[Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 ||...
13
[27] Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics in Living Mammalian Cell Nuclei By Diana A. Stavreva and James G. McNally Introduction Fluorescence recovery after photobleaching (FRAP) is an optical tech- nique used to measure the temporal dynamics of fluorescently tagged molecules. FRAP is a relatively old technique, 1–5 but its application to the study of proteins in living cells is more recent. This renaissance is driven largely by the advent of confocal microscopy and the introduction of green fluorescent protein (GFP) as an endogenous protein marker. After GFP (or another fluorophore) is covalently attached to the pro- tein of interest, its cellular distribution can be visualized at low light inten- sities that do not damage cellular processes. Photobleaching is the irreversible destruction of fluorescence in a region within the sample by brief exposure to high light intensities. After bleaching a region, the recov- ery of fluorescence over time there can be recorded to measure the rate of redistribution of fluorescent molecules. This rate of fluorescence redistribution provides information about the processes involved in the movement of the molecule. The movement re- flects diffusion, which can be retarded if the fluorescently tagged molecule binds to other molecules. In the latter case, the rate of redistribution of bleached and unbleached molecules will contain information about the strength of the binding interaction. Thus, FRAP is a valuable technique to study molecular dynamics in live cells. Designing a FRAP Experiment The most commonly available instrumentation for FRAP is the laser scanning confocal microscope, the scanning capability of which provides 1 M. Poo and R. A. Cone, Nature 247, 438 (1974). 2 P. A. Liebman and G. Entine, Science 185, 457 (1974). 3 M. Edidin, Y. Agyansky, and T. J. Lardner, Science 191, 466 (1976). 4 J. Schlessinger, D. E. Koppel, D. Axelrod, K. Jacobson, W. W. Webb, and E. L. Elson, Proc. Natl. Acad. Sci. USA 73, 2409 (1976). 5 D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson, and W. W. Webb, Biophys. J. 16, 1055 (1976). [27] FRAP for visualizing protein dynamics 443 Copyright 2004, Elsevier Inc. All rights reserved. METHODS IN ENZYMOLOGY, VOL. 375 0076-6879/04 $35.00
Transcript of [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 ||...
![Page 1: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/1.jpg)
[27] FRAP for visualizing protein dynamics 443
[27] Fluorescence Recovery after Photobleaching (FRAP)Methods for Visualizing Protein Dynamics in Living
Mammalian Cell Nuclei
By Diana A. Stavreva and James G. McNally
Introduction
Fluorescence recovery after photobleaching (FRAP) is an optical tech-nique used to measure the temporal dynamics of fluorescently taggedmolecules. FRAP is a relatively old technique,1–5 but its application tothe study of proteins in living cells is more recent. This renaissance is drivenlargely by the advent of confocal microscopy and the introduction of greenfluorescent protein (GFP) as an endogenous protein marker.
After GFP (or another fluorophore) is covalently attached to the pro-tein of interest, its cellular distribution can be visualized at low light inten-sities that do not damage cellular processes. Photobleaching is theirreversible destruction of fluorescence in a region within the sample bybrief exposure to high light intensities. After bleaching a region, the recov-ery of fluorescence over time there can be recorded to measure the rate ofredistribution of fluorescent molecules.
This rate of fluorescence redistribution provides information about theprocesses involved in the movement of the molecule. The movement re-flects diffusion, which can be retarded if the fluorescently tagged moleculebinds to other molecules. In the latter case, the rate of redistribution ofbleached and unbleached molecules will contain information about thestrength of the binding interaction. Thus, FRAP is a valuable techniqueto study molecular dynamics in live cells.
Designing a FRAP Experiment
The most commonly available instrumentation for FRAP is the laserscanning confocal microscope, the scanning capability of which provides
1 M. Poo and R. A. Cone, Nature 247, 438 (1974).2 P. A. Liebman and G. Entine, Science 185, 457 (1974).3 M. Edidin, Y. Agyansky, and T. J. Lardner, Science 191, 466 (1976).4 J. Schlessinger, D. E. Koppel, D. Axelrod, K. Jacobson, W. W. Webb, and E. L. Elson,
Proc. Natl. Acad. Sci. USA 73, 2409 (1976).5 D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson, and W. W. Webb, Biophys. J. 16, 1055
(1976).
