
Molecular design of the N-methyl-D-aspartate receptor binding site ...
5
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 8021-8025, August 1995 Neurobiology Molecular design of the N-methyl-D-aspartate receptor binding site for phencyclidine and dizolcipine (protein structure/synaptic plasticity/ion channels/membrane proteins/glutamate receptor) ANTONIO V. FERRER-MONTIEL, WILLIAM SUN, AND MAURICIO MONTAL Department of Biology, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0366 Communicated by S. J. Singer, University of California, San Diego, CA, May 9, 1995 ABSTRACT TheN-methyl-D-aspartate receptor (NMDAR), a pivotal entity for synaptic plasticity and excitotoxicity in the brain, is a target of psychotomimetic drugs such as phencyclidine (PCP) and dizolcipine (MK-801). In contrast, a related gluta- mate receptor, the a-anuno-3-hydroxy-5-methyl-4-isoxazole pro- pionate/kainate receptor GluRl, is weakly sensitive to these drugs. Three point mutations on GluRl, mimicking homologous residues on the NMDAR, confer the PCP and MK-801 blockade properties that are characteristic of the NMDAR-namely, high potency, voltage dependence, and use dependence. The molecular determinants that specify the PCP block appear confined to the putative M2 transmembrane segment, whereas the sensitivity to MK-801 requires an interplay between residues from M2 and M3. Given the plausible involvement of the NMDAR in the etiology of several neurodegenerative diseases and in excitotoxic neuronal cell death, tailored glutamate receptors with specific properties may be models for designing and screening new drugs targeted to prevent glutamate-mediated neural damage. Ionotropic glutamate receptors (GluRs) play a fundamental role in the function (1) and dysfunction (2-6) of the brain. These receptors are classified in two major classes according to their physiological and pharmacological properties: the N-methyl-D-aspartate receptor (NMDAR) and non-NMDAR (1, 7, 8). NMDARs are highly permeable to calcium and are considered crucial for induction of synaptic plasticity and neurotoxicity (1-8). NMDARs, in contrast to non-NMDARs, are targets of potent psychotropic agents such as the dissocia- tive anesthetic phencyclidine (PCP) and the anticonvulsant and anxiolytic dizolcipine (MK-801), collectively known as noncompetitive NMDA antagonists (1, 7-14). PCP and MK- 801, open channel blockers of the NMDAR that transiently occlude the pore, act with nanomolar inhibition constants and in a voltage- and use-dependent manner (1, 9-21). Their potency as NMDAR blockers has made this class of agents a promising strategy to prevent glutamate-mediated neuronal cell death and associated disorders such as stroke, epilepsy, and Huntington disease (2-6). Although a wealth of information on the molecular mech- anism of NMDAR blockade by PCP and MK-801 has accrued (1, 9-21), the structural determinants that specify the receptor affinity are still elusive. Here we report the molecular design of PCP and MK-801 high-affinity binding sites on a cloned human brain non-NMDAR, GluRl, one of the a-amino-3- hydroxy-5-methyl-4-isoxazole propionate (AMPA)/kainate (KA) receptors that is weakly sensitive to these drugs (22). To create the drug binding pocket, we use the highly sensitive NMDAR1 (23) as a template for the identification of struc- tural components that are required to establish the specificity of action by PCP and MK-801. Site-selective mutants were functionally expressed and assayed as homooligomers in am- phibian oocytes. Three point mutations on the weakly sensitive GluRl were sufficient to transform it into a highly sensitive receptor, endowed with features that approximate those that are characteristic of the NMDAR. A preliminary account of this research appeared elsewhere (24). MATERIALS AND METHODS Site-Directed Mutagenesis and cRNA Preparation. HBGR1 is a cDNA clone encoding a functional AMPA/KA receptor, here denoted as GluRl (22). hNR1 is a cDNA clone for a functional NMDAR, here denoted as NMDAR1 (23). Site- directed mutagenesis was performed on single-stranded DNA (25) or by a combination of polymerase chain reaction (PCR) and subcloning. For the latter, the mutagenic primer was one of a pair of PCR primers used to amplify a fragment of the GluRl cDNA, which was then subcloned back into the GluRl cDNA. Mutants were screened and confirmed by dideoxy sequencing (22, 23). cRNA was synthesized from linearized cDNA using the mMessage mMachine kit from Ambion. Electrophysiological Characterization of Receptor Mutants in Xenopus Oocytes. cRNA (20 ng) from GluRl or its mutants was injected into Xenopus oocytes, and whole-cell currents were recorded with a conventional two-microelectrode volt- age-clamp amplifier (22). Additional details concerning re- cording and procedures are given in ref. 26. Ionic currents were activated by application of 8-s pulses of 500 ,tM KA in the absence or presence of increasing concentrations of channel blockers. For the PCP dose-response of the L577W/Q582T, L577W/Q582N/S610A, and L577W/Q582T/S610A mutants, oocytes were exposed to 18-s KA pulses because of the slower blockade kinetics exhibited by these mutant channels. Re- sponses were normalized with respect to that evoked in the absence of channel blockers. Dose-response curves were fitted to a Michaelis-Menten binding isotherm (11-14, 22, 23): I/Imax = 1/(1 + [blocker]sloPe/IC50), where IC50 denotes the channel blocker concentration that inhibits half of the response ob- tained in its absence (Imax) and slope indicates the steepness of the inhibition curve. Each value represents the mean ± SEM with n : 9. Experiments were carried out in Ba2+-Ringer's solution at 19 ± 1°C. Immunodetection of Expressed Receptors in Xenopus Oo- cytes. For immunoblot analysis, 10 oocytes injected with each mutant cRNA were homogenized in 0.1 ml of extraction buffer (50 mM Tris, pH 7.4/0.1 M NaCl/10 mM EDTA/1.5% Triton X-100) supplemented with 1 ,ug phenylmethylsulfonyl fluoride and 0.2 ,ug pepstatin. The homogenate was incubated on ice for 30 min and then sedimented in a Microfuge for 20 min at 4°C. The supernatants were subjected to SDS/PAGE (27) using 7.5% mini Protean gels (Bio-Rad). Proteins were electrotrans- ferred onto poly(vinylidene difluoride) membranes (Bio-Rad). Membranes were incubated with 0.5 mg of rabbit anti-GluRl Abbreviations: AMPA, a-amino-3-hydroxy-5-methyl-4-isoxazole pro- pionate; GluR, glutamate receptor; KA, kainic acid; MK-801, dizol- cipine; NMDAR, N-methyl-D-aspartate receptor; PCP, phencyclidine; Vh, holding potential. 8021 The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Transcript of Molecular design of the N-methyl-D-aspartate receptor binding site ...

