






mTOR/RICTOR is the Ser473 Kinase for Akt/PKB in 3T3-L1 ... · the proposed candidate kinases are...
26
1 mTOR/RICTOR is the Ser473 Kinase for Akt/PKB in 3T3-L1 Adipocytes * Richard C. Hresko and Mike Mueckler § From the Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri 63110 Running Title: Phosphorylation of Akt/PKB Ser-473 by mTOR/RICTOR The insulin-signaling pathway leading to the activation of Akt/PKB has been well- characterized except for a single step, the phosphorylation of Akt at Ser473. Double- stranded DNA -dependent p rotein k inase (DNA- PK). a taxia t elangiectasia m utated (ATM) gene product, I ntegrin-l inked k inase (ILK), P rotein K inase Cα (PKCα ) and m ammalian t arget of R apamycin (mTOR) when complexed to r apamycin i nsensitive c ompanion of mTOR (RICTOR) have all been identified as playing a critical role in Akt-Ser473 phosphorylation. However, the apparently disparate results reported in these studies are difficult to evaluate, given that different stimuli and cell types were examined and that all of the candidate proteins have never been systematically studied in a single system. Additionally, none of these studies were performed in a classical insulin-responsive cell- type or tissue such as muscle or fat. We therefore examined each of these candidates in 3T3-L1 adipocytes. In vitro kinase assays, using different subcellular fractions of 3T3-L1 adipocytes, revealed that phosphatidylinositol 3,4,5-trisphosphate (PIP3)-stimulated Ser473 phosphorylation correlated well with the amount of DNA-PK, mTOR, and RICTOR but did not correlate with levels of ATM, ILK, and PKCα . PKCα was completely absent from compartments with Ser473 phosphorylation activity. Although purified DNA-PK could phosphorylate a peptide derived from Akt that contains amino acid Ser473, it could not phosphorylate full-length Akt2. Vesicles immunoprecipitated from low-density microsomes (LDM) using antibodies directed against mTOR or RICTOR had PIP3- stimulated Ser473 activity that was sensitive to wortmannin but not staurosporine. In contrast, immunopurified LDM vesicles containing ILK could not phosphorylate Akt on Ser473 in vitro. Small interference RNA (siRNA)-knockdown of RICTOR, but not DNA- PK, ATM, or ILK, suppressed insulin-activated Ser473 phosphorylation and to a lesser extent Thr308 phosphorylation in 3T3-L1 adipocytes. Based on our cell-free kinase and siRNA results, we conclude mTOR complexed to RICTOR is the Ser473 kinase in 3T3-L1 adipocytes. Insulin increases glucose transport in muscle and fat by stimulating the translocation of the glucose transporter isoform Glut 4 from intracellular storage pools to the cell surface,(1,2) a process that requires the activation of the serine/threonine kinase Akt (PKB)(3-8). Although the molecular events that occur after Akt activation are poorly understood, the early insulin- signaling pathway has been well characterized except for one step, the phosphorylation of Akt at Ser-473. The signaling cascade is initiated when insulin binds to specific receptors at the plasma membrane (PM), triggering the autophosphorylation of several critical intracellular tyrosine residues and thereby activating an intrinsic tyrosine kinase that can phosphorylate cellular substrates, most notably the insulin receptor substrate (IRS) proteins(9-11). Tyrosine-phosphorylated IRS proteins can recruit and activate phosphoinositide 3-kinase (PI 3- kinase), which generates phosphatidylinositol 3,4,5-trisphosphate (PIP3) using inositol- containing phospholipids in the PM as substrates(12). The activity of Akt is markedly stimulated in a PI 3-kinase-dependent manner(13). This phenomenon predominately relies on the phosphorylation of Akt on two of its amino acid residues, 1) threonine 308 in the activation loop of the kinase catalytic domain and 2) serine 473 in the “hydrophobic motif” (HM) carboxy-terminal domain(14). The phosphorylations of both of these regulatory sites occur after the recruitment of Akt to the PM through the binding of its pleckstrin http://www.jbc.org/cgi/doi/10.1074/jbc.M508361200 The latest version is at JBC Papers in Press. Published on October 11, 2005 as Manuscript M508361200 Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc. by guest on May 16, 2019 http://www.jbc.org/ Downloaded from
Transcript of mTOR/RICTOR is the Ser473 Kinase for Akt/PKB in 3T3-L1 ... · the proposed candidate kinases are...

1
mTOR/RICTOR is the Ser473 Kinase for Akt/PKB in 3T3-L1 Adipocytes*
Richard C. Hresko and Mike Mueckler§ From the Department of Cell Biology and Physiology, Washington University
School of Medicine, St. Louis, Missouri 63110 Running Title: Phosphorylation of Akt/PKB Ser-473 by mTOR/RICTOR
The insulin-signaling pathway leading to the activation of Akt/PKB has been well-characterized except for a single step, the phosphorylation of Akt at Ser473. Double-stranded DNA-dependent protein kinase (DNA-PK). ataxia telangiectasia mutated (ATM) gene product, Integrin-linked kinase (ILK), Protein Kinase Cα (PKCα ) and mammalian target of Rapamycin (mTOR) when complexed to rapamycin insensitive companion of mTOR (RICTOR) have all been identified as playing a critical role in Akt-Ser473 phosphorylation. However, the apparently disparate results reported in these studies are difficult to evaluate, given that different stimuli and cell types were examined and that all of the candidate proteins have never been systematically studied in a single system. Additionally, none of these studies were performed in a classical insulin-responsive cell-type or tissue such as muscle or fat. We therefore examined each of these candidates in 3T3-L1 adipocytes. In vitro kinase assays, using different subcellular fractions of 3T3-L1 adipocytes, revealed that phosphatidylinositol 3,4,5-trisphosphate (PIP3)-stimulated Ser473 phosphorylation correlated well with the amount of DNA-PK, mTOR, and RICTOR but did not correlate with levels of ATM, ILK, and PKCα . PKCα was completely absent from compartments with Ser473 phosphorylation activity. Although purified DNA-PK could phosphorylate a peptide derived from Akt that contains amino acid Ser473, it could not phosphorylate full-length Akt2. Vesicles immunoprecipitated from low-density microsomes (LDM) using antibodies directed against mTOR or RICTOR had PIP3-stimulated Ser473 activity that was sensitive to wortmannin but not staurosporine. In contrast, immunopurified LDM vesicles containing ILK could not phosphorylate Akt on
Ser473 in vitro. Small interference RNA (siRNA)-knockdown of RICTOR, but not DNA-PK, ATM, or ILK, suppressed insulin-activated Ser473 phosphorylation and to a lesser extent Thr308 phosphorylation in 3T3-L1 adipocytes. Based on our cell-free kinase and siRNA results, we conclude mTOR complexed to RICTOR is the Ser473 kinase in 3T3-L1 adipocytes.
Insulin increases glucose transport in
muscle and fat by stimulating the translocation of the glucose transporter isoform Glut 4 from intracellular storage pools to the cell surface,(1,2) a process that requires the activation of the serine/threonine kinase Akt (PKB)(3-8). Although the molecular events that occur after Akt activation are poorly understood, the early insulin-signaling pathway has been well characterized except for one step, the phosphorylation of Akt at Ser-473. The signaling cascade is initiated when insulin binds to specific receptors at the plasma membrane (PM), triggering the autophosphorylation of several critical intracellular tyrosine residues and thereby activating an intrinsic tyrosine kinase that can phosphorylate cellular substrates, most notably the insulin receptor substrate (IRS) proteins(9-11). Tyrosine-phosphorylated IRS proteins can recruit and activate phosphoinositide 3-kinase (PI 3-kinase), which generates phosphatidylinositol 3,4,5-trisphosphate (PIP3) using inositol-containing phospholipids in the PM as substrates(12). The activity of Akt is markedly stimulated in a PI 3-kinase-dependent manner(13). This phenomenon predominately relies on the phosphorylation of Akt on two of its amino acid residues, 1) threonine 308 in the activation loop of the kinase catalytic domain and 2) serine 473 in the “hydrophobic motif” (HM) carboxy-terminal domain(14). The phosphorylations of both of these regulatory sites occur after the recruitment of Akt to the PM through the binding of its pleckstrin
http://www.jbc.org/cgi/doi/10.1074/jbc.M508361200The latest version is at JBC Papers in Press. Published on October 11, 2005 as Manuscript M508361200
Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

