
Messa a punto dell’analisi traslocazione RET/PTC · paraffin‐embedded tissue of patients’...
56
1 SSMT Locarno 2012/2013 Lavoro di diploma per il corso di Tecnici in Analisi Biomediche Presso l’Istituto Cantonale di Patologia di Locarno Messa a punto dell’analisi della traslocazione RET/PTC Lavoro di Diploma di AMBRA CATTANI Responsabili Dott.ssa Francesca Molinari Dott. Milo Frattini
Transcript of Messa a punto dell’analisi traslocazione RET/PTC · paraffin‐embedded tissue of patients’...

1
SSMT Locarno 2012/2013
Lavoro di diploma per il corso di Tecnici in Analisi Biomediche
Presso l’Istituto Cantonale di Patologia di Locarno
Messa a punto dell’analisi della
traslocazione RET/PTC
Lavoro di Diploma di AMBRA CATTANI
Responsabili Dott.ssa Francesca Molinari
Dott. Milo Frattini

Ambra Cattani TAB3 SSMT Locarno 2012/2013
2
Sommario
1. Abstract ......................................................................................................................................................... 4
2. Abbreviazioni ................................................................................................................................................. 7
3. Introduzione .................................................................................................................................................. 8
3.1 I tumori tiroidei ........................................................................................................................................ 8
3.2 Alterazioni molecolari del carcinoma tiroideo ........................................................................................ 9
3.3 Alterazioni molecolari del carcinoma papillare ..................................................................................... 10
3.3.1 La mutazione del gene BRAF .......................................................................................................... 10
3.3.2 La traslocazione RET/PTC ............................................................................................................... 11
4. Scopo del lavoro .......................................................................................................................................... 15
5. Pazienti, materiali e metodi......................................................................................................................... 16
5.1 Pazienti .................................................................................................................................................. 16
5.2 Materiali e metodi ................................................................................................................................. 16
5.2.1 Estrazione degli acidi nucleici ......................................................................................................... 16
5.2.2 Quantificazione degli acidi nucleici estratti dai tessuti .................................................................. 17
5.2.3 Amplificazione del DNA genomico tramite PCR ............................................................................. 17
5.2.4 Analisi dello stato mutazionale del gene BRAF .............................................................................. 18
5.2.5 Elettroforesi su gel di agarosio ....................................................................................................... 19
5.2.6 Purificazione del DNA amplificato .................................................................................................. 19
5.2.7 PCR di sequenza (cyclesequencing) ................................................................................................ 20
5.2.8 Purificazione del DNA dopo PCR di sequenza ................................................................................ 20
5.2.9 Il sequenziamento .......................................................................................................................... 20
5.2.10 Retrotrascrizione dell’RNA a cDNA ............................................................................................... 21
5.2.11 Messa a punto dell’analisi delle traslocazioni RET/PTC tramite metodica di RT‐PCR .................. 23
5.2.12 Scelta dei primers e PCR a gradiente di temperatura .................................................................. 23
5.2.13 Validazione della metodica di analisi di traslocazione RET/PTC ................................................... 26
5.2.14 Amplificazione dei geni di controllo PGK1 e PAX8 ....................................................................... 27
5.2.15 Analisi dei prodotti di amplificazione tramite elettroforesi capillare .......................................... 28
6. Risultati ........................................................................................................................................................ 31
6.1 Messa a punto delle reazioni di PCR per l’analisi delle traslocazioni RET/PTC1 e RET/PTC3 ................ 31
6.2 Validazione della metodica per l’analisi dei frammenti delle traslocazioni RET/PTC1 e RET/PTC3,
tramite RT‐PCR ed analisi dei frammenti .................................................................................................... 36
6.3 Analisi dello stato mutazionale del gene BRAF ..................................................................................... 39

Ambra Cattani TAB3 SSMT Locarno 2012/2013
3
7. Discussione .................................................................................................................................................. 42
8. Conclusione ................................................................................................................................................. 45
9. Bibliografia ................................................................................................................................................... 46
10. Ringraziamenti ........................................................................................................................................... 48
11. Allegati ....................................................................................................................................................... 49
11.1 Estrazione di DNA genomico da tessuti fissati in formalina ed inclusi in paraffina (FFPE) ................. 49
11.2 Estrazione RNA da tessuto FFPE .......................................................................................................... 50
11.3 Quantificazione degli acidi nucleici ..................................................................................................... 51
11.4 Costituzione della miscela di reazione per la PCR ............................................................................... 52
11.5 Elettroforesi su gel di agarosio ............................................................................................................ 53
11.6 Purificazione del DNA amplificato ....................................................................................................... 54
11.7 PCR di sequenza (cyclesequencing) ..................................................................................................... 54
11.8 Purificazione del DNA dopo PCR di sequenza ..................................................................................... 55

Ambra Cattani TAB3 SSMT Locarno 2012/2013
4
1. Abstract
Introduzione
I tumori tiroidei rappresentano il principale
tipo di tumore endocrino, con una prevalenza
dei tumori papillari (circa l’80%). Il metodo di
indagine diagnostica preoperatorio più
diffuso e accurato per la valutazione della
natura benigna o maligna dei noduli tiroidei è
l’esame citologico su aspirazione bioptica con
ago sottile. Tuttavia, nel 15‐20% circa delle
analisi citologiche non è possibile giungere a
diagnosi certa. In questi casi le analisi delle
alterazioni di marcatori molecolari specifici
del carcinoma tiroideo possono essere
d’aiuto nel discriminare i campioni patologici
e aiutare nelle scelte terapeutiche. Nei
carcinomi papillari tiroidei le alterazioni più
frequenti e specifiche di tale tumore sono
rappresentate da mutazioni puntiformi del
gene BRAF e dalle traslocazioni RET/PTC,
ovvero riarrangiamenti del gene RET con geni
eterologhi espressi ubiquitariamente. Le due
forme più frequenti di riarrangiamento sono
RET/PTC1 e RET/PTC3, determinate dalla
fusione del gene RET con i geni CCDC6 e
NCOA4 rispettivamente.
Scopo del lavoro
Lo scopo di questo lavoro di diploma è quello
di ottimizzare un protocollo di analisi delle
traslocazioni RET/PTC tramite RT‐PCR ed
analisi dei frammenti nel Laboratorio di
Patologia Molecolare dell’Istituto Cantonale
di Patologia di Locarno.
Materiali e metodi
Per la messa a punto del protocollo delle
analisi RET/PTC sono stati usati due costrutti
plasmidici contenenti le traslocazioni
RET/PTC1 e RET/PTC3. I protocolli di PCR,
specifici per individuare le traslocazioni
RET/PTC1 e RET/PTC3, sono stati messi a
punto tramite PCR a gradiente di
temperatura e validati su 17 campioni di RNA
estratto da tessuti fissati in formalina ed
inclusi in paraffina di pazienti affetti da
tumore papillare tiroideo. I campioni di RNA
analizzati sono stati estratti tramite un kit
commerciale e retrotrascritti a cDNA tramite
protocolli già in uso presso l’ICP. I cDNA
ottenuti sono stati analizzati tramite PCR
seguendo il protocollo messo a punto sui
costrutti plasmidici e gli amplificati ottenuti
sono stati analizzati al sequenziatore. Inoltre,
per ottenere un quadro molecolare più
completo, è stata valutata la mutazione
dell’esone 15 del gene BRAF tramite PCR e
sequenziamento diretto, partendo da DNA
genomico dei pazienti analizzati, seguendo i
protocolli già in uso presso l’ICP.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
5
Risultati e conclusione
I risultati ottenuti dalle PCR a gradiente di
temperatura e dalla successiva analisi dei
frammenti effettuati su costrutti plasmidici, si
sono rilevati ottimi a tutte le temperature
provate, e pertanto è stata stabilita come
temperatura di ibridazione la temperatura
media del gradiente pari a 57°C. Due
campioni di tessuto tumorale su 17 (11,8%), a
fronte di nessuno dei tessuti sani, sono
risultati positivi per le traslocazioni RET/PTC,
risultato concordante con la letteratura.
L’analisi del gene BRAF ha identificato una
mutazione in 10 campioni su 17 (58,8%), una
percentuale alta, ma comunque concordante
con diversi testi presenti in letteratura. In
conclusione si può dire che le metodiche
messe a punto sono state validate con
successo e possono essere introdotte in
diagnosi.
Background
Thyroid tumors are the main type of
endocrine tumor, with a prevalence of
papillary tumors (about 80%). The most
popular and accurate method of
preoperative diagnostic evaluation of benign
or malignant thyroid nodules is the cytologic
examination of fine needle aspiration biopsy.
However, in 15‐20% of cytological analysis it
is not possible to arrive at a diagnosis. In
these cases the analysis of alterations of
specific molecular markers of thyroid
carcinoma may be helpful in discriminating
the pathological samples and help with
therapeutic choices. In papillary thyroid
carcinomas, the most frequent and specific
alterations of this tumor are represented by
point mutations of the BRAF gene and
RET/PTC translocations, or rearrangements of
the RET gene with heterologous genes
ubiquitously expressed. The two most
frequent forms of rearrangement are
RET/PTC1 and RET/PTC3 and determined by a
fusion of the RET gene with the genes NCOA4
and CCDC6 respectively.
The aim of this study
The aim of this diploma work is to optimize a
protocol analysis of RET/PTC translocations
using RT‐PCR and fragments analysis in the
Molecular Pathology Laboratory of Locarno.
Materials and methods
For the elaboration of the Protocol of the
RET/PTC analysis, two plasmid constructs
containing translocations RET/PTC1 and
RET/PTC3 were used. PCR Protocols, specific
to identifying translocations RET/PTC1 and
RET/PTC3, were developed by means of
temperature gradient PCR and tested on 17
RNA samples retrieved from formalin‐fixed
paraffin‐embedded tissue of patients’ thyroid
papillary cancer. The RNA samples analyzed

Ambra Cattani TAB3 SSMT Locarno 2012/2013
6
were extracted using a commercial kit and
retrotranscripted to cDNA using protocols
already in use at the ICP. The cDNA obtained
were analyzed by PCR using the Protocol
developed on plasmid constructs and the
amplified samples obtained were analyzed on
the sequencer. In addition, to obtain a more
complete molecular framework, genomic
DNA of the analyzed patients, was evaluated
for the mutation of the Exon 15 of the BRAF
gene by PCR and direct sequencing, following
the protocols already in use at the ICP.
Results and conclusion
The results obtained from temperature
gradient PCR and subsequent analysis of the
fragments performed on plasmid constructs,
were found excellent at all temperatures
tested, and thus the average temperature
gradient of 57°C was established as
hybridization temperature. Two samples of
tumor tissue in 17 (11.8%), compared with
none of the healthy tissues, were positive for
translocations RET/PTC, results consistent
with the literature. The BRAF gene analysis
has identified a mutation in 10 samples out
of 17 (58.8%), a high percentage, but still
consistent with several texts presented in
literature. In conclusion we can say that the
methods developed were validated
successfully and can be introduced in
diagnosis.
.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
7
2. Abbreviazioni
bp: paia di basi
ddNTPs: dideossinucleotidi trifosfati
dNTPs: deossinucleotidi trifosfati
FFPE: tessuti fissati in formalina e inclusi in paraffina
FTC: tumore follicolare tiroideo
Fw: forward
ICP: Istituto Cantonale di Patologia
PCR: Polymerase Chain Reaction
PTC: tumore papillare tiroideo
RTK: recettori tirosin chinasici
RT‐PCR: Reverse Transcription Polimerase Chain Reaction
Rw: reverse
Tm: temperature di melting
WT: Wild‐Type

Ambra Cattani TAB3 SSMT Locarno 2012/2013
8
3. Introduzione
3.1 I tumori tiroidei
I carcinomi della tiroide rappresentano la neoplasia più frequente del sistema endocrino e la loro
incidenza è in costante crescita negli Stati Uniti e nella maggior parte degli altri paesi (Nikiforov,
2011). L’aumento complessivo dell’incidenza del tumore è maggiore nelle donne rispetto agli
uomini ed è maggiore nei soggetti di età compresa tra i 45 e i 50 anni. Uno dei principali fattori di
rischio predisponenti allo sviluppo del tumore tiroideo è rappresentato dall’esposizione a
radiazioni ionizzanti. Tale associazione è stata accertata da diversi studi condotti dopo il disastro di
Chernobyl del 1986, che hanno evidenziato un aumento dell’incidenza di neoplasie tiroidee nei
soggetti che al momento dell’esposizione avevano un’età compresa tra i 5 e i 10 anni. Altre
condizioni predisponenti includono una familiarità per tumore tiroideo, una preesistente patologia
tiroidea benigna, fattori ormonali e gravidanze, aumenti di peso ed un insufficiente apporto di
iodio nella dieta (Romei et al, 2012).
Più del 95% dei tumori tiroidei originano dalle cellule dell’epitelio follicolare e comprendono
adenomi, carcinomi ben differenziati, carcinomi poco differenziati e carcinomi anaplastici, mentre
il 3,2% dei tumori tiroidei sono derivati dalle cellule C o parafollicolari (Chien et al, 2012). I
carcinomi ben differenziati rappresentano circa l’80% di tutti i tumori tiroidei e comprendono due
tipi principali: il carcinoma papillare (PTC) e il carcinoma follicolare (FTC). Il PTC è il tipo più
comune e rappresenta l’80% di tutte le neoplasie tiroidee. Il PTC è generalmente indolente e
curabile, ma questo tumore può comunque diffondere localmente ai linfonodi e molto comuni
sono la persistenza della malattia e le recidive. Il FTC rappresenta il 15% di tutte le neoplasie
tiroidee ed è il secondo tipo più comune di carcinoma ben differenziato originato dall’epitelio
follicolare della tiroide ed è anch’esso caratterizzato da un fenotipo indolente (Nikiforov, 2009). I
carcinomi poco differenziati e i carcinomi anaplastici sono invece molto aggressivi e possono
insorgere de novo o da preesistenti carcinomi follicolari o papillari (Kondo et al, 2006).
Importanti fattori sia da un punto di vista prognostico sia da quello decisionale terapeutico
risultano essere: l’età alla diagnosi, il tipo istologico e l’estensione della neoplasia. Tale estensione
è definita dal sistema di stadiazione TNM che classifica il tumore in base alle dimensioni e al grado
di infiltrazione (T), all’implicazione dei linfonodi (N) e alla presenza di metastasi a distanza (M).
I noduli tiroidei sono molto frequenti nella popolazione adulta con una frequenza negli USA del 4‐
7%, ma solo una minima percentuale di questi noduli (5‐10%) risulta essere maligna (Romei et al,

