
Modeling Fluorescently Tagged DNA and RNA Oligonucleotides...
12
+ Modeling Fluorescently Tagged DNA and RNA Oligonucleotides for Direct Comparison to FRET Experiments Amy Speelman Hope College Advisor: Dr. Brent Krueger MU3C Online Conference February 9-11, 2010
-
Upload
doankhuong -
Category
Documents
-
view
218 -
download
0
Transcript of Modeling Fluorescently Tagged DNA and RNA Oligonucleotides...

+Modeling
Fluorescently Tagged
DNA and RNA
Oligonucleotides for
Direct Comparison to
FRET Experiments
Amy Speelman
Hope College
Advisor: Dr. Brent Krueger
MU3C Online Conference
February 9-11, 2010

+Introduction
The goal of our research is to combine computational modeling with experimental studies to come to a better understanding of structural dynamics than is possible with either technique individually.
Experimental technique: Fluorescence-Detected Resonance Energy Transfer (FRET)
In a FRET experiment, a molecule of interest is tagged with two fluorescent dyes: a donor and an acceptor. When the donor is excited, it can relax to the ground state by fluorescence or by transferring energy to the acceptor, which can then relax to its ground state by fluorescence. The rate of energy transfer is distance-dependent, and so the ratio of donor to acceptor fluorescence (FRET efficiency) provides information about the structure of the molecule of interest.
Computational technique: Molecular Dynamics Simulations
We perform MD simulations on the same systems we study experimentally. By evaluating the electronic coupling between the dyes throughout the MD trajectory, we can generate simulated fluorescence data for direct comparison to experimental results.

+Systems Under Study
The systems under study are short DNA or RNA sequences based on the hairpin ribozyme. Each system is tagged with a donor (Cy3) and an acceptor (Cy5).
Cy3 is attached to the oligonucleotidethrough a 5’ phosphate
Cy5 is attached at one of three positions, either through a 5’ phosphate or through a thymine linker
Duplex A DNA Loop A RNA
G
CTC
CA
A TC
G
G
G
A
T
a
A
TCC
GT
AGG
C
d
5’
CA
GT
a3’
a
G
CUC
CA
A UC
G
G
G
A
U
a
A
UCC
GU
AGG
C
d
5’
CA
GU
a3’
a
A
UCC
GU
AGG
C
C
CAC
AG
GUG
U
A
AA
GCUG
A
d
a
a
5’
3’
a
Duplex A RNAHN
O
N
N
-O3S
SO3-
NH
O
ON
O
HO
HH
HH
OP-O
O-
O
HN
O
NH
O
ON
O
HO
HH
HH
OPO
O-
O
NH
O
HN
O
5’ phosphate linker Thymine linker
Dyes:
n = 1 Cy3
n = 2 Cy5
( )n

+Parametrization
We run our molecular dynamics simulations using AMBER. In AMBER, the energy of a system is calculated as the sum of bonded and nonbonded contributions:
Sets of parameters (bond, angle, and dihedral parameters, atomic charges) exist for nucleic acids and proteins, but we must define parameters for the dyes and linkages. This is done by deriving charges using the RESP charge fitting procedure and assigning bond, angle, and dihedral parameters by analogy to existing parameters.1
1 Leland, B. A.; Paul, D. A.; Krueger, B. P.; Walker, R. C.,“AMBER Advanced Tutorials A1: Setting up an advanced system (including charge derivation).” http://ambermd.org/tutorials/advanced/tutorial1/ AMBER Tutorials Website (not peer reviewed) Originally posted June 2008.
N
1i
N
1ij ij0
jiN
1i
N
1ij
6
ij
ij
12
ij
ij
ij
torsions
n
angles
2
ij,0ij
bonds
2
ij,0ij
ijr,
r4π
r
σ
r
σ4ε
γ))cos(nω(12
V)θ(θ
2
k)r(r
2
kE

+Parametrization
Before beginning, the system is divided into fragments that can be parametrizedindividually.
Dividing the system into fragments is convenient because it makes the QM calculations easier and allows us to use the same fragment several times.
NH
O
ON
O
HO
HH
HH
OPO
O-
O
NH
O
HN
N
SO3-
N
-O3S
O
NH
O
ONH
NH
O
HN
O
O
HO
HH
HH
OPO
O-
O
N
SO3-
N
-O3S
HN
O

