and efficiency of calculation inherent infragment descriptors is also retained, which makes it
possible to use multilevel model networks in high-per-formance virtual screening.
It should be noted that the multilevel approach notonly improves the quality of prediction but also is ableto overcome disadvantages sometimes called disadvan-tages of the fragmental approach, namely the lack ofphysicochemical interpretation and the problem of“missing fragments” [4]. First of all, since the interme-diate models give at the output experimentally measur-able physical quantities, the final model itself has clearphysicochemical interpretation in terms of these quan-tities. For such interpretation with the use of neural net-work models, a method suggested in [6] can be used. Asfor the missing fragments, which are absent in the train-ing set but present in the test set, this problem is miti-gated since these fragments can be present in the chem-ical structures included in the set of considerably largersize used for training the models of previous layers.
Let us consider two examples that demonstrate theadvantages of using the multilevel approach. In thefirst case, on the basis of published data [7], weformed set 1 containing logarithms of soil sorptioncoefficients (log
K
oc
) for 568 organic compounds. In thesecond case, to form set 2, logarithms of solubility(log
S
) of fullerene C
60
in 165 organic solvents, includ-ing alkane, benzene, and naphthalene derivatives andnitrogen-, oxygen-, chlorine-, and bromine-containingcompounds, were taken from [8, 9]. In constructingquantitative structure–property models in the frame-work of the single-level approach, chemical com-pounds were described by means of sets of fragmentdescriptors [10] containing up to six non-hydrogenatoms. Preliminary selection of descriptors was per-formed by the fast stepwise multiple linear regression(FSMLR) method [11]. The selected descriptor setswere used for constructing neural network structure–property models by means of multilayer perceptrons[12]. When constructing models in the framework ofthe two-level approach, the models of the first levelwere obtained in precisely the same manner with theuse of fragment descriptors and a combination of theFSMLR method and multilayer perceptrons. Thesemodels made it possible to predict the lipophilicitylog
P
and four Abraham constants A, B, E, and S, whichcharacterize, respectively, the acidity and basicity withrespect to hydrogen bonding, excess molar refraction,and dipolarity or polarizability. For the lipophilicitymodel, set 3 was used containing 7805 compounds[13]; and for the Abraham constants, we used set 4 of457 compounds reported in [14].
The statistical characteristics of the first-level mod-els are presented in Table 1. In the second step, theresults of prognosis obtained with the use of the first-level models for the corresponding sets of the loga-rithms of soil sorption coefficients of organic com-pounds and the logarithms of solubilities of fullereneC
60
in organic solvents were used as descriptors in con-structing neural network models of the second level for
Table 1.
Statistical characteristics of the first-level struc-ture–property models for calculation of lipophilicity log
P
and Abraham constants, A, B, E, and S for organic com-pounds contained in data sets 3 and 4, respectively [13, 14]
Property
Numberof com-
pounds in the set
Correlationcoefficient
RMSE
for the training set
for the test set (1/10 of
the set)
log
P
7805 0.980 0.345 0.395
A 457 0.983 0.051 0.058
B 457 0.971 0.066 0.081
E 457 0.997 0.040 0.074
S 457 0.987 0.072 0.137
Table 1.
Statistical characteristics of the first-level struc-ture–property models for calculation of lipophilicity log
P
and Abraham constants, A, B, E, and S for organic com-pounds contained in data sets 3 and 4, respectively [13, 14]
Property
Numberof com-
pounds in the set
Correlationcoefficient
RMSE
for the training set
for the test set (1/10 of
the set)
log
P
7805 0.980 0.345 0.395
A 457 0.983 0.051 0.058
B 457 0.971 0.066 0.081
E 457 0.997 0.040 0.074
S 457 0.987 0.072 0.137
Table 2.
Comparative statistical characteristics of the struc-ture–property models for calculation of the logarithm of soilsorption coefficient of organic compounds (set 1, 568 com-pounds) and the logarithm of solubility of fullerene C
60
in or-ganic solvents (set 2, 165 solvents) obtained in the frame-work of the single-level and multilevel QSPR approaches
Property
Single-levelapproach
Multilevelapproach
RMSE
DCV
RMSE
DCV
log
K
oc
0.598 0.759 0.800 0.534
log
S
for fullerene C
60
0.448 0.912 0.637 0.739
QDCV2 QDCV
2

DOKLADY CHEMISTRY
Vol. 427
Part 1
2009
MULTILEVEL APPROACH TO THE PREDICTION OF PROPERTIES 175
calculation of these properties. In all cases, the predic-tive power of the models was estimated by means of the5
×
4-fold double cross validation procedure [11]. TheQSPR models were constructed with the NASAWIN
program package [15]. The values and the root-mean-square error of prediction
RMSE
DCV
for the mod-els obtained with the use of the single-level and multi-level approaches to the calculation of the logarithm ofsoil sorption coefficient of organic compounds and thelogarithm of solubility of fullerene
ë
60
are summarizedin Table 2. As is seen, the predictive power of the QSPRmodels obtained in the framework of the multilevelapproach significantly exceeds the predictive power ofsingle-level models, although all models were con-structed on the basis of the same sets of fragmentdescriptors by means of the same machine-learningmethod. The diagrams of the experimental and calcu-lated and values obtained on the basisof the multilevel neural network models are shown inFig. 2.