Copyright 2004, Elsevier Inc.All rights reserved.
METHODS IN ENZYMOLOGY, VOL. 375 0076-6879/04 $35.00
![Page 2: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/2.jpg)
444 molecular cytology of chromatin functions [27]
flexibility in defining the bleach and imaging regions. The confocal micro-scope must also have the ability to rapidly change the laser illumination in-tensity and collect a time-lapse series of images. Conventional microscopeconfigurations with charge-coupled device (CCD) cameras can also be usedfor FRAP.6
After identifying a region of interest, the strategy in a typical experimentis to (1) collect a set of images before the bleach (to be used for normaliza-tion of the collected intensities after the bleach), (2) bleach the selected areaby scanning it with high laser power, and (3) monitor the recovery of fluor-escence by scanning with low laser power over time until the fluorescent andphotobleached molecules redistribute to equilibrium. The time course ofintensity changes reflects the fluorescence recovery, and this generates aFRAP curve that provides quantitative information about the mobility ofthe fluorescent molecules (Fig. 1). Analyses of the FRAP curve can showthe presence of an immobile or a transiently immobilized fraction, and also,in principle, provide information about both the diffusion coefficient andthe strength of binding interactions occurring within the bleached region.Here we focus on FRAP, because it is the most widely used photobleachingstrategy, but other photobleaching approaches are sometimes valuable. Thescanning capabilities of the confocal microscope enable two relatively newapproaches known as fluorescence loss in photobleaching (FLIP) andinverse FRAP (IFRAP). Here we briefly describe these two approaches.For more information, consult Cole et al.7 and McNally and Smith.8
FLIP is a technique in which fluorescence is bleached at one site in thecell, and loss of fluorescence is monitored at another site in the cell. Its chiefvalue is to assess whether there is exchange of molecules between two com-partments. For example, if a protein is localized to nucleoli, it may be ofinterest to know whether there is exchange of the protein between individ-ual nucleoli, or whether once targeted to the nucleolus, the protein remainsthere. FLIP can be used to assess this by bleaching fluorescence in one nu-cleolus and then monitoring whether there is loss of fluorescence in othernucleoli. If there is no exchange between different nucleoli, then bleachingone nucleolus will not affect the fluorescence in other nucleoli. On theother hand, if there is some exchange between nucleoli, then bleachingone nucleolus will eventually reduce fluorescence in other nucleoli. Todetect this loss of fluorescence in other nucleoli, it is often necessary to
6 A. S. Verkman, Methods Enzymol. 360, 635 (2003).7 N. B. Cole, C. L. Smith, N. Sciaky, M. Terasaki, M. Edidin, and J. Lippincott Schwartz,
Science 273, 797 (1996).8 J. G. McNally and C. L. Smith, in ‘‘Confocal and Two-Photon Microscopy: Foundations,
Applications, and Advances’’ (A. Diaspro, ed.), p. 525. Wiley-Liss, New York, 2002.
![Page 3: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/3.jpg)
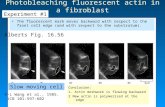
Fig. 1. FRAP of the GFP-tagged glucocorticoid receptor (GFP–GR) at its regulatory sites.
Cell line 3617, stably containing 200 tandem repeats of a 9-kb element composed of the
MMTV promoter followed by ras and BPV genes stably expressing GFP-tagged GR under
the control of a tetracycline-off system [D. Walker, H. Htun, and G. L. Hager, Methods 19,
386 (1999)], was used. (A) After hormone induction, a population of the cells shows a visible
bright spot in the nucleus corresponding to the MMTV array. (B) Series of images showing
the recovery of the GFP–GR after photobleaching. For faster imaging, only a strip of the
nucleus including the structure of interest was imaged. Resulting individual FRAP curves are
rather noisy (C) but the average of 12 individual curves (D) gives a good representative FRAP
curve.
[27] FRAP for visualizing protein dynamics 445
repeat the photobleach in the initial nucleolus as it recovers fluorescence.This gradually depletes the shared pool of fluorescent molecules, finallyleading to a clear loss of fluorescence in all of the other nucleoli.
