
Microarray Normalization Xiaole Shirley Liu STAT115 / STAT215.
28
Microarray Normalization Xiaole Shirley Liu STAT115 / STAT215
-
Upload
sharlene-flynn -
Category
Documents
-
view
226 -
download
3
Transcript of Microarray Normalization Xiaole Shirley Liu STAT115 / STAT215.

Microarray Normalization
Xiaole Shirley Liu
STAT115 / STAT215

2
Affymetrix Microarray Imagine Analysis
• Affymetrix GeneChip Operating System (GCOS)– Gridding: based on spike-in DNA– cel file
X Y MEAN STDV NPIXELS
701 523 311.0 76.5 16
702 523 48.0 10.5 16
– cdf file• Which probe at (X,Y) corresponds to which probe
sequence and targeted transcript• MM probes always (X,Y+1) PM

Normalization
• Try to preserve biological variation and minimize experimental variation, so different experiments can be compared
• Assumption: most genes / probes don’t change between two conditions
• Normalization can have larger effect on analysis than downstream steps (e.g. group comparisons)
3

4
Median Scaling
• Linear scaling– Ensure the different arrays have the same
median value and same dynamic range
– X' = (X – c1) * c2
array2 array2
arra
y1
arra
y1

5
LOESS
• LOcally WEighted Scatterplot Smoothing, more general form is LOESS
• Fit a smooth curve– Use robust local linear fits– Effectively applies different scaling factors at
different intensity levels– Y = f(X)– Transform X to X' = f(X)– Y and X' are comparable

6
Quantile Normalization
Probes
Experiments Mean
• Bolstad et al Bioinformatics 2003– Currently considered the best normalization method
– Assume most of the probes/genes don’t change between samples
• Calculate mean for each quantile and reassign each probe by the quantile mean
• No experiment retain original value, but all experiments have exact same distribution

