
MOLECULAR DYNAMICS ON CYCLIN-DEPENDENT KINASES
152
MOLECULAR DYNAMICS ON CYCLIN-DEPENDENT KINASES Iveta Bártová
Transcript of MOLECULAR DYNAMICS ON CYCLIN-DEPENDENT KINASES

MOLECULAR DYNAMICS ON CYCLIN-DEPENDENT
KINASES
Iveta Bártová


MASARYK UNIVERSITY BRNO
FACULTY OF SCIENCE
NATIONAL CENTRE FOR BIOMOLECULAR RESEARCH
MOLECULAR DYNAMICS
ON CYCLIN-DEPENDENT KINASES
PH. D. THESIS
BRNO 2006 IVETA BÁRTOVÁ


Supervisor: Prof. RNDr. Jaroslav Koča, DrSc. National Centre for Biomolecular Research Faculty of Science, Masaryk University Brno Supervisor-specialist: Mgr. Zdeněk Kříž, PhD. National Centre for Biomolecular Research Faculty of Science, Masaryk University Brno

Enzymes are things invented by biologists that explain things which otherwise require harder thinking.
Jerome Lettvin

ACKNOWLEDGEMENTS
I would like to thank professor Jaroslav Koča for giving me the chance to spend four academic years in the Laboratory of Computational Chemistry and for great leadership, many valuable discussions, and support.
Michal Otyepka and Zdeněk Kříž, I thank you very much for your kindness, support, advices and all help during my work in the world of molecular modelling and computational chemistry. My deep thanks are addressed to Jiří Damborský for help and encouragement. Petr Kulhánek, I would like to thank you for your invaluable help, advices, suggestions, and for developing programs that were very useful for me. All NCBR members, I wish to thank for forming the friendly environment during my postgraduate study.
Thanks go also to Supercomputing Centre Brno for providing me computer time.
Last but not least, I would like to thank my parents, my sister, and my friend for their kind support and patience during my study.


List of Abbreviations ATP adenosine-5’-triphosphate cA Cyclin A CAK CDK-activating kinase CBF cyclin box fold CDK2 cyclin-dependent kinase-2 CDK5 cyclin-dependent kinase-5 CDKs cyclin-dependent kinases CKII casein kinase II CKI CDK inhibitor Clk CDK-like family CTD carboxy-terminal domain ERK extracellular signal-regulated protein kinase ESP electrostatic potential G-loop Glycine-rich loop GSK3 glycogen synthase kinase-3 GTP guanosine triphosphate HCMV human cytomegalovirus HIV human immunodeficiency virus HF Hartree-Fock HSV herpes simplex viruses KAP kinase associated phosphatase IR infrared LJ Lennard-Jones MAPK mitogen-activated protein kinase MAT1 metastasis-associated protein 1 MD molecular dynamics MM molecular mechanics NMR nuclear magnetic resonance PES potential energy (hyper)surface QM quantum mechanics RESP restrained electrostatic potential RNA ribonucleic acid vdW van der Waals


Table of Contents 1. Motivation ……………………………………………………………… 11 2. Summary ………………………………………………………………. 12 3. Shrnutí …………………………………………………………………. 14 I. THEORY 4. The Cell Cycle …………………………………………………………. 19 5. Overview of the Protein Kinase Family ……………………………… 22 6. Cyclin-Dependent Kinases (CDKs) …………………………………... 25 6.1 Cyclin-Dependent Kinase-2 (CDK2) .………………………….…. 27 6.2 Cyclin-Dependent Kinase-5 (CDK5) .………………………….…. 33 7. Computational Chemistry …………………………………………….. 36 7.1 Molecular Mechanics .………………………………………….….. 37 7.1.1 Force Field Parameter Development ……………………….. 38 7.2 Simulation Techniques …………………………………………….. 39 7.2.1 Molecular Dynamics .………………………………………. 39 II. RESULTS 8. Synopsis of Results …………………………………………………….. 45 8.1 Interactions of CDK2 with Water Molecules ……………………… 45 8.2 Mechanisms of the CDK2 Regulation …………………………….. 47 8.3 Dynamics of Human CDK5; Comparison to CDK2 ………………. 49 III. RESULTS - APPENDIX Analysis of CDK2 Active-Site Hydration:
A Method to Design New Inhibitors ..……………………………….….
55 Activation and Inhibition of Cyclin-Dependent Kinase-2
by Phosphorylation; A Molecular Dynamics Study Reveals the Functional Importance of the Glycine-Rich Loop ..………………………………………………
79

The Mechanism of Inhibition of the Cyclin-Dependent Kinase-2 as Revealed by the Molecular Dynamics Study on the Complex CDK2 with the Peptide Substrate HHASPRK ..………………………………...
95 Different Mechanisms of CDK5 and CDK2 Activation
as Revealed by CDK5/p25 and CDK2/Cyclin A Dynamics ...…………..
109 Bibliography …………………………………………………………… 133 Curriculum Vitae ……………………………………………………… 146 List of Publications ……………………………………………………. 148 List of Presentations …………………………………………………… 149

11
1. Motivation
Cyclin-dependent kinases (CDKs) and their regulatory subunits cyclins are the key molecules that control and coordinate DNA-synthesis, chromosome separation, neuronal differentiation, apoptosis and cell division. CDK and cyclin together drive the cell from one cell cycle phase to the next. To understand the fundamental mechanisms of the cell cycle control, we must understand the structure and regulation of the CDKs.
This thesis summarizes studies on cyclin-dependent kinases (CDKs), their activation and inhibition by theoretical and simulation methods.
The majority of all living organisms consist of cells, which divide and multiply. Except for bacteria, viruses and blue-green algae, all organisms are made up of eukaryotic cells, which have their genetic information, in chromosomes, located in a nucleus separated from the rest of the cell, the cytoplasm. During cell division, while the nucleus goes through a cycle, cytoplasm divides by constriction in animals and by the formation of a membrane in plants. Yeasts and amoebae are unicellular, plants and animals are multi-cellular. An adult human being, for instance, is made
up of about a billion cells per gram of tissue, all of which have originated from a single fertilised egg cell. There are also cells in an adult organism that continuously divide and replace dying ones. The molecular basis of cell cycle and cell division, which has come to be understood fully in recent years, is highly conserved in evolution and operates in the same way in all eukaryotic organisms [1-3].

12
2. Summary
The cyclin-dependent kinases (CDKs) (EC 2.7.1.37) are the catalytic subunits of a large family of heterodimeric serine/threonine protein kinases, which are important for regulation of many biologically critical processes in eukaryotic cell (for example CDK1, CDK2, CDK4). CDK5, one of CDK family members having also the non-cell-cycle role, is expressed in post-mitotic cells of the central nervous system and is required during neuronal differentiation. Therefore, CDKs belong to promising biological targets in human medicine for design new inhibitors due to observation of their deregulation in series cancer. CDKs inhibitors are tested for treatment of many serious human diseases e.g., cancer, neurodegenerative disorders (e.g. Alzheimer’s disease, amyotrophic lateral sclerosis and stroke), diabetes, cardiovascular disorders (e.g. atherosclerosis and restenosis), viral infections (e.g. HCMV, HIV and HSV) etc.
The presented PhD thesis is focused on study of cyclin-dependent kinase 2 (CDK2) and cyclin-dependent kinase 5 (CDK5) and describes structure, dynamics, ligand binding and interaction, hydration, and molecular interactions of CDKs with using computational techniques. The activity of these enzymes is controlled by reversible protein phosphorylation and synthesis and degradation of activator and inhibitor subunits. Consequently, a special attention is paid to the activation and inhibition of CDKs by phosphorylation and interaction of CDKs with regulatory subunits (cyclin A and p25 are regulatory subunits of CDK2 and CDK5, respectively). In eukaryotes, protein phosphorylation is probably the most important regulatory event. Many enzymes are switched “on” or “off” by phosphorylation and dephosphorylation. The insight of mechanism of cell cycle regulation is important for understanding deregulation and origin of relevant diseases. These studies provided interpretation of the different mechanisms of CDK2 and CDK5 regulation.
Analysis of the CDK2 active site hydration was used as a method for design new inhibitors. The molecular dynamics (MD) simulations of the CDK2/ATP (native substrate of CDK2) complex and CDK2 in the complex with roscovitine and isopentenyladenine (purine-like inhibitors of CDK) were studied. A number of water molecules that were in contact with the protein for the whole trajectory were assigned. The positions of structural water molecules1 were compared with the positions of ligand (inhibitor) polar groups and water molecules found by X-ray analysis. The structural water molecules in the free CDK2, which occupy the same binding sites as polar groups of ligand molecules, were found. The behavior of water molecules interacting with amino acids in the enzyme active
1 A key characteristics of structural water molecules is the interaction time of the water molecule with the protein atom. The water molecules with interaction time larger than 950 ps within 1000 ps long MD simulation were considered as structural.

13
site, their interaction energies, and location, provides information that is useful for the rational drug design of new potent and selective inhibitors.
Nanoseconds long MD trajectories of differently active complexes of human CDK2 (inactive, semiactive, fully active, and inhibited CDK2) and CDK2 (fully active and inhibited form) in complex with peptide substrate HHASPRK were produced. The mechanism of the CDK2 inhibition by phosphorylation on inhibitory sites in the glycine-rich loop (G-loop) was studied in detail. The MD simulations results of CDK2 inhibition by phosphorylation on inhibitory sites provide insight into the structural aspects of CDK2 deactivation. The catalytic function of CDKs is the covalent phosphorylation of substrate protein via transfer of the γ-phosphoryl group of ATP (the ATP is bound as a complex with the Mg2+ ion) to threonine or serine of the target protein substrate. Therefore, the correct coordination of the Mg2+ ion and appropriate conformation and orientation of the ATP phosphate group are important for ATP terminal phospho-group transfer to the CDK2 substrate. The position of the ATP γ-phosphate relative to the phosphorylation site of the peptide substrate in the active CDK2 was described and compared with inhibited forms of CDK2. The inhibitory phosphorylation causes ATP misalignment for phosphorylation, changes in the Mg2+ ion coordination sphere, and G-loop conformational change (its shift away from the ATP binding site), which leads to the opening of the CDK2 substrate binding box and substrate destabilization. All mentioned effects explain the lost of kinase activity after inhibitory phosphorylation in the G-loop.
The interactions of the specific residues for substrate HHASPRK binding to CDK2 were studied with MD simulations. The results clearly provided an explanation previously not known as to why a basic residue (R/K) is preferred at the P2 peptide substrate position2. The R2 interacts with the ATP phosphate moiety and, therefore, it can play a role in appropriate ATP alignment before the reaction.
The dynamics and mechanisms of activation and inhibition by phosphorylation of CDK5 and CDK5/roscovitine were studied and compared to CDK2 regulation. Energy decomposition analysis was used to gain a detailed interaction scheme for roscovitine with CDK5 (unphosphorylated and phosphorylated CDK5) and also for CDK2 for comparison. The same method was also used to determine regions with the highest energy contributions to the interaction energy between CDK5 and p25, and, also between CDK2 and cyclin A and revealed that p25 binding is sufficient to stabilize the extended active T-loop conformation of CDK5, while cyclin A binding to CDK2 is not able to gain a biologically fully active system, this needing further phosphorylation of the T160 in the activation segment (T-loop). Differences between both studied systems, preference of CDK5 for p25 and CDK2 for cyclin A, and their different mechanisms of regulation, were investigated and discussed with respect to their specific biological function. 2 Subscripts denote amino acid positions in the substrate numbered from the phosphorylation residue with increasing numbers toward the C-terminus.

14
3. Shrnutí
Cyklin-dependentní kinasy (CDK) (EC 2.7.1.37) přísluší do katalytické podjednotky heterodimerních serin/threonin protein kinas (patří zde např. CDK1, CDK2, CDK4), které jsou v eukaryotických buňkách důležité pro regulaci mnoha kritických biologických procesů. Role CDK5, jenž náleží také mezi CDK enzymy, je rovněž nebuněčná. CDK5 se nachází v post-mitotických buňkách centrálního nervového systému a je vyžadována během nervového dělení. Z důvodu, že deregulace CDK byla prokázána v řadě nádorů, patří tyto enzymy v lékařství mezi slibné biologické terče pro návrh nových inhibitorů. CDK inihibitory jsou testovány pro léčbu mnoha závažných onemocnění, mezi které patří např. rakovina, neurodegenerativní poruchy (např. Alzheimerova choroba, amyotrofní laterální skleróza, mrtvice), cukrovka, kardiovaskulární poruchy (např. arterioskleróza, restenóza), virové nákazy (např. HCMV, HIV, HSV) atd.
Disertační práce byla zaměřena na studium cyklin-dependentní kinasy-2 (CDK2) a cyklin-dependentní kinasy-5 (CDK5), kdy v jejím průběhu byla s pomocí počítačových metod popsána struktura, dynamika, vazba a interakce ligandů k CDK, hydratace a molekulové interakce CDK. Aktivita těchto enzymů je kontrolována reverzibilní fosforylací, syntézou a degradací aktivačních a inhibičních podjednotek. Speciální pozornost byla proto také věnována studiu aktivace a inhibice CDK fosforylací a interakcím CDK s jejich regulačními podjednotkami (např. cyclin A je jedním z proteinů, který aktivuje CDK2 a p25, který patří mezi regulační podjednotky proteinu CDK5).
Fosforylace proteinů v eukaryotech patří mezi nejdůležitější funkce regulace buňky, kdy enzymy jsou aktivovány a deaktivovány fosforylací a defosforylací. Pro pochopení deregulace a původu závažných nemocí je důležité porozumění mechanismu regulace buněčného cyklu. Proto je dalším přínosem této disertační práce vysvětlení rozdílných regulačních mechanismů CDK2 a CDK5.
Analýza hydratace aktivního místa CDK2 byla použita pro návrh nových inhibitorů. Byly připraveny molekulové dynamické simulace CDK2 s ATP, který je přirozeným substrátem CDK, a simulace komplexů CDK2 s roskovitinem a isopentenyladeninem, které patří mezi purinové inhibitory CDK. S využitím těchto simulací bylo nalezeno několik molekul vod, které se vyskytovaly v kontaktu s proteinem po celou dobu simulace. Pozice nalezených stabilních molekul vod byly porovnány s pozicí polárních skupin ligandů a pozicí molekul vod získaných RTG analýzou. Touto analýzou byly nalezeny stabilní molekuly vod ve volné CDK2, které zaujímají stejné vazebné místo jako polární skupiny sledovaných ligandů. Chování molekul vod interagujících s aminokyselinami v aktivním místě enzymu, jejich interakční energie a informace o vazebném místě poskytují cenné výsledky, které mohou být využity pro návrh a vývoj selektivních inhibitorů.
Pomocí molekulové dynamiky bylo na nanosekundové časové škále sledováno několik různě aktivních forem CDK2 (neaktivní, částečně aktivní,

15
aktivní a inhibované formy proteinu) a CDK2 (aktivní a inhibované formy) v komplexu se substrátem HHASPRK. Detailně byl studován mechanismus inhibice CDK2 po fosforylaci na inhibičních místech proteinu v glycinové smyčce (G-smyčce). Výsledky studia inhibice CDK2 pomocí fosforylace na inhibičních místech proteinu poskytují dříve neznámé strukturní detaily deaktivace tohoto proteinu. Katalytickou funkcí CDK je přenos γ-fosfátové skupiny z ATP (ATP je vázáno v komplexu s Mg2+ kationtem) k serinu, případně threoninu cílových proteinů. Z tohoto důvodu je pro přenos koncové fosfátové skupiny z ATP k substrátu CDK2 důležitá správná koordinace Mg2+ iontu a vhodná konformace a orientace koncové fosfátové skupiny ATP. Pozice a orientace γ-fosfátové skupiny ATP vzhledem k serinu, fosforylačnímu místu substrátu HHASPRK, byla sledována v aktivní CDK2 a porovnána k jejímu umístění a orientaci, kterou získala po inhibiční fosforylaci CDK2. Inhibiční fosforylace způsobila změnu konformace a orientace ATP, která byla vhodná pro přenos fosfátové skupiny. Zároveň dochází také ke změně koordinační sféry Mg2+ iontu a odklonu inhibičního segmentu (G-smyčky), což způsobilo otevření vazebného boxu pro substrát a tím vedlo k destabilizaci substrátu. Všechny výše zmíněné efekty jasně vysvětlují ztrátu aktivity enzymu po inhibiční fosforylaci v G-smyčce.
S využitím počítačových simulací byly také sledovány interakce specifických residuí pro vazbu substrátu HHASPRK k CDK2, které objasnily dříve neznámý fakt, proč je v pozici P2 substrátu preferované bazické residuum (R/K). R2 residuum interaguje s fosfátovou částí ATP, což může hrát důležitou roli v reorientaci ATP do pozice, která je vhodná pro katalytickou reakci.
Dále byla studována dynamika enzymu CDK5 a komplexu CDK5 s roskovitinem a mechanismus jeho aktivace a inhibice fosforylací. Dosažené výsledky byly porovnány s mechanismem regulace CDK2. K detailnímu popisu interakčních energií roskovitinu k CDK5 (u nefosforylované a fosforylované formy CDK5 v G-smyčce) byla použita metoda energetické dekompoziční analýzy. Pro srovnání byla tato metoda také použita u komplexů CDK2. Stejný přístup byl využit k nalezení regionů proteinu s nejvyššími energetickými příspěvky k interakční energii mezi CDK5 a p25 a také mezi CDK2 a cyklinem A. Použitá metoda ukázala, že vazba p25 k CDK5 je dostačující pro stabilizaci aktivní konformace T-smyčky v CDK5, zatímco interakce cyklinu A k CDK2 není schopna CDK2 plně aktivovat. Tento enzym dále pro svou aktivaci požaduje fosforylaci T160 v aktivačním segmentu (T-smyčce). Rozdíly mezi oběma studovanými systémy, preference CDK5 pro p25 a CDK2 pro cyklin A a jejich regulační mechanismy byly podrobně zkoumány a diskutovány s ohledem k jejich specifické biologické funkci.

16

17
I. THEORY

18

19
4. The Cell Cycle
The Nobel Prize for Physiology and Medicine was awarded for seminal discoveries concerning the control of the cell cycle in 2001. Leland Hartwell, Timothy Hunt, and Paul Nurse have enabled an understanding at the molecular level how the cell is driven from one phase to the next during division.
The cell cycle entails the orchestration of virtually all cellular processes: metabolism, protein synthesis, secretion, DNA replication, organelle biogenesis, cytoskeletal dynamics and chromosome segregation [4].
The cell cycle progression is divided into 4 distinct phases. In response to mitogenic signals, cells progress from the resting phase, G0, to G1, during which they become committed to progression through the cell cycle. From G1, they enter S phase, when the chromosomes are duplicated once and only once. After a second gap or growth phase, G2, they enter mitosis when the cell divides into two daughter cells. The duration of the cycle is dependent on the cell type. In most mammalian cells it ranges from 10 to 30 hours.
It is essential that the different phases are precisely coordinated and controlled so that one phase is completed before the next one can begin. Errors in coordination can lead to chromosomal aberrations. Chromosomes or their parts can be lost, rearranged or distributed unequally between the daughter cells. This type of alteration is often seen in cancer cells. Therefore, an understanding of the regulation of cell division, how cells determine when and how to multiply or otherwise develop, and how that process can go awry, is fundamental to understanding how cancer cells mutate and to developing approaches that predict, prevent or reverse the alteration.
Using yeast as a model organism, Hartwell demonstrated that DNA replication and nuclear events are coordinated with cytoplasmic events at a point, which he called START, and this crucial point was controlled by a gene, which he designated CDC28. This gene was actually the first CDK gene identified, which has formed the basis for major advances in the understanding of cell cycle in the last 15 years. Following Hartwell, Paul Nurse used a different yeast, Schizzosaccharomyces pombe, which is distantly related to baker's yeast, as a model organism and discovered the gene CDC2 which had a key role in the control of the transition from the G2 phase to mitosis [5]. Later Nurse found that CDC2 had a more general function. In 1987 Nurse isolated the corresponding gene in humans, later called CDK1 [6]. The gene encodes a protein that belongs to a family cyclin dependent kinases, CDKs. Nurse showed that activation of CDK is dependent on reversible phosphorylation. This finding has formed the basis for the identification of several other CDK molecules in human organisms.
Cyclin-dependent kinases are dependent not only on reversible phosphorylation but also on cyclin binding. Following the initial discovery of cyclin B in sea urchin eggs by Evans et al. [7], it was also shown that cyclin B homologues were present in most eukaryotes. Cyclins are proteins formed and

20
degraded during each cell cycle. The CDK levels, by contrast, are constant, at least in the simplest cell cycles. But the cyclins bind to the CDK molecules, thereby regulating CDK activity and selecting the proteins to be phosphorylated. Periodic protein degradation is thus the key control mechanism (Figure 1).
The CDK and cyclin together drive the cell from one phase to the next in the cell cycle. Hunt later discovered cyclins in other species as well and found that cyclins were conserved during evolution. Today we know of 10 different cyclins in humans.
While the findings are of significance to all biomedical research, they have a crucial bearing on the treatment of cancer.
Unveiling the molecular mechanisms that control the cell cycle is critical to understand the molecular bases of cancer. It is likely that chromosomal aberrations are owing to defective cell cycle control. It has been suggested that genes for CDK molecules and cyclins function as oncogenes, the cancer-causing genes. Increased levels of CDK and cyclins are sometimes found in human tumours. CDK molecules and cyclins have also been shown to collaborate with the products of tumour-suppressor genes during the cell cycle. These discoveries are about to be applied to tumour diagnostics.
The above Nobel Prize winning research may give rise to new principles for cancer therapy. Inhibitors of CDK molecules are already undergoing trials as anti-cancer drugs.
An ordered sequence of cyclin-CDK activities trigger most of the events of the cell cycle. During G1 phase, CDK activity is reduced to a minimum by CDK inhibitors (CKIs), cyclin proteolysis, and decreased cyclin gene transcription. When environmental conditions are favourable, G1- (CDK2,4,6) and G1/S-CDKs (CDK2) increase in concentration, overcoming these inhibitory barriers in late G1 and triggering the activation of S-CDK (CDK2). The S-CDK phosphorylates proteins at DNA replication origins, initiating DNA synthesis through a mechanism that ensures that the DNA is duplicated only once per cell cycle. Once S phase is completed, the activation of M-CDK (CDK1) leads to the events of early mitosis, whereby the cell assembles a mitotic spindle and prepares for segregation of the duplicated chromosomes-which consist of sister chromatids glued together. Anaphase is triggered by the destruction of the proteins that hold the sisters together. The M-CDK is then inactivated by cyclin proteolysis, which leads to cytokinesis and the end of M phase. Progression through the cell cycle is regulated precisely by various inhibitory mechanisms that arrest the cell cycle at specific checkpoints when events are not completed successfully, when DNA damage occurs, or when extracellular conditions are unfavourable (Figure 1).

21
Figure 1. A simplified view of the core of the cell-cycle control system. The two larger phases of the cell cycle are mitosis and interphase. Mitosis is the part of the cell cycle when the cell prepares for and completes cell division. Interphase is made up of three distinct phases: G1, S phase, and G2. The G1 and G2 phases serve as checkpoint for the cell to make sure that it is ready to proceed in the cell cycle. If it is not, the cell will use this time to make proper adjustments that can include cell growth, correction or completion of DNA synthesis, and duplication of intracellular components. S phase involves the replication of chromosomes. The critical CDK/cyclin complexes for the cell division are CDK2/cyclin E, driving the cell across the G1/S-phase border; CDK2/cyclin A, mediating DNA replication during the S phase; and CDK1/cyclin B, controlling the entry into mitosis [8] (adopted from [9]).

22
5. Overview of the Protein Kinase Family
The protein kinase family, that includes cyclin-dependent kinases (CDKs), is one of the largest in the human genome, comprising about 500 genes [10] (Figure 2).
Figure 2. Overview of the human protein kinases (kinome). Kinome: A subset of the genome consisting of the protein kinase genes. The complete complement of over 500 protein kinases constitutes one of the largest of all human gene families (adopted from [11]).

23
It has been estimated, however, that there may exist in excess of 2,000 human proteins that utilise adenosine-5’-triphosphate (ATP) [12, 13].
The majority of kinases contain a 250-300 amino acid residue catalytic domain whose structure and in particular key catalytic residues are highly or absolutely conserved in the protein kinase family. This domain comprises a binding pocket for ATP (less frequently GTP – guanosine-5’-triphosphate) (Figure 3).
Figure 3. View of the adenosine-5’-triphosphate (sticks model) binding pocket of CDK2. The secondary structure of CDK2 is shown in red (β-sheet) and in blue (α-helix). The surface of binding pocket for ATP (the ATP is bound in complex with Mg2+) is shown in transparent blue.
The common catalytic function of protein kinases is the covalent
phosphorylation of substrate proteins via transfer of the γ-phosphoryl group of ATP to threonine, serine (S/T-specific kinases), or tyrosine residues (Y-specific kinases) of the substrate [14]. The phosphate donor is always bound as a complex with a divalent ion (usually Mg2+ or Mn2+) (see Figure 3). Important function of the catalytic domain is the binding and orientation for phosphoryl transfer of the macromolecular substrate [13].

24
In eukaryotes, protein phosphorylation is probably the most important regulatory event. Many enzymes and receptors are switched “on” or “off” by phosphorylation and dephosphorylation. Phosphorylation is catalysed by various specific protein kinases, whereas phosphatases dephosphorylate. Phosphorylation was first identified as a mechanism for regulating protein activity in the 1950s by Fischer and Krebs [15].
Indeed, kinases are involved in signal transference pathways and in the regulation of cell growth, cell division, cell mobility, metabolism, membrane transport, gene expression, learning and memory [16].

25
6. Cyclin-Dependent Kinases (CDKs)
The global protein network must embody the main features of cell cycle control, as dictated primarily by protein synthesis, degradation and phosphorylation.
Hanks and Quinn [17] have provided the following expert multiple alignment of 390 kinase sequences. As a result is organization of the known members of the eukaryotic protein kinase superfamily into distinct families that share basic structural and functional properties. The primary criterion used in the development of this classification scheme is similarity in catalytic domain amino acid sequence. The key catalytic domain of CDKs occurs in the CMGC group (comprises the cyclin-dependent kinases (CDKs), the Erk (MAP) kinases, the glycogen synthase kinase-3 (GSK3) family, the casein kinase II (CKII), and the CDK-like (Clk) family) of the protein kinases [17] (Figure 2).
Thus kinases having closely-related catalytic domains tend also to: 1) be similar in overall structural topology, 2) have similar modes of regulation, and 3) have similar substrate specificities.
The discovery of more than 10 CDC2-related proteins in vertebrates led initially to speculation that higher eukaryotic cell cycle control involved immensely complex combinations of CDKs and cyclins (Figure 4).
CDK family members generally contain about 300 residues and are 35-65% identical to the prototypes CDC2 and CDC28 (molecular weight: 33-40 kDa). The catalytic domain of kinases is highly conserved. Human CDK1 and CDK2 are functionally homologous to yeast CDC2 and CDC28 (60-65% identity in amino-acid sequence) and are clearly involved in central cell cycle functions.
The CDK2 interacts with cyclin E at the beginning of S phase to induce the initiation of DNA synthesis, and then binds cyclin A throughout S phase, when it plays some poorly defined role in progression through DNA synthesis. The CDC2/cyclin B complex initiates mitosis. Complexes between CDC2 and cyclin A may also contributed to the preparation for mitosis (Figure 1).
The CDK3 is very closely related to CDC2 and CDK2. The CDK3 protein is not readily detected in mammalian cells, and its cyclin partner has not been identified. The ability of overexpressed dominant-negative CDK3 mutants to slow progression through G1 may indicate a role for this kinase in cell cycle control [18].

26
Figure 4. Cell Cycle Regulators. Cell division is controlled by an intricate balance of cell cycle regulatory proteins: 1) positive regulators: the cyclins and the cyclin dependent kinases (CDKs) and 2) negative regulators: the cyclin kinase inhibitors (CKIs) (adopted from [19]).
Cell proliferation in vertebrates often requires the presence of external
growth factors. A key response to growth factors in many cell types is the activation of CDK4 or its close relative CDK6 by members of the cyclin D family. These complexes are required for progression through G1 phase.
Complete activation of CDKs requires more than just cyclin binding; the CDK subunit must also be phosphorylated by the CDK-activating kinase (CAK). The majority candidate for CAK in higher eukaryotes is itself a CDK complex containing CDK7 and cyclin H. The CDK7 with its cyclin partner cyclin H and a third subunit MAT1 has been implicated in positive regulation of the other CDK-cyclins in vitro [20]. MAT1 stabilizes the association between CDK7 and cyclin H.

27
However, recent studies have identified CDK7/cyclin H as having a non-cell-cycle role because it also possesses kinase activity for the carboxy-terminal domain (CTD) of RNA polymerase II [21, 22].
Further diversity of the CDK family, was the subsequent observation that a molecule that binds to the HIV tat gene product also facilitates phosphorylation of the RNA polymerase II CTD by activating a kinase – CDK9 – that has obvious similarity to the CDKs [23].
The CDK/cyclin pair from vertebrates and flies, known as CDK8/cyclin C, associates with RNA polymerase II and possesses also CTD kinase activity (phosphorylates the carboxyl-terminal domain) [18].
CDK5, one of CDK family members having also the non-cell-cycle role, is expressed in post-mitotic cells of the central nervous system and is required during neural differentiation [18, 24, 25].
Due to their cellular role, the CDKs are promising biological targets in human medicine. Recent results confirm this fact and CDKs inhibitors are tested for treatment of many serious human diseases e.g., cancer, neurodegenerative disorders (e.g. Alzheimer’s disease, amyotrophic lateral sclerosis and stroke), diabetes, cardiovascular disorders (e.g. atherosclerosis and restenosis), viral infections (e.g. HCMV, HIV and HSV) etc. [13, 26].
6.1. Cyclin-Dependent Kinase-2 (CDK2)
In human cell, cell cycle progression is tightly controlled by the activity of cyclin-dependent kinases (S/T kinases; EC 2.7.1.37). Cyclin-dependent kinase-2 (CDK2) is one of CDK family members controlling the eukaryotic cell division cycle.
CDK2 is inactive as monomer (Scheme 1). An activation segment, region of the protein called T-loop (residues 152-170), blocks the active site.
The binding of cyclin A or cyclin E causes the T-loop to move out of the active site, resulting in partial activation of the CDK2. Cyclin also decreases the flexibility of the T-loop, which can explain observations that phosphorylation at T160 is cyclin-dependent in vitro [27].
Phosphorylation of CDK2 by CDK-activating kinase (CAK; CDK7/cyclin H complex) at a specific threonine residue in the T-loop (T160 in CDK2) fully activates the enzyme by changing the shape of the T-loop, improving the ability of the enzyme to bind its protein substrate [18, 28] (Figure 5).

28
Scheme 1. Scheme of CDK2 regulation. Inactive form CDK2/ATP (I) binds to cyclin and may be phosphorylated at Y15 by WEE1 kinase. Inhibited complex pY15-CDK2/cyclin A/ATP (II) is phosphorylated by CAK at T160 and pY15,pT160-CDK2/cyclin A/ATP complex (III) is activated at the pY15 site by dephosphorylation by CDC25. The fully active complex pT160-CDK2/cyclin A/ATP (IV) after cyclin is lost is dephosphorylated by PP2C or KAP at pT160 (adopted from [29]).
Recently, the other regulatory subunit of CDK2 (also CDK1) was found. A Xenopus protein named RINGO [30] or Speedy [31] plays an important role in the regulation of the meiotic cell cycle in oocytes. The RINGO/Speedy exhibits no amino acid sequence homology to cyclins but can directly activate CDK1 and CDK2 (independent of cyclin binding) and even bypassing the requirement for phosphorylation in the activation loop. A human homologue of RINGO/Speedy has been reported, which has been proposed to play a role in the G1/S transition [32].

29
Figure 5. The structural basis of CDK2 activation. The CDK activation is a two-step process that requires cyclin binding and phosphorylation in the activation segment (known also as the “T-loop”) (adopted from [9]).
The function of the CDK2 is to catalyze the phosphoryl transfer of the
adenosine-5’-triphosphate (ATP) γ-phosphate to the serine or threonine hydroxyl in the protein substrate.
CDK2 contains the classic bi-lobal kinase fold [18, 27]. The N-terminal domain is composed mainly of β-sheet, containing five anti-parallel β-strands, and one α-helix (the C-helix) bearing PSTAIRE motif. The larger C-terminal domain is predominantly α-helical, and is linked to the N-terminal domain by a flexible hinge. The ATP-binding site is located in the deep cleft between the two lobes. The adenine base of ATP binds deep within the cleft in a hydrophobic pocket, while the ATP ribose/phosphate moiety is an extensive solvent-exposed pocket formed in part by the flexible glycine-rich loop (G-loop; CDK2 residues 11-18) [27]. The γ-phosphate of ATP points to the mouth of the cleft where the activation loop (T-loop; CDK2 residues 152-170, containing the T160 phosphorylation site) is located, and where protein substrates bind (Figure 6) [33]. The ATP is always bound as a complex with a usually Mg2+ ion and the correct the ATP phosphate orientation and the magnesium coordination is thought to be critical for catalysis.

30
Figure 6. The picture of fully active CDK2 (pT160-CDK2/cyclin A/ATP) complex represents the CDK2/cyclin A secondary structure (CDK2 is predominantly shown in blue and cyclin A is represented in grey). The pT160 (highlighted) activation site is located in the T-loop (red colored secondary structure). G-loop, that forms the ATP (highlighted) binding site at partly, is shown in yellow.
The cyclins are a remarkably diverse family of proteins. Sequence
homology tends to be concentrated in a 100-residue section known as the cyclin box fold (CBF) [34], which is necessary for CDK binding and activation [18, 35]. The cyclin box fold constitutes a compact domain consisting of five α-helices that are tandemly repeated in the C-terminal domain [36].
An important finding from a comparison of the structures of free and bound cyclin A is that cyclin A undergoes no significant structural change on association with CDK2 [36]. In striking contrast, CDK2 undergoes a significant conformational response on complex formation that results in activation of the enzyme.
The cyclin A-CDK2 interface is formed by PSTAIRE helix, the T-loop, portions of the N-terminal and C-terminal lobe from CDK2, and helices α3, α4, and α5 from the first repeat, as well as the N-terminal helix from cyclin A [28].