Ambra Cattani TAB3 SSMT Locarno 2012/2013
9
2012). Poiché la maggioranza dei noduli tiroidei sono benigni e poiché la maggior parte dei tumori
tiroidei sono curabili con tecniche chirurgiche se identificati precocemente, è molto importante
distinguere tra i noduli benigni e maligni in modo tale che il paziente possa ricevere un
trattamento appropriato, minimizzando il rischio di interventi non necessari. Attualmente la
citologia su aspirazione bioptica con ago sottile (FNA), sotto controllo ecografico, è il metodo di
indagine preoperatorio più diffuso che permette di diagnosticare accuratamente la maggior parte
delle lesioni benigne o maligne, indirizzando quindi le decisioni terapeutiche o i follow‐up. In
generale nel 15% dei casi si ottiene un campione non diagnostico e in circa il 20% dei casi i
campioni sono classificati nella categoria diagnostica di proliferazione follicolare di significato
indeterminato per malignità (TIR3). In questa categoria, solo il 20% dei casi risultano poi essere
carcinomi all’esame istologico, eseguito dopo emitiroidectomia diagnostica (Nelva et al, 2011).
Il trattamento d’elezione per il carcinoma differenziato della tiroide prevede l’asportazione totale
della tiroide. Tale trattamento viene completato somministrando radio‐iodio a scopo ablativo di
eventuali residui tiroidei e successiva terapia ormonale sostitutiva a vita con L‐tiroxina (LT4) con lo
scopo di correggere l’ipotiroidismo indotto e di sopprimere i livelli ematici di TSH di secrezione
ipofisaria.
3.2 Alterazioni molecolari del carcinoma tiroideo
Numerose alterazioni genetiche sono coinvolte nello sviluppo del tumore tiroideo derivante
dall’epitelio follicolare. Tali alterazioni sono rappresentate prevalentemente da mutazioni
puntiformi e traslocazioni che portano all’attivazione oncogenica di importanti vie di trasduzione
del segnale, in particolare la via delle MAPK (Mitogen‐Activated Protein Kinase) che regola
processi chiave della proliferazione, differenziamento e apoptosi cellulare, tramite l’attivazione
costitutiva di oncogeni come RET, NTRK‐1, MET, BRAF e KRAS o il silenziamento di oncosoppressori
come p53, PPARγ e PTEN. Alcune di queste alterazioni genetiche sono specifiche di alcuni tipi di
carcinoma tiroideo (Tabella 1) e possono essere utilizzate come importante strumento per
migliorare la diagnosi delle neoplasie tiroidee (Hunt et al, 2002). Quattro tipi di alterazioni
genetiche costituiscono la maggioranza delle mutazioni che incorrono nei tumori papillari e
follicolari e costituiscono importanti marcatori diagnostici e prognostici. Le più frequenti
alterazioni nei PTC sono rappresentate da mutazioni puntiformi nei geni BRAF e RAS (riscontrate
nel 40% e nel 10% dei casi rispettivamente) e riarrangiamenti RET/PTC (riscontrati nel 10‐20% dei
casi), e quelli che contengono il gene TRK (meno del 5% dei casi). Nel complesso, queste

Ambra Cattani TAB3 SSMT Locarno 2012/2013
10
alterazioni identificano il 70% dei PTC e sono generalmente mutuamente esclusive. Le più
frequenti alterazioni che incorrono negli FTC sono rappresentate invece da mutazioni di RAS (nel
45% dei casi) e da riarrangiamenti PAX/PPAγ (nel 35% dei casi) che sono anch’esse mutuamente
esclusive. Mutazioni di RAS e in un minor misura traslocazioni PAX8/PPARγ sono riscontrate anche
negli adenomi follicolari con frequenze del 20‐40% e del 2‐13%, rispettivamente. Diversi studi
hanno mostrato che la valutazione preoperatoria di questi marcatori può migliorare l’accuratezza
della diagnosi citologica fatta su FNA per pazienti con diagnosi di significato indeterminato o con
sospetto di malignità per identificare le lesioni maligne e aiutare quindi nella scelta terapeutica
(Nikiforov, 2011).
Tabella 1: alterazioni molecolari del carcinoma tiroideo (tratta da Nikiforov, 2011)
Carcinoma papillare della tiroide Prevalenza (%)
Mutazione BRAF 40‐45
Traslocazione RET/PTC 10‐20
Mutazione RAS 10‐20
Traslocazione TRK <5
Carcinoma follicolare della tiroide Prevalenza (%)
Mutazione RAS 40‐50
Traslocazione PAX8/PPARy 30‐35
3.3 Alterazioni molecolari del carcinoma papillare
3.3.1 La mutazione del gene BRAF
L’alterazione più comune dei PTC è la mutazione del gene BRAF (presente nel 40‐45% dei casi).
BRAF è una proteina serin treonina chinasi che appartiene alla famiglia delle proteine RAF,
effettori intracellulari della cascata delle MAPK. Le mutazioni principali di BRAF nel PTC sono
mutazioni puntiformi che coinvolgono il nucleotide 1799 e che portano alla sostituzione di una
valina con un acido glutammico al codone 600 (V600E). Tale mutazione comporta la costitutiva
attivazione della chinasi BRAF con conseguente attivazione cronica della via metabolica delle
MAPK e successiva deregolazione della crescita e della proliferazione cellulare. La mutazione
V600E di BRAF è tipica del PTC con istologia classica e della variante a cellule alte, mentre è rara
nella variante follicolare. Le mutazioni di BRAF non sono state trovate nei FTC e nei noduli tiroidei
benigni e, rappresentano pertanto un marcatore specifico del PTC e, quindi, un utile supporto per

Ambra Cattani TAB3 SSMT Locarno 2012/2013
11
migliorare l’accuratezza della diagnosi citologica dei noduli tiroidei (Nikiforov, 2011). Diversi studi
indicano che le mutazioni di BRAF si associano ad una maggiore aggressività del tumore in termini
di invasione, stadio clinico e rischio di ripresa dalla malattia (Xing et al, 2005). Inoltre tali mutazioni
sono associate alla perdita della capacità di captare lo iodio 131 e quindi ad una scarsa risposta
terapeutica. Per questi motivi la valutazione preoperatoria dello stato mutazionale di BRAF
rappresenterebbe un utile parametro per la scelta di un miglior approccio chirurgico e terapeutico.
3.3.2 La traslocazione RET/PTC
Il proto‐oncogene RET codifica per un recettore tirosin chinasico (RTK) di membrana coinvolto nel
controllo della differenziazione cellulare e della proliferazione. Normalmente il gene RET è
espresso nelle cellule parafollicolari o cellule C, ma non nelle cellule follicolari dove, invece, può
essere attivato solo in caso di presenza di un riarrangiamento del gene RET. Il recettore RET è una
proteina transmembrana che presenta una porzione extracellulare formata da quattro domini
simili alle caderine (cadherin‐like) e una parte intracellulare che presenta dei domini ricchi in
tirosina (Figura 1). In presenza di specifici ligandi quali neururina, artimina, persepina e i fattori di
derivazione dalle cellule gliali, il recettore RET, presente nella membrana cellulare sotto forma di
monomero, dimerizza con un altro recettore. Tale dimerizzazione porta all’attivazione del
recettore tramite un processo di autofosforilazione dei domini tirosinchinasici (TK) e quindi
all’attivazione delle cascate metaboliche a valle (MAPK e via di PI3K‐AKT). Tali cascate intracellulari
sevono a propagare i segnali del recettore RET di membrana all’interno del nucleo attraverso una
serie di proteine adattatore e chinasi intracitoplasmatiche (tra le quali RAS e BRAF) che portano
alla regolazione della trascrizione di geni coinvolti nella differenziazione, nella proliferazione e
nella sopravvienza cellulare (Figura 2). Nel PTC l’attivazione di questa via metabolica è causata
principalmente da mutazioni puntiformi di RAS e BRAF o da riarrangiamenti RET/PTC e tale
attivazione costitutiva risulta essere responsabile dell’insorgenza e della progressione tumorale
(Nikiforov, 2011).
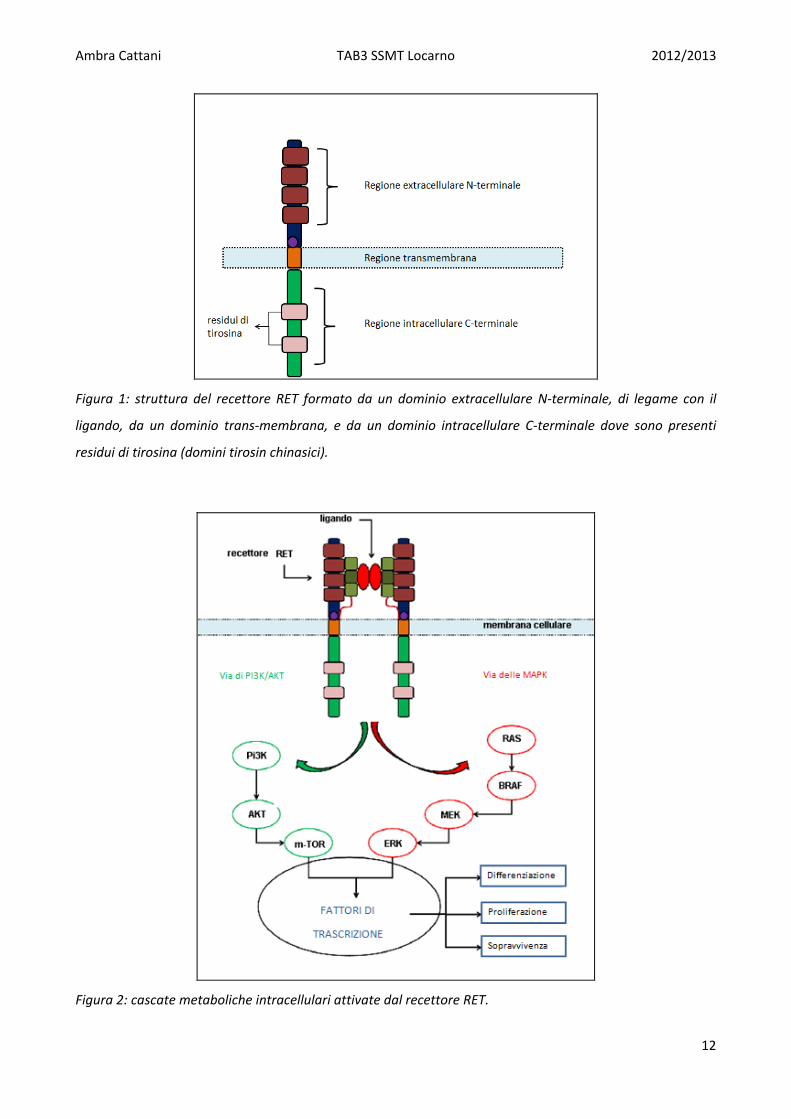
Ambra Cattani TAB3 SSMT Locarno 2012/2013
12
Figura 1: struttura del recettore RET formato da un dominio extracellulare N‐terminale, di legame con il
ligando, da un dominio trans‐membrana, e da un dominio intracellulare C‐terminale dove sono presenti
residui di tirosina (domini tirosin chinasici).
Figura 2: cascate metaboliche intracellulari attivate dal recettore RET.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
13
I riarrangiamenti a carico del gene RET (RET/PTC), rappresentano la seconda alterazione più
comune nei PTC. Tali riarrangiamenti sono presenti in circa il 35% dei casi di PTC sebbene la
percentuale sia variabile tra il 3 e l’85% a seconda degli studi, in funzione dell’area geografica di
provenienza dei tumori, delle differenti metodologie di analisi utilizzate e dell’eterogeneità
tumorale. La distribuzione del riarrangiamento RET/PTC all’interno di un tumore può essere infatti
eterogenea e può essere presente o nella maggior parte delle cellule neoplastiche
(riarrangiamento RET/PTC di tipo clonale), oppure in una piccola frazione di cellule tumorali
(riarrangiamento RET/PTC di tipo non clonale) (Santoro et al, 2006). Questo aspetto è da tenere in
considerazione per le scelte delle differenti metodologie di analisi utilizzate: l’analisi di questa
traslocazione può infatti essere effettuata sia con RT‐PCR (Reverse Transcription Polimerase Chain
Reaction), tecnica migliore per tessuti freschi o congelati con una sensibilità che non dovrebbe
essere maggiore all’1% per evitare di rilevare mutazioni non clonali e quindi senza rilevanza clinica,
oppure mediante FISH, tecnica più performante in caso di tessuti FFPE ma la cui valutazione
richiede la determinazione di livelli di cut‐off corretti affinché non si incorra in risultati falsi positivi
(Nikiforov, 2011). I riarrangiamenti clonali sono descritti in circa il 20% dei PTC mentre i
riarrangiamenti non clonali sono stati identificati non solo nei PTC, ma anche nel 10‐45% degli
adenomi tiroidei e altre lesioni non neoplastiche (Santoro et al, 2006). I riarrangiamenti RET/PTC
risultano essere più frequenti nei pazienti esposti a radiazioni (50‐80%) e nei carcinomi papillari
dei bambini o adolescenti (40%‐70%) (Nikiforov, 2011). Tali riarrangiamenti sono specifici delle
lesioni papillari maligne e generalmente non sono riscontrati (se non con metodologie di analisi
molto sensibili) negli adenomi e nelle lesioni benigne. I riarrangiamenti RET/PTC portano alla
formazione di oncogeni chimerici che risultano dalla fusione tra la porzione 3’ del gene RET e la
porzione 5’ di geni eterologhi ubiquitariamente espressi e caratterizzati dalla presenza di sequenze
nucleotidiche codificanti per proteine che formeranno domini coiled coil che permettono la
dimerizzazione costitutiva del dominio tirosin chinasico di RET. Le proteine di fusione contengono
il dominio tirosin chinasico del recettore RET intatto e risultano costitutivamente attivate
portando quindi all’attivazione costitutiva della cascata delle MAPK. Tale processo determina una
proliferazione incontrollata delle cellule follicolari caratterizzate dalla presenza del
riarrangiamento RET/PTC e quindi allo sviluppo successivo del tumore. Sono state descritte più di
11 varianti, ma i due tipi di riarrangiamento più frequenti sono RET/PTC1 (nel 60‐70% dei casi) e
RET/PTC3 (nel 20‐30% dei casi). RET/PTC1 è formato dalla fusione del gene RET con il gene CCDC6
(H4) e RET/PTC3 dalla fusione del gene RET con il gene NCOA4 (ELE1). Entrambe le traslocazioni