How to Visualize Microarray Normalization?
7

8
Dilution Series
• RNA sample in 5 different concentrations
• 5 replicates scanned on 5 different scanners
• Before and after quantile normalization
9
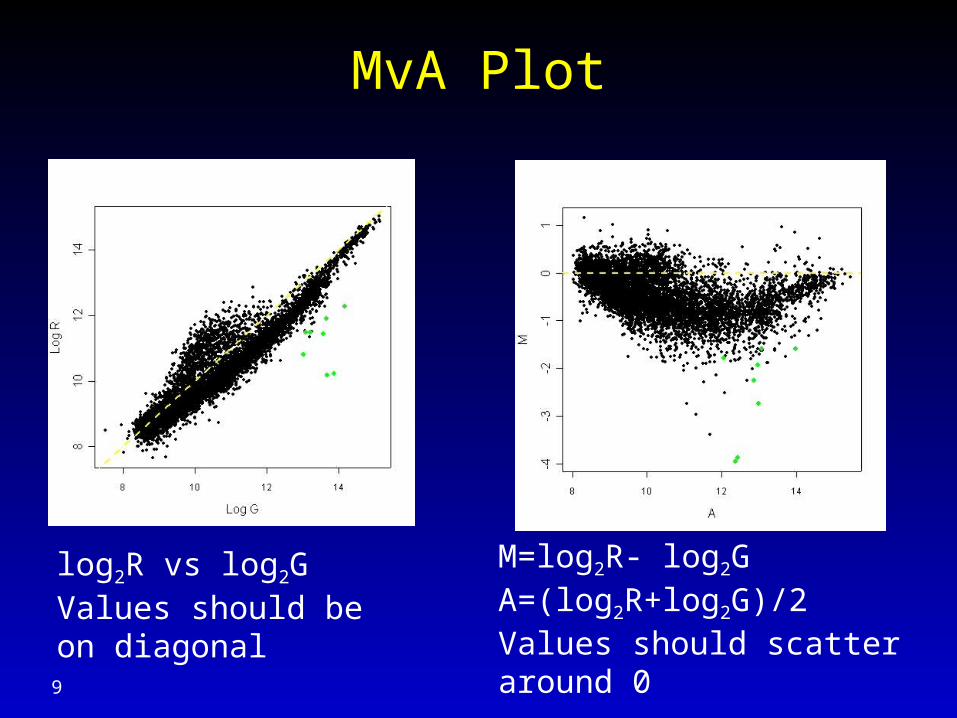
MvA Plot
log2R vs log2G Values should be on diagonal
M=log2R- log2GA=(log2R+log2G)/2Values should scatter around 0
10
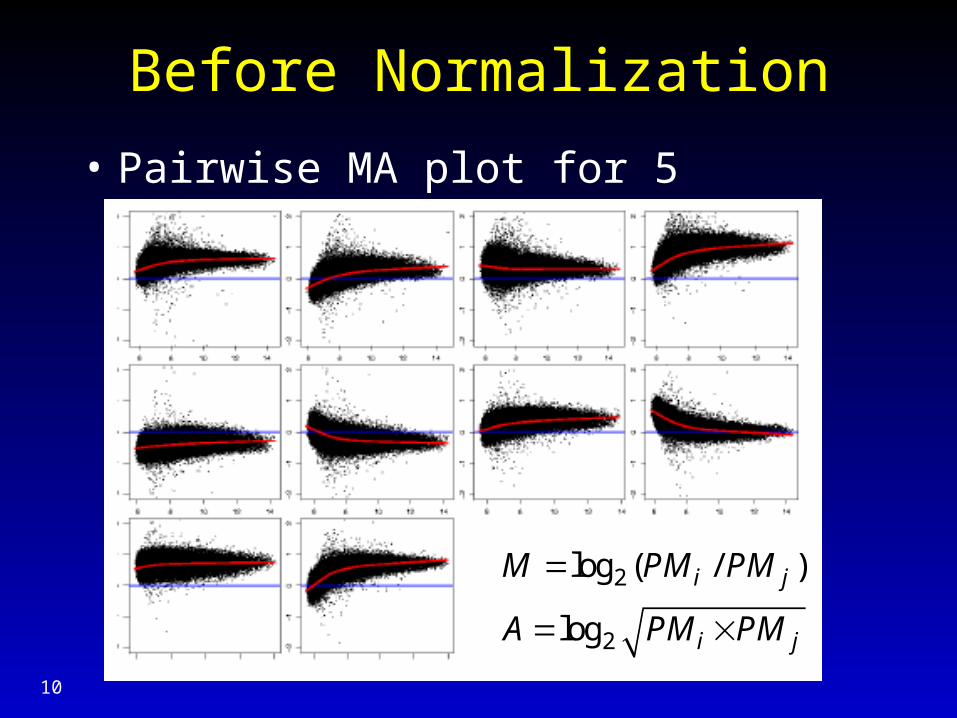
Before Normalization
• Pairwise MA plot for 5 arrays, probe (PM)
2
2
log ( / )
log
i j
i j
M PM PM
A PM PM
11
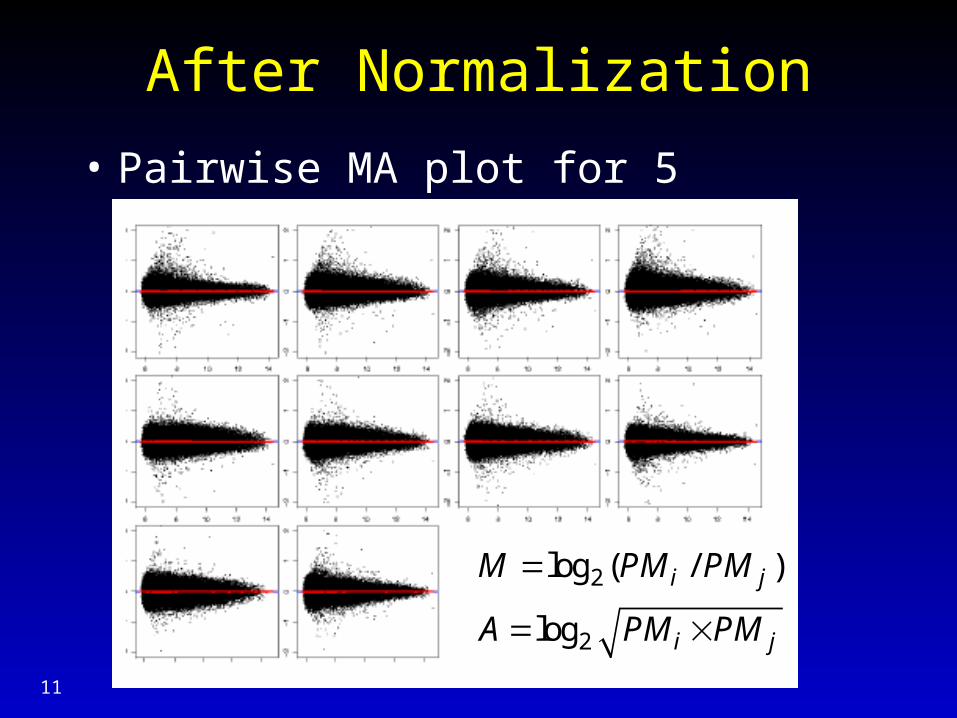
After Normalization
• Pairwise MA plot for 5 arrays, probe (PM)
2
2
log ( / )
log
i j
i j
M PM PM
A PM PM