Proc. Natl. Acad. Sci. USAVol. 92, pp. 8021-8025, August 1995Neurobiology
Molecular design of the N-methyl-D-aspartate receptor bindingsite for phencyclidine and dizolcipine
(protein structure/synaptic plasticity/ion channels/membrane proteins/glutamate receptor)
ANTONIO V. FERRER-MONTIEL, WILLIAM SUN, AND MAURICIO MONTALDepartment of Biology, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0366
Communicated by S. J. Singer, University of California, San Diego, CA, May 9, 1995
ABSTRACT TheN-methyl-D-aspartate receptor (NMDAR),a pivotal entity for synaptic plasticity and excitotoxicity in thebrain, is a target ofpsychotomimetic drugs such as phencyclidine(PCP) and dizolcipine (MK-801). In contrast, a related gluta-mate receptor, the a-anuno-3-hydroxy-5-methyl-4-isoxazole pro-pionate/kainate receptor GluRl, is weakly sensitive to thesedrugs. Three point mutations on GluRl, mimicking homologousresidues on the NMDAR, confer the PCP and MK-801 blockadeproperties that are characteristic of the NMDAR-namely, highpotency, voltage dependence, and use dependence. The moleculardeterminants that specify the PCP block appear confined to theputative M2 transmembrane segment, whereas the sensitivity toMK-801 requires an interplay between residues from M2 andM3. Given the plausible involvement of the NMDAR in theetiology of several neurodegenerative diseases and in excitotoxicneuronal cell death, tailored glutamate receptors with specificproperties may be models for designing and screening new drugstargeted to prevent glutamate-mediated neural damage.
Ionotropic glutamate receptors (GluRs) play a fundamentalrole in the function (1) and dysfunction (2-6) of the brain.These receptors are classified in two major classes accordingto their physiological and pharmacological properties: theN-methyl-D-aspartate receptor (NMDAR) and non-NMDAR(1, 7, 8). NMDARs are highly permeable to calcium and areconsidered crucial for induction of synaptic plasticity andneurotoxicity (1-8). NMDARs, in contrast to non-NMDARs,are targets of potent psychotropic agents such as the dissocia-tive anesthetic phencyclidine (PCP) and the anticonvulsantand anxiolytic dizolcipine (MK-801), collectively known asnoncompetitive NMDA antagonists (1, 7-14). PCP and MK-801, open channel blockers of the NMDAR that transientlyocclude the pore, act with nanomolar inhibition constants andin a voltage- and use-dependent manner (1, 9-21). Theirpotency as NMDAR blockers has made this class of agents apromising strategy to prevent glutamate-mediated neuronalcell death and associated disorders such as stroke, epilepsy,and Huntington disease (2-6).Although a wealth of information on the molecular mech-
anism ofNMDAR blockade by PCP and MK-801 has accrued(1, 9-21), the structural determinants that specify the receptoraffinity are still elusive. Here we report the molecular designof PCP and MK-801 high-affinity binding sites on a clonedhuman brain non-NMDAR, GluRl, one of the a-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA)/kainate(KA) receptors that is weakly sensitive to these drugs (22). Tocreate the drug binding pocket, we use the highly sensitiveNMDAR1 (23) as a template for the identification of struc-tural components that are required to establish the specificityof action by PCP and MK-801. Site-selective mutants werefunctionally expressed and assayed as homooligomers in am-phibian oocytes. Three point mutations on the weakly sensitive
GluRl were sufficient to transform it into a highly sensitivereceptor, endowed with features that approximate those thatare characteristic of the NMDAR. A preliminary account ofthis research appeared elsewhere (24).
MATERIALS AND METHODSSite-Directed Mutagenesis and cRNA Preparation. HBGR1
is a cDNA clone encoding a functional AMPA/KA receptor,here denoted as GluRl (22). hNR1 is a cDNA clone for afunctional NMDAR, here denoted as NMDAR1 (23). Site-directed mutagenesis was performed on single-stranded DNA(25) or by a combination of polymerase chain reaction (PCR)and subcloning. For the latter, the mutagenic primer was oneof a pair of PCR primers used to amplify a fragment of theGluRl cDNA, which was then subcloned back into the GluRlcDNA. Mutants were screened and confirmed by dideoxysequencing (22, 23). cRNA was synthesized from linearizedcDNA using the mMessage mMachine kit from Ambion.
Electrophysiological Characterization of Receptor Mutantsin Xenopus Oocytes. cRNA (20 ng) from GluRl or its mutantswas injected into Xenopus oocytes, and whole-cell currentswere recorded with a conventional two-microelectrode volt-age-clamp amplifier (22). Additional details concerning re-cording and procedures are given in ref. 26. Ionic currents wereactivated by application of 8-s pulses of 500 ,tM KA in theabsence or presence of increasing concentrations of channelblockers. For the PCP dose-response of the L577W/Q582T,L577W/Q582N/S610A, and L577W/Q582T/S610A mutants,oocytes were exposed to 18-s KA pulses because of the slowerblockade kinetics exhibited by these mutant channels. Re-sponses were normalized with respect to that evoked in theabsence of channel blockers. Dose-response curves were fittedto a Michaelis-Menten binding isotherm (11-14, 22, 23): I/Imax= 1/(1 + [blocker]sloPe/IC50), where IC50 denotes the channelblocker concentration that inhibits half of the response ob-tained in its absence (Imax) and slope indicates the steepness ofthe inhibition curve. Each value represents the mean ± SEMwith n : 9. Experiments were carried out in Ba2+-Ringer'ssolution at 19 ± 1°C.Immunodetection of Expressed Receptors in Xenopus Oo-
cytes. For immunoblot analysis, 10 oocytes injected with eachmutant cRNA were homogenized in 0.1 ml of extraction buffer(50 mM Tris, pH 7.4/0.1 M NaCl/10 mM EDTA/1.5% TritonX-100) supplemented with 1 ,ug phenylmethylsulfonyl fluorideand 0.2 ,ug pepstatin. The homogenate was incubated on ice for30 min and then sedimented in a Microfuge for 20 min at 4°C.The supernatants were subjected to SDS/PAGE (27) using7.5% mini Protean gels (Bio-Rad). Proteins were electrotrans-ferred onto poly(vinylidene difluoride) membranes (Bio-Rad).Membranes were incubated with 0.5 mg of rabbit anti-GluRl
Abbreviations: AMPA, a-amino-3-hydroxy-5-methyl-4-isoxazole pro-pionate; GluR, glutamate receptor; KA, kainic acid; MK-801, dizol-cipine; NMDAR, N-methyl-D-aspartate receptor; PCP, phencyclidine;Vh, holding potential.