Ambra Cattani TAB3 SSMT Locarno 2012/2013
14
sono inversioni paracentriche poiché sia il gene RET che i geni CCDC6 ed NCOA4 si trovano sul
braccio lungo del cromosoma 10 (Nikiforov, 2003). RET/PTC1 si trova con maggiore frequenza nei
tumori sporadici e nelle varianti classiche del PTC, ma può essere anche trovato nei
microcarcinomi papillari, mentre RET/PTC3 è maggiormente presente nei carcinomi papillari
indotti da radiazioni e nelle varianti solide dei PTC (Santoro et al, 2006). La correlazione tra la
presenza delle traslocazioni RET/PTC e la prognosi non sono attualmente chiarite; vi sono evidenze
che indicano un’associazione tra la presenza di un riarrangiamento RET/PTC1 a un decorso più
favorevole della malattia, rispetto ai pazienti con presenza di traslocazione RET/PTC3, associata ad
un comportamento clinico più aggressivo. Altri studi hanno suggerito che i casi positivi per la
presenza di un riarrangiamento RET/PTC sembrano mostrare un’estensione più ampia della
malattia e un tasso di coinvolgimento linfonodale maggiore rispetto ai casi negativi per le
traslocazioni. Tuttavia altri studi hanno confutato tali considerazioni (Romei et al, 2012). Dal punto
di vista diagnostico lo studio della traslocazione RET/PTC ha poco significato nel caso in cui vi sia
un prelievo istologico, perché i tumori caratterizzati dalla traslocazione RET/PTC normalmente
hanno un’architettura di tipo papillare, che non crea grosse difficoltà diagnostiche. Al contrario
tale analisi potrebbe risultare particolarmente utile nelle diagnosi preoperatorie effettuate
mediante FNA dove sovente si hanno difficoltà a livello diagnostico soprattutto nei casi che hanno
una citologia di significato indeterminato (TIR3) (Nikiforov, 2011).

Ambra Cattani TAB3 SSMT Locarno 2012/2013
15
4. Scopo del lavoro Negli ultimi anni l’acquisizione di conoscenze sempre più approfondite e specifiche riguardanti le
alterazioni genetiche proprie delle neoplasie tiroidee ha aperto la possibilità di applicare
metodiche molecolari per l’analisi di tali deregolazioni alla diagnostica citologica delle patologie
tiroidee. Le principali alterazioni molecolari specifiche dei carcinomi tiroidei sono i riarrangiamenti
PAX8/PPARγ e mutazioni RAS per i carcinomi follicolari, e riarrangiamenti RET e mutazioni BRAF
per i carcinomi papillari.
Nel Laboratorio di Patologia Molecolare dell’ICP di Locarno sono già in uso diagnostico le analisi di
PAX8/PPARG, RAS e di BRAF per coadiuvare l’analisi citologica dei tumori tiroidei. L’obiettivo del
mio lavoro sarà quello di mettere a punto l’analisi delle traslocazioni RET/PTC con lo scopo di
completare il pannello delle principali analisi molecolari per la diagnostica del tumore tiroideo.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
16
5. Pazienti, materiali e metodi
5.1 Pazienti
Per la validazione dell’analisi di traslocazione RET/PTC sono stati analizzati 17 pazienti affetti da
PTC con diagnosi istologica effettuata presso l’ICP di Locarno tra il 2010 e il 2012. I tessuti di ogni
paziente, fissati in formalina ed inclusi in paraffina (FFPE) sono disponibili presso gli archivi
dell’ICP. Per ogni paziente l’analisi della traslocazione RET/PTC è stata effettuata sia su tessuto
tumorale sia sul tessuto normale della tiroide. Sul tessuto tumorale è stata effettuata inoltre
l’analisi dello stato mutazionale del gene BRAF, per ottenere un quadro completo delle alterazioni
molecolari più frequenti nel PTC.
5.2 Materiali e metodi
Per eseguire le metodiche descritte nel presente lavoro di diploma, è stato necessario lavorare in
sterilità e nelle migliori condizioni che evitassero il più possibile le contaminazioni: i banconi di
lavoro, le pipette e gli strumenti utilizzati sono stati puliti con ipoclorito di sodio per le lavorazioni
con acidi nucleici (DNA e RNA) e con RNAsi Zap (Invitrogen) per le lavorazioni con RNA, i guanti
sono stati cambiati ad ogni passaggio e la fase di estrazione degli acidi nucleici è stata effettuata
sotto cappa chimica precedentemente irradiata con raggi UV.
5.2.1 Estrazione degli acidi nucleici
Il DNA e l’RNA sono stati estratti da tessuti FFPE dopo la valutazione delle sezioni istologiche,
colorate mediante ematossilina‐eosina, da un medico patologo. Per l’analisi RET/PTC l’RNA è stato
estratto sia da tessuto tumorale sia da tessuto sano. L’analisi dello stato mutazionale di BRAF è
stata effettuata solo su DNA genomico estratto da tessuto tumorale. Per l’estrazione di DNA e RNA
da tessuto tumorale, nei casi in cui l’area di tumore selezionato fosse composta da più del 70% di
cellule tumorali, sono state poste 6‐7 sezioni istologiche da 7 µm (per il DNA) o 2‐4 sezioni da 10
µm (per l’RNA) in provette Eppendorf da 2ml. Per i tessuti in cui la quantità di cellule tumorali era
inferiore al 70% è stata effettuata una macrodissezione: l’area selezionata dal patologo è stata
prelevata con bisturi monouso da sezioni in bianco su vetrino (dello stesso spessore delle sezioni
poste in provetta) e posta in provette Eppendorf da 2 ml. Per l’analisi del tessuto sano sono stati
scelti reperi comprendenti il 100% di cellule sane. Per l’estrazione del DNA e dell’RNA da FFPE

Ambra Cattani TAB3 SSMT Locarno 2012/2013
17
sono stati usati kit commerciali: il QIAmp DNA MiniKIT e l’RNAeasi MiniKit (Qiagen, Chatsworth,
CA, USA) in cui sono presenti reagenti e materiali necessari per l’estrazione. I protocolli prevedono
una fase di lisi cellulare che permetta il rilascio degli acidi nucleici e una purificazione degli acidi
nucleici tramite l’assorbimento su una membrana di silice della colonnina presente nei kit. In
seguito è prevista una fase di eluizione in cui gli acidi nucleici vengono rilasciati dalla membrana
nel tampone di eluizione o in acqua DNAasi e RNAasi free. I procedimenti sono descritti
dettagliatamente negli allegati 11.1 e 11.2.
5.2.2 Quantificazione degli acidi nucleici estratti dai tessuti
La quantificazione degli acidi nucleici è stata effettuata tramite uno spettrometro UV‐visibile
Nanodrop 1000 (Witec, Littau, CH) che stabilisce la concentrazione di acidi nucleici misurando
l’assorbanza dei campioni in volumi di 2 µl. Il procedimento è descritto in dettaglio nell’allegato
11.3.
5.2.3 Amplificazione del DNA genomico tramite PCR
Il DNA genomico estratto è stato amplificato tramite reazione di PCR (Polymerase Chain Reaction),
reazione che permette l’amplificazione in vitro degli acidi nucleici. La reazione di PCR inizia con
una denaturazione di 10 minuti a 95°C, in seguito alla quale vengono ripetuti tre passaggi per 35‐
45 volte con tempi e temperature definite. Questi passaggi sono la denaturazione (92‐98°C) che
permette di separare le due catene complementari di DNA, l’Ibridazione (40‐60°C), passaggio in
cui i primers si legano a specifiche sequenze complementari del DNA stampo e la cui temperatura
è determinata sperimentalmente, e l’estensione (70‐72°C), passaggio in cui la DNA polimerasi
sintetizza il nuovo filamento di DNA. La reazione avviene all’interno di termociclatori, strumenti
che mantengono i campioni in incubazione, variando le temperature a seconda dell’impostazione
del protocollo.
50 ng di DNA (2 µl di una diluizione a 25 ng/µl) vengono aliquotati in provette da 0,2 ml,
aggiungendo un controllo positivo, che consiste in un campione di DNA già amplificato
precedentemente con successo, e un controllo bianco, cioè acqua sterile, privo di DNA, per
valutare la presenza di contaminazioni. A questi verranno aggiunti 23 µl di una miscela di reazione,
la cui costituzione è descritta nell’allegato 11.4.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
18
5.2.4 Analisi dello stato mutazionale del gene BRAF
L’analisi dello stato genico dell’esone 15 di BRAF è stata eseguita mediante PCR e sequenziamento
diretto seguendo i protocolli già in uso presso l’ICP per l’attività diagnostica. Per l’analisi dello
stato mutazionale del gene BRAF è stata amplificata, tramite primers specifici, una regione
dell’esone 15 comprendente il codone 600, codone in cui avvengono la quasi totalità delle
mutazioni oncogeniche del gene BRAF. Per l’analisi dell’esone 15 del gene BRAF è stato utilizzato il
seguente protocollo (tabelle 2 e 3) con la seguente coppia di primers, con la relativa temperatura
di ibridazione e lunghezza dell’amplificato:
‐ Primer BRAF esone 15 Forward: 5’‐TCATAATGCTTGCTCTGATAGGA‐3’
‐ Primer BRAF esone 15 Reverse: 5’‐ GGCCAAAAATTTAATCAGTGGA‐3’
‐ Temperatura di annealing: 52°C
‐ Dimensione dell’amplificato: 250 bp
Tabella 2: composizione della miscela di reazione per la reazione di PCR dell’esone 15 del gene BRAF
Concentrazione iniziale Conc./quantità finale Volume/campione
DNA 25,0 ng/µl 50 ng 2,0 µl
PCR Buffer 10X 1X 2,5 µl
MgCl2 25 mM 3 mM 3,0 µl
dNTPs 10 mM 0,2 mM 0,5 µl
dUTP 10 mM 0,04 mM 0,1 µl
Primer BRAF Fw 10 µM 0,5 µM 1,25 µl
Primer BRAF Rw 10 µM 0,5 µM 1,25 µl
Taq Gold 5 U/µM 2, U/campione 0,4 µl
Acqua sterile 14 µl

Ambra Cattani TAB3 SSMT Locarno 2012/2013
19
45 cicli
Tabella 3: protocollo di amplificazione dell’esone 15 del gene BRAF
Temperature Tempo di reazione
Stabilizzazione reazione 50°C 2’
Denaturazione iniziale 95°C 10’
Denaturazione 95°C 15’’
Ibridazione 52°C 30’
Estensione 72°C 30’’
Estensione finale 72°C 3’
Fine PCR 10°C ∞
5.2.5 Elettroforesi su gel di agarosio
Al fine di verificare l’ottenimento di un buon prodotto di amplificazione per tutti i campioni
analizzati si è ricorso alla corsa su gel di agarosio. Si tratta di un metodo che permette di separare
le molecole di DNA in base alle loro dimensioni: molecole piccole infatti migrano più velocemente
rispetto a quelle grandi. La tecnica sfrutta la carica elettrica negativa del DNA per permettere la
sua migrazione in un campo elettrico, verso il polo positivo. La migrazione avviene attraverso un
gel di agarosio che funge da setaccio molecolare, rallentando la corsa delle molecole. La
concentrazione del gel può variare a seconda delle necessità: se le molecole da separare hanno
dimensioni poco diverse l’una dall’altra, occorrerà avere maglie più strette e perciò la
concentrazione del gel dovrà essere maggiore. Se, come nel nostro caso, i prodotti di
amplificazione sono di dimensioni di circa 100‐300 bp, è sufficiente utilizzare un gel all’1,8%. La
dimensione degli amplificati ottenuti è stata poi valutata grazie ad un marker, una miscela di
frammenti di DNA a lunghezza nota che viene fatta correre nello stesso gel. I procedimenti di
preparazione del gel di agarosio, della corsa elettroforetica e di visualizzazione dei risultati sono
descritti in dettaglio nell’allegato 11.5.
5.2.6 Purificazione del DNA amplificato
Prima di procedere alla PCR di sequenza è necessario purificare il DNA amplificato, per eliminare i
reagenti in esubero della PCR rimasti in soluzione (primers, nucleotidi non incorporati e la Taq
Polimerasi), che potrebbero compromettere il corretto sequenziamento del DNA. Per questo
scopo è stato usato il kit MSB®SPIN PCRapace (STRATEC Molecular, Gmbh, Berlino, Germania), che