Gene Expression Index
13
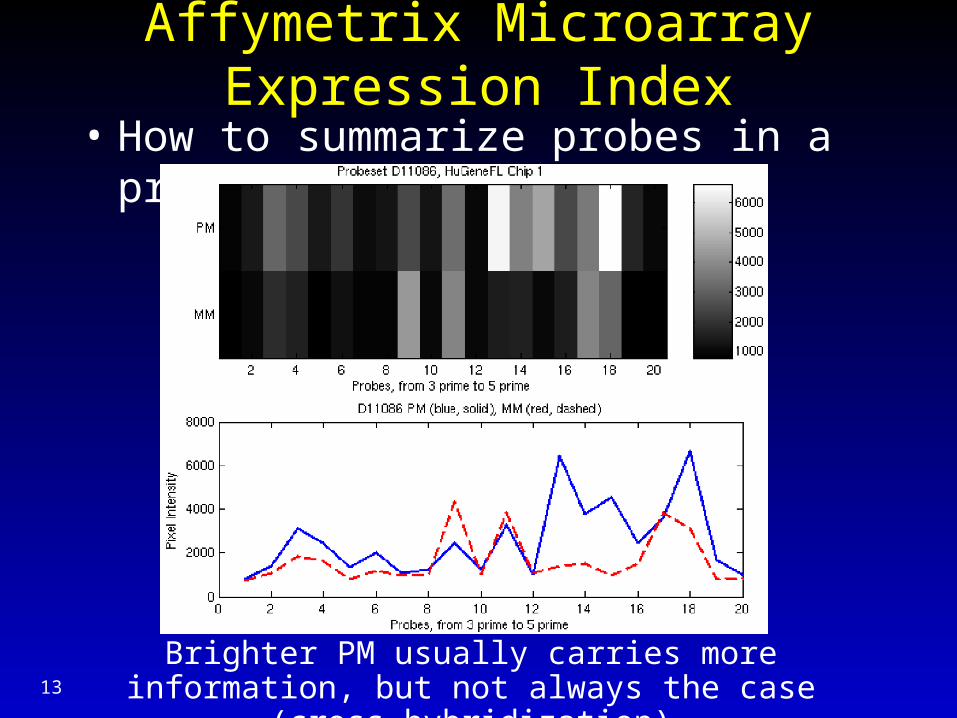
Affymetrix Microarray Expression Index
• How to summarize probes in a probeset?
Brighter PM usually carries more information, but not always the case (cross-hybridization)

14
MAS4• GeneChip® older software Microarray Analysis
Software 4.0 uses AvgDiff
• A: a set of suitable pairs chosen by software– Remove highest/lowest– Calculate mean, sd from remaining probes– Eliminate probes more than 3 sd from mean
• Drawback (naïve algorithm):– Can omit 30-40% probes – Can give negative values
j
jj MMPMAvgDiff )(1