+Charge Derivation
Quantum mechanical calculations
A geometry optimization at the B3LYP/6-31G(d) level is performed on each low-energy conformation of the fragment
An electrostatic potential is derived at the HF/6-31G(d) level, which mimics charge distribution in solution
RESP (Restrained Electrostatic Potential) Fitting1
The RESP charge fitting method uses the ESP to determine charges
RESP fits are performed in two stages
First stage: all charges (except those the user constrains) are allowed to change
Second stage: rotationally degenerate atoms are constrained to have the same charge
In doing RESP fitting, we must make sure the charges fulfill certain criteria
The charges at the junctions between fragments should be similar
After the entire system has been built, it must have integer charge
1 J. Phys. Chem. 1993, 97, 10269-10280.
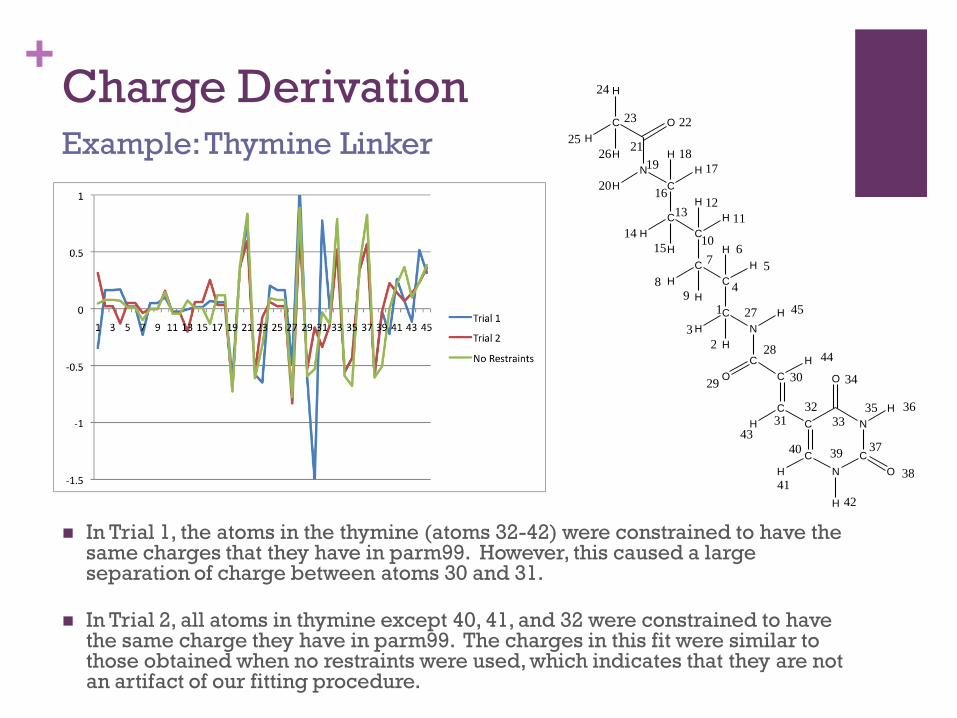
+Charge Derivation
In Trial 1, the atoms in the thymine (atoms 32-42) were constrained to have the same charges that they have in parm99. However, this caused a large separation of charge between atoms 30 and 31.
In Trial 2, all atoms in thymine except 40, 41, and 32 were constrained to have the same charge they have in parm99. The charges in this fit were similar to those obtained when no restraints were used, which indicates that they are not an artifact of our fitting procedure.
Example: Thymine Linker
C
C C
N
O
O
C
N
C
C
N
C
C
C
C
C
C
N
C O
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
38
37
35 36
34
33
39
42
41
40
32
4331
30
44
O29
28
27 45
2
3
1
5
6
4
7
89
10
11
1213
14
15
16
17
1819
20
21
2223
24
25
26