Ambra Cattani TAB3 SSMT Locarno 2012/2013
20
contiene colonne con un filtro che trattiene il DNA, mentre i contaminanti vengono eliminati
tramite un lavaggio. In seguito il DNA viene eluito con un buffer in una provetta Eppendorf da 1,5
ml. Il procedimento di purificazione è descritto in dettaglio nell’allegato 11.6.
5.2.7 PCR di sequenza (cyclesequencing)
I filamenti del prodotto amplificato, a seguito della purificazione, vengono marcati e amplificati
nella reazione di sequenziamento (cyclesequencing). Si tratta di una particolare PCR effettuata in
presenza di un solo primer (forward o reverse), dei quattro dNTPs e di quattro dideossinucleotidi
trifosfati (ddNTPs) marcati con quattro fluorocromi differenti che fungono da terminatori della
reazione. La reazione avviene normalmente, ma in alcune catene vengono introdotti casualmente i
ddNTPs che bloccano la reazione siccome sono privi del gruppo ossidrile (‐OH) all’estremità 3’ che
permetterebbe l’incorporamento di ulteriori nucleotidi. Ciò permette la formazione di frammenti
di lunghezze diverse, marcate a seconda del nucleotide che blocca la reazione. Questi frammenti
verranno rilevati grazie ad un raggio laser di un sequenziatore automatico e interpretati da un
programma apposito. Il dettaglio è descritto nell’allegato 11.7.
5.2.8 Purificazione del DNA dopo PCR di sequenza
Prima di poter sequenziare i frammenti ottenuti mediante la PCR di sequenza è necessario
eliminare i dNTPs, i ddNTPs e i primers che non sono stati incorporati nella reazione. Per tale
scopo viene usato il kit G50 Dye Terminator Removal kit (RBC Bioscience) che fornisce delle
colonne contenenti sfere di gel pre‐idratate. Il principio è quello della cromatografia per gel‐
filtrazione, dove i contaminanti vengono separati dal DNA sfruttando il differente peso molecolare
e le differenti dimensioni. Il DNA attraversa velocemente il gel e viene recuperato all’interno di
una eppendorf di 1,5 ml. Il procedimento è scritto in dettaglio nell’allegato 11.8.
5.2.9 Il sequenziamento
Il sequenziamento avviene in uno strumento in grado di effettuare l’elettroforesi capillare, dotato
da un raggio laser (3130 Genetic Analyzer, Applied Biosystems). Esso è in grado di separare due
frammenti di DNA che differiscono solo di 1 bp distinguendoli grazie ai fluorocromi marcati. Per
ottenere le sequenze dell’amplificato del gene d’interesse, i pellet ottenuti a seguito della seconda
purificazione sono stati risospesi in 15 µl di formammide (Hi‐Di Formammide, Applied Biosystems)

Ambra Cattani TAB3 SSMT Locarno 2012/2013
21
e messi in un termoblocco a 95°C per 5 minuti, al fine di denaturare il DNA. I filamenti, di cui uno
marcato, vengono separati in base alle loro dimensioni tramite migrazione capillare in un polimero
(3130 POP‐7 Performancy Optimized Polymer, Applied Biosystems) immerso in un campo
elettrico. Un raggio laser posto al termine del capillare eccita i fluorocromi coniugati all’ultimo
didessossiribonucleotide incorporato. Lo strumento rileva e analizza la fluorescenza riemessa dalle
molecole e il programma apposito (Sequencing Analysis Software v5.2, Applied Biosystems)
rielabora le fluorescenze rilevate in un elettroferogramma, cioé un grafico che rappresenta la
sequenza di basi visualizzata tramite una successione di picchi i cui colori corrispondono ai diversi
fluorocromi e quindi alle diverse basi.
5.2.10 Retrotrascrizione dell’RNA a cDNA
La retrotrascizione è una metodica che serve a sintetizzare una molecola di DNA a partire da RNA
ad opera dell’enzima trascrittasi inversa. Il DNA ottenuto, complementare al filamento stampo di
RNA messaggero, prende il nome di cDNA. L’enzima trascrittasi inversa per poter iniziare la
reazione di retrotrascrizione dell’RNA necessita di brevi sequenze nucleotidiche d’innesco che
possono essere rappresentate o da un oligonucleotide costituito da una serie di timine (oligo dT)
complementari alle adenine presenti nelle code di 150‐200 bp di adenine (catene poli‐A) presenti
all’estremità 3’ dell’RNA messaggero, oppure da primers a sequenza casuale (random hexamers)
che si appaiano casualmente alle sequenze di RNA. Nel nostro caso dato che l’RNA estratto da
tessuti FFPE può essere degradato ad opera della formalina, utilizzeremo sia gli oligo dT che i
random hexamers, per rendere più efficiente la reazione di retrotrascrzione,. La retrotrascrizione è
stata eseguita utilizzando protocolli e reagenti forniti da Invitrogen (Carlsbad, California, USA). 500
ng di RNA estratto da tessuto FFPE (diluito in un volume finale di 7,5 µl con acqua RNAasi free)
vengono aliquotati in provette da 0,2 µl, aggiungendo un controllo positivo che consiste in una
miscela di RNA totale umano (Applied Biosystems) per verificare che la reazione di
retrotrascrizione avvenga correttamente, e un controllo bianco, cioé acqua sterile, per valutare la
presenza di eventuale contaminazione. A questi viene aggiunta una prima miscela di reazione
(Tabella 4) costituita da:
‐ Random hexamers (Invitrogen): corti oligonucleotidi che si legano casualmente all’RNA,
‐ Oligo dT (Invitrogen): oligonucleotidi che si appaiano alla coda poli‐A dell’RNA messaggero

Ambra Cattani TAB3 SSMT Locarno 2012/2013
22
I campioni vengono poi posti nel termociclatore a 70°C per 5 minuti (protocollo di retrotrascrizione
fase I, tabella 5). In seguito ai campioni viene aggiunta una seconda miscela di reazione (Tabella 6)
composta da:
‐ Miscela equimolare di dNTPs (Amersham, Buckinghamshire, UK) 10 mM
‐ DTT (ditiotreitolo)(Invitrogen), forte agente riducente che ottimizza il lavoro della
trascrittasi inversa prevenendone l’ossidazione
‐ Buffer FS (Invitrogen), buffer di retrotrascrizione
‐ Rnase Inhibitor (Invitrogen), enzima che inibisce l’attività della RNAasi
‐ Acqua sterile RNAasi free
‐ SuperScript (Invitrogen), enzima stabile ad alte temperature proveniente da un gene
modificato di un retrovirus (Moloney Murine Leukemia Virus) che è in grado di sintetizzare
cDNA a partire da un mRNA stampo.
Si procede quindi con l’incubazione a 37°C per un’ora affinché l’enzima retrotrascriva l’RNA a
cDNA, in seguito la temperatura viene portata a 95°C per 5 minuti per disattivare l’enzima
(protocollo di retrotrascrizione fase II, tabella 7). Il cDNA così ottenuto può essere conservato a ‐
20°C e utilizzato per le successive reazioni di PCR per l’analisi delle traslocazioni RET/PTC.
Tabella 4: composizione della miscela di reazione di retrotrascrizione fase I
Conc. iniziale Quantità/Conc. finale Volume/campione
Campione RNA ‐ 500 ng 7,5 µl
R. hexamers 50 µM 5 µM 2,0 µl
Oligo dT 50 µM 5 µM 2,0 µl
Tabella 5: protocollo di retrotrascrizione fase I
temperatura tempo
Ibridazione 70°C 5’
Fine programma 25°C ∞
Tabella 6: composizione della miscela di reazione di retrotrascrizione fase II
Conc. iniziale Conc. finale Volume/campione
dNTPs 10 mM 5 mM 1 µl
DTT 0,1 M 0,01 M 2 µl

Ambra Cattani TAB3 SSMT Locarno 2012/2013
23
Buffer FS 5X 1X 4 µl
Rnase Inibitor 20 U/µl 0,5 U/µl 0,5 µl
Superscript 200 U/µl 10 U/µl 1 µl
Tabella 7: protocollo di retrotrascrizione fase II
temperatura tempo
Ibridazione 60°C 60’
Disattivazione enzima 95°C 5’
Fine programma 4°C ∞
5.2.11 Messa a punto dell’analisi delle traslocazioni RET/PTC tramite metodica di RT‐
PCR
L’analisi della traslocazione RET/PTC prevede, a seguito dell’estrazione di RNA e successiva
retrotrascrizione a cDNA, l’amplificazione del gene di fusione determinato dalla traslocazione del
dominio tirosin chinasico del gene RET a regioni 5’ terminali di geni eterologhi che portano
all’attivazione costitutiva dell’oncogene RET/PTC. In particolare sono state allestite due reazioni di
PCR per l’amplificazione delle due forme principali di traslocazione: RET/PTC1 (determinato dalla
fusione del gene RET con il gene CCDC6) e RET/PTC3 (determinato dalla fusione del gene RET con il
gene NCOA4), utilizzando due coppie di primers fiancheggianti i punti di fusione dei geni di fusione
RET/PTC1 e RET/PTC3. I primers forward di ogni coppia sono stati marcati con due fluorocromi
diversi in modo tale da permettere la visualizzazione dei prodotti di PCR tramite l’analisi dei
frammenti amplificati, eseguita mediante elettroforesi capillare in un sequenziatore automatico
(3130 Genetic Analyzer, Applied Biosystems).
5.2.12 Scelta dei primers e PCR a gradiente di temperatura
Per mettere a punto la metodica di PCR per l’analisi dei geni di fusione RET/PTC1 e RET/PTC3 è
stato necessario cercare le sequenze dei geni di interesse (RET, CCDC6 e NCOA4), tramite l’uso di
siti internet specializzati nel settore quali National Center for Biotecnology Information (NCBI,
www.ncbi.nlm.nih.gov/). La scelta dei primers adatti all’amplificazione genica dei due geni di
fusione, è avvenuta tramite la consultazione della letteratura scientifica proposta da PubMed
inerente l’analisi tramite RT‐PCR delle traslocazioni RET/PTC1 e RET/PTC3. Per ogni gene di fusione
analizzato è stata scelta una coppia di primers di cui è stata verificata la posizione di appaiamento

Ambra Cattani TAB3 SSMT Locarno 2012/2013
24
sulle sequenze geniche di interesse, verificando in particolare che fossero disegnati sulle regioni
fiancheggianti il punto di fusione tra due geni coinvolti nella traslocazione (esone 12 di RET, esone
1 di CCDC6 ed esone 10 di NCOA4). Sono stati scelti i primers i cui prodotti di amplificazione non
superassero le 250 bp per ridurre il rischio che i frammenti traslocati non venissero amplificati, in
quanto gli acidi nucleici provenienti da tessuto FFPE sono molto frammentati. Per ogni coppia di
primers è stata calcolata la temperatura di melting (Tm) teorica tramite l’utilizzo di un software
reperibile in rete che si basa su un algoritmo matematico (Oligo Calculator
(http://mcb.berkeley.edu/labs/krantz/tools/oligocalc.html). In seguito sono state stabilite le
temperature di ibridazione ottimali mediante una PCR a gradiente di temperatura, scegliendo, per
le reazioni di amplificazione, cinque temperature di ibridazione che ricadevano nel range calcolato
dalla Tm media, tra i primers forward e reverse, ±5°C. Lo scopo era quello di definire la
temperatura migliore di ibridazione, alla quale la reazione di PCR risultasse maggiormente
efficiente e alla quale l’analisi dei frammenti fosse di buona qualità. Dato che non avevamo a
disposizione campioni di pazienti o linee cellulari recanti la traslocazione RET/PTC1 e RET/PTC3,
per mettere a punto la metodica sono stati utilizzati dei costrutti plasmidici comprendenti le
regioni genomiche coinvolte nei riarrangiamenti RET/PTC1 e RET/PTC3 (Figura 3) .
Figura 3: a) sequenza di parte del gene di fusione RET/PTC1 inserita nel costrutto plasmidico; in rosso
l’esone 12 del gene RET, in nero parte dell’esone 1 del gene CCDC6. b) Sequenza del gene di fusione
RET/PTC3 inserita nel costrutto plasmidico; in rosso l’esone 12 del gene RET, in nero l’esone 10 del gene
NCOA4.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
25
I costrutti sono stati diluiti a 10 ng/µl e a 1 ng/µl, e utilizzati 5 µl di DNA per le PCR a gradiente. È
stato aggiunto per ogni temperatura del gradiente un controllo negativo (acqua distillata) allo
scopo di mostrare l’assenza di contaminazioni. Per l’amplificazione delle traslocazioni RET/PTC1 e
RET/PTC3 sono state eseguite le reazioni di PCR a gradiente con il seguente protocollo (tabelle 8 e
9), utilizzando le seguenti coppie di primers, con le relative temperature di annealing e lunghezza
attesa dell’amplificato:
‐ Primers RET/PTC1 Forward: 5’‐6FAM‐GGAGACCTACAAACTGAAGTGCAA‐3’
Tm= 56°C
‐ Primers RET/PTC1 Reverse: 5’‐CCCTTCTCCTAGAGTTTTTCCAAGA‐3’
Tm=56°C
Dimensioni dell’amplificato: 136 bp
‐ Primers RET/PTC3 Forward: 5’‐NED‐CCAGTGGTTATCAAGCTCCTTACA‐3’
Tm=57°C
‐ Primers RET/PTC3 Reverse: 5’‐ GGGAATTCCCACTTTGGATCCTC‐3’
Tm=56°C
Dimensioni dell’amplificato: 106 bp
I primers forward sono fluorocromati (RET/PTC1 con 6‐FAM di colore blu e RET/PTC3 con NED di
colore giallo), allo scopo di evidenziare i prodotti amplificati nell’analisi dei frammenti mediante
elettroforesi capillare su un sequenziatore automatico (3130 Genetic Analyser, Applied
Biosystems).
Tabella 8: composizione della miscela di reazione usata per la messa a punto dell'analisi delle traslocazioni
RET/PTC1 e RET/PTC3
Conc. iniziale Quantità/Conc. finale Volume/campione
Costrutti RET/PTC1 e RET/PTC3 10 ng/ µl; 1 ng/ µl 10 ng/ µl; 1 ng/ µl 5 µl
PCR Buffer II 10X 1X 2,5 µl
MgCl2 25 mM 2 mM 2 µl
dNTPs 10 mM 0,2 mM 0,5 µl
dUTP 10 mM 0,04 mM 0,1 µl
Primer Forward (RET/PTC1 o
RET/PTC3)
10 µM 0,2 µM 0,5 µl

