
MOLECULAR DESIGNING AND DOCKING STUDIES ON SOME …
18
www.wjpps.com Vol 9, Issue 5, 2020. 1726 1726 Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences MOLECULAR DESIGNING AND DOCKING STUDIES ON SOME NOVEL 5-ARYL 1,3,4-THIADIAZOLE DERIVATIVES AS INSULYSIN INHIBITORS Sherin Abdul Hameed* 1 , Joyamma Varkey 2 and Jayasekhar Parameswaran Nair 3 1 College of Pharmaceutical Sciences, Government Medical College, Thiruvananthapuram. 2 College of Pharmaceutical Sciences, Government Medical College, Alappuzha. 3 College of Pharmacy, National University of Science and Technology, Sultanate of Oman. ABSTRACT Diabetes mellitus (DM) is a leading non-communicable disease which affects more than100 million people worldwide and is considered as one of leading diseases which causes death in the world. Type-2 diabetes mellitus (T2DM) is a chronic metabolic disorder that results from defects in both insulin secretion and insulin action. Insulin degrading enzyme (IDE, Insulysin), a structurally distinctive zinc metallopeptidase, is a best characterized mediator of insulin catabolism. IDE deficit increases the abundance and signalling of pancreatic hormones insulin, amylin and glucagon. Therefore, agents that inhibit the activity of IDE would be a viable approach towards treatment of T2DM. In the present study, a series of 5-aryl-1,3,4 -thiadiazole derivatives were designed and subjected to in silico screening for drug likeness and desired molecular properties compared with probe ML345. Those analogues having best molecular parameters and good PASS score are selected for molecular docking studies using the docking software Discovery studio 2018. Only 12 of them showed significant interaction with the target protein. PA33 and PA34 have good docking score and more hydrogen bonding interactions with active site residues of Insulysin protein target (PDB ID: 2G47 and 3E4A). From the docking studies, the best pose for the compounds were obtained with least energy value. With all these results, it can be hypothesized that these compounds can be considered as potential inhibitors of Insulysin (IDE) that could hold therapeutic value for the treatment of diabetes. WORLD JOURNAL OF PHARMACY AND PHARMACEUTICAL SCIENCES SJIF Impact Factor 7.632 Volume 9, Issue 5, 1726-1743 Research Article ISSN 2278 – 4357 Article Received on 19 March 2020, Revised on 07 April 2020, Accepted on 27 April 2020 DOI: 10.20959/wjpps20205-16155 *Corresponding Author Asst. Prof. Sherin Abdul Hameed College of Pharmaceutical Sciences, Government Medical College, Thiruvananthapuram.
Transcript of MOLECULAR DESIGNING AND DOCKING STUDIES ON SOME …

www.wjpps.com Vol 9, Issue 5, 2020.
1726
1726
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
MOLECULAR DESIGNING AND DOCKING STUDIES ON SOME
NOVEL 5-ARYL 1,3,4-THIADIAZOLE DERIVATIVES AS INSULYSIN
INHIBITORS
Sherin Abdul Hameed*1, Joyamma Varkey
2 and Jayasekhar Parameswaran Nair
3
1College of Pharmaceutical Sciences, Government Medical College, Thiruvananthapuram.
2College of Pharmaceutical Sciences, Government Medical College, Alappuzha.
3College of Pharmacy, National University of Science and Technology, Sultanate of Oman.
ABSTRACT
Diabetes mellitus (DM) is a leading non-communicable disease which
affects more than100 million people worldwide and is considered as
one of leading diseases which causes death in the world. Type-2
diabetes mellitus (T2DM) is a chronic metabolic disorder that results
from defects in both insulin secretion and insulin action. Insulin
degrading enzyme (IDE, Insulysin), a structurally distinctive zinc
metallopeptidase, is a best characterized mediator of insulin
catabolism. IDE deficit increases the abundance and signalling of
pancreatic hormones insulin, amylin and glucagon. Therefore, agents
that inhibit the activity of IDE would be a viable approach towards
treatment of T2DM. In the present study, a series of 5-aryl-1,3,4
-thiadiazole derivatives were designed and subjected to in silico screening for drug likeness
and desired molecular properties compared with probe ML345. Those analogues having best
molecular parameters and good PASS score are selected for molecular docking studies using
the docking software Discovery studio 2018. Only 12 of them showed significant interaction
with the target protein. PA33 and PA34 have good docking score and more hydrogen
bonding interactions with active site residues of Insulysin protein target (PDB ID: 2G47 and
3E4A). From the docking studies, the best pose for the compounds were obtained with least
energy value. With all these results, it can be hypothesized that these compounds can be
considered as potential inhibitors of Insulysin (IDE) that could hold therapeutic value for the
treatment of diabetes.
WORLD JOURNAL OF PHARMACY AND PHARMACEUTICAL SCIENCES
SJIF Impact Factor 7.632
Volume 9, Issue 5, 1726-1743 Research Article ISSN 2278 – 4357
Article Received on
19 March 2020,
Revised on 07 April 2020,
Accepted on 27 April 2020
DOI: 10.20959/wjpps20205-16155
*Corresponding Author
Asst. Prof. Sherin Abdul
Hameed
College of Pharmaceutical
Sciences, Government
Medical College,
Thiruvananthapuram.