31
The critical CDK/cyclin complexes for the cell cycle function are CDK2/cyclin E, driving a cell across the G1/S-phase border and CDK2/cyclin A, mediating DNA replication (see Figure 4).
The contact site of the CDK2 and cyclin A is the α1 helix of CDK2, which contains the PSTAIRE sequence motif characteristic of the CDKs family. After cyclin A binding to CDK2, this helix rotates about its axis and moves several angstroms into the catalytic cleft, compared to free CDK2 [28]. Cyclin A binding induces another conformational changes within CDK2. The significant feature is the reconfiguration of the ATP binding site into a conformation that favours its nucleophilic attack by the substrate and a positional switch of the T-loop, which opens the catalytic cleft, affects the orientation of the putative substrate binding site of CDK2, and leads to appropriate exposure of T160.
Phosphorylation of T160 by CAK induces further conformational changes in the T-loop and in the C-terminal lobe of CDK2 and stabilizes the substrate binding site [37-39]. After this conformational change in the T-loop the binding pocket for substrate is created (conformation typical of active proline-directed kinases). CDKs display an absolute requirement for proline in the substrate P+1 position3, hence the common reference to these enzymes as proline-directed kinases [40].
The peptide libraries’ analyses for CDK2 substrate preference have detected the sequence X–1(S/T)0P1X*2(K/R)3 to be an optimal substrate. Here, S/T are phosphorylation residues (serine or threonine), X is any amino acid, but K/R are favoured at X* position [41, 42]. The structure of the fully active CDK2 (pT160-CDK2/cyclin A/ATP) in complex with the substrate peptide (HHASPRK) comprising the optimal motif explains the basis of the peptide binding to CDK2 (PDB code 1QMZ) [40] (Figure 7).
The substrate peptide binds in an extended conformation across the catalytic site and its binding site is located close to the active site on the C-terminal lobe surface. Some parts of the binding site are formed by the G-loop and T-loop. The T-loop forms a suitably shaped pocket to accept the substrate proline P1 residue, next to the phosphorylation residue (S0). The lysine K3 is H-bonded to the pT160 phosphate and this interaction explains the specificity for basic residues at this position.
3 Subscripts denote amino acid positions in the substrate numbered from the phosphorylation residue with increasing numbers toward the C-terminus.

32
Figure 7. View on fully active CDK2 (pT160-CDK2/cyclin A/ATP) in complex with the substrate peptide (HHASPRK). CDK2 is predominantly coloured blue and cyclin A is represented in grey. Substrate peptide HHASPRK (shown as sticks) binding site is formed by the G-loop (yellow) and T-loop (red) at partly.
Generally, protein kinases are inactivated by modulation of one or more of four conserved structural elements. These modulatory mechanisms comprise: 1) blocking the ATP binding pocket, 2) distorting the glycine-rich loop or flap, 3) altering the position of C-helix (corresponding to the “PSTAIRE” helix), and 4) altering the conformation of the activation segment, usually via phosphorylation [14].
Deactivation of the CDK2 can include reversal of the activation steps, such as dissociation (and degradation) of cyclin, blockage of the substrate of ATP binding site, or additional modifications.
The activity of a CDK2 complex can be inhibited by phosphorylation at the T14 and Y15 residues in the inhibition segment (G-loop) (Scheme 1). Phosphorylation of T14 and Y15 in the glycine-rich loop, the ‘ceiling’ of the ATP binding site, is an important element of CDK regulation [43], whose structural aspects for CDK2 were recently proposed using molecular dynamics [29, 44]. Phosphorylation of Y15 by the Wee1 kinase negatively regulates CDK1 activity

33
and is a master switch in the control of the G2/M transition of the cell cycle [43]. The amino acid residue Y15 and to a lesser extent T14 of CDK2 are phosphorylated by human Wee1Hu [45], which inhibits CDK2 activity, while dephosphorylation of these sites by a phosphatase known as CDC25 increases CDK2 activity [46, 47]. Recently, the phosphorylation mechanisms of the cell were revisited with the finding that pY15-CDK2 dephosphorylation by CDC25 is an important regulation mechanism of correct cell cycle timing [48].
CDK/cyclin complexes can also be regulated by the binding of CDK inhibitor proteins (CKIs) (see Figure 4). There are a variety of CKI proteins, and they are primarily employed in the control of G1 and S phase. The three-dimensional structure of a CDK/cyclin/CKI complex reveals that CKI binding dramatically rearranges the structure of the CDK active site, rendering it inactive [49-51].
Dephosphorylation of monomeric CDK2 at phospho-T160 residue in the activation segment (T-loop) deactivates CDK2. Kinase – associated phosphatase (KAP) catalyze dephosphorylation of the threonine in the activation segment [52].
CDK2 can be inhibited by interaction with the small molecules. Many types of CDK inhibitors have been found: aminopyrazoles, aminopyrimidines, indirubines, quanine, paullones, purine-based inhibitors [13, 53-59] and others. The some purine-like inhibitors occupy the same binding site as the ATP. However, not all inhibitors from this category bind the enzyme in the same way as the ATP does. For example, one of the effective purine-like inhibitors, roscovitine [2-(R)-(1-ethyl-2-hydroxy-ethylamino)-6-benzylamino-9-isopropylpurine], occupies the same active site but the binding mode is different than for the ATP. Another inhibitor, isopentenyladenine [6-(3,3-dimethylallyl-amino)purine), is also positioned in the same active site but it occupies a different binding mode than both ATP and roscovitine [55]. Most kinase inhibitor molecules currently being developed are targeted at the ATP-binding site, a ubiquitous ‘receptor’ in nature. Concern about monitoring of the selectivity of inhibitors was discovered due to similarity between the ATP-binding sites of different kinases. Most kinase inhibitors mimic mainly the adenine moiety of ATP [13].
6.2. Cyclin-Dependent Kinase-5 (CDK5)
Although most CDKs have been implicated in the regulation of the cell division cycle, emerging evidence indicates that certain members of this family are involved in other processes. An important example is represented by CDK5. Albeit the CDK5 is widely expressed in many tissues and cell, CDK5 kinase activity is restricted to neuronal cells [60-62]. This specificity for neuronal tissue is the results of the CDK5 activator proteins p35 and p39. CDK5 deregulation is injected by p35 cleave by calpain producing active fragment p25

34
(p25 is a proteolytic product consisting of residues 99-307 of p35 and containing the essential CDK5 activation domain). The p25 fragment looses membrane localization of p35 and induces CDK5 hyperactivation. CDK5/p25 translocates from the plasma membrane to the cytosol and nucleus, where novel substrates become hyperphosphorylated leading to neuronal cell death [63]. Since deregulation of CDK5 has been implicated in neurodegenerative diseases, there is a strong interest in new chemical inhibitors of CDK5 to treat these serious brain illnesses [64].
Increased CDK5 kinase activity has been implicated in Alzheimer’s disease [62, 65] and amyotrophic lateral sclerosis [64]. Since deregulation of CDK5 has been implicated in Alzheimer’s disease, there is strong interest in chemical inhibitors of CDK5 that could be used as anti-Alzheimer’s disease agents in clinic.
Several CDKs (CDK1, CDK2, CDK4 and CDK6) show a dual mechanism of activation based on cyclin binding and phosphorylation of the activation loop (T-loop) [18] (see chapter above). This model of activation, however, does not apply to CDK5, despite sequence identities running near 60% for CDK2-CDK5 pairs in different species. In fact, CDK5 binds to cyclins D and E, but these fail to ignite its kinase activity [61, 64]. Instead, CDK5 activity is triggered by p35 and p39, homologous proteins whose expression is limited to neurons and to a few other cell types [64]. Furthermore, CDK5 does not seem to be activated by phosphorylation of the activation loop, even if this contains a potential site of phosphorylation (Ser159, equivalent to Thr161 or Thr160 of CDC2 or CDK2, respectively) [61].
The structure of the unphosphorylated CDK5/p25 complex (PDB code 1H4L) [61] confirmed that a cyclin box fold (CBF) is embedded in p25, p35 and p39 (Figure 8). The CBF is the approximately 100-residue structural motif used by cyclins to interact with their CDK partners [36, 64, 66]. While cyclin A contains 2 consecutive CBF domains (of which only the first binds CDK2), only a single CBF is found in p25, and a comparison of p25 structure with p35 and p39 suggests that all three proteins contain a single CBF motif [61]. The p25 CBF binds CDK5 around the αC helix, which contains the PSAALRE sequence motif, and the activation loop. Overall aspect of the CDK5/p25 complex is very similar to the CDK2/cyclin A complex [61]. However, some important structural and regulatory differences between the CDK5/p25 and CDK2/cyclin A complex activation are noticeable. A relevant difference is that even in the absence of a phosphate group on Ser159 the CDK5 activation loop adopts correct conformation for substrate binding i.e. conformation typical of active proline-directed kinases. This conformation is almost identical to that observed in the phospho-CDK2/cyclin A complex, establishing that p25 acts as a one-step activator of CDK5, whose binding to CDK5 introduces in a single step the conformational changes CDK2 undergoes via 2 independent regulatory events such as cyclin A binding and T160 phosphorylation [61, 64].

35
Figure 8. The picture of active CDK5 (CDK5/p25) complex represents the CDK5/p25 secondary structure (CDK5 is predominantly coloured blue and p25 is shown in grey). The pS159 (highlighted) inhibitory site is located in the T-loop (red colored secondary structure). G-loop is shown in yellow.
In vitro, CDK5 is phosphorylated on T14 and inhibited by an unidentified kinase activity, but the significance of T14 in vivo remains uncertain. The Y15 phosphorylation of CDK5, on the other hand, occurs both in vitro and in vivo. Phosphorylation of Y15 on CDK5 by Abelson (Abl) tyrosine kinase is stimulatory [61, 67], while phosphorylation of Y15 and T14 by Wee1 family kinases is inhibitory for CDK1 and CDK2 [61].

36
7. Computational Chemistry
Computational chemistry encompasses quantum mechanics, molecular mechanics, simulations, conformational analysis and other computer-based methods. All these theoretical or computational techniques are used for understanding and predicting behaviour of molecular systems. The majority of computational chemistry methods involve description of the intra- and inter- molecular interactions in the system.
Two most common models used are quantum mechanics (QM) and molecular mechanics (MM). These models determine how the energy of the system varies as the positions of the atoms (nuclei) change. The other stage is the calculation itself, such as energy minimisation, molecular dynamics or Monte Carlo simulation, or conformational search [68, 69]. Computational methods and techniques, which were predominantly used during studies on CDKs, are in detail described below.
The relationship between geometry of studied molecular system and its energy is the basic relationship for all computational chemistry methods. Changes in the energy of a system can be considered as movements on a multidimensional ‘surface’ called the Potential Energy (hyper)Surface (PES) (Figure 9). The potential energy functions determine energy as a function of atomic positions. There are two main approaches within computational chemistry devoted to the energy determination and quantification: quantum mechanics and molecular mechanics [69].
Figure 9. The potential energy surface (PES) is often represented by illustrations like this one. Each point corresponds to the specific values of the structural variables, and thus represents a particular molecular structure, with the height of the surface at that point corresponding to the energy of that structure.

37
A force field is an empirical fit to the potential energy surface. We shall be particularly interested in stationary points on the energy surface, where the first derivatives of the energy with respect to all coordinates are zero. Points with minimum energy are of one type of stationary point, where Hessian H has none negative eigenvalue; these correspond to stable structures. An attention is also paid to stationary points, where Hessian exhibits just one negative eigenvalue; these are called transition states according to the transition state theory [68, 70].
7.1. Molecular Mechanics
Force field methods (also known as molecular mechanics) do not involve explicitly electrons in calculations and evaluate the energy of a system as a function of the nuclear positions only [71].
Molecular mechanics is based upon a rather simple model of the interactions within a system with contributions of processes such as the stretching of bonds, the opening and closing of angles, and the rotations about single bonds.
Transferability is a key attribute of a force field, which means that a set of parameters developed and tested on a relatively small number of cases could be applied to a much wider range of problems. Moreover, parameters developed from data on small molecules can be used to study much larger molecules such as polymers and biomacromolecules. Molecular mechanics is thus invariably used to perform calculations on systems containing large numbers of atoms.
The force fields contain terms that describe relationship of the changes between energy and geometry of the system in specific internal coordinates such as bond lengths, angles, and the rotation of bonds or movements of atoms relative to each other. One functional form for such a force field (e.g. parm99 force field [72] as implemented in the AMBER software package [73]) that was used in this thesis to model single molecules or assemblies of atoms and/or molecules is described by equation 7.1.
( ) ( )
( )( ) ∑∑∑
∑ ∑
= +=
+
−
+−++
−+−=
N
i
N
ij ij
ji
ij
ij
ij
ijij
torsions
iiii
in
bonds
i
angles
iii
iii
ilN
rqq
rrn
V
kll
kV
1 1 0
612
0,,
20,
,20,
,
44cos1
2
22)r(
πεσσ
εγω
θθθ
(7.1)
there, V denotes the potential energy, which is a function of the positions r of N atoms. The first term in equation (7.1) represents interactions between bonded
)( Nr

38
atoms, modelled by a harmonic potential. The second term is a summation over all valence angles in the molecule, again modelled using a harmonic potential. The third term is a torsional potential that models how the energy changes as the bond rotates. The fourth contribution is the non-bonded term. In a simple force field the non-bonded term is usually modelled using the Coulomb potential term for electrostatic interactions and the Lennard-Jones (LJ) potential for van der Waals interactions. In equation (7.1), l is the bond length; θ, is the valence angle; ω, is the dihedral or torsion angle; and rij is the distance between atoms i and j.
Parameters, that represent actual force field, include the bond force constant and equilibrium distance, kl,i and li,0, respectively; the valence angle force constant and equilibrium angle, kθ, and θi,0, respectively; the dihedral force constant, multiplicity and phase angle, Vn,i, ni, and γi, respectively. Non-bonded parameters between atoms i and j include the partial atomic charges, qi, and the LJ well-depth, εij, and minimum interaction radius, σij, used to treat the van der Waals interactions. This is very important as the two main features that define the quality of a given force field are the functional form and the parameters derived as constant in the potential function.
Similar terms evaluating the energy in equation (7.1) are currently used in other biomolecular force fields, including, for example, CHARMM [74], GROMOS [75], and OPLS [76].
7.1.1. Force Field Parameter Development
Molecular mechanical models (force field methods models), that are useful for simulations of conformational energies and noncovalent interactions of complex molecular systems, are composed of two parts: an analytic energy function and parameters. There are a wide variety of function forms as well as many different approaches to derive the parameters for molecular mechanical models. Force fields are constructed by parameterising the potential function using the either experimental data (e.g. X-ray and electron diffraction, NMR and IR spectroscopy) or ab initio and semi-empirical quantum mechanical calculations.
The simplest function form of the force fields is the harmonic one used by AMBER and OPLS, DREIDING, etc (see chapter 7.1.). In order to accurately fit conformational and nonbonded energies, one should use restrained electrostatic potential (RESP) [77, 78] charges for calculation of the partial atomic charge, qi. The AMBER force field applies RESP charges derived from quantum mechanical electrostatic potential (ESP) to calculate the electrostatic energy [79] in this way: first, quantum-mechanical optimizations are performed for non-standard residues using the Hartree-Fock method with polarizable double-zeta basis set with appropriate (e.g. GAUSSIAN) software package [80]. Electrostatic potential is then calculated using Merz-Sighn-Kollman scheme at HF/6-31G(d) level for the

39
minimized geometry. For each compound, RESP charges are derived using only the electrostatic potential of the lower energy conformer.
Parmscan [79], an automatic force field parameter optimization program can be used to derive new torsional parameters. Parmscan primarily attempts to find the best Fourier series and force constants so as to reproduce precisely the energy differences of the training set. The main purpose of parmscan is to change the force constants systematically, with a certain step, to find the optimum torsional parameters that give the smallest absolute error of molecular mechanical (MM) energy differences when compared with experimental or ab initio data [72].
7.2. Simulation Techniques
The computational chemistry may be used to understand and to predict the properties of liquids, solutions and solids, to study complex processes such as the absorption of molecules onto surfaces and into solids and to investigate the behaviour of macromolecules, which have many closely separated minima. Computer simulation methods enable the time-dependent behaviour (with exception Monte Carlo method) of atomic and molecular systems to be followed, providing a detailed picture of the way in which a system changes from one conformation or configuration to another. Most computer simulation studies utilize a mathematical model that represents a potential energy surface for the molecules of interest. A variety of simulation techniques are available to study energetic, structural, and dynamical aspects of molecular systems. Simulation techniques are also widely used in some experimental procedures, such as the determination of protein structures from X-ray crystallography and NMR method.
The two most common simulation techniques used in molecular modelling are molecular dynamics (MD) and Monte Carlo methods. These methods, mapping the phase space, reflect real behaviour of the system and allow us also to gain information about important thermodynamical characteristics, i.e., entropy and Gibbs free energy.
7.2.1. Molecular Dynamics
Molecular dynamics calculates the evolution of the system in time, from which time averages of properties can be calculated. Sets of atomic positions are derived in sequence by applying Newton’s equations of motion.

40
Molecular dynamics is a deterministic method. In the MD, the state of the system at any future time can be predicted from its current state4. The Newton’s equations of motion (7.2) are integrated by dividing the calculation into a series of very short time steps. The time step used in MD calculations must be approximately one order of magnitude smaller than the highest frequency motion of the system. Usually, these are bond stretch vibrations (≈ 1014 s-1), which limit MD time steps to approximately 1 fs.
i
Nii
mdtd ),...,,( 21
2
2 rrrFr= (7.2)
These equations describe the motion of a particle of mass mi with the position ri. The force on atom i at time t is easily computed as the negative gradient of the potential energy function.
)( N
ii V
rrF
∂∂
−= (7.3)
At each step, the forces Fi on the atoms are computed and combined with the current positions and velocities to generate new positions and velocities a short time ahead. The force acting on each atom is assumed to be constant during the time interval. The atoms are then moved to the new position, an updated set of forces is computed, and so on.
Frequently, some internal degrees of freedom, especially bond stretching modes, are constrained in MD simulations. The most common constraint methodology is the SHAKE procedure [81], which applies additional forces to keep bond lengths fixed at equilibrium values. The constraint of high frequency modes like bond vibration permits a slightly larger integration time step in MD simulations.
In this way molecular dynamics generates a trajectory that describes how the system changes in time.
4 limited only by numerical precision during computer calculation

41

42

43
II. RESULTS

44

45
8. Synopsis of Results The cell cycle is the central process for cell growth and cell division. The
initiation, progression and completion of the cell cycle events are governed by the cyclin-dependent kinases (CDKs) whose activities are controlled by reversible protein phosphorylation and synthesis and degradation of activator and inhibitor subunits. These results provided detail insight into structure, dynamics, ligand binding, hydration, and molecular interactions of cyclin-dependent kinases (CDKs) on the nanosecond time scale. The activation and inhibition of CDKs by phosphorylation and interaction of CDKs with regulatory subunits of CDKs were described within the framework of the thesis. These studies were directed to investigation of the different mechanisms of CDK2 and CDK5 regulation. Mechanism of cell cycle regulation is important for understanding deregulation and, eventually, the origin relevant diseases. That is the main reason why regulation of CDKs was investigated.
The interaction energies between CDK (especially CDK2 and CDK5) and small molecules were investigated and compared with to reveal and understand structural differences. The small molecules (inhibitors of CDKs) are important for medicinal chemistry and pharmacology, where some of them are used in clinical tests. For example, the below discussed inhibitor (R)-roscovitine (CYC202) is now entering phase II clinical trials against cancer and phase I clinical tests against glomerulonephritis, following encouraging results obtained in preclinical tests [82].
8.1. Interactions of CDK2 with Water Molecules We have performed a very detailed analysis of solvent behaviour over
4 molecular dynamics (MD) trajectories on free CDK2 and its substrate/inhibitor complexes. Molecular dynamics simulations of the CDK2-native substrate adenosine triphosphate (ATP) complex and CDK2 in the complex with two purine-like inhibitors (roscovitine and isopentenyladenine) were employed for the study.
Protein region with a high density of water molecules, as well as structural water molecules, were determined by using MD simulations. As structural water molecules were considered water molecules with interaction time larger than 950 ps within 1000 ps long MD simulations. A number of water molecules that were in contact with the protein for the whole trajectory were assigned. The 39, 27, 49, and 32 water molecules bound to the protein were found for trajectories of the free CDK2, CDK2/ATP, CDK2/roscovitine, and CDK2/isopentenyladenine complexes, respectively. Two stable water molecules in the trajectory of the free CDK2 were found that occupy the same position as the nitrogens N3 and N9 of the

46
isopentenyladenine or N1 and N6 nitrogens of the ATP (Figure 10). The positions of structural water molecules were compared with the position of substrate polar groups and water molecules found by X-ray analysis. Appropriate replacement of the aforementioned water molecules in the active site leads to more selective inhibitors as demonstrated by the replacement of stable water molecules by polar groups of inhibitor.
Figure 10. Superimposition of CDK2/isopentenyladenine complex with structural
water molecules found for free CDK2. It is seen that structural water molecules in the free CDK2 occupy the same positions as polar groups of ligand molecule in the protein.
The MD method provides information about interaction energies between
molecules and is also helpful in the case of uncompleted X-ray data. In our case, the MD simulation detected a larger number of stable water
molecules in the active site than X-ray crystallography. Moreover, it provided us with information about changes in the configuration of water molecules in the CDK2 active site.
The behaviour of water molecules interacting with amino acids in the enzyme active site, and their interaction energies, provides information that is useful for the rational drug design of new potent and selective inhibitors. It was concluded that analysis of the enzyme active site hydration and tracing tightly bound water molecules may substantially help in designing new inhibitors. Results were published in Proteins: Structure, Function, and Bioinformatics (Proteins: Struct., Funct., Bioinf. 55, 2004, 258-274).

47
8.2. Mechanisms of the CDK2 Regulation
The CDK2 activation and inhibition were investigated because the mechanism of cell cycle regulation is important for understanding deregulation and origin relevant diseases.
Nanoseconds long molecular dynamics simulation (MD) trajectories of differently active complexes of human cyclin-dependent kinase-2 (inactive CDK2/ATP, semiactive CDK2/cyclin A/ATP, fully active pT160-CDK2/cyclin A/ATP, and inhibited pT14-; pY15-; and pT14,pY15,pT160-CDK2/cyclin A/ATP) were produced and compared.
The MD simulations emphasized the known fact that CDK2/cyclin A association stabilizes the three-dimensional structure of CDK2 and the activating phosphorylation of Thr160 stabilizes T-loop (activation segment) conformation for substrate binding and phosphoryl transfer. The MD simulations results of CDK2 inhibition by phosphorylation at Thr14 and/or Tyr15 sites in the Glycine-rich loop (G-loop) provide insight into the structural aspects of CDK2 deactivation. Phosphorylation of Thr14 and both inhibitory sites Thr14 and Tyr15 together causes ATP misalignment for phosphorylation and G-loop conformational change, which leads to the opening of the CDK2 substrate binding box. Such changes might decrease CDK2 affinity to its substrate. The phosphorylated Tyr15 residue negatively affects substrate binding or its correct alignment for ATP terminal phospho-group transfer to the CDK2 substrate.
To verify hypotheses from studies on differently active forms of CDK2, ten nanosecond-long simulations of the fully active CDK2 in a complex with a short peptide (HHASPRK) substrate and of CDK2-HHASPRK complex inhibited by phosphorylation of Thr14 and/or Tyr15 were produced.
The HHASPRK peptide is tightly bound to the substrate binding box during simulation of the fully active form of CDK2 with the peptide substrate. While the substrate HHA-residues are the most flexible ones, the K3 residue is very rigid, staying tightly bound to phosphate moiety of pThr160 residue. Subscripts denote amino acid positions in the substrate numbered from the phosphorylation residue (S0) with increasing numbers toward the C-terminus.
The preference of CDK2 for basic residue (R/K)2 observed from kinetic experiments cannot be deduced from the crystal structure, because R2 makes no contact with the protein, having its side chain oriented to the bulk solvent. In contrast, in the MD simulation the R2 side chain changed conformation and therefore offers a simple explanation for the above preference. It is based on the idea that R2 interacts with the ATP phosphate moiety and, consequently, it can also play a role in appropriate ATP alignment before the reaction.
The phosphorylation of the Tyr15 residue (or both residues altogether) causes the R2 positively charged side chain to interact preferably with this group and the interaction with ATP phosphates is lost.

48
On the other hand, the inhibitory phosphorylations at Thr14 and/or Tyr15 do not affect interaction of K3 with the pThr160 side chain.
The phosphorylated Tyr15 residue negatively affects substrate binding or its correct alignment for ATP terminal phospho-group transfer to the CDK2 substrate. The simulations of CDK2 with HHASPRK substrate peptide inhibited by phosphorylation show that the phosphorylation in all cases causes the ATP phosphate moiety misalignment, changes in the Mg2+ ion coordination sphere, namely the loss of Asn132, (a residue conserved in all protein kinases), coordination and G-loop shift (~5 Å) away from the ATP binding site, which leads to the substrate binding box opening. The ATP misalignment resulting in terminal phospho-group reconformation was demonstrated by increasing the SO-Oγ…Pγ-ATP distance. The lengths of occurrences of the distance SO-Oγ…Pγ-ATP below the value of 3.90 Å were calculated (see Table 1). The distance remains lower than the threshold during 87.7%, 9.6%, 5.8%, and 18.1% of simulation times of the fully active CDK2 and CDK2 inhibited by phosphorylation at Tyr15, Thr14, and Thr14/Tyr15 residues, respectively. All mentioned effects clearly explain the lost of kinase activity after inhibitory phosphorylation of the CDK2 G-loop, because correct coordination of the Mg2+ ion and appropriate orientation and conformation of the ATP phosphate moiety are crucial for the phospho-group transfer to the serine (S0) hydroxyl from the peptide substrate.
Table 1. The occurrence of distance S0-Oγ … Pγ-ATP below the value of 3.90 Å,
and the mean value during the simulation. This distance between the ATP terminal phospho-group and the phosphorylation serine hydroxyl group of the peptide substrate is equal to the 3.7 Å in the X-ray crystal structure.
System Mean distance
in Å Distance ≤ 3.9 Å
(% of time) X-ray structure 3.70 pT160-CDK2/cA/HHASPRK/ATP 3.63 ± 0.27 87.7 pT14,pT160-CDK2/cA/HHASPRK/ATP 6.58 ± 1.50 5.8 pY15,pT160-CDK2/cA/HHASPRK/ATP 8.81 ± 2.20 9.6 pT14,pY15,pT160-CDK2/cA/HHASPRK/ATP 4.83 ± 1.27 18.1
The G-loop movement that seems to be periodical was noticed during 15-
nsec long MD simulation of the fully active CDK2. The G-loop flexibility is facilitated by its primary sequence, which includes three highly, conserved glycine residues (GxGxxG) in protein kinases. The primary function of each G-loop motif residue can be deduced from comparison of presented results with the most extensive investigated member of the protein kinase family, the cAMP-dependent kinase (PKA) [83]. We conclude that the conserved motif GxGxxG is an evolutionarily optimized one because it guarantees G-loop flexibility, good accessibility of the active site, and order due to formation of the secondary

49
structure. The results were published in two Protein Science papers (Protein Sci. 13, 2004, 1449-1457 and Protein Sci. 14, 2005, 445-451).
8.3. Dynamics of Human CDK5; Comparison to CDK2
Nanoseconds long molecular dynamics (MD) simulations of CDK5/p25
and CDK5/p25/roscovitine, and these complexes phosphorylated at Tyr15 and Ser159 residues were produced. Mechanisms of CDK5 activation and inhibition were investigated and compared to mechanisms of CDK2 regulation. Energy decomposition analysis was used to description a detailed interaction scheme for roscovitine with CDK5 and CDK2 and for description interaction energies of CDK5 and CDK2 to regulatory subunits (p25 and cyclin A, respectively).
The CDK5/p25 starting structure was taken from PDB (PDB code: 1H4L), and CDK5/p25/roscovitine was created by superposition of CDK5/p25 with CDK2/roscovitine complex. The G-loop position and conformation were not affected by presence of roscovitine inhibitor in the active site but the inhibitor lowered the G-loop flexibility. On the other hand, the insertion of the roscovitine to the CDK5 active site increases the T-loop flexibility, which is in agreement with behaviour of these loops in CDK2 after roscovitine insertion to the CDK2 active site.
To quantify roscovitine interaction with CDK5 or CDK2, the interaction energies between inhibitor and protein kinase were calculated using the AMBER force field (parm99) and averaged over MD simulation. The mean interaction energies between CDK5 or CDK2 and roscovitine are equal to –54.4 ± 0.1 and –50.3 ± 0.1 kcal.mol–1, respectively, and correlate with IC50 roscovitine values against CDK5 (0.16 µM) [62] and CDK2 (0.70 µM) [84]. Energy decomposition analysis was also used to quantify differences between CDK2 and CDK5. To identify contributions of inhibitor moieties to interaction energy between CDK and inhibitor, the interaction energies of seven fragments of the inhibitor were calculated. These interaction energies are almost identical with exception of two differences in N7 and C8 interactions. The differences are caused by Cys83/Leu83 and Leu133/Leu134 in CDK5/CDK2 by electrostatics and by the van der Waals term, respectively. The interaction energies of the Cys83/Leu83 and Leu133/Leu134 with roscovitine are lower for CDK2 due to side chain reconformation of these residues in CDK2 and its shift away from the roscovitine. The van der Waals contributions to the interaction energy document that roscovitine fits into the CDK5 active site better because the vdW interaction energy between CDK5 and roscovitine is lower by 2.9 kcal.mol-1 compared to the interaction energy between CDK2 and roscovitine. Understanding of the interaction energy pattern is fundamental for rational drug design and it is useful in the design of more selective inhibitors.

50
The simulation of pY15-CDK5/p25/roscovitine complex shows that Tyr15 phosphorylation leads to Tyr15 exposure to solvent and also to shift of G-loop (~8 Å). The analysis of interaction energies between roscovitine and CDK5 (unphosphorylated and phosphorylated CDK5 at Tyr15 residue) shows that Tyr15 phosphorylation has an almost negligible influence on roscovitine binding in according to experimental observations [85]. These interaction energies are equal to –54.4 ± 0.1 kcal.mol-1 and –51.4 ± 0.1 kcal.mol-1 for CDK5/p25/roscovitine and pY15-CDK5/p25/roscovitine, respectively.
In spite of the absence of the phosphate group on Ser159 (equivalent to Thr160 of CDK2), the CDK5 activation loop adopts an extended conformation typical of active proline-directed kinases (see Figure 11 and also Figure 5).
Figure 11. Superposition of CDK5/p25 and pT160-CDK2/cyclin A complexes.
CDK5 is predominantly shown in blue and p25 in green. A part of the CDK5 activation loop (residues: 152-157) is represented in red. CDK2 is shown in orange and its regulatory subunit (cyclin A) in gray.
To gain information about differences in Ser159 position (CDK5) with
respect to Thr160 (CDK2), the distances between Ser159 and Thr160 Cα atoms were calculated after superposition of the averaged structures from the ends of MD simulations of CDK5/p25 and differently active forms of CDK2 (semi-active and fully active complexes of CDK2). The Cα atom of Ser159 is only 1.9 Å away from the Cα atom of the phosphorylated Thr160 in pT160-CDK2/cyclin A/HHASPRK complex (fully active CDK2). The Val163 adopts a left-handed orientation typical for active loop conformation that is stabilized by H-bonds network. These structural aspects imply that the CDK5 does not need activation by

51
phosphorylation of the activation segment, even if this contains a potential site for phosphorylation on Ser159. The detailed interaction energy analysis proved that the T-loop active conformation in the CDK5/p25 complex is stabilized by interactions with the p25. The residues 152-157 from the CDK5 activation loop have different position in comparison with their position in the CDK2 and this part of T-loop contributes to CDK5-p25 interaction and stabilization (Figure 11).
Energy decomposition analysis was also used to determine regions with the highest energy contributions to the interaction energy between CDK5 and p25, and, for comparison, also between CDK2 and cyclin A. The detail analysis of interaction patterns helps to understand the specificity of binding between CDK and the regulation subunit.
The simulation of CDK5/p25 complex phosphorylated at Ser159 was performed to confirm the idea that CDK5 does not need phosphorylation in the T-loop for activation. The Ser159 was phosphorylated in silico, and as a starting pSer159 conformation pThr160 (CDK2) conformation was used. No significant changes in structure were observed during MD simulation and the activation segment remained in the same conformation as found in CDK5/p25 complex.
The results are described in paper accepted for publication (J. Biol. Chem., 2006).

52

53
III. RESULTS – APPENDIX

54

55
Analysis of CDK2 Active-Site Hydration: A Method
to Design New Inhibitors
Zdeněk Kříž, Michal Otyepka, Iveta Bártová, and Jaroslav Koča
Proteins: Struct., Funct., Bioinf. 55, 2004, 258-274

56

57
Analysis of CDK2 Active-Site Hydration: A Method to Design New Inhibitors Zdeněk Kříž,
1
Michal Otyepka,2
Iveta Bártová,1
and Jaroslav Koča1*
1
National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Brno, Czech Republic 2
Department of Physical Chemistry, Faculty of Science, Palacky University, Olomouc, Czech Republic
ABSTRACT The interactions between the protein and the solvent were analyzed, and protein
regions with a high density of water molecules, as well as structural water molecules, were determined by using molecular dynamics (MD) simulations. A number of water molecules that were in contact with the protein for the whole trajectory were determined. Their interaction energies and hydrogen bonds with protein residues were analyzed. Altogether, 39, 27, 49, and 32 water molecules bound to the protein were found for trajectories of the free CDK2, CDK2/ATP, CDK2/roscovitine, and CDK2/isopentenyladenine complexes, respectively. Positions of observed water molecules were compared with X-ray crystallography data. Special attention was paid to water molecules in the active site of the enzyme, and especially to the deep pocket, where the N9 roscovitine side-chain is buried. Exchange of active-site water molecules with bulk water through the tunnel from the pocket was observed. In the CDK2/isopentenyladenine complex simulation, two water molecules that arrange interaction between the inhibitor and the enzyme via an H-bond were observed. Two stable water molecules in the trajectory of the free CDK2 were found that occupy the same position as the nitrogens N3 and N9 of the isopentenyladenine or N1 and N6 nitrogens of the adenosine triphosphate (ATP). The positions of structural water molecules were compared with the positions of substrate polar groups and crystallographic water molecules found in the Brookhaven Protein Data Bank for various CDK2 complexes. It was concluded that tracing tightly bound water molecules may substantially help in designing new inhibitors. Proteins 2004;55:258–274.
© 2004 Wiley-Liss, Inc.
Key words: cyclin-dependent kinase; ATP; roscovitine; isopentenyladenine; molecular dynamics; hydration of proteins; structural water molecules
Grant sponsor: Grant Agency of the Czech Republic; Grant number: 201/98/K041. *Correspondence to: Jaroslav Koča, National Centre for Biomolecular Research, Faculty of Science, Masaryk
University, Kotlářská 2, 611 37 Brno, Czech Republic. E-mail: [email protected] Received 9 June 2003; Accepted 14 September 2003 Published online 27 February 2004 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/prot.20026
© 2004 WILEY-LISS, INC.