IFRAP is a photobleaching strategy in which all regions of the cellexcept the region of interest are bleached. Loss of fluorescence from theregion of interest is then monitored. This approach is especially useful asa control to test whether loss of fluorescence in FRAP is an artifact due tobleaching the molecules there. In IFRAP, these molecules are not bleached,so if they still leave the site, this loss cannot arise from damage due to photo-bleaching. As long as the distribution of fluorescence is at equilibriumbefore bleaching, the loss of fluorescence from the site in IFRAP shouldoccur at the same rate as the recovery of fluorescence at the site in FRAP.
![Page 4: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/4.jpg)
446 molecular cytology of chromatin functions [27]
Cells and Cell Culture Conditions
The first step in a FRAP experiment is to demonstrate that the fluores-cently tagged protein of interest is functional and associates with the appro-priate compartments within the cell. Either cell lines stably expressing thefluorescently tagged protein or transiently transfected cells can be used.
It is preferable to use stable cell lines in which the protein under study isnot significantly overexpressed. Overexpression can lead to an unnaturalsituation in which nonspecific binding reactions may arise that alter themobility of the protein. Thus, stable cell lines should be selected for mod-erate expression levels. Alternatively, expression of the fluorescentlytagged protein can be placed under the control of a regulated promoter,such as the tetracycline-regulated system.
To prepare cells for a FRAP experiment, they must be grown in achamber suitable for live cell imaging. The basic requirements are thatthe cells grow on a glass surface equivalent to a standard no. 1.5 coverslipfor which all microscope objectives are designed. In addition, the chamberideally should enable temperature and pH control by CO2. A number ofsuch systems are commercially available [Bioptechs (Butler, PA); CarlZeiss (Thornwood, NY); binomics chamber [20/20 Technology, Wilming-ton, NC); Harvard Apparatus (Holliston, MA)], ranging in cost from$3000 to $20,000. A less expensive alternative that offers less precise con-trol of environmental conditions is a live cell coverslip chamber (cham-bered coverglass system; Nalge Nunc International, Naperville, IL). Byheating the microscope stage with an air stream stage incubator (Nevtek,Burnsville, VA) the temperature in these chambers can be regulated, whilepH can be controlled by using a buffer that does not require CO2.
For any of these chambers, cells must first be transferred to the cham-ber in phenol red-free growth medium. Phenol red is fluorescent; thus,removing it from the medium reduces background fluorescence. If theGFP-tagged protein is under tetracycline control (positive or negative),then the antibiotic must be added or removed to induce expression. Cellsare typically incubated overnight in the appropriate chamber to permitattachment. If the cells are not adherent, then it is essential to induce at-tachment by growth on a modified surface such as polylysine, becausefluorescence recovery must be measured from a stationary cell. It is recom-mended that the concentration of suspended cells giving 60 to 70% cellconfluence the next day be experimentally determined. If the cells arenot confluent, it takes more time to find them and to perform enoughFRAP measurements. When the cells are too nearly confluent, it becomesdifficult to distinguish individual cells, and overgrowth may also lead to un-healthy conditions that could alter the function and mobility of the protein.
![Page 5: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/5.jpg)
[27] FRAP for visualizing protein dynamics 447
If transiently transfected cells are used, they can be examined 18–24 hposttransfection. Again, it is essential to perform FRAP experiments oncells with low expression levels that mirror the endogenous distributionof the protein. Immunofluorescence of the endogenous protein can be usedas a control.
For reproducibility and reliability of collected data, optimal growthconditions should be maintained during the FRAP experiment. FRAP isa sensitive technique and for some proteins even small deviations fromthe optimal growth state can lead to slower fluorescence recovery. Keepingcells at 37
�by using stage and objective heaters and maintaining the proper
pH (using 5% CO2 when necessary) is especially critical when the mobilityof the protein is energy dependent. It is also a useful control to compareFRAP curves measured in the first few minutes on the microscope stagewith those measured at later times. If these differ, then conditions formaintaining the cells on the microscope become suspect.
Setting up the Microscope
Different proteins have specific functions, binding partners, and inter-actions of different strength. As a result, mobilities differ from protein toprotein, and so for each protein under investigation, specific experimentalconditions (frame rate, bleach intensity, and duration of measurement)must be established.