www.wjpps.com Vol 9, Issue 5, 2020.
1727
1727
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
KEYWORDS: Insulysin, insulin degrading enzyme, 1,3,4-thiadiazole, diabetes mellitus,
molecular docking.
INTRODUCTION
Diabetes mellitus is a diverse and complicated disorder that is characterized by persistent
hyperglycemia. Type 2 diabetes affects approximately 200 million people worldwide,
including more than a quarter of elderly living in developed countries. It is also defined as a
condition in which the pancreas no longer produce enough insulin or cells stop responding to
insulin that is produced, so that glucose in the blood cannot be absorbed in to the cells of the
body. When not properly controlled, complications include neuropathy, coronary artery
disease, and renal failure may have resulted. Diabetes is a major cause of blindness, kidney
failure, heart attacks, stroke and lower limb amputation. In 2016, an estimated 1.6 million
deaths were directly caused by diabetes. WHO estimates that diabetes was the seventh
leading cause of death in 2016.[1]
Several drugs are approved for diabetes treatment; however,
their use is associated with side effects and lack of efficacy in attenuating the development of
long-term complications. [2]
Hypoglycemic medication is used to lower the blood sugar level
in the body or to treat other severe symptoms of diabetes mellitus. These medications can be
categorized into insulin and insulin preparations, which are used only parenterally, and
hypoglycemic medicine, which can be administered orally.[3], [4]
Global prevalence of diabetes
and its rising frequency makes it a key area of research in drug discovery programs.
Insulin-degrading enzyme (IDE) is an allosteric Zn+2
metalloprotease involved in the
degradation of many peptides including amyloid-b, and insulin that play key roles in
Alzheimer‟s disease (AD) and type 2 diabetes mellitus (T2DM), respectively. It is the key
enzyme responsible for the cellular proteolysis of insulin. Like insulin, IDE is structurally
distinctive, consisting of two bowl-shaped halves connected by a flexible linker that can
switch between „„open‟‟ and „„closed‟‟ states. In its closed state, IDE completely encapsulates
its substrates within an unusually large internal cavity that appears remarkably well-adapted
to accommodate insulin.[5]
IDE deficit increases the abundance and signalling of the
pancreatic hormones insulin, amylin, and glucagon. Increased insulin improves glucose
tolerance, and increased amylin levels slow postprandial gastric emptying. [6]
In principle, it
should be possible to enhance insulin signalling within cells by inhibiting IDE-mediated
insulin catabolism. A viable approach towards the treatment of T2DM is thus evolved by
designing IDE inhibitors.

www.wjpps.com Vol 9, Issue 5, 2020.
1728
1728
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
It is known that heterocyclic scaffolds possess a leading role in designing novel class of
chemotypes as drug candidates. Thiadiazole is a significant heterocyclic nucleus with two
carbon atoms, two hydrogens, two nitrogens and one sulphur. The molecular structural
formula is C2H2N2S. They occur in four isomeric forms viz 1,2,3-thiadiazole; 1,2,5-
thiadiazole and 1,3,4-thiadiazole. [7]
1, 3, 4 - Thiadiazoles have diversified biocidal activities
probably by virtue of a toxophoric – N=C-S- Grouping. Strong aromaticity of the ring system
gives greater stability and possess low toxicity for higher vertebrates. The sulfur atom of the
thiadiazole imparts improved liposolubility, and the mesoionic nature of thiadiazoles makes
these compounds better able to cross cellular membranes.[8]
Literature survey revealed that
various thiadiazoles have resulted in many potential drugs and are known to exhibit a broad
spectrum of pharmacological properties. 1,3,4-thiadiazole and its derivatives possesses wide
range of therapeutic activities like antimicrobial[9]
antifungal,[10]
antitubercular,[11], [12]
anti-
inflammatory,[13]
diuretics, antiulcer, antidiabetic,[14]
anticonvulsant,[15]
anticancer[16]
anti-
leshmanial, carbonic anhydrase inhibitors, antidepressant, antioxidant, radio-protective, and
antiviral activities.[17]
Datar and Deokule proposed a series of thiadiazolimines exhibiting
antidiabetic activity via peroxisome proliferator-activated receptor gamma (PPARgamma)
inhibition. An isothiazole derivative, ML345 (5-Fluoro-2- (2-morpholino-5-
(morpholinosulfonyl) phenyl) benzo[d] isothiazol-3(2H)-one) is an IDE inhibitor molecular
probe, which targets a specific cysteine residue (Cys819) in IDE. So far, modification of the
thiadiazole ring have proven highly effective with improved potency and lesser toxicity.
The application of computational technology during drug discovery and development offers
considerable potential for reducing the number of experimental studies required for
compound selection and development and for improving the success rate. Computational
methods are required because the amount of biological data has increased and manual
screening against such data requires much time and human resources.[18]
In silico approaches
are being used today in drug discovery to assess the ADME properties of compounds at the
early stages of discovery and development. ACD Lab Chemsketch, PASS (Prediction of
Activity Spectra for Substances), Osiris property calculator, Pre-ADMET prediction,
Molinspiration and SwissADME are some of the software which help pharmaceutical
scientists to select the best candidates for development as well as to reject those with a low
probability of success.[19]

www.wjpps.com Vol 9, Issue 5, 2020.
1729
1729
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
The virtual screening approach for docking small molecules into a known protein structure is
a powerful tool for drug design that has become an integral part of the drug discovery process
in recent years.[20]
In silico virtual screening has become a reliable, cost effective and time-
saving technique that is complementary to in vitro screening for the discovery and
optimization of potent lead and hit compounds. There are two broad categories of screening
techniques, the ligand-based virtual screening and receptor-based virtual screening, to select
candidate compounds that are likely to interact favourably with the target binding sites from a
chemical database.[21]
The three-dimensional structure of protein or protein-ligand complex is
helpful in lead identification using molecular modelling. Discovery studio, AUTODOCK,
Glide, and SwissDOCK are some of the software providing docking analysis. Discovery
Studio is a suite of software for simulating small molecule and macromolecule systems. It is
developed and distributed by Dassault Systemes BIOVIA (formerly Accelrys). Quantitative
structure-activity relationship (QSAR), pharmacophore and biological assays can be helpful
to optimize and design new leads.
Literature survey prompted us to propose some compounds with 1,3,4-thiadazole as core
molecule with an expectation of antidiabetic activity by inhibition of insulin catabolism. In
our present study, a series of 5-aryl-1,3,4-thiadiazole derivatives were designed and screened
and compared with insulysin inhibitor probe ML345, for their drug liking, molecular and
pharmacological parameters towards insulin degrading enzyme. Biological activity, selected
pharmacokinetic and toxicity properties were predicted using different software. Docking
studies were included to find the affinity and interaction of proposed derivatives with the
protein target. Figure 1 shows the schematic representation of insulin degrading enzyme.
Figure 1.