Ambra Cattani TAB3 SSMT Locarno 2012/2013
26
40 cicli
Primer Reverse (RET/PTC1 o
RET/PTC3)
10 µM 0,2 µM 0,5 µl
Taq Gold 5 U/µM 2 U/campione 0,4 µl
Acqua sterile 13,5 µl
Tabella 9: protocollo di amplificazione a gradiente di temperatura usato per la messa a punto dell'analisi
delle traslocazioni RET/PTC1 e RET/PTC3
Temperature Tempo di reazione
Stabilizzazione reazione 50°C 2’
Denaturazione iniziale 95°C 10’
Denaturazione 95°C 30’’
Ibridazione (annealing) 56°C (Tm)±5°C 30’’
Estensione 72°C 30’’
Estensione finale 72°C 7’
Fine PCR 4°C ∞
5.2.13 Validazione della metodica di analisi di traslocazione RET/PTC
Per la validazione della metodica di analisi delle traslocazioni RET/PTC1 e RET/PTC3 è stato
estratto l’RNA di 17 pazienti e retrotrascritto a cDNA. In seguito è stata e seguita la reazione di
PCR per le traslocazioni RET/PTC1 e RET/PTC3 secondo i protocolli messi a punto. Per verificare la
qualità e l’integrità dell’RNA estratto, l’adeguatezza del campione analizzato e l’efficienza della
reazione di retrotrascrizione, per ogni campione di cDNA (compresi i controlli bianchi e positivi di
retrotrascrizione) sono stati amplificati due geni di controllo, PGK1 e PAX8, secondo protocolli già
in uso diagnostico presso l’ICP (paragrafo 5.2.14). Per ogni reazione di PCR sono stati aggiunti un
controllo negativo (acqua sterile) e come controlli positivi i costrutti plasmidici diluiti a 1 ng/µl.
L’analisi RET/PTC1 e RET/PTC3 è stata eseguita per ogni paziente sia su tessuto sano che su tessuto
tumorale, al fine di mostrare che l’eventuale presenza di una traslocazione fosse presente
solamente nel tessuto tumorale. Con lo scopo di avere un quadro maggiormente completo delle
alterazioni genetiche dei pazienti presi in esame, è stata effettuata l’analisi dell’esone 15 del gene
BRAF (paragrafo 5.2.4).

Ambra Cattani TAB3 SSMT Locarno 2012/2013
27
5.2.14 Amplificazione dei geni di controllo PGK1 e PAX8
Il gene PGK1 è espresso ubiquitariamente in tutti i tessuti umani e per la sua amplificazione
tramite PCR sono stati utilizzati primers costruiti sull’esone 9 e sull’esone 11, che permettono
quindi di amplificare soltanto il cDNA e non il DNA genomico. La lunghezza dell’amplificato
ottenuto è di 225 bp circa e quindi l’avvenuta amplificazione di questo gene nei campioni di RNA
estratto è indice di buona qualità e di integrità dell’RNA stesso. Il gene PAX8 è invece espresso
soltanto dalle cellule epiteliali follicolari tiroidee e quindi la sua amplificazione tramite PCR nei
campioni in analisi indica non solo l’integrità dell’RNA, ma anche la presenza di cellule tiroidee nel
materiale analizzato. I primers utilizzati per la reazione di PCR sono costruiti sugli esoni 5 e 6 del
gene PAX8. Entrambi i primers forward dei due geni di controllo sono marcati con il fluorocromo
VIC. Le reazioni di PCR dei geni di controllo sono allestite in una singola miscela di reazione
eseguendo una PCR duplex. Per l’analisi dei geni di controllo è stato utilizzato il seguente
protocollo allestito sulla base della pubblicazione di Algeciras‐Schimnich (Algeciras‐Schimnich et al,
2010) (Tabelle 10 e 11) con le seguenti coppie di primers, con le relative temperature di
ibridazione e lunghezza degli amplificati:
Sequenza dei primers:
‐ Primer PAX8 Forward: 5’‐VIC ‐TCAACCTCCCTATGGACAGC‐3’
‐ Primer PAX8 Reverse: 5’‐GGAGTAGGTGGAGCCCAGG‐3’
‐ Primer PGK1 Forward: 5’‐VIC ‐CAGTTTGGAGCTCCTGGAAG‐3’
‐ Primer PGK1 Reverse: 5’‐GAGCTGAGATGCTGTGCAAC‐3’
‐ Temperatura di annealing: 65°C
‐ Dimensione dell’amplificato: PAX 8: 122 bp
PGK1: 224 bp
Tabella 10: composizione della miscela di reazione per la reazione di PCR dei geni di controllo PAX8 e PGK1
Concentrazione iniziale Quantità/Conc. finale Volume/campione
cDNA 25 ng/µl 10 ng 5 µl
PCR Buffer 10X 1X 2,5 µl
MgCl2 25 mM 2 mM 2 µl
dNTPs 10 mM 0,2 mM 0,5 µl
dUTPs 10 mM 0,04 mM 0,1 µl
Primer PAX8 Fw 10 µM 0,2 µM 0,5 µl

Ambra Cattani TAB3 SSMT Locarno 2012/2013
28
40 cicli
Primer PAX8 Rw 10 µM 0,2 µM 0,5 µl
Primer PGK1 Fw 10 µM 0,2 µM 0,5 µl
Primer PGK1 Rw 10 µM 0,2 µM 0,5 µl
Taq Gold 5 U/µM 2 U/campione 1 µl
Acqua sterile 11.9 µl
Tabella 11: protocollo di amplificazione per i geni di controllo PAX8 e PGK1
Temperature Tempo di reazione
Stabilizzazione reazione 50°C 2’
Denaturazione iniziale 95°C 10’
Denaturazione 95°C 30’’
Ibridazione 65°C 30’’
Estensione 72°C 30’’
Estensione finale 72°C 7’
Fine PCR 4°C ∞
5.2.15 Analisi dei prodotti di amplificazione tramite elettroforesi capillare
L’elettroforesi capillare è una metodica che sfrutta un campo elettrico per separare i frammenti di
DNA in base alle loro dimensioni. Tramite la migrazione attraverso un polimero (3130 POP‐7
Performancy Optimized Polymer, Applied Biosystems), i frammenti di DNA vengono analizzati alla
fine della migrazione da un raggio laser che rileva i fluorocromi legati ai primers utilizzati per
l’amplificazione dei geni di interesse. I prodotti ottenuti dall’amplificazione di RET/PTC1 e
RET/PTC3 e dei geni di controllo PGK1 e PAX8 sono stati diluiti in acqua sterile e caricati al
sequenziatore per l’elettroforesi capillare. Dato che i prodotti RET/PTC1 e RET/PTC3 sono marcati
con fluorocromi diversi e i prodotti PGK1 e PAX8, che pur essendo marcati con lo stesso
fluorocromo hanno dimensioni diverse, essi possono essere miscelati e diluiti nella stessa provetta
ed essere analizzati simultaneamente nello stesso capillare. È quindi stato stabilito il seguente
schema per la combinazione e la diluizione dei diversi prodotti prima della corsa elettroforetica
(tabella 12):

Ambra Cattani TAB3 SSMT Locarno 2012/2013
29
Tabella 12: composizione della miscela dei prodotti di PCR per l'analisi dei frammenti
Prodotti di amplificazione volume
RET/PTC1 4 µl
RET/PTC1 4 µl
PAX8‐PGK1 2 µl
H2O 20 µl
1 µl di questa miscela è stato posto in provette da 0,2 ml e dopo aver aggiunto 0.25 µl di
marcatore Genescan™‐500 Liz Size Standard (Applied Biosystems) e 10 µl di formammide (Hi‐Di
Formamide, Applied Biosystems), i campioni sono stati posti in un termociclatore a 95°C per 2
minuti: in questo modo è avvenuta la denaturazione dei frammenti di DNA (processo che
determina la formazione di due filamenti separati, di cui uno marcato con il fluorocromo). A
questo punto i campioni sono pronti per essere aliquotati nella piastra e analizzati con il
sequenziatore automatizzato. Mediante l’uso dell’apposito programma Gene Mapper Software
version 3 (Applied Biosystems), viene visualizzato l’elettroferogramma dei campioni analizzati. La
migrazione contemporanea, nello stesso capillare, dei prodotti di PCR e del marker, consente di
valutare precisamente le dimensioni dei prodotti di PCR e di visualizzarli contemporaneamente
nello stesso elettroferogramma. La valutazione della presenza di un riarrangiamento di RET/PTC1 e
RET/PTC3 può essere effettuata solo nel caso in cui, nel campione analizzato, siano presenti i
prodotti di amplificazione dei geni di controllo PAX8 e PGK1, identificati da picchi verdi delle
dimensioni di 122 bp e 224 bp rispettivamente. Se presente solo PAX8 la valutazione può essere
effettuata perché i prodotti RET/PTC1 e RET/PTC3 hanno dimensioni molto vicine a quelle di PAX8
(136 bp e 106 bp rispettivamente), ma è importante considerare che l’assenza di PGK1 significa
che l’RNA è degradato e questo potrebbe comportare dei risultati falsi negativi. Se presente solo
PGK1, il campione non è valutabile in quanto la presenza di PAX8 è necessaria per indicare la
presenza di tireociti. Se entrambi i geni di controllo sono assenti, il campione non è valutabile. In
presenza di controlli positivi rilevati correttamente, le presenza di un riarrangiamento RET/PTC1 è
rilevata con un picco blu a 130 bp circa, mentre la presenza di un riarrangiamento RET/PTC3 è
rilevata con un picco nero a 100 bp circa. Al fine di confermare la presenza di riarrangiamenti
RET/PTC1 e RET/PTC3, l’analisi è stata ripetuta riamplificando il cDNA per i campioni in cui è stata
rilevata la presenza di un riarrangiamento. Le sequenze nucleotidiche dei costrutti plasmidici
positivi e i campioni positivi per la presenza di un riarrangiamento sono state analizzate tramite

Ambra Cattani TAB3 SSMT Locarno 2012/2013
30
sequenziamento diretto secondo i protocolli descritti precedentemente utilizzando come primer di
sequenza il primer reverse sia per RET/PTC1 che per RET/PTC3.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
31
6. Risultati
6.1 Messa a punto delle reazioni di PCR per l’analisi delle traslocazioni
RET/PTC1 e RET/PTC3 Uno dei passaggi fondamentali per la messa a punto dell’analisi delle traslocazioni RET/PTC1 e
RET/PTC3 è stato quello di ricercare nella letteratura scientifica inerente tali analisi e specifica in
particolar modo per le metodologie e le tecniche utilizzate, le sequenze e le porzioni dei geni RET,
CCDC6 e NCOA4 coinvolte nel riarrangiamento e nella formazione dei geni di fusione RET/PTC1 e
RET/PTC3. In particolare gli articoli scientifici sui quali ci siamo basati per la messa a punto della
metodologia sono stati la pubblicazione di Nikiforov e colleghi del 2009 (Nikiforov et al, 2009), per
la scelta dei primers, e quella di Ferraz e colleghi del 2011 (Ferraz et al, 2011) per la scelta della
metodica d’analisi dei prodotti di PCR (analisi di frammenti tramite elettroforesi capillare). Dallo
studio della letteratura scientifica è emerso che il gene di fusione RET/PTC1 è determinato dalla
fusione dell’esone 1 del gene CCDC6 con la porzione TK del gene RET comprendente l’esone 12,
mentre il gene di fusione RET/PTC3 è determinato dal riarrangiamento tra la porzione del gene
NCOA4 compresa tra l’esone 1 e l’esone 10 e il dominio TK del gene RET. Sono state quindi
valutate le posizioni di appaiamento dei primers scelti (vedi materiali e metodi, paragrafo 5.2.12),
verificando che si legassero alle regioni fiancheggianti il punto di fusione del gene traslocato
(Figure 4 e 5). Il passaggio successivo è stato quello di valutare i primers mettendo a punto una
reazione di PCR. A questo scopo uno dei parametri principali da ottimizzare è la scelta della
temperatura di ibridazione dei primers, la quale viene stabilita sperimentalmente mediante una
PCR a gradiente di temperatura (vedi materiali e metodi, paragrafo 5.2.12).