Controlling Laser Illumination Intensity
There are three general ways to change the illumination intensity: byvarying the output of the laser, by inserting the neutral density filter infront of the laser, or by using an acousto-optical modulator (AOM) oracousto-optic tunable filter (AOTF). Acousto-optical devices are includedwith more recent confocal microscopes from Zeiss, Leica (Bannockburn,IL), and Bio-Rad (Hercules, CA). These devices allow 1000-fold variationin illumination intensity within a few seconds, as well as graded controlover laser illumination. The rapid switch in intensity levels is critical, espe-cially for studying the mobility of rapidly exchanging molecules to preventtheir redistribution into the bleached region before the first postbleachimage is taken.
To determine the conditions for photobleaching, fixed cells can be used.Cells are grown on coverslips and fixed for 15 min with 3.5% paraformal-dehyde in phosphate-buffered saline (PBS) with shaking, washed in PBS(three times, 5–10 min each), and mounted on a slide for microscopy.Ideally, conditions should be set to bleach to background levels. In
![Page 6: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/6.jpg)
448 molecular cytology of chromatin functions [27]
practice, this may require multiple iterations of the bleach, which takeslonger to perform. This is a drawback in the case of a fast-moving proteinthat may redistribute extensively during the bleach. Naturally, laser powershould be set at high levels, because this leads to a deeper bleach in lesstime, and also because the laser is more stable in this mode than at lowoutput levels. Another important factor in the rate of bleaching is the zoomsetting on the confocal microscope Photobleaching increases with thesquare of the zoom,9 so the scan area at zoom 5 is 1/25th the area at zoom1 and the rate of bleaching is 25 times faster.
However, if the laser power or zoom is set too high, it may lead to sub-stantial bleaching as the recovery is monitored, even if the AOTF is set atlow levels. Too low a transmission setting for the AOTF (<1%) may alsolead to unstable transmission levels, so a compromise must be found be-tween laser power levels, AOTF levels, and zoom settings that leads torapid and deep bleaching during the photobleach but minimal bleachingwhile monitoring the recovery. In the actual FRAP experiment, a bleachdepth that is 50% of starting intensity is often sufficient.
Defining Scan and Bleach Regions
The most sophisticated microscopes allow the user to define regions tobe bleached that can be any shape or size. This is a considerable advantagebecause many biological structures have unusual shapes and thereforebleaching can be restricted to precisely that structure, or to specific subdo-mains of the structure. This feature also enables IFRAP, in which thebleach region invariably has an irregular shape encompassing the wholefluorescent pool except a small region of interest. These arbitrary bleachingpatterns are achieved by using an AOM or AOTF to blank the beam atdefined times during the scan.
In selecting a region to be imaged, several considerations arise. For rap-idly moving fluorescent molecules, it is advisable to scan as rapidly as pos-sible to enable high temporal resolution for the recovery. Rapid bleachingis achieved by using a high transmission objective (e.g., a Fluar or Planapoobjective) with high numerical aperture (NA), by using only one iterationof the bleach, and by scanning a narrow section from the cell nucleus in-cluding the region or structure of interest, rather than scanning the wholecell. The changes in fluorescence outside the bleached region within thenarrow section are used later for correcting for the loss of intensity dueto the scanning (Fig. 1B).
9 V. Centonze and J. B. Pawley, in ‘‘Handbook of Biological Confocal Microscopy’’
(J. B. Pawley, ed.), p. 549. Plenum Press, New York, 1995.
![Page 7: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/7.jpg)
[27] FRAP for visualizing protein dynamics 449
For both better reproducibility and easier comparison of FRAP curvesgenerated on different days or under different conditions (mutant proteinor drug treatment), it is critical to use the same parameters for all experi-ments (magnification, bleach spot size, laser power, scan speed, number ofiterations, time lapse intervals, and duration). Despite this, in our experi-ence, some small day-to-day differences between the FRAP curves arisewhen using well-standardized experimental conditions. As a consequence,to permit valid comparisons for each experimental treatment, controlexperiments must always be performed in parallel.