www.wjpps.com Vol 9, Issue 5, 2020.
1730
1730
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
MATERIAL AND METHODS
ACD Lab Chemsketch 2018.2.5. All derivatives were drawn using this software. molecular
parameters like parachor, index of refraction, surface tension, log p, molecular weight, molar
refractivity, molar volume density and polarizability.[22]
PASS (Prediction of Activity Spectra for Substances) provides simultaneous predictions of
many types of biological activity based on the structure of organic compounds.[23,24,25]
Structures of the title compounds were drawn through Chem Sketch software, submitted to
the PASS online program and predicted the possible mechanisms of action as well as
biological activities.
Osiris property calculator[26]
is chemical structural database that calculates various
molecular parameters like clogp, solubility, Molecular weight, Drug likeness. Molinspiration
offers SMILES and SDfile conversion, RO5 compliance, normalization of molecules,
generation of tautomers, molecule fragmentation, calculation of various molecular parameters
and bioactivity towards different receptors needed in QSAR, molecular modelling and drug
design.[27,28]
Pre-ADMET prediction software provided toxicological properties and ADME parameters.
Docking Analysis[29]
of the selected targets with synthetic ligands were analyzed using the
docking software Biovia Discovery studio 2018. Library docking is performed for identifying
the binding affinity with the targets using CHARMM as force field. The data regarding the
protein targets PDB ID: 2G47 and 3E4A were obtained from Protein Data Bank (PDB). The
list of software used for in silico screening in this study is given in table 1. ML345[30]
is the
insulysin inhibitor probe of which molecular parameters were compared.
www.wjpps.com Vol 9, Issue 5, 2020.
1731
1731
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
Table 1: List of software used.
Software Usage
PASS Online To predict the insulysin inhibitory action of proposed moiety
ACD Lab Chemsketch 10.00 3-D drawing, optimizing and calculating various physiochemical
descriptors of proposed molecules
Molinspiration To calculate log P values, Lipinski‟s rule of five and drug likeness
Open Babel 2.3.2l To interconvert file formats and data for the analysis of docking results
Osiris Property Explorer To predict ADME and toxicity
PreADMET Prediction
To predict permeability for Caco-2 cell, MDCK cell and BBB(blood-
brain barrier), HIA(human intestinal absorption), skin permeability and
plasma protein binding
PreADMET Toxicity Prediction To predict toxicological properties from chemical structures, such as
mutagenicity and carcinogenicity
Biovia Discovery studio 2018 To analyse Docking interactions of proposed derivatives.
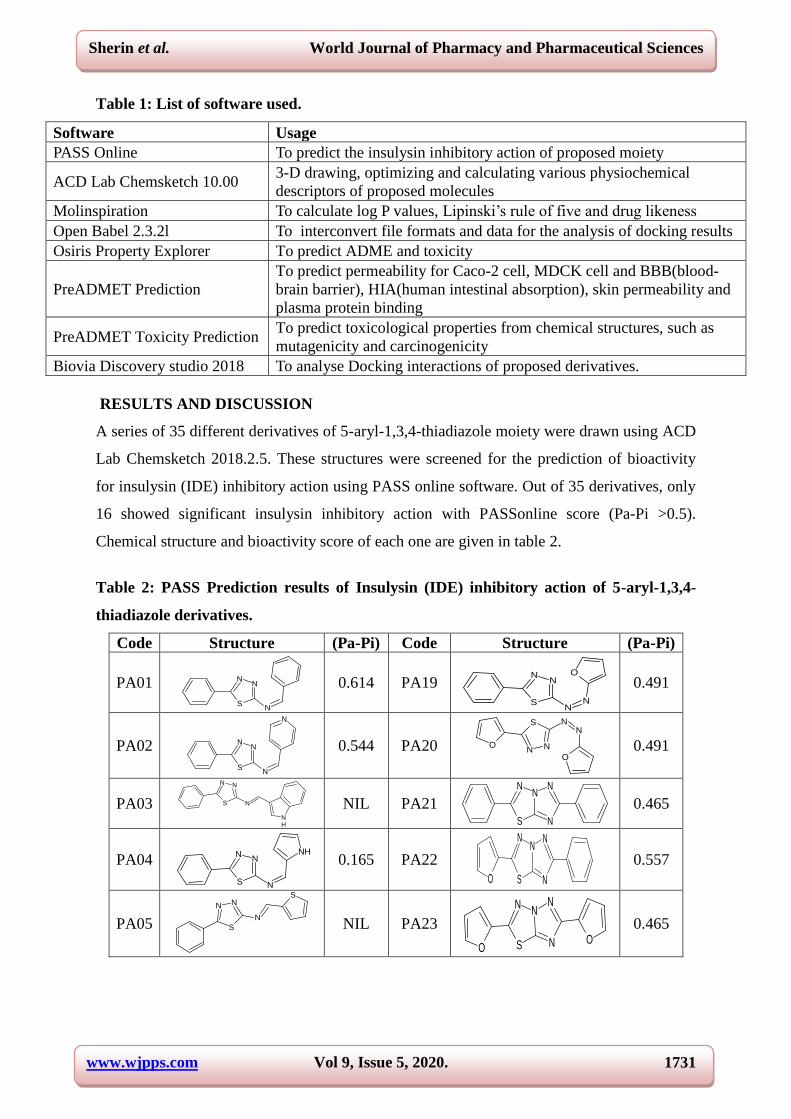
RESULTS AND DISCUSSION
A series of 35 different derivatives of 5-aryl-1,3,4-thiadiazole moiety were drawn using ACD
Lab Chemsketch 2018.2.5. These structures were screened for the prediction of bioactivity
for insulysin (IDE) inhibitory action using PASS online software. Out of 35 derivatives, only
16 showed significant insulysin inhibitory action with PASSonline score (Pa-Pi >0.5).
Chemical structure and bioactivity score of each one are given in table 2.
Table 2: PASS Prediction results of Insulysin (IDE) inhibitory action of 5-aryl-1,3,4-
thiadiazole derivatives.
Code Structure (Pa-Pi) Code Structure (Pa-Pi)
PA01 N
S
N
N
0.614 PA19 N
S
N
NN
O
0.491
PA02 N
S
N
N
N
0.544 PA20 ON
S
N
NN
O
0.491
PA03
N
S
N
N
NH
NIL PA21
N
S
NN
N
0.465
PA04 N
S
N
N
NH
0.165 PA22
O
N
S
NN
N
0.557
PA05
N
S
N
N
S
NIL PA23
O
N
S
NN
N O
0.465