15
MAS5• GeneChip® newest version
• Tukey Biweight down-weights points far from the estimated center of the data scatter, robust statistics resistant to outliers
• CT* (change threshold) a version of MM that is never bigger than PM– If MM<PM, CT* = MM– If MM>PM, estimate typical case (Tukeybiweight)
MM for PM (~70% PM)– If typical MMs > PM for, set CT* = PM -
• Works OK but ad hoc
)}{log( *jj CTPMghtTukeyBiweisignal
16
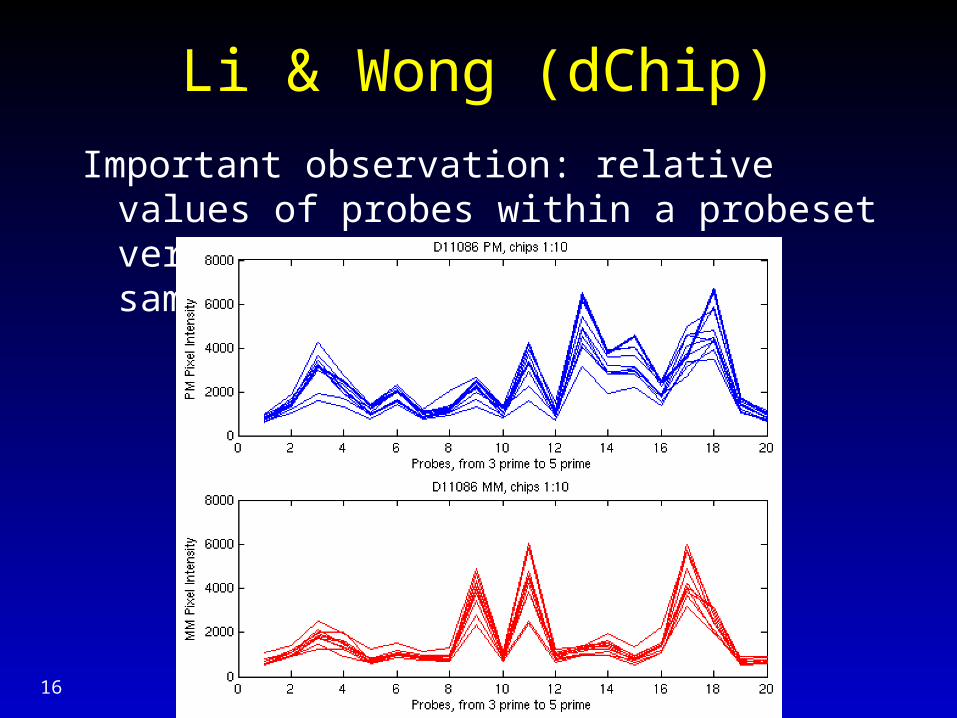
Li & Wong (dChip)
Important observation: relative values of probes within a probeset very stable across multiple samples.

17
Model-Based Expression Index
• Look at multiple samples at a time, give different probes a different weight
• Each probe signal is proportional to – Amount of target sample:
– Affinity of specific probe sequence to the target: j
1
2
Probes 1 2 3
sample 1
sample 2
18
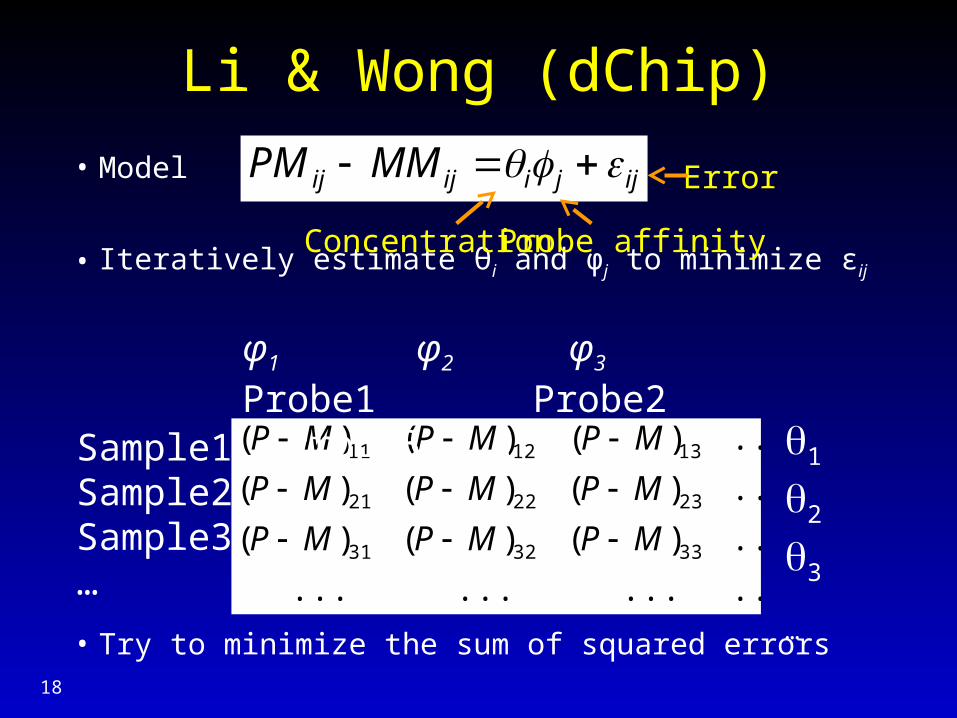
Li & Wong (dChip)
• Model
• Iteratively estimate θi and φj to minimize εij
• Try to minimize the sum of squared errors
ijjiijij MMPM
............
...)()()(
...)()()(
...)()()(
333231
232221
131211
MPMPMP
MPMPMP
MPMPMP
Sample1
Sample2Sample3…
φ1 φ2 φ3
Probe1 Probe2 Probe3 …1
2
3
…
Concentration Probe affinity
Error