58
INTRODUCTION Water molecules play an essential role in the structure and dynamics of biological systems. In
particular the solvation of proteins is involved in most biological processes [86]. They act as a bridge between secondary structural elements [87]. Water is known to contribute significantly to the stability of biomacromolecules and to play a crucial role in molecular association. Buried water molecules are involved in local structural stabilization of proteins and are therefore termed “structural water” [88].
They are usually characterized by long residence time and very restricted movement [89]. Water molecules in the binding interface can provide useful information for drug design. Strategies for the design of new inhibitors, which take into consideration structural water molecules, include either saturating the water position with appropriate hydrogen bond donors and acceptors to achieve maximum complementarity or replacing it to gain in affinity through entropic effect [90].
X-ray crystallography, NMR spectroscopy, and neutron diffraction are typical experimental methods to analyze water molecules at the atomic level. However, high-resolution structures are usually preferred for a reliable analysis of structural aspects of water associated with a host protein [91]. In the absence of high-resolution experimental data, theoretical studies, such as molecular dynamics simulations, represent complementary methods of locating water positions and understanding the dynamics and energetics of these water molecules [92-95].
The cyclin-dependent kinases (CDKs) play a significant role in eucaryotic cell cycle regulation. Their sequential activation ensures the correct timing and ordering of events required for cell cycle progression. The CDKs activate host proteins through phosphorylation on serine or threonine using adenosine triphosphate (ATP) as a phosphate donor. The activity of CDKs is extensively regulated by association with regulatory subunits (cyclins) and by specific phosphorylation at a positive site (Thr160 in CDK2) or negative sites (Thr14 and/or Tyr15 in CDK2) or by association with native inhibitors [18]. The CDK2 associates with cyclin E to promote progression through the G1 phase and subsequently relocates into a complex with cyclin A that is essential for entry into the S phase. Selective CDK inhibitors would be invaluable tools for structural, cell, and molecular biologists working in the cell cycle field. Furthermore, the characterization and development of low molecular weight CDK inhibitors, which could restore normal cell cycle control in tumor cells, would also have considerable potential in cancer therapeutics [96].
To date, only structures of CDK2, CDK5 and CDK6 solved by X-ray crystallography have been reported. The X-ray structures of the CDK2 complexed with ATP [27], purvalanol [97], staurosporine [98], olomoucine and isopentenyladenine [99], roscovitine [100], flavopiridol [101], quinazoline [102], indirubin [103] and many others have been published. Comparison of the X-ray structures for CDK2/cyclin A/ATP [99], CDK2/cyclin A/p27Kip1(native protein inhibitor) [104], CDK2/cyclin A and T160pCDK2/cyclin A complexes [105] and CDK2 alone [27] reveals only limited differences in the vicinity of the ATP adenine binding site between different activation states of CDK2. This suggests that information derived from monomeric CDK2/inhibitor structures can help us to design inhibitors against the active binary complex.
All known eucaryotic protein kinases exhibit a conserved catalytic core domain with an adenosine 5’-triphosphate (ATP) binding site, which is often targeted in drug discovery programs. It is well known how the ATP binding site of CDK2 can accommodate structurally diverse inhibitor types. However, as ATP is common to kinases and other proteins, inhibitors of CDK2 competitive with ATP are also active in other proteins, which are reflected by the toxicity of these drugs. For the design of a selective inhibitor with high activity it is necessary to describe all specific interactions of the ligand in the active site including those with solvent molecules [106]. Information obtained from MD simulations, especially calculated interaction energies and positions of structural water molecules in the active site, helps us to understand the specificities of the protein-ligand binding.
The aim of this work is to present analyses of structural water molecules determined from molecular dynamics (MD) studies performed on free CDK2 and its three complexes: with native

59
substrate ATP, with the non-selective inhibitor isopentenyladenine, and with the highly selective inhibitor roscovitine. Molecular dynamics can supplement information from X-ray crystallography mainly concerning the dynamics of the system and its behavior in solution. Calculated interaction energies based on MD results are more realistic than energies based on calculations from the X-ray structure due to relaxation of the system in its native environment and the averaging of energies over many single frames. It is an effort of this paper to open a different insight into the CDK2 active site and to broaden the scope for rational drug design. METHODS
All molecular dynamics simulations presented in this work were carried out using the AMBER-6.0 program package [107] and the Cornell et al. force field [108] with the TIP3P water model. The X-ray structures were used as starting data for MD simulations. The free CDK2 and CDK2/ATP crystal structures were obtained from the Brookhaven Protein Data Bank (PDB codes 1hcl and 1hck). The X-ray structures of the cdk2/roscovitine and cdk2/isopentenyladenine complexes were kindly provided by Prof. S.-H. Kim (University of California, Berkeley CA, U.S.A.). The resolutions of X-ray data were 1.8 Å (free CDK2), 1.9 Å (CDK2/ATP), 2.4 Å (CDK2/roscovitine), and 1.8 Å (CDK2/isopentenyladenine), respectively. Molecular Dynamics Simulation
First, the hydrogen atoms of unionizable residues were added to the X-ray structures using the PROTONATE program [107]. The protonation states of ionizable residues were set and hydrogen atoms were added by WHATIF [109] program suite together with full optimization of hydrogen bond network. The used method is implemented in HB2NET module [110]. The method is based on special force field (H-bond force field). It places the hydrogen atom onto ionizable residues and optimizes its position to create maximum number of H-bond between residues. It is also able to “flip” the terminal dihedral angle of asparagine and glutamine to optimize the possibility of H-bond. The side chain of histidine is also able to “flip” within this force field. However, in all the above changes a penalty is put on energy value. In our case, all basic residues on the surface that may interact with water molecules were protonated and also all acidic residues on the surface were deprotonated. This should well mimic the situation at physiological pH and, therefore, it is likely that our results are realistic. Also all histidines on the surface of the protein were fully protonated, considering internal histidines, only HIS 125 was set as δ-protonated. Crystallographic water molecules were kept in the structure.
The all-atom model was neutralized by adding thirteen chloride counter ions and was immersed in a rectangular TIP3P water box. This was carried out using the EDIT program of the AMBER package [107]. The initial box size was 82×75×72 Å for free CDK2 simulation and nearly the same for the simulations of CDK2 complexes. The system was at initially relaxed and slowly heated before trajectory production. First, the position of bulk water molecules and counterions was optimized using minimization procedure of SANDER program followed by 3 ps molecular dynamics on these residues. Then the optimization of hydrogen atoms of the protein was performed. The next procedure was to relax the position of protein side chain and backbone using series of minimizations with decreased restrains applied on the backbone atoms. The last procedure was the 50 ps length molecular dynamics simulation with slow heating of the system from 10 K to 298 K. The unrestrained molecular dynamics was then run on the system. The MM energy, density and volume of the system and RMSD of the protein to X-ray structure were monitored. The production phase was started after the density of the system became balanced around 1.0 g.cm-3, temperature balanced around 298 K, and calculated RMSD was also stable. The size of the simulation box was 81×72×69 Å for free CDK2 simulation at this time. Detailed description of the trajectory preparation can be found in our recent paper [55]. After the equilibration phase, a 1 ns trajectory was produced for each system. The coordinates were saved each picosecond. All energy minimization and molecular dynamics simulations were carried out using the SANDER program of the AMBER package.

60
The MD simulations were performed using the following conditions. A 2 fs time step was used in all simulations and the particle-mesh Ewald (PME) [111] method to treat electrostatic interactions was employed. The simulations were run under the periodic boundary conditions in the NPT ensemble at 298 K with Berendsen temperature coupling and constant pressure (1 atm) with isotropic molecule-based scaling [112]. The SHAKE algorithm [113], with a tolerance of 10-5, was applied to fix all bonds containing a hydrogen atom, and the non-bond pair list was update every 10 steps. A 9.0 Å cut-off was applied. Calculation of Solvent Density Distribution
All rotational and translational movements of the simulation box were removed before analyses. The water molecule coordinates were transformed, taking the periodic boundaries into account. The coordinates of water oxygen atoms were mapped onto the three-dimensional rectangular grid with a 0.5 Å grid step, producing an average three-dimensional density distribution. This was carried out using the PTRAJ program [107]. The particular choice of the grid step is a compromise between the uncertainty in location of the density features and statistical error in the local density value that arises due to a lower number of counts in each grid cell. This methodology was developed in M. Pettitt’s group (and is described, for example, in the paper) [114]. The water density map was fitted onto the average structure and regions with a density of larger than 140% of the bulk water density were localized. This was performed using the MIDAS PLUS program [115]. Localization of Structural Water Molecules
Here, we were interested in water molecules that have longer contact with the protein (called structural water molecules). By definition [89], these water molecules are strictly bonded to protein and show small fluctuations. One descriptor of fluctuations is mean square displacement (MSD) value of which being smaller than 1Å2 is a characteristic for structural water molecules. Since the PTRAJ and MIDAS procedures localize also regions with high fluctuation of water molecules, we had to employ the new programs RESTIME and SURFTIME [116] for the detection of structural water molecules. Moreover, our inspection is somewhat extended as it is based on an analysis of van der Waals contacts between water molecules and protein atoms. It means, that also water molecules with much larger mean square displacement will results from our analysis. We will show in this article that also these water molecules may serve to a local stabilization of proteins and, therefore, may be called structural as well.
A key characteristics of structural water molecules is the interaction time Ti (w,j) of the water molecule w with the protein atom j. In time t, it is calculated using equation (1).
0( , ) ( , )
Nsteps
i tT w j step P w j= ⋅ ∑ (1)
Here, the binary function, P(w,j)t is assigned by the value of 1 if the distance between the water molecule w oxygen atom and the protein atom j is smaller than the sum of vdw radii of the oxygen atom and the atom j increased by a parameter RFACTOR. Othervise, P(w,j)t is set to zero. The parameter step is a period by which snapshots of the trajectory are saved (1 ps in our case) and Nstep is the number of snapshots.
The RFACTOR was set to 2 Å that allows for some limited fluctuations of water molecules around protein atoms. As structural were considered water molecules with interaction time larger than 950 ps within 1000 ps long trajectories. The value was selected because there was a gap for lower values up to 800 ps. We have also compared this value with the residence times of water molecules in the first hydration shell of ions. We have found out that these are in the case of monovalent ions in the range from 10 to 30 ps at 300 K [117, 118]. In our case, the residence times of structural water molecules are taken larger than 950 ps, which is in general agreement with experimental data for proteins [119].

61
The time scale at which water exchanges with other solvent molecules has been observed in the range of 10 - 50 ps for water bound in protein surface crevices, to 0.1 – 1.0 ns for strongly bound waters, and ns to ms for interior waters [89, 120, 121].
These water molecules were then analyzed using standard AMBER procedures. The H-bond analyses were carried out using the CARNAL program of the AMBER-6.0 package [107]. The structural water molecules in the active site of the enzyme were then analyzed from the interaction energy point of view. The energy decomposition between these molecules and amino acids of the active site was calculated using the ANAL program of the AMBER-6.0 package [107].
All MD simulations were carried out on two 700 MHz processors of the SGI 1200 PC cluster. All analyses were performed on SGI Indigo workstations and PC’s running Unix operating systems. RESULTS AND DISCUSSION
The stability of produced trajectories and conformational behavior were discussed in our previous paper [55]. The basic data about the number of water molecules is summarized in Table I. TABLE I. Statistical Data Concerning Water Molecules in the Molecular Dynamics Simulations
Number of tightly bound water molecules
Trajectory Number of crystallographic water molecules
Total number of water molecules
in simulations Total In the active site(a)
Free CDK2 180 12075 39 16 (23)
CDK2/ATP 108 11743 27 12(10)
CDK2/ROSC 82 12525 49 15 (9)
CDK2/IPA 99 12020 32 11 (8)
aNumbers in parentheses refer to the number of water molecules in the CDK2 active site, detected by X-ray crystallography.
The data in Table I shows that the total number of water molecules included in each simulation is comparable. The number of water molecules detected by X-ray crystallography depends on the structure hydration model used. As seen from Table I, the number of structural water molecules in the active site of the enzyme is comparable for simulations of the CDK2 complexes. For the free CDK2 simulation, the cavity is more opened than in the crystal and will therefore be filled by more structural water molecules. Hydrated Regions of the CDK2
The maps of regions with a high density of water molecules are depicted in Figure 1 (for notation see also Scheme 1).

62
A
B
C
D
Fig. 1. Stereo view of regions with more than 140% bulk water density in the CDK2: (A) free CDK2, (B) CDK2/ATP, (C) CDK2/roscovitine, (D) CDK2/isopentenyladenine complexes.
The maps demonstrate that the hydrated regions are very similar for all simulations and the presence of the ligand in the active site does not significantly affect the behavior of solvent around the enzyme. The figure shows the three most hydrated regions. The first region is in the CDK2 active site (around the αL12 helix, residues 148 – 151) (region A). The second region (region B) surrounds the loop between the 3/10-1, α3 (residues 122 – 130), and the 3/10-2 helixes (residues 182 – 198). The third region is around the loop between the β6 and β7 strands (residues 135 – 141) (region C). As expected, the polar phosphate group of the ATP and the polar side chain at the C2 position of the roscovitine inhibitor increase remarkably the density of water molecules in the active site. The differences between maps in the case of free cdk2 and cdk2/X complexes in the active site part could be explained by presence of polar atoms of ligands and by reorientation of amino acid side chains in the active site. The ligand in the active site affects loops near the active site (G-loop and T-loop). It causes a partial opening of the active site so more water molecules can enter the cavity. Additionally, reorientation of the side chains in regions C and B (Scheme 1) of the free cdk2 and cdk2/ligand complexes can also cause different solvation.

63
Scheme 1. Scheme of the CDK2 secondary structure with highlighted solvated regions. Structural Water Molecules
The localization of structural water molecules found in all MD simulations is depicted in Figure 2, and comparison of their positions with positions of X-ray detected water molecules is shown in Figure 3.
Large clusters of structural water molecules mirror the previous results obtained from the maps of hydrated regions, especially in the case of the area surrounding the loop between the 3/10-1, α3, and 3/10-2 helixes (region C). A large cluster of structural waters was found, as expected, in ribose and phosphate pockets of the active site. The detailed analysis also uncovers other regions with just a few structural water molecules. One of them is located between the β4, B5 strands and the PSTAIRE motif (region D).
There are many regions with only one structural water molecule, which is usually important for the structural stability. Such water molecules cannot be found by procedures based on tracing regions with a high density of water. There are also some areas (see Fig. 1) that are occupied by one or two structural water molecules frequently exchanged within a network of surrounding water molecules. The structural water molecules detected by MD are in similar positions to the X-ray detected water molecules. The best correspondence was found for free CDK2. The water molecules found on the surface of the protein by X-ray crystallography are relatively unstable and bulk water molecules frequently exchange them. The number of X-ray detected water molecules is quite different in the case of CDK2/roscovitine and CDK2/isopentenyladenine complexes in comparison with free CDK2 and the CDK2/ATP complex. This discrepancy could be explained either by using different refinement procedures in the X-ray data analysis or by possible differences between the X-ray and solvent relaxed structures.
A detailed inspection of Figure 3 shows that the region between the α1 helix and the loop between the β4 and β5 strands (region D) is hydrated by three water molecules in the case of free CDK2, two water molecules in the complex CDK2/ATP, and four water molecules and two water molecules in the cases of complexes CDK2/Roscovitine and CDK2/Isopentenyladenine, respectively. X-ray crystallography detects only one or no water molecule (CDK2/Isopentenyladenine complex) here. The reason could be that the X-ray structures show high temperature factors for atoms in this region, and in some structures, the region was even excluded from the refinement due to the low electronic density found by X-ray.

64
A
B
C
D
Fig. 2. The location of structural water molecules found by MD simulations: (A) free CDK2, (B) CDK2/ATP,
(C) CDK2/roscovitine, (D) CDK2/isopentenyladenine complexes.

65
A
B
C
D
Fig. 3. The comparison of structural water molecule positions in the X-ray structure (gray) and in the
average structures from equilibrated parts of MD trajectories (black). Water molecules are represented by the position of oxygen atoms. (A) free CDK2, (B) CDK2/ATP, (C) CDK2/roscovitine, (D) CDK2/isopentenyladenine complexes.

66
Structural Water Molecules in the CDK2 Active Site Free CDK2 Water molecules location. A detailed picture of the location of structural water molecules in the active site is shown in Figure 4. A comparison of the location of X-ray and MD structural water molecules is shown in Figure 5.
Fig. 4. The location of X-ray and MD structural water molecules in the CDK2 active site. The active site of
CDK2 is modeled as a solvent-accessible surface; water molecules are in the CPK model (top). For better clarity, the same situation is pictured without the surface, and the positions of water molecules are represented by oxygen atoms (balls; bottom). Water molecules numbered as 480 or lower are those detected also by X-ray crystallography.
The analysis of the MD simulation data revealed 16 structural water molecules in the free CDK2 active site. Six of them are concentrated in the ribose-pocket of the active site (waters 319, 320, 363, 377, 394, 6795). Only one of them (6795) is a bulk water molecule. Four water molecules (316, 317, 323, 418) create the cluster between the LYS 33 side chain and the glycine-rich loop (G-loop, residues 11 – 16). These water molecules were detected by X-ray crystallography but only two of them keep their original position (316, 418). The position of the remaining ones (317, 323) changes during the simulation to create a stable cluster with stronger H-bonds to each other and to amino acids in the active site.
The next water cluster (348, 403, 412, 427) in the free CDK2 active site is positioned in a shallow cavity created by LEU54, LYS56, ILE63, VAL64, ALA144, ASP145 and PHE146. It is interesting that this cluster is located in proximity of hydrophobic residues. This is because of its stabilization by interactions with the polar backbone of the protein. The above-mentioned water molecules were also detected by X-ray crystallography and their positions are relatively stable for the whole trajectory. The last well solvated region of the active site lies in the purine pocket (the hinge region created by residues 81 – 84) and is filled by two water molecules (315, 6690). This area is solvated only in free CDK2, because the nitrogens of the purine rings replace the positions of these water molecules in CDK2/purine inhibitor complexes. These water molecules create hydrogen bonds with the NH group of the LEU 83 (6690) and carbonyl oxygen of the GLU 81 backbone (315). The positions of these two water molecules are the same as the positions of the N6 and N1 ATP nitrogens or N3 and N9 isopentenyladenine nitrogens in corresponding CDK2 complexes (Fig. 6). The positions of these two water molecules are also very close to the positions of the N2’, N3, and N9 atoms of roscovitine. The observation that structural water molecules in the free CDK2 are replaced by polar groups of ligand molecules in the complexed protein implies that the information about the location of structural water molecules is useful for rational drug design.
A detailed analysis of the X-ray structure shows 3 water molecules in this region. Only one of them (315) was detected as structural in the MD simulation. The second one was replaced by a bulk water

67
molecule (6690) in the relaxation phase of the simulation and it remained stable for the whole trajectory. The position of the third X-ray water molecule was not detected as a position of a structural water molecule. Interaction energies. The calculated interaction energies between stable water molecules and CDK2 active site residues together with the results of the H-bond analysis are summarized in Table II. Some water molecules (315, 323, 403, 427, 6690) interact with only one CDK2 residue during the whole trajectory. Several water molecules interact with two residues (316, 317, 377, 394, 412, 6795). The remaining water molecules move slightly along neighboring residues and create alternate hydrogen bonds with a number of residues. Representatives of this category are waters 319, 320, 348, 363, and 418.
The hinge region (residues 81 – 84) is solvated by water molecules of the first category (interacting with just one atom). The interaction energies of water molecules in this region of the active site are –7.1 kcal/mol (water 315 and GLU 81) and –4.8 kcal/mol (water 6690 and LEU 83). The ribose pocket is filled by 4 water molecules 316, 317, 323, and 418. The detailed analysis shows that the water 316 creates a bridge between the backbone of the TYR 15 and GLY 16 and contributes to stability of the enzyme structure. Water molecule 317 interconnects the LYS 33 and ASP 145 side chains. The high interaction energies calculated for this water molecule are caused by interactions with the charged side chains of lysine and aspartic acid.
The phosphate pocket of the active site is solvated by a cluster of four water molecules (320, 363, 377 and 6795). A similar situation as for water 317 was observed for water 320, which stabilizes the orientation of the ASP 127 and HIS 125 side chains. Its weak interactions with other CDK2 residues was also observed. The water molecule 363 moves along residues HIS 125, ASP 145, GLY 127 and ALA 149 during the simulation.
Detailed analysis of the interactions within the hydrophobic pocket of the active site (348, 403, 412, 427) shows that hydrogen bonds are created between these water molecules and the backbone atoms of the amino acids. CDK2/ATP complex Water molecules location. The presence of the substrate is reflected by the lower number of structural water molecules in the active site. The arrangement of these water molecules is shown in Figure 7.
Fig. 5. Comparison of the locations of crystallographic and MD structural water molecules for free CDK2 (X-ray, gray; X-ray structural water molecules in MD simulation, red; structural water molecules from bulk, blue).
The ATP ribose is solvated by five water molecules (314, 315, 340, 379, 381), while another water
molecule (313) interacts with the glycine-rich loop. The ATP phosphate part is solvated by five water molecules (312, 330, 391, 396, and 7327). Comparison of the location of X-ray and MD structural water molecules is shown in Figure 8. Only two bulk water molecules became structural water molecules in the active site. One of them (3987) exchanged position with X-ray water and interacted with the ASP 86 side chain and with ATP-ribose oxygen. The second one (7327) interacted with the ATP beta phosphate. It was also observed that the water cluster around the ATP gamma phosphate was restructured.

68
A
B
C D
Fig. 6. Superimposition of CDK2/ATP complex (A) and CDK2/isopentenyladenine complex (B) with structural water molecules found for free CDK2. It is seen that structural water molecules in the free CDK2 occupy the same positions as the oxygen or nitrogen atoms of the ligands in the protein. (C) The position of the key residues of the CDK2 active site. (D) The detailed view of two structural water molecules (315 and 6690) bonded to Glu81 and Leu83 within the free CDK2 active site. Numbers in parenthesis show mean H-bond distances.
Fig. 7. The location of structural water molecules in the CDK2 active site found for CDK2/ATP complex. ATP is represented by the tube model, the active site of the enzyme by the solvent accessible surface, and water molecules by the CPK model (top). For better clarity, the same situation is pictured without the surface, and the positions of water molecules are represented by oxygen atoms (balls; bottom). Water molecules numbered as 400 or lower are those detected by X-ray crystallography.

69
Interaction energies. The interaction energies and results of the H-bond analyses are summarized in Table III. TABLE II. Interaction Energies and H-Bond Analyses Calculated for MD Structural Water Molecules in the Free CDK2 Trajectory
Interaction energy [kcal/mol] Residue Occupancy [%]
Distance [Å]
H-bond angle [degree] Electrostatic VdW Total
Water 315 GLU 81 (O) 99.2 2.8±0.2 12.9±7.4 -8.1±1.7 1.0±1.1 -7.1±1.1
Water 316 TYR 15 (N-H) 99.4 2.9±0.2 18.6±7.9 -4.3±1.3 0.3±0.9 -3.9±1.0 GLY 16 (N-H) 83.8 3.3±0.3 37.4±11.5 -0.4±0.9 -0.3±0.3 -0.7±0.8
Water 317 ASP 145 (OD1) ASP 145 (N-H)
95.6 31.6
2.8±0.33.4±0.3
15.1±10.628.1±10.2
-12.8±4.5 1.7±1.6 -11.1±3.7
LYS 33 (NZ-HZ) 88.0 3.3±0.3 31.1±15.6 -5.0±5.9 0.3±1.0 -4.7±5.3 Water 319
ASP 127 (OD2) 52.9 2.9±0.4 19.4±15.3 -6.8±5.9 0.4±1.3 -6.4±4.9 TYR 15 (OH-HH) 51.8 2.9±0.3 16.5±13.9 -2.4±3.7 0.4±1.1 -2.0±2.9 ASP 145 (O) 43.1 2.7±0.2 11.9±7.6 -4.5±4.1 0.5±1.0 -4.0±3.4 ALA 149 (N-H) 27.6 3.4±0.3 25.9±15.4 -1.4±1.4 -0.2±0.4 -1.6±2.2
Water 320 ASP 127 (OD2) ASP 127 (O)
97.1 33.5
2.7±0.13.6±0.2
9.9±6.246.8±8.8
-14.0±4.0 1.5±1.4 -12.5±3.5
HIS 125 (NE2) 94.6 2.9±0.2 15.7±8.8 -5.0±2.0 0.7±1.0 -4.3±1.6 ASN 132 (ND2-H) 37.6 3.4±0.3 12.9±7.4 -1.6±0.6 -0.2±0.1 -1.4±0.6 TYR 15 (OH-HH) 25.5 3.1±0.5 24.7±15.9 -2.2±2.6 0.1±0.9 -2.1±1.9
Water 323 ASP 145 (N-H) ASP 145 (OD1)
50.5 40.6
3.4±0.42.7±0.1
37.0±12.213.9±8.0
-3.9±7.4 0.7±1.5 -3.2±6.2
Water 348 LEU 55 (O) 99.9 2.8±0.1 14.8±8.9 -6.4±1.6 0.8±1.1 -5.6±0.9 ASN 59 (OD1) 67.4 2.8±0.2 13.9±8.5 -4.4±3.3 0.5±1.0 -3.9±2.6 LEU 66 (N-H) 51.8 3.7±0.2 30.7±9.6 -0.5±0.2 -0.3±0.1 -0.8±0.3
Water 363 ALA 149 (N-H) 64.4 3.4±0.3 26.0±13.5 -1.8±1.5 -0.2±0.1 -2.0±1.5 ASP 145 (O) 60.8 2.9±0.4 19.2±16.1 -5.1±4.0 0.6±1.1 -4.5±3.2 GLY 147 (N-H) 38.2 3.3±0.3 42.0±8.5 -3.1±2.1 0.7±0.9 -2.4±1.5 HIS 125 (O) HIS 125 (NE2)
38.3 26.3
2.9±0.23.4±0.3
17.3±10.439.3±15.4
-3.7±2.1 -0.4±0.2 -3.3±1.6
Water 377 ASP 145 (OD2) 76.3 2.7±0.2 12.1±10.6 -12.6±4.9 1.5±1.5 -11.1±4.1 THR 14 (OG1-HG1) 57.1 2.9±0.3 15.8±13.8 -4.3±3.7 0.7±1.3 -3.6±2.9
Water 394 GLY 147 (O) 81.9 2.8±0.2 13.0±8.6 -4.1±2.6 0.8±1.0 -3.3±2.1 ALA 151 (N-H) 65.1 3.3±0.3 34.2±12.4 -1.4±1.4 -0.1±0.1 -1.3±1.2
Water 403 VAL 64 (O) 100.0 2.7±0.1 11.3±6.2 -7.6±1.2 1.3±1.1 -6.3±0.7
Water 412 PHE 146 (N-H) 98.2 3.3±0.3 42.0±8.5 -1.6±0.2 -0.3±0.1 -1.9±0.2 LEU 143 (O) 98.0 2.9±0.2 16.4±9.7 -5.9±1.6 0.5±0.9 -5.4±1.0
Water 418 LYS 34 (O) 100.0 2.8±0.2 14.7±9.1 -4.3±1.5 0.8±1.0 -3.5±1.7 VAL 17 (O) 71.8 3.1±0.3 22.8±14.4 -3.2±2.3 -1.1±0.1 -4.3±2.0 GLY16 (O) 20.1 3.1±0.3 28.9±15.3 -0.6±0.3 -0.4±0.2 -1.0±0.4
Water 427 PHE 146 (O) 100.0 2.8±0.2 16.9±10.1 -4.9±1.4 0.7±1.1 -4.2±1.2
Water 6690 LEU 83 (O) LEU 83 (N-H)
79.4 57.1
2.8±0.23.3±0.3
16.8±10.624.9±12.5
-5.4±3.3 0.6±1.1 -4.8±2.8

70
Water 6795 ASN 132 (ND2-HD22) 62.0 3.0±0.2 14.3±9.4 -3.1±3.3 -0.3±0.2 -3.4±2.8 THR 14 (OG1-HG1) THR 14 (OG1)
33.1 21.4
3.5±0.43.6±0.3
42.0±13.841.3±12.7
-0.6±0.8 -0.1±0.6 -0.7±0.7
Two water molecules, which interact with only one residue of the protein, were found. Structural
water 314 solvates the GLN 131 backbone, and water 315 interacts with the ASP 145 side chain. Moreover, these two water molecules weakly interact with the nitrogen N7 of the purine ring and water 314 also has short contact with O3* oxygen of the ATP ribose. The ATP ribose is also weakly solvated by water molecule 3987, which creates a hydrogen bond with the ASP 86 side chain. Water 313 creates a bridge between the TYR 15 and GLY 16 backbones, and the phosphate group of the ATP. Similar behavior is exhibited by water 312, which interacts with the ASP 127 and ASN 132 side chains and the gamma phosphate group of the ATP. Water 330 solvates the ASP 127 side chain and for a significant part of the trajectory creates bridges between the THR 14 and LYS 129 side chains and the gamma phosphate group of the ATP. The calculated strong repulsion of water 330 with the LYS 129 side chain is balanced by attractive interactions with other residues. Water molecule 391 contributes to the tertiary structure stabilization of the CDK2 due to the bridge between the ALA 144 backbone and the HIS 125 side chain. Similar behavior is seen for water 396, which creates a bridge between the TYR 15 backbone and the HIS 125 side chain. High interaction energies are caused by interactions of charged phosphate group with water molecules. Water 3987 exhibits strong interaction with the ASP 86 side chain. The H-bond between the water and ASP 86 lasts only for 17% of the trajectory, but the interaction energy is large with large deviations due to interaction with charged residue. This water molecule also exhibits repulsive interaction with the ATP and a short interaction with the LYS 89 side chain. The large interaction energy deviations indicate the high mobility of this water. Structural water 7327 contributes to the stability of the ATP phosphate side chain and THR 14 side chain. High interaction energies are caused by strong interactions between charged atoms of the ATP and the oxygens of the water molecules.
Fig. 8. Comparison of the locations of crystallographic and MD structural water molecules for CDK2/ATP complex (X-ray, gray; X-ray structural water molecules in MD simulation, red; structural water molecules from bulk, blue). CDK2/Roscovitine Complex Water molecules location. In this case the water molecules create a large cluster stabilized by a network of H-bonds, which is remarkably more stable than the clusters in the other simulations. The cluster of 8 water molecules is positioned between the LYS 33 side chain and polar phosphate pocket of the CDK2 active site (residues THR 14, TYR 15, LYS 129, ASN 132, ASP 145). The situation in the active site is shown in Figure 9.

71
TABLE III. Interaction Energies and H-Bond Analyses Calculated for MD Structural Water Molecules in the CDK2/ATP Trajectory
Interaction energy [kcal/mol] Residue Occupancy [%]
Distance [Å]
H-bond angle [degrees] Electrostatic VdW Total
Water 312 ASP 127 (OD) 38.7 2.8±0.3 14.1±11.6 -2.1±1.7 -0.7±0.1 -2.3±1.7 ASN 132 (ND-HD) 91.5 3.0±0.3 23.8±10.9 -1.8±1.4 -1.8±0.8 -2.3±1.3 ATP (OG) 100.0 2.9±0.3 24.4±14.8 -24.7±5.3 1.8±2.0 -23.6±5.0
Water 313 TYR 15 (N-H) 98.0 3.1±0.2 22.8±10.6 -2.7±1.5 -0.1±0.5 -2.8±1.3 GLY 16 (N-H) 99.3 3.0±0.1 20.5±9.4 -3.1±1.1 0.4±0.8 -2.7±0.9 ATP (OA) 99.8 2.7±0.1 13.6±6.3 -31.9±3.1 3.8±1.9 -28.5±2.1 ATP (OG) 100.0 2.6±0.1 8.9±5.1
Water 314 GLN 131 (O) 99.0 2.7±0.2 14.4±8.1 -6.8±1.6 1.4±1.4 -5.4±1.4
Water 315 ASP 145 (OD1) 100.0 2.8±0.2 13.3±8.3 -2.1±0.4 -0.1±0.1 -2.1±0.3
Water 330 THR 14 (OG1) 74.0 3.2±0.3 31.2±12.7 -0.9±1.1 -0.1±0.3 -1.1±1.1 ASP 127 (OD) 100.0 2.7±0.2 13.4±9.0 -10.4±9.2 1.5±1.6 -8.8±8.1 LYS 129 (NZ-HZ) 58.6 3.3±0.4 29.8±14.2 3.9±8.0 0.1±0.1 3.9±7.5 ATP (OG) 60.5 2.7±0.2 16.6±9.3 -15.4±10.3 0.7±1.3 -14.6±9.5
Water 340 LEU 55 (O) 69.2 3.1±0.3 18.3±9.9 -5.0±2.2 0.2±1.1 -4.8±1.5 ASN 59 (OD1) 48.5 3.0±0.3 27.3±14.7 -4.9±2.1 0.2±0.9 -4.7±1.5 VAL 64 (O) 57.3 2.9±0.2 17.1±10.8 -2.8±3.7 0.1±0.2 -2.6±3.2 LEU 66 (N-H) 58.5 3.4±0.4 27.2±12.8 -0.6±1.6 -0.3±0.5 -0.9±1.3
Water 379 ASP 145 (OD) 48.8 3.2±0.6 21.9±15.6 -1.4±5.4 -0.1±0.1 -1.4±4.9 ATP (N7) 69.5 2.8±0.1 14.3±7.5 -5.3±3.8 1.7±1.7 -3.6±2.4
Water 381 VAL 64 (O) 20.2 3.0±0.3 22.7±12.0 -3.1±2.3 -0.1±0.1 -3.1±1.9 PHE 146 (N-H) 64.7 3.3±0.3 25.6±11.6 -1.9±1.3 -0.3±0.2 -2.2±1.3
Water 391 ALA 144 (O) 100.0 2.8±0.2 13.7±8.1 -5.7±1.5 1.0±1.0 -4.7±0.9 HIS 125 (ND-HD) 100.0 2.9±0.1 19.2±7.6 -6.4±1.5 1.1±1.1 -5.3±1.1
Water 396 TYR 15 (N-H) 97.7 2.9±0.2 10.0±7.2 -5.8±2.4 1.3±1.4 -4.5±1.6 HIS 125 (NE) 79.7 3.1±0.3 20.5±12.3 -4.7±3.8 -0.2±0.7 -4.9±3.4
Water 3987 LYS 89 (NZ-H) 20.0 3.0±0.2 21.9±10.5 -3.7±0.5 0.0±0.1 -3.7±0.5 ATP (O2*-H) 18.7 3.7±0.2 42.8±5.2 1.4±3.6 -0.4±0.2 1.0±3.6 ATP (O2*) 11.7 3.7±0.2 34.1±11.5 ASP 86 (OD) 16.7 2.9±0.3 19.9±12.5 -11.8±7.9 1.1±1.5 -10.7±7.2
Water 7327 THR 14 (OG) 82.0 2.9±0.2 15.7±8.7 -1.2±1.8 0.4±0.9 -0.8±1.5 ATP (OB) 100.0 2.6±0.1 8.8±5.1 -21.7±5.4 2.4±1.8 -19.4±4.8 ATP (OG) 29.7 3.7±0.2 42.5±6.1
One water molecule (881) has a strong contact with the phenylene ring of the roscovitine. The calculated interaction energy of the water molecule is -4.9±1.4 kcal/mol. The standard AMBER H-bond analysis did not indicate any strong hydrogen bond between this water molecule and roscovitine atoms. This is because the interaction with the aromatic ring can be considered as a X-H…π-hydrogen bond [122]. Water molecule 881 has a strong hydrogen bond with the ASP 86 residue. This water molecule creates a bridge between the roscovitine phenylene ring and ASP 86. The isopropyl substituent on the N9 roscovitine atom induces a conformational change of the LYS 33 side chain. The change opens a deep cavity from the purine pocket through the whole enzyme to the bulk. The cavity entrance is created by hydrophobic amino acids (VAL 64, PHE 80, ALA 144, and

72
LEU 148). However, the interior of the cavity is hydrophilic and it is filled by five water molecules (332, 336, 341, 344, 357). These water molecules interact with polar groups of the LEU 55, ASP 145, and PHE 146 backbone. Although water molecules 332, 336 and 341 are positioned far from the roscovitine molecule, we have considered them in our analyses, because these water molecules show large fluctuations during the simulation involving the roscovitine molecule. In the cases of the free CDK2 and CDK2/IPA complex MD simulations, the cavity is smaller. An exchange of the positions of water molecules in the cavity and the penetration of the bulk water molecules behind the roscovitine into the cavity were observed. The exchange time of the positions of water molecules in the cavity is from 200 to 300 ps. The comparison of the positions of structural water molecules detected by MD simulations with crystallographic water molecules is shown in Figure 10.
Major changes in water molecules location in comparison with other trajectories are visible in the
purine base pocket. The cluster in the phosphate pocket of the CDK2 active site is reorganized to create a configuration with a larger number of hydrogen bonds between water molecules and the enzyme. Partial exchange of X-ray water molecules by bulk MD waters in the phosphate part of the CDK2 active site is observed. Interaction energies. The interaction energies calculated from MD simulations between water molecules and amino acids in the CDK2 active site are summarized in Table IV.
Fig. 9. The location of structural water molecules in the enzyme active site found for CDK2/roscovitine complex. Roscovitine is represented by the tube model, active site of the CDK2 is modeled by the solvent accessible surface, the water molecules by the CPK model (top). For better clarity, the same situation is pictured without the surface, and the positions of water molecules are represented by oxygen atoms (balls; bottom). Water molecules numbered as 394 or lower are those detected also by X-ray crystallography.
Fig. 10. Comparison of the locations of crystallographic and MD structural water molecules for the CDK2/roscovitine complex (X-ray structure, gray; X-ray structural water molecules in MD simulation, red; structural water from bulk, blue).