www.wjpps.com Vol 9, Issue 5, 2020.
1732
1732
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
PA06 N
S
N
N
O
0.628 PA24 O N
S
N
N
CH3
CH3
0.604
PA07 N
S
N
N
NIL PA25 O
N
S
N
N
0.451
PA08 N
N
S
N
N
0.605 PA26
O
N
S
N
N
NCH3
CH3
0.715
PA09 N
S
N
NNH
NIL PA27 O
N
S
NN
Cl
Cl
0.496
PA10 N
S
N
NNH
0.373 PA28
O
N
S
N
N
OO
CH3
CH3
O
CH3
0.651
PA11 N
S
N
N
S
0.311 PA29
O
N
S
N
N
OH
0.709
PA12 N
S
N
N
O
0.674 PA30 O
N
S
N
N
N+
O–
O
0.423
PA13 O
N
S
N
N
0.260 PA31
N N
SN
N
S
N
NH2NH2
0.263
PA14 O
N
S
N
N
N
0.622 PA32
N
S
N
NH
0.544
PA15
O
N
S
N
N
O
0.567 PA33 N
NN
O
S
O
0.683
PA16
O
N
S
N
N
NH
0.336 PA34
OH
N
NN
O
S
O
0.674

www.wjpps.com Vol 9, Issue 5, 2020.
1733
1733
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
PA17 O
N
S
N
N
S
0.097 PA35 ON
NN
O
S
O
0.641
PA18
O
N
S
N
N
NH
S
O
O
0.184 ML345 O
F
S N
S
O
O
N ONO
0.936
Molecular properties of derivatives having a PASSonline value (Pa-Pi) > 0.5 were calculated
using Molinspiration online property calculation toolkit. Among the 35 derivatives, only 16
showed good bioactivity score as insulin degrading enzyme inhibitor. The standard values of
molecular properties were based on Lipinskis rule of 5 which stated as molecular weight less
than 500 Dalton, Log p values less than 5, not more than 10 H bond acceptors and not more
than 5 H bond donors. All the derivatives were found to obey Lipinski‟s rule of 5 (RO5)
which is evaluated by Molinspiration software. Some selected pharmacokinetic properties,
which are of high importance to drug discovery process were predicted using Chemsketch v
12.0. Molecular Formula, number of atoms Molar Refractivity, Molar Volume, Parachor,
Surface Tension and Polarizability of 16 selected compounds were detailed in table 3. Molar
Refractivity is an important descriptor in the study of molecular modeling, which relates the
dispersive forces of London present in the drug-receptor interaction. It is found that the
derivatives PA33 and PA34 having high molar refractivity than others and was near to the
value of ML345.To improve the predictions of druglikeness, the rules have spawned many
extensions like Ghose filter and Veber rule. Ghose filter rule stated that compounds with
Partition coefficient log P in (−0.4 to +5.6) range, Molar refractivity from 40 to 130,
Molecular weight from 180 to 480 and Number of atoms from 20 to 70 (includes H-bond
donors [e.g. OHs and NHs] and H-bond acceptors [e.g. Ns and Os]) give more druglikeness
character. While Veber rule stated that for a compound predicted to have good oral
bioavailability then the rotatable bond count should be less than or equal to 10 and polar
surface area should be less than or equal to 140. All of the proposed compounds and insulysin
inhibitor probe ML345 have the said parameters within the range. Lipophilicity affects drug
absorption, bioavailability, hydrophobic drug-receptor interactions, metabolism of molecules,
as well as their toxicity. All the proposed molecules have log p greater than that of ML345
but less than 5. The number of rotatable bonds indicated that all derivatives are flexible. All
derivatives have enough rotatable bonds comparable to that of ML345. TPSA is a very useful