2
homology (PH) domain to PIP3(15) and are completely ablated in vivo by the PI 3-kinase inhibitor wortmannin(16). The protein kinase responsible for phosphorylating Akt on Thr308 is phosphoinositide-dependent kinase 1 (PDK1)(17-19). The identity of the kinase responsible for phosphorylating Akt on Ser473 is controversial.
Akt is part of the protein kinase AGC (cAMP-dependent, cGMP-dependent, and protein kinase C) subfamily whose members are activated in a manner similar to Akt(20). All members possess a phosphorylation site in the activation loop equivalent to Thr308 while only some require phosphorylation at the HM site. Some AGC members have a negatively charged acidic residue instead of a S/T in the C-terminal hydrophobic domain(21). Phosphorylation of the HM site or substitution with an acidic residue provides a docking site to recruit PDK1 and stimulate the phosphorylation of Thr308(20). X-ray crystallographic studies have suggested that in the inactive state the activation loop of Akt adopts a disordered structure that prevents the binding of ATP and protein substrates(22). Phosphorylation of Ser473 results in a disorder to order transition allowing interaction between the HM domain and the N-terminal lobe leading to the activation of the kinase(23).
At least ten kinases have been proposed to function as the hydrophobic motif domain protein kinase (HMD-PK) that phosphorylates Akt on Ser473, including Integrin-linked kinase (ILK), Protein Kinase Cα (PKCα), double-stranded DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia mutated (ATM) gene product, and the mammalian target of Rapamycin (mTOR)(24). It has also been suggested that Akt can undergo autophosphorylation at Ser473(25), however, this is most likely not the mechanism by which insulin stimulates Ser473 phosphorylation(15,16,26,27). Of all the proposed candidates, evidence supporting DNA-PK and mTOR are the most compelling. Based on the observation that membrane localization is sufficient to activate Akt,(15) Hemmings and coworkers used a biochemical approach to purify a constitutively active membrane kinase from HEK 293 cells capable of phosphorylating Akt on Ser473(28). The kinase, DNA-PK, was found to associate and co-localize with Akt at the PM as well as phosphorylate and activate Akt 10-fold in
vitro(29). These results were surprising since DNA-PK has been described as a nuclear protein involved in DNA replication, gene transcription, and DNA repair(30). Nevertheless, knockdown studies with small interference RNA (siRNA) and experiments using DNA-PK-deficient glioblastoma cells revealed that insulin-stimulated Ser473 phosphorylation was greatly impaired in both cases(29). Like DNA-PK, mTOR is a member of the family of PI 3-kinase related kinases whose activities are strongly inhibited by wortmannin and LY294002(31,32). mTOR can regulate cell growth and proliferation through its activation by growth factors and nutrients(33). It is also the best-characterized HM-kinase, capable of phosphorylating the hydrophobic motif of p70S6 Kinase when mTOR is complexed with GβL(34) and regulatory associated protein of mTOR (RAPTOR)(35). Phosphorylation at the HM site of p70S6K(32) but not Akt(36,37) is inhibited by rapamycin. It is for this reason that mTOR was initially ruled out as being the HMD-PK for Akt. Recently, however, Sabatini and coworkers have shown that mTOR can associate with GβL and another protein called rapamycin insensitive companion of mTOR (RICTOR) forming a rapamycin-insensitive complex capable of phosphorylating Akt at Ser473(31,38). RNAi experiments in Drosophila and human cell lines as well as several in vitro studies provide intriguing evidence that the mTOR/RICTOR complex can phosphorylate the HM site of Akt(31).
It is still not exactly clear which if any of the proposed candidate kinases are the HMD-PK for Akt in an insulin-responsive tissue such as muscle and fat. In fact it has been suggested that phosphorylation of Ser473 may be due to multiple kinases that are cell-type- or signaling pathway- specific(24). Several years ago we characterized the phosphorylation of Akt at Ser-473 using a cell-free insulin-signaling assay we developed using subcellular fractions from 3T3-L1 adipocytes(27,39). We found that insulin-stimulated Akt phosphorylation at Ser-473 was PI 3-kinase-dependent and due to a kinase localized in both low-density microsomes (LDM) and a subpopulation of the PM fraction enriched in proteins associated with the actin cytoskeleton (Ext-HiP)(27). Utilizing the cell-free system as well as in vivo siRNA knockdown studies, we addressed whether ILK. PKCα, DNA-PK. ATM,
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

3
or mTOR were involved in the insulin-stimulated phosphorylation of Akt at Ser473 in 3T3-L1 adipocytes. Our results indicate that the mTOR/RICTOR complex is the HMD-PK for Akt in fat cells.
EXPERIMENTAL PROCEDURES
Cell Culture of 3T3-L1 Adipocytes – 3T3-L1 preadipocytes obtained from the American Type Culture Collection were grown to confluence and 48 h later subjected to differentiation as described previously(40). 3T3-L1 adipocytes were used 10 – 14 days after initiating differentiation except where otherwise noted. In Vitro Assay – Mature 3T3-L1 adipocytes were serum-starved overnight and then fractionated in the basal state as described previously except that 1 mM dithiothreitol (DTT) was included in HES (50 mM Hepes, pH 7.4, 255 mM sucrose, 1 mM EDTA, and protease inhibitors) and IC (20 mM Hepes, pH 7.4, 140 mM potassium glutamate, 5 mM NaCl, 1 mM EGTA, and protease inhibitors) buffers(39,41). Protease inhibitors (Sigma) were as described(39). Ext-HiP and salt-washed plasma membranes (PM(SW)) were isolated in the manner described previously(27). In vitro reactions were prepared by mixing various combinations of cytosol (CYT), PM, nuclear (NUC)-enriched fraction, LDM, and Ext-HiP with 16 nM Akt2 (Upstate) and 10 µM PIP3 (Calbiochem), prepared in a sonicated mixture of 100 µM phosphatidylcholine (Avanti Polar Lipids) and 100 µM phosphatidylserine (Avanti Polar Lipids). Reactions (typically 100 µl final volume) were initiated with the addition of an ATP-regenerating system (final reaction concentrations, 1 mM ATP, 8 mM creatine phosphate, 30 units/ml creatine phosphokinase, and 5 mM MgCl2) and then incubated with rotation at 37oC for the indicated period of time. Assays were quenched by the addition of an equal volume of buffer A (50 mM Hepes, pH 7.4, 150 mM NaCl, 1 mM sodium vanadate, 100 mM NaF, 10 mM sodium pyrophosphate, and protease inhibitors) containing 2 % SDS. Immunoblot Analysis – Protein samples were subjected to SDS-PAGE and transferred to nitrocellulose. Akt phospho-specific and PKCα (2056) antibodies were obtained from Cell
Signaling Technology. DNA-PKcs (H-163) and ATM (5C2) antibodies were from Santa Cruz Biotechnologies. Two mTOR antibodies were used for immunoblot analysis, a polyclonal antibody mTOR (mTab2) from Upstate and a monoclonal antibody RAFT1 (611132) from BD Transduction Laboratories. RAPTOR antibodies were either from Cell Signaling Technology (4978) or from Abcam (ab 5454). RICTOR antibodies were a kind gift from Dr. David M. Sabatini (Whitehead Institute) and were also obtained from Bethyl Laboratories. Immunoblots visualized by ECL Detection (Amersham) were quantified using NIH Image 1.62f. 6 and 12 µg of control proteins were run on each gel to establish the linearity of the autoradiogram. Immunoprecipitation of LDM – 500 µg of LDM in 500 µl of IC buffer (with 1 mM DTT) were precleared with 40 µl of protein A-agarose for 30 min. After centrifugation for 10 min at 4oC in a microfuge, the supernatant was rotated overnight with 4 µg of primary antibody. Samples were then centrifuged for 5 min at 4oC in a microfuge to remove nonspecific aggregates that formed overnight. 30 µl of protein-A agarose were added to the supernatants for 2 h at 4oC. Pellets were washed six times with IC buffer, resuspended in IC buffer, and then assayed in vitro for HMD-PK activity. ILK (3862) polyclonal antibodies (Cell Signaling Technology), mTOR (mTab1) polyclonal antibodies (Upstate), and RICTOR antibodies (D.M. Sabatini) were used in the immunoprecipitaion studies. HMD-PK peptide Assay – HMD-PK activity was assayed using a modified version of Hill et al(28). 50 µl reactions were incubated for 20 min at 30oC in a buffer containing IC buffer, 1 mM DTT, 10 mM MgCl2, 1 µM PKI, 100 µM ATP, 1 mCi [γ-32P)] ATP, 0.5 mg/ml substrate (RRPHFPQFSYSASSTA) or control (RRPHFPQFAYSASSTA) peptide. Peptides were phosphorylated using 25 units DNA-PK (Promega), 0.75 mg/ml LDM, or 0.4 mg/ml Ext-HiP. 10 µg/ml salmon sperm DNA were added in some cases. Reactions were quenched by adding 5 µl of 100 % (w/v) trichloroacetic acid and then centrifuged for 10 min in a microfuge at room temperature. 35 µl of the supernatant were spotted on P81 paper (Whatmann), washed extensively in 0.1 % phosphoric acid, and then analyzed by
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