73
Fig. 11. The location of structural water molecules in the enzyme active site found for CDK2/ isopentenyladenine complex. Isopentenyladenine is represented by the tube model; active site of the CDK2 is modeled by the solvent accessible surface, water molecules by the CPK model (top). For better clarity, the same situation is pictured without the surface, and the positions of water molecules are represented by oxygen atoms (balls; bottom). Water molecules numbered as 411 or lower are those detected also by X-ray crystallography.
Fig. 12. Comparison of the locations of crystallographic and MD structural water molecules for the CDK2/isopentenyladenine complex (X-ray structure, gray; X-ray structural water molecules in MD simulation, red; structural water from bulk, blue).
Fig. 13. Superimposition of structural water molecules found in free CDK2 MD simulations (blue) with the polar sulphonate group of CDK2/indirubin-5-sulphonate complex (color). [X-ray obtained from the Brookhaven protein crystallographic database (PDB code: 1e9h).]
Fig. 14. Superimposition of structural water molecules found in free CDK2 MD simulations (blue) with the polar group of CDK2/hymenialdisine complex (color). [X-ray obtained from the Brookhaven protein crystallographic database (PDB code: 1dm2).]

74
TABLE IV. Interaction Energies and H-Bond Analyses Calculated for MD Structural Water Molecules in the CDK2/Roscovitine Trajectory
Interaction energy [kcal/mol] Residue
Occupancy [%]
Distance [Å]
H-bond angle [degrees] Electrostatic VdW Total
Water 323 ASP 145 (OD2) 82.9 2.7±0.1 10.7±6.3 -10.3±7.0 1.3±1.6 -9.0±6.0LYS 129 (NZ-HZ) 49.3 3.3±0.3 40.2±10.8 -4.9±4.5 -0.2±0.5 -5.1±4.2ASP 127 (OD2) 33.3 2.9±0.4 13.4±13.8 -4.5±7.2 0.3±1.4 -4.2±6.1
Water 332 ASP 145 (N-H) 59.9 3.1±0.2 23.3±13.8 -1.7±1.0 -0.3±0.3 -2.0±1.0
Water 336 PHE 146 (O) 99.9 2.8±0.2 18.0±11.0 -5.9±1.5 0.7±1.1 -5.2±1.0
Water 341 ASN 59 (ND2-HD2) ASN 59 (OD1)
75.8 18.7
3.0±0.22.8±0.2
15.0±9.118.5±11.6
-6.3±2.3 0.6±1.0 -5.7±1.8
LEU 55 (O) 73.5 2.9±0.2 13.8±9.0 -3.9±2.5 0.4±0.9 -3.5±2.1Water 344
ASP 145 (OD1) ASP 145 (N-H)
74.1 45.8
2.6±0.13.5±0.4
10.3±6.332.1±11.4
-9.5±6.9 1.9±1.9 -7.6±5.6
LYS 33 (NZ-HZ) 54.2 3.2±0.4 28.5±12.6 -3.0±4.4 0.1±0.8 -2.9±4.0Water 357
ASP 145 (N-H) 37.1 3.2±0.4 28.7±15.7 -1.5±1.3 -0.2±0.2 -1.7±1.3VAL 64 (O) 32.6 3.0±0.3 15.9±10.8 -2.0±3.0 -0.2±0.2 -2.2±2.6
Water 369 THR 14 (N-H) 57.8 3.1±0.2 15.5±7.5 -1.9±2.1 0.1±0.1 -1.8±1.5
Water 370 TYR 15 (N-H) 100.0 3.0±0.2 12.8±7.2 -3.9±0.9 0.3±0.9 -3.6±0.8ASP 145 (OD2) 99.5 2.7±0.1 13.3±7.7 -14.2±3.1 2.2±1.5 -12.0±2.3LYS 33 (NZ-HZ) 91.8 3.0±0.2 24.3±13.2 -4.3±5.2 0.6±0.9 -3.7±4.8THR 14 (OG1) THR 14 (N-H)
44.4 21.1
3.1±0.33.1±0.2
13.2±8.950.6±6.0
-1.7±1.8 -0.1±0.7 -1.8±1.4
GLY 16 (N-H) 40.6 3.7±0.2 34.4±10.7 0.3±0.8 -0.2±0.1 0.1±0.8Water 373
ASP 127 (OD2) ASP 127 (O)
100.0 85.5
2.7±0.12.9±0.2
11.6±7.126.9±11.3
-16.6±6.7 2.3±2.0 -14.3±5.4
HIS 125 (ND1-HD1) 53.9 3.4±0.4 40.8±14.6 -0.2±0.2 -0.1±0.1 -0.3±0.2LYS 129 (NZ-HZ) 26.2 3.3±0.3 32.6±17.8 -0.3±0.4 -0.2±0.5 -0.5±0.5
Water 881 ASP 86 (OD2) ASP 86 (N-H)
87.6 56.4
2.7±0.13.4±0.3
10.6±6.325.9±11.2
-13.5±4.8 1.7±1.6 -11.9±4.0
GLN 85 (NE2-HE22) 21.9 3.4±0.4 34.3±18.3 -2.0±2.9 -0.2±0.5 -2.2±2.7Water 4036
GLY 13 (O) GLY 13 (N-H)
41.6 20.0
2.9±0.33.6±0.2
19.2±11.536.9±12.1
-2.0±2.5 0.2±0.7 -1.8±2.1
THR 158 (OG1-HG1) 27.4 3.3±0.5 30.6±17.0 -1.5±2.7 0.1±0.8 -1.4±2.1TYR 159 (OH) 27.3 3.5±0.3 42.7±11.4 -0.5±0.7 -0.3±0.4 -0.8±0.8
Water 4431 LYS 33 (NZ-HZ) 91.3 2.9±0.2 23.5±12.3 -12.3±5.7 1.1±1.3 -11.2±5.0GLU 12 (O) 74.8 3.0±0.3 38.1±15.9 -4.4±3.2 -0.1±0.4 -4.5±2.9GLY 16 (O) 55.5 2.8±0.2 13.5±8.9 -3.8±3.2 0.8±1.2 -3.0±2.5
Water 6157 LYS 129 (NZ-HZ) 37.8 3.0±0.3 28.5±12.5 -3.8±5.6 0.2±0.8 -3.6±5.1THR 14 (N-H) 26.0 3.2±0.2 17.8±6.9 -1.0±1.2 -0.1±0.3 -1.1±1.2LYS 33 (NZ-HZ) 15.2 2.8±0.1 33.3±12.4 -2.1±5.5 0.2±0.7 -1.9±5.0GLY 16 (O) 10.4 2.7±0.1 12.3±7.2 -0.9±2.3 0.2±0.7 -0.7±1.7GLU 12 (O) 10.0 3.1±0.4 27.7±12.0 -1.1±2.0 0.0±0.3 -1.1±1.9
Water 7274 THR 14 (O) THR 14 (N-H)
48.9 16.7
2.8±0.23.1±0.2
12.8±8.416.9±9.6
-3.3±2.9 0.4±1.0
-2.9±2.3
LYS 129 (NZ-HZ) 17.0 3.1±0.4 33.1±13.4 -2.0±3.9 0.0±0.4 -2.0±3.7

75
Water 9376 ASP 86 (OD1) 29.6 2.8±0.3 13.7±9.9 -2.6±5.8 0.2±0.8 -2.4±5.2ASP 145 (OD2) 15.2 2.8±0.3 15.7±12.4 -2.1±5.0 0.3±0.8 -1.8±4.4
As in the previous case, the highest interaction energies generate interactions between charged side chains of polar amino acids and stable water molecules. The residues in the active site interacting with only one structural water molecule are ASP 145, PHE 146, and THR 14. Water molecule 323 creates a bridge between the ASP 145 side chain and the LYS 129 or ASP 127 side chains. Water molecule 341 stabilizes interaction between the LEU 55 and ASN 59 side chains. The large number of interactions found for water molecules 370, 373, 4036 indicates the positional fluctuations of these water molecules.
During the relaxation phase of the MD simulation, some bulk water molecules entered the active site and remained stable there for the rest of the trajectory. This is the case with water molecules 4431 and 7274. Water 4431 stabilizes the position of the LYS 33 side chain with respect to the GLU 12 and GLY 16 backbone. Water 7274 hydrates the THR 14 backbone and exhibits also a short interaction with the LYS 129 side chain. The cluster of water molecules in the phosphate part of the active site is enlarged by three other water molecules (4036, 6157, 9376). These water molecules joined the cluster during the production phase of the simulation. This is reflected by high deviations in interaction energies and in the number of peptide residues that are in contact with these water molecules, and also by low H-bond occupancy of observed hydrogen bonds. CDK2/Isopentenyladenine Complex Water molecules location. The configuration of 11 structural water molecules in the active site found in the CDK2/isopentenyladenine MD simulation is depicted in the Figure 11. One water molecule interacting with the purine ring (water 7067) was found at a similar position as water 881 in the case of CDK2/roscovitine complex. The orientation of the isopentenyladenine purine ring in the CDK2 active site differs from the orientation of the roscovitine purine ring and is more favorable for interactions with water molecules. The hydrophobic isopentenyladenine side chain interacts with only one water molecule (320). The cavity formed by VAL 64, PHE 80, ALA 144, and LEU 148 behind the purine pocket is smaller than in the case of CDK2/roscovitine and it is filled by 4 water molecules (320, 400, 404, 409). Water molecules, as in the previous cases, also hydrate the ASP 145 side chain.
The phosphate pocket of the active site is filled by just four water molecules. The smaller number of water molecules could be explained by the influence of the hydrophobic side chain of isopentenyladenine. The comparison of the positions of the structural water molecules found by MD with crystallographic water molecules is shown in Figure 12.
A small reorientation of water molecules is seen in the phosphate pocket. In this case, bulk water molecules replaced three X-ray structural water molecules.
Interaction energies. Calculated interaction energies are summarized in Table V.
As in the previous cases, one water molecule (313) stabilizes the position of the ASP 145 side
chain relative to the backbone of the TYR 15 or GLY 16. Water 320 arranges the interaction between the ASP 145, LYS 33 side chains and the C6 isopentenyladenine side chain. The interaction analysis of water molecules 400, 404 and 405 indicates the positional exchange of these waters in the active site. Water 405 stabilizes the orientation of the ASP 145 side chain to the GLY 16 backbone. Similar stabilization is seen in the case of water molecule 409 (ASN 59 side chain to the LEU 55 backbone). Bulk water molecule 5089 stabilizes the position of the THR 14, LYS 129 and ASN 132 side chains. Water molecule 7067 creates a bridge between the LEU 83 backbone and the isopentenyladenine molecule. There are also some water molecules (5089, 5091, 5165) found that create a cluster with some of the above mentioned water molecules and stabilize the entire system.

76
TABLE V. Interaction Energies and H-Bond Analyses Calculated for MD Structural Water Molecules in the CDK2/Isopentenyladenine Trajectory Residue Occupancy Distance H-bond angle Interaction energy [kcal/mol] [%] [Å] [degree] Electrostatic VdW Total
Water 313ASP 145 (OD1) 97.8 2.8 ±0.2 12.4±9.2 -13.4±3.7 1.2±1.3 -12.2±3.0TYR 15 (N-H) 77.4 3.1±0.2 16.0±7.4 -2.5±1.2 -0.1±0.7 -2.6±1.5GLY 16 (N-H) 49.9 3.5±0.3 39.5±10.8 0.1±0.9 -0.3±0.2 -0.2±0.8
Water 320ASP 145 (OD1) 100.0 2.8±0.4 17.7±13.2 -5.8±7.5 0.9±1.6 -4.9±6.3LYS 33 (NZ-H) 93.7 3.0±0.2 28.3±15.3 -8.3±5.6 0.8±1.1 -7.5±5.0IPA 299 (N6-H) 35.8 3.3±0.3 16.7±7.9 -0.7±2.0 -0.2±0.6 -0.9±1.7
Water 400ASP 145 (OD2) 54.1 2.7±0.1 13.0±7.4 -6.3±6.3 1.0±1.5 -5.3±5.3LYS 33 (NZ-H) 25.2 3.1±0.3 38.1±13.5 -3.5±4.4 0.2±0.7 -3.3±4.0ASN 59 (ND-H) 22.8 3.2±0.3 20.5±13.5 -1.6±2.9 0.2±0.7 -1.4±2.6
Water 404PHE 146 (O) 60.1 2.9±0.3 12.7±7.6 -3.9±2.9 0.1±0.1 -3.8±2.6ASP 145 (O) 41.7 3.2±0.3 28.0±16.0 -2.6±0.9 0.1±0.1 -2.5±0.9
Water 405GLY 147 (N-H) 73.4 3.2±0.3 34.8±12.2 -6.2±4.2 -0.4±0.1 -6.6±3.5LEU 148 (N-H) 31.1 3.3±0.3 50.1±6.1 -2.2±1.4 -0.4±0.2 -2.6±1.2TYR 15 (OH) 31.0 3.1±0.3 24.1±14.2 -2.5±3.1 0.4±1.2 -2.1±2.2HIS 125 (NE2) 26.0 3.3±0.3 32.7±16.5 -2.7±2.1 -0.2±0.8 -2.9±1.6
Water 407GLY 16 (O) 78.1 2.9±0.2 16.7±11.1 -4.0±2.7 0.6±1.1 -3.5±2.1ASP 145 (OD2) 51.0 3.2±0.5 32.5±18.5 -1.5±6.9 0.1±0.8 -1.4±6.3
Water 409ASN 59 (OD1) 100.0 2.8±0.2 17.2±10.5 -5.8±2.6 0.6±1.1 -5.2±2.0LEU 55 (O) 75.9 3.0±0.3 18.2±11.3 -3.7±2.5 0.1±0.8 -3.6±2.1
Water 5089LYS 129 (NZ-H) 78.8 3.0±0.2 28.8±13.9 -11.3±5.1 0.6±1.2 -10.7±4.3THR 14 (OG1) 58.8 2.9±0.3 20.9±14.2 -2.1±2.7 0.2±0.8 -1.9±2.1ASN 132 (ND2-H) 39.3 3.1±0.2 20.5±11.3 -2.1±2.6 0.1±0.6 -2.0±2.3
Water 5091THR 14 (N-H) 28.8 3.2±0.3 24.7±10.8 -0.6±1.2 -0.1±0.4 -0.7±1.0ASN 132 (ND2-H) 27.9 3.1±0.4 19.7±11.6 -1.4±2.7 -0.1±0.7 -1.5±2.3ASP 145 (OD2) 26.5 2.8±0.2 12.3±8.5 -5.2±7.8 0.6±1.2 -4.6±7.0
Water 5165THR 14 (OG1-H) 46.3 3.3±0.3 34.4±12.3 -0.1±2.0 -0.2±0.6 -0.3±1.6ASN 132 (ND2-H) 56.3 3.2±0.3 33.4±15.2 -1.8±2.6 -0.1±0.7 -1.9±2.3ASP 127 (OD2) 35.9 2.8±0.2 11.9±9.2 -5.8±7.5 0.6±1.3 -5.2±6.6HIS 125 (NE2) 32.9 3.0±0.2 14.4±8.5 -2.6±3.0 0.2±0.6 -2.4±2.6
Water 7067IPA 299 (N1) 92.9 3.1±0.2 22.8±12.2 -2.7±1.6 -0.1±0.7 -2.8±1.3LEU 83 (O) 88.2 2.9±0.3 16.5±10.3 -4.5±2.3 0.3±0.8 -4.2±1.9
The statistical analysis of binding energy data introduced in above tables shows that the correlation coefficients between the occupancies of H-bond and interaction energies as calculated by the force field are between 0.4 and 0.7, which indicates correlation on 95 % probability level. We have also calculated the correlation between energies and H-bond distances or H-bond angles. The calculated correlation coefficients between the energies and H-bond distances are in range of 0.6 to 0.7. It also indicates correlation on 95 % probability level. The correlation between the energies and H-bond angles is not so significant (calculated between 0.3 and 0.6). It is caused by predomination of electrostatic term in interaction energy calculation.

77
Comparison With Other X-Ray Structures From the PDB Database The positions of MD structural water molecules in the free CDK2 active site were compared with the positions of polar groups of known nonpeptidic inhibitors. For this was used X-ray data of 17 complexes of CDK2 with nonpeptidic inhibitors stored in the Brookhaven Protein Data Bank. As expected, water molecules interacting with the GLU 81 and LEU 83 backbone found in MD were replaced by a polar group of inhibitors. These H-bond interactions are important for substrate binding to the active site [59]. Inhibitors that have a polar group oriented to the ribose pocket have this group oriented in the same direction as structural water molecules that were found. This is illustrated in Figure 13 and Figure 14 for the inhibitors hymenialdisine and indirubin-5-sulphonate, respectively. CONCLUSIONS
We have presented a very detailed analysis of solvent behavior over four molecular dynamics trajectories on CDK2 and its substrate/inhibitor complexes. The data obtained were compared with X-ray information. The presented method is complementary to that of X-ray crystallography. The static picture of structure from X-ray data is completed by information about the dynamics of molecules. The MD method also brings information about interaction energies between molecules and is also helpful in the case of uncompleted X-ray data. In our case, the MD simulation detected a larger number of stable water molecules in the active site than X-ray crystallography. Moreover, it provided us with information about changes in the configuration of water molecules in the CDK2 active site.
The behavior of water molecules interacting with amino acids in the enzyme active site, and their interaction energies, brings information useful for the rational drug design of new potent and selective inhibitors. The structural water molecules found for free CDK2 were in the same positions as the nitrogens of the purine ring of the ATP and inhibitors. Appropriate replacement of the afore-mentioned water molecule in the active site leads to more selective inhibitors as demonstrated by the replacement of stable water molecules by polar groups of inhibitor.
The interaction of inhibitor with Asp 86 was found for many compounds [59]. Therefore one can suppose the replacing of the π-interacting water molecule by a hydrophilic group results in an increase in inhibitor activity. This assumption is demonstrated by higher activities of -OH substituted purine-like inhibitors [123]. The inclusion of the substrate into the active site evokes the conformational changes in this part, which allows the opening of the cavity through the enzyme and movement of the water molecules to the active site. The detailed analysis of solvent behavior and interaction energy information may be used as a tool to design new inhibitors because it indicates where the polar groups should be oriented. ACKNOWLEDGMENTS Our thanks to the Supercomputer Center Brno for providing us with computer time, and to Roger Turland for language corrections.

78

79
Activation and inhibition of cyclin-dependent
kinase-2 by phosphorylation; a molecular
dynamics study reveals the functional importance
of the glycine-rich loop
Iveta Bártová, Michal Otyepka, Zdeněk Kříž, and Jaroslav Koča
Protein Sci. 13, 2004, 1449-1457

80

81
Activation and inhibition of cyclin-dependent kinase-2 by phosphorylation; a molecular
dynamics study reveals the functional importance of the glycine-rich loop
IVETA BÁRTOVÁ,1 MICHAL OTYEPKA,2 ZDENĚK KŘÍŽ,1 AND JAROSLAV KOČA1 1National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Brno, Czech Republic 2Department of Physical Chemistry, Palacky University, Olomouc, Czech Republic (RECEIVED December 17, 2003; FINAL REVISION February 20, 2004; ACCEPTED February 20, 2004) Abstract Nanoseconds long molecular dynamics (MD) trajectories of differently active complexes of human cyclin-dependent kinase 2 (inactive CDK2/ATP, semiactive CDK2/Cyclin A/ATP, fully active pT160-CDK2/ Cyclin A/ATP, inhibited pT14-; pY15-; and pT14,pY15,pT160 CDK2/Cyclin A/ATP) were compared. The MD simulations results of CDK2 inhibition by phosphorylation at T14 and/or Y15 sites provide insight into the structural aspects of CDK2 deactivation. The inhibitory sites are localized in the glycine-rich loop (G-loop) positioned opposite the activation T-loop. Phosphorylation of T14 and both inhibitory sites T14 and Y15 together causes ATP misalignment for phosphorylation and G-loop conformational change. This conformational change leads to the opening of the CDK2 substrate binding box. The phosphorylated Y15 residue negatively affects substrate binding or its correct alignment for ATP terminal phospho-group transfer to the CDK2 substrate. The MD simulations of the CDK2 activation process provide results in agreement with previous X-ray data. Keywords: cell cycle; CDK regulation; phosphorylated tyrosine; threonine
Dedicated to Professor Milan Kratochvíl on the occasion of his 80th birthday. Reprint requests to: Michal Otyepka, Department of Physical Chemistry, Palacky University, tr. Svobody 26,
771 46 Olomouc, Czech Republic; e-mail: [email protected], fax: 420-585634420; or Jaroslav Koča, National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic; e-mail: [email protected]; fax: 420-541129506.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03578504. Protein Science (2004), 13:1449–1457. Published by Cold Spring Harbor Laboratory Press. Copyright © 2004 The Protein Society

82
Cyclin-dependent kinases (CDK) play a central role in the control of the eukaryotic cell division cycle. The function of CDK is to catalyze the phosphoryl transfer of the adenosine-5’-triphosphate (ATP) γ-phosphate to serine or threonine hydroxyl in the protein substrate. The CDK activity is stringently controlled by cyclin binding, phosphorylation, and binding of protein inhibitors. The CDK activation is a two-step process that requires cyclin binding and phosphorylation in the activation loop (known also as the ‘T-loop‘) [8, 18, 124]. The critical CDK/cyclin complexes for the cell division are CDK2/cyclin E, driving a cell across the G1/S-phase border; CDK2/cyclin A, mediating DNA replication; and CDK1/cyclin B, controlling the entry into mitosis [8]. The CDKs are very intensively studied enzymes mainly as targets for medical and molecular biological applications [26] and also as exemplary biochemical models of nonautoinhibitory regulation [8].
A detailed description of the activation pathway of p34cdc2 (CDC2, CDK1) came from studies of Xenopus egg extracts and it is believed that the mechanism is conserved over all eukaryotic organisms [125]. The CDK2 activation diagram (see Scheme 1) can be extrapolated from the best understood CDK1 regulation model [18, 126]. Monomeric CDK2 (Scheme 1, I) is inactive and for its activation requires binding to a cyclin (cyclin E at the G1/S transition, cyclin A during the S phase). The CDK2/cyclin complex (Scheme 1, IIa and IIb) is recognized by multiple protein kinases and it results in phosphorylations on T14, Y15, and T160 (in CDK2). The amino acid residue Y15 and to a lesser extent T14 are phosphorylated by human Wee1Hu [45]. This inhibitory phosphorylation is independent of previous cyclin binding [48]. Inhibitory phosphorylation likely precedes the activating T160 phosphorylation by CAK (CDK7/Cyclin H) because activatory phosphorylation requires cyclin binding. The over-phosphorylated complex (Scheme 1, III) is inactive and subsequent dephosphorylation of T14 and Y15 by CDC25 [46, 47] results in activation. Recently, the phosphorylation mechanisms of the cell were revisited with the finding that pY15-CDK2 dephosphorylation by CDC25 is an important regulation mechanism of correct cell cycle timing [48]. The importance of inhibitory sites was also probed by site-directed mutagenesis of T14 (T14A) and Y15 (Y15F). Such mutations stimulate kinase activity [127] but the expression of mutated protein (T14A,Y15F)CDK2 is cytotoxic [128]. The fully active CDK2/cyclin complex (Scheme 1, IV) is phosphorylated only at T160. Feedback from the active form of the pT160-CDK2/cyclin complex stimulates CDC25 activity and inhibits Wee1 activity. Such an ‘autocatalytic’ activation loop leads to a rapid activation of CDK2. Two phosphatases, KAP [52] and PP2C [129, 130] were found to be dephosphorylating monomeric CDK2 rather then CDK2/cyclin complex.
CDK2 has the typical bilobal kinase fold (Figure 1). The active site is positioned between two lobes, the smaller N-terminal and the bigger C-terminal. The smaller lobe is primarily composed of β-sheet with one α-helix, the C-helix, whose correct orientation is important for catalysis. The helix includes the conserved PSTAIRE motif (residues 45-51; this helix is also denoted as PSTAIRE helix) important for cyclin binding. The CDK2 activation site of the T-loop is located at T160. Close to the activation segment is a functionally opposite segment, the inhibitory loop (residues 11-18), named the glycine-rich loop (G-loop) because its primary sequence includes three highly conserved glycine residues (CDK2: 11-GEGTYG) [17]. The G-loop includes two possible inhibitory sites, T14 and Y15. The phosphorylation of any of these residues leads to the loss of kinase activity.

83
Scheme 1. Scheme of CDK2 regulation. Inactive form CDK2/ATP (I) binds to Cyclin and may be phosphorylated at Y15 by WEE1 kinase. Inhibited complex pY15-CDK2/Cyclin/ATP (II) is phosphorylated by CAK at T160 and pY15,pT160–CDK2/Cyclin/ATP complex (III) is activated at the pY15 site by dephosphorylation by CDC25. The fully active complex pT160–CDK2/Cyclin/ATP (IV) after Cyclin is lost is dephosphorylated by PP2C or KAP at pT160.
Two recent articles studying the process of CDK2/Cyclin A complex formation and T160
phosphorylation [38, 131] have concluded that the CDK2/Cyclin A complex formation is two-step process; the first step is a CDK2/Cyclin A associate formation followed by CDK2/Cyclin A complex activation by conformational change of the T-loop. The association of unphosphorylated CDK2 with Cyclin A serves to configure the active site for ground-state binding of both ATP and a protein substrate and aligns ATP in the transition state for phosphoryl transfer. The optimization of the mentioned ATP alignment and stabilization of substrate binding is the principal role of phosphorylation at T160. Both articles broaden the knowledge gained from previous X-ray studies on inactive, semiactive, and fully active CDK2 forms [27, 28, 39, 99]. Cook et al. [132] deduce that in CDK2, the shift in the T-loop upon phosphorylation creates a pocket that can accept the substrate prolyl side chain, and the substrate basic group (at the P+3 position) to make contact with the pT160 (phospho-threonine 160) phosphate [40, 133]. The serine of the peptide substrate is hydrogen bonded to the ATP γ-phosphate oxygen, to the catalytic aspartate D127, and to the conserved lysine K129 [132].
The mechanism of inhibition by phosphorylation is not yet well understood from the
structural point of view. It has been suggested that pY15 perturbs the binding of protein substrate at the catalytic site through sterical hindrance [54, 124]. However, to our best knowledge this fact has not yet been clearly confirmed. The aim of this article is a detailed study on CDK2 inhibition and activation by phosphorylation using molecular dynamics simulations.

84
Figure 1. View of CDK2/ATP (1HCK coordinates taken from PDB database) complex is shown in tube representation. The T160 (shown in gray-colored licorice representation) activation site is located on the T-loop. The G-loop (black-colored tube representation) includes two possible inhibitory sites, T14 (gray-colored licorice representation) and Y15 (black-colored licorice representation).
Results
All the trajectories analyzed below (for a brief summary, see Table 1) were stable during the whole production part of the molecular dynamics (MD) simulation. The trajectory stabilities were confirmed by analysis of secondary structure elements, radius of gyration, root-mean-square deviation to the crystal structure, and total energy (Table 1).
Enhanced thermal movements observed on the CDK2/ATP complex are localized on the C-helix (PSTAIRE helix, residues 46-56), on residues around T26, E73, L96, Y179, and residues between P228 and L255 (Figure 2A). The B-factor curves for the CDK2/Cyclin A/ATP system display similar sharp peaks corresponding to the same residues (Figure 2B). Only the high peak corresponding to the enhanced mobility of the PSTAIRE helix disappeared due to the presence of the bound cyclin that interacts with the PSTAIRE helix and stabilizes its secondary structure fold. The phosphorylation of T160 residue in the CDK2/Cyclin A/ATP complex increases the mobility of the G-loop (Figure 2B, C), in agreement with experimental X-ray data [28, 39]. Phosphorylations in the G-loop lead to an additional increase in the G-loop thermal fluctuations (19, 28, 29, and 34 are the mean B-factors of G-loop residues for pT160-CDK2/Cyclin A/ATP, pT14,pT160-CDK2/Cyclin A/ATP, pY15,pT160-CDK2/Cyclin A/ATP, and pT14,pY15,pT160-CDK2/Cyclin A/ATP complexes, respectively) (Figure 2C-F). The experimental B-factor values are in all cases (Figure 2A-C) larger than those B-factors calculated from MD simulations. This observation agrees with results described by Philippopoulos and Lim [134].

85
Table 1. Summary of trajectories production part characteristics
System Used PDB structure
E 105 kcal·mol-1
Rg (MD) Å
Rg (X-ray) Å
RMSD Å
CDK2/ATPa 1HCK -0.93 ± 0.01 20.14 ± 0.07 19.85 1.72 ± 0.11
CDK2/Cyclin A/ATPb 1FIN -1.50 ± 0.01 25.54 ± 0.08 25.37 1.27 ± 0.09
pT160-CDK2/Cyclin A/ATPc 1JST -1.43 ± 0.01 25.84 ± 0.10 25.31 1.33 ± 0.07
pY15,pT160-CDK2/Cyclin A/ATPd 1JST* -1.39 ± 0.01 25.94 ± 0.07 - 1.57 ± 0.15
pT14,pT160-CDK2/Cyclin A/ATPd 1JST* -1.39 ± 0.01 26.20 ± 0.13 - 1.50 ± 0.09
pT14,pY15,pT160-CDK2/Cyclin A/ATPd
1JST* -1.40 ± 0.01 25.99 ± 0.09 - 1.53 ± 0.10
E, mean total energy; Rg (MD), mean radius of gyration; Rg (X-ray), radius of gyration of the X-ray structure; RMSD, root-mean-square deviation of backbone atoms compared with initial X-ray crystal structures. a The following were modified by in silico phosphorylation of either Y15 or T14, and both T14,Y15 residues: b Inactive CDK2. c Semiactive CDK2.
d Fully active CDK2.
e CDK2 inhibited by phosphorylation.
Remarkable differences in secondary structure behavior were noticed between
monomeric CDK2 and complexes of CDK2/Cyclin A. The PSTAIRE helix fold is unstable during CDK2/ATP simulation corresponding to our previous results obtained by simulations of CDK2/purine inhibitor complexes. The PSTAIRE helix fold is destabilized by ligand presence in the CDK2 active site [55]. All simulations of CDK2/Cyclin A complexes reveal enhanced stability of the PSTAIRE helix and surrounding secondary structure elements (Figure 3). These were also stable during all simulations with systems inhibited by phosphorylation (data not present). Hence, the Cyclin binding to CDK2 remarkably stabilizes the PSTAIRE helix fold in agreement with crystalographic experiments [28].
Scheme 2. Structural formula of adenosine 5’-triphosphate (ATP) molecule with atom labeling used in this article.

86
Figure 2. Comparison of temperature B-factors calculated from the last 500 psec of MD simulations (solid line); free CDK2/ATP (A); CDK2/Cyclin A/ATP (B); pT160–CDK2/Cyclin A/ATP (C); pY15,pT160–CDK2/Cyclin A/ATP (D); pT14,pT160–CDK2/Cyclin A/ATP (E); and pT14,pY15,pT160–CDK2/Cyclin A /ATP complex (F). B-factor values of CDK2/ATP (A), CDK2/Cyclin A/ATP (B), and pT160–CDK2/Cyclin A/ATP (C) complexes determined by X-ray experiments are added to the plots (dotted line).
Analysis of the ATP dihedral angle C8-N9-C1*-C2* (Scheme 2) between the adenine and ribose moieties reveals that the angle changed during the early stage of CDK2/ATP simulation in comparison with its conformation in the crystal structure (Figure 4A). This torsion angle was stable for the other studied systems and comparable with its value found in the crystal structures. The conformation of the ATP phosphate moiety was stable during simulation of inactive CDK2 but it changed during the early stage of semi- (CDK2/Cyclin A/ATP) and fully active (pT160-CDK2/Cyclin A/ATP) complex simulations (Figure 4B, C).
Dramatic changes were observed in ATP conformation and/or position after inhibitory phosphorylations at T14 and/or Y15. The shift of the ATP Pγ group after T14 phosphorylation (Figure 5B, C) towards the adenine base is especially remarkable. This ATP reorientation also causes a shift in the Mg2+ ion position and consequently affects the Mg2+ ion coordination sphere (Table 2, Figure 5).