www.wjpps.com Vol 9, Issue 5, 2020.
1734
1734
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
parameter for the prediction of drug transport properties and is defined as a sum of surfaces
of polar atoms (usually oxygens, nitrogens and attached hydrogens) in a molecule. PSA is a
commonly used medicinal chemistry metric for the optimization of a drug's ability to
permeate cells. Molecules with a polar surface area of greater than 140 angstroms squared
tend to be poor at permeating cell membranes. All the proposed derivatives have TPSA value
less than140 angstroms. This parameter has been shown to correlate very well with the
human intestinal absorption, Caco-2 monolayer‟s permeability, and blood brain barrier
penetration. Percentage of absorption (% ABS) was estimated using the equation: %
ABS=109−(0.345×TPSA), according to Zhao et al. Table 4 represents a calculated
percentage of absorption (% ABS), topological polar surface area (TPSA) and Lipinski
parameters of the compounds.
Table 3: Molecular descriptors of selected 5-aryl-1,3,4-Thiadiazole derivatives using
Chemsketch software.
Code Molecular
Formula
No of
atoms
Mol. Ref
Cm3
Parachor
Cm3
Surface tension
Dyne /cm
Polarizability
10-24
cm3
ML345 C21H22FN3O5S2 54 119.18± 0.4 908.5± 6.0 62.0 ± 3.0 47.24± 0.5
PA01 C15H11N3S 30 81.00 ± 0.5 577.8± 8.0 50.1 ± 7.0 32.11± 0.5
PA02 C14H10N4S 29 79.44 ± 0.5 559.0 ±8.0 54.4 ± 7.0 31.49± 0.5
PA06 C13H9N3OS 27 73.17 ± 0.5 519.8± 8.0 54.2 ± 7.0 29.00± 0.5
PA08 C14H10N4S 29 79.44 ± 0.5 559.0± 8.0 54.4 ± 7.0 31.49± 0.5
PA12 C13H9N3OS 27 73.17 ± 0.5 519.8± 8.0 54.2 ± 7.0 29.00± 0.5
PA14 C12H8N4OS 26 71.61 ± 0.5 500.9± 8.0 59.8 ± 7.0 28.39± 0.5
PA15 C11H7N3O2S 24 65.34 ± 0.5 461.7 ±7.0 60.1 ± 7.0 25.90± 0.5
PA22 C13H8N4OS 27 74.57 ± 0.5 499.1± 8.0 68.0 ± 7.0 29.56± 0.5
PA24 C11H11N3OS 27 64.48 ± 0.5 441.9± 8.0 53.2 ± 7.0 25.56± 0.5
PA26 C15H14N4OS 35 85.97 ± 0.5 616.1± 8.0 49.1 ± 7.0 34.08± 0.5
PA28 C16H15N3O4S 39 90.61 ± 0.5 670.6± 8.0 46.6 ± 7.0 35.92± 0.5
PA29 C13H9N3O2S 28 74.02 ± 0.5 525.5± 8.0 60.0 ± 7.0 29.34± 0.5
PA32 C14H11N3S 29 74.78 ± 0.3 545.4 ±8.0 59.1 ± 3.0 29.64± 0.5
PA33 C19H13N3O2S 38 99.66 ± 0.5 715.8± 8.0 52.4 ± 7.0 39.51 ± 0.5
PA34 C19H13N3O3S 39 100.51±0.5 721.5± 8.0 56.4 ± 7.0 39.84 ± 0.5
PA35 C17H11N3O3S 35 91.83 ± 0.5 657.8± 8.0 56.1 ± 7.0 36.40 ± 0.5

www.wjpps.com Vol 9, Issue 5, 2020.
1735
1735
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
Table 4: Molecular parameters predicted via Molinspiration online property calculation
toolkit.
Insulin degrading enzyme is a type of protease enzyme. The bioactivity score of proposed
derivatives on different receptors were shown in table 5. It is found that derivatives like
PA32, PA33, PA34, PA35 have more protease inhibiting activity compared to others with
PA33 and PA34 have its maximum value which also supports the docking results. The
preADMET server was used to calculate parameters such as human intestinal absorption,
cellular permeability Caco-2 in vitro, cell permeability Maden Darby Canine Kidney
(MDCK), skin permeability, plasma protein binding, and penetration of the blood brain
barrier, carcinogenicity and mutagenicity were listed in table 6 and table 7 while aqueous
solubility, drug likeness score and drug score were calculated used Osiris property explorer
online and were given in table 8.
Code Mol. Wt Log P nHAc nHDo nrot b TPSA %ABS
ML345 479.5 1.78 8 0 4 81.09 81.02
PA01 265.33 4.11 3 0 3 38.15 95.8
PA02 266.32 2.82 4 0 3 51.04 91.4
PA06 255.30 3.36 4 0 3 51.29 91.3
PA08 266.32 2.82 4 0 3 51.04 91.4
PA12 255.30 3.25 4 0 3 51.29 91.3
PA14 256.28 1.96 5 0 3 64.18 86.9
PA15 245.26 2.50 5 0 3 64.43 86.8
PA22 268.29 3.34 5 0 2 56.23 89.6
PA24 233.29 3.80 4 0 1 43.33 94.1
PA26 298.36 3.35 5 0 4 54.53 90.2
PA28 345.37 2.88 7 0 6 78.99 81.8
PA29 271.29 3.19 5 1 3 71.52 84.3
PA32 253.32 4.56 3 1 3 37.81 96.0
PA33 347.39 4.93 5 0 5 64.43 86.8
PA34 363.39 4.69 6 1 5 84.66 79.8
PA35 337.35 4.00 6 0 5 77.57 82.2

