
Modern Variational Inference - Broad Institute · Modern Variational Inference David M. Blei...
19
Modern Variational Inference David M. Blei Departments of Computer Science and Statistics Data Science Institute Columbia University
Transcript of Modern Variational Inference - Broad Institute · Modern Variational Inference David M. Blei...

Modern Variational Inference
David M. BleiDepartments of Computer Science and StatisticsData Science InstituteColumbia University

Communities discovered in a 3.7M node network of U.S. Patents
[Gopalan and Blei, PNAS 2013]

ST01CH10-Blei ARI 4 December 2013 17:0
Game
Second
SeasonTeam
Play
Games
Players
Points
Coach
Giants
1
House
Bush
Political
Party
ClintonCampaign
Republican
Democratic
SenatorDemocrats
6
School
Life
Children
Family
Says
Women
HelpMother
ParentsChild
11
StreetSchool
House
Life
Children
FamilySays
Night
Man
Know
2
Percent
Street
House
Building
Real
SpaceDevelopment
SquareHousing
Buildings
7
Percent
Business
Market
Companies
Stock
Bank
Financial
Fund
InvestorsFunds
12
Life
Says
Show
ManDirector
Television
Film
Story
Movie
Films
3
Game
SecondTeam
Play
Won
Open
Race
Win
RoundCup
8
Government
Life
WarWomen
PoliticalBlack
Church
Jewish
Catholic
Pope
13
House
Life
Children
Man
War
Book
Story
Books
Author
Novel
4
Game
Season
Team
RunLeague
GamesHit
Baseball
Yankees
Mets
9
Street
Show
ArtMuseum
WorksArtists
Artist
Gallery
ExhibitionPaintings
14
Street
House
NightPlace
Park
Room
Hotel
Restaurant
Garden
Wine
5
Government
Officials
WarMilitary
Iraq
Army
Forces
Troops
Iraqi
Soldiers
10
Street
YesterdayPolice
Man
CaseFound
Officer
Shot
Officers
Charged
15
Figure 5Topics found in a corpus of 1.8 million articles from the New York Times. Modified from Hoffman et al. (2013).
a particular movie), our prediction of the rating depends on a linear combination of the user’sembedding and the movie’s embedding. We can also use these inferred representations to findgroups of users that have similar tastes and groups of movies that are enjoyed by the same kindsof users.
Figure 4c illustrates the graphical model. This model is closely related to a linear factor model,except that each cell’s distribution is determined by hidden variables that depend on the cell’s rowand column. The overlapping plates show how the observations at the nth row share its embeddingwn but use different variables γm for each column. Similarly, the observations in the mth columnshare its embedding γm but use different variables wn for each row. Casting matrix factorization
214 Blei
Ann
ual R
evie
w o
f Sta
tistic
s and
Its A
pplic
atio
n 20
14.1
:203
-232
. Dow
nloa
ded
from
ww
w.a
nnua
lrevi
ews.o
rgby
Prin
ceto
n U
nive
rsity
Lib
rary
on
01/0
9/14
. For
per
sona
l use
onl
y.
Topics found in 1.8M articles from the New York Times
[Hoffman, Blei, Wang, Paisley, JMLR 2013]

Adygei BalochiBantuKenyaBantuSouthAfricaBasque Bedouin BiakaPygmy Brahui BurushoCambodianColombianDai Daur Druze French Han Han−NChinaHazaraHezhenItalian Japanese Kalash KaritianaLahu Makrani Mandenka MayaMbutiPygmyMelanesianMiaoMongola Mozabite NaxiOrcadianOroqen Palestinian Papuan Pathan Pima Russian San Sardinian She Sindhi Surui Tu TujiaTuscanUygurXibo Yakut Yi Yoruba
prob
pops1234567
Adygei BalochiBantuKenyaBantuSouthAfricaBasque Bedouin BiakaPygmy Brahui BurushoCambodianColombianDai Daur Druze French Han Han−NChinaHazaraHezhenItalian Japanese Kalash KaritianaLahu Makrani Mandenka MayaMbutiPygmyMelanesianMiaoMongola Mozabite NaxiOrcadianOroqen Palestinian Papuan Pathan Pima Russian San Sardinian She Sindhi Surui Tu TujiaTuscanUygurXibo Yakut Yi Yoruba
prob
pops1234567
Population analysis of 2 billion genetic measurements
[Gopalan, Hao, Blei, Storey, submitted]

Neuroscience analysis of 220 million fMRI measurements
[Manning et al., PLOS ONE 2014]

Analysis of 1.7M taxi trajectories, in Stan
[Kucukelbir et al., 2016]