87
The G-loop conformation was changed during the early stage of inactive (CDK2/ATP), partly active (CDK2/Cyclin A/ATP), and fully active (pT160-CDK2/Cyclin A/ATP) CDK2 simulations in comparison with its conformation as found in the crystal structures (Figure 4, Figure 6). The G-loop moves away from the ATP phosphate moiety binding site after the interaction of CDK2 with Cyclin A and again after CDK2/Cyclin A/ATP complex phosphorylation at the T160 site (Figure 6A). The shift of the G-loop is equal to 3.5 Å (CDK2/Cyclin A/ATP) and 8.6 Å (pT160-CDK2/Cyclin A/ATP) in comparison with the G-loop position found in the CDK2/ATP system. The G-loop conformation remains stable (low thermal fluctuations) after the shift described above (Figure 7A). The G-loop backbone possesses similar conformation in the pT160-CDK2/Cyclin A/ATP and pY15,pT160-CDK2/Cyclin A/ATP complex simulations (Figure 5A) but the pY15 residue reorients its side chain towards a bulk solvent due to hydration of the phospho-group. The Y15 phosphorylation causes an increase in the G-loop thermal fluctuations (Figure 7B). The G-loop conformation changes remarkably during the simulations of pT14,pT160-CDK2/Cyclin A/ATP and pT14,pY15,pT160-CDK2/Cyclin A/ATP complexes. In these simulations the G-loop moves away from the ATP phosphate binding pocket. Displacement of the G-loop is equal to 4.41 Å (pT14,pT160-CDK2/Cyclin A/ATP) and 5.94 Å (pT14,pY15,pT160-CDK2/Cyclin A/ATP) compared to its position in the fully active CDK2 (Figure 5B, C). In general, the G-loop phosphorylation increases its thermal fluctuations (Figure 7A-D) due to the insertion of the bulky hydrophilic group. Discussion
The Cyclin A binding stabilizes the PSTAIRE helix remarkably and decreases thermal movements in this region by direct interaction of the α1-helix with Cyclin A. This stabilization is also well known from previous crystallographic studies [28] demonstrating that molecular dynamics simulations results are in agreement with experimental data. The X-ray studies [39] as well as the kinetic experiments [33] conclude that the phosphorylation of T160 residue causes the correct orientation of the T-loop, the creation of a substrate binding box, and also the correct alignment of ATP for phospho-group transfer.
Figure 3. Secondary structure elements (α-helix and β-sheet in blue and in red, respectively) determined during the whole production part of MD simulations of CDK2/ATP, CDK2/Cyclin A/ATP, and pT160–CDK2/Cyclin A/ATP complexes. Stabilization of the PSTAIRE helix (denoted as α1-helix) after Cyclin A binding to CDK2 is clearly seen.

88
Moreover, it was suggested that the phosphorylation of T160 leads to substrate stabilization due to the interaction of pT160 with substrate basic residue at the P+3 position [40, 42, 133]. The MD simulations emphasize the known fact that CDK2/Cyclin A association stabilizes the three-dimensional structure of CDK2 and the activating phosphorylation of T160 stabilizes T-loop conformation for substrate binding and phosphoryl transfer. The molecular mechanism of inhibition by phosphorylation remains until now unclear. Our simulations shed light on the mechanism and suggest a model for how the process of inhibition by phosphorylation may work.
It can be deduced from the simulations and from alignments of X-ray crystal structures or averaged MD structures to X-ray structures of fully active CDK2 with peptide substrate (1QMZ, 1GY3) that the inhibitory sites utilize different mechanisms of action. T14 inhibitory phosphorylation leads to G-loop shift and ATP misalignment for phospho-group transfer due to the shift of the terminal ATP phospho-group towards the ATP adenine base moiety in the pT14,pT160-CDK2/Cyclin A/ATP complex (3.8 Å) and also in the pT14,pY15,pT160-CDK2/Cyclin A/ATP structure (3.8 Å). The G-loop shift causes dramatic changes in substrate binding box conformation. Such changes might decrease CDK2 affinity to its substrate. The G-loop shift and substrate box opening is much more remarkable in the pT14,pY15,pT160-CDK2/Cyclin A/ATP complex.
The Y15 residue is buried in an active pT160-CDK2/Cyclin A/ATP complex. After phosphorylation or substrate binding the conformation of Y15 changes leading to the exposure of the pY15 side chain to solvent (Figure 5A) and to an increase in G-loop flexibility (Figure 7B). Conformation of the Y15 residue in the crystal structure of fully active CDK2 with peptide substrate (1QMZ) [40] is very similar to the conformation of pY15 in the pY15,pT160-CDK2/Cyclin A/ATP complex. The distance between the pY15 phospho-group and the substrate arginine (P+2 substrate position) terminal groups is equal to 4.5 Å (Figure 8). One can deduce that Y15 phosphorylation affects substrate binding or its correct alignment for phosphorylation, because pY15 can directly interact with the substrate and also decreases substrate interaction with the pT160 residue due to competition. These findings correspond with previously published articles [54, 124] where the authors deduced from the not yet published crystal structure of pY15,pT160-CDK2/Cyclin A/ATP complex that the phosphorylation of Y15 does not significantly alter the overall structure of the complex nor does it prevent ATP binding. The authors also suggest that phosphorylation of Y15 may perturb the protein substrate binding at the catalytic site through a steric hindrance.
Table 2. Coordination of the Mg2+ ion was monitored during the entire simulation period of all trajectories: the results show the magnesium ion was hexacoordinated during all simulations System Atoms coordinating Mg2+ CDK2/ATP
ATP (Oα1) ATP (Oβ2) ATP (Oγ1) N132 (Oδ1) D145 (Oδ2) WAT314 (O)
CDK2/Cyclin A/ATP ATP (Oα2) ATP (Oβ1) N132 (Oδ1) D145 (Oδ2) WAT4896 (O) WAT8293 (O) pT160-CDK2/ Cyclin A/ATP
ATP (Oα1)
N132 (Oδ1)
D145 (Oδ2)
WAT7781 (O)
WAT7803 (O)
WAT7831 (O)
pY15,pT160-CDK2/ Cyclin A/ATP
ATP (Oα1)
N132 (Oδ1)
D145 (Oδ2)
WAT1707 (O)
WAT2232 (O)
WAT5368 (O)
pT14,pT160-CDK2/ Cyclin A/ATP
ATP (Oβ1)
ATP (Oγ3)
D145 (Oδ2)
WAT2413 (O)
WAT2560 (O)
WAT8065 (O)
pT14,pY15,pT160- CDK2/Cyclin A/ATP
ATP (Oα2)
ATP (Oγ3)
D145 (Oδ2)
WAT7647 (O)
WAT7693 (O)
WAT8053 (O)

89
Figure 4. Stereoview of selected residues (D127, K129), ATP and G-loop conformation; comparison of structures determined by MD simulations (gray) and X-ray experiments (black); CDK2/ATP (A), CDK2/Cyclin A/ATP (B), and pT160–CDK2/Cyclin A/ATP (C).
The glycine-rich loop (G-loop) enables protein kinase to adopt a wide range of backbone conformations. The significance of this domain is demonstrated by the fact that substitution of the glycine residues in the glycine-rich loop, particularly the first and the second glycine (GxGxxG) with either alanine or serine results in a dramatic decrease in cAMP-dependent protein kinase catalytic (cAPK) activity. The functional importance of the glycine-rich loop has been described in detail for cAPK [83, 135-137] but its importance for CDK regulation has not been yet discussed. It is believed that the glycine-rich loop catalytic function, i.e.

90
correct ATP binding and alignment, is the same as its function in cAPK, but it exhibits a new inhibitory function for CDK.
Figure 5. Stereoview of the superimposition of pT160–CDK2/Cyclin A/ATP (black) and complexes phosphorylated in the G-loop (gray); at Y15 residue (A), at T14 residue (B), and at T14/Y15 residues (C). ATP, Mg2+ ion, and D127 and K129 residues are shown in licorice representation. Inhibitory sites at T14 and Y15 residues are depicted in licorice representation, and G-loop in tube representation.
The functionally important flexibility of the G-loop is assured by its primary sequence where the inhibitory sites T14 and/or Y15 are bordered by conserved glycine residues 11-

91
GEGTYG (conservation profiles for all protein kinases catalytic domains show the G-G--G motif as one of the most conserved protein kinase motifs [17], also known as the nucleotide binding loop or P-loop). This sequence has been found in human CDK1 (CDC2), CDK2, CDK3, and CDK5 and also in human CDK10. A very similar sequence has also been found in CDK8 (GRGTYG). The remaining cyclin-dependent kinases have mutated inhibitory sites. The CDK4 (GVGAYG) and CDK6 (GEGAYG) have mutated threonine to alanine, CDK9 (GQGTFG) has changed tyrosine to phenylalanine and CDK7 (GEGQFA) has mutated both sites and one of the very conserved glycines is also mutated to alanine. The GEGTYG motif and all its aforementioned variations occur only in the CMGC group of protein kinases (classification according to [17]). Important is the fact that the function of these sites can differ among cyclin-dependent kinases. While T and/or Y residues serve as inhibitory sites in CDK1 homology kinases (CDK1, CDK2, and probably in CDK3), Y serves as an activatory site for CDK5/p35 [67]. In contrast, Y17 phosphorylation of CDK4 (and perhaps CDK6) is specifically used to initiate control cell cycle start from quiescence but not for G1 traverse [138]. Hence, Y17 phosphorylation can also be considered as inhibitory because phosphorylation of CDK4 Y17 residue occurs during entry into quiescence and dephosphorylation must occur sometime during cell cycle start. These findings emphasize the fact that the G-loop plays a very important biological role in CDKs.
Figure 6. Stereoview of G-loop and ATP conformations in CDK2/ATP (thin black), CDK2/Cyclin A/ATP (thick gray), and pT160–CDK2/Cyclin A/ATP (thick black) as determined by MD simulations (A) and X-ray experiments (B).

92
Figure 7. Stereoview of the superimposition of the snapshots taken every 200 psec from the production parts of molecular dynamics simulations of pT160–CDK2/Cyclin A/ATP (A) and complexes phosphorylated in the inhibition segment (G-loop) at Y15 (B), T14 (C), and T14/Y15 together (D).

93
Figure 8. Stereoview of G-loop and pY15 residue conformations in the pY15,pT160–CDK2/Cyclin A/ATP complex (gray) and crystal structure of fully active CDK2 with peptide substrate (black; 1QMZ; [40]). Only P–P + 3 peptide substrate residues are depicted for clarity. Materials and methods
Molecular dynamics simulations of six systems (CDK2/ATP, CDK2/Cyclin A/ATP, pT160-CDK2/Cyclin A/ATP, pT14,pT160-CDK2/Cyclin A/ATP, pY15,pT160-CDK2/Cyclin A/ATP, and pT14,pY15,pT160-CDK2/Cyclin A/ATP; see also Table 1) were carried out using the SANDER module of the AMBER 6.0 software package [73] with the parm99 force field [72]. The starting geometries for simulation were prepared using X-ray structures obtained from the Brookhaven protein databank (www.pdb.org). MD simulations of free CDK2 and CDK2/purine inhibitor complexes [55] were used for comparison in the discussion part.
Starting structures for the molecular dynamics simulations were prepared according to
standard procedures. At first, the protonation states of histidines were checked by WHATIF [109] and then all hydrogens were added using Xleap from the AMBER 6.0 package. The structures were neutralized by adding 11, 16, 15, 13, 13, and 11 chloride counter ions for inactive, semi-, and fully-active CDK2, and fully active CDK2 phosphorylated at Y15, T14, and T14/Y15 residues, respectively. Such systems were inserted in a rectangular water box where the layer of water molecules was equal to 10 Ǻ. All systems were minimized prior to the production part of molecular dynamics in this way. The protein was frozen and the solvent molecules with counterions were allowed to move during a 1000 step minimization and a 2 ps long molecular dynamics run. Then, the side chains were relaxed by several following minimizations with a decreasing force constant applied to the backbone atoms. After the relaxation, the system was heated to 250 K in 10 ps and then to 298.15 K in 40 ps. The production part of CDK2/ATP took 1.2 ns, of CDK2/Cyclin A/ATP 2 ns, of pT160-CDK2/Cyclin A/ATP 2.5 ns, of all inhibited systems 3 ns. The length of the simulations was chosen as compromise between the quality of configuration space sampling and the size of the studied systems (~60.000 atoms). The 2 fs time integration step and particle-mesh Ewald (PME) methods for treating electrostatic interaction were used. All simulations were run under periodic boundary condition in the NpT ensemble at 298.16 K and a constant pressure of 1 atm. The SHAKE algorithm with a tolerance of 10-5, was applied to fix all bonds containing hydrogen atoms. The 8.0 Ǻ cutoff was applied to treat non-bonding interactions. Coordinates were stored every 2 psec.
All analyses of MD simulations were carried out by CARNAL and PTRAJ modules of
AMBER-6.0 [73], by GROMACS [139], and by gOpenMol [140] program packages. Parametrization of the phosphorylated tyrosine residue (Table 3) was done according to the

94
standard Cornell et al. scheme [108]. The Gaussian98 program package [80] was used for all necessary ab-initio calculations at HF/6-31G(d) level. Partial atomic charges for ATP-Mg2+ and for phosphorylated threonine and tyrosine residues were prepared using the restrained electrostatic potential (RESP) procedure [108]. Table 3. Torsion angle parameters used in this study for phosphorylated tyrosine residue (pY15)
Dihedral angle IDIVF PK(Vn/2) [kcal⋅mol-1] Phase [°] PN
O2-P-OS-CA 3 4.7 162.2 3
P-OS-CA-CA 2 11.6 0.0 -2
P-OS-CA-CA 2 6.4 180.0 4
These parameters were determined using the standard Cornell et al. Scheme (1995). IDIVF, total number of torsion angles about a single bond that the potential applies to; PK, one-half of the barrier magnitude; Phase, phase shift of the torsion function; PN, periodicity of the torsion function. Acknowledgments We thank the MetaCenter (http://meta.cesnet.cz) for computer time. This work was supported by the Ministry of Education of the Czech Republic (I.B., Z.K., J.K.; Grant LN00A016) and the Grant Agency of the Czech Republic (M.O.; 301/02/0475). Their financial support is gratefully acknowledged. The authors thank P. Banáš for the parametrization of phospho-tyrosine. Our thanks are also addressed to Dr. Jiří Damborský (Brno) for valuable discussions on the paper. L.H. Jones (UK) and R. Turland (UK) are gratefully acknowledged for English corrections.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.

95
The mechanism of inhibition of the cyclin-
dependent kinase-2 as revealed by the molecular
dynamics study on the complex CDK2 with the
peptide substrate HHASPRK
Iveta Bártová, Michal Otyepka, Zdeněk Kříž, and Jaroslav Koča
Protein Sci. 14, 2005, 445-451

96

97
The mechanism of inhibition of the cyclin-dependent kinase-2 as revealed by the
molecular dynamics study on the complex CDK2 with the peptide substrate HHASPRK
IVETA BÁRTOVÁ,1 MICHAL OTYEPKA,2,3 ZDENĚK KŘÍŽ,1 AND JAROSLAV KOČA1 1National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Brno, Czech Republic 2Department of Physical Chemistry, Palacky University, 771 46 Olomouc, Czech Republic 3Laboratory of Growth Regulators, Palacky University and Institute of Experimental Botany AS CR, 783 71 Olomouc, Czech Republic (RECEIVED June 29, 2004; FINAL REVISION October 13, 2004; ACCEPTED October 18, 2004) Abstract Molecular dynamics (MD) simulations were used to explain structural details of cyclin dependent kinase-2 (CDK2) inhibition by phosphorylation at T14 and/or Y15 located in the glycine-rich loop (G-loop). Tennanosecond-long simulations of fully active CDK2 in a complex with a short peptide (HHASPRK) substrate and of CDK2 inhibited by phosphorylation of T14 and/or Y15 were produced. The inhibitory phosphorylations at T14 and/or Y15 show namely an ATP misalignment and a G-loop shift (<5 Å) causing the opening of the substrate binding box. The biological functions of the G-loop and GxGxxG motif evolutionary conservation in protein kinases are discussed. The position of the ATP γ-phosphate relative to the phosphorylation site (S/T) of the peptide substrate in the active CDK2 is described and compared with inhibited forms of CDK2. The MD results clearly provide an explanation previously not known as to why a basic residue (R/K) is preferred at the P2 position in phosphorylated S/T peptide substrates. Keywords: cell cycle; CDK inhibition; phosphorylated tyrosine and threonine; glycine-rich loop; GxGxxG motif Reprint requests to: Michal Otyepka, Department of Physical Chemistry, Palacky University, tr. Svobody 26, 771 46 Olomouc, Czech Republic; e-mail: [email protected]; fax: +420 585634425; or Jaroslav Koča, National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Kotlarska 2, 611 37 Brno, Czech Republic; e-mail: [email protected]; fax +420 549492556. Abbreviations: p denotes phosphorylation, i.e., pT160 is phosphothreonine 160; G-loop, glycine-rich loop (CDK2 residues 11–18); JST, pT160-CDK2/Cyclin A/ATP; QMZ, pT160-CDK2/Cyclin A/HHASPRK/ATP; pT14-QMZ, pT14,pT160-CDK2/Cyclin A/HHASPRK/ATP; pY15-QMZ, pY15,pT160-CDK2/Cyclin A/HHASPRK/ATP; pT14,pY15-QMZ, pT14,pY15,pT160-CDK2/Cyclin A/HHASPRK/ATP. Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04959705. Protein Science (2005), 14:445–451. Published by Cold Spring Harbor Laboratory Press. Copyright © 2005 The Protein Society

98
Cyclin-dependent kinase 2 (CDK2) is a member of the eukaryotic S/T protein kinase family and its function is to catalyze the phosphoryl transfer of ATP γ-phosphate to serine or threonine hydroxyl (denoted as S0/T0) in a protein substrate. CDK2 participates in eukaryotic cell cycle regulation at the G1/S boundary. CDK2 deregulation has been proved to occur in tumor cells, evoking a strong interest in artificial [55, 82, 141, 142] and native [59] inhibitors. CDK2 activity is tightly regulated by a complex mechanism including a positive regulatory subunit binding, and phosphorylations at positive and/or negative regulatory sites [18]. For activation it requires a binding to Cyclin A or Cyclin E and phosphorylation of the T160 residue in the activation segment (T-loop) [28].
The structure of CDK2 exhibits the classical bi-lobal kinase fold [18], where the N-terminal domain is composed mainly of the β-sheet, containing five anti-parallel β-strands, and one α-helix (the C-helix). The larger C-terminal domain is predominantly α-helical, and it is linked to the N-terminal domain by a flexible hinge. The catalytic cleft which natively binds ATP is located between both domains (Figure 1). The ATP phosphate binding pocket is partly formed by the G-loop and it is believed that the primary function of the G-loop is to help in correct alignment of the ATP phosphate moiety for reaction. The G-loop motif (GxGxxG) is conserved in many kinases [17]. In the CDK2/substrate complexes, the substrate S/T is directly hydrogen bonded to the catalytic aspartate, D127, and to the conserved lysine, K129 [40, 132]. The first and the most important step of the ATP γ-phosphate transfer to the substrate is the correct alignment of both molecules, i.e., the peptide and ATP-Mg2+, to favor an in-line mechanism for phosphoryl transfer.
The peptide libraries’ analyses for CDK2 substrate preference have detected the sequence
X–1(S/T)0P1X*2(K/R)3 to be an optimal substrate. Here, S/T are phosphorylation residues (serine or threonine), X is any amino acid, but K/R are favored at X* position. Subscripts denote amino acid positions in the substrate numbered from phosphorylation residue with increasing numbers toward the C-terminus [41, 42]. The published structure of pT160-CDK2/Cyclin A/ATP (JST) in complex with the substrate peptide comprising the optimal motif (HHASPRK; PDB Code 1QMZ; [40]) explains the basis of the peptide binding to CDK2. The substrate peptide binds in an extended conformation across the catalytic site and its binding site is located close to the active site on the C-terminal lobe surface. Some parts of the binding site are formed by the G-loop and T-loop (Figure 1). The T-loop forms a suitably shaped pocket to accept the substrate proline P1 residue, next to the phosphorylation residue (S0). The arginine R2 makes no contact with the protein and is directed to the solvent. The lysine K3 is H-bonded to the pT160 phosphate and to the Cyclin A I270 main chain oxygen. Interaction of the pT160 phosphate with lysine K3 explains the specificity for basic residues at this position. Histidine H-2 makes one H-bond from Nε to the main chain of G205 [40], the glycine of the conserved CDK motif GDSEID of the CMGC protein kinase group [17].

99
Figure 1. The picture of pT160-CDK2/Cyclin A/HHASPRK/ATP (QMZ) (coordinates taken from the PDB database: 1QMZ code) represents the CDK2/Cyclin secondary structure. The T160 (shown in black colored licorice representation) activation site is located in the T-loop. The G-loop includes two possible inhibitory sites, T14 and Y15 (black colored licorice representation). Peptide substrate (HHASPRK), and ATP-Mg2+
are shown in licorice representation.
CDK2 can be deactivated by phosphorylation of Y15 and T14 residues in the glycine-rich loop (G-loop). This process is known from kinetics experiments [48] but the structural aspects of inhibition remain unclear. Endicott and Johnson deduced with a not yet published crystal structure of the pY15,pT160-CDK2/Cyclin A/ATP (pY15-JST) complex that the phosphorylation of the Y15 residue may perturb the protein substrate binding at the catalytic site through a steric hindrance [54, 124]. The structural aspects of the inhibitory phosphorylation of the pT160-CDK2/Cyclin A/ATP (JST) complex were recently studied by molecular dynamics simulations [29], concluding that the inhibitory sites utilize a different mechanism of action. Phosphorylation of either T14 residue or both inhibitory sites’ T14 and Y15 residues together causes an ATP misalignment for phosphorylation and a G-loop conformational change, which leads to the opening of the CDK2 substrate binding box. On the other hand, the Y15 residue phosphorylation can lead to an incorrect ATP terminal phospho-group alignment for transfer to the CDK2 substrate. Consequently, we have proposed, similarly as Endicott and Johnson, that the phosphorylated Y15 residue can negatively affect substrate binding [54, 124].
The objective of this work is to study the dynamics of the QMZ system, i.e., fully active
CDK2 in complex with HHASPRK (an optimal peptide substrate), namely interactions of CDK2 with peptide substrate and the dynamics of the G-loop. These findings are also compared with the behavior of QMZ systems inhibited by phosphorylation in the G-loop.
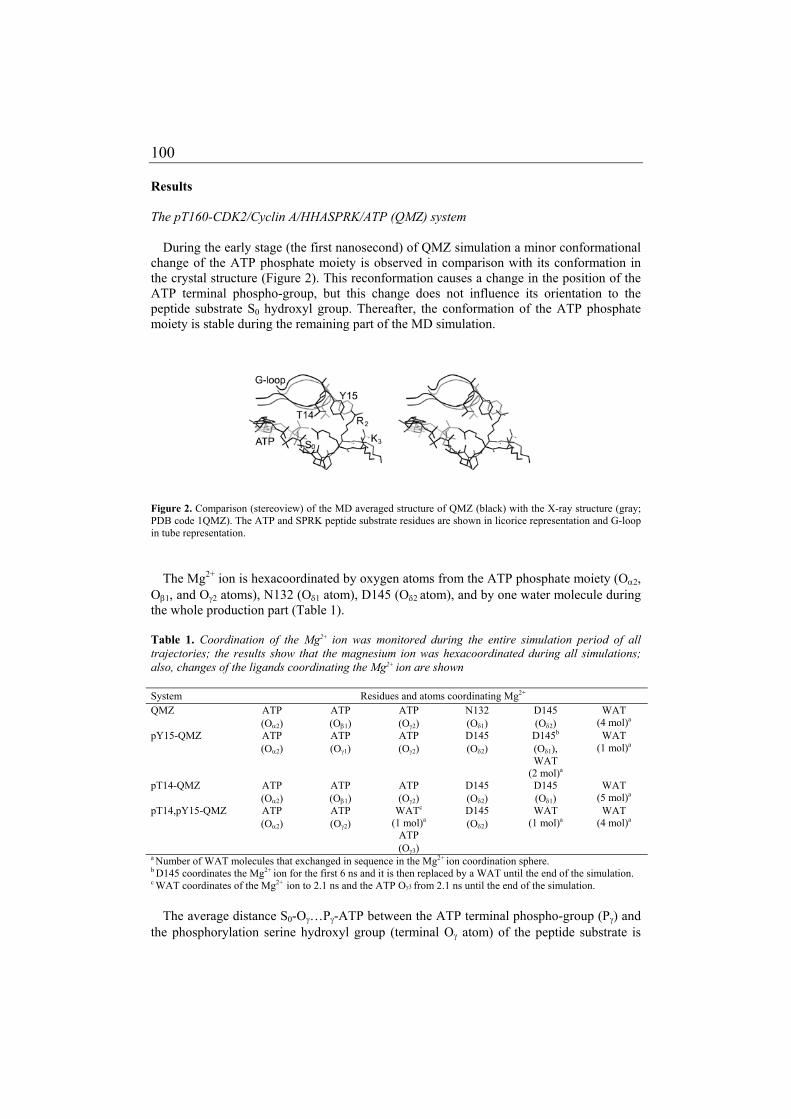
100
Results The pT160-CDK2/Cyclin A/HHASPRK/ATP (QMZ) system
During the early stage (the first nanosecond) of QMZ simulation a minor conformational change of the ATP phosphate moiety is observed in comparison with its conformation in the crystal structure (Figure 2). This reconformation causes a change in the position of the ATP terminal phospho-group, but this change does not influence its orientation to the peptide substrate S0 hydroxyl group. Thereafter, the conformation of the ATP phosphate moiety is stable during the remaining part of the MD simulation.
Figure 2. Comparison (stereoview) of the MD averaged structure of QMZ (black) with the X-ray structure (gray; PDB code 1QMZ). The ATP and SPRK peptide substrate residues are shown in licorice representation and G-loop in tube representation.
The Mg2+ ion is hexacoordinated by oxygen atoms from the ATP phosphate moiety (Oα2, Oβ1, and Oγ2 atoms), N132 (Oδ1 atom), D145 (Oδ2 atom), and by one water molecule during the whole production part (Table 1). Table 1. Coordination of the Mg2+ ion was monitored during the entire simulation period of all trajectories; the results show that the magnesium ion was hexacoordinated during all simulations; also, changes of the ligands coordinating the Mg2+ ion are shown System Residues and atoms coordinating Mg2+ QMZ ATP
(Oα2) ATP (Oβ1)
ATP (Oγ2)
N132 (Oδ1)
D145 (Oδ2)
WAT (4 mol)a
pY15-QMZ ATP (Oα2)
ATP (Oγ1)
ATP (Oγ2)
D145 (Oδ2)
D145b (Oδ1), WAT
(2 mol)a
WAT (1 mol)a
pT14-QMZ ATP (Oα2)
ATP (Oβ1)
ATP (Oγ2)
D145 (Oδ2)
D145 (Oδ1)
WAT (5 mol)a
pT14,pY15-QMZ ATP (Oα2)
ATP (Oγ2)
WATc (1 mol)a
ATP (Oγ3)
D145 (Oδ2)
WAT (1 mol)a
WAT (4 mol)a
a Number of WAT molecules that exchanged in sequence in the Mg2+ ion coordination sphere.
b D145 coordinates the Mg2+ ion for the first 6 ns and it is then replaced by a WAT until the end of the simulation. c WAT coordinates of the Mg2+ ion to 2.1 ns and the ATP Oγ3 from 2.1 ns until the end of the simulation.
The average distance S0-Oγ…Pγ-ATP between the ATP terminal phospho-group (Pγ) and the phosphorylation serine hydroxyl group (terminal Oγ atom) of the peptide substrate is

101
3.75 ± 0.70 Å, and this is close to the X-ray crystal structure value (3.7 Å). This distance sharply increases up twice to 8.5 Å (1420 – 1870 ps, 10750 – 11200 ps) due to the movement of the N-terminal substrate peptide moiety (HHASPRK) away from the substrate binding box to the solvent. When the two above periods are excluded from the calculation then the mean distance S0-Oγ…Pγ-ATP is equal to 3.63 ± 0.27 Å. Table 2 collects the lengths of occurrences of the distance S0-Oγ…Pγ-ATP below the value of 3.90 Å (obtained as sx + , i.e. 3.63 + 0.27 Å) during the simulation. The distance remains lower than the threshold during 87.7 % of simulation time. Table 2. The occurrence of distance S0-Oγ. . . Pγ-ATP below the value of 3.90 Å, and the mean value during the simulation System Distance ≤ 3.90 Å (% of time) Mean distance in Å
QMZ 87.7 3.63 ± 0.27
pY15-QMZ 9.6 8.81 ± 2.20
pT14-QMZ 5.8 6.58 ± 1.50
pT14,pY15-QMZ 18.1 4.83 ± 1.27
The V164 residue exhibits a left-handed conformation (ϕ = 60 ± 9, ψ = 130 ± 11) during the whole simulation that agrees well with the X-ray study [40]. This unfavourable conformation is stabilized by two H-bonds to R169 and R126. The K3 residue is strongly H-bonded to the pT160 phosphate group during the simulation. The R2 side chain moves towards the ATP phosphate moiety at 2.8 ns of the MD simulation (Figure 2). The H–2 makes a strong H-bond to the D206 side chain of the conserved CDK GDSEID motif.
The G-loop, bearing residues crucial for appropriate ATP alignment for phosphorylation [17], moves periodically during the QMZ simulation causing opening and closing of the substrate binding box (Figure 3). The G-loop shift is remarkable on five occasions during the simulation. Each shift away-and-back lasts approximately ~1.2-1.8 ns and occurs every 2-4 ns (Figure 4). The pT14-QMZ, pY15-QMZ, and pT14,pY15-QMZ systems
The phosphorylation at inhibitory sites in the G-loop affects the ATP phosphate moiety orientation. It causes its misalignment due to the ATP phospho-group conformational changes (the pT14-QMZ and pY15-QMZ complexes) or due to the shift of the ATP terminal phospho-group towards the ATP adenine base (the pT14,pY15-QMZ complex) (Figure 5). These changes lead to an increase in the S0-Oγ…Pγ-ATP distance and a significant decrease of the optimal distance occurrences (Table 2).

102
Figure 3. Stereoview of superimposed snapshots taken every 1 nsec from MD simulations of QMZ (A) and inhibited systems pT14-QMZ (B), pY15-QMZ (C), and pT14,pY15-QMZ (D).

103
Inhibitory phosphorylations affect also the Mg2+ ion coordination sphere (Table 1) resulting in the loss of Mg2+ ion coordination by N132 (Oδ1). This ligand is replaced by D145 (Oδ1) in pT14-QMZ during the whole simulation. In pY15-QMZ the replacement takes 6 ns, then D145 (Oδ1) is replaced by a water molecule. The phosphorylations cause N132 side chain reconformation leading to a change of the H-bonds network within the CDK2 active site. The N132 (Oδ1) creates a new H-bond to the conserved K129 (Nξ) residue and the N132 (Nδ2) makes a new H-bond to the catalytic aspartate D127 (Oδ1(δ2)) residue. The Mg2+ ion coordination by N132 (Oδ1) is replaced by an oxygen atom from the water molecule (during the whole simulation) in the pT14,pY15-QMZ system (Table 2). The Mg2+ ion coordination by water is replaced by the ATP (Oγ3) at 2.1 ns of the MD simulation in the pT14,pY15-QMZ in relation to a shift of the ATP terminal phospho-group towards the ATP adenine base.
Figure 4. The time dependence of distance between the G-loop (represented as the mass center of residues E12, G13, and T14) and the mass center of active site residues (D127, N132, and D145) for the QMZ (A) and phosphorylated systems pT14-QMZ (B), pY15-QMZ (C), and pT14,pY15-QMZ (D) simulations. Distances are calculated during equilibration and the production part of the molecular dynamics simulations.

104
Figure 5. Stereoview of the superimposition of QMZ (black) and complexes phosphorylated in the G-loop (gray); pT14-QMZ (A), pY15-QMZ (B), and pT14,pY15-QMZ (C). The ATP, phosphorylated residues (T14 and/or Y15), and SPRK peptide substrate residues are shown in licorice representation and the G-loop is shown in tube representation.
The interaction of the K3 residue with pT160 phosphate is not significantly affected by inhibitory phosphorylations in the G-loop. However, the R2 residue in pT14-QMZ moves toward the ATP phosphate moiety in the some way as in the QMZ system and its positively charged terminal group exhibits electrostatic interaction with the negatively charged pT14 terminal phospho-group. The Y15 phosphorylation causes that the R2 residue interacts strongly with pY15 phospho-group blocking the R2 motion towards the ATP (Figure 5). In

105
the pT14,pY15-QMZ system, the R2 residue forms two H-bonds to phospho-groups of both pT14 and pY15. The inhibitory phosphorylations decrease interaction between H–2 (Nδ1) and the D206 terminal carboxyl group. The phosphorylations cause a remarkable shift of the G-loop during the first 2 ns of the MD simulations in all studied systems (pT14-, pY15-, and pT14,pY15-QMZ) (Figure 4, Figure 5). The distances between mean G-loop positions (calculated for snapshots between 200ps and the end of MD simulations) in the QMZ structure and in the pT14-, pY15-, and pT14,pY15-QMZ systems were equal to 5.3, 5.5, and 6.2 Å, respectively. Discussion The pT160-CDK2/Cyclin A/HHASPRK/ATP (QMZ) system
The 15 ns long MD simulation of the QMZ system reveals some new aspects not known from the previous simulations on the JST system. Particularly, interesting is the G-loop movement that seems to be periodical. This motion is only observable at ns or longer time-scales. This is the reason why it was not detected in our previous JST simulation lasting only 3 ns [29]. The G-loop flexibility is facilitated by its primary sequence, which includes three highly conserved glycine residues (GxGxxG) in protein kinases. The GxGxxG motif is the part of the protein kinase ATP PROSITE motif (PS00107) and majority of known protein kinases bear it [17]. The primary function of each G-loop motif residue can be deduced from comparison of presented results with the most extensively investigated member of the protein kinase family, the cAMP-dependent kinase (PKA) [83]. Similarly to S53-PKA [137], the primary function of T14 is not anchoring the ATP γ-phosphate transition state but predominantly regulation, i.e., T14 serves as inhibitory site. The G11 and G13 are H-bonded with V18 and G16, respectively, making the secondary structure of the N-terminal β-sheet (β-strand-turn-β-strand) structure. Any mutation of G11 and G13 must lead to improper orientation of ATP due to sterical hindrance between side chains and ATP ribose (G11) or ATP β-phosphate (cf. [135, 143]). A mutational study results show a gradation of functional importance for glycines of the GxGxxG motif, the last G16 being the least important and the first G11 being the most important. Some amino acids introduced to G16 can be accepted due to indirect influence on ATP through interaction of their side chains with K33, but residues larger than S cannot be readily accommodated [144]. We conclude that the conserved motif GxGxxG is evolutionary optimized one because it guarantees G-loop flexibility, good accessibility of the active site and order due to formation of the secondary structure.
The HHASPRK peptide is tightly bound to the substrate binding box during the entire
simulations. While the HHA-residues are the most flexible ones, the K3 residue is very rigid, staying tightly bound to pT160 residue. The preference of CDK2 for (R/K)2 (either R or K residues at the P2 position) observed from kinetic experiments [42] cannot be deduced from the 1QMZ crystal structure [40], because R2 makes no contact with the protein, having its side chain oriented to the bulk solvent. In contrast, the MD simulation offers a simple explanation for the above preference. It is based on the idea that R2 interacts with the ATP phosphate moiety and, consequently, it can also play a role in appropriate ATP alignment before the reaction.