19
RMA = Robust Multi-chip Analysis
• Irizarry & Speed, 2003
• 1: Probe intensity background adjustment
• 2: Quantile normalize the Log transformed background adjusted PM
• 3: Robust probe summary
20
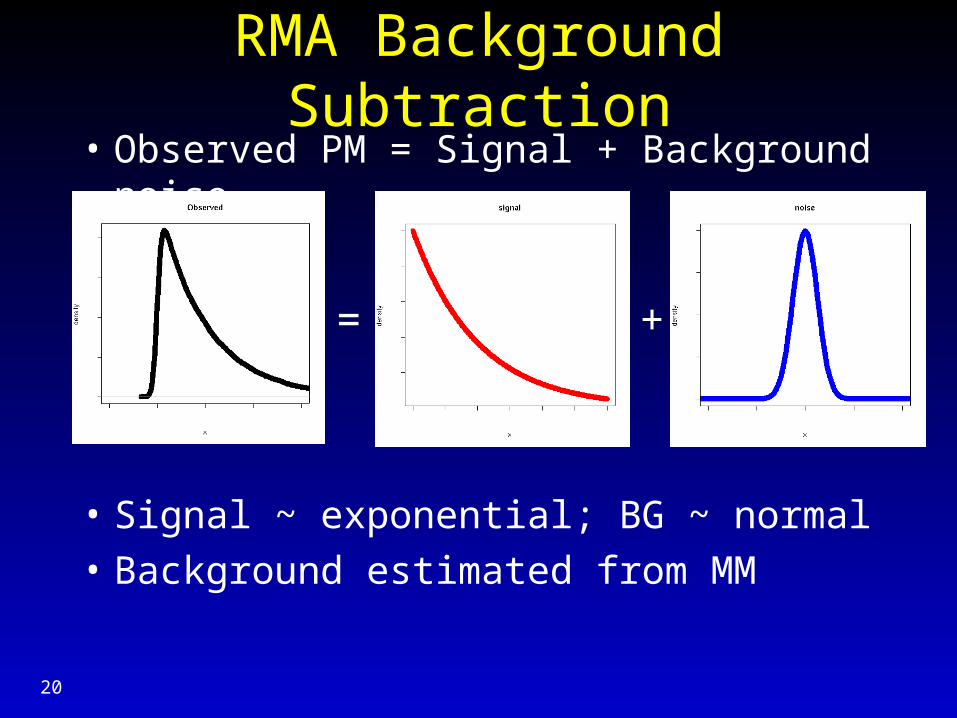
RMA Background Subtraction• Observed PM = Signal + Background noise
• Signal ~ exponential; BG ~ normal
• Background estimated from MM
+=
21
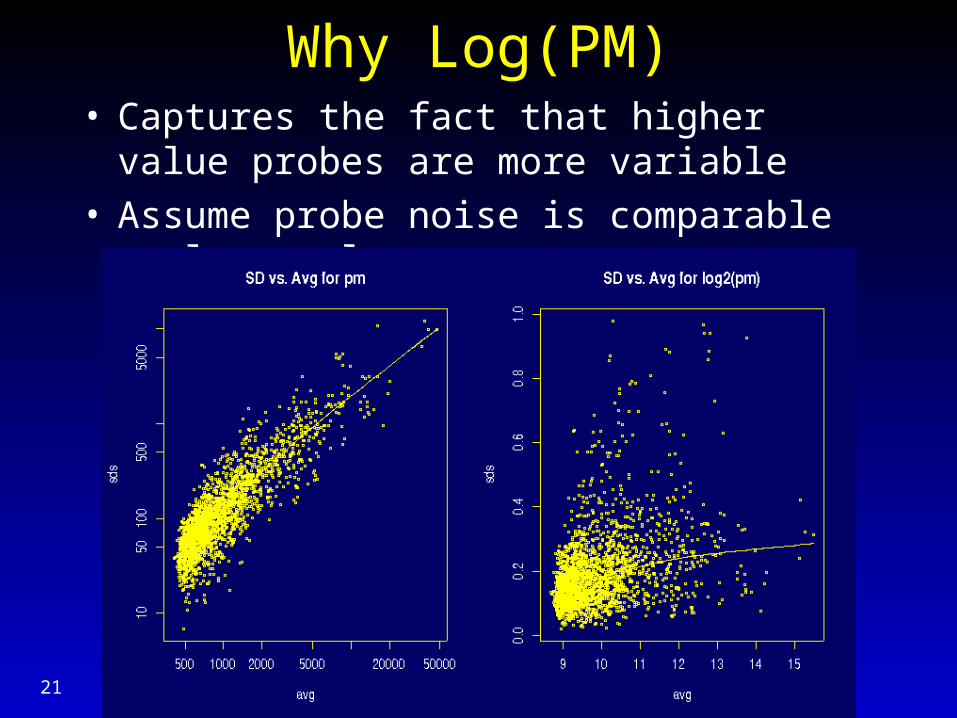
Why Log(PM)• Captures the fact that higher value probes are
more variable• Assume probe noise is comparable on log scale