www.wjpps.com Vol 9, Issue 5, 2020.
1736
1736
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
Table 5: Molinspiration bioactivity score.
Code GPCR
ligand
Ion channel
modulator
Kinase
inhibitor
Nuclear
receptor
Protease
inhibitor
Enzyme
inhibitor
ML345 -0.04 -0.39 -0.10 -0.47 -0.26 0.02
PA01 -0.92 -1.16 -0.46 -0.85 -1.03 -0.44
PA02 -0.86 -1.08 -0.30 -0.89 -0.99 -0.35
PA06 -1.07 -1.33 -0.86 -1.17 -1.31 -0.66
PA08 -0.87 -1.08 -0.29 -0.90 -1.00 -0.37
PA12 -1.17 -1.45 -0.94 -1.34 -1.48 -0.67
PA14 -1.10 -1.36 -0.77 -1.38 -1.44 -0.57
PA15 -1.29 -1.54 -1.28 -1.61 -1.64 -0.81
PA22 -0.69 -0.71 -0.53 -1.34 -1.20 -0.56
PA24 -1.05 -1.18 -1.43 -1.50 -1.89 -0.49
PA26 -0.89 -1.28 -0.64 -1.00 -1.18 -0.56
PA28 -0.81 -1.20 -0.59 -0.95 -1.03 -0.53
PA29 -1.03 -1.46 -0.83 -1.09 -1.28 -0.58
PA32 -0.67 -0.82 -0.59 -0.95 -0.84 -0.28
PA33 -0.56 -0.97 -0.83 -1.09 -0.80 -0.38
PA34 -0.53 -0.94 -0.16 -0.70 -0.77 -0.35
PA35 -0.66 -1.18 -0.63 -0.65 -0.90 -0.43
Table 6: Pharmacokinetic parameters obtained from PreADMET Prediction software.
Code PPB BBB HIA% Caco2 MDCK SP
ML345 93.16 0.18 99.79 21.70 0.201 -3.91
PA01 95.99 3.22 98.26 34.89 197.26 -2.76
PA02 90.39 0.52 99.08 33.20 191.34 -3.36
PA06 97.04 1.56 98.76 32.01 188.15 -3.43
PA08 91.39 0.52 99.08 32.90 191.34 -3.36
PA12 95.86 1.56 98.76 31.50 239.62 -3.42
PA14 87.31 0.21 96.99 27.40 246.66 -3.95
PA15 86.90 0.64 95.20 29.95 209.74 -3.89
PA22 89.80 1.69 99.09 46.60 149.30 -3.77
PA24 100 2.43 98.51 49.82 292.40 -3.58
PA26 91.86 1.21 99.12 36.53 20.81 -3.47
PA28 90.68 0.43 96.99 27.18 2.93 -3.94
PA29 96.00 0.81 95.57 10.17 78.69 -3.66
PA32 91.82 1.45 95.96 38.48 199.34 -2.91
PA33 94.72 1.98 99.43 28.60 92.33 -2.86
PA34 93.71 0.85 97.81 27.42 91.94 -3.05
PA35 94.69 1.02 98.41 26.83 150.64 -3.43

www.wjpps.com Vol 9, Issue 5, 2020.
1737
1737
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
Table 7: Toxicological parameters.
Code Rat Mouse Ames Test hERG Inhibition
ML345 Negative Negative Mutagen Medium Risk
PA01 Negative Negative Mutagen Medium Risk
PA02 Negative Negative Mutagen Medium Risk
PA06 Positive Negative Mutagen Medium Risk
PA08 Negative Negative Mutagen Medium Risk
PA12 Positive Negative Mutagen Medium Risk
PA14 Positive Negative Mutagen Medium Risk
PA15 Positive Negative Mutagen Medium Risk
PA22 Positive Negative Mutagen Medium Risk
PA24 Positive Positive Mutagen Low Risk
PA26 Positive Negative Mutagen Medium Risk
PA28 Positive Negative Mutagen Medium Risk
PA29 Positive Negative Mutagen Medium Risk
PA32 Negative Negative Mutagen Medium Risk
PA33 Positive Negative Mutagen Medium Risk
PA34 Positive Positive Mutagen Medium Risk
PA35 Positive Negative Mutagen Medium Risk
Table 8: Drug score values predicted using Osiris property explorer.
Code C log P Aqueous solubility TPSA Drug-likeness Drug score
ML345 1.65 -4.47 113.0 2.25 0.36
PA01 3.70 -3.66 66.38 3.24 0.76
PA02 2.70 -2.87 79.27 3.04 0.86
PA06 2.89 -3.34 79.52 3.16 0.83
PA08 2.70 -2.87 79.27 2.90 0.86
PA12 2.93 -3.01 79.52 2.97 0.84
PA14 1.93 -2.22 92.41 2.83 0.90
PA15 2.12 -2.69 92.66 2.92 0.89
PA22 2.60 -2.78 84.46 3.35 0.70
PA24 2.64 -4.26 66.40 4.20 0.78
PA26 2.82 -3.05 82.76 2.20 0.49
PA28 2.72 -3.07 107.2 6.30 0.83
PA29 2.58 -2.72 99.75 3.02 0.87
PA32 3.86 -3.70 66.05 1.70 0.71
PA33 3.83 -4.59 92.66 1.70 0.61
PA34 3.48 -4.30 112.8 3.06 0.68
PA35 3.02 -4.28 105.8 2.77 0.43
Among the 16 ligands, only 12 have showed docking score more than 60. Since the insulin
degrading enzyme acquires a big cavity appearance, the dimensions of derivatives PA01,
PA02, PA08 and PA32 may be less to have interaction with the active site residues of the
selected protein. Docking studies were done with two protein targets (PDB ID: 2G47, 3E4A).
The 12 ligands were docked at the active site residues of PDB ID: 2G47. Active site residues