Collecting Data
Before starting the experiments, it is necessary to empirically determinethe frame rate and duration of measurement. Most nuclear proteins recovermore slowly than expected for free diffusion, that is, with a total recoveryperiod on the order of tens of seconds, to minutes. However, there is usuallya substantial change in the first few seconds often accounting for 50–80% ofthe total recovery. Thus, it is usually necessary to obtain high temporal reso-lution in the first few seconds of the recovery phase. This can be achieved byusing the AOM or AOTF and high-throughput objectives as described inControlling Laser Illumination Intensity (see previously). In addition, a shortinterval between the bleach and the first time point is important. This intervalis a function of the instrumentation and software in use, and should be con-sidered when selecting a specific microscope. Rapid bleaching is importantto provide a more accurate determination of the first time point’s intensity.This is achieved by using high laser power, using a high transmission object-ive, selecting a small bleach area, and using only one iteration of the bleach.
To determine how much time is needed to record the recovery, a pilotexperiment should be performed to measure the recovery at later timepoints (after at least 5–10 s or even minutes). The recovery curve will flat-ten at some time point, so enough data must be collected to observe thisflattening. The time point at which flattening occurs may change with dif-ferent mutants or drug treatments, so different experiments will requiredifferent durations.
In our experiments, we typically acquire �100 data points during therecovery phase. The number of collected data points is a compromise be-tween acquiring enough time points to generate a smooth recovery curveand minimizing the photobleaching during recovery. As a rule, this photo-bleaching should not be greater than 5–10%; otherwise, any errors in cor-recting for this bleaching will contribute substantially to the final FRAPcurve. When both steep and shallow components are present in a curve,it is preferable to acquire images at highest scan speed during the steep
![Page 8: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/8.jpg)
450 molecular cytology of chromatin functions [27]
phase, and then increase the time interval between images during theshallow phase. This reduces bleaching during the recovery.
The parameters used for imaging the recovery are important. Allrecorded intensity values should be within the detector range (no satur-ation). For a 12-bit photomultiplier tube, this means that no pixel valueshould be at 4095. This ensures that the FRAP curve is quantitative. An-other parameter that needs to be set is the pinhole diameter, which impactsthe signal-to-noise ratio. A larger pinhole improves the signal-to-noiseratio, but also increases the optical section thickness. Thicker opticalsections may yield recovery data from undesired regions above or belowthe structure of interest.
In our experiments, the area monitored during recovery is typicallylarger than the bleached region. This makes it possible to simultaneouslycollect data from the same cell to correct for bleaching due to imagingand also makes it possible to verify that the specimen remains in focusduring the experiment. When data are collected in a strip image thatextends beyond the bleach spot along the scanning axis, data collectiontime is not increased on a Zeiss 510 confocal microscope. This is becausethe microscope always scans the full length of the axis anyway, whetheror not a subregion has been selected.
To generate a FRAP recovery curve from the image data, average in-tensities at each time point must be calculated for the bleached region(on the bleach spot), for the region to be used for the bleach correctiondue to monitoring (off the bleach spot), and for the background signalin the image (bgd). Care should be exercised in selecting the region forbleach correction so that it is far enough away from the bleach spot so asnot to be influenced by the intensity changes occurring near the bleachspot. Most confocal microscope software packages enable calculation ofthese three values by drawing the three regions of interest on the first timepoint of the image sequence. The software then computes the averageintensity at every time point for each of the three regions. For these meas-urements to be accurate, it is critical to ensure that the cell has remained infocus and not drifted during the course of the experiment. Thus, the imagesequence should be examined carefully with particular attention paid towhether the bleach spot region appears to be shifting in any way. Thenumbers generated from the region measurements can then be importedinto a spreadsheet program. After the data are tabulated, the next step isto subtract the background level intensity from all of the measurements(on � bgd and off � bgd). The background-corrected data are furthercorrected for bleaching due to monitoring (I ¼ on � bgd divided byoff � bgd). Computed intensities (I) are further normalized to the averageintensity of the prebleached region [Inorm ¼ I divided by a constant
![Page 9: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/9.jpg)
[27] FRAP for visualizing protein dynamics 451
(average intensity of prebleached I values)]. Typically, we acquire atleast 5–10 prebleach images to provide a good estimate of the prebleachvalues.