22
• For each probe set, PMij = ij
• Fit the model:
– aj is expression index, bj is probe effect– Log2n() stands for logarithm after quantile
normalization of n samples
RMA
)log()log()(log jiijPM
ijjiij baPM )bg(nlog2

RMA
• Examples…• Iteratively refit aj and bj using median
polish– Alternately remove (subtract) row and column
medians until sum of absolute residuals converges
– For complex data structures, can efficiently find a “general picture” of the data
– Robust to outliers in large data sets
• Similar to dChip, but minimize error at logPM, so less weight on large PMs
23

Gene Expression IndexMethod Comparison

25
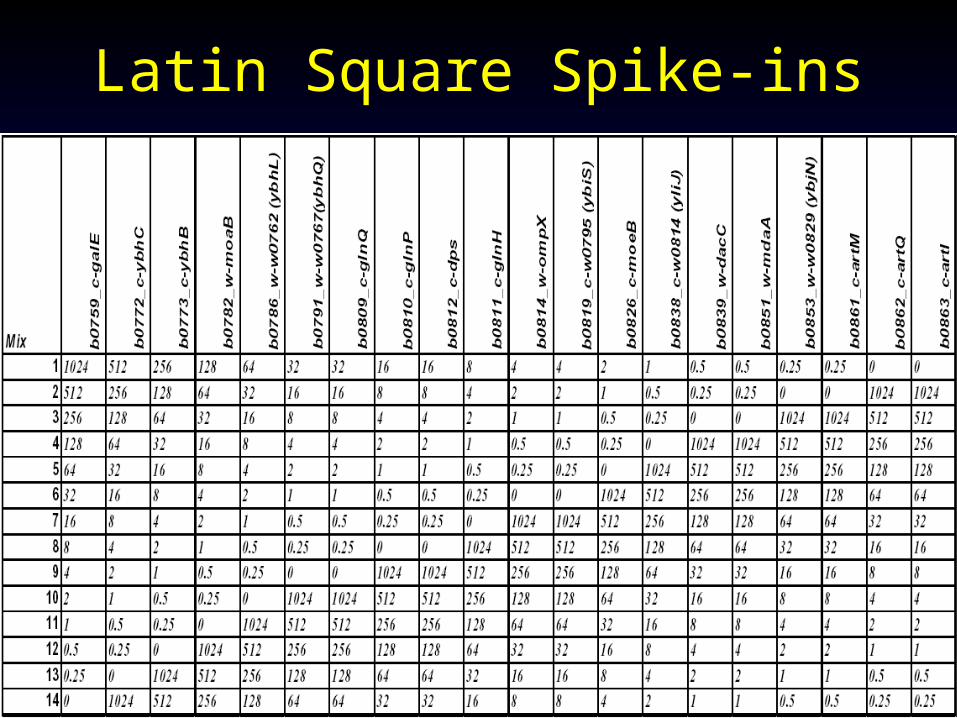
Method Comparison Standard• Spike-ins: introduce markers with known
concentration (intensity) to RNA samples– Should cover a broad range of concentrations– Run two samples with and without spike-in, see
whether algorithm can detect the spike-in (differential expression)
• Dilutions: – Serial dilutions: 1:2, 1:4, 1:8…
• Latin square spike-in captures both approaches above
• Compare both accuracy qualitatively and expression index quantiatively
26
Latin Square Spike-ins

Method Comparison of Spike-in
27
MAS4 MAS 5
dChip RMA
Red numbers indicate spikedgenes

Summary
• Cel file and cdf file. • Array normalization: Loess, qnorm
– Assumptions
• Normalization visualization: MA plots• Gene Expression Index
– RMA models probe effect in expression arrays
– Use MM to correct background
– Qnorm log (PM)
– Median polish, model probe behavior to get expression index
• Method comparison28


