The probabilistic pipeline
Make assumptions Discover patterns
DATA
Predict & Explore
KNOWLEDGE
R A. 7Aty
This content downloaded from 128.59.38.144 on Thu, 12 Nov 2015 01:49:31 UTCAll use subject to JSTOR Terms and Conditions
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIH
pops1234567
K=7
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIH
pops12345678
K=8
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIHpops
123456789
K=9
Figure S2: Population structure inferred from the TGP data set using the TeraStructure algorithmat three values for the number of populations K. The visualization of the ✓’s in the Figure showspatterns consistent with the major geographical regions. Some of the clusters identify a specificregion (e.g. red for Africa) while others represent admixture between regions (e.g. green for Eu-ropeans and Central/South Americans). The presence of clusters that are shared between differentregions demonstrates the more continuous nature of the structure. The new cluster from K = 7 toK = 8 matches structure differentiating between American groups. For K = 9, the new cluster isunpopulated.
28
I Customized data analysis is important to many fields.
I Pipeline separates assumptions, computation, application
I Eases collaborative solutions to statistics problems

The probabilistic pipeline
Make assumptions Discover patterns
DATA
Predict & Explore
KNOWLEDGE
R A. 7Aty
This content downloaded from 128.59.38.144 on Thu, 12 Nov 2015 01:49:31 UTCAll use subject to JSTOR Terms and Conditions
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIH
pops1234567
K=7
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIH
pops12345678
K=8
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIHpops
123456789
K=9
Figure S2: Population structure inferred from the TGP data set using the TeraStructure algorithmat three values for the number of populations K. The visualization of the ✓’s in the Figure showspatterns consistent with the major geographical regions. Some of the clusters identify a specificregion (e.g. red for Africa) while others represent admixture between regions (e.g. green for Eu-ropeans and Central/South Americans). The presence of clusters that are shared between differentregions demonstrates the more continuous nature of the structure. The new cluster from K = 7 toK = 8 matches structure differentiating between American groups. For K = 9, the new cluster isunpopulated.
28
I Inference is the key algorithmic problem.
I Answers the question: What does this model say about this data?
I Our goal: General and scalable approaches to inference

The probabilistic pipeline
Make assumptions Discover patterns
DATA
Predict & Explore
KNOWLEDGE
R A. 7Aty
This content downloaded from 128.59.38.144 on Thu, 12 Nov 2015 01:49:31 UTCAll use subject to JSTOR Terms and Conditions
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIH
pops1234567
K=7
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIH
pops12345678
K=8
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIHpops
123456789
K=9
Figure S2: Population structure inferred from the TGP data set using the TeraStructure algorithmat three values for the number of populations K. The visualization of the ✓’s in the Figure showspatterns consistent with the major geographical regions. Some of the clusters identify a specificregion (e.g. red for Africa) while others represent admixture between regions (e.g. green for Eu-ropeans and Central/South Americans). The presence of clusters that are shared between differentregions demonstrates the more continuous nature of the structure. The new cluster from K = 7 toK = 8 matches structure differentiating between American groups. For K = 9, the new cluster isunpopulated.
28
I Variational methods: inference as optimization [Jordan et al., 1999]
I Scale up with stochastic variational inference (SVI)
I Generalize with black box variational inference (BBVI)

GLOBAL HIDDEN STRUCTURE
Subsampledata
Infer local structure
Update global structure
MASSIVEDATA
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIH
pops1234567
K=7
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIH
pops12345678
K=8
LWK YRI ACB ASW CDX CHB CHS JPT KHV CEU FIN GBR IBS TSI MXL PUR CLM PEL GIHpops
123456789
K=9
Figure S2: Population structure inferred from the TGP data set using the TeraStructure algorithmat three values for the number of populations K. The visualization of the ✓’s in the Figure showspatterns consistent with the major geographical regions. Some of the clusters identify a specificregion (e.g. red for Africa) while others represent admixture between regions (e.g. green for Eu-ropeans and Central/South Americans). The presence of clusters that are shared between differentregions demonstrates the more continuous nature of the structure. The new cluster from K = 7 toK = 8 matches structure differentiating between American groups. For K = 9, the new cluster isunpopulated.
28
“Stochastic variational inference” [Hoffman et al., 2013, JMLR]

REUSABLE VARIATIONAL
FAMILIES
BLACK BOX VARIATIONAL INFERENCE
p.ˇ; z j x/ANY MODEL
REUSABLE VARIATIONAL
FAMILIESREUSABLE
VARIATIONAL FAMILIES
MASSIVEDATA
“Black box variational inference” [Ranganath et al., 2014, AISTATS]

Edward: A library for probabilistic modeling, inference, and criticism
github.com/blei-lab/edward
(lead by Dustin Tran)