Ambra Cattani TAB3 SSMT Locarno 2012/2013
32
Figura 4: sequenza del gene di fusione RET/PTC1. In nero l’esone 1 di CCDC6; in rosso l’esone 12 del gene
RET; evidenziate in giallo le posizioni delle sequenze in cui si appaiano i primers RET/PTC1 forward (Fw) e
reverse (Rw).
Figura 5: sequenza del gene di fusione RET/PTC3. In nero l’esone 10 di NCOA4; in rosso l’esone 12 del gene
RET; evidenziate in azzurro le posizioni delle sequenze in cui si appaiano i primers RET/PTC3 forward (Fw) e
reverse (Rw).
Per mettere a punto le due reazioni di PCR per l’analisi delle traslocazioni RET/PTC1 e RET/PTC3
sono stati utilizzati due costrutti plasmidici contenenti i due geni di fusione (vedi materiali e
metodi, paragrafo 5.2.12). La temperatura di melting media per entrambe le coppie di primers

Ambra Cattani TAB3 SSMT Locarno 2012/2013
33
RET/PTC1 e RET/PTC3 è stata calcolata pari a 56°C e le 5 temperature di ibridazione scelte per
eseguire le PCR a gradiente per testare le due coppie di primers sono elencate nella tabella 13:
Tabella 13: temperature di ibridazione per le PCR a gradiente di temperatura (T1‐T2‐T3‐T4‐T5) per la messa
a punto dell’analisi delle traslocazioni RET/PTC1 e RET/PTC3
T1 T2 T3 T4 T5
51,9°C 54,2°C 56,6°C 59,0°C 61,0°C
A seguito della PCR, i prodotti amplificati sono stati valutati tramite elettroforesi su gel di agarosio,
per verificare a quali temperature ci fosse una buona qualità di prodotto PCR e l’assenza di bande
aspecifiche. A tutte le temperature scelte per il gradiente (da 52°C a 61°C) e per entrambe le
concentrazioni di partenza dei costrutti, la reazione di amplificazione dei costrutti RET/PTC1 e
RET/PTC3 è risultata efficiente e specifica (Figure 6 e 7) e la lunghezza dei prodotti di
amplificazione ottenuti è risultata uguale a quella attesa (circa 130 bp per RET/PTC1 e 100 bp per
RET/PTC3).
Figura 6: Elettroforesi su gel di agarosio del prodotto di PCR a gradiente di temperatura per l’analisi della
traslocazione RET/PTC1. M: Marker, 1: costrutto plasmidico RET/PTC1 diluito a 10 ng/µl, 2: costrutto
plasmidico RET/PTC1 diluito a 1 ng/µl, 3: controllo negativo. T1‐T5: temperature di ibriazione scelte (vedi
Tabella 13).

Ambra Cattani TAB3 SSMT Locarno 2012/2013
34
Figura 7: Elettroforesi su gel di agarosio del prodotto di PCR a gradiente di temperatura per l’analisi della
traslocazione RET/PTC3. M: Marker, 1: costrutto plasmidico RET/PTC3 diluito a 10 ng/µl, 2: costrutto
plasmidico RET/PTC3 diluito a 1 ng/µl, 3: controllo negativo. T1‐T5: le temperature di ibridazione scelte
(vedi tabella 13).
Tutti i campioni amplificati sono stati in seguito analizzati tramite elettroforesi capillare in un
sequenziatore automatico per valutare il rilevamento dei prodotti amplificati tramite l’acquisizione
del segnale di fluorescenza ottenuto dal fluorocromo coniugato ai primers forward di entrambe le
coppie di primers. A tutte le temperature scelte per il gradiente e per entrambe le concentrazioni
di partenza dei costrutti si sono ottenuti segnali ottimi e delle dimensioni attese per entrambi i
costrutti RET/PTC1 e RET/PTC3 (Figure 8 e 9).

Ambra Cattani TAB3 SSMT Locarno 2012/2013
35
Figura 8: esempio di elettroferogramma del prodotto di amplificazione del costrutto RET/PTC1 (picco blu a
130 bp) rilevabile grazie al fluorocromo 6‐FAM coniugato al primer forward di RET/PTC1.
Figura 9: esempio di elettroferogramma del prodotto di amplificazione del costrutto RET/PTC3 (picco nero a
100 bp) rilevabile grazie al fluorocromo NED coniugato al primer forward di RET/PTC3.
Dimensione dell’amplificato (bp)
Dimensione dell’amplificato (bp)

Ambra Cattani TAB3 SSMT Locarno 2012/2013
36
Dato che i risultati ottenuti per entrambe le PCR sono ottimi a tutte le temperature di ibridazione
analizzate, è stata stabilita come temperatura di ibridazione sia per RET/PTC1 che per RET/PTC3 la
temperatura intermedia del gradiente pari a 57°C. I prodotti di amplificazione dei costrutti
RET/PTC1 e RET/PTC3 ottenuti a 57°C sono stati amplificati in una PCR di sequenza con i primers
reverse (non fluorocromato) per visualizzare la sequenza del gene di fusione che si è mostrata
uguale a quella attesa.
6.2 Validazione della metodica per l’analisi dei frammenti delle
traslocazioni RET/PTC1 e RET/PTC3, tramite RT‐PCR ed analisi dei
frammenti La validazione dei protocolli per l’analisi delle traslocazioni RET/PTC1 e RET/PTC3 è stata eseguita
mediante l’analisi di campioni di RNA estratti da tessuti FFPE (sia tumorali che sani) di 17 pazienti
con PTC, diagnosticato tra il 2010 e il 2012 presso l’ICP di Locarno. La casistica comprende 14
donne e 3 uomini, di età compresa tra i 20 e i 70 anni. In due casi (PC12 e PC18) l’RNA è stato
estratto solo da tessuto tumorale perché il corrispondente tessuto sano non era disponibile. L’RNA
estratto è stato retrotrascritto a cDNA secondo i protocolli già in uso presso l’ICP di Locarno. I
cDNA ottenuti sono stati analizzati per RET/PTC1 e RET/PTC3 con i protocolli di PCR messi a punto
sui costrutti plasmidici. Inoltre si sono effettuate, su ogni campione di cDNA, delle amplificazioni
per i geni di controllo PAX8 e PGK1 per verificare la buona qualità dell’RNA di partenza e accertare
che il corrispondente cDNA fosse amplificabile. In tutti i campioni analizzati il gene di controllo
PAX8 si è amplificato, indicando quindi l’adeguatezza del materiale di partenza (presenza di
tireociti) e assenza di degradazione dell’RNA in frammenti inferiori alle dimensioni dell’amplificato
PAX8 (122 bp). In un solo caso (PC4) il gene di controllo PGK1 non si è amplificato, indicando
quindi una parziale degradazione dell’RNA (Figura 11). Tuttavia, dal momento che i prodotti di PCR
per le forme traslocate hanno dimensioni di 100‐130 bp, paragonabili quindi alle dimensioni del
prodotto di amplificazione del gene PAX8, tutti i campioni analizzati possono essere valutati in
maniera certa e attendibile per la presenza o meno della traslocazione.
In tutti i campioni di tessuto normale analizzati, non si è rilevata la presenza di un riarrangiamento
(Figura 10). L’analisi dei campioni tumorali ha rilevato la presenza di un riarrangiamento in due
casi su 17 (11,8%). In particolare un caso è caratterizzato dalla presenza della traslocazione
RET/PTC1 (PC6, Figura 12) e un caso è caratterizzato dalla presenza del riarrangiamento RET/PTC3
(PC16, Figura 13).

Ambra Cattani TAB3 SSMT Locarno 2012/2013
37
Figura 10: esempio di elettroferogramma di un campione negativo per la presenza delle traslocazioni
RET/PTC1 e RET/PTC3. In verde i due picchi corrispondenti ai geni di controllo PAX8 (a 122 bp) e PGK1 (a 224
bp).
Figura 11: esempio di elettroferogramma di un campione (PC4) negativo per la presenza della traslocazione
RET/PTC in cui è avvenuta l’amplificazione del gene PAX8 ma non del gene PGK1, indice di una degradazione
parziale dell’RNA.
Dimensione dell’amplificato (bp)
Dimensione dell’amplificato (bp)

Ambra Cattani TAB3 SSMT Locarno 2012/2013
38
Figura 12: Esempio di un elettroferogramma di un campione (PC6) positivo per la traslocazione RET/PTC1
(picco blu a 130 bp). In verde i due picchi corrispondenti ai geni di controllo PAX8 (122 bp) e PGK1(224 bp).
Figura 13: Esempio di un elettroferogramma di un campione (PC16) positivo per la traslocazione RET/PTC3
(picco nero a 100 bp). In verde i due picchi corrispondenti ai geni di controllo PAX8 (122 bp) e PGK1(224 bp).
Dimensione dell’amplificato (bp)
Dimensione dell’amplificato (bp)

Ambra Cattani TAB3 SSMT Locarno 2012/2013
39
6.3 Analisi dello stato mutazionale del gene BRAF
Oltre all’analisi delle traslocazioni RET/PTC1 e RET/PTC3 è stata eseguita l’analisi dello stato
mutazionale dell’esone 15 del gene BRAF tramite sequenziamento diretto. Dei 17 pazienti
analizzati, 10 casi su 17 (58,8%) hanno mostrato la mutazione V600E determinata dalla
sostituzione di una timina con un’adenina (GTGGaG) nella seconda base del codone 600 che
porta al cambiamento amminoacidico di una valina con acido glutammico (Figura 14b).
Figura 14: a) elettroferogramma della sequenza WT del codone 600 del gene BRAF (esone 15)
b) elettroferogramma della sequenza dell’esone 15 del gene BRAF recante la mutazione puntiforme TA
che comporta il cambiamento nel codone GTGGaG e seguente generazione della mutazione V600E
In un paziente (PC16) è stata rilevata la presenza sia della traslocazione RET/PTC3 sia della
mutazione puntiforme del gene BRAF. Nello specifico, in questo caso, le presenze delle
traslocazioni è stata confermata in 2 analisi effettuate su 4, mentre dalla valutazione
dell’elettroferogramma il picco corrispondente alla mutazione di BRAF risultava particolarmente
basso e riconfermato in due analisi distinte.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
40
Tabella 14: risultati delle analisi delle traslocazioni RET/PTC1 e RET/PTC3 e dello stato mutazionale di BRAF
per i pazienti con PTC. Legenda: T: tumore, N: sano, WT: wild‐type, NV: non valutabile
caso sesso Età/diagnosi RET/PTC1 e RET/PTC3 BRAF
PC1 N F 66
assenza di traslocazione
PC1 T assenza di traslocazione WT
PC2 N F 41
assenza di traslocazione
PC2 T assenza di traslocazione WT
PC3 N F 64
assenza di traslocazione
PC3 T assenza di traslocazione V600E
PC4 N F 81
assenza di traslocazione
PC4 T assenza di traslocazione V600E
PC5 N F 40
assenza di traslocazione
PC5 T assenza di traslocazione V600E
PC6 N F 45
assenza di traslocazione
PC6 T presenza della traslocazione RET/PTC1 WT
PC9 N F 61
assenza di traslocazione
PC9 T assenza di traslocazione WT
PC10 N M 43
assenza di traslocazione
PC10 T assenza di traslocazione WT
PC12 N M 47
NV
PC12 T assenza di traslocazione V600E
PC13 N F 74
assenza di traslocazione
PC13 T assenza di traslocazione V600E
PC14 N F 48
assenza di traslocazione
PC14 T assenza di traslocazione V600E
PC16 N F 47
assenza di traslocazione
PC16 T presenza della traslocazione RET/PTC3 V600E
PC17 N F 22
assenza di traslocazione
PC17 T assenza di traslocazione V600E
PC18 N M 50
NV
PC18 T assenza di traslocazione V600E
PC19 N F 29
assenza di traslocazione
PC19 T assenza di traslocazione WT
PC 20 N F 42 assenza di traslocazione

Ambra Cattani TAB3 SSMT Locarno 2012/2013
41
PC 20 T assenza di traslocazione WT
PC21 N F 31
assenza di traslocazione
PC21 T assenza di traslocazione V600E

Ambra Cattani TAB3 SSMT Locarno 2012/2013
42
7. Discussione I noduli della tiroide rappresentano la patologia endocrina più frequente la cui prevalenza può
arrivare fino al 70% nella popolazione generale. Sebbene la maggior parte dei noduli tiroidei siano
benigni è sempre necessario escluderne la malignità. Molto importante risulta quindi essere la
diagnosi preoperatoria del nodulo tiorideo affinché il paziente possa ricevere tempestivamente la
terapia corretta in caso di malignità o possa evitare di subire un intervento chirurgico non
necessario nel caso in cui la lesione fosse benigna. Attualmente il metodo di esame preoperatorio
d’elezione per la diagnosi di un nodulo tiroideo è l’aspirazione con ago sottile e la seguente
valutazione citologica del campione, la quale, tuttavia, in circa il 30‐35% dei casi può dare un
risultato non diagnostico (in caso di inadeguatezza del materiale) o un risultato di significato
indeterminato di malignità (TIR3). Per cercare di risolvere tali problemi e migliorare quindi
l’accuratezza della diagnosi citologica fatta su FNA si sono sviluppate nuove analisi di diagnostica
molecolare che si basano sulla valutazione dello stato mutazionale di alcuni geni, le cui alterazioni
sono caratteristiche e specifiche per un particolare tipo di tumore tiroideo. Il tumore tiroideo più
diffuso è il carcinoma papillare (80% dei casi) caratterizzato, a livello molecolare, da due principali
alterazioni genetiche: le mutazioni puntiformi del gene BRAF e i riarrangiamenti RET/PTC. Il
secondo tipo di tumore tiroideo più frequente è il carcinoma follicolare (15% dei casi)
caratterizzato, a livello molecolare, dalla traslocazione PAX8/PPARy e da mutazioni dei geni RAS.
La valutazione dei marcatori molecolari sopra citati è raccomandata dalle linee guida
dell’associazione americana sulla tiroide (2009 Revised ATA management Guidelines) per
migliorare l’accuratezza diagnostica dei campioni citologici indeterminati. Ad eccezione dell’analisi
della traslocazione RET/PTC, tutte le altre (mutazioni di BRAF, RAS e la traslocazione PAX8/PPARy)
sono disponibili presso l’ICP di Locarno. Lo scopo del presente lavoro di diploma è stato quindi
quello di mettere a punto una metodica per l’analisi delle traslocazioni RET/PTC, avvalendosi di
dati pubblicati da lavori scientifici e protocolli standard presenti all’ICP. La ricerca dei
riarrangiamenti RET/PTC prevede una prima fase di estrazione dell’RNA da materiale istologico o
citologico adeguato, la retrotrascrizione dell’RNA a cDNA, un secondo passaggio di amplificazione
del cDNA e una seguente analisi dei frammenti in un sequenziatore automatico. I protocolli di
estrazione dell’RNA e retrotrascizione ed analisi dei frammenti erano già disponibili presso l’ICP,
quindi la messa a punto ha riguardato principalmente la preparazione di due protocolli di PCR per
la valutazione delle due forme più comuni di riarrangiamento RET/PTC1 e RET/PTC3. La sequenza