8021
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement" inaccordance with 18 U.S.C. §1734 solely to indicate this fact.
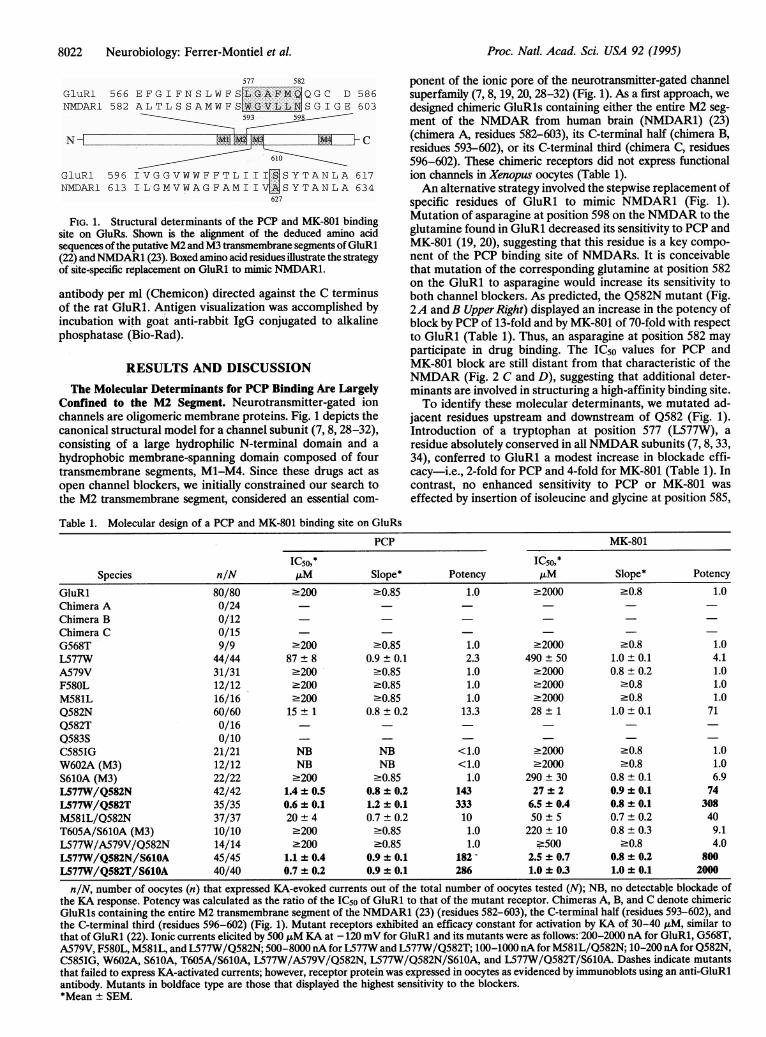
8022 Neurobiology: Ferrer-Montiel et al.
577 582
GluRl 566 EFGIFNSLWFSLGAFM QGC D 586NMDAR1 582 ALTLSSAMWFSWGVLL SGIGE 603
593
N M C~~~~~~~~~610=GluRl 596 IVGGVWWFFTL1II[ISYTANLA 617NMDARI 613 ILGMVWAGFAMIIVWMSYTANLA 634
627
FIG. 1. Structural determinants of the PCP and MK-801 bindingsite on GluRs. Shown is the alignment of the deduced amino acidsequences of the putative M2 and M3 transmembrane segrnents ofGluRl(22) andNMDAR1 (23). Boxed amino acid residues illustrate the strategyof site-specific replacement on GluRl to mimic NMDAR1.
antibody per ml (Chemicon) directed against the C terminusof the rat GluRl. Antigen visualization was accomplished byincubation with goat anti-rabbit IgG conjugated to alkalinephosphatase (Bio-Ra,d).