www.wjpps.com Vol 9, Issue 5, 2020.
1738
1738
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
of the selected target were Asn139, Glu 189, Arg824, and Tyr831. The ligands such as PA24,
PA29, PA33, and PA34 showed better interaction at critical residues at Arg 824. PA33 and
PA34 have good docking score and more hydrogen bonding interaction with the active sites.
(Table 9)
Table 9: Docking score of derivatives with PDB ID: 2G47.
SL:No. Code Docking score Interacting Residue of PDB ID: 2G47
1 PA06 73.5505 No H Bond Interaction
2 PA12 102.195 His108
3 PA14 101.281 His108 whb:Gln111
4 PA15 67.9659 Asn139
5 PA22 94.7679 No H Bond Interaction
6 PA24 87.8602 Arg824,Thr825
7 PA26 79.0343 Whb:Ser138, Glu182
8 PA28 93.0204 Thr825,WHB: Ser138,Glu182,Ser128,Glu124
9 PA29 76.691 Arg824, Thr825
10 PA33 105.05 Arg824, Thr825
11 PA34 110.147 Arg824, Thr825
12 PA35 95.8804 His112,whb: Glu189,Ser138,Gln111
The 12 ligands were docked against protein target PDB ID: 3E4A also. Asn139, Glu 189,
Arg824, and Tyr831 are the active site residues. The proposed derivatives such as PA15,
PA33 and PA34 showed exact binding interaction at the critical residue Arg824. From the
docking result with PDB ID: 3E4A, the derivative PA34 have a dock score of about 112.241
with three H- bonds interaction against Insulin degrading enzyme (Table 10).
Table 10: Docking score of derivatives with PDB ID: 3E4A.
SL:no. Name of the ligand Docking score Interacting Residue of PDB ID: 3E4A
1 PA06 77.3707 Gly136 whb: Ser128,Ser138
2 PA12 96.9876 His108,Asn139, whb:Gln111,Thr142
3 PA14 96.3918 His108,Asn139 whb:Gln111,Thr142
4 PA15 74.3331 Arg824 whb:Ser128
5 PA22 95.5868 whb:Ser138, Arg824
6 PA24 89.694 Ser128
7 PA26 81.7984 Whb: Gly136,Glu124,Ser128
8 PA28 80.782 Whb:Glu817,Ser128, Ser138
9 PA29 82.2773 Gly136, Whb:Ser128, Ser138
10 PA33 107.399 Arg824,Thr825
11 PA34 112.241 Arg824,Thr825,Tyr831
12 PA35 105.166 Gln111,whb:Glu189,Gln111,Ser128
From the docking studies, it is found that the derivatives PA34 and PA33 have shown
maximum docking score with both protein targets. The bioactivity score for protease

www.wjpps.com Vol 9, Issue 5, 2020.
1739
1739
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
inhibition is maximum for PA33 and PA34. Both of them have 5-furyl 1,3,4-thiadiazole
moiety linked with 1-(furan-2-yl)-3-phenylprop-2-en-1-one (chalcone). Two furyl ring and
one phenyl ring present in their structure may contribute much to their interaction with the
proteins. Both of them are having similar structural dimensions of that of ML345 with a bond
length (between two extreme atoms in 3d optimised structure) more than 13 angstroms. The
insulysin probe ML345 is also having bulk structure which would help to occupy the big
cavity of insulysin for effective blocking. It might help them to have more surface area to
interact more with the Insulin Degrading Enzyme. The completely wrapping of the molecules
PA33 and PA34 by amino acid residues at the active site pocket region of PDB ID 2G47
were displayed in Figure 2 (a, b), 3 (a, b) while that of PDB ID 3E4A in figure 4 (a, b), and 5
(a, b).
(a) (b)
Figure 2: 2G47 interaction with PA33
(a) (b)
Figure 3: 2G47 interaction with PA34.

www.wjpps.com Vol 9, Issue 5, 2020.
1740
1740
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
(a) (b)
Figure 4: 3E4A interaction with PA33.
(a) (b)
Figure 5: 3E4A interaction with PA34.
CONCLUSION
Using computational and bioinformatics tools, the study identified, designed, and proposed
twelve novel lead molecules from among 35 different derivatives and were compared with
insulysin inhibitor probe ML345. Docking analysis, molecular parameters and bioactivity
score states that PA33 (1-(furan-2-yl)-N-[5-(furan-2-yl)-1,3,4-thiadiazol-2-yl]-3-phenylprop-
2-en-1-imine) and PA34 (3-(furan-2-yl)-3-{[5-(furan-2-yl)-1,3,4-thiadiazol-2-yl] imino}
prop-1-en-1-yl] phenol) these analogues can be considered as the most probable Insulysin
Inhibitors which can be used for the treatment of diabetes mellitus by reducing our
dependency on insulin. Thus this paper proves to be significant for further research work on
the bioactive thiadiazole ring as antidiabetic agent. Further wet lab assessment of these
derivatives has to be performed to establish their insulin mimicking activity.
Conflict of interests
The authors declare that there is no conflict of interests regarding the publication of this
paper.

www.wjpps.com Vol 9, Issue 5, 2020.
1741
1741
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
ACKNOWLEDGEMENT
The authors are thankful to the authorities of all software used in this study for providing free
access and also to Department of Computational Biology & Bioinformatics, University of
Kerala for performing Docking Analysis.
REFERENCES
1. World Health Organization, Fact sheet, 2018.
2. Goodman LS. The pharmacological Basis of Therapeutics, McGraw Hill, New York,
1996; 1.
3. Meneses MJ, Silva BM, Sousa M, Rosalia S, Oliveirl PF and Alves MG. Antidiabetic
Drugs: Mechanisms of Action and Potential Outcomes on Cellular Metabolism. Current
Pharmaceutical Design, 2015; 21(25): 3606-20.
4. Lorenzati B, Zucco C, Miglietta S, Lamberti F and Bruno G. Oral Hypoglycemic Drugs:
Pathophysiological Basis of Their Mechanism of Action. Pharmaceuticals, 2010; 3:
3005-20 doi:10.3390/ph3093005.
5. Leissring MA, Malito E, Hedocin S, Reinstatler L, Sahara T. Et al. Designed Inhibitors of
Insulin Degrading Enzyme Regulate the Catabolism and Activity of Insulin. PLoS ONE,
2010; 5(5): e110504. Doi.10.1371/journal.pone.0010504.
6. Safia C and Peter CB. Insulin-Degrading Enzyme Inhibition, a Novel Therapy for Type 2
Diabetes? Cell Metabolism, 2014; 5: 201-203.
7. Shavkar SD and Sattu H. Synthesis and biological evaluation of imine linked thiadiazole
derivatives. Indo American J. Pharm. Research., 2015; 5(08): 2715-21.
8. Jalhan S, Anil J, Avneet G and Hemraj. Synthesis, biological activities and chemistry of
thiadiazole derivatives and Schiff bases. Asian J Pharm and Clinical Res., 2012; 5:
199-208.
9. Serban G, Stanasel O, Eugenia S and Bota S. 2-Amino-1,3,4-thiadiazole as a potential
scaffold for promising antimicrobial agents. Drug Design, Development and Therapy,
2018; 12: 1545–66
10. Shrivastava K, Purohit S and Singhal S. Studies On Nitrogen and Sulphur Containing
Heterocyclic Compound: 1,3,4 - thiadiazole. Asian Journal of Biomedical and
Pharmaceutical Sciences, 2013; 3(21): 6-23.
11. Karigar AA, Himaja M, Malisunil V and Jagadeesh PK. One pot synthesis and
antitubercular activity of 2-amino-5-aryl-5H-thiazolo (4,3-b) 1,3,4- thiadiazoles. IRJP,
2011; 2(1): 153-158.