Example: The admixture model of Pritchard et al. (2000)
Copyright 2000 by the Genetics Society of America
Inference of Population Structure Using Multilocus Genotype Data
Jonathan K. Pritchard, Matthew Stephens and Peter DonnellyDepartment of Statistics, University of Oxford, Oxford OX1 3TG, United Kingdom
Manuscript received September 23, 1999Accepted for publication February 18, 2000
ABSTRACTWe describe a model-based clustering method for using multilocus genotype data to infer population
structure and assign individuals to populations. We assume a model in which there are K populations(where K may be unknown) , each of which is characterized by a set of allele frequencies at each locus.Individuals in the sample are assigned (probabilistically) to populations, or jointly to two or more popula-tions if their genotypes indicate that they are admixed. Our model does not assume a particular mutationprocess, and it can be applied to most of the commonly used genetic markers, provided that they are notclosely linked. Applications of our method include demonstrating the presence of population structure,assigning individuals to populations, studying hybrid zones, and identifying migrants and admixed individu-als. We showthat the method can produce highlyaccurate assignments using modest numbers of loci—e.g.,seven microsatellite loci in an example using genotype data from an endangered bird species. The softwareused for this article is available from http:// www.stats.ox.ac.uk/ zpritch/ home.html.
IN applications of population genetics, it is often use- populations based on these subjective criteria representsa natural assignment in genetic terms, and it would beful to classify individuals in a sample into popula-
tions. In one scenario, the investigator begins with a useful to be able to confirm that subjective classificationsare consistent with genetic information and hence ap-sample of individuals and wants to say something about
the properties of populations. For example, in studies propriate for studying the questions of interest. Further,there are situations where one is interested in “cryptic”of human evolution, the population is often considered
to be the unit of interest, and a great deal of work has population structure—i.e., population structure that isdifficult to detect using visible characters, but may befocused on learning about the evolutionary relation-
ships of modern populations (e.g., Caval l i et al. 1994) . significant in genetic terms. For example, when associa-tion mapping is used to find disease genes, the presenceIn a second scenario, the investigator begins with a set
of predefined populations and wishes to classify individ- of undetected population structure can lead to spuriousassociations and thus invalidate standard tests (Ewensuals of unknown origin. This type of problem arises
in many contexts ( reviewed by Davies et al. 1999) . A and Spiel man 1995) . The problem of cryptic populationstructure also arises in the context of DNA fingerprint-standard approach involves sampling DNA from mem-
bers of a number of potential source populations and ing for forensics, where it is important to assess thedegree of population structure to estimate the probabil-using these samples to estimate allele frequencies inity of false matches (Bal d ing and Nich ol s 1994, 1995;each population at a series of unlinked loci. Using theFor eman et al. 1997; Roeder et al. 1998) .estimated allele frequencies, it is then possible to com-Pr it ch ar d and Rosenber g (1999) considered howpute the likelihood that a given genotype originated in
genetic information might be used to detect the pres-each population. Individuals of unknown origin can beence of cryptic population structure in the associationassigned to populations according to these likelihoodsmapping context. More generally, one would like to bePaet kau et al. 1995; Rannal a and Mount a in 1997) .able to identify the actual subpopulations and assignIn both situations described above, a crucial first stepindividuals (probabilistically) to these populations. Inis to define a set of populations. The definition of popu-this article we use a Bayesian clustering approach tolations is typically subjective, based, for example, ontackle this problem. We assume a model in which therelinguistic, cultural, or physical characters, as well as theare K populations (where K may be unknown) , each ofgeographic location of sampled individuals. This subjec-which is characterized by a set of allele frequencies attive approach is usually a sensible way of incorporatingeach locus. Our method attempts to assign individualsdiverse types of information. However, it maybe difficultto populations on the basis of their genotypes, whileto know whether a given assignment of individuals tosimultaneously estimating population allele frequen-cies. The method can be applied to various types ofmarkers [e.g., microsatellites, restriction fragment
Corresponding author: Jonathan Pritchard, Department of Statistics, length polymorphisms (RFLPs) , or single nucleotideUniversity of Oxford, 1 S. Parks Rd., Oxford OX1 3TG, United King-dom. E-mail: [email protected] polymorphisms (SNPs)] , but it assumes that the marker
Genetics 155: 945–959 ( June 2000)
TeraStructure: Fast inference for the PSD model.