RESULTS AND DISCUSSION
The Molecular Determinants for PCP Binding Are LargelyConfined to the M2 Segment. Neurotransmitter-gated ionchannels ,are oligomeric membrane proteins. Fig. 1 depicts thecanonical structural model for a channel subunit (7, 8, 28-32),consisting of a large hydrophilic N-terminal domain and a
hydrophobic membrane-spanning domain composed of fourtransmembrane segments, M1-M4. Since these drugs act as
open channel blockers, we initially constrained our search tothe M2 transmembrane segment, considered an essential com-
ponent of the ionic pore of the neurotransmitter-gated channelsuperfamily (7, 8, 19, 20, 28-32) (Fig. 1). As a first approach, wedesigned chimeric GluRls containing either the entire M2 seg-
ment of the NMDAR from human brain (NMDAR1) (23)(chimera A, residues 582-603), its C-terminal half (chimera B,residues 593-602), or its C-terminal third (chimera C, residues596-602). These chimeric receptors did not express functionalion channels in Xenopus oocytes (Table 1).An alternative strategy involved the stepwise replacement of
specific residues of GluRl to mimic NMDAR1 (Fig. 1).Mutation of asparagine at position 598 on the NMDAR to theglutamine found in GluRl decreased its sensitivity to PCP andMK-801 (19, 20), suggesting that this residue is a key compo-
nent of the PCP binding site of NMDARs. It is conceivablethat mutation of the corresponding glutamine at position 582on the GluRl to asparagine would increase its sensitivity toboth channel blockers. As predicted, the Q582N mutant (Fig.2A and B Upper Right) displayed an increase in the potency ofblock by PCP of 13-fold and by MK-801 of 70-fold with respectto GluRl (Table 1). Thus, an asparagine at position 582 mayparticipate in drug binding. The IC50 values for PCP andMK-801 block are still distant from that characteristic of theNMDAR (Fig. 2 C and D), suggesting that additional deter-minants are involved in structuring a high-affinity binding site.To identify these molecular determinants, we mutated ad-
jacent residues upstream and downstream of Q582 (Fig. 1).Introduction of a tryptophan at position 577 (L577W), a
residue absolutely conserved in all NMDAR subunits (7, 8, 33,34), conferred to GluRl a modest increase in blockade effi-cacy-i.e., 2-fold for PCP and 4-fold for MK-801 (Table 1). Incontrast, no enhanced sensitivity to PCP or MK-801 was
effected by insertion of isoleucine and glycine at position 585,
Table 1. Molecular design of a PCP and MK-801 binding site on GluRs
PCP MK-801
IC50,* IC50,*Species n/N FM Slope* Potency ,uM Slope* Potency
GluRl 80/80 2200 >0.85 1.0 .2000 .0.8 1.0Chimera A 0/24Chimera B 0/12Chimera C 0/15G568T 9/9 .200 >0.85 1.0 .2000 .0.8 1.0L577W 44/44 87 ± 8 0.9 ± 0.1 2.3 490 ± 50 1.0 ± 0.1 4.1A579V 31/31 2200 >0.85 1.0 22000 0.8 ± 0.2 1.0F580L 12/12 .200 >0.85 1.0 .2000 .0.8 1.0M581L 16/16 2200 >0.85 1.0 22000 20.8 1.0Q582N 60/60 15 ± 1 0.8 ± 0.2 13.3 28 ± 1 1.0 ± 0.1 71Q582T 0/16 - -Q583S 0/10 - - -C585IG 21/21 NB NB <1.0 .2000 .0.8 1.0W602A (M3) 12/12 NB NB <1.0 .2000 .0.8 1.0S610A (M3) 22/22 .200 >0.85 1.0 290 + 30 0.8 ± 0.1 6.9L577W/Q582N 42/42 1.4 ± 0.5 0.8 ± 0.2 143 27 + 2 0.9 + 0.1 74L577W/Q582T 35/35 0.6 ± 0.1 1.2 ± 0.1 333 6.5 ± 0.4 0.8 ± 0.1 308M581L/Q582N 37/37 20 ± 4 0.7 ± 0.2 10 50 ± 5 0.7 + 0.2 40T605A/S610A (M3) 10/10 .200 >0.85 1.0 220 ± 10 0.8 ± 0.3 9.1L577W/A579V/Q582N 14/14 .200 >0.85 1.0 -500 -0.8 4.0L577W/Q582N/S610A 45/45 1.1 ± 0.4 0.9 ± 0.1 182 - 2.5 + 0.7 0.8 ± 0.2 800L577W/Q582T/S610A 40/40 0.7 ± 0.2 0.9 ± 0.1 286 1.0 ± 0.3 1.0 ± 0.1 2000
n/N, number of oocytes (n) that expressed KA-evoked currents out of the total number of oocytes tested (N); NB, no detectable blockade ofthe KA response. Potency was calculated as the ratio of the IC5o of GluRl to that of the mutant receptor. Chimeras A, B, and C denote chimericGluRls containing the entire M2 transmembrane segment of the NMDAR1 (23) (residues 582-603), the C-terminal half (residues 593-602), andthe C-terminal third (residues 596-602) (Fig. 1). Mutant receptors exhibited an efficacy constant for activation by KA of 30-40 ,uM, similar tothat of GluRi (22). Ionic currents elicited by 500 F&M KA at - 120 mV for GluRl and its mutants were as follows:'200-2000 nA for GluRl, G568T,A579V, F580L, M581L, and L577W/Q582N; 500-8000 nA for L577W and L577W/Q582T; 100-1000 nA for M581L/Q582N; 10-200 nA for Q582N,C585IG, W602A, S610A, T605A/S610A, L577W/A579V/Q582N, L577W/Q582N/S610A, and L577W/Q582T/S610A. Dashes indicate mutantsthat failed to express KA-activated currents; however, receptor protein was expressed in oocytes as evidenced by immunoblots using an anti-GluRlantibody. Mutants in boldface type are those that displayed the highest sensitivity to the blockers.*Mean ± SEM.