106
The pT14-QMZ, pY15-QMZ, and pT14,pY15-QMZ systems
Our previous study on systems inhibited by phosphorylation (pT14-, pY15-, pT14,pY15-JST) suggested a mechanism for inhibitory phosphorylation. It was proposed that inhibitory phosphorylation of either T14 residue or both inhibitory sites T14 and Y15 together, causes ATP misalignment and a G-loop shift resulting in the opening of the substrate binding box. On the other hand, Y15 inhibitory phosphorylation did not affect the G-loop position but it led to ATP phosphate moiety misalignment. It was speculated that the interaction of pY15 with (R/K)2 peptide residue may result in substrate misalignment [29].
The simulations of QMZ systems inhibited by phosphorylation show that the
phosphorylation in all cases causes ATP phosphate moiety misalignment, changes in the Mg2+ ion coordination sphere, namely the loss of N132, (a residue conserved in all protein kinases), coordination and G-loop shift away from the ATP binding site. However, it is a positive fact that the previously predicted interaction (from simulations with JST) between pY15 phospho-group and R2 residue is observed in the pY15-QMZ system. The ATP misalignment resulting in terminal phospho-group reconformation is demonstrated in Table 2 by increasing the S0-Oγ…Pγ-ATP distance. All mentioned effects clearly explain the lost of kinase activity after inhibitory phosphorylation of the CDK2 G-loop, because correct coordination of the Mg2+ ion and appropriate orientation and conformation of the ATP phosphate moiety are crucial for the phospho-group transfer to the serine S0 hydroxyl from the peptide substrate. However, it is not possible to decide whether the ATP misalignment and changes in Mg2+ ion coordination sphere are caused by G-loop shift or by electrostatic repulsion between two negatively charged phosphate groups at pT14/pY15 and ATP. It seems that both effects are involved. The insertion of one negatively charged phosphate group at T14 position causes that the R2 positively charged side chain interacts with this group and the interaction with ATP phosphates is significantly weaker. The phosphorylation of Y15 residue (or both residues altogether) causes that the R2 positively charged side chain interacts preferably with this group and the interaction with ATP phosphates is lost. On the other hand, the inhibitory phosphorylations at T14 and/or Y15 do not affect interaction of K3 with pT160 side chain.
The biological consequences and importance for CDK2 regulation of GEGTYG G-loop motif, namely the presence of TY inhibitory sites, have been sketched elsewhere [29]. The function of all GEGTYG motif residues in CDK2 can be summarized as follows: (1) G11, G13, and G16 form a structural motif ensuring the primary function, (2) T14 and Y15 serve as inhibitory sites, (3) Y15 interacts with substrate backbone and preferred basic residue at +2 substrate position. The G-loop functions, generalized for all protein kinases, may include: (1) nucleotide alignment, (2) phosphorylation site with regulatory function, (3) formation of substrate binding box, and (4) conveying specificity for phosphorylation. Materials and methods
Molecular dynamic simulations of QMZ, and QMZ phosphorylated at T14 and/or Y15 G-loop inhibitory sites pT14-QMZ, pY15-QMZ, and pT14,pY15-QMZ (cf. Table 3) were carried out using the SANDER module of the AMBER 6.0 software package [73] with the parm99 force field [72]. The starting geometries for simulation were prepared from the X-ray structure (PDB ID code: 1QMZ) and the T14, Y15, T14/Y15 residues were

107
phosphorylated ”in silico” using InsightII. The MD simulation protocol was used as follows. At first, the protonation states of histidines were checked by WHATIF [109], H-3 was ε-protonated and H-2 was double protonated to create an optimal H-bonds network. All hydrogens were added using the Xleap program from the AMBER 6.0 package. The structures were neutralized by adding 17, 15, 15, and 13 Cl– counter ions for QMZ, pT14-QMZ, pY15-QMZ, and pT14,pY15-QMZ, respectively. Each system was inserted in a rectangular water box where the layer of the water molecules was equal to 10 Å. The optional closeness parameter, which is used to control how close the solvent atoms can come to the solute atoms, was reduced from the default value of 1.0 to 0.5 Å. This parameter helps to reduce “vacuum” shell between the solute and the water box and to increase the initial density (from ~0.86 to ~0.95 g.cm–3 in our cases). Then, each system was energy minimized prior to the production part of the molecular dynamics run in the following way. The protein was frozen and the solvent molecules with counterions were allowed to move during a 1000 step minimization and a 2 ps long molecular dynamics run under NpT conditions. Then, the side chains were relaxed by several consequent minimizations with decreasing force constants applied to the backbone atoms. After the relaxation, the system was heated to 250 K during 10 ps and then to 298.15 K during 40 ps. The production parts were run for 15 ns for QMZ and 10 ns for all inhibited systems. The size of the studied systems was ~60 000 atoms. The simulation period was chosen as a compromise between the quality of configuration space sampling and the calculation length. The 2 fs time integration step and particle-mesh Ewald (PME) methods for treating electrostatic interaction were used. All simulations were run under periodic boundary conditions in the NpT ensemble at 298.16 K and at a constant pressure of 1 atm. The SHAKE algorithm with a tolerance of 10-5 Å, was applied to fix all bonds containing hydrogen atoms. The 8.0 Ǻ cutoff was applied to treat non-bonding interactions. Coordinates were stored every 2 picoseconds. All analyses of the MD simulations were carried out by the CARNAL and PTRAJ modules of AMBER-6.0 [73], by GROMACS [139], and by the program Retinal (Masaryk University, Czech Republic), for methodology see Kříž 2004 [145]. Parametrization of the phosphorylated tyrosine residue was done according to the standard Cornell et al. scheme [108] and is published elsewhere [29].
All the discussed trajectories were stable during the whole production part of the molecular dynamics (MD) simulation. The trajectory stability was monitored and confirmed by the analysis of secondary structure elements (data not present), radius of gyration, root-mean-square deviation (for backbone atoms) from the crystal structure as a function of time, and total energy (Table 3). Table 3. Summary of trajectories’ characteristics.
System T
(ns)
E
(105 kcal·mol-1)
Rg (MD)
(Å)
Rg (X-ray)
(Å)
RMSD
(Å) QMZ 15 –1.540 ± 0.001 25.99 ± 0.23 25.40 1.51 ± 0.14
pY15-QMZ 10 –1.540 ± 0.001 26.18 ± 0.24 1.72 ± 0.18
pT14-QMZ 10 –1.544 ± 0.001 25.83 ± 0.11 1.82 ± 0.22
pT14,pY15-QMZ 10 –1.542 ± 0.001 26.04 ± 0.16 1.54 ± 0.15
(T) Duration of the simulation production part. (E) Mean total energy. (Rg (MD)) Mean radius of gyration. (Rg (X-ray)) Radius of gyration of the X-ray structure. (RMSD) Root mean squared deviation of backbone atoms compared to the initial X-ray crystal structure. (QMZ) pT160-CDK2/Cyclin A/HHASPRK/ATP complex.

108
Acknowledgments We thank MetaCenter (meta.cesnet.cz) for computer time. This work was supported by the Ministry of Education of the Czech Republic (Grant LN00A016). This financial support is gratefully acknowledged. Pavel Banáš (Olomouc, CZ) is also gratefully acknowledged for phosphotyrosine parametrization. Our thanks are also addressed to R. Turland (UK) for language corrections.

109
Different Mechanisms of CDK5 and CDK2
Activation as Revealed by CDK5/p25
and CDK2/Cyclin A Dynamics
Michal Otyepka, Iveta Bártová, Zdeněk Kříž, and Jaroslav Koča
J. Biol. Chem., 2006 (accepted for publication)

110

111
DIFFERENT MECHANISMS OF CDK5 AND CDK2 ACTIVATION AS REVEALED BY CDK5/P25 AND
CDK2/CYCLIN A DYNAMICS* Michal Otyepka*1, Iveta Bártová1,2, Zdeněk Kříž2, and Jaroslav Koča*2 From the 1Department of Physical Chemistry and Center for Biomolecules and Complex Molecular Systems, Palacký University, tr. Svobody 26, 771 46 Olomouc, Czech Republic and the 2National Centre for Biomolecular Research, Faculty of Science, Masaryk University Brno, Kamenice 5, A-4, 625 00 Brno, Czech Republic.
Running Title: CDK5/p25 Binding and Dynamics Address correspondence to: Michal Otyepka, Department of Physical Chemistry and Centre for Biomolecules and Complex Molecular Systems, Palacky University, tr. Svobody 26, 771 46 Olomouc, Czech Republic, Tel. +420 5856374756; Fax. +420 585634425, E-mail: [email protected] or Jaroslav Koča, National Centre for Biomolecular Research, Masaryk University Brno, Kamenice 5, A-4, 625 00 Brno, Czech Republic, Tel. +420 549494947; Fax. +420 549492556; E-mail: [email protected].
A detailed analysis is presented of the dynamics of human CDK5 in complexes with the protein activator p25 and the purine-like inhibitor roscovitine. These, and other findings related to the activation of CDK5 are critically reviewed from a molecular perspective. In addition, the results obtained on the behavior of CDK5 are compared to data on CDK2 to assess the differences and similarities between the two kinases in terms of (i) roscovitine binding, (ii) regulatory subunit association, (iii) conformational changes in the T-loop following CDK/regulatory subunit complex formation and (iv) specificity in CDK/regulatory subunit recognition. An energy decomposition analysis, used for these purposes, revealed why the binding of p25 alone is sufficient to stabilize the extended active T-loop conformation of CDK5, whereas the equivalent conformational change in CDK2 requires both the binding of Cyclin A and phosphorylation of the Thr160 residue. The interaction energy of the CDK5 T-loop with p25 is about 26 kcal.mol-1 greater than that of the CDK2 T-loop with Cyclin A. The binding pattern between CDK5 and p25 was compared to that of CDK2/Cyclin A to find specific regions involved in CDK/regulatory subunit recognition. The analyses performed revealed that the αNT-helix of Cyclin A interacts with the α6-α7 loop and the α7 helix of CDK2, but these regions do not interact in the CDK5/p25 complex. Further differences between the CDK5/p25 and CDK2/Cyclin A systems studied are discussed with respect to their specific functionality. Cyclin-dependent kinases (CDKs) control the progression of the cell cycle [18] and participate in a subset of apoptosis programs [18, 146, 147]. CDKs consist of two subunits one catalytic and the other regulatory. The catalytic subunit is an S/T-kinase from the CMGC kinase family [17]; the associated regulatory proteins are called cyclins [18]. Cyclins are highly specific for individual kinases, resulting in the formation of distinct complexes, e.g., CDK1/Cyclin B1 and CDK2/Cyclin E1. The primary biological function of these complexes is related to regulation of the cell cycle. However, certain CDKs do not participate in cell cycle control, instead being involved in controlling cell differentiation

112
in neuronal and muscle cells. This class of CDK is exemplified by CDK5, which plays a critical role during neuronal development [60, 61, 63, 64].
Several CDKs (CDK1-4 and CDK6) show a dual mechanism of activation based on the binding of the cyclin box fold (CBF) region of the regulatory subunit to the kinase and phosphorylation of the activation loop (also known as the T-loop) of the kinase [18]. However, this mode of activation is not observed in CDK5, despite sequence identities of almost 60% for CDK2-CDK5 pairs in different species (Scheme 1). CDK5 is a unique member of the CDK family, since it is not activated by a cyclin. It binds to Cyclins D and E, but they fail to induce its kinase activity [61, 64]. Instead, CDK5 activity is triggered by p35ncka and p39nckai (henceforth referred to as p35 and p39, respectively; see Scheme 2) – proteins expressed only in neurons and a few other cell types [64, 148]. Neither p35 nor p39 exhibit any detectable similarity to cyclins. The association of CDK5 with p35 or p39 is sufficient to induce full activation of CDK5. Furthermore, CDK5 is not activated by phosphorylation of the activation loop although the loop contains a residue (Ser159, equivalent to Thr160 of CDK2) that could potentially be phosphorylated [61, 149, 150, 151], and from pers. comm. from Shin-ichi Hisanaga, Tokyo, Metropolitan University, Japan).
Scheme 1. A dendrogram of selected human cyclin-dependent kinases showing the high similarity between CDK5 and CDK2. CDKs belong to class of the CMGC group of protein kinases [17]; the complete human kinome has been published in Science [152].
Scheme 2. The activation of CDK2 involves a two-step process, i.e., cyclin binding and then phosphorylation of the activation T-loop (at Thr160). By contrast, association with either the p35 or p39 activator proteins alone is sufficient to fully induce CDK5 activity.

113
Although CDK5 is expressed in a number of tissues, its activity is restricted to neurons by the localization of its activators. Unregulated CDK5 activity has been implicated in the pathology of many neurodegenerative diseases. Deregulation of CDK5 activity is induced by the proteolytic cleavage of p35 by calpain, forming the active fragment p25. The p25 fragment induces hyperactivation of CDK5. Furthermore, unlike p35, it is not localised in the cell membrane. The CDK5/p25 complex can thus translocate from the plasma membrane to the cytosol and nucleus, where it hyperphosphorylates a number of substrates, leading to neuronal cell death [63, 148, 153]. Since the deregulation of CDK5 has been implicated in neurodegenerative diseases, there is significant interest in the development of new chemical inhibitors of CDK5 to treat these serious brain illnesses [64, 154].
Structural aspects of CDK5 activation by p25 have been revealed by analysis of the crystal structure of the CDK5/p25 complex [61]. The structure of the unphosphorylated complex confirms that the cyclin box fold (CBF) is a structural feature of p25. p25 has eight α helices, five of which (α1 to α5) adopt a topology similar to that of the CBF (Fig. 1A). The p25 CBF binds to the activation loop and the Cα helix of CDK5, the latter of which contains the PSAALRE sequence motif (residues 45-51). Overall, the structural features of the CDK5/p25 complex are similar to those of the CDK2/Cyclin A complex (Fig. 1B) [28, 61]. However, there are also some noteworthy structural and regulatory differences between the two complexes. One such difference is that even in the absence of a phosphate group on Ser159, the CDK5 activation loop adopts the correct conformation for substrate binding (a conformation typical of active proline-directed kinases) [64]. This conformation (Fig. 1C, D) resembles that observed in the fully active pT160-CDK2/Cyclin A complex [39]. Thus, p25 acts as a one-step activator in that its binding to CDK5 induces in a single step the conformational changes which CDK2 undergoes only after two independent regulatory events; Cyclin A binding and the phosphorylation of Thr160 [61, 64] (see Scheme 2).
The pThr160 (phosphothreonine) of CDK2 acts as an organizing center that interacts with three neighbouring arginine residues (Arg50, Arg126 and Arg150), stabilizing the active, extended conformation of the T-loop [39, 40]. In the CDK5/p25 complex, the structural requirement for phosphorylation is bypassed by an extensive network of interactions between p25 and the CDK5 activation loop – a feature that is not observed in the phospho-CDK2/Cyclin A complex. The phosphorylation of the activation loop (Ser159) may even have an inhibitory effect on CDK5/p35 interaction. This is suggested by the fact that substituting Ser159 with glutamic acid impairs the interaction of CDK5 with p35, and even the apparently conservative substitution of Ser159 by threonine causes a significant decrease in p35-binding ability [61], whereas the Ser159Ala-CDK5/activator complex is fully active.
The phosphorylation of Thr14 and Tyr15 in the glycine-rich loop (G-loop; residues 11-16) that forms the ‘ceiling’ of the ATP-binding site is important in the regulation of CDK activity [43]. Certain structural aspects of CDK2 were recently proposed on the basis of molecular dynamics simulations [29, 44]. Inhibitory phosphorylation of specific residues (Thr14, Tyr15) in CDK2 results in ATP misalignment and a shift of ~5 Å in the position of the G-loop, "opening" the substrate binding site. Phosphorylation of Thr14 of CDK5 by an unidentified kinase results in similar inhibition. However, this has only been observed in vitro, and the significance of Thr14 phosphorylation in vivo remains unclear. The phosphorylation of Tyr15 of CDK5, on the other hand, occurs both in vitro and in vivo. The Tyr15 is phosphorylated by Abl tyrosine kinase, and this increases CDK5 activity to some extent [61, 67, 155]. By contrast, the phosphorylation of Tyr15 and Thr14 by kinases of the

114
Wee1 family inhibits CDK1 and CDK2 [43]. It is not yet known why the phosphorylation of Tyr15 in CDK5 has an opposite effect to that of phosphorylation of the adjacent Thr14.
Fig. 1. A: stereofigure showing secondary structure elements of the CDK5/p25 complex; CDK5 (blue) and p25 (green), G-loop (red) and ‘T-loop’ (yellow). The Ser159 is shown in stick representation. B: stereofigure showing the pT160-CDK2/Cyclin A complex; CDK2 (blue), Cyclin A (green), G-loop (red), T-loop (yellow) and pThr160 (stick). C: Val163 adopts a left-handed (L) conformation in CDK5/p25 (blue). The left-handed (L) conformation, which is one of the structural aspects characterizing the active conformation of the CDK activation loop, is adopted by Val164 in the fully active form of CDK2 (pT160-CDK2/Cyclin A) (grey) in contrast to its right-handed (R) conformation in the CDK2 inactive form (CDK2/Cyclin A) (green). D: superposition of the T-loop conformation of CDK5/p25 (blue), CDK2/Cyclin A (green), pT160-CDK2/Cyclin A (grey), CDK5-Ser159 and pThr160, both shown in stick representation. The conformation of the CDK5 loop (blue) resembles that of the T-loop found in the active CDK2 form (grey). As the key regulatory enzymes of the cell cycle, CDKs have become important pharmaceutical targets for inhibitor design. The purine derivative roscovitine (2-(1-ethyl-2-hydroxy-ethylamino)-6-benzyl-amino-9-isopropylpurine; Scheme 3) has been shown to be a potent inhibitor of CDKs, with IC50 values of 0.16 µM for CDK5 [62] and 0.70 µM for CDK2 [84]. The crystal structure of the CDK2/roscovitine complex indicates that the inhibitor binds in the ATP-binding pocket of CDK2 but that its mode of binding is different from that of ATP [55, 84, 145]. Recently, Mapelli et al. [85] described the crystal structure

115
of CDK5D144N/p25/roscovitine. The (R) enantiomer (CYC202) of the inhibitor roscovitine, which is discussed in this paper, is particularly interesting because of its importance in medicinal chemistry: it is now entering phase II cancer clinical trials and phase I clinical tests against glomerulonephritis, following encouraging results from preclinical tests [82].
Scheme 3. Roscovitine structural formula, the grey labels denote atoms discussed in the results section.
METHODS
Molecular dynamics simulations of CDK5/p25 and CDK5/p25/roscovitine complexes and of versions of these complexes phosphorylated “in silico” on the Tyr15 residue of the G-loop were carried out using the program PMEMD-3.01 [156] with the parm99 force field [72]. The CDK5/p25 starting structure was taken from PDB (PDB code: 1H4L), and the starting structure for CDK5/p25/roscovitine was created by superposition of the CDK5/p25 and CDK2/roscovitine complexes. The residues not observed (residues: 1, 11-14, and 39-42) in the crystal structure (1H4L) were added to the simulated proteins using Insight II (using the CDK2/roscovitine X-ray crystal structure as a template structure [84]) and annealed and minimized before running the MD simulation protocol with constraints on those residues whose position was specified in the crystal. The missing terminal residues of CDK5 (residues 288-292) were not included in the simulations. The MD simulation protocol applied was as follows. First, the protonation states of all histidines were checked using WHATIF [109] to create an optimal H-bond network. All hydrogens were added using the Xleap program from the AMBER 6.0 package [73]. The structures were neutralized by adding 6 Cl– counter ions to the unphosphorylated systems and 4 Cl– counter ions to the systems phosphorylated at Tyr15. Each system was solvated in a rectangular water box with a layer of water molecules 10 Å thick. The energy of each system was then minimised as follows prior to the main molecular dynamics simulation run. The protein was frozen and the solvent molecules and counterions were allowed to move during a 1000 step minimization and a 2 ps long molecular dynamics run under NpT conditions. The side chains were then relaxed using several sequential minimizations with the force constants applied to the backbone atoms being decreased in each run. Following this relaxation, the

116
system was heated to 250 K for 10 ps and then to 298.15 K for 40 ps. The production phases were run for 10 ns for both unphosphorylated systems and for 5 ns for the Tyr15-phosphorylated systems. Each individual system studied comprised ~55 000 atoms. The simulation period was chosen as a compromise between the quality of configuration space sampling and the calculation length. Time integration steps of 2 fs were used, together with particle-mesh Ewald (PME) methods for electrostatic interactions. All simulations were run under periodic boundary conditions in the NpT ensemble at 298.16 K and at a constant pressure of 1 atm. The SHAKE algorithm with a tolerance of 10-5 Å was applied to fix all bonds containing hydrogen atoms. Non-bonding interactions were subject to a 10.0 Ǻ cutoff. Coordinates were stored every 2 picoseconds. All analyses of the MD simulations were carried out using the CARNAL, ANAL, and PTRAJ modules of AMBER-6.0 [73], and with GROMACS [139]. The method of Cornell et al. [108] was used for the parameterization of roscovitine and the phosphorylated tyrosine [29] and threonine residues. The results obtained were compared to previously obtained data on the fully active form of CDK2 (pT160-CDK2/Cyclin A/ATP) and the fully active form of CDK2 complexed with the peptide substrate HHASPRK (pT160-CDK2/Cyclin A/ATP/HHASPRK) [29, 44]. The results of an MD simulation of the CDK2/roscovitine complex [55] were used to compare the interactions of roscovitine with CDK5 to those with CDK2.
RESULTS
Table 1 shows global characteristics of the MD trajectories. The CDK5/p25 and CDK5/p25/roscovitine complexes were stable during the production runs of the molecular dynamics simulations, as confirmed by analysis of the root-mean-square deviation (RMS) from the X-ray crystal structure, radius of gyration (Rg), secondary structure elements (data not shown) and total energy. Table 1 Global characteristics of the trajectories from the whole production part of the MD simulations: root-mean-square deviations (RMS) of backbone atoms compared to initial X-ray crystal structures; mean radius of gyration (Rg) and mean total energy (E). Trajectory t [ns] RMS [Å]
(CDK5) RMS [Å] (G-loop)
Rg [Å] * E [105 kcal · mole-1]
Cdk5/p25 10 1.50 ± 0.15 1.26 ± 0.16 23.60 ± 0.08 -1.308 ± 0.001 Cdk5/p25/roscovitine 10 1.35 ± 0.13 1.23 ± 0.16 23.84 ± 0.08 -1.320 ± 0.001 pY15-Cdk5/p25/roscovitine † 8 1.56 ± 0.14 1.34 ± 0.10 23.94 ± 0.15 -1.310 ± 0.001 * Rg for Cdk5/p25 X-ray structure (1H4L) and CDK5/p25/roscovitine equals 23.47 A † the pY15-Cdk5/p25 trajectory was also produced for comparison
The overall protein fold was stable throughout the simulations, except for the G-loop, which relaxed in both the CDK5/p25 and CDK5/p25/roscovitine complexes at an early stage of the simulations. In both cases, the simulated relaxed G-loop conformation agreed well with that in the recently-published X-ray crystal structure of the CDK5D144N/p25/roscovitine complex [85]. The position and conformation of the G-loop were not affected by the presence of roscovitine in the active site, but the flexibility of the

117
G-loop was reduced somewhat in the presence of the inhibitor; this was confirmed by analysis of the temperature B-factors and visual inspection.
Interactions of Roscovitine with CDK5
Four hydrogen bonds (H-bonds) were observed between CDK5 and roscovitine in the CDK5/p25/roscovitine complex (Fig. 2). The network of H-bonds involves the residues Cys83 (which is involved in two H-bonds), Ile10, Glu12, and Gln130. The Cys83 amide N-H interacts with the roscovitine N7 atom, while the Cys83 carbonyl C=O forms an H-bond with the roscovitine N6 atom. In the CDK2/roscovitine complex, Leu83 also forms H-bonds with the N6 and N7 atoms of roscovitine (see Scheme 3 for atom numbering). The hydroxyl group of the chiral hydroxyethyl substituent of (R)-roscovitine is H-bonded to the Glu12 carbonyl oxygen at the beginning of the CDK5/p25/roscovitine simulation, but shifts to the Gln130 carbonyl oxygen during the course of the simulation. By contrast, in the CDK2/roscovitine simulation, the roscovitine hydroxyl group alternately formed bonds with the Ile10 carbonyl oxygen, the Asp86 Oδ atom and Gln131 carboxyl oxygen at various points during the molecular dynamics run. The amino group containing the N2 atom of (R)-roscovitine (i.e. the amino group of the C2 substituent) forms an H-bond with the Ile10 carbonyl oxygen of CDK5 (Table 2).
Fig. 2. Stereoview of roscovitine hydrogen bonds with the CDK5 active site; roscovitine is H-bonded to the G-loop (Ile10, Glu12) and the hinge region (Cys83) residues. Roscovitine, an effective purine-like inhibitor of CDK undergoing phase II of leukemia clinical trials, is positioned in the CDK5 active site but its binding mode differs from that of ATP, the 'natural' CDK substrate. Table 2 Hydrogen bonds between the roscovitine (ROC) and CDK5. Trajectory Donor Acceptor Duration
[%] Mean Distance
[Å] Mean Angle
[degree] CDK5/p25/roscovitine Cys83 N ROC N7 50.4 3.18 ± 0.15 19.78 ± 7.04 ROC N6 Cys83 O 88.6 2.92 ± 0.13 20.20 ± 5.78 ROC N2 Ile10 O 39.0 3.17 ± 0.18 14.62 ± 7.04 ROC O1 Glu12 O 15.3 2.90 ± 0.20 16.04 ± 7.13 ROC O1 Gln130 O 5.6 2.91 ± 0.20 15.11 ± 6.72 pY15-CDK5/p25/roscovitine Cys83 N ROC N7 43.7 3.24 ± 0.14 20.02 ± 7.01 ROC N6 Cys83 O 85.5 2.95 ± 0.15 20.32 ± 5.72 ROC O1 Asp86 OD1 7.4 2.78 ± 0.18 11.46 ± 6.34 ROC O1 Gln130 O 9.3 2.79 ± 0.15 14.25 ± 6.83
To quantify the interactions between roscovitine and CDK5/CDK2, the energies of interaction between the inhibitor and the respective kinase were calculated using the

118
AMBER force field (parm99) and averaged over the MD trajectories (Table 3). The AMBER force field appears, on average, to reproduce satisfactorily the interaction energies calculated using highly accurate quantum mechanical methods (MP2/Complete-Basis-Set-Limit with CCSD(T) correction) [157, 158] and is therefore a suitable method for semi-quantitative comparisons. The AMBER force field used evaluates two terms for noncovalent interactions, (i) an electrostatic term, Eels, described by Coulomb’s law and (ii) a van der Waals term, Evdw, evaluated as a Lennard-Jones potential. The van der Waals interaction term provides a good approximation of dispersion forces and repulsive interactions [159].
Fig. 3. A: plot representing contributions of different roscovitine moieties to the interaction energy between CDK5/CDK2 and the inhibitor. B: plot of the interaction energies of CDK5/CDK2 residues with roscovitine

119
showing that only ten residues contribute significantly to inhibitor binding (Etot < –1.5 kcal·mol-1). The phosphorylation of Tyr15 (CDK5) does not significantly affect the interaction of roscovitine with CDK5. IC50 values are taken from the literature (see text for detailed discussion). Asterisks denote residues at which the primary sequences of CDK5 and CDK2 differ. Table 3 The total interaction energy Etot in kcal·mol-1 and its electrostatic (Eels) and van der Waals (Evdw) components between roscovitine and CDK2/CDK5. An εr (dielectric constant) of 4 was used when calculating the electrostatic interactions. Trajectory Eels [kcal·mol-1] Evdw [kcal·mol-1] Etot [kcal·mol-1] IC50 [µM] CDK5/p25/roscovitine –4.0 ± 0.1 –50.4 ± 0.1 –54.4 ± 0.1 0.16 CDK2/roscovitine –2.8 ± 0.1
–3.5a –47.5 ± 0.1
–48.5a –50.3 ± 0.1
–52.0a
–65.8b
0.70
a from the literature [55], b from the literature [158] – MP2/aug-cc-pVDZ calculation To assess the contributions of different structural moieties of the inhibitor to the
interaction energy of the inhibitor with the CDKs, the interaction energies of seven fragments of the inhibitor were calculated (see Figure 3). The interaction patterns of roscovitine with CDK5 and CDK2 are almost identical except for two minor differences in the interactions formed by N7 and C8 (Fig. 3A). The differences in the interactions of roscovitine with CDK5 or CDK2 arise from the facts that the position of Cys83 in CDK5 is occupied by Leu83 in CDK2, and that where the inhibitor interacts with Leu133 in CDK5, it interacts with Leu134 in CDK2 (Fig. 3B).
Conformational Behavior and Energetics of pY15-CDK5
Phosphorylation of the Tyr15 residue of the G-loop of the CDK5/p25/roscovitine complex causes the position of the G-loop to shift relative to that in the unphosphorylated complex, and also increases the flexibility of the loop. The loop moves away from the roscovitine binding site, but the pTyr15 (phosphotyrosine) side chain still points towards the C2 group of roscovitine. Tyr15 phosphorylation shifts the position of the G-loop by about ~8 Å compared to that in the unphosphorylated CDK5/p25/roscovitine complex (the distance quoted is that between the relative positions of the Cα atoms of Thr14 in the two complexes) (Fig. 4A). However, Tyr15 phosphorylation in the CDK5/p25 and CDK5/p25/roscovitine complexes does not affect the secondary structure elements of CDK5 (results not shown).
The interaction energies of roscovitine with the unphosphorylated and phosphorylated CDK5/p25 complexes are equal to –54.4 ± 0.1 kcal·mol-1 and –51.4 ± 0.1 kcal·mol-1, respectively. The mean interaction energies differ by about 5.5 %, reflecting the experimental finding that Tyr15-CDK5 phosphorylation does not significantly influence roscovitine binding to CDK5 [85]. The H-bond network formed between CDK5 and roscovitine in the unphosphorylated complex is similar to that formed after Tyr15 phosphorylation.

120
Fig. 4. A: stereoview of the CDK5 G-loop shift (grey tube representation) after Tyr15 phosphorylation (thin black tube). Tyr15 and pTyr15 are shown in stick representation. B: a comparison of G-loop positions at the end of MD simulations of pT160-CDK2/Cyclin A (thick black), and pT160-CDK2/Cyclin A/HHASPRK (thick grey), where the presence of the substrate peptide causes reorientation of the Tyr15 (stick) side chain. Positions of the pTyr15 side chain do not differ in the pY15,pT160-CDK2/Cyclin A (thin black) and pY15,pT160-CDK2/Cyclin A/HHASPRK (thin grey) complexes.
Conformational Behavior and Energetics of the Activation Segment Like the X-ray structural analysis [61], analysis of the interaction energy strongly suggests that interactions with p25 effectively stabilise the active conformation of the T-loop in the CDK5/p25 complex (Fig. 5A). The mean interaction energy between T-loop residues (residues 150-165) and p25 is –86 kcal·mol-1, while the equivalent interaction energy between CDK2 and Cyclin A is lower, at –69 kcal·mol-1 and –60 kcal·mol-1 for CDK2/Cyclin A/ATP and pT160-CDK2/Cyclin A/ATP, respectively. The lower interaction energy between CDK2 and Cyclin A in the fully active CDK2 complex reflects known aspects of the stabilisation of the active T-loop conformation in CDK2. The effect of the phosphorylation of the Thr160 side chain is not to increase the strength of the interaction between CDK2 and Cyclin A, but rather to increase the strength of the interactions between T-loop residues and specific CDK2 arginine residues (Arg50, Arg150 and Arg126). The interaction energy patterns of Thr160, pThr160 (CDK2/Cyclin, pT160-CDK2/Cyclin A) and Ser159 (CDK5/p25) were further analyzed to further elucidate the mechanism of the conformational change in the T-loop. The CDK5 Ser159 forms favorable interactions with other CDK5/p25 residues, namely A160, Y158, E240, E161, R125, Y179, V162, L177, N239, C157, I241, R149, and D192 (the underlined residues come from p25). The total (non-bonded) interaction energies of Ser159 with CDK5 and p25 are –16 and –7 kcal.mol–1, respectively. The interaction energy of Thr160 with CDK2 residues is equal to –17 kcal.mol–1, while that of Thr160 with Cyclin A is negligible. The strength of the interaction between pThr160 and CDK2 is −151 kcal.mol−1, but the phosphorylation of Thr160 also gives rise to repulsive interactions with Cyclin A that sum to +28 kcal.mol–1! Thus, an energetic price is paid in converting the T-loop of CDK2 to its active conformation. The pThr160 residue also forms stabilizing interactions with the substrate peptide (HHASPRK) that sum to −36 kcal.mol–1. Specifically, there is a favourable interaction involving the K residue at the P+3 position of the peptide (numbered from the site of phosphorylation, i.e. S is at position zero, P0) equal to –24 kcal.mol–1 (Fig. 5B) [44].