+Atom Type Assignment
The bond, angle, and dihedral parameters are assigned by analogy to pre-existing sets of parameters
We draw from two sets of parameters: parm991 (parameters for nucleic acids and proteins) and GAFF (general set of parameters for organic molecules)2
Parameters are defined based on atom type
For example, the carbonyl C-O bond is described as a bond between an atom of type C (carbonyl carbon) and an atom of type O (carbonyl oxygen) and has a force constant and equilibrium value that is used for all carbonyl bonds. These values are different for other types of C-O bonds, such as the C-O bond in an alcohol (type CT – type OH).
For each atom, we determine whether it can be described by an atom type in parm99. If it cannot, we look in GAFF for an atom type that describes that atom. We then create a new atom type and define all parameters containing this atom type by analogy to values in parm99 or GAFF.
1 J. Am. Chem. Soc. 1995, 117, 5179-5197. 2 J. Comput. Chem. 2004, 25, 1157-1174.

+Atom Type Assignment
All atoms except the three in red were assigned the same type as they have in thymine. There are no parameters in parm99 for non-aromatic conjugated systems, so parameters from GAFF were used.
The new atom types CE and CF were assigned to these atoms and parameters for every bond, angle, and dihedral containing CE and CF were assigned by analogy to either parm99 or GAFF.
For example, the parameters for the CM-CF bond are the same as the CM-CM parameters from parm99 and the parameters for the CE-CF bond are the same as the ce-cf parameters from GAFF
Example: Thymine linker
CM
CM C
NA
C
O
O
(HC)3CT
N*
H
H4
c2
ce
ce
cf
cf
c2CF
CM C
NA
C
O
O
CF
N*
CE
H
HA
HA
H4
parm99 atom types
C: carbonyl carbon
CM: sp2 carbon
CT: sp3 carbon
H: H attached to N
H4: aromatic H on C bonded to one electron-withdrawing group
HA: H attached to aromatic carbon
HC: H attached to aliphatic carbon
NA: aromatic nitrogen
N*: sp2 nitrogen
O: carbonyl oxygen
GAFF atom types
c2: sp2 carbon (aliphatic)
ce/cf: inner sp2 carbon in conjugated chain systems
Thymine (parm99)
Conjugated chain (GAFF)
Thymine linker
C NH
O
O
C
N
C
H
H

+Setup of MD Simulations
The parameters are saved into a LEaPlibrary file (charges, atom types) or a forcemod (bond, angle, dihedral parameters) which are used when creating input files for MD simulations.
The oligo-dye constructs are created by manually attaching the dyes to the oligonucleotide and minimizing the resulting structure.
Na+ ions are added to neutralize the charge of the system. Excess Na+ and Cl- is added to give [NaCl] = 10 mM, which mimics cellular ionic strength. This system is then explicitly solvated in a truncated octahedral water box (MD is run with periodic boundary conditions).
MD simulations are ongoing. We will obtain at least 100 ns of simulation for each system before attempting comparison to experiment in order to give the system time to sample multiple conformations.

+Current and Future Work
The structural information from the MD simulation will be used to generate simulated experimental results by modeling the electronic coupling between the donor and acceptor throughout the MD trajectory. This can be done by approximating the electronic coupling as transition dipole – transition dipole coupling or more rigorously by using QM methods to calculate the electronic coupling.1
We are attempting to generate three types of simulated experimental data
Donor decay (for comparison to bulk time-resolved fluorescence data)
Average FRET efficiency (for comparison to steady-state FRET experiments)
Histogram of FRET efficiencies (for comparison to single-molecule FRET experiments)
We are currently working with simulations generated by previous group members Darren VanBeek (simulation of the protein hen egg-white lysozyme (HEWL) tagged with DACM and eosin) and Katie Hinkle (simulations of DNA tagged with Pacific Blue and Fluorescein) to determine how to sample the MD trajectory.
1 Biophys. J. 2009, 96, 4779-4788

+Acknowledgements
Dr. Krueger Past and present Krueger group members, especially David
Paul, Bryan Leland, Darren VanBeek, and Katie Hinkle Mr. Paul Van Allsburg Dr. Ross Walker (UCSD)
Hope College NSF HHMI ACS-PRF NIH
MU3C Teragrid


