Proc. Natl. Acad. Sci. USA 92 (1995)
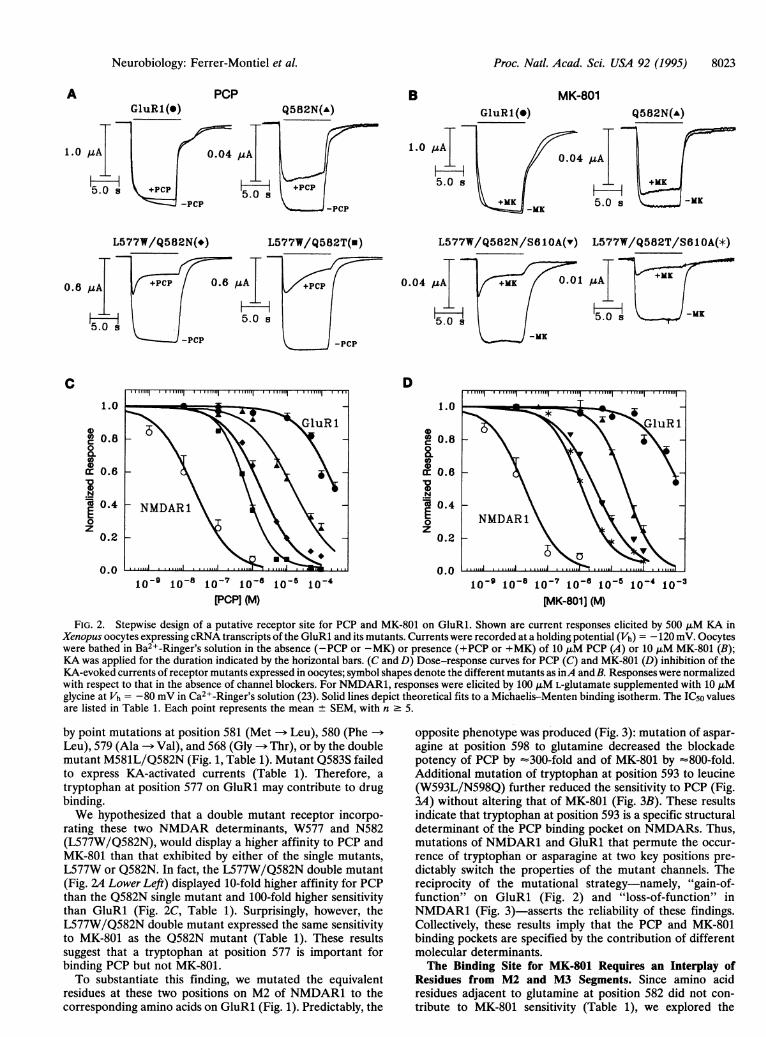
Proc. Natl. Acad. Sci. USA 92 (1995) 8023
GluR 1 (°)PCP
Q582N(A)B MK-801
Q582N(A)
1.0
L577W/Q582N(-) L577W/Q582T(u) L577W/Q582N/S61OA(v) L577W/Q582T/S61OA(*)
0.04 uA .01 /4A
-j- ~ ~ H-5.0 ~~~~~ 5.0 s -MK
-MK
D1.0
0a 0.8
0o0a: 0.6
.Ncc 0.4
z0.2
0.01o-9 10-18 10-71[-6 10-5
PCP] (M)10-91 10 10-7 18-1 10-51(-4 10-3
[AK-801] (M)
FIG. 2. Stepwise design of a putative receptor site for PCP and MK-801 on GluRl. Shown are current responses elicited by 500 ,uM KA inXenopus oocytes expressing cRNA transcripts of the GluRl and its mutants. Currents were recorded at a holding potential (Vh) -120 mV. Oocyteswere bathed in Ba2 -Ringer's solution in the absence (-PCP or -MK) or presence (+PCP or +MK) of 10 ,xM PCP (A) or 10 ,uM MK-801 (B);KA was applied for the duration indicated by the horizontal bars. (C and D) Dose-response curves for PCP (C) and MK-801 (D) inhibition of theKA-evoked currents of receptor mutants expressed in oocytes; symbol shapes denote the different mutants as inA and B. Responses were normalizedwith respect to that in the absence of channel blockers. For NMDAR1, responses were elicited by 100 JIM L-glutamate supplemented with 10 JIMglycine at Vh = -80 mV in Ca2+-Ringer's solution (23). Solid lines depict theoretical fits to a Michaelis-Menten binding isotherm. The IC50 valuesare listed in Table 1. Each point represents the mean ± SEM, with n 5.
by point mutations at position 581 (Met -- Leu), 580 (Phe ->
Leu), 579 (Ala -- Val), and 568 (Gly -> Thr), or by the double
mutant M581L/Q582N (Fig. 1, Table 1). Mutant Q583S failedto express KA-activated currents (Table 1). Therefore, a
tryptophan at position 577 on GluRl may contribute to drugbinding.We hypothesized that a double mutant receptor incorpo-
rating these two NMDAR determinants, W577 and N582(L577W/Q582N), would display a higher affinity to PCP andMK-801 than that exhibited by either of the single mutants,L577W or Q582N. In fact, the L577W/Q582N double mutant(Fig. 24 Lower Left) displayed 10-fold higher affinity for PCPthan the Q582N single mutant and 100-fold higher sensitivitythan GluRl (Fig. 2C, Table 1). Surprisingly, however, theL577W/Q582N double mutant expressed the same sensitivityto MK-801 as the Q582N mutant (Table 1). These resultssuggest that a tryptophan at position 577 is important forbinding PCP but not MK-801.To substantiate this finding, we mutated the equivalent
residues at these two positions on M2 of NMDAR1 to thecorresponding amino acids on GluRl (Fig. 1). Predictably, the
opposite phenotype was produced (Fig. 3): mutation of aspar-agine at position 598 to glutamine decreased the blockadepotency of PCP by '300-fold and of MK-801 by '800-fold.Additional mutation of tryptophan at position 593 to leucine(W593L/N598Q) further reduced the sensitivity to PCP (Fig.3A) without altering that of MK-801 (Fig. 3B). These resultsindicate that tryptophan at position 593 is a specific structuraldeterminant of the PCP binding pocket on NMDARs. Thus,mutations of NMDAR1 and GluRl that permute the occur-rence of tryptophan or asparagine at two key positions pre-dictably switch the properties of the mutant channels. Thereciprocity of the mutational strategy-namely, "gain-of-function" on GluRl (Fig. 2) and "loss-of-function" inNMDAR1 (Fig. 3)-asserts the reliability of these findings.Collectively, these results imply that the PCP and MK-801binding pockets are specified by the contribution of differentmolecular determinants.The Binding Site for MK-801 Requires an Interplay of
Residues from M2 and M3 Segments. Since amino acidresidues adjacent to glutamine at position 582 did not con-tribute to MK-801 sensitivity (Table 1), we explored the
A
1.0
0.6
C1.0
0°a 0.800.0
t: 0.6
eN@ 0.40z
0.2
0.0
Neurobiology: Feffer-Montiel et al.