121
Fig. 5. A: plot of the interaction energies of the CDK5 activation segment (T-loop) residues with the whole p25 (light grey), and the T-loop residues of pT160-CDK2 (black) or CDK2 (dark grey) with Cyclin A (cA). B: the most significant interactions between CDK5 residues and p25, and between CDK2 residues and Cyclin A (cA). In the X-ray structure, no H-bond between Ser159 and p25 was observed. The CDK5 Ser159 Oγ atom remained H-bonded to the Glu240 Oε1/2 for the entire MD simulation of the CDK5/p25 complex (Table 4). Table 4 Hydrogen bonds between the CDK5 activation segment and p25 Trajectory Donor Acceptor Duration
[%] Mean Distance
[Å] Mean Angle
[degree] CDK5/p25 Ile153 N Asn276 Oδ1 58.8 3.13 ± 0.19 11.95 ± 6.37 Cys157 Sγ Asn239 O 19.0 3.25 ± 0.13 16.29 ± 7.03 Ser159 Oγ Glu240 Oξ1 40.1 2.71 ± 0.14 10.96 ± 5.83 Ser159 Oγ Glu240 Oξ2 42.9 2.69 ± 0.14 10.54 ± 5.45 CDK5/p25/roscovitine Asn239 Nδ2 Cys157 O 49.6 2.88 ± 0.13 11.61 ± 6.10 Ser159 N Asn239 O 52.8 3.00 ± 0.17 16.02 ± 7.08
Ile153 N Asn276 Oδ1 58.9 3.15 ± 0.19 13.11 ± 6.81

122
The Ile153 backbone amide, at the bending tip of the activation loop, forms an H-bond with the Asn276 Oδ1 atom in both CDK5/p25 and CDK5/p25/roscovitine complexes. The Cys157 Sγ atom is H-bonded to the Asn239 backbone carbonyl oxygen in the CDK5/p25 complex (Fig. 6).
Fig. 6. Stereoview of the T-loop (dark grey) H-bonds with p25 (light grey) in the CDK5/p25 complex. The Ile153, Cys157, and Ser159 residues of CDK5 are H-bonded to the Asn276, Asn239, and Glu240 residues of p25 (all residues shown in stick representation), respectively.
The Val163 residue adopts a left-handed conformation with φ = 53.3° ± 14.9°, ψ = 155.9° ± 38.2° and φ = 61.8° ± 9.9°, ψ = 130.3° ± 11.6° in the CDK5/p25 and CDK5/p25/roscovitine complexes, respectively. The unusual Val conformation is stabilized by the H-bond network, in which the Val163 backbone carbonyl oxygen forms an H-bond with the guanidine group of the Arg168 side chain (NH1 group) while the Val163 backbone amide forms an H-bond with the Arg125 backbone carbonyl oxygen in the CDK5/p25 complex. The left-handed Val163 conformation is also stabilized by two H-bonds with the Arg125 and Arg168 side chains in the CDK5/p25/roscovitine complex. The left-handed Val164-CDK2 conformation is one of the two characteristic structural features of the active conformation of the activation loop [39]. The conformation of Val163 in the CDK5/p25 complex parallels that of the Val164 conformation in pT160-CDK2/Cyclin A, i.e., it adopts the conformation of the fully activated complex (Fig. 1C).
The Ser159 residue in the activation loop of CDK5 is a potential phosphorylation site, comparable to Thr160 in the T-loop of CDK2. Differences in the positioning of the Thr160 residues of the various CDK2 complexes relative to that of Ser159 in CDK5/p25 were assessed by superimposing the averaged structure of each protein obtained at the end of their respective MD runs and then calculating the distance between the Cα atoms of the relevant residues in each complex. The separations thus obtained were 7.5 Å, 2.9 Å, and 1.9 Å, respectively, for CDK2/Cyclin A, pT160-CDK2/Cyclin A, and pT160-CDK2/Cyclin A/HHASPRK. The presence of roscovitine in the active site of CDK5 changed these values to 6.3 Å, 2.0 Å, and 1.9 Å, respectively. From these data, it seems that the conformation of Ser159 in CDK5/p25 most closely matches that of the pThr160 side chain in the fully active CDK2 complex.

123
CDK5-p25 Interactions; Kinase/Cyclin Recognition Energy decomposition analysis was also used to determine the extent to which different regions of the complex contributed to the interaction energy between CDK5 and p25, and, for comparative purposes, to that between CDK2 and Cyclin A (see Supplementary Material). Such detailed analysis of interaction patterns can provide useful insights into the origins of the specificity of the binding between the CDK and the regulatory subunit, which remains a mystery of cell biology. The key structural elements of CDK5 in terms of p25 binding were found to be the Cα-helix (the PSTAIRE helix) and the following loops: Cα-β4 (residues 50-60), β4-β5 (residues 71-75), α3-β6 (residues 116-123), the T-loop (residues 150-161), and the loop after the short α4-helix (residues 177-181). The key binding elements of p25 were found to be the α1-α2 and α3-α4 loops, and the α3 (C-terminus), α4, α5, and α6 helices. Similar regions were identified in the CDK2/Cyclin A complex. In general, the regions of CDK2 that interact with Cyclin A are similar to those of CDK5 that interact with p25 (cf. Fig. 5B); in addition to these, it was found that the α6-α7 loop and α7 helix of CDK2 forms significant interactions with the αNT-helix of Cyclin A. As expected, more significant differences were observed between Cyclin A and p25. The αNT, α2 (N-terminus), α3, α5, and α6 helices of Cyclin A contribute significantly to its interactions with CDK2, as do the α2-α3, α3-α4, and α5-α6 loops (Fig. 7). These findings broaden and quantify previously published experimental data suggesting that residues 150-200 of p25 are responsible for CDK5 binding and, in addition, residues 279-291 are required for CDK5 activation [149].
Figure 5B highlights several significant differences in the interactions between residues of CDK5 or CDK2 and those of p25 and Cyclin A, respectively. It was found that residues Glu57, His71, and Lys177 of CDK5 formed more favorable interactions with p25 than the corresponding residues of CDK2 did with Cyclin A. On the other hand, the Asp73 and Asn121 residues of CDK5 interacted more favourably with one another than did the equivalent residues in the CDK2/Cyclin A complex. Glu57 (CDK5) makes a favorable contact with the close α6 helix of p25, while in CDK2 Glu57 binds to Arg122 (mutation in CDK5 has replaced this residue with Asn121), but its interaction with Cyclin A is weak. His71 (CDK5) forms a salt bridge with the residues Asp259 or Glu255 in the N-terminal part of the α5 helix of p25, but there is no equivalent salt bridge acceptor in the CDK2/Cyclin A complex. The Asp73 (Glu73 in CDK2) repels the acidic residues of the N-terminal part of α5 of p25. In contrast, the corresponding part of α5 (CDK2) contains basic residues (KKQVLRMEHLVLK). The Lys177 side chain interacts with the T-loop backbone in CDK5 but not in CDK2. The strong attraction between Arg122 (CDK2) and Cyclin A is probably caused by favorable electrostatic interactions with acidic residues of the αNT-helix. Such interactions are not observed in CDK5 due to the Arg/Asn mutation in its primary sequence. A difference in the interactions of CDK5 with p25 and of CDK2 with Cyclin A centres on the Arg50 residue, which is discussed in some detail in Barrett’s recent article [160]. In the fully-activated CDK2 complex, Arg50 (together with Arg126 and Arg150) 'anchors' pThr160. In the CDK2/Cyclin A complex, it interacts with the α3-α4 loop of Cyclin A; the magnitude of this interaction is reduced in the pT160-CDK2/Cyclin A complex relative to that in unphosphorylated CDK2/Cyclin A (cf. Fig. 5B). It should be noted that while our investigations did not suggest there to be any differences between the interaction energies of the three arginine residues (50, 126, 150) with pThr160, such differences are discussed in Barrett’s article [160]. The interaction energies between pThr160 (Thr160 in the unphosphorylated complex) and Arg50, Arg126, and Arg150 were, respectively, –41.5, –40.9, and –42.7 kcal.mol–1 in pT160-CDK2/Cyclin A; –40.8, –41.3,

124
and –42.8 kcal.mol–1 in pT160-CDK2/Cyclin A/HHASPRK; and 0.0, –0.2, –0.1 kcal.mol–1
in CDK2/Cyclin A.
Fig. 7. Comparison of the interaction energies of CDK5 and p25 (part A) residues with the interaction energies of CDK2 with residues of Cyclin A (part B) identifies subunit spots responsible for interaction.
DISCUSSION
CDK5/CDK2-Roscovitine Interactions Roscovitine belongs to the class of very effective, selective, and therapeutically potent purine-like inhibitors of CDKs [55, 82, 85] and competes with ATP for occupation of the CDK active site. Roscovitine binds in the same position and orientation to both CDK5 and CDK2, and its position during MD simulations does not significantly differ from that implied by the crystallographic data. The mean CDK5/roscovitine and CDK2/roscovitine interaction energies are equal to –54.4 ± 0.1 and –50.3 ± 0.1 kcal.mol–1, respectively. These values are consistent with roscovitine's IC50 values of 0.16 µM for CDK5 and 0.70 µM for CDK2. Energy decomposition analysis was used to quantify differences between the interaction footprints of the inhibitor with CDK5 and CDK2. The mean interaction energies differ significantly only for Cys83 and Leu133 (which correspond to Leu83 and Leu134, respectively, in CDK2). The difference between Cys83/Leu83 is attributed to the different electrostatic properties of these residues, while the difference between interactions with

125
Leu133 (CDK5) and Leu134 (CDK2) arises from the van der Waals term. The interaction energies for these residues with roscovitine are –4.2 kcal·mol-1 and –3.3 kcal·mol-1 for Cys83 (CDK5) and Leu83 (CDK2) respectively, while those for Leu133 (CDK5) and Leu134 (CDK2) are –5.4 kcal·mol-1 and –4.2 kcal·mol-1, respectively. In both cases, the energies of interaction between roscovitine and CDK2 are lower than those with CDK5 due to the differences in side chain conformations between the two proteins; in CDK2, the side chains are directed away from the bound roscovitine. The van der Waals contributions to the interaction energy suggest that roscovitine fits into the active site of CDK5 better than it does in that of CDK2 because the van der Waals interaction energy between CDK5 and roscovitine is 2.9 kcal.mol-1 lower than that between CDK2 and roscovitine (Table 3). In addition, as also shown in Table 3, van der Waals (i.e. dispersion) interactions are responsible for about 94% of the total interaction energy of roscovitine with CDK2/CDK5. This finding illustrates the importance of the dispersion energy in the binding of purine-like inhibitors to CDKs, and is in agreement with recent correlated ab-initio calculations [158].
An understanding of the interaction energy pattern is essential in rational drug design and is useful in the design of new, more selective inhibitors. In this context, it is surprising that only ten residues (I10, V18, F80, F82, L/C83, H/D84, Q85, D86, Q131/130, L133/134) contribute significantly to the total interaction energy of roscovitine with CDK2/CDK5 and that contributions from other residues are negligible. This finding suggests that it may be necessary to force new inhibitors to interact with these “unimportant” residues in order to obtain stronger and more selective binding. Such forcing may be achieved by synthesising roscovitine derivatives with extended (longer and bulkier) substituents. From another perspective, the interaction energy patterns for roscovitine with CDK5 are not significantly different to those with CDK2, implying that the inhibitor will exhibit very limited selectivity between the two CDKs. This opens up the possibility of designing purine derivatives with longer substituents capable of binding to the kinases via different binding modes or even using different structures, in order to increase their selectivity for CDK5 or CDK2. An excellent study describing the use of approaches such as those discussed above for the rational design of potent and selective (non-purine) compounds targeting CDK5 has been recently published [161].
G-loop Dynamics and Interactions A G-G--G consensus sequence pattern has been identified in proteins with a preference for dinucleotide binding [162, 163] and this motif is one of the most highly conserved in protein kinases [17]. The functionality of the motif in protein kinases has been investigated in several studies [44, 137, 164]. Phosphorylation of the Thr and/or Tyr residues of the G-loop depresses CDK1 and CDK2 functionality; by contrast, Tyr15 phosphorylation increases CDK5/p35 activity [67]. The structural mechanism of activation by Tyr15 has been considered by Mapelli et al. [85] in the context of roscovitine binding. Simulations of the pY15-CDK5/p25/roscovitine complex suggest that Tyr15 phosphorylation leads to a G-loop shift which results in the side chain of pTyr15 being directed towards the bound roscovitine (Fig. 4A). Analysis of interaction energies between roscovitine and CDK5/pY15-CDK5 reveals that the influence of Tyr15 phosphorylation on roscovitine binding is almost negligible (Fig. 4B), a finding supported by experimental observations [85]. Tyr15 phosphorylation in CDK2 results in the phosphate group being exposed to the solvent, along with a shift of the G-loop [44]. The binding of the substrate peptide also prompts changes in the conformations of the Tyr15 side chain (cf. Fig. 4B). However, these results do not explain why phosphorylation is inhibitory in CDK2 but stimulatory in CDK5. Since molecular dynamics simulations of the protein alone have not provided any clear

126
explanations of this phenomenon, it has been suggested that it may be necessary to consider the effects of phosphorylation in the presence of the substrate. CDK2 prefers basic substrates at the P+3 position (S/TPRK is the favored substrate motif [42]); the origins of this preference were revealed with the publication of the crystal structure of the pT160-CDK2/Cyclin A/HHASPRK complex. The structure provided evidence of a direct interaction between K3 and pThr160 [40], the magnitude of which was found to be –24 kcal.mol–1 [44]. CDK5 also exhibits a preference for basic residues at the P+2-P+4 positions despite the lack of phosphorylation on Ser159 [165-167]. It has been proposed that the Glu240 residue of p25 interacts with basic P+3 substrate residues, which fulfil the role of pThr160 in CDK2 [64], but analysis of a superposition of CDK5/p25 and pT160-CDK2/CyclinA/HHASPRK suggested that interactions with two additional acidic residues, Glu161 (the Glu161Ala mutation leads to a loss of CDK5 activity towards H1 derived peptide [167]), and Glu42 (CDK5), should also be taken into account. Consequently, pTyr15 may serve as an ideal partner interacting with the basic substrate residues. This hypothesis is also supported by the fact that a direct interaction between R+2 and pTyr15 has been observed in simulations of pY15,pT160-CDK2/Cyclin A/HHASRPK complex [44] and the presence of peptide substrates results in changes in the conformation of the Tyr15 side chain, as observed in X-ray structures [40] (Fig. 4B).
The experimental results suggest that phosphorylation of Thr14 is inhibitory in CDK5, while the phosphorylation of Tyr15 is stimulatory [67]. Activation by Tyr15 phosphorylation is discussed above. By contrast, the mechanism of inhibition by Thr14 phosphorylation may be similar to the mechanism of CDK2 inhibition by Thr14 phosphorylation, which results in significant misalignment of ATP in the active site due to electrostatic repulsion between two adjacent negatively charged groups, i.e., the phosphate moiety of ATP and that of pThr14 [29, 44]. Consequently, the misaligned ATP γ-phosphate cannot be transferred to the substrate Ser/Thr.
T-loop Dynamics and Interactions In spite of the absence of a phosphate group on Ser159, the CDK5 activation loop (T-loop) adopts an extended conformation typical of active proline-directed kinases. This conformation is almost identical to that observed in the fully active CDK2 (pT160-CDK2/Cyclin A/HHASPRK) complex (Fig. 1D). Superposition of the averaged structures of the fully activated CDK2/Cyclin A/HHASPRK complex and the CDK5/p25 complex obtained during MD simulations reveals that the Cα atom of Ser159 is only 1.9 Å away from the Cα atom of pThr160 in the pT160-CDK2/Cyclin A/HHASPRK complex. The Val163 residue of CDK5/p25 adopts a left-handed orientation typical of the active loop conformation (Fig. 1C). These structural aspects imply that CDK5 may be activated without phosphorylation of the activation loop, although its activation loop contains a potential phosphorylation site in Ser159.
The binding patterns of Thr160, pThr160 (CDK2) and Ser159 (CDK5) were analyzed in order to determine why T-loop phosphorylation is necessary for CDK2 to adopt the active T-loop conformation. The interaction energy between Ser159 and other residues of CDK5 and p25 is equal to –16 and –7 kcal.mol–1, respectively. That between Thr160 and other CDK2 residues is –17 kcal.mol–1, but the interaction energy of Thr160 with Cyclin A residues is 0 kcal.mol–1. Phosphorylation of Thr160 increases the Thr160/CDK2 interaction energy to –151 kcal.mol–1, but also results in a repulsive interaction between Thr160 and Cyclin A of +28 kcal.mol–1. The interaction energy between the whole T-loop (residues 150-165) of CDK5 and p25 is –86 kcal·mol-1 while the equivalent interaction energy between CDK2 and Cyclin A is −69 kcal·mol−1, and –60 kcal·mol−1 in the cases

127
of CDK2/Cyclin A/ATP and pT160-CDK2/Cyclin A/ATP, respectively. These results support the conclusion drawn from studies of the complexes' X-ray structures, namely that pThr160 (CDK2) acts as an organizing center, interacting with three neighboring Arg residues (Arg50, Arg126 and Arg150) and stabilizes the active, extended conformation of the T-loop [39, 40]. Furthermore, the results reported in this study enable these interactions to be quantified. It is also observed that pThr160 forms stabilizing interactions with the substrate peptide (HHASPRK), and that the magnitude of these interactions is equal to −36 kcal.mol–1. The interaction with the P+3 residue is particularly noteworthy, contributing –24 kcal.mol–1 [44]. The −17 kcal.mol–1 difference in interaction energy between CDK5/p25 and CDK2/Cyclin A is sufficient to stabilize the active conformation of the T-loop in the former complex. This fact illustrates the complex interplay between CDK/regulatory subunit recognition and regulation of CDK activity. Analysis of the interaction energy between CDKs and regulatory subunits reveals considerable differences between CDK5 and CDK2, with Ile153, Cys157, and Ser159 forming H-bonds to p25 in the former case (Fig. 6). However, in general, it is not easy to relate differences in interaction patterns to one or a few residues, and consequently it seems that the processes and interactions underpinning CDK/regulatory subunit recognition and specificity are complex and cooperative.
Insofar as we are aware, there is no evidence suggesting that CDK5 activity requires the phosphorylation of Ser159. However, a simulation of the pS159-CDK5/p25 complex was performed to investigate the extent (if any) to which phosphorylation affects the structure of the activation segment. Ser159 was phosphorylated in silico, using the conformation of pThr160 (CDK2) as a starting point for the pS159-CDK5 complex, with the phosphate group directed towards three anchoring arginines. No significant changes in structure were observed during the simulation and the activation segment retained the conformation observed in the CDK5/p25 complex. However, decomposition analysis suggested that the interaction energy profile of pS159-CDK5/p25 resembles that of pT160-CDK2/Cyclin A in that phosphorylation of Ser159 reduces the strength of the binding interactions with p25 but compensates by forming favorable interactions with three anchoring arginines (Arg50, Agr127 and Arg151).
CDK5/p25 and CDK2/Cyclin A Interaction Patterns
The energy decomposition analysis revealed some important differences in the interactions between CDK5 and p25 and those between CDK2 and Cyclin A. These differences may underpin the preferential binding of CDK5 to p25 and that of CDK2 to Cyclins A or E. The surface of p25 interacting with CDK5 is smaller than the surface of Cyclin A interacting with CDK2, due to the different positions of the αNT-helix in p25 and Cyclin A (Fig. 8A-E). The αNT-helix of Cyclin A contributes significantly to its interaction energy with CDK2, and thus plays a significant role in CDK/Cyclin recognition. By contrast, the surface of p25 in contact with the T-loop adopts a different shape to that of Cyclin A, and seems to be more negatively charged (Fig. 8D-E). Certain specific interactions with p25 that stabilise the T-loop of CDK5 in its active conformation are discussed above, but in general adoption of the active T-loop conformation appears to be a consequence of cooperative interactions of all the T-loop residues with the p25 surface, which provides a good geometric match for the active conformation of the T-loop (Fig. 5A). The p25 and Cyclin A surfaces also differ in the N-terminal parts of the α5-helix, which is positively charged in the case of Cyclin A (as mentioned in the results section) but which bears a negative charge (in the form of the Glu255 residue) in p25 (Fig. 8D-E). The CDK5 and CDK2 surfaces that interact with p25 or Cyclin A (Fig. 8F-H) are, as expected, very

128
similar. One significant difference between the two is highlighted in Fig. 8 – Lys278 in CDK2 is positively charged (indicated by a yellow circle) and forms a salt bridge with the αNT-helix of Cyclin A. The same region of CDK5 contains two acidic residues, Glu278 and Glu279, and is thus negatively charged. All these differences are important in CDK/Cyclin recognition. A more extensive study will be necessary to identify all the regions involved in CDK/Cyclin recognition and specificity. Unfortunately, little is known about the structures of some biologically-important complexes, e.g. CDK1, and this makes such studies very difficult to perform. However, there has recently been some encouraging progress in this direction (e.g. the determination of the CDK2/Cyclin E structure [168]). In this context, further encouragement may be derived from the fact that the interaction maps for CDK/Cyclin complexes calculated from minimized X-ray structures agree well with maps constructed using data from averaged MD simulations (results not shown).
CDK2/Cyclin A substrate specificity is conferred in part by a recruitment site [169, 170] localized on the Cyclin A surface. The comparison of the CDK5/p25 and CDK2/Cyclin A surfaces in Figure 8 (I-K) shows differences between both complexes. The CDK2 recruitment site touches two spots, which are oppositely charged (Fig. 8K), but the region of p25 thought to correspond to this site is predominantly negatively charged. Such differences contribute to substrate discrimination and give rise to CDK/Cyclin specific functionality, i.e., to the different biological roles of specific complexes [171].

129
A
D
F
I
5
α
αNT
α5
α6
α1
α2
α3
α4α7
B
G
J
0
T-loop
αNT
C
E
H
K
9
α1
α5
α2
α3α4
recruit.O
K28
α1-β4-loop β4-β5-loopE25
C
α6-α7-loop
α7α2-loop
E24

130
Fig. 8. Differences between the CDK5/p25 and CDK2/Cyclin A interactions. A: p25 (red; showing its α-helices), B: superposition of p25 and Cyclin A in the configurations they adopt when bound to CDK5 and CDK2, respectively. C: Cyclin A, showing its α-helices. D: p25 electrostatic surface (negatively and positively charged regions are indicated in red and blue, respectively) with CDK5 (green) and CDK2 (orange) regions (shown by labels) with significant interaction with CDK, the positions of Glu240 and Glu255 residues of p25 are shown by bold labels, the positions of p25 or Cyclin A residues emphasize the regions where the electrostatic potentials of p25 and Cyclin A are notably different, E: Cyclin A electrostatic surface showing regions that significantly interact with CDK5 (green) and CDK2 (orange), and the position of its Lys289 residue. F: Electrostatic surface of CDK5 in the same orientation as in G, where CDK5 (green) and CDK2 (orange) are superimposed. H: electrostatic surface of CDK2 in the same orientation as in G; the yellow circle denotes the spot where CDK2 interacts with the αNT-helix and the preceding loop. The last three images show: I, the electrostatic surface of the CDK5/p25 complex in the same orientation as in J, in which CDK5/p25 and CDK2/Cyclin A are superimposed, and (K) electrostatic surface of the CDK2/Cyclin A complex with a small yellow coil showing the position of a peptide in the Cyclin A recruitment site [169].
FOOTNOTES
*The CDK5 activation loop corresponding to the T-loop of CDK2 is referred to as the T-loop in this article even though it contains Ser instead of Thr at the phosphorylation site.
Used abbreviations: CDK – Cyclin-Dependent Kinase, CMGC – a class of protein kinases according Hanks and Quinn nomenclature [17] containing Cyclin-dependent kinases, Mitogen-activated protein kinases, Glycogen synthase kinases and CDK-like kinases, CBF – cyclin box fold, MD – molecular dynamics.
The authors thank the Ministry of Education, Youth and Sports (projects MSM0021622413 (IB, ZK, and JK), MSM6198959216 (MO), and LC512 (MO)) for financial support. We also thank MetaCenter (meta.cesnet.cz) for computer time, R. Turland (UK), J. and D. Blackwell (UK) for linguistic corrections and M. Strnad (CZ) for valuable discussions.

131
SUPPLEMENTARY MATERIAL
Interaction energy matrix for all the residues of CDK5/p25 and CDK2/Cyclin A as averaged during MD simulations. The most significant regions discussed in this article are highlighted. The black/gray points indicate favorable interactions between corresponding residues. This matrix is related to Figs. 5 and 7, representing row and column sums, and Fig. 8 where the electrostatics are projected onto the structures’ surfaces.

132

133
Bibliography
1. Tyson, J.J. and Novak, B., Regulation of the eukaryotic cell cycle:
molecular antagonism, hysteresis, and irreversible transitions. Journal of Theoretical Biology, 2001, 210: 249-263.
2. Mitchison, T.J. and Salmon, E.D., Mitosis: a history of division. Nature Cell Biology, 2001, 3: E17-E21.
3. Cooper, S., On the proposal of a G0 phase and the restriction point. FASEB Journal, 1998, 12: 367-373.
4. Tyers, M., Cell cycle goes global. Current Opinion in Cell Biology, 2004, 16: 602-613.
5. Nurse, P. and Bissett, Y., Gene required in G1 for commitment to cell cycle and in G2 for control of mitosis in fission yeast. Nature, 1981, 292: 558 - 560.
6. Lee, M.G. and Nurse, P., Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature, 1987, 327: 31 - 35.
7. Evans, T., Rosenthal, E.T., Youngblom, J., Distel, D., and Hunt, T., Cyclin: A Protein Specified by Maternal mRNA in Sea Urchin Eggs That Is Destroyed at Each Cleavage Division. Cell, 1983, 33: 389-396.
8. Lew, J., MAP Kinases and CDKs: Kinetic Basis for Catalytic Activation. Biochemistry, 2003, 42: 849-856.
9. Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P., Molecular Biology of the Cell. 2002: Garland Science.
10. Manning, G., Whyte, D.B., Martinez, R., Hunter, T., and Sudarsanam, S., The Protein Kinase Complement of the Human Genome. Science, 2002, 298: 1912-1934.
11. Human Protein Kinases, Cell Signaling Technology, Inc., http://www.cellsignal.com/reference/kinase/tree.asp.
12. Cohen, P., The development and therapeutic potential of protein kinase inhibitors. Current Opinion in Chemical Biology, 1999, 3: 459-465.
13. Fischer, P.M., The Design of Drug Candidate Molecules as Selective Inhibitors of Therapeutically Relevant Protein Kinases. Current Medicinal Chemistry, 2004, 11: 1563-1583.
14. Engh, R.A. and Bossemeyer, D., The protein kinase activity modulation sites: : mechanisms for cellular regulation — targets for therapeutic intervention. Advances in Enzyme Regulation, 2001, 41: 121-149.
15. Fischer, E.H. and Krebs, E.G., Conversion of phosphorylase b to phosphorylase a in muscle extracts. Journal of Biological Chemistry, 1955, 216: 121-132.
16. Johnson, L.N., Lowe, E.D., Noble, M.E.M., and Owen, D.J., The structural basis for substrate recognition and control by protein kinases. FEBS Letters, 1998, 430: 1-11.

134
17. Hanks, S. and Quinn, A.M., Protein Kinase Catalytic Domain Sequence Database: Identification of Conserved Features of Primary Structure and Classification of Family Members. Methods in Enzymology, 1991, 200: 38-62.
18. Morgan, D.O., Cyclin-dependent Kinases: Engines, Clocks, and Microprocessors. Annual Review of Cell and Developmental Biology, 1997, 13: 261-291.
19. Cancer, http://www.fhcrc.org/education/courses/cancer_course/basic/img/regulation.gif.
20. Kaldiss, P., Sutton, A., and Solomon, M.J., The Cdk-activating kinase (CAK) from budding yeast. Cell, 1996, 86: 553-564.
21. Feaver, W.J., Svejstrup, J.Q., Henry, N.L., and Kornberg, R.D., Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK. Cell, 1994, 79: 1103-1109.
22. Serizawa, H., Makela, T.P., Conaway, J.W., Weinberg, R.A., and Young, R.A., Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature, 1995, 374: 280-282.
23. Sausville, E.A., Complexities in the development of cyclin-dependent kinase inhibitor drugs. Trends in Molecular Medicine, 2002, 8: S32-S37.
24. Ohshima, T., Ward, J.M., Huh, C.G., and Longenecker, G., Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proceedings of the National Academy of Sciences USA, 1996, 93: 11173-11178.
25. Chae, T., Kwon, Y.T., Bronson, R., Dikkes, P., Li, E., and Tsai, L.H., Mice lacking p35, a neuronal-specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron, 1997, 18: 29-42.
26. Knockaert, M., Greengard, P., and Meijer, L., Pharmacological inhibitors of cyclin-dependent kinases. Trends in Pharmacological Sciences, 2002, 23: 417-425.
27. De Bondt, H.L., Rosenblatt, J., Jancarik, J., Jones, H.D., Morgan, D.O., and Kim, S.H., Crystal structure of cyclin-dependent kinase 2. Nature, 1993, 363: 595-602.
28. Jeffrey, P.D., Russo, A.A., Polyak, K., Gibbs, E., Hurwitz, J., Massague, J., and Pavletich, N.P., Mechanism of cdk activation revealed by the structure of a cyclin A - cdk2 complex. Nature, 1995, 376: 313-320.
29. Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., Activation and Inhibition of Cyclin-Dependent Kinase-2 by Phosphorylation; A Molecular Dynamics Study Reveals the Functional Importance of the Glycine-Rich Loop. Protein Science, 2004, 13: 1449-1457.
30. Ferby, I., Blazquez, M., Palmer, A., and Eritja, R., A novel p34(cdc2)-binding and activating protein that is necessary and sufficient to trigger G(2)/M progression in Xenopus oocytes. Genes and Development, 1999, 13: 2177-2189.

135
31. Lenormand, J.L., Dellinger, R.W., Knudsen, K.E., Subramani, S., and Donoghue, D.J., Speedy: a novel cell cycle regulator of the G2/M transition. EMBO Journal, 1999, 18: 1869-1877.
32. Porter, L.A., Kong-Beltran, M., and Donoghue, D.J., Spy1 interacts with p27Kip1 to allow G1/S progression. Molecular Biology of the Cell, 2003, 14: 3664-3674.
33. Hagopian, J.C., Kirtley, M.P., and Stevenson, L.M., Kinetic Basis for Activation of CDK2/Cyclin A by Phosphorylation. Journal of Biological Chemistry, 2001, 276: 275-280.
34. Minshull, J., Pines, J., Golsteyn, R., Standart, N., Mackie, S., Colman, A., Blow, J., Ruderman, J.V., Wu, M., and Hunt, T., The role of cyclin synthesis, modification and destruction in the control of cell division. Journal of Cell Biology, 1989, 12: 77-97.
35. Kobayashi, H., Stewart, E., Poon, R., Adamczewski, J.P., Gannon, J., and Hunt, T., Identification of the domains in cyclin A required for binding to, and activation of, p34cdc2 and p32cdk2 protein kinase subunits. Molecular Biology of the Cell, 1992, 3: 1279-1294.
36. Brown, N.R., Noble, M.E.M., Endicott, J.A., Garman, E.F., Wakatsuki, S., Mitchell, E., Rasmussen, B., Hunt, T., and Johnson, L.N., The crystal structure of cyclin A. Structure, 1995, 3: 1235-1247.
37. Brown, N.R., Noble, M.E.M., Lawrie, A.M., Morris, M.C., Tunnah, P., Divita, G., Johnson, L.N., and Endicott, J.A., Effects of Phosphorylation of Threonine 160 on Cyclin-dependent Kinase 2 Structure and Activity. Journal of Biological Chemistry, 1999, 274: 8746-8756.
38. Morris, M.C., Gondeau, C., Tainer, J.A., and Divita, G., Kinetic Mechanism of Activation of the Cdk2/Cyclin A Complex. Journal of Biological Chemistry, 2002, 277: 23847-23853.
39. Russo, A.A., Jeffrey, P.D., and Pavletich, N.P., Structural basis of cyclin-dependent kinase 2 activation by phosphorylation. Nature Structural Biology, 1996, 3: 696-700.
40. Brown, N.R., Noble, M.E.M., Endicott, J.A., and Johnson, L.N., The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nature Cell Biology, 1999, 1: 438-443.
41. Songyang, Z., Blechner, S., Hoagland, N., Hoekstra, M.F., Piwnica- Worms, H., and Cantley, L.C., Use of an oriented peptide library to determine the optimal substrates of protein kinases. Current Biology, 1994, 4: 973-982.
42. Holmes, J.K. and Solomon, M.K., A predictive Scale for Evaluation Cyclin-dependent Kinase Substrates; A comparison of p34cdc2 and p33cdk2. Journal of Biological Chemistry, 1996, 271: 25240-25246.
43. Morgan, D.O., Principles of CDK regulation. Nature, 1995, 374: 131-134. 44. Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., The mechanism of
inhibition of the cyclin-dependent kinase-2 as revealed by the molecular dynamics study on the complex CDK2 with the peptide substrate HHASPRK. Protein Science, 2005, 14: 445-451.