4
scintillation counting. Specific counts were calculated from the difference between the Ser473 peptide and the control peptide. siRNA Duplexes – Complementary sense and antisense strands of RNA oligonucleotides for DNA-PK, ATM, and ILK were synthesized, annealed, and purified (PAGE) by Ambion. Negative Control #1 siRNA (Ambion) and the following mouse sequences of strands of siRNA were used: DNA-PK sense, AGGGCCAAGCUAUCAUUCUtt; DNA-PK antisense, AGAAUGAUAGCUUGGCCCUtt; ATM sense, CAUACUACUCAAAGACAUUtt; ATM antisense, AAUGUCUUUGAGUAGUAUGtt; ILK sense, UGUUAAGUUUUCUUUCCAGUGtt; ILK antisense, CACUGGAAAGAAAACUUAACAtt. siGENOME Smartpool siRNA for RICTOR (M-064598-00, mouse 4921505C17RIK) and siCONTROL Non-Targeting siRNA #1 were from Dharmacon RNA Technologies. siRNA Electroporation of 3T3-L1 Adipocytes – 3T3-L1 adipocytes were electroporated according to the procedure of Jiang et al(6). Five days after differentiation, 3T3-L1 adipocytes were detached from cell culture plates with 1 mg/ml collagenase and 0.05 % trypsin / 0.02 % EDTA. Cells were washed three times with ice-cold PBS and resuspended in PBS prior to electroporation. Approximately 5 million cells (half of the cells on a 150 mm dish) were resuspended in 0.5 ml PBS, mixed with 20 nmol siRNA duplexes, and electroporated at the setting of 0.18 kV and 950 microfarads using the BioRad Gene Pulser Xcell Electroporation System. Cells were immediately incubated with fresh media for 10 min before reseeding. Experiments were conducted 72 h after electroporation.
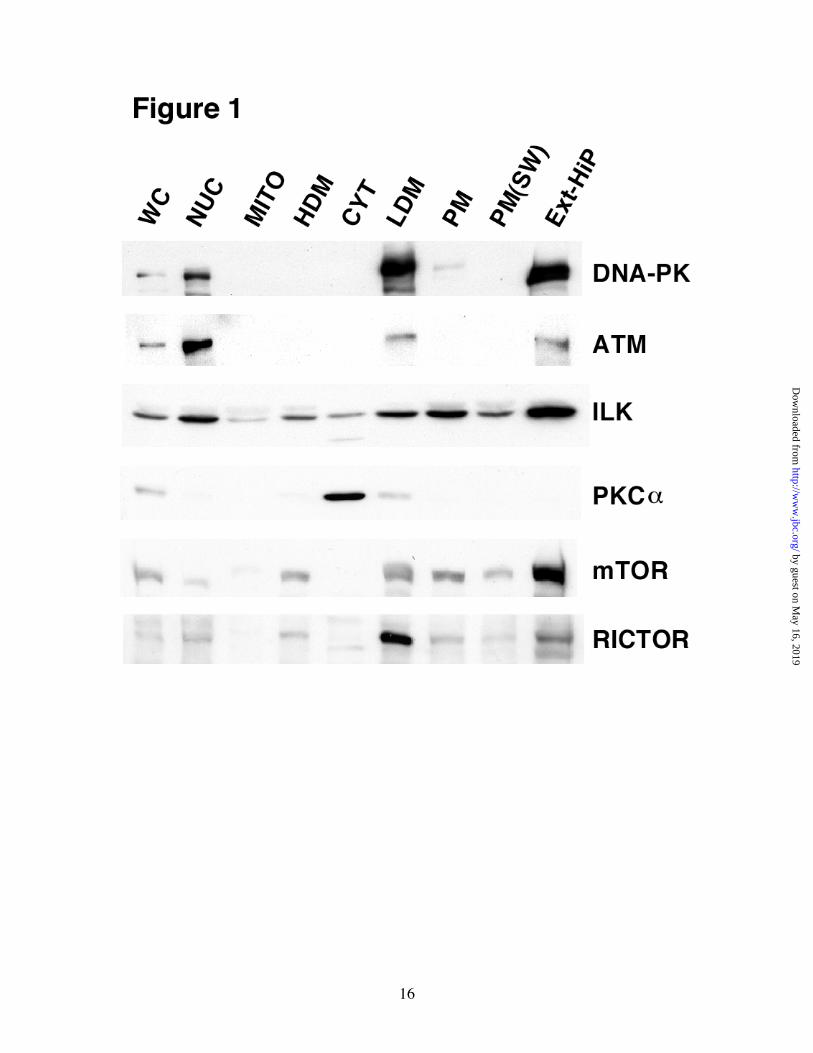
RESULTS Subcellular Localizations of DNA-PK and mTOR/RICTOR Correlate with HMD-PK Activity – ILK, PKCα, DNA-PK, ATM, and mTOR have all been proposed to play a major role in Akt-Ser473 phosphorylation based on experiments carried out on a variety of different cell-types and stimuli(24). We have decided to directly compare the functions of these five proteins in terms of insulin-stimulated Akt-Ser473 phosphorylation in a single cell-type, 3T3-L1 adipocytes. Previously,
we have shown that LDM and Ext-HiP, purified from 3T3-L1 adipocytes, are greatly enriched in insulin-stimulated HMD-PK activity(27). To determine if any of these proteins are enriched in LDM and Ext-HiP, we carried out immunoblot analyses on subcellular fractions prepared from serum-starved 3T3-L1 adipocytes (Fig. 1). DNA-PK was found in the NUC fraction as expected due to its well-documented nuclear functions(30). It was, however, more greatly enriched in LDM and Ext-HiP but absent (or below detection) in high-density microsomes (HDM), mitochondrial, and CYT fractions. HDM is enriched in endoplasmic reticulum and LDM contains Golgi markers(42). ATM, another member of the PI 3-kinase-related kinase family, had the same distribution as DNA-PK but was more abundant in NUC than LDM and Ext-HiP as compared with DNA-PK. ILK had a ubiquitous distribution but was slightly more enriched in NUC, LDM, and ExtiHiP fractions. PKCα was found almost exclusively in CYT in the basal state. PKCs are cytosolic proteins that translocate to membranes upon hormone stimulation(43). mTOR was greatly enriched in Ext-HiP but was also found in NUC, HDM, and LDM fractions. mTOR has been previously reported to be associated with the endoplasmic reticulum,(44) Golgi,(44) and in the nucleus(45). RICTOR, mTOR’s binding protein that is necessary for Ser473 phosphorylation(31), was very abundant in LDM, less abundant in Ext-HiP, but was also present in NUC and HDM fractions. Next, we wanted to directly correlate HMD-PK activity with the amount of each candidate protein using the cell-free assay. We have previously shown that PIP3-dependent Akt-Ser473 phosphorylation in our in vitro assay was entirely dependent on exogenous ATP and the time of reaction incubation(27,39). In vitro reactions were performed by combining CYT, NUC, PM, and LDM fractions with purified recombinant Akt2 and ATP in the absence and presence of PIP3. The amount of the particular fraction and the time of incubation were such that the reaction activities were in the linear range (data not shown). Assays were quenched and examined by immunoblot analyses (Fig. 2). The PIP3-stimulated Ser473 activity in these particular reactions was highest in LDM, followed by NUC, and then PM. As we previously observed, no
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