10-4
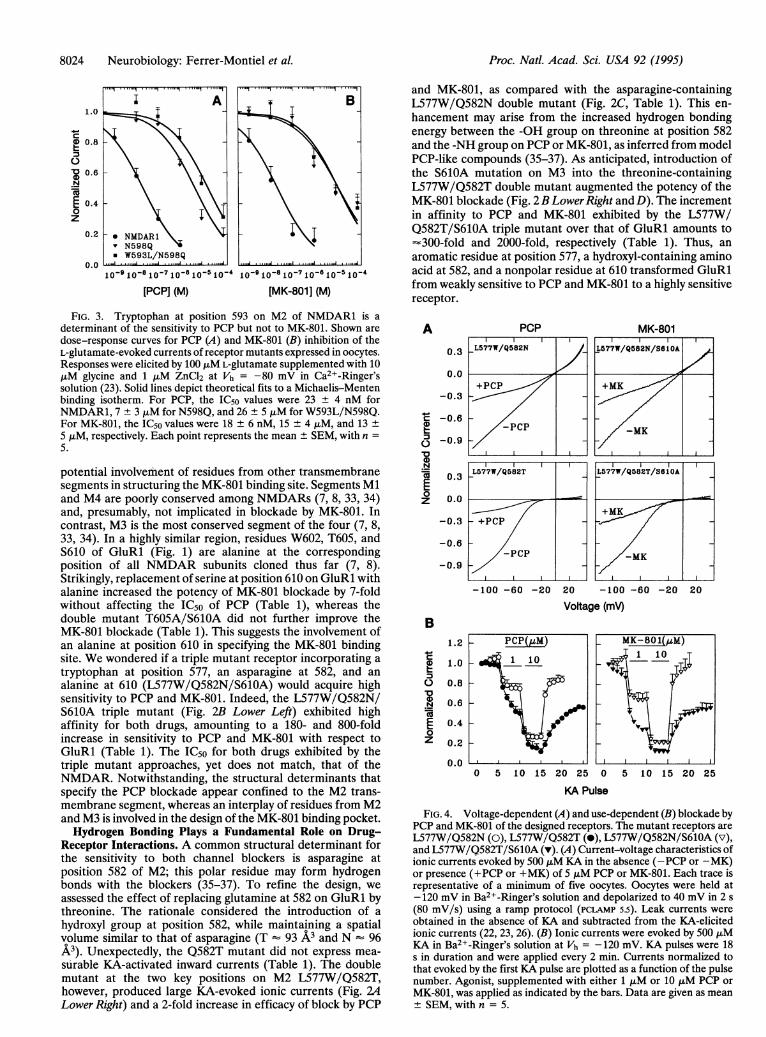
8024 Neurobiology: Ferrer-Montiel et al.
I ~A1.0
0.8
0.N
E 0.4
o
z
0.2 *NMDARIv N598Q* W593L/N598Q
0.0 O ...-A10-910-810-710-810 510 '4 10-910-5 10-710-6 10 510 4
[PCPI (M) [MK-801] (M)
Proc. Natl. Acad. Sci. USA 92 (1995)
and MK-801, as compared with the asparagine-containingL577W/Q582N double mutant (Fig. 2C, Table 1). This en-hancement may arise from the increased hydrogen bondingenergy between the -OH group on threonine at position 582and the -NH group on PCP or MK-801, as inferred from modelPCP-like compounds (35-37). As anticipated, introduction ofthe S610A mutation on M3 into the threonine-containingL577W/Q582T double mutant augmented the potency of theMK-801 blockade (Fig. 2BLowerRight andD). The incrementin affinity to PCP and MK-801 exhibited by the L577W/Q582T/S610A triple mutant over that of GluRl amounts to-300-fold and 2000-fold, respectively (Table 1). Thus, an
aromatic residue at position 577, a hydroxyl-containing aminoacid at 582, and a nonpolar residue at 610 transformed GluRlfrom weakly sensitive to PCP and MK-801 to a highly sensitivereceptor.
FIG. 3. Tryptophan at position 593 on M2 of NMDAR1 is adeterminant of the sensitivity to PCP but not to MK-801. Shown are
dose-response curves for PCP (A) and MK-801 (B) inhibition of theL-glutamate-evoked currents of receptor mutants expressed in oocytes.Responses were elicited by 100 JIM L-glutamate supplemented with 10,iM glycine and 1 jiM ZnCl2 at Vh = -80 mV in Ca2+-Ringer'ssolution (23). Solid lines depict theoretical fits to a Michaelis-Mentenbinding isotherm. For PCP, the IC50 values were 23 ± 4 nM forNMDAR1, 7 + 3 ,uM for N598Q, and 26 ± 5 ,uM for W593L/N598Q.For MK-801, the IC50 values were 18 ± 6 nM, 15 ± 4 ,uM, and 13 +
5 jiM, respectively. Each point represents the mean ± SEM, with n =
5.
potential involvemnent of residues from other transmembranesegments in structuring the MK-801 binding site. Segments Mland M4 are poorly conserved among NMDARs (7, 8, 33, 34)and, presumably, not implicated in blockade by MK-801. Incontrast, M3 is the most conserved segment of the four (7, 8,33, 34). In a highly similar region, residues W602, T605, andS610 of GluRl (Fig. 1) are alanine at the correspondingposition of all NMDAR subunits cloned thus far (7, 8).Strikingly, replacement of serine at position 610 on GluRl withalanine increased the potency of MK-801 blockade by 7-foldwithout affecting the IC50 of PCP (Table 1), whereas thedouble mutant T605A/S610A did not further improve theMK-801 blockade (Table 1). This suggests the involvement ofan alanine at position 610 in specifying the MK-801 bindingsite. We wondered if a triple mutant receptor incorporating a
tryptophan at position 577, an asparagine at 582, and analanine at 610 (L577W/Q582N/S610A) would acquire highsensitivity to PCP and MK-801. Indeed, the L577W/Q582N/S610A triple mutant (Fig. 2B Lower Left) exhibited highaffinity for both drugs, amounting to a 180- and 800-foldincrease in sensitivity to PCP and MK-801 with respect toGluRl (Table 1). The IC50 for both drugs exhibited by thetriple mutant approaches, yet does not match, that of theNMDAR. Notwithstanding, the structural determinants thatspecify the PCP blockade appear confined to the M2 trans-membrane segment, whereas an interplay of residues from M2and M3 is involved in the design of the MK-801 binding pocket.Hydrogen Bonding Plays a Fundamental Role on Drug-
Receptor Interactions. A common structural determinant forthe sensitivity to both channel blockers is asparagine atposition 582 of M2; this polar residue may form hydrogenbonds with the blockers (35-37). To refine the design, weassessed the effect of replacing glutamine at 582 on GluRl bythreonine. The rationale considered the introduction of a
hydroxyl group at position 582, while maintaining a spatialvolume similar to that of asparagine (T - 93 A3 and N 96
A3). Unexpectedly, the Q582T mutant did not express mea-surable KA-activated inward currents (Table 1). The doublemutant at the two key positions on M2 L577W/Q582T,however, produced large KA-evoked ionic currents (Fig. 2A4Lower Right) and a 2-fold increase in efficacy of block by PCP
A
CD
N
E0
z
0.3
0.0
-0.3
-0.6
-0.9
0.3
0.0
-0.3
-0.6
-0.9
PcP
L577W/Q582N
+PCP
-PCP
L577W/Q582T
- +PCP
-PCP
-100 -60 -20
B
4._
0
z
1.2
1.0
0.8
0.6
0.4
0.2
0.0
MK-801L577;W/Q582N/S81OA
+MK
-MK
I
L577W/Q582T/S61OA
+MK
MK
20 -100 -60 -20 20
Voltage (mV)
0 5 10 15 20 25 0 5 10 15 20 25
KA Pulse
FIG. 4. Voltage-dependent (A) and use-dependent (B) blockade byPCP and MK-801 of the designed receptors. The mutant receptors are
L577W/Q582N (0), L577W/Q582T (e), L577W/Q582N/S610A (v),and L577W/Q582T/S610A (v). (A) Current-voltage characteristics ofionic currents evoked by 500 ,uM KA in the absence (-PCP or -MK)or presence (+PCP or +MK) of 5 ,uM PCP or MK-801. Each trace isrepresentative of a minimum of five oocytes. Oocytes were held at-120 mV in Ba2+-Ringer's solution and depolarized to 40 mV in 2 s