Oracle
TeraStructure
1M individuals1,000,000 individuals
10,000 individualsOracle
TeraStructure
ADMIXTURE
fastSTRUCTURE
10,000 individualsOracle
TeraStructure
100,000 individuals100,000 individuals
Figure 2: TeraStructure recovers the underlying per-individual population proportions on the sim-ulated data sets generated via Scenario A. Each panel shows a visualization of the simulationparameters ✓⇤
i and the inferred EŒ✓i j O✓i ç for all individuals in a data set. The current state-of-the-artalgorithms cannot complete their analyses of 100,000 and 1,000,000 individuals. TeraStructure isable to analyze data of this size and gives highly accurate estimates.
25
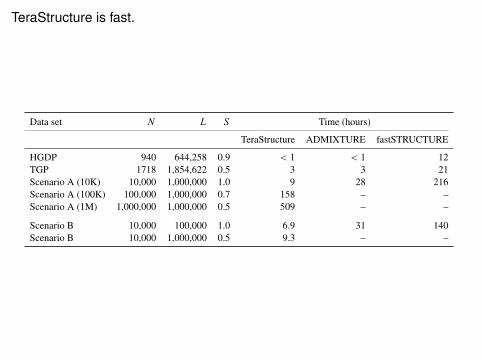
TeraStructure is fast.
Data set N L S Time (hours)
TeraStructure ADMIXTURE fastSTRUCTURE
HGDP 940 644,258 0.9 < 1 < 1 12TGP 1718 1,854,622 0.5 3 3 21Scenario A (10K) 10,000 1,000,000 1.0 9 28 216Scenario A (100K) 100,000 1,000,000 0.7 158 – –Scenario A (1M) 1,000,000 1,000,000 0.5 509 – –
Scenario B 10,000 100,000 1.0 6.9 31 140Scenario B 10,000 1,000,000 0.5 9.3 – –
Table 1: The running time of all algorithms on both real and simulated data. TeraStructure is theonly algorithm that can scale beyond N D 10; 000 individuals to the simulated data sets withN D 100; 000 individuals and N D 1; 000; 000 individuals. S is the fraction of SNP locationssubsampled, with repetition, during training; L is the number of SNP locations. The TeraStruc-ture and ADMIXTURE algorithms were run with ten parallel threads, while fastSTRUCTURE,which does not have a threading option, was run with a single thread. Even under the best-caseassumption of ten times speedup due to parallel computation, the TeraStructure algorithm is twiceas fast as both ADMIXTURE and fastSTRUCTURE algorithms on the data set with N D 10; 000individuals. On the real data sets, TeraStructure is as fast as the other algorithms. In contrast toother methods that iterated multiple times over the entire data set, TeraStructure iterated over theSNP locations at most once on all data sets.
27

TeraStructure is accurate.
Data set Replication N L Median per-individual KL divergence
TeraStructure ADMIXTURE fastSTRUCTURE
Scenario A (10K) 1 10,000 1,000,000 0.016 0.020 6.68Scenario A (10K) 2 10,000 1,000,000 0.009 0.019 5.15Scenario A (10K) 3 10,000 1,000,000 0.020 0.022 4.49Scenario A (100K) 1 100,000 1,000,000 0.006 – –Scenario A (100K) 2 100,000 1,000,000 0.013 – –Scenario A (100K) 3 100,000 1,000,000 0.009 – –Scenario A (1M) 1 1,000,000 1,000,000 0.015 – –
Scenario B 1 10,000 100,000 0.21 0.42 7.21Scenario B 2 10,000 100,000 0.27 0.42 7.97Scenario B 3 10,000 100,000 0.26 0.42 7.68Scenario B 1 10,000 1,000,000 0.16 – –Scenario B 2 10,000 1,000,000 0.23 – –Scenario B 3 10,000 1,000,000 0.25 – –
Table S2: The accuracy of the algorithms on simulated data generated via Scenario A. TeraStruc-ture is the only algorithm that was able to complete its analysis on the simulated data sets withN D 100; 000 individuals and N D 1; 000; 000 individuals. On these massive data sets, TeraS-tructure found a highly accurate fit to the data (see also Figure 2). On smaller simulated data,TeraStructure finds a fit to the data that is closer to the simulation model than either of the othermethods. The number of ancestral populations is set to the number of ancestral populations usedin the simulation: K=6.
32

Figures and Tables
Subsample a SNP Infer ancestral population frequencies for the SNP Update population proportions
Oracle
TeraStructure
1M individuals
0 1 2 1 0 1 1 0
Fitted population proportions
SNPs
individuals
Massive genotype data
TeraStructure
Randomly initialize population proportions
Check for convergence
Figure 1: A schematic diagram of TeraStructure, stochastic variational inference for the Pritchard-Stephens-Donnelly (PSD) model. The algorithm maintains an estimate of the latent populationproportions for each individual. At each iteration it samples SNP measurements from the largedatabase, infers the per-population frequencies for that SNP, and updates its idea of the populationproportions. This is much more efficient than algorithms that must iterate across all SNPs at eachiteration.
24

Why does TeraStructure work?

1. Introduction to variational inference
2. Coordinate ascent variational inference (CAVI)
3. CAVI for the PSD model
4. Scalable inference with stochastic variational inference (SVI)
5. SVI for the PSD model
6. Black box variational inference


