5
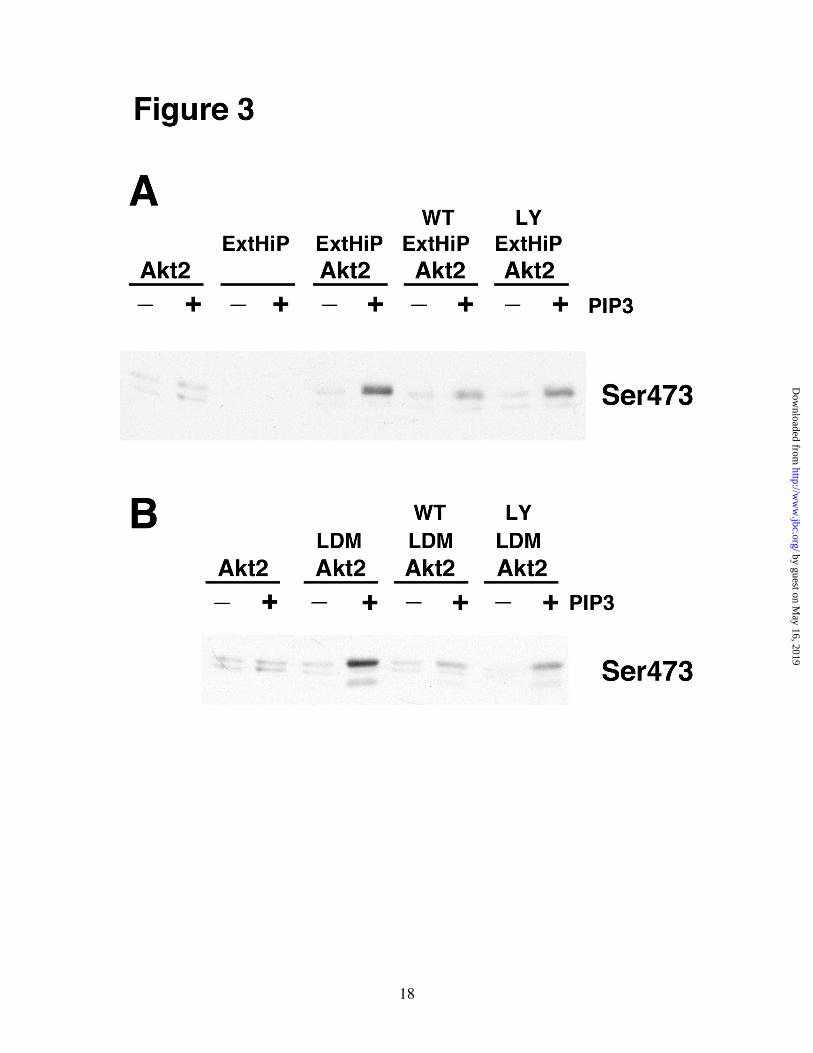
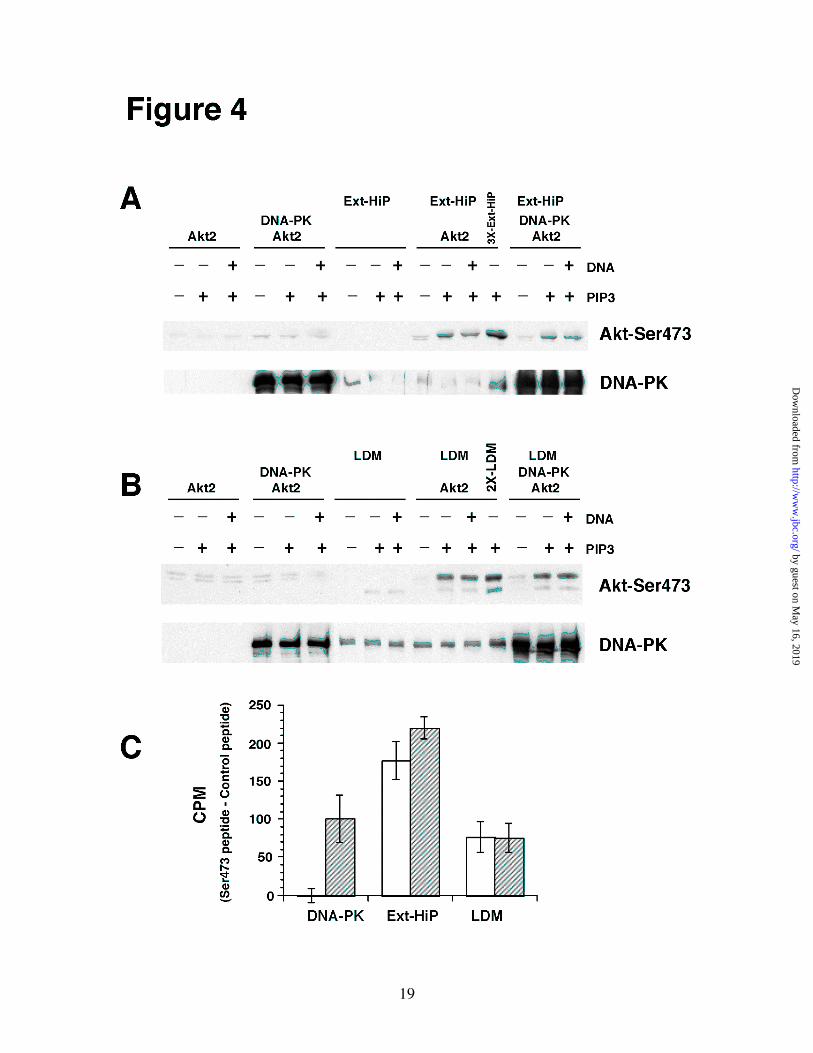
HMD-PK activity was found in CYT(27). The Ser473 activity correlated well with the amount of DNA-PK, mTOR, and RICTOR but did not correlate with levels of ATM, ILK, and PKCα. Based on the fact that PKCα was almost exclusively localized in CYT, which had no HMD-PK activity, we concluded that PKCα was not the HMD-PK in 3T3-L1 adipocytes. As previously discussed, DNA-PK, ATM, and mTOR are members of the PI 3-kinase related family of kinases(30). If one of these proteins is indeed HMD-PK in 3T3-L1 adipocytes, then our in vitro Ser473 phosphorylation activity should be directly sensitive to PI 3-kinase inhibitors. As shown in Fig. 3, PIP3-stimulated Ser473 activities in both Ext-HiP and LDM were inhibited by wortmannin and LY294002. Purified DNA-PK does not Phosphorylate Full-length Akt2 in a PIP3-dependent Manner - Feng et al(29) has shown that purified DNA-PK (Promega) can phosphorylate a C-terminal Akt peptide containing Ser473 and two truncated versions of Akt (GST-AKT1419-480, and �PH -Akt2T309P) lacking the pleckstrin homology (PH) domain. Only the peptide phosphorylation was DNA-dependent. Since it was never shown, we wanted to verify that purified DNA-PK could phosphorylate full-length Akt2 in a PIP3-dependent manner. In vitro reactions, conducted using various combinations of DNA-PK, Akt2, PIP3, and double-stranded DNA, were analyzed by immunoblot analysis (Fig. 4). Control reactions were also performed containing Ext-HiP and LDM. Full-length purified Akt2 was phosphorylated on Ser473 in a PIP3-dependent manner with either Ext-HiP (Fig. 4A) or LDM (Fig. 4B). The phosphorylation was not stimulated by DNA. No Ser473 phosphorylation was observed with Akt2 or Ext-HiP alone. Reactions containing LDM, Akt2, and PIP3 generated two Ser473 bands of different molecular weights. We have found that the lower band corresponds to endogenous Akt present in LDM, and the upper band is due to the His-tagged recombinant Akt2 (data not shown). In the absence of exogenous Akt2, only the lower Ser473 band was observed in reactions containing LDM and PIP3. Surprisingly, purified DNA-PK could not phosphorylate full-length Akt2 to any discernible degree in either the presence or
absence of PIP3 and DNA. When reactions were analyzed for DNA-PK content, the amount of exogenous DNA-PK greatly exceeded the amount found in Ext-HiP or LDM. To verify that the commercial DNA-PK was active, assays were carried out with the same Ser473 peptide used by Feng et al(29) (Fig. 4C). As was reported previously, purified DNA-PK phosphorylated the Ser473 peptide in a DNA-dependent manner(29). Ext-HiP and LDM could also phosphorylate the peptide, however, the presence of DNA had no statistical effect on the peptide phosphorylation. These results indicate that purified DNA-PK by itself cannot phosphorylate full-length Akt2. It is possible that DNA-PK requires an accessory protein, found in Ext-HiP or LDM, to phosphorylate full-length Akt. The holoenzyme of DNA-PK consists of a 465-kDa catalytic subunit and two Ku antigen subunits (Ku70/Ku80)(30). All three subunits, however, were found in purified DNA-PK, Ext-HiP, and LDM (data not shown). If an accessory protein is required, it is unlikely Ku70/80. To determine whether an accessory protein was present in Ext-HiP and LDM and was in excess relative to DNA-PK, we added purified DNA-PK to in vitro reactions containing Ext-HiP/Akt2 (Fig. 4A) or LDM/Akt2 (Fig. 4B) and compared the measured PIP3-stimulated Ser473 activity with reactions containing Ext-HiP/Akt2 or LDM/Akt2 without exogenous DNA-PK. We found that adding exogenous DNA-PK had no effect. Control reactions containing 3 times the amount of Ext-HiP (3X-Ext-HiP) or twice the amount of LDM (2X-LDM) verified the reactions were not limited in substrate. Since purified DNA-PK could not phosphorylate full-length Akt2 in a PIP3-dependent manner, either DNA-PK is not HMD-PK or an accessory protein is required but is limiting in LDM or Ext-HiP relative to the amount of DNA-PK. HMD-PK Activity is not Associated with ILK in LDM – Since Glut 4 vesicles can be easily immunoisolated from LDM of 3T3-L1 adipocytes(46), we attempted to immunoprecipitate DNA-PK, ATM, or ILK from LDM and measure PIP3-stimulated Ser473 activity in the immunoprecipitates using an in vitro assay. We tested five antibodies for both DNA-PK and ATM and failed to find one that
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

6
recognized the mouse form of the proteins and that could immunoprecipitate. ILK, however, was successfully immunoisolated from LDM. In vitro assays were then conducted using supernatants and pellets from ILK or control immunoprecipitation reactions. Although we brought down approximately 50 % of ILK from LDM, we observed no change in the Ser473 activity remaining in the supernatant nor could we measure PIP3-stimulated Ser473 activity associated with ILK in the pellets (Fig. 5). siRNA-mediated Depletion of DNA-PK, ATM, or ILK had no Effect on Insulin-stimulated Akt-Ser473 Phosphorylation in 3T3-L1 Adipocytes - Next we wanted to determine whether suppressing the expression of DNA-PK, ATM, or ILK by siRNA had any effect on insulin-activated Akt-Ser473 phosphorylation in intact 3T3-L1 adipocytes. Using the electroporation method described by Czech and collegues(6), we could substantially reduce the expressions of DNA-PK (88.7 % ± 6.6 %), ATM (87 % ± 3.5 %), and ILK (64 % ± 2.6 %) (Fig. 6A). Serum-starved cells were then stimulated with insulin and tested for Akt phosphoryation using phospho-specific antibodies. We observed no change in Akt phosphorylation at either Ser473 or Thr308 in cells depleted of DNA-PK or ATM compared with cells tested with a negative control siRNA (Figs. 6B and 6C). In ILK-depleted cells, we consistently observed a 25 % increase in insulin-induced Ser473 phosphorylation but saw no change in insulin-stimulated Thr308 phosphorylation (Figs. 6B and 6C). The observed increase in Ser473 phosphorylation was surprising since we had expected a decrease in insulin-stimulated Ser473 phosphorylation if ILK was indeed the HMD-PK for Akt. Therefore, based on these results in intact adipocytes and in conjunction with our cell-free experiments, we conclude that DNA-PK, ATM, ILK, and PKCα are most likely not the HMD-PK in 3T3-L1 adipocytes. PIP3-stimulated HMD-PK Activity was Associated with mTOR/RICTOR Complexes from LDM – Next, we focused on mTOR/RICTOR by immunoisolating mTOR complexes from LDM and measuring Ser473 activity with an in vitro assay. PIP3-stimulated Ser473 activity was found
in mTOR complexes immunoprecipitated with antibodies directed against mTOR or RICTOR (Fig. 7A). mTOR, RICTOR, and RAPTOR were found in the pellets using the mTOR antibody while the RICTOR antibody brought down mTOR and RICTOR but not RAPTOR. We found no commercial RAPTOR antibody that could successfully immunoprecipitate RAPTOR. Nevertheless, our results indicate that complexes containing mTOR and RICTOR are capable of phosphorylating Akt on Ser473 in a PIP3-dependent manner. The in vitro reactions were also probed for the presence of DNA-PK, ATM, and ILK. We found no detectable amounts of these three proteins in the mTOR or RICTOR immunoprecipitation pellets even though all three were present in total LDM (Fig. 7B). To help rule out the possibility that Akt was phosphorylated on Ser473 by a kinase other than mTOR that happens to co-immunoprecipitate in our system, we immunoprecipitated mTOR from LDM and then measured HMD-PK activity in the presence of PI 3-kinase inhibitors and staurosporine (Fig. 8). PIP3-stimulated Ser473 activity was inhibited with wortmannin and LY294002 but not staurosporine. Our results are consistent with those of Sabatini and colleagues for mTOR(31). siRNA-mediated Depletion of RICTOR Inhibited Insulin-stimulated Akt-Ser473 Phosphorylation in 3T3-L1 Adipocytes - Unfortunately, we were unsuccessful at silencing mTOR expression in 3T3-L1 adipocytes using 6 different commercially available siRNAs. Regardless, it has been reported that almost complete knockdown of mTOR is required to observe any effect on insulin-stimulated Akt-Ser473 phosphorylation (31), a result most likely due to the feed-back inhibition of insulin signaling caused by serine phosphorylation of IRS-1 by mTOR/RAPTOR(36). To circumvent this problem, we attempted to suppress RICTOR expression in order to affect insulin-stimulated Akt–Ser473 phosphorylation in vivo. We were able to substantially silence RICTOR expression (85 % ± 3.5 %) using electroporated siRNA with no effect on mTOR levels (Fig. 9A). Serum-starved adipocytes were stimulated with insulin and then analyzed for Akt phosphorylation using phospho-specific antibodies (Fig. 9B and 9C).
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