Ambra Cattani TAB3 SSMT Locarno 2012/2013
43
dei primers utilizzata è stata prelevata da articoli scientifici (Ferraz et al, 2011) presenti su PubMed
(http://www.ncbi.nlm.nih.gov/pubmed) e le temperature di annealing sono state determinate
sperimentalmente tramite PCR a gradiente di temperatura. Ciò è stato fatto poiché la temperatura
di ibridazione dei primers è uno degli aspetti fondamentali di una reazione di PCR e, in alcuni casi,
una temperatura non ottimale potrebbe inficiare l’analisi. La messa a punto dei protocolli di PCR è
stata effettuata su dei costrutti plasmidici sintetici comprendenti le regioni genomiche coinvolte
nei riarrangiamenti RET/PTC1 e RET/PTC3 poiché non erano a disposizione campioni di pazienti o
linee cellulari recanti tali traslocazioni. L’amplificazione dei costrutti e la successiva analisi dei
frammenti amplificati ha dato dei risultati ottimi a tutte le temperature testate e per entrambi i
costrutti.
Un passaggio fondamentale per analizzare i protocolli messi a punto è stato quello della loro
validazione tramite l’analisi dell’RNA estratto da tessuti FFPE di pazienti affetti da PTC, valutando
sia la componente tumorale che quella normale. Dal punto di vista tecnico i risultati ottenuti dalla
valutazione dei geni di controllo PAX8 e PGK1 hanno evidenziato l’adeguatezza del materiale di
partenza (ossia RNA estratto da tessuti FFPE) all’analisi di geni espressi dalla ghiandola tiroidea.
Nello specifico, l’amplificazione in tutti i campioni del gene PAX8, gene espresso soltanto dalle
cellule tiroidee, indica non solo la presenza di tireociti in tutti i campioni analizzati, ma anche la
possibilità di amplificare geni specifici e costitutivi della ghiandola tiroidea e l’integrità dell’RNA
estratto. La conferma della presenza di tireociti tramite l’analisi di PAX8 è un aspetto molto
importante, non tanto per la valutazione dei campioni istologici (resezioni chirurgiche della
tiroide), quanto per i campioni citologici ottenuti da FNA, in cui spesso le cellule tiroidee sono
poche e frammiste a sangue e ad altri tipi di cellule. Per quanto riguarda PGK1, l’amplificazione è
avvenuta con successo in 16 casi su 17 analizzati (sia campione sano che tumorale), indicando
pertanto l’integrità dell’RNA analizzato e quindi la possibilità di applicare correttamente
metodiche di valutazione dell’RNA anche partendo da tessuti FFPE in cui, data la fissazione in
formalina, gli acidi nucleici, in particolare l’RNA, potrebbero essere fortemente degradati. Due casi
su 17 (11,8%) analizzati sono risultati traslocati per RET/PTC (uno per RET/PTC1 e uno per
RET/PTC3), risultato in linea con quanto riportato in letteratura (Nikiforov et al, 2011). In entrambi
i campioni è stato rilevato il riarrangiamento solamente nel tessuto tumorale e non nel
corrispondente tessuto sano, evidenziando e confermando quindi che tale traslocazione è tumore
specifica. Per avere un quadro più completo delle alterazioni presenti nella casistica PTC utilizzata
per la validazione, è stata effettuata l’analisi di BRAF sui tessuti tumorali di tutti i pazienti. La

Ambra Cattani TAB3 SSMT Locarno 2012/2013
44
mutazione di BRAF è stata trovata in 10 casi su 17 (58,8%), mostrando come tale mutazione sia
l’anomalia genetica dominante nei PTC, la percentuale risulta piuttosto alta rispetto a quanto
riportato mediamente dalla letteratura, ma la review di Xing (Xing, 2005) riporta vari studi che
mostrano la possibilità di avere un range più alto. Un caso è risultato positivo sia per la presenza
della mutazione BRAF sia per la presenza della traslocazione RET/PTC. Tale risultato non si accorda
con la maggior parte dei testi presenti in letteratura che vorrebbe tali anomalie mutuamente
esclusive. Tuttavia, uno studio condotto da Xiulong e colleghi (Xiulong et al, 2003) studia una
possibile cooperazione tra la mutazione BRAF e le traslocazioni RET/PTC nella tumoreogenesi del
PTC e anche un abstract presentato al National Cancer Research Institute‐Cancer Conference nel
2012 (Guerra et al, 2012), mostra la possibilità di una coesistenza della traslocazione RET/PTC e
della mutazione di BRAF, soprattutto in presenza di metastasi. Tale considerazione può risultare
rilevante in associazione al fatto che il campione analizzato è una metastasi linfonodale di un PTC.
È bene inoltre tenere in considerazione che in tale campione, per quanto riguarda l’analisi di BRAF,
il picco corrispondente alla mutazione, sebbene confermato in due analisi distinte, è risultato
molto basso rispetto al picco WT, suggerendo quindi che non tutte le cellule tumorali analizzate
erano caratterizzate dalla presenza di tale mutazione. Per quanto riguarda l’analisi RET/PTC, la
traslocazione RET/PTC3 è stata identificata in due esperimenti condotti su 4. Questi risultati
potrebbero far pensare ad una possibile eterogeneità tumorale , quindi alla presenza di diverse
popolazioni cellulari, alcune recanti la mutazione di BRAF e altre la traslocazione RET/PTC3.
Per confermare l’ipotesi e la possibilità di una coesistenza delle mutazioni di BRAF e delle
traslocazioni di RET, sarebbe necessario analizzare una casistica più ampia di pazienti.
Per quanto concerne la metodologia, la metodica per l’analisi della traslocazione RET/PTC è stata
messa a punto e valutata con successo. È necessario tenere presente che l’analisi messa a punto
identifica solo le due traslocazioni RET/PTC1 e RET/PTC3 (presenti nel 60‐70% e nel 20‐30% dei casi
traslocati rispettivamente, Zhu et al, 2006), ma in letteratura sono descritte più di 15 varianti
diverse la cui identificazione richiederebbe la messa a punto di altrettante reazioni di PCR.
Nel futuro sarà necessario indagare la sensibilità analitica della metodica tramite l’analisi di RNA
positivi per la traslocazione diluiti a varie concentrazioni con campioni di RNA negativi per il
riarrangiamento. Inoltre, dato che la metodica è stata validata su campioni istologici, ma sarà
maggiormente utilizzata per la valutazione degli FNA citologici, sarà necessario nel futuro
analizzare anche campioni citologici disponibili dei pazienti indagati per confermare la presenza,
anche in questo tipo di materiale, delle alterazioni riscontrate a livello istologico.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
45
8. Conclusione La messa a punto della metodica per l’analisi dei frammenti delle traslocazioni RET/PTC1 e
RET/PTC3 è stata conclusa con successo e quindi i protocolli per la reazione di PCR sono ottimizzati
e disponibili per l’utilizzo diagnostico. Non si sono verificati problemi né nel testare i primers, né
nell’utilizzo di tutti i protocolli durante la validazione. Inoltre i risultati delle validazioni,
dimostrano la specificità della metodica utilizzata. Previa validazione citologica, prevista in un
prossimo futuro, la metodica va a completare il pannello di diagnosi molecolare del tumore
tiroideo, disponibile presso l’ICP di Locarno.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
46
9. Bibliografia Reviews
1) Nikiforov, Molecular Diagnostics of thyroid Tumors, 569‐577, Archives of Pathology
and Laboratory Medicine, vol 135, 2011
2) Romei et al, RET/PTC translocations and clinic‐pathological features in human papillary
thyroid carcinoma, art. 54, Frontiers in Endocrinology, 2012.
3) Chien et al, Molecular Biology of Thyroid Cancer, Endocrine Updates 30, Springer
Science+Buisiness Media, 2012
4) Kondo et al, Pathogenic mechanism in thyroid follicular‐cell neoplasia, 292‐306, Nature
Reviews Cancer, 2006
5) Nelva, Linee di indirizzo interne per la patologia nodulare tiroidea presso l’ASL BI, Gruppo
Interdisciplinare Tiroide‐Epidemiologia della patologia nodulare tiroidea nel Biellese, 2011
6) Hunt, Molecular Mutations in Thyroid Carcinogenesis, 116‐127, Journal of Clinical
Pathology, 2002
7) Santoro et al, RET/PTC activation in papillary thyroid carcinoma, 645‐653, European Journal
of Endocrinolgy Prize Lecture, 2006
8) Xing, BRAF mutation in thyroid cancer, 245‐262, Endocrine‐Related Cancer, 2005
Articoli
9) Nikiforov et al, Molecular Testing for Mutations in Improving the Fine‐Needle Aspiration
Diagnosis of Thyroid Nodules, 2092‐2098, The Journal of Clinical Endocrinology, 2009
10) Xing et al, BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer,
6373‐6379, The Journal of Clinical Endocrinology, 2006
11) Algeciras‐Schimnich et al, Evaluation of the PAX8/PPARG Translocation in Follicular Thyroid
Cancer with a 4‐Color Reverse‐Transcription PCR Assay and Automated High‐Resolution
Fragment Analysis, 391‐398, Clinical Chemistry, 2010
12) Ferraz et al, Detection of PAX8/PPARG and RET/PTC Rearrangements are Feasible in
Routine Air Dried Fine Needle Aspiration (FNA) Smears, 1‐22, Official Journal of the
American Thyroid Association, 2011
13) Xiulong et al, High Prevalence of BRAF Gene Mutation in Papillary Thyroid Carcinomas and
Thyroid Tumor Cell Lines, 4561‐4567, Cancer Research, 2003

Ambra Cattani TAB3 SSMT Locarno 2012/2013
47
14) Zhu et al, Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: effects of
the detection methods and genetic heterogeneity, 3603‐3610, the Journal of Clinical
Endocrinology,2006
15) Cooper et al, Revised American Thyroid Association Management Guidelines for Patients
with Thyroid Nodules and Differentiated Thyroid Cancer, 1167‐1214, The American Thyroid
Association (ATA), 2009
Libri
16) Nikiforov, Recent Developpements in the Molecular biology of the Thyroid, 191‐209,
Endocrine Pathology, 2003
Conferenze
17) Guerra et al, Simultaneous occurrence of BRAF mutation and RET/PTC rearrangement is
frequent in papillary thyroid carcinoma, 1787,Endocrine Abstracts, 2012
Siti internet
18) http://www.ncbi.nlm.nih.gov/pubmed
19) http://mcb.berkeley.edu/labs/krantz/tools/oligocalc.html