Once the corrected and normalized recovery data are generated, theyshould be plotted as a function of time, and the resultant FRAP curveexamined. In some cases, there may be large spikes in the curve due to afluctuation in laser intensity or a sudden movement of the cell. Such datashould be discarded. In addition, sometimes the FRAP curve will risesteadily, but then start to gradually decline in intensity at later time points.This is physically impossible, and indicates either that the cell is drifting outof focus, or that there is some problem with the bleach correction. In othercases, the recovery curve will increase steadily and become significantlylarger than one. This is also physically impossible, and again indicates prob-lems with either cell focus or the bleach correction. All such anomalousdata should be discarded. Individual FRAP curves are often rather noisy(Fig. 1C) due to the low light levels used for imaging, and possibly to smalljiggling motions in the live cell under study. In addition, there may be sub-stantial cell-to-cell variability in FRAP curves, reflecting not only thisnoise, but also possibly subtle physiological differences among cells. Toobtain a good, representative FRAP curve, about 10 individual FRAPmeasurements should be averaged and plotted as a function of time(Fig. 1D). When comparing FRAP curves obtained from controls withthose from mutant proteins or drug-treated cells, differences may be smallbut real. This reflects the fact that diffusion often contributes significantlyto the FRAP recovery, and does not change even though binding inter-actions do. To detect small differences, the experiment should be per-formed on at least three separate days to determine whether the shiftbetween the two curves is reproducible.
Experiments with FRAP
The FRAP technique is not limited only to studies of protein mobilityunder normal physiological conditions. It can also be used in different ex-periments in which the GFP fusion protein is altered either by mutations orspecific drug treatments. By comparing the resultant fluorescence recoverycurve with the control recovery curve, useful information about changes inprotein mobility and binding affinity can be obtained. Using FRAP, it ispossible to determine whether protein mobility is an energy-dependentprocess. To answer that question, FRAP experiments must be performedat 37
�to generate a control FRAP curve and compared with curves pro-
duced either at 22�
or in the presence of an ATP inhibitor (e.g., sodiumazide). If these conditions do not alter the FRAP recovery, then protein
![Page 10: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/10.jpg)
Fig. 2. After treatment with an ATP inhibitor (sodium azide), the resulting FRAP curve is
slower, which indicates involvement of energy-dependent factors in the mobility of the
protein.
452 molecular cytology of chromatin functions [27]
mobility is not energy dependent. If changes are observed, then energydependence is indicated. Typically, for energy dependence, fluorescencerecovery will be much slower than the control and exhibit an immobilefraction (Fig. 2).
When performing FRAP experiments, it is advisable to limit the timethat a specimen remains on the microscope stage. For energy-independentprotein mobility, 2 h is the recommended maximum; 1 h or less is recom-mended for either energy-dependent mobility or after any form ofdrug treatment. These limits need to be assessed by several controls. Thesimplest is a test for cell viability after the FRAP experiment, for example,by trypan blue exclusion. In addition, it is critical to compare FRAP re-covery curves obtained immediately after the cells are placed on themicroscope with those obtained later. If there are differences, this canindicate problems with temperature or pH control on the stage, or simplynonspecific effects arising from prolonged imaging of the sample.
A second control is needed to assure that a drug treatment or other dis-ruption has not changed the mobility of all proteins in the nucleus. Asimple approach here is to transfect cells with GFP only, and then treatwith the same concentration of test drug or with vehicle. GFP is uniformlydistributed in the whole cell and its dynamics are governed by pure
![Page 11: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/11.jpg)
Fig. 3. Double-bleach experimental approach. After the first bleach, the same region of
interest is bleached again (A) and the resulting FRAP curves are compared (B). In the case of
an immobile fraction, the second bleach leads to a complete recovery because the immobile
molecules are now invisible.
[27] FRAP for visualizing protein dynamics 453
diffusion.10 The recovery of the GFP-expressing, drug-treated cells shouldbe compared with the recovery of the control cells (treated only withvehicle). No change in GFP recovery rates indicates that the drug treat-ment has not generically changed nuclear mobilities. It is also possible tolook at another functional protein unrelated to the protein under study,and show that its mobility is unaffected by the drug treatment.