7
Insulin-activated Akt phosphorylation of Ser473 and Thr308 were suppressed 87 % ± 3.2 % and 55 % ± 9.35 % respectively when RICTOR expression was reduced with siRNA. These results were also confirmed using a pool of two independent RICTOR siRNAs (Qiagen, target regions 470-490 and 903-923) whose sequences were different from the ones used above (data not shown). The correlation between RICTOR expression levels and the amount of insulin-stimulated Akt-Ser473 phosphorylation was further demonstrated by a RICTOR siRNA dose response experiment (Fig. 9D). Based on our cell-free and siRNA results, we conclude mTOR complexed with RICTOR is the HMD-PK in 3T3-L1 adipocytes.
DISCUSSION DNA-PK(29), ATM,(47) ILK(48,49), PKCα(50) and mTOR(31) have all been recently identified as candidates for the HMD-PK for Akt. These studies, however, were all conducted in different cell-types using a variety of stimuli. Our report is the first to directly address all five candidates in a single cell-type, 3T3-L1 adipocytes, activated using a single stimulus, insulin. We selected 3T3-L1 adipocytes because these cells are very responsive in terms of insulin-stimulated glucose transport(51), a process in which Akt activation is a critical step(3-5,8).
The original identification of DNA-PK as the Ser473 kinase was very surprising since DNA-PK was already well documented to possess several DNA-related functions that take place in the nucleus(30). To carry out these functions, double-stranded DNA is required to co-localize substrates with DNA-PK through their interactions with Ku proteins and to activate DNA-PK. It is possible that our apparent enrichment of DNA-PK in LDM and Ext-HiP is entirely due to the disruption of nuclei that may occur during the homogenization process (Fig. 1). What argues against this is that Feng et al have shown by confocal microscopy that a significant fraction of DNA-PK is found in the cytosol and at the membrane in addition to the nucleus in 3T3-L1 fibroblasts and M059K cells(29). The strongest data that supported the idea that DNA-PK can function as a Ser473 kinase were siRNA knockdown experiments in HEK293 cells, the lack
of significant insulin-induced Ser473 phosphorylation in DNA-PK-deficient M059J glioblastoma cells, and rescue experiments with the M059J/Fus1 cells(29). The residual amount of insulin-activated Ser473 phosphorylation in M059J cells has been recently shown to be due to mTOR/RICTOR(31). Nevertheless, mTOR/RICTOR cannot explain why Ser473 phosphorylation is so low in the absence of DNA-PK in M059J cells. It is possible that in certain cell-types under certain conditions, DNA-PK does function as the primary HMD-PK for Akt. Recently, DNA-PK has been implicated in the activation of Akt by CpG-DNA(52). Stimulation of Ser473 phosphorylation by CpG-DNA is markedly reduced in bone marrow-derived macrophages from mice lacking DNA-PK. Our results, however, indicated that DNA-PK could not phosphorylate full-length Akt2 even though it could phosphorylate truncated versions of Akt ((29), Fig. 4). Therefore if DNA-PK can really phosphorylate Ser473 in cells, it most likely would require an accessory factor. The necessity of an accessory factor for DNA-PK to phosphorylate Akt would not be unexpected since its related family member mTOR must form a complex with either RAPTOR or RICTOR to phosphorylate p70S6 kinase(35) or Akt(31) respectively. In the case of DNA-PK, this accessory factor would be necessary to shift its phosphorylation preference from S/TQ motifs(53) to the FXXF(S/T)(Y/F) found in Akt(20). In fact, it was recently shown that purified DNA-PK could phosphorylate Akt2 in the presence of a synthetic CpG oligonucleotide (CpG-OGN) but not in its absence(52). It is clear, however, from the lack of an effect on insulin-stimulated Akt-Ser473 phosphorylation when DNA-PK was knocked down by almost 90 % with siRNA in 3T3-L1 adipocytes, that DNA-PK does not significantly contribute to HMD-PK activity in fat cells in response to insulin (Fig. 6). Our results, however, do not address the function of DNA-PK in other cell types. ATM, like DNA-PK, is a kinase involved in DNA repair(54) that has also been implicated in Akt-Ser473 phosphorylation(47). It has been reported that overexpression of ATM robustly increased Ser473 phosphorylation while knockdown by siRNA significantly decreased it. Thr308 phosphorylation was not affected in either case. ATM, however, cannot directly
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

8
phosphorylate Akt on Ser473 in vitro suggesting that it may play an indirect role in Ser473 phosphorylation. Our results in 3T3-L1 adipocytes indicated that ATM most likely has little if any role in Ser473 phosphorylation, direct or indirect. ATM was not very abundant in LDM or Ext-HiP, the two fractions that possess the highest Ser473 activity (Fig. 1 and 2). In addition, a greater than 85 % reduction in the expression of ATM by siRNA had no effect on the phosphorylation of Ser473 in response to insulin (Fig. 6). Dedhar and coworkers have published multiple reports providing evidence that ILK can function as the Akt-Ser473 kinase(48,49,55). Recombinant ILK can phosphorylate Akt at Ser473 in vitro(55). A kinase-dead ILK has been shown to act as a dominant negative suppressing endogenous Ser473 phosphorylation activity(49). Knockdown experiments by siRNA and knockouts using the Cre-LoxP system markedly reduced Ser473 phosphorylation(48). Despite these data, there are other studies that indicate ILK may not be the physiological Akt-Ser473 kinase. It was reported that in ILK-deficient fibroblasts(56) and chondrocytes(57), Ser473 phosphorylation was normal. Other groups have not been able to phosphorylate Akt at the HM site in vitro(28,58,59). There is even uncertainty that ILK is a functional kinase, as several critical residues normally found in the catalytic domain of protein kinases are not conserved(59). Thus, what role ILK has in terms of the phosphorylation of Akt on Ser473 may be indirect. In 3T3-L1 adipocytes, our data indicated that ILK was not the HMD-PK for Akt. We saw no change in the level of PIP3-stimulated Akt-Ser473 phosphorylation activity in LDM after removing more than half the ILK and saw no corresponding appearance of activity in the pellets after immunoprecipitation (Fig. 5). Similarly, after reducing the expression of ILK by approximately 65 % with siRNA, we actually observed a 25 % increase rather than a decrease in insulin-activated Akt-Ser473 phosphorylation (Fig. 6). Further study is necessary to determine whether the 25 % increase in Ser473 phosphorylation is of physiological significance. PKCα is activated by diacylglycerol and calcium(43) and has been implicated in Ser473 phosphorylation(50). This PKC isoform can
phosphorylate Akt on Ser473 in vitro in the presence of lipid and calcium as well as undergo IGF-1 stimulated translocation to membranes in endothelial cells. Overexpression of PKCα increased Ser473 phosphorylation while dominant-negative PKCα constructs or reducing the expression by siRNA decreased Ser473 phosphorylation. We found that in 3T3-L1 adipocytes, PKCα was almost entirely cytosolic and absent from compartments with Ser473 phosphorylation activity (Figs. 1 and 2). In addition, our cell-free assays were all conducted in the absence of calcium since 1 mM EGTA was included in the reaction buffer. Our conclusion that PKCα is not involved in the Ser473 phosphorylation of Akt in adipocytes is in agreement with an earlier report that Akt phosphorylation at Ser473 actually increased when PKCα was knocked out in fat and muscle(60). The authors of the knock-out study concluded that PKCα inhibits insulin signaling by phosphorylating IRS proteins on serine residues resulting in the suppression of insulin-stimulated tyrosine phosphorylation of IRS proteins by the insulin receptor. Interesting with regard to PKCα, Sarbassov et al(38) reported that mTOR/RICTOR complex can regulate the organization of the actin cytoskeleton and they have proposed that this regulation is mediated via the phosphorylation of PKCα. PKCα does not, however, form a stable complex with mTOR/RICTOR in detergent solubilized cells(31). In addition, PKCα immunoprecipitates prepared from detergent solubilized cells did not phosphorylate Akt-Ser473 in an in vitro kinase assay. Based on our results, we have concluded that mTOR/RICTOR is the HMD-PK for Akt in 3T3-L1 adipocytes. mTOR/RICTOR was greatly enriched in fractions that contain the highest Ser473 activity (Figs. 1 and 2). mTOR/RICTOR complexes, purified by immunoprecitation of LDM with mTOR or RICTOR antibodies, had PIP3-stimulated Ser473 phosphorylation activity (Fig. 7A) and did not contain DNA-PK, ATM, or ILK (Fig.7B). In addition, the Ser473 activity associated with mTOR/RICTOR complexes was sensitive to PI 3-kinase inhibitors but not staurosporine (Figs. 8), the same drug-sensitivity has been shown for insulin-stimulated Akt-Ser473
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