www.wjpps.com Vol 9, Issue 5, 2020.
1742
1742
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
12. Syed MA, YRP Reddy and Chandrasekhar KB. Design, one pot synthesis and biological
evaluation of imidazo [2,1-b] [1,3,4]-thiadiazole derivatives for their antitubercular and
antifungal activity. J of Applied Pharm. Sci., 2018; 8(07): 021-027.
13. Pattan SR, Kekare P, Dighe NS, Nirmal SA, Musmade DS, Parjane SK Et al. Synthesis
and biological evaluation of some 1, 3, 4-thiadiazoles. Journal of Chemical and
Pharmaceutical Research, 2009; 1(1): 191-198.
14. Datar PA, and Deokule TA. Design and Synthesis of Thiadiazole Derivatives as
Antidiabetic Agents. Med chem., 2014; 4: 390-399.
15. Raj V, Rai A, Singh M, Kumar R, Kumar A, Kumar S, and Sharma SK. Recent Update
on 1,3,4-Thiadiazole Derivatives: As Anticonvulsant Agents. American Research Journal
of Pharmacy, 2015; 1(1): 34-61.
16. Malleshappa NN, Patel HM, Singh N, Gadad AK, Cameotra SS and Badiger A. Synthesis
and anticancer evaluation of novel 2-cyclo propyl imidazo[2,1-b]1,3,4-thiadiazole
derivatives. Eur J of Med Chem, 2011; 49(9): 4411-18.
17. Sherin AH, Joyamma V, Jayasekhar P. Schiff bases and Bicyclic derivatives comprising
1, 3, 4-thiadiazole moiety- A Review on their Pharmacological activities. Asian J. Pharm.
Res., 2019; 9(4): 299-306.
18. Rishipathak DD, Patil KV, Wajpeyi PS and Daryani MJ: Design and molecular docking
studies of some 1,3,4- thiadiazole derivatives. Int J Pharm Sci Res., 2016; 7(12):
5044-51.doi: 10.13040/IJPSR.0975-8232. 7(12). 5044-51.
19. Bibi S and Sakata. K Current Status of Computer-Aided Drug Design for Type 2
Diabetes. Current Computer-Aided Drug Design., 2016; 12: 167-177.
20. Oprea TI. Virtual Screening in Lead Discovery: A Viewpoint. Molecules., 2002; 7:
51–62.
21. Emmanuel SM, Matheus NDR, Francisco RDSM, Emanuelle MM, Filho CLDM and
Marinho MM. In Silico Study of Pharmacokinetics and Toxicity of Tryperpenoid
Glycosidic Asparagalin A. World Journal of Pharmacy and Pharmaceutical Sciences,
2020; 9 (4): 1-33.
22. ACD labs Chem sketch. ver 12.0 for Windows, Advanced Chemistry Development Inc,
Toronto, Canada, http://www.acdlabs.com.
23. “PASS (Prediction of Activity Spectra for Substances) program”.,
http://www.way2drug.com/passonline.

www.wjpps.com Vol 9, Issue 5, 2020.
1743
1743
Sherin et al. World Journal of Pharmacy and Pharmaceutical Sciences
24. Wadapurkar RM, Shilpa MD, Katti AKS and Sulochana MB. In silico drug design for
staphylococcus aureus and development of host pathogen interaction network.
Informatics in Medicine Unlocked, 2018; 10: 58–70.
25. Tharmatt A, Mehta M, Das R, Mahajan S, Anand A Et al. Identification of molecular
targets of potent antidiabetic drugs using prediction of activity spectra for substances and
molecular docking. International Journal of Green Pharmacy. LPU Conference Apr-Jun
Special Issue, 2018; 1-10.
26. “Osiris Property Explorer.”: ‹http://www.organic-chemistry.org/ prog/peo/›
27. Zhao YH, Abraham MH, Le J, Hersey A, Luscombe CN, Beck G Et al. Rate-Limited
Steps of Human Oral Absorption and QSAR Studies. Pharmaceutical Research, 2002;
19(10): 1446-57.
28. Rajasekhar KK. Abdrrahman SS, Mekonnen YT, Padmavathamma M, Ranganayakulu D
and Shankarananth V. In Silico Prediction of Biological Activity, Selected
Pharmacokinetic and Toxicity Profile of Some 2,4,6-Trisubstituted Pyrimidines Derived
from Guanabenz and Guanfacine. International Journal of Innovative Pharmaceutical
Research, 2015; 6(2): 468-77.
29. Ravi KK. Giri A and Nadendla RR. In silico Adme Profiling of Cdk9 Inhibitors. J Sci Res
Pharm, 2018; 7(3): 30-34.
30. Bannister TD, Wang H, Abdul-Hay SO, Masson A, Madoux F, Ferguson J Et al. ML345,
A Small-Molecule Inhibitor of the Insulin-Degrading Enzyme (IDE). SRIPPS Probe
Reports from the NIH Molecular Libraries Program [Internet], 2014: 1-32.


