(80 mV/s) using a ramp protocol (PCLAMP 5.5). Leak currents were
obtained in the absence of KA and subtracted from the KA-elicitedionic currents (22, 23, 26). (B) Ionic currents were evoked by 500 JIMKA in Ba2+-Ringer's solution at Vh = -120 mV. KA pulses were 18s in duration and were applied every 2 min. Currents normalized tothat evoked by the first KA pulse are plotted as a function of the pulsenumber. Agonist, supplemented with either 1 ,iM or 10 AiM PCP or
MK-801, was applied as indicated by the bars. Data are given as mean± SEM, with n = 5.
PCP(/IM)_ I 10
MK-801(,M)
1jII I~~~~LiL

Proc. Natl. Acad. Sci. USA 92 (1995) 8025
Drug-Induced Blockade of the Designed Receptors MimicsThat of the NMDAR The restructured receptors qualitativelyreproduced the two hallmarks of the NMDAR blockadeexerted by PCP and MK-801-namely, voltage and use de-pendence (1, 15-18, 21). Fig. 4,4 shows that PCP and MK-801blocked the mutant channels in a voltage-dependent manner,inhibiting at negative membrane potentials but not at depo-larizing potentials. Fig. 4B demonstrates that repetitive pulsesof KA, supplemented with 1 ,uM or 10 p,M blocker, progres-sively augmented the block of the KA-evoked ionic current ofthe L577W/Q582T mutant (-). Such blockade was exclusivelyobserved when the blocker was simultaneously applied withKA (see also Fig. 2A and B Lower). Recovery of the KA-elicitedcurrent after removal of the channel blockers from the agonist-containing solution was also prolonged. The extent and therate of recovery increased with more depolarizing voltages.This pattern of use dependence reflects the slow dissociationof the drug from its engineered binding site, as reported for theNMDAR (21). The nature of the residue at position 582appears crucial to endow the mutant receptors with use-dependent blockade. Asparagine mutants do not display use-dependent blockade by either drug (Fig. 4B, o, v), consistentwith the higher IC5o of these mutant receptors for the channelblockers (Table 1). In contrast, the efficacy and use depen-dency of block of the threonine mutants was more powerful:the L577W/Q582T/S610A mutant shows use-dependentblockade by PCP to the same extent as the L577W/Q582Tmutant (data not shown) and, in addition, displays remarkableuse-dependent blockade by MK-801 (Fig. 4B, v). Further,block was effectively irreversible at hyperpolarized potentials,a distinctive property of these drugs at the NMDAR (21).Thus, just three point mutations on GluRl, L577W/Q582T/S610A, created a receptor channel that exhibits the high-potency, voltage-dependent, and use-dependent blockadeproperties by PCP and MK-801 that are characteristic of theNMDAR.
Designing and restructuring a putative receptor site havetaken us from a GluR that is weakly sensitive to PCP andMK-801 to a receptor that is blocked by both drugs withsubmicromolar inhibition constants, emulating the featuresthat are characteristic of the NMDAR. The newly acquiredsensitivity to PCP and MK-801 suggests that the triple mutantmay structurally mimic the pore-lining and the drug-bindingpocket of the NMDAR. Taken together, these resultsstrengthen the notion that the M2 segment is an essentialstructural component of the ionic pore in the GluR super-family and indicate a role of M3 in modulating aspects of porefunction. Having identified key structural determinants in thechannel lining of GluRs that interact with noncompetitiveantagonists may facilitate molecular modeling studies of thepore-forming structure and the binding sites for blockers.Specifically, the implication of residues from M3 in the actionof open channel blockers may require revision of the prevailingmodels of the receptor pore-lining structure based exclusivelyon M2 (28-32) and of the overall folding topology of the GluRsuperfamily (38-40).
A.V.F.-M. and W.S. contributed equally to this work and are listedalphabetically. We thank R. Doolittle, J. Schroeder, M. Saier, M.Oblatt-Montal, R. Planells-Cases, A. Atkinson, G. Fernandez-Ballester, S. Ferroni, and C. D. Patten for valuable comments. Thiswork was supported by National Institutes of Health Grant GM49711,Research Scientist Award MH00778 from the Alcohol, Drug Abuseand Mental Health Administration, Department of the Army MedicalResearch Grant DAMD 17-93-C-3100, and Office of the NavalResearch Grant N00014-89-J-1469.
1. Collingridge, G. L. & Lester, R. A. J. (1989) Pharmacol. Rev. 40,143-210.
2. Choi, D. W. & Rothman, S. M. (1990) Annu. Rev. Neurosci. 13,171-182.
3. Olney, J. W. (1990) Annu. Rev. Pharnacol. Toxicol. 30, 47-71.4. Foster, A. C., Gill, R., Iversen, L. L., Kemp, J. A., Wong, E. H. F. &
Woodruff, G. N. (1990) Prog. Clin. Biol. Res. 361, 301-329.5. Choi, D. W. (1992) Science 258, 241.6. Nakanishi, S. (1992) Science 258, 597.7. Nakanishi, S. & Masu, M. (1994) Annu. Rev. Biophys. Biomol. Struct.