9
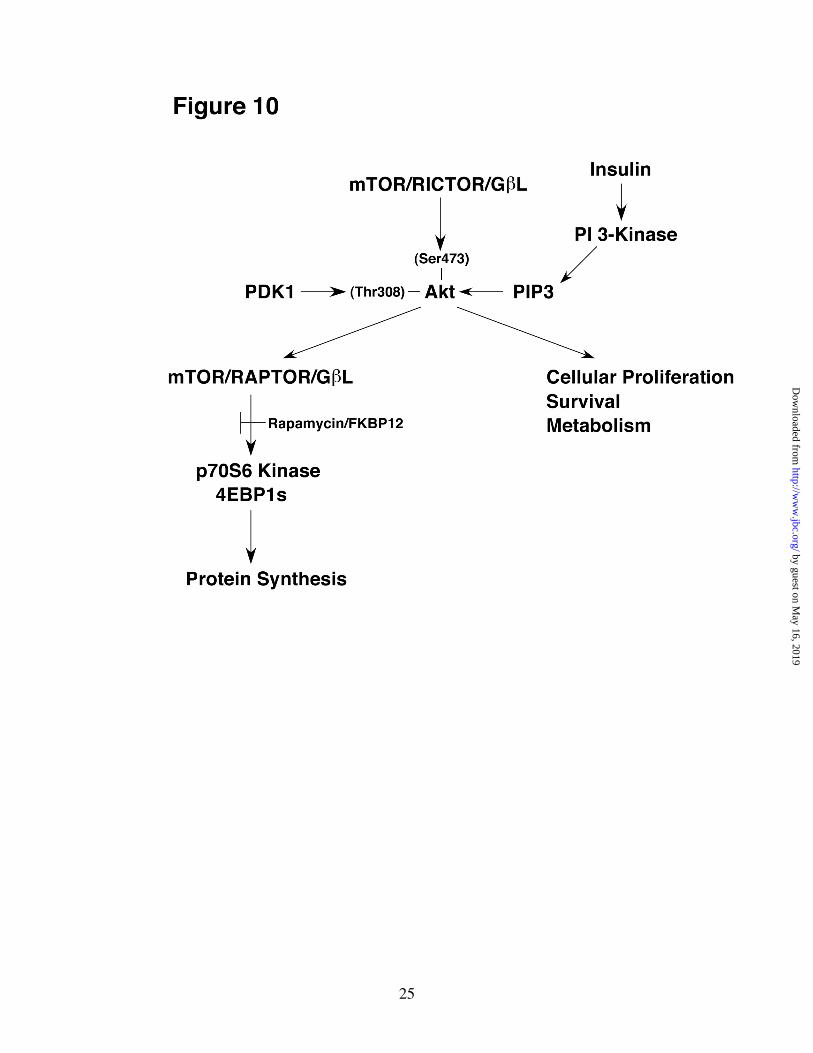
phosphorylation(16,61). Decreasing the expression of RICTOR by siRNA substantially inhibited insulin-stimulated Akt-Ser473 and to a lesser extent Thr308 phosphorylation in intact adipocytes (Fig. 9). Effects on both Ser473 and Thr308 phosphorylations are in agreement with the model that the phosphorylation of Ser473 provides a docking site to recruit PDK1 in order to phosphorylate Thr308 (20). It should also be pointed out that the basal activity we observed in the Ser473 peptide assays conducted with Ext-HiP or LDM was most likely due to mTOR/RICTOR complexes in these fractions and not due to DNA-PK (Fig. 4C). All of our results are entirely consistent with those published by Sabatini and colleagues(31). It has also been recently reported that treating 3T3-L1 adipocytes with rapamycin actually increases insulin-stimulated Akt-Ser473 phosphorylation(36). This result can be fully explained in terms of feed-back inhibition of insulin signaling caused by serine phosphorylation of IRS-1. Rapamycin inhibited insulin activation of p70S6 kinase, decreased Ser636/639 IRS-1 phosphorylation, increased PIP3 levels, and resulted in an increase in Akt-Ser473 phosphorylation(36). The model proposed by Sabatini and colleagues(31) also explains how mTOR can be found both upstream and downstream of Akt in the insulin-stimulated PI 3-kinase pathway (Fig. 10). mTOR, when complexed to RICTOR and GβL acts as HMD-PK activating Akt through the phosphorylation of Ser473. Akt also activates mTOR/RAPTOR, either directly or through the phosphorylation of the tumor suppressor, TSC2, leading to the stimulation of the p70S6 Kinase/4EBP1 pathway(62).
Interestingly, Sabatini and colleagues have also reported that the Ser473 kinase activity of mTOR/RICTOR was markedly stimulated by
serum in vivo(31). Kinase assays conducted on immunoprecipitates isolated from 0.3 % CHAPS-solubilized serum-treated HeLa cells were significantly higher than those isolated from basal cells. Activation of HMD-PK by serum would disagree with the assumption by Hemminngs that the Ser473 kinase is constitutively active at the plasma membrane(15,63,64). It is thought that binding of Akt to PIP3, formed in PM in response to insulin, has two effects, it co-localizes Akt with a constitutively active HMD-PK(15,63) and also induces a conformational change in Akt that allows for Ser473 and Thr308 phosphorylation(65), (Fig. 10). Therefore, insulin enhances Ser473 phosphorylation through effects on Akt itself as opposed to effects on HMD-PK. We, in fact, have observed no difference in HMD-PK activity in cell-free assays conducted using Ext-HiP (or LDM) purified from basal and insulin-treated adipocytes (unpublished data), which tend to support the idea that insulin does not activate HMD-PK directly. One explanation for the apparent discrepancy is that the observed serum-induced increase in HMD-PK activity reported by Sabatini and coworkers is actually due to the presence of PIP3 in the immunoprecipitates when mTOR complexes were isolated from serum-treated cells. 0.3 % CHAPS is not a high enough detergent concentration to completely solubilize membranes. In fact, these same authors have reported that the mTOR/RICTOR complex was not stable under conditions (1 % Triton X-100) typically used to solubilize membranes. In summary, our results indicate that mTOR/RICTOR, is responsible for insulin-induced Akt-Ser473 phosphorylation in 3T3-L1 adipocytes. Our results do not address whether DNA-PK, ATM, ILK, or PKCα can function as HMD-PK for Akt in other cell-types under certain conditions.
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

10
REFERENCES
1. Cushman, S. W., and Wardzala, L.J. (1980) J. Biol. Chem. 255, 4758-4762
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