Thus, integration of even a small number of modelsinto a network can lead to a noticeable improvement ofthe predictive power of higher level models since thelatter take into account the information contained inadditional databases used for constructing lower levelmodels. We believe that the multilevel approach canconsiderably improve the prediction performance ofthe models used for predicting not only the physico-chemical properties, as was shown above for two exam-ples, but also the biological activity, which is the focusof our further work in this area. It can be foreseen thatin further development of the QSAR/QSPR methodol-ogy, isolated and unrelated single-level structure–prop-erty/structure–activity models will be replaced by thenetwork of interrelated models organized in the form ofthe “chemical brain” accommodating considerable vol-umes of experimental data and knowledge, which willserve to improve the quality of prediction of variousproperties of organic compounds.
REFERENCES
1. Varnek, A., Gaudin, C., Marcou, G., et al.,
J. Chem. Inf.Model.,
2009, vol. 49, pp. 133–144.2. Baskin, I.I., Palyulin, V.A., and Zefirov, N.S.,
Dokl.Akad. Nauk
, 1993, vol. 332, no. 6, pp. 713–716.3. Zefirov, N.S. and Palyulin, V.A.,
J. Chem. Inf. Comput.Sci.
, 2002, vol. 42, pp. 1112–1122.4. Baskin, I.I. and Varnek, A.,
ChemoinformaticsApproaches to Virtual Screening
, Varnek, A. and Trop-sha, A., Eds., Cambridge: RSC Publ., 2008, pp. 1–43.
5. Baskin, I.I., Skvortsova, M.I., Stankevich, I.V., andZefirov, N.S.,
J. Chem. Inf. Comput. Sci.,
1995, vol. 35,pp. 527–531.
6. Baskin, I.I., Ait, A.O., Halberstam, N.M., Palyulin, V.A.,and Zefirov, N.S.,
SAR QSAR Environ. Res
, 2002,vol.
13, no. 1, pp. 35–41.7. Huuskonen, J.,
J. Chem. Inf. Comput. Sci.,
2003, vol. 43,pp. 1457–1462.
8. Danauskas, S.M. and Jurs, P.C.,
J. Chem. Inf. Comput.Sci.,
2001, vol. 41, pp. 419–424.9. Marcus, Y.,
J. Phys. Chem. B
, 2001, vol. 105, pp. 2499–2506.
10. Artemenko, N.V., Baskin, I.I., Palyulin, V.A., andZefirov, N.S.,
Dokl. Chem.
, 2001, vol. 381, nos. 1–3,pp.
317–320 [
Dokl. Akad. Nauk,
2001, vol. 381, no. 2,pp. 203–206].
11. Zhokhova, N.I., Baskin, I.I., Palyulin, V.A., Zefirov,
A.N.,and Zefirov, N.S.,
Dokl. Chem.
, 2007, vol. 417, part 2,pp. 282–284.
12. Baskin, I.I., Palyulin, V.A., and Zefirov, N.S,
Ross.Khim. Zh.
, 2006, vol. 50, no. 2, pp. 86–96.13. Artemenko, N.V., Palyulin, V.A., and Zefirov, N.S.,
Dokl. Chem
., 2002, vol. 383, nos. 4–6, pp. 114–116[
Dokl. Akad. Nauk
, 2002, vol. 383, no. 6, pp. 771–773].14. Jover, J., Bosque, R., and Sales, J.,
J. Chem. Inf. Comput.Sci.
, 2004, vol. 44, pp. 1098–1106.15. Baskin, I.I., Halberstam, N.M., Artemenko, N.V., et al.,
in
EuroQSAR 2002, Designing Drugs and Crop Pro-tectants: Processes, Problems and Solutions
, Mel-bourne: Blackwell, 2003, pp. 260–263.
QDCV2
Koclog Slog


