23, 319-348.8. Hollmann, M. & Heinemann, S. F. (1994) Annu. Rev. Neurosci. 17,
31-108.9. Wong, E. H. F., Kemp, J. A., Priestley, T., Knight, A. R., Woodruff,
G. N. & Iversen, L. L. (1986) Proc. Natl. Acad. Sci. USA 83, 7104-7108.
10. Mayer, L. M., Vyklicky, L., Jr., & Sernagor, E. (1989) Drug Dev. Res.17, 263-280.
11. Moriyoshi, K., Masu, M., Ishii, T., Shigemoto, R., Mizuno, N. &Nakanishi, S. (1991) Nature (London) 354, 31-37.
12. Mishina, M., Mori, H., Araki, K., Kushiya, E., Meguro, H., Kutsu-wada, T., Kashiwabuchi, N., Ikeda, K., Nagasawa, M., Yamazaki, M.,Masaki, H., Ymakura, T. & Sakimura, K. (1993)Ann. N.Y Acad. Sci.707, 136-152.
13. Masu, M., Nakajima, Y., Moriyoshi, K, Ishii, T., Akazawa, E. &Nakanishi, S. (1993) Ann. N.Y Acad. Sci. 707, 153-164.
14. Durand, G. M., Gregor, P., Zheng, X., Bennet, M. V. L., Uhl, G. R.& Zukin, R. S. (1992) Proc. Natl. Acad. Sci. USA 89, 9359-9363.
15. Honey, C. R., Miljkovic, Z. & McDonald, J. F. (1985) Neurosci. Lett.61, 135-139.
16. Davies, S. N., Alford, S. T., Coan, E. J., Lester, R. A. J. & Col-lingridge, G. L. (1988) Neurosci. Lett. 92, 213-217.
17. Lerma, J., Zukin, J. R. S. & Bennett, M. V. L. (1991) Neurosci. Lett.123, 187-191.
18. McDonald, J. F., Barlett, M. C., Mody, I. & Pahapill, P. (1991) J.Physiol. 432, 483-508.
19. Sakurada, K., Masu, M. & Nakanishi, S. (1993) J. Biol. Chem. 268,410-415.
20. Yamakura, T., Mori, H., Masaki, H., Shimoji, K. & Mishina, M. (1993)NeuroReport 4, 687-690.
21. Huettner, J. E. & Bean, B. P. (1988) Proc. Natl. Acad. Sci. USA 85,1307-1311.
22. Sun, W., Ferrer-Montiel, A. V., Schinder, A. F., McPherson, J. P.,Evans, G. A. & Montal, M. (1992) Proc. Natl. Acad. Sci. USA 89,1443-1447.
23. Planells-Cases, R., Sun, W., Ferrer-Montiel, A. V. & Montal, M.(19293) Proc. Natl. Acad. Sci. USA 90, 5057-5061.
24. Sun, W., Ferrer-Montiel, A. V. & Montal, M. (1994) Soc. Neurosci.Abstr. 20, 235.
25. Kunkel, T. A., Roberts, J. D. & Zakour, R. A. (1987) Methods Enzy-mol. 154, 367-382.
26. Ferrer-Montiel, A. V. & Montal, M. (1994) Methods Comp. MethodsEnzymol. 6, 60-69.
27. Laemmli, U. K. (1970) Nature (London) 227, 680-682.28. Unwin, N. (1993) Cell 72/Neuron 10, 31-41.29. Galzi, J.-L., Devillers-Thiery, A., Hussy, N., Bertrand, S., Changeux,
J.-P. & Bertrand, D. (1992) Nature (London) 359, 500-505.30. Dingledine, R., Hume, R. L. & Heinemann, S. F. (1992) J. Neurosci.
12, 4080-4087.31. Burnashev, N., Shoeopfer, R., Monyer, H., Ruppersberg, J. P.,
Gunther, W., Seeburg, P. H. & Sakmann, B. (1992) Science 257,1415-1419.
32. Ferrer-Montiel, A. V. & Montal, M. (1993) FEBS Lett. 324, 185-190.33. Ishii, T., Moriyoshi, K, Sugihara, H., Sakurada, K., Kadotani, M. Y.,
Akazawa, C., Shigemoto, R., Mizuno, N., Masu, M. & Nakanishi, S.(1993) J. Biol. Chem. 268, 2836-2843.
34. Monyer, H., Sprengel, R., Schoepfer, R., Herb, A., Higuchi, M.,Lomeli, H., Burnashev, N., Sakmann, B. & Seeburg, P. H. (1992)Science&256, 1217-1221.
35. Leeson, P. D., Carling, R. W., James, K., Smith, J. D., Moore, K. W.,Wong, E. H. F. & Baker, R. (1990) J. Med. Chem. 33, 1296-1305.
36. Manallack, D. T., Wong, M. G., Costa, M., Andrews, P. R. & Beart,P. M. (1988) Mol. Pharmacol. 34, 863-879.
37. Kozikowski, A. P. & Pang, Y.-P. (1990) Mol. Pharmacol. 37,352-375.38. Wo, A. G. & Oswald, R. E. (1994) Proc. Natl. Acad. Sci. USA 91,
7154-7158.39. Hollmann, M., Maron, C. & Heinemann, S. (1994) Neuron 13,
1331-1343.40. Bennett, J. A. & Dingledine, R. (1995) Neuron 14, 373-384.
Neurobiology: Ferrer-Montiel et aL
![Evidence-based Clinical Practice Guideline for ...sydney.edu.au/medicine/cdpc/documents/resources/deprescribing... · methyl-D-aspartate (NMDA) receptor antagonist, memantine [3].](https://static.fdocuments.in/doc/165x107/5bbf717c09d3f2e13b8bface/evidence-based-clinical-practice-guideline-for-methyl-d-aspartate-nmda.jpg)

