11
2. Suzuki, K., and Kono, T. (1980) Proc. Natl. Acad. Sci. USA 77, 2542-2545 3. Kohn, A. D., Summers, S.A., Birnbaum, M.J. and Roth, R.A. (1996) J. Biol.
Chem. 271, 31372-31378 4. Hill, M. M., Clark, S.F., Tucker, D.F., Birnbaum, M.J., James, D.E., and
Macaulay, S.L. (1999) Mol. Cell. Biol. 19, 7771-7781. 5. Bae, S. S., Cho, H., Mu, J. and Birnbaum, M.J. (2003) J. Biol. Chem. 278, 49530-
49536 6. Jiang, Z. Y., Zhou,Q.L., Coleman, K.A., Chouinard, M., Boese, Q., and Czech,
M.P. (2003) Proc. Natl. Acad. Sci. USA 100, 7569-7574 7. Zeigerer, A., McBrayer, M.K., and McGraw, T.E. (2004) Mol. Biol. Cell 15,
4406-4415 8. Welsh, G. I., Hers, I., Berwick, D.C., Wherlock, M., Birkin, R., Leney, S., and
Tavare, J.M. (2005) Biochem. Soc. Trans. 33, 346-349 9. Saltiel, A. R., and Kahn, C.R. (2001) Nature 414, 799-806. 10. White, M. F., and Kahn, C.R. (1994) J. Biol. Chem. 269, 1-4 11. White, M. F. (1998) Mol. Cell. Biochem. 182, 3-11 12. Shepherd, P. R., Withers, D.J., and Siddle, K. (1998) Biochem. J. 333, 471-490 13. Lawlor, M. A., and Alessi, D.R. (2001) J. Cell Sci. 114, 2903-2910 14. Vanhaesebroeck, B., and Alessi, D.R. (2000) Biochem. J. 346, 561-576 15. Andjekkovic, M., Alessi, D.R., Meier, R., Fernandez, A., Lamb, N.J.C., Frech,
M., Cron, P., Cohen, P., Lucocq, J.M. and Hemming, B.A. (1997) J. Biol. Chem. 272, 31515-31524.
16. Alessi, D. R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P., and Hemmings, B.A. (1996) EMBO J. 15, 6541-6551
17. Alessi, D. R., James, S.R., Downes, C.P., Holmes, A.B.; Gaffney, P.R., Reese, C.B., and Cohen, P. (1997) Curr. Biol. 7, 261-269
18. Stephens, L., Anderson, K., Stokoe, D., Erdjument-Bromage, H., Painter, G.F., Holmes, A.B., Gaffney, P.R., Reese, C.B., McCormick, F., Tempst, P., Coadwell, J. and Hawkins, P.T. (1998) Science 279, 710-714
19. Alessi, D. R., Deak, M., Casamayor, A., Caudwell, F.B., Morrice, N., Norman, D.G., Gaffney, P., Reese, C.B., MacDougall, C.N., Harbison, D., Ashworth, A. and Bownes, M. et al. (1997) Curr. Biol. 7, 776-789
20. Frodin, M., Antal, T.L., Dummler, B.A., Jensen. C.J., Deak, M., Gammeltoft, S. and Biondi, R.M. (2002) EMBO J. 21, 5396-5407
21. Parekh, D. B., Ziegler, W. and Parker, P.J. (2000) EMBO J. 19, 496-503 22. Huang, X., Begley, M., Morgenstern, K.A., Gu, Y., Rose, P., Zhao, H., and Zhu,
X. (2003) Structure (Camb) 11, 21-30 23. Yang, J., Cron,P., Good, V.M., Thompson, V., Hemmings, B.A., and Barford, D.
(2002) Nat. Struct. Biol. 9, 940-944 24. Dong, L. Q., and Liu, F. (2005) Am. J. Physiol. Endocrinol. Metab. 289, E187-
196. 25. Toker, A., and Newton, A.C. (2000) J. Biol. Chem. 275, 8271-8274 26. Wick, M. J., Dong, L.Q., Riojas, R.A., Ramos, F., and Liu, F. (2000) J. Biol.
Chem. 275, 40400-40406 27. Hresko, R. C., Murata, H., and Mueckler, M. (2003) J. Biol. Chem. 278, 21615-
21622
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

12
28. Hill, M. M., Feng, J., and Hemmings, B.A. (2002) Curr. Biol. 12, 1251-1255. 29. Feng, J., Park, J., Cron, P., Hess, D., and Hemmings, B.A. (2004) J. Biol. Chem.
279, 41189-41196 30. Smith, G. C., and Jackson, S.P. (1999) Genes Dev. 13, 916-934 31. Sarbassov., D. D., Guertin, D.A., Ali, S.M., Sabatini, D.M. (2005) Science 307,
1098-1101 32. Chung, J., Kuo, C.J., Crabtree, G.R., and Blenis, J. (1992) Cell 69, 1227-1236 33. Fingar, D. C., and Blenis, J. (2004) Oncogene 23, 3151-3171 34. Kim, D.-H., Sarbassov, D.D., Ali, S.M., Latek, R.R., Guntur, K.V.P., Erdjument-
Bromage, H., Tempst, P., and Sabatini, D.M. (2003) Mol. Cell 11, 895-904 35. Hara, K., Maruki, Y., Long, X., Y., K.-i., Oshiro, N., Hidayat, S., Tokunaga, C.,
Avruch, J., and Yonezawa, K. (2002) Cell 110, 177-189 36. Tremblay, F., Gagnon, A., Veilleux, A., Sorisky, A., and Marette, A. (2005)
Endocrinology 146, 1328-1337 37. Burgering, B. M., and Coffer, P.J. (1995) Nature 376, 599-602 38. Sarbassov., D. D., Ali, S.M., Kim, D.-H., Guertin, D.A., Latek, R.R., Erdjument-
Bromage, H., Tempst, P., and Sabatini, D.M. (2004) Curr. Biol. 14, 1296-1302 39. Murata, H., Hresko, R.C., and Mueckler, M. (2003) J. Biol. Chem. 278, 21607-
21614 40. Tordjman, K. M., Leingang, K. A., James, D. E., and Mueckler, M. M. (1989)
Proc. Natl. Acad. Sci. USA 86, 7761-7765 41. Piper, R. C., Hess, L. J., and James, D. E. (1991) Am. J. Physiol. 260, C570-C580 42. Simpson, A. A., Yver, D.R., Hissin, P.J., Wardzala, L.J., Karnieli, E., Salans,
L.B., and Cuchman, S.W. (1983) Biochim. Biophys. Acta 763, 393-407 43. Liu, W. S., and Heckman, C.A. (1998) Cell. Signal. 10, 529-542 44. Drenan, R. M., Liu, X., Bertram, P.G., and Zheng, X.F.S. (2004) J. Biol. Chem.
279, 772-778 45. Kim, J. E., and Chen, J. (2000) Proc. Natl. Acad. Sci. USA 97, 14340-14345 46. Mastick, C. C., Aebersold, R., and Liendard, G.E. (1994) J. Biol. Chem. 269,
6089-6092 47. Viniegra, J. G., Martinez, N., Modirassari, P., Losa, J.H., Cobo, C.P., Sanchez-
Arevalo Lobo, V.J., Luquero, C.I.A., Alvarez-Vallina, L., Ramon y Cajal, S., Rojas, J.M., and Sanchez-Prieto, R. (2005) J. Biol. Chem. 280, 4029-4036
48. Troussard, A. A., Mawji, N.M., Ong, C., Mui, A., St-Arnaud, R., and Dedhar, S. (2003) J. Biol. Chem. 278, 22374-22378
49. Persad, S., Attwell, S., Gray, V., Mawji, N., Deng, J.T., Leung, D., Yan, J., Sanghera, J., Walsh, M.P., and Dedhar, S. (2001) J. Biol. Chem. 276, 27462-27469.
50. Partovian, C., and Simons, M. (2004) Cell. Signal. 16, 951-957 51. Calderhead, D. M., Kitagawa, K., Tanner, L. I., Holman, G. D., and Lienhard, G.
E. (1990) J. Biol. Chem. 265, 13801-13808 52. Dragoi, A.-M., Fu, X., Ivanov, S., Zhang, P., Sheng, L., Wu, D., Li, G.C., and
Chu, W.-M. (2005) EMBO J. 24, 779-789 53. Chan, D. W., Ye, R., Veillette, C.J., and Lees-Miller, S.P. (1999) Biochemistry
38, 1819-1828
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

13
54. Canman, C. E., Lim, D.S., Cimprich, K.A., Taya, Y., Sakaguchi, K., Appella, E., Kastan, M.B., and Siliciano, J.D. (1998) Science 281, 1677-1679
55. Delcommenne, M., Tan, C., Gray, V., Ruel,L., Woodgett, J., and Dedhar, S. (1998) Proc. Natl. Acad. Sci. USA 95, 11211-11216.
56. Sakai, T., Li, S., Docheva, D., Grashoff, C., Sakai, K., Kostka, G., Braun, A., Pfeifer,A., Yurchenco, P.D., and Fassler, R. (2003) Genes Dev. 17, 926-940
57. Grashoff, C., Asxodi, A., Sakai, T., Hunziker, E.B., and Fassler, R. (2003) EMBO Rep. 4, 432-438
58. Balendran, A., Casamayor, A.,Deak, M., Paterson, A., Gaffney, P., Currie, R., Downes, C.P., and Alessi, D.R. (1999) Curr. Biol. 9, 393-404
59. Lynch, D. K., Ellis, C.A., Edwards, P.A.W., and Hiles, I.D. (1999) Oncogene 18, 8024-8032.
60. Leitges, M., Plomann, M., Standaert, M.L., Bandyopadhyay, G., Sajan, M.P., Kanoh, Y., and Farese, R.V. (2002) Mol. Endocrinol. 16, 847-858
61. Hill, M. M., Andjelkovic, M., Brazil, D.P., Ferrari, S., Fabbro, D., and Hemming, B.A. (2001) J. Biol. Chem. 276, 25643-25646
62. Manning, B. D. (2004) J. Cell Biol. 167, 399-403 63. Andjekkovic, M., Maira, S.-M., Cron, P., Parker, P.J. and Hemming, B.A. (1999)
Mol. Cell. Biol. 19, 5061-5072. 64. Meier, R., Alessi, D.R., Cron, P., Andjelkovic, M., and Hemmings, B.A. (1997) J.
Biol. Chem. 272, 30491-30497 65. Sable, C. L., Filippa, N., Filloux, C., Hemmings, B.A., and Van Obberghen, E.
(1998) J. Biol. Chem. 273, 29600-29606 FOOTNOTES * We thank Dr. David Sabatini for his generous gift of antibodies to RICTOR. This work was supported in part by National Institute of Health Grant R01 DK067229 and a research grant from the American Diabetes Association. § To whom all correspondence should be addressed: Dept. of Cell Biology and Physiology, Washington University School of Medicine, 660 S. Euclid Ave. St. Louis, MO 63110. Tel.: 314-362-4160; Fax: 314-362-7463; E-mail: [email protected]. 1The abbreviations used are: ATM, ataxia telangiectasia mutated gene product; CYT, cytosol; DNA-PK, double-stranded DNA-dependent protein kinase; DTT, dithiothreitol; Ext, extract; HDM, high-density microsomes; HiP, high speed pellet; HMD-PK, hydrophobic motif domain protein kinase; ILK, integrin-linked kinase; LDM, low-density microsomes; mTOR, mammalian target of rapamycin; NUC, nuclear; PDK1, phosphoinositide-dependent kinase 1; PIP3; phosphatidylinositol 3,4,5-trisphosphate; PKC, protin kinase C; PM, plasma membrane; PM(SW), salt-washed PM; RAPTOR, regulatory associated protein of mTOR; RICTOR, rapamycin insensitive companion of mTOR; siRNA, small interference RNA.
FIGURE LEGENDS
Fig. 1. Subcellular localization of HMD-PK candidate proteins in 3T3-L1 adipocytes. Fully differentiated 3T3-L1 adipocytes were fractionated by differential centrifugation as described under “Experimental Procedures.” Whole cell lysates, (WC), NUC, mitochondrial-enriched
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