136
45. Watanabe, N., Broome, M., and Hunter, T., Regulation of the human Wee1Hu Cdk tyrosine 15-kinase during the cell-cycle. EMBO Journal, 1995, 14: 1878-1891.
46. Sebastian, B., Kakizuka, A., and Hunter, T., Cdc25M2 activation of cyclin-dependent kinases by dephosphorylation of threonine-14 and tyrosine-15. Proceedings of the National Academy of Sciences USA, 1993, 90: 3521-3524.
47. Rudolph, J., Epstein, D.M., Parker, L., and Eckstein, J., Specifity of Natural and Artificial Substrates for Human CDC25A. Analytical Biochemistry, 2001, 289: 43-51.
48. Coulonval, K., Bockstaele, L., Paternot, S., and Roger, P.P., Phosphorylations of cyclin-dependent kinase 2 revisited using two-dimensional gel electrophoresis. Journal of Biological Chemistry, 2003, 278: 52052-52061.
49. Shintania, S., Mihara, M., Nakahara, Y., Kiyota, A., Ueyama, Y., Matsumura, T., and Wong, D.T.W., Expression of cell cycle control proteins in normal epithelium, premalignant and malignant lesions of oral cavity. Oral Oncology, 2002, 38: 235–243.
50. Go, J.H., Expressions of the CIP/KIP Family of CDK Inhibitor Proteins in Primary Intestinal Large B-cell Lymphomas: Correlation with Clinical Outcomes. Pathology - Research and Practice, 2002, 198: 741-746.
51. Nishi, K., Schnier, J.B., and Bradbury, E.M., The Accumulation of Cyclin-Dependent Kinase Inhibitor p27kip1 Is a Primary Response to Staurosporine and Independent of G1 Cell Cycle Arrest. Experimental Cell Research, 1998, 243: 222–231.
52. Poon, R.Y.C. and Hunter, T., Dephosphorylation of Cdk2 Thr160 by the Cyclin-Dependent Kinase-Interacting Phosphatase KAP in the Absence of Cyclin. Science, 1995, 270: 90-93.
53. Fischer, P.M. and Lane, D.P., Inhibitors of cyclin-dependent kinases as anticancer therapeutics. Current Medicinal Chemistry, 2000, 7: 1213-1245.
54. Endicott, J.A., Noble, M.E.M., and Tucker, J.A., Cyclin-dependent kinases: inhibition and substrate recognition. Current Opinien in Cell Biology, 1999, 9: 738-744.
55. Otyepka, M., Kříž, Z., and Koča, J., Dynamics and Binding Modes of Free cdk2 and its Two Complexes with Inhibitors Studied by Computer Simulations. Journal of Biomolecular Structure & Dynamics, 2002, 20: 141-154.
56. Crews, C.M. and Mohan, R., Small-molecule inhibitors of the cell cycle. Current Opinion in Chemical Bioliology, 2000, 4: 47-53.
57. McInnes, C., Wang, S., Anderson, S., O'Boyle, J., Jackson, W., Kontopidis, G., Meades, C., Mezna, M., Thomas, M., and Wood, G., Structural Determinants of CDK4 Inhibition and Design of Selective ATP Competitive Inhibitors. Chemistry & Biology, 2004, 11: 525-534.

137
58. Davies, T.G., Bentley, J., Arris, C.E., Boyle, T.F., Curtin, N.J., Endicott, J.A., Gibson, A.E., Golding, B.T., Griffin, R.J., Hardcastle, I.R., Jewsbury, P., Johnson, L.N., Mesguiche, V., Newell, D.R., Noble, M.E.M., Tucker, J.A., Wang, L., and Whitfield, H.J., Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor. Natural Structural Biology, 2002, 9: 745-749.
59. Davies, T.G., Pratt, D.J., Endicott, J.A., Johnson, L.N., and Noble, M.E.M., Structure-based design of cyclin-dependent kinase inhibitors. Pharmacology & Therapeutics, 2002, 93: 125-133.
60. Smith, D.S., Greer, P.L., and Tsai, L.H., Cdk5 on the Brain. Cell Growth & Differentiation, 2001, 12: 277-283.
61. Tarricone, C., Dhavan, R., Peng, J., Areces, L.B., Tsai, L.H., and Musacchio, A., Structure and Regulation of the CDK5-p25nck5a Complex. Molecular Cell, 2001, 8: 657-669.
62. De Azevedo, W.F., Gaspar, R.T., Canduri, F., Camera, J.C., and da Silveira, N.J.F., Molecular model of cyclin-dependent kinase 5 complexed with roscovitine. Biochemical and Biophysical Research Communications, 2002, 11: 1154-1158.
63. Dhavan, R. and Tsai, L.H., A Decade of CDK5. Nature Reviews Molecular Cell Biology, 2001, 2: 749-759.
64. Mapelli, M. and Musacchio, A., The Structural Perspective on CDK5. Neurosignals, 2003, 12: 164-172.
65. Alvarez, A., Toro, R., Cárceres, A., and Maccioni, R.B., Inhibition of tau phosphorylating protein kinase CDK5 prevents b-amyloidinduced neuronal death. FEBS Letters, 1999, 459: 421-426.
66. Gibson, T.J., Thompson, J.D., Blocker, A., and T., K., Evidence for a protein domain superfamily shared by the cyclins, TFIIB and RB/p107. Nucleic Acids Research, 1994, 22: 946-952.
67. Zukerberg, L.R., Patrick, G.N., Nikolic, M., Humbert, S., Wu, C.L., Lanier, L.M., Gertler, F.B., Vidal, M., Van Etten, R.A., and Tsai, L.H., Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron, 2000, 26: 543-544.
68. Grant, G.H. and Richards, W.G., Computational Chemistry. 1995, Oxford University Press: New York.
69. Leach, A.R., Molecular Modelling: Principles and Applications. 2001: Pearson Education EMA.
70. Jensen, F., Introduction to Computational Chemistry. 1999, England: Wiley-Interscience.
71. Cramer, C.J., Essentials of Computational Chemistry: Theories and Models. 2002: Wiley-Interscience.
72. Wang, J.M., Cieplak, P., and Kollman, P.A., How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Journal of Computational Chemistry, 2000, 21: 1049-1074.

138
73. Case, D.A., Pearlman, D.A., Caldwell, J.W., Cheatham III., T.E., Ross, W.S., Simmerling, C.L., Darden, T.A., Merz, K.M., Stanton, R.V., Cheng, A.L., Vincent, J.J., Crowley, M., Tsui, V., Radmer, R.J., Duan, Y., Pitera, J., Massova, I., Seibel, G.L., Singh, U.C., Weiner, P.K., and Kollman, P.A., AMBER 6. 1999: University of California, San Francisco.
74. Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M., CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of Computational Chemistry, 1983, 4: 187-217.
75. Scott, W.R.P., Hunenberger, P.H., Tironi, I.G., Mark, A.E., Billeter, S.R., Fennen, J., Torda, A.E., Huber, T., Kruger, P., and van Gunsteren, W.F., The GROMOS Biomolecular Simulation Program Package. Journal of Physical Chemistry A, 1999, 103: 3596-3607.
76. Jorgensen, W.L. and Tirado-Rives, J., The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. Journal of the American Chemical Society, 1988, 110: 1657 - 1666.
77. Cornell, W.D., Cieplak, P., and Bayly, C.I., Application of Resp Charges to Calculate Conformational Energies, Hydrogen-Bond Energies, and Free-Energies of Solvation. Journal of the American Chemical Society, 1993, 115: 9620-9631.
78. Bayly, C.I., Cieplak, P., and Cornell, W.D., A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges - The Resp Model. Journal of Physical Chemistry, 1993, 97: 10269-10280.
79. Wang, J. and Kollman, P.A., Automatic parameterization of force field by systematic search and genetic algorithms. Journal of Computational Chemistry, 2001, 22: 1219-1228.
80. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Zakrzewski, V.G., Montgomery, J.A., Stratmann, R.E., Burant, J.C., Dapprich, S., Millam, J.M., Daniels, A.D., Kudin, K.N., Strain, M.C., Farkas, O., Tomasi, J., Barone, V., Cossi, M., Cammi, R., Mennucci, B., Pomelli, C., Adamo, C., Clifford, S., Ochterski, J., Petersson, G.A., Ayala, P.Y., Cui, Q., Morokuma, K., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Cioslowski, J., Ortiz, J.V., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Gomperts, R., Challacombe, M., Gill, P.M.W., Johnson, B.G., Chen, W., Wong, M.W., Andres, J.L., Head-Gordon, M., Replogle, E.S., and Pople, J.A., Gaussian98. 1998, Gaussian, Inc.: Pittsburgh, PA.
81. Kräutler, V., van Gunsteren, W.F., and Hünenberger, P.H., A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. Journal of Computational Chemistry, 2001, 22: 501-508.

139
82. Meijer, L. and Raymond, E., Roscovitine and other purines as kinase inhibitors. From starfish oocytes to clinical trials. Accounts of Chemical Research, 2003, 36: 417-425.
83. Johnson, D.A., Akamine, P., Radzio-Andzelm, E., Madhusudan, and Taylor, S.S., Dynamics of cAMP-Dependent Protein Kinase. Chemical Review, 2001, 101: 2243-2270.
84. De Azevedo, W.F., Leclerc, S., Meijer, L., Havlíček, L., Strnad, M., and Kim, S.H., Inhibition of cyclin-dependent kinases by purine analogues - Crystal structure of human cdk2 complexed with roscovitine. European Journal of Biochemistry, 1997, 243: 518-526.
85. Mapelli, M., Massimiliano, L., Crovace, C., Seeliger, M.A., Tsai, L.H., Meijer, L., and Musacchio, A., Mechanism of CDK5/p25 Binding by CDK Inhibitors. Journal of Medicinal Chemistry, 2005, 48: 671-679.
86. Brooks III, C.L. and Karplus, M., Solvent Effects on Protein Motion and Protein Effects on Solvent Motion. Journal of Molecular Biology, 1989, 208: 159-181.
87. Thanki, N., Thornton, J.M., and Goodfellow, J.M., Influence of secondary structure on the hydration of serine, threonine and tyrosine residues in proteins. Protein Engineering, 1990, 3: 495-508.
88. Ni, H., Sotriffer, C.A., and McCammon, J.A., Ordered Water and Ligand Mobility in the HIV-1 Integrase-5CITEP Complex: A Molecular Dynamics Study. Journal of Medicinal Chemistry, 2001, 44: 3043-3047.
89. Garcia, A.E. and Hummer, G., Water penetration and escape in proteins. PROTEINS: Structure, Function, and Genetics, 2000, 38: 261-272.
90. Ladbury, J.E., Just add water! The effect of water on the specificity of protein-ligand binding sites and its potential application to drug design. Chemistry and Biology, 1996, 3: 973-980.
91. Koellner, G., Kryger, G., Millard, C.B., Silman, I., Sussman, J.L., and Steiner, T., Active site gorge and buried water molecules in crystal structures of Acetylcholinesterase from Torpedo California. Journal of Molecular Biology, 2000, 296: 713-735.
92. Likic, V.A. and Prendergast, F.G., Dynamics of Internal Water in Fatty Acid Binding Protein: Computer Simulations and Comparison With Experiments. PROTEINS: Structure, Function, and Genetics, 2001, 43: 65-72.
93. Lau, E.Y. and Bruice, T.C., The Active Site Dynamics of 4-chlorbenzoyl-CoA Dehalogenase. Proceedings of the National Academy of Sciences USA, 2001, 98: 9527-9532.
94. Naidoo, K.J. and Kuttel, M., Water Structure about the Dimer and Hexamer Repeat Units of Amylose from Molecular Dynamics. Computer Simulations. Journal of Computational Chemistry, 2001, 22: 445-456.
95. Bellissent-Funel, M.-C., Hydration in Protein Dynamics and Function. Journal of Molecular Liquids, 2000, 84: 39-52.

140
96. Meijer, L., Leclerc, S., and Leost, M., Properties and Potential Applications of Chemical Inhibitors of Cyclin-Dependent Kinases. Pharmacology & Therapeutics, 1999, 82: 279 - 284.
97. Gray, N.S., Wodicka, L., Thunnissen, A.M.W.H., Norman, T.C., Kwon, S.J., Espinoza, F.H., Morgan, D.O., Barnes, G., LeClerc, S., Meijer, L., Kim, S.H., Lockhart, D.J., and Schultz, P.G., Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science, 1998, 281: 533-538.
98. Lawrie, A.M., M., N.M.E., Tunnah, P., Brown, N.R., Johnson, L.N., and A., E.J., Protein kinase inhibition by staurosporine revealed in details of the molecular interaction with CDK2. Nature Structural Biology, 1997, 4: 796-801.
99. Schulze-Gahmen, U., Brandsen, J., Jones, H.D., Morgan, D., Meijer, L., Veselý, J., and Kim, S.-H., Multiple modes of ligand recognition: Crystal structures of cyclin-dependent protein kinase 2 in complex with ATP and two inhibitors, olomoucine and isopenthenyladenine. PROTEINS: Structure, Function, and Genetics, 1995, 22: 378-391.
100. De Azevedo, W.F., LeClerc, S., Meijer, L., Havlíček, L., Strnad, M., and Kim, S.-H., Inhibition of cyclin-dependent kinases by purine analogues. European Journal of Biochemistry, 1997, 243: 518-526.
101. De Azevedo, W.F., Mueller-Dieckman, H.J., Schulze-Gahmen U., Worland, P.J., Sausville, E.A., and Kim, S.H., Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proceedings of the National Academy of Sciences USA, 1996, 93: 2735-2740.
102. Sielecki, T.M., Johnson, T.L., Liu, J., Muckelbauer, J.K., Grafstrom, R.H., Cox, S., Boylan, J.F., Burton, C.R., Chen, H., Smallwood, A., Chang, C.-H., Boisclair, M., Benfield, P.A., Trainor, G.L., and Seitz, S.P., Quinazolines as Cyclin Dependent Kinase Inhibitors. Bioorganic & Medicinal Chemistry Letters, 2001, 11: 1157-1160.
103. Hoessel, R., Leclerc, S., Endicott, J.A., Noble, M., Lawrie, A., Tunnah, P., Leost, M., Damiens, E., Domonique, M., Marko, D., Niederberger, E., Tang, W., Eisenbrand, G., and Meijer, L., Indirubin, the active constituent of a chinese antileukemia medicine inhibits cyclin-dependent kinases. Nature Cell Biology, 1999, 1: 60-67.
104. Russo, A.A., Jeffrey P.D., Patten, A.K., Massagué, J., and N.P., P., Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-CDK2 complex. Nature, 1996, 382: 325-331.
105. Davies, T.G., Endicott, J.A., M., N.M.E., Johnson, L.N., Ch.E., A., Bentley, J., Boyle, T.F., Calvert, A.H., Curtin, N.J., Jewsbury, P.J., Gibson, A.E., Golding, B.T., Griffin, R.J., Hardcastle, I., Mesguiche, V., Parsons, R., Whitfield, H., and Newell, D.R., Structural and Thermodynamic Validation of Inactive CDK2 as a Template for Structure-Based Drug design. Cellular & Molecular Biology Letters, 2001, 6: 514-515.

141
106. Naumann, T. and Matter, H., Structural Classification of Protein Kinases Using 3D Molecular Interaction Field Analysis of Their Ligand Binding Sites: Target Family Landscapes. Journal of Medicinal Chemistry, 2001.
107. Case, D.A., Pearlman, D.A., Caldwell, J.W., Cheatham III., T.E., Ross, W.S., Simmerling, C.L., Darden, T.A., Merz, K.M., Stanton, R.V., Cheng, A.L., Vincent, J.J., Crosley, M., Ferguson, D.M., Radmer, R.J., Seibel, G.L., Singh, U.C., Weiner, P.K., and Kollman, P.A., AMBER 5. 1997: University of California, San Francisco.
108. Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, J.K.M., Ferguson, D.M., Spellmeyer, D.C., Fox , T., Caldwell, J.W., and Kollman, P.A., A 2nd Generation Force-Field for Simulation of Proteins, Nucleic-Acids and Organic-Molecules. Journal of American Chemical Society, 1995, 117: 5179 - 5197.
109. Vriend, G., WHAT IF. 1997, EMBL: Heidelberg. 110. Hooft, R.W.W., Sander, C., and Vriend, G., Positioning Hydrogen Atoms
by Optimizing Hydrogen-Bond Network in Protein Structures. PROTEINS: Structure, Function, and Genetics, 1996, 26: 363-376.
111. Essmann, U., Perera, L., Berkowitz, M.L., Darden, T.A., Lee, H., and Pedersen, L.G., A Smooth Particle Mesh Ewald Method. Journal of Chemical Physics, 1995, 103: 8577-8593.
112. Berendsen, H.C., Postma, J.P.M., van Gunsteren, W.F., DiNola, A., and R., H.J., Molecular-Dynamics with Coupling to an External Bath. Journal of Computational Physics, 1984, 81: 3684 - 3690.
113. Ryckaert, J.P., Ciccotti, G., and Berendsen, H.C., Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-alcanes. Journal Computational Physics, 1977, 23: 327 - 341.
114. Makarov, V.A., Andrews, B.K., Smith, P.E., and Pettitt, M.B., Residence Times of Water Molecules in the Hydration Sites of Myoglobin. Biophysical Journal, 2000, 79: 2966-2974.
115. Ferrin, T.E., Huang, C.C., Jarvis, L.E., and Langridge, R., The MIDAS display system. Journal of Molecular Graphics, 1988, 6: 13 - 27.
116. Rittenhouse, R., Unpublished software. 2000. 117. Burgess, J., Ions in Solution - Basic Principles of Chemical Interactions.
Ellis Horwood Series in Inorganic Chemistry. 1988. 118. Lynden-Bell, R.M., Rasaiah, J.C., and Noworyta, J.P., Using simulation to
study solvation in water. Pure and Applied Chemistry, 2001, 73: 1721-1731.
119. Pal, S.K., Peon, J., and Zewail, A.H., Biological water at the protein surface: Dynamical solvation probed directly with femtosecond resolution. Proceedings of the National Academy of Sciences USA, 2002, 99: 1763-1768.
120. Denisov, V.P. and Halle, B., Protein Hydration Dynamics in Aqueous Solution: A Comparison of Bovine Pancreatic Trypsin Inhibitor and

142
Ubiquitin by Oxygen-17 Spin Relaxation Dispersion. Journal of Molecular Biology, 1995, 245: 682-697.
121. Denisov, V.P. and Halle, B., Hydrogen Exchange and Protein Hydration: The Deuteron Spin Relaxation Dispersions of Bovine Pancreatic Trypsin Inhibitor and Ubiquitin. Journal of Molecular Biology, 1995, 245: 698-709.
122. Desiraju, G.R. and Steiner, T., The Weak Hydrogen Bond In Structural Chemistry and Biology. 1999, Oxford: Oxford University Press.
123. Krystof, V. and Strnad, M., Inhibitors of Cyclin-dependent Kinases. Chemicke listy, 2001, 95: 295-301.
124. Johnson, L.N. and Lewis, R.J., Structural Basis for Control by Phosphorylation. Chemical Review, 2001, 101: 2209-2242.
125. Pagano, M., Cell Cycle Control. 1998, New York: New York University School of Medicine.
126. Morgan, D.O., The dynamics of cyclin dependent kinase structure. Current Opinien in Cell Biology, 1996, 8: 767-772.
127. Gu, Y., Rosenblatt, J., and Morgan, D.O., Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO Journal, 1992, 11: 3995-4005.
128. Chow, J.P., Siu, W.Y., Ho, H.T., Ma, K.H., Ho, C.C., and Poon, R.Y.C., Differential contribution of inhibitory phosphorylation of CDC2 and CDK2 for unperturbed cell cycle control and DNA integrity checkpoints. Journal of Biological Chemistry, 2003, 278: 40815-40828.
129. Cheng, A., Ross, K.E., Kaldis, P., and Solomon, M.J., Dephosphorylation of cyclin-dependent kinases by type 2C protein phosphatases. Genes and Development, 1999, 13: 2946-2957.
130. Cheng, A., Kaldis, P., and Solomon, M.J., Dephosphorylation of human cyclin-dependent kinases by protein phosphatase type 2C alpha and beta 2 isoforms. Journal of Biological Chemistry, 2000, 275: 34744-34749.
131. Stevenson, L.M., Deal, M.S., Hagopian, J.C., and Lew, J., Activation Mechanism of CDK2: Role of Cyclin Binding versus Phosphorylation. Biochemistry, 2002, 41: 8528-8534.
132. Cook, A., Lowe, E.D., Chrysina, E.D., Skamnaki, V.T., Oikonomakos, N.G., and Johnson, L.N., Structural Studies on Phospho-CDK2/Cyclin A Bound to Nitrate, a Transition State Analogue: Implications for the Protein Kinase Mechanism. Biochemistry, 2002, 41: 7301-7311.
133. Holmes, J.K. and Solomon, M.J., The role of Thr160 phosphorylation of Cdk2 in substrate recognition. European Journal of Biochemistry, 2001, 268: 4647-4652.
134. Philippopoulos, M. and Lim, C., Molecular Dynamics Simulation of E. coli Ribonuclease H1 in Solution: Correlation with NMR and X-ray Data and Insights into Biological Function. Journal of Molecular Biology, 1995, 254: 771-792.

143
135. Hemmer, W., McGlone, M., Tsigelny, I., and Taylor, S.S., Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. Journal of Biological Chemistry, 1997, 272: 16946-16954.
136. Tsigelny, I., Greenberg, J.P., Cox, S., Nichols, W.L., Taylor, S.S., and Ten Eyck, L.F., 600 ps molecular dynamics reveals stable substructures and flexible hinge points in cAMP dependent protein kinase. Biopolymers, 1999, 50: 513-524.
137. Aimes, R.T., Hemmer, W., and Taylor, S.S., Serine-53 at the tip of the glycine-rich loop of cAMP-dependent protein kinase: Role in catalysis, P-site specificity, and interaction with inhibitors. Biochemistry, 2000, 39: 8325-8332.
138. Jinno, S., Hung, S.C., and Okayama, H., Cell cycle start from quiescence controlled by tyrosine phosphorylation of Cdk4. Oncogene, 1999, 18: 565-571.
139. Spoel, D.V.D., Buuren, A.R.V., Apol, E., Meulenhoff, P.J., Tieleman, P.D., Sijbers, A.L.T.M., Hess, B., Feenstra, K.A., Lindhal, E., Drunen, R.V., and Berendsen, H.J.C., GROMACS. 1991-2002, University of Groningen: Groningen.
140. Laaksonen, L., A graphics program for the analysis and display of molecular dynamics trajectories. Journal of Molecular Graphics, 1992, 10: 33-34.
141. Otyepka, M., Kryštof, V., Havlíček, L., Siglerová, V., Strnad, M., and Koča, J., Docking-based development of purine-like inhibitors of cyclin-dependent kinase-2. Journal of Medicinal Chemistry, 2000, 43: 2506-2513.
142. Fischer, P.M., Endicott, J., and Meijer, L., Cyclin-dependent kinase inhibitors. Progress in Cell Cycle Research, 2003, 5: 235-248.
143. Grant, B.D., Tsigelny, I., Adams, J.A., and Taylor, S.S., Examination of an active-site electrostatic node in the cAMP-dependent protein kinase catalytic subunit. Protein Science, 1996, 5: 1316-1324.
144. Odawara, M., Kadowaki, T., Yamamoto, R., Shibasaki, Y., Tobe, K., Accili, D., Bevins, C., Mikami, Y., Matsuura, N., and Akanuma, Y., Human diabetes associated with a mutation in the tyrosine kinase domain of the insulin receptor. Science, 1989, 245: 66-68.
145. Kříž, Z., Otyepka, M., Bártová, I., and Koča, J., Analysis of CDK2 Active-Site Hydration: A Method to Design New Inhibitors. Proteins: Structure, Function, and Bioinformatics, 2004, 55: 258-274.
146. Golsteyn, R.M., Cdk1 and Cdk2 complexes (cyclin dependent kinases) in apoptosis: a role beyond the cell cycle. Cancer Letters, 2005, 217: 129-138.
147. Murray, A.W., Revisiting the cell cycle: cyclins revisited. Cell, 2004, 116: 221-234.
148. Kesavapany, S., Li, B.S., Amin, N., Zheng, Y.L., Grant, P., and Pant, H.C., Neuronal cyclin-dependent kinase 5: role in nervous system function and its specific inhibition by the Cdk5 inhibitory peptide. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics, 2004, 1697: 143-153.

144
149. Poon, R.Y.C., Lew, J., and Hunter, T., Identification of Functional Domains in the Neuronal Cdk5 Activator Protein. Journal of Biological Chemistry, 1997, 272: 5703 - 5708.
150. Qi, Z., Huang, Q.Q., Lee, K.Y., Lew, J., and Wang, J.H., Reconstitution of Neuronal Cdc2-like Kinase from Bacteria-expressed Cdk5 and an Active Fragment of the Brain-specific Activator. Journal of Biological Chemistry, 1995, 270: 10847 - 10854.
151. Sakaue, F., Saito, T., Sato, Y., Asada, A., Ishiguro, K., Masato, H., and Hisinaga, S., Phosphorylation of FTDP-17 mutant tau by cyclin-dependent kinase 5 complexed with p35, p25, or p39. Journal of Biological Chemistry, 2005, 280: 31522-31529.
152. Whyte, D.B., Martinez, R., Hunter, T., and Sudarsanam, S., The Protein Kinase Complement of the Human Genome G Manning. Science, 2002, 298: 1912-1934.
153. Maccioni, R.B., Otth, C., and Concha, I., The protein kinase Cdk5 - Structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. European Journal of Biochemistry, 2001, 268: 1518-1527.
154. Weishaupt, J.H., Neusch, C., and Bahr, M., Cyclin-dependent kinase 5 (CDK5) and neuronal cell death. Cell and Tissue Research, 2003, 312: 1-8.
155. Smith, D.S. and Tsai, L.H., CDK5 behind the wheel. Trends in Cell Biology, 2002, 12: 28-36.
156. Duke, R.E. and Pedersen, L.G., PMEMD 3. 2003: University of North Carolina-Chapel Hill.
157. Jurečka, P., Šponer, J., and Hobza, P., Potential Energy Surface of the Cytosine Dimer: MP2 Complete Basis Set Limit Interaction Energies, CCSD(T) Correction Term, and Comparison with the AMBER Force Field. Journal of Physical Chemistry B, 2004, 108: 5466-5471.
158. Dobes, P., Otyepka, M., Strnad, M., and Hobza, P., Interaction energies of purine inhibitor - roscovitine with cyclin-dependent kinase 2: correlated ab initio quantum chemical, DFT and empirical calculations. Chemistry - A European Journal, 2005, (Submitted).
159. Muller-Dethlefs, K. and Hobza, P., Noncovalent interactions: A challenge for experiment and theory. Chemical Review, 2000, 100: 143-167.
160. Barrett, C.P. and Noble, M.E.M., Molecular motions of human cyclin-dependent kinase 2. Journal of Biological Chemistry, 2005, 280: 13993-14005.
161. Ahn, J.S., Radhakrishnan, M.L., Mapelli, M., Choi, S., Tidor, B., Cuny, G.D., Musacchio, A., Yeh, L.A., and Kosik, K.S., Defining CDK5 ligand chemical space with small molecule inhibitors of tau phosphorylation. Chemistry and Biology, 2005, 12: 811-823.
162. Bossemeyer, D., The glycine-rich sequence of protein kinases: a multifunctional element. Trends in Biochemical Sciences, 1994, 19: 201-205.

145
163. Schulz, G.E., Binding of nucleotides by proteins : Current opinion in structural biology. Current Opinion in Structural Biology, 1992, 2: 61-67.
164. Cheng, Y., Zhang, Y., and McCammon, J.A., How does the cAMP-dependent protein kinase catalyze the phosphorylation reaction: an ab initio QM/MM study. Journal of American Chemical Society, 2005, 127: 1553-1562.
165. Beaudette, K.N., Lew, J., and Wang, J.H., Substrate specificity characterization of a cdc2-like protein kinase purified from bovine brain. Journal of Biological Chemistry, 1993, 268: 20825-20830.
166. Songyang, Z., Lu, K.P., Kwon, Y.T., Tsai, L.H., Filhol, O., Cochet, C., Brickey, D.A., Soderling, T.R., Bartleson, C., Graves, D.J., DeMaggio, A.J., Hoekstra, M.F., Blenis, J., Hunter, T., and Cantley, L.C., A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Molecular and Cellular Biology, 1996, 16: 6486-6493.
167. Sharma, P., Steinbach, P.J., Sharma, M., Amin, N.D., Barchi, J.J., and Pant, H.C., Identification of Substrate Binding Site of Cyclin-dependent Kinase 5. Journal of Biological Chemistry, 1999, 274: 9600-9606.
168. Honda, R., Lowe, E.D., Dubinina, E., Skamnaki, V., Cook, A., Brown, N.R., and Johnson, L.N., The structure of cyclin E1/CDK2: implications for CDK2 activation and CDK2-independent roles. The EMBO Journal, 2005, 24: 452-463.
169. Lowe, E.D., Tews, I., Cheng, K.Y., Brown, N.R., Gul, S., Noble, M.E.M., Gamblin, S.J., and Johnson, L.N., Specificity determinants of recruitment peptides bound to phospho-CDK2/cyclin A. Biochemistry, 2002, 41: 15625-15634.
170. Schulman, B., Lindstrom, D.L., and Harlow, E., Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proceedings of the National Academy of Sciences of the USA, 1998, 95: 10453-10458.
171. Loog, M. and Morgan, D.O., Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature, 2005, 434: 104-108.

146
Curriculum Vitae
Personal Identification Name and surname Iveta Bártová Title MSc. Date and place of birth 24 February 1978, Bílovec Nationality Czech Marital status Single Address National Centre for Biomolecular Research
(NCBR), Faculty of Science, Masaryk University Brno, Kamenice 5, A-4, 625 00 Brno, Czech Republic
Department of Physical Chemistry,
Faculty of Science, Palacký University, tř. Svobody 26, 771 46 Olomouc, Czech Republic
Phone +420 585634757 Fax +420 585634425 E-mail [email protected] Permanent Residence Address Nad Střelnicí 890/62, 743 01 Bílovec, Czech
Republic Education From 2001 Doctoral degree programme: National Centre for
Biomolecular Research, Faculty of Science, Masaryk University Brno, Czech Republic
1996-2001 Master’s degree programme: Department of Physical Chemistry, Faculty of Science, Masaryk University Brno, Czech Republic
1992-1996 Secondary Chemical School acad. Heyrovsky, Ostrava, Czech Republic
Study field: Analytical Chemistry

147
Molecular Modelling Skills Molecular mechanics AMBER, GROMACS Quantum mechanics GAUSSIAN Molecular graphics GOPENMOL, INSIGHT II, MOIL-VIEW,
MOLDEN, PYMOL, RASMOL, SPARTAN, TRITON, VMD, XLEAP
Attended Courses 08/2002 Summer School of Theoretical and Computational
Chemistry, Czech Academy of Science, Prague, Czech Republic
10/2004 Course: Databases of molecular structures – tools for chemistry and biology (Dr. Bohdan Schneider, Center for Complex Molecular Systems and Biomolecules and Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Prague).
Teaching Experience 2002-2004 Practical course focusing on utilizing of Unix
environment in Computational chemistry at the Masaryk University, Brno, Czech Republic
2005 Practical course of Molecular Modelling at the Palacký University, Olomouc, Czech Republic.
Appreciation by Scientific Community 2005 Award of Dean of Faculty of Science of Masaryk
University, Brno, Czech Republic.

148
List of Publications
Kříž, Z., Otyepka, M., Bártová, I., and Koča, J., Analysis of CDK2 Active-Site Hydration: A Method to Design New Inhibitors. Proteins: Struct., Funct., Bioinf. 55, 2004, 258-274. Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., Activation and Inhibition of Cyclin-Dependent Kinase-2 by Phosphorylation; A Molecular Dynamics Study Reveals the Functional Importance of the Glycine-Rich Loop. Protein Sci. 13, 2004, 1449-1457. Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., The Mechanism of Inhibition of the Cyclin-Dependent Kinase-2 as Revealed by the Molecular Dynamics Study on the Complex CDK2 with the Peptide Substrate HHASPRK. Protein Sci. 14, 2005, 445-451. Otyepka, M., Bártová, I., Kříž, Z., and Koča, J., Different Mechanisms of CDK5 and CDK2 Activation as Revealed by CDK5/p25 and CDK2/Cyclin A Dynamics. J. Biol. Chem., 2006 (accepted for publication).

149
List of Presentations
Otyepka, M., Bártová, I., Kříž, Z., and Koča, J., Dynamika regulačních enzymů buněčných procesů CDK2 a CDK5 studovaná molekulovou dynamikou, Chem Zi 1/1. 57. zjazd chemických, Tatranské Matliare, 2005, p. 277 (poster, presented by Otyepka, M.). Otyepka, M., Bártová, I., Kříž, Z., and Koča, J., Dynamics of human CDK2 and CDK5 studied by computer simulations. Cellular and Molecular Biology Letters. 4th International Conference Inhibitors of Protein Kinases, Warsaw, 2005, p. 116-117 (poster, presented by Otyepka, M.). Kříž, Z., Otyepka, M., Bártová, I., and Koča, J., Molecular dynamics study of protein-ligand interactions. Cellular and Molecular Biology Letters. 4th International Conference Inhibitors of Protein Kinases, Warsaw, 2005, p. 111-112 (poster, presented by Kříž, Z.). Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., Molecular dynamics simulations on Cyclin Dependent Kinase 2. Solvent Behavior Analysis and Activation and Inhibition by Phosphorylation - Tools to Design New Inhibitors and Reveal Mechanism, Modelling Structure and Reactivity. Watoc 2005, 7th Congress of the World Association of Theoretically Oriented Chemists, Cape Town, 2005, p.83 (lecture, presented by Koča, J.). Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., A Molecular Dynamics Study of the Cyclin-Dependent Kinase-2 (CDK2) with Substrate Peptide (HHASPRK) Inhibition by Phosphorylation. Acta Univ. Palacki. Olomouc., Fac. Rer. Nat., Chemica 43S. XIX. Meeting of CSSBMB, Olomouc, 2004, p. 260-261 (lecture). Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., A Molecular Dynamics Study of the Cyclin-Dependent Kinase-2 (CDK2) with Substrate Peptide (HHASPRK), Inhibition of CDK2 by Phosphorylation. Materials in Structure Chemistry, Biology, Physics and Technology. III. Setkání českých a slovenských strukturních biologů, Nové Hrady, 2004, p. 42-43 (poster). Kříž, Z., Otyepka, M., Bártová, I., and Koča, J., Detailed study of interactions between small molecules and proteins using molecular modeling. Materials in Structure Chemistry, Biology, Physics and Technology. III. Setkání českých a slovenských strukturních biologů, Nové Hrady, 2004, p. 47 (poster, presented by Kříž Z.).

150
Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., Computer Simulations of Cyclin-Dependent Kinase-2 Notes about Inhibition. Workshop on Modeling Interactions in Biomolecules, Nové Hrady, 2003, p. 8 (lecture). Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., Cyclin-Dependent Protein Kinase-2 Regulation by Phosphorylation, A Molecular Dynamics Study. Cellular and Molecular Biology Letters. 3rd International Conference Inhibitors of Protein Kinases, Warsaw, 2003, p. 574-575 (poster). Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., CDK2 Activation and Inhibition by Phosphorylation, A Molecular Dynamics Study. Materials in Structure Chemistry, Biology, Physics and Technology. II. Setkání českých a slovenských strukturních biologů, Nové Hrady, 2003, p. 45 (lecture). Bártová, I., Otyepka, M., Kříž, Z., and Koča, J., Molecular Dynamics Simulations of CDK2/ATP complex. Chemické listy 6. 54. Sjezd chemických společností, Brno, 2002, p. 427 (poster).


















![The regulation of SIRT2 function by cyclin-dependent kinases ......916JCB • VOLUME 180 • NUMBER 5 • 2008 with recombinant baculoviral cyclin E – Cdk2 and -[ 32 P]ATP ( Fig.](https://static.fdocuments.in/doc/165x107/60d8933f6f7c6259ee7c52cd/the-regulation-of-sirt2-function-by-cyclin-dependent-kinases-916jcb-a.jpg)