A variation of FRAP known as a double bleach is useful to confirm thepresence of an immobile fraction. In this approach, a region of interest isbleached and, after a defined recovery time, a second bleach is performedat the same place (Fig. 3A). Recovery of fluorescence after the secondbleach is compared with recovery after the first bleach (Fig. 3B). In the caseof an immobile fraction, the first bleach renders the bound molecules ‘‘in-visible’’ in the recovery after the second bleach. Thus, the fluorescence
10 R. Swaminathan, C. P. Hoang, and A. S. Verkman, Biophys. J. 72, 1900 (1997).
![Page 12: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/12.jpg)
454 molecular cytology of chromatin functions [27]
available for the second bleach should be fully mobile and recover to100%. If it does not, then the fraction is not truly immobile. The time delaybetween the first and second bleach can be varied to determine how longthe molecules remain ‘‘immobilized.’’
Quantitative Analysis of FRAP Data
At a minimum, FRAP recovery data contain information about the dif-fusion rate of the protein under study. However, in many cases, the proteinnot only diffuses but also interacts with other molecules. These bindinginteractions slow the rate of fluorescence recovery.
As a first step in obtaining estimates of diffusion and binding constants,the recovery of the protein should be compared with the recovery of thefluorescent protein tag alone (e.g., GFP only for a GFP-tagged protein).If under the same bleach conditions the recoveries are nearly identical,then this indicates little or no retardation due to binding, and so the recov-ery reflects principally diffusion of the tagged protein. (The larger size ofthe tagged protein compared with GFP alone has a minimal effect on itsdiffusion constant, because this goes as the cube root of the mass.) Thus,if the protein is not binding, then its recovery will be rapid, because diffu-sion is fast. For a 1-�m-diameter bleach spot, recovery is complete within�1 s for a freely diffusing protein.
If the fluorescence recovery is slower than that of GFP alone, then thisindicates that binding interactions occur. The longer the interaction withother molecules, the slower is the resulting FRAP curve. In the extremecase, when the molecules are completely immobile, the fluorescence ofthe bleached spot does not recover at all and the resulting FRAP curveresembles the curves from a fixed specimen.
For extraction of on and off rates of binding, it is critical to determinewhether diffusion contributes to the recovery curve. In some cases, diffu-sion and binding are uncoupled. This occurs when diffusion is fast com-pared with the binding interactions. In this scenario, diffusion can beignored because it occurs so rapidly, and thus the resultant FRAP curve re-flects only binding interactions. In all other cases, diffusion and binding areinextricably coupled, and this must be considered in extracting estimates ofthe on and off rates.
To determine whether diffusion contributes to the FRAP recovery,bleaches of different spot sizes are performed (Fig. 4). Two different spotsare used: one smaller and one larger than the typical spot used for a FRAPexperiment. If diffusion is extremely rapid compared with binding, then theFRAP recovery will be identical for different spot sizes. However, to detecta difference, it is important to determine an average value over at least 10
![Page 13: [Methods in Enzymology] Chromatin and Chromatin Remodeling Enzymes, Part A Volume 375 || Fluorescence Recovery after Photobleaching (FRAP) Methods for Visualizing Protein Dynamics](https://reader043.fdocuments.in/reader043/viewer/2022030117/5750a1d71a28abcf0c969a99/html5/page/13.jpg)
Fig. 4. Spot size dependence of fluorescence recovery. When diffusion plays a role in the
mobility of the protein under study, a larger spot size [compare (A) and (B)] will lead to a
slower recovery curve (C).
[27] FRAP for visualizing protein dynamics 455
bleaches for each spot size. If there is a difference in recovery as a functionof spot size, then diffusion contributes to the recovery and must beincluded in the model for fluorescence recovery. Because the diffusionconstant can be estimated using GFP only, this value can be substitutedinto the model describing the FRAP recovery, which will depend on thisdiffusion constant and the on and off rates of binding.
We are developing a systematic protocol for fitting FRAP data, includ-ing a set of model equations for the range of possible behaviors observedwith either a single binding reaction or multiple, independent binding reac-tions.11 These protocols can be consulted for detailed instructions, and aWeb version of this software will soon be available.
11 B. L. Sprague, R. L. Pego, D. A. Stavreva, and J. G. McNally, submitted (2003).