14
(MITO), HDM, CYT, LDM, PM, PM(SW), and Ext-HiP fractions were separated by SDS-PAGE (50 µg of protein) and analyzed by immunoblot analysis using each of the designated primary antibodies. Fig. 2. DNA-PK and mTOR/RICTOR protein levels correlate with HMD-PK activity. In vitro reactions containing 3 mg/ml CYT, 1 mg/ml NUC, 1 mg/ml PM, or 0.37 mg/ml LDM were premixed on ice with 16 nM Akt2 and 10 µM PIP3 (sonicated in a mixture of 100 µΜ phosphatidylcholine and 100 µΜ phosphatidylserine). Reactions were initiated with ATP, incubated for 10 min at 37oC, and then subjected to immunoblot analysis using the designated antibodies. Fig. 3. HMD-PK activities in Ext-HiP and LDM are sensitive to PI 3-Kinase inhibitors. In vitro reactions containing different combinations of 0.2 mg/ml Ext-HiP, 0.75 mg/ml LDM,16 nM Akt2,10 µΜ PIP3 (sonicated in a mixture of 100 µM phosphatidylcholine and 100 µM phosphatidylserine), 2 µM wortmannin (Calbiochem), and 15 µM LY294002 (Calbiochem) were mixed and preincubated on ice for 15 min. A, Ext-HiP reactions were initiated with ATP and incubated for 15 min at 37oC. B, LDM reactions were initiated with ATP and incubated for 5 min at 37oC. Samples were quenched and subjected to immunoblot analysis using Akt-Ser473 phospho-specific antibodies. Fig. 4. Purified DNA-PK does not phosphorylate full-length Akt2 in a PIP3-dependent manner. In vitro reactions containing different combinations of 0.2 mg/ml Ext-HiP, 0.75 mg/ml LDM, 100 units DNA-PK (Promega),16 nM Akt2,10 µM PIP3 (sonicated in a mixture of 100 µM phosphatidylcholine and 100 µM phosphatidylserine), and10 µg/ml salmon sperm DNA were mixed on ice. A, Ext-HiP reactions were initiated with ATP and incubated for 15 min at 37oC. 3X-Ext-HiP refers to reactions in which three times the amount of Ext-HiP (0.6 mg/ml) was used along with Akt2 and PIP3. B, LDM reactions were initiated with ATP and incubated for 5 min at 37oC. 2X-LDM refers to reactions in which twice the amount of LDM (1.5 mg/ml) was used along with Akt2 and PIP3. Samples in A and B were quenched and subjected to immunoblot analysis using Akt-Ser473 phospho-specific or DNA-PK antibodies. C, HMD-PK peptide assays were carried out as described in “Experimental Procedures.” Results were quantified as the mean ± S.E. of three independent experiments. White bars represent results from basal cells and hatched bars denote that of insulin-stimulated cells. Fig. 5. HMD-PK activity is not associated with ILK in LDM. 500 µg of LDM were immunoprecipitated with control (CON) or ILK-specific polyclonal antibodies as described in “Experimental Procedures.” In vitro reactions were carried out with either 20 % of the material that remained in the supernatant after the immunoprecipitation (LDM-SUP) or with the entire material found associated with the immunoprecipitation pellet (LDM-PELLET). After adding Akt2 (16 nM), ± 10 µM PIP3 (sonicated in a mixture of 100 µM phosphatidylcholine and 100 µM phosphatidylserine), reactions containing LDM-SUP and LDM-PELLETS were incubated with ATP for 5 min and 10 min at 37oC, respectively. Samples were quenched and subjected to immunoblot analysis using Akt-Ser473 monoclonal phospho-specific (Cell Signaling) or monoclonal ILK (Upstate) antibodies. Fig. 6. siRNA-mediated depletion of DNA-PK, ATM, or ILK has no effect on insulin-stimulated Akt-Ser473 phosphorylation in 3T3-L1 adipocytes. Control, DNA-PK, ATM, and ILK siRNAs were electroporated in differentiated 3T3-L1 adipocytes (day 5) as described in “Experimental Procedures.” 72h after electroporation, cells were serum-starved for 2h, activated or not with 100 nM insulin for 1.5 min, and then harvested in Buffer A containing 1 % SDS. Equal amounts of
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

15
lysates were subjected to immunoblot analysis using the designated primary antibodies. A, immunoblot analysis using DNA-PK, ATM, and ILK specific antibodies. B, immunoblot analysis using Akt-Ser473 and Akt-Thr308 phospho-specific antibodies. C, quantification of the mean ± S.E. of three independent experiments. White bars represent results from basal cells and hatched bars denote that of insulin-stimulated cells. Phosphorylations were normalized to (+ insulin) controls. *, Phosphorylations differed significantly (p < 0.05) compared to insulin-stimulated control samples. An unpaired Student’s t test was used in all statistical analyses. Fig. 7. PIP3-stimulated HMD-PK activity associates with mTOR/RICTOR complexes from LDM. 500 µg of LDM were immunoprecipitated with control (CON), mTOR, or RICTOR specific polyclonal antibodies as described in “Experimental Procedures.” In vitro reactions containing pellets from the immunoprecipitates, Akt2, and ± 10 µM PIP3 (sonicated in a mixture of 100 µM phosphatidylcholine and 100 µM phosphatidylserine) were mixed with ATP and incubated for 10 min at 37oC. A, Reactions were quenched and subjected to immunoblot analysis using the designated antibodies. B, reactions from A were also subjected to immunoblot analysis using DNA-PK, ATM, and ILK specific antibodies. Total LDM provides a positive control for the DNA-PK, ATM, and ILK antibodies. Fig. 8. PIP3-stimulated HMD-PK activity associated with mTOR was sensitive to PI 3-kinase inhibitors but resistant to staurosporine. 500 µg of LDM were immunoprecipitated with mTOR specific polyclonal antibodies as described in “Experimental Procedures.” In vitro reactions containing pellets from the immunoprecipitates, Akt2, and ± 10 µM PIP3 (sonicated in a mixture of 100 µM phosphatidylcholine and 100 µM phosphatidylserine) were preincubated with wortmannin (2 µM), LY294002 (15 µM), or staurosporine (5 µM) for 30 min. Reactions were initiated with ATP, incubated for 10 min at 37oC, quenched, and subjected to immunoblot analysis using Akt-Ser473 monoclonal and mTOR monoclonal antibodies. Fig. 9. siRNA-mediated depletion of RICTOR inhibits insulin-stimulated Akt-Ser473 phosphorylation in 3T3-L1 adipocytes. Control (20 nmol) and RICTOR (50 nmol) siRNAs were electroporated in differentiated 3T3-L1 adipocytes (day 5) as described in “Experimental Procedures.” 72h after electroporation, cells were serum-starved for 2h, activated or not with 100 nM insulin for 1.5 min, and then harvested in Buffer A containing 1 % SDS. Equal amounts of lysates were subjected to immunoblot analysis using the designated primary antibodies. A, immunoblot analysis using RICTOR, mTOR, and Akt specific antibodies. B, immunoblot analysis using Akt-Ser473 and Akt-Thr308 phospho-specific antibodies. C, Results in panel B were quantified as the mean ± S.E. of three independent experiments. White bars represent results from basal cells and hatched bars denote that of insulin-stimulated cells. Phosphorylations were normalized to (+ insulin) controls. *, Phosphorylations differed significantly (p < 0.05) compared to insulin-stimulated control samples. An unpaired Student’s t test was used in all statistical analyses. D, RICTOR siRNA dose response on insulin-stimulated Akt-Ser473 phosphorylation. The amount of control and RICTOR siRNAs are indicated in parentheses. Immunoblot analyses using RICTOR antibodies and Akt-Ser473 phospho-specific antibodies were quantified as the mean ± S.E. of three independent experiments. Fig. 10. Role of two mTOR protein complexes in insulin signaling. Signal transduction pathway for Akt activation by insulin stimulates protein synthesis, cellular proliferation, survivial, and metabolism. Arrows indicate activation; bars indicate inhibition.
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Richard C. Hresko and Mike MuecklermTOR/RICTOR is the Ser473 kinase for Akt/PKB in 3T3-L1 adipocytes
published online October 11, 2005J. Biol. Chem.
10.1074/jbc.M508361200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on May 16, 2019
http://ww
w.jbc.org/
Dow
nloaded from


