Ambra Cattani TAB3 SSMT Locarno 2012/2013
48
10.RingraziamentiRingrazio vivamente tutto il personale del laboratorio di Patologia Molecolare dell’ICP di Locarno
dove ho avuto l’opportunità di svolgere il mio lavoro di diploma. In particolar modo ringrazio il Dr.
Milo Frattini e la Dr.ssa Francesca Molinari che mi hanno insegnato le basi del lavoro di laboratorio
di biologia molecolare e mi hanno seguita durante tutte le fasi dello svolgimento del lavoro,
dandomi consigli e aiutandomi nei momenti di difficoltà.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
49
11.Allegati11.1 Estrazione di DNA genomico da tessuti fissati in formalina ed inclusi
in paraffina (FFPE)
Trattamento con proteinasi k
‐ Aggiungere 180 µl di tampone di lisi tissutale ATL del kit e 20 µl di Proteinasi k (0,02 mg/µl)
(Sigma‐Aldrich, St.Louis, MO, USA) ai campioni di tessuto FFPE.
‐ Vortexare e sigillare con parafilm
‐ Incubare alla temperatura di 70°C su termoblocco in agitazione, per tutta la notte. A tale
temperatura la paraffina si scioglie e l’enzima è attivo per la lisi cellulare.
Trattamento di lisi cellulare
‐ Centrifugare le provette a 13000 g per 10 secondi e aggiungere 200 µl di tampone di lisi
cellulare AL del kit, vortexare per 15 secondi e incubare a 70°C per 10 minuti, centrifugare
alla velocità massima.
‐ Aggiungere 200 µl di etanolo al 100% (Sigma‐Aldrich), vortexare e centrifugare alla velocità
massima.
Purificazione del DNA su colonne QIAmp DNA Mini kit (Qiagen)
‐ Applicare la miscela ottenuta su una colonna QIAamp fornita dal kit con membrana
costituita da una resina in silice. La resina, a scambio ionico, è carica positivamente e
trattiene per interazione elettrostatica il materiale dotato di carica negativa, tra cui il DNA.
Centrifugare a 8000 g per 1 minuto.
‐ Trasferire la colonna su una provetta vuota ed eliminare la provetta contenente il filtrato.
Aggiungere 500 µl di tampone di lavaggio AW1, un tampone ad elevata concentrazione
salina che permette l’eluizione delle molecole rimaste attaccate in modo aspecifico alla
resina. Centrifugare a 8000 g per 1 minuto.
‐ Trasferire la colonna su una provetta vuota ed eliminare la provetta contenente il filtrato.
Aggiungere 500 µl di tampone di lavaggio AW2, un tampone a concentrazione salina
inferiore del precedente che permette la completa rimozione di proteine o altri
contaminanti legati in modo aspecifico alla membrana. Centrifugare a 14000 g per 1
minuto.
‐ Trasferire la colonna su una provetta vuota ed eliminare la provetta contenente il filtrato.
Centrifugare a 14000 g per 1 minuto.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
50
‐ Trasferire la colonna QIAmp in una Eppendorf sterile da 1,5 ml e aggiungere 50 µl di
tampone di eluizione AE (10 mM Tris‐HCl, 0,5 mM EDTA, pH 9,0) in modo da recuperare il
DNA legato elettrostaticamente al supporto della colonna, nell’eluato. Incubare a
temperatura ambiente per 5 minuti, centrifugare a 8000 g per 1 minuto.
‐ Al fine di aumentare la resa dell’estrazione, riprendere l’eluato centrifugato nel punto
precedente e caricarlo di nuovo sulla colonna QIAmp. Incubare a temperatura ambiente
per 5 minuti, centrifugare a 8000 g per 1 minuto.
‐ Eliminare la colonna QIAmp e tenere la provetta Eppendorf con il DNA estratto, che verrà
quantificato con lo spettrofotometro.
11.2 Estrazione RNA da tessuto FFPE
Per l’estrazione dell’RNA da tessuto FFPE è stato usato il kit RNAeasy MiniKit (Qiagen, Chatsworth,
CA, USA). Prima di ogni operazione pulire le superfici, le pipette e i materiali d’uso, con RNAsi Zap
(Invitrogen, Carlsbad, CA, USA)
‐ Posizionare le sezioni in una provetta Eppendorf da 1.5 ml.
‐ Aggiungere 160 μl di Deparaffinization Solution, miscelare con il vortex per 10 secondi e
centrifugare brevemente.
‐ Incubare per 3 minuti a 56°C.
‐ Aggiungere 150 μl di buffer PKD e miscelare con il vortex.
‐ Centrifugare per 1 minuto a 10000 rpm (11000 g).
‐ Aggiungere 10 μl di proteinasi K alla fase sottostante più chiara. Miscelare delicatamente
utilizzando la pipetta.
‐ Incubare per 15 minuti a 56°C, poi per 15 minuti a 80°C.
‐ Trasferire la fase sottostante, più chiara in una nuova eppendorf da 2 ml.
‐ Incubare in ghiaccio per 3 minuti. Centrifugare per 15 minuti a 13500 rpm (20000 g).
‐ Trasferire il surnatante ad una nuova provetta Eppendorf prestando attenzione a non
disgregare il pellet.
‐ Aggiungere 16 μl di DNase Booster Buffer e 10 μl di DNase I. Miscelare invertendo la
provetta. Centrifugare brevemente.
‐ Incubare per 15 minuti a RT.
‐ Aggiungere 320 μl di Buffer RBC e miscelare il lisato accuratamente.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
51
‐ Aggiungere 720 μl di etanolo (100%) al campione e miscelare bene con la pipetta. NON
centrifugare.
‐ Trasferire 700 μl del campione, inclusi i precipitati che possono essere formatisi, sulla
colonna RNeasy MinElute precedentemente posizionata in un collection tube da 2 ml
(fornito dal kit). Chiudere il tappo delicatamente e centrifugare per 15 secondi a 10000 rpm
(8000g). Eliminare lo scarto.
‐ Ripetere il passaggio precedente in modo da caricare tutto il campione sulla colonna,
utilizzando lo stesso collection tube.
‐ Aggiungere 500 μl di Buffer RPE alla colonna. Chiudere il tappo delicatamente e
centrifugare per 15 secondi a 10000 rpm (8000g). Eliminare lo scarto.
‐ Aggiungere 500 μl di Buffer RPE alla colonna. Chiudere il tappo delicatamente e
centrifugare per 2 minuti a 10000 rpm (8000g) per lavare la membrana della colonna.
Eliminare lo scarto. NB: fare attenzione che, rimuovendo la colonna, questa non venga a
contatto con lo scarto.
‐ Posizionare la colonna in un nuovo collection tube da 2 ml. Aprire il coperchio della colonna
e centrifugare alla max velocità per 5 minuti. Eliminare lo scarto.
‐ Porre la colonna in un nuovo collection tube da 1.5 ml (fornito dal kit). Aggiungere 20 μl di
acqua RNase free direttamente sulla membrana all’interno della colonna. Chiudere
delicatamente il tappo e centrifugare per 1 minuto alla max velocità per eluire l’RNA. NB: il
volume di acqua RNase free può variare tra 14 e 30 μl. Un minor volume corrisponde ad
una maggiore concentrazione dell’RNA totale ma ad una minore quantità di prodotto.
‐ Quantificare l’RNA estratto con lo spettrofotometro nanodrop.
11.3 Quantificazione degli acidi nucleici
La quantificazione degli acidi nucleici viene effettuata utilizzando un apposito spettrofotometro
(Nanodrop 1000) in grado di utilizzare solo 2 µl di soluzione. Il Nanodrop è uno spettrofotometro
UV‐sensibile che rileva la tensione superficiale che piccoli volumi di soluzioni esercitano quando
sono collocati tra due superfici vicine per misurare la densità ottica del campione. Prima di iniziare
a quantificare, lo strumento viene tarato analizzando un bianco che, nel caso di quantificazione del
DNA genomico si tratta del tampone di eluizione AE, in cui sono stati eluiti i DNA genomici, mentre
nel caso dell’RNA si tratta di acqua RNAase free. A seguito della taratura dell’apparecchio,
vengono pipettatati 2 µl di campione sull’apposito sito di lettura in modo da creare una colonna di

Ambra Cattani TAB3 SSMT Locarno 2012/2013
52
liquido a diretto contatto con le due fibre ottiche, permettendo una rapida analisi del campione.
Lo spettrofotometro Nanodrop è in grado di eseguire contemporaneamente letture a diverse
lunghezze d’onda, perciò è possibile determinare, oltre che il valore d’estinzione specifico degli
acidi nucleici ad una lunghezza d’onda di 260 nm, anche il loro livello di purezza. Una soluzione di
acidi nucleici pura alla lunghezza d’onda di 230 nm (che corrisponde al picco di assorbimento degli
zuccheri e degli alcoli come xilolo e etanolo) e alla lunghezza d’onda di 280 nm (che corrisponde al
picco di assorbimento delle proteine) ha un valore di assorbanza pari alla metà di quello misurato
a 260 nm. L’apparecchio, oltre che a fornire la concentrazione degli acidi nucleici, calcola
automaticamente i rapporti 260/230 nm e 260/280 nm, quando questi sono inferiori a 1,8‐2
significa che vi è presenza di contaminanti oltre gli acidi nucleici. A seguito della quantificazione
degli acidi nucleici si provvede alla conservazione del materiale: la soluzione madre di DNA è stata
conservata a ‐20°C, mentre le diluizioni di lavoro effettuate (ottenendo una concentrazione finale
di 25 ng/µl), sono state conservate a 4°C. L’RNA è stato retro‐trascritto in giornata, diluendolo in
modo da ottenere una concentrazione finale di cDNA pari a 25 ng/µl, conservato a una
temperatura di ‐20°C, mentre l’RNA non retro‐trascritto è stato messo a ‐80°C.
11.4 Costituzione della miscela di reazione per la PCR
‐ Tampone di reazione PCR buffer II 10x (Applied Biosystems, Foster City, CA, USA) composto
da Tris‐HCl 100 mM, pH 8,3 e da KCl 500 mM, che permette di garantire il pH ottimale per
l’attivazione della Taq polimerasi alle varie temperature e di favorire l’ibridazione tra
primer e templato.
‐ Cloruro di magnesio (25 mM MgCl2, Applied Biosystems). La cui concentrazione influisce
sulla specificità e sull’efficienza di reazione.
‐ Enzima Taq Gold 5 U/µl (Applied Biosystems), enzima DNA polimerasi stabile ad alte
temperature.
‐ Miscela equimolare di deossinucleotiditrifosfati (dNTPs; Amersham, Buckinghamshire, UK)
10 mM che vengono incorporati dalla Taq polimerasi per sintetizzare un nuovo filamento,
composta da:
o Deossiadenintrifosfato (dATP),
o Deossitiminatrifosfato (dTTP),
o Deossicitosinatrifosfato (dCTP),
o Deossiguaninatrifosfato (dGTP)

Ambra Cattani TAB3 SSMT Locarno 2012/2013
53
‐ Deossiuridinatrifosfato (dUTP) 10 mM (Amersham), nucleotide che permette di evitare le
contaminazioni di templati amplificati in precedenza. Questo nucleotide è infatti
incorporato nel prodotto PCR e viene riconosciuto dall’enzima UNG che taglia il DNA
amplificato in corrispondenza di tale nucleotide.
‐ Primers 10 µM (Invitrogen), sono corti segmenti di DNA (senso‐forward e antisenso
reverse) che ibridano il DNA stampo a fianco delle regioni di interesse e permettono l’avvio
della reazione di polimerizzazione del nuovo filamento di DNA da parte della DNA
polimerasi, poiché fungono da punto d’innesco della reazione.
‐ Acqua sterile.
11.5 Elettroforesi su gel di agarosio Preparazione del gel di agarosio 1,8%
‐ Pesare 1,8 grammi di agarosio (PeqGOLD Universal Agarose; Biotechnologie GmbH,
Erlangen, Germania) in una beuta.
‐ Portare a volume a 100 ml con tampone TBE 1X, ottenuto per diluizione del Tris‐Borato‐
EDTA 10X (Sigma‐Aldrich)
‐ Sciogliere la polvere di agarosio in modo omogeneo utilizzando un forno a microonde a 200
W per 10‐12 minuti.
‐ Aggiungere al gel 0,2 µl GelRed (Biotium’s GelRed Nucleic Acid Gel Stain; Biotium, Hayward,
CA, USA). Il colorante si intercala nelle molecule di DNA durante la corsa elettroforetica.
‐ Quando la soluzione raggiunge circa la temperatura di 60°C versarla nel supporto della cella
elettroforetica dopo aver inserito un pettine che permette di ottenere i pozzetti nel gel in
cui caricare i campioni.
‐ Lasciare raffreddare il gel a temperatura ambiente, poi estrarre il pettine e posare il gel
nella cella elettroforetica, immergendolo nel tampone TBE 1X
Preparazione dei campioni e corsa elettroforetica
‐ Preparazione del marker: miscelare 1 µl di DNA marker PeqGOLD 50 bp DNA‐Leiter (0,5 µg
totali) (Biotechnologie GmbH) con 9 µl di tampone 1X e 2 µl di una soluzione di Blu di
Bromofenolo (BBF), precedentemente preparata sciogliendo 0,25 grammi di BBF polvere
(Sigma‐Aldrich) che mantiene sotto controllo la migrazione dei campioni e 40 grammi di
saccarosio (Merck, Whitehouse Station, Nj, USA) che addensa i campioni nei pozzetti in 100
ml di acqua sterile.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
54
‐ Preparazione dei campioni: miscelare 5 µl del prodotto di PCR con 2 µl della soluzione BBF
‐ Caricare il marker e i campioni nei pozzetti del gel di agarosio e avviare a corsa
elettroforetica a 100 V costanti, per circa 35‐40 minuti.
Visualizzazione dei risultati
‐ Porre il gel nel transilluminatore (Image Master, Pharmacia, Stoccolma, Svezia). La
radiazione UV del transilluminatore eccita il colorante GelRed intercalato nella doppia elica
di DNA che riemette luce nella lunghezza d’onda nel visibile, permettendo di riconoscere le
bande (composte da DNA amplificato o frammenti di marker) che appaiono fluorescenti su
sfondo scuro. Questo permette di vedere se l’amplificazione del gene d’interesse sia
riuscita e verificare che il controllo negativo sia relamente negativo e che il controllo
positivo si sia amplificato come i campioni. Dopodiché si può procedere con le analisi
successive.
11.6 Purificazione del DNA amplificato
È stato utilizzato il kit MSB®SPIN PCRapace (STRATEC Molecular, Gmbh, Berlino, Germania), con i
seguenti passaggi condotti a temperatura ambiente:
‐ Aggiungere 200 µl di Binding Buffer al campione, miscelando bene con la pipetta.
‐ Trasferire completamente il prodotto PCR con il Binding Buffer sulla colonna provvista di
spin filter.
‐ Centrifugare i campioni a 13200 g per 5 minuti.
‐ Eliminare il fltrato e porre il filtro (Spin Filter) in un nuovo Collection Tube da 1,5 ml.
‐ Aggiungere 20 µl di Eluition Buffer nella colonna e incubare per 1 minuto a temperatura
ambiente.
‐ Centrifugare i campioni a 9300 g per 1 minuto
‐ Eliminare la colonna e il DNA eluato è pronto per l’uso nella provetta.
11.7 PCR di sequenza (cyclesequencing)
Preparare una miscela costituita da:
‐ Primer forward o reverse. I primers sono gli stessi utilizzati nella reazione di amplificazione
del DNA genomico.
‐ Sequencing Buffer: Big Dye Terminator v1.1/3.1 Sequencing buffer 5X (Applied Biosystems)

Ambra Cattani TAB3 SSMT Locarno 2012/2013
55
25 cicli
‐ Miscela di terminatori, deossinucleotidi e Taq Polmerasi: ABI PRISM Big Dye Terminator
v1.1 Ready Reaction mix (Applied Biosystems). Questo componente viene aggiunto per
ultimo alla miscela di reazione per ridurre il tempo di esposizione dei terminatori alla luce,
data la loro fotosensibilità.
‐ Acqua sterile
Aliquotare 0,4 µl di DNA purificato in provette da 0,2 ml e aggiungere 9,6 µl della miscela di
reazione descritta nella tabella 15. Inserire le provette nel termociclatore e dare avvio al
programma. Il protocollo di amplificazione della PCR di sequenza è sempre lo stesso, riportato
nella a tabella 11.
Tabella 15: composizione della miscela di reazione per la reazione di PCR di sequenza
Conc. iniziale Quantità/Conc. finale Volume/campione
DNA purificato 0,4 µl
Sequ. Buffer 5X 0,75 X 1,5 µl
TRR mix 2,5X 0,25 X 1,0 µl
Primer Fw o Rw 10 µM 0,5 µM 0,5 µl
Acqua sterile 6,6 µl
Tabella 16: protocollo di amplificazione delle PCR di sequenza
Temperature Tempo di reazione
Denaturazione iniziale 96°C 1’
Denaturazione 96°C 10’
Ibridazione 50°C 5’
Estensione 60°C 4’
Fine PCR 4°C ∞
11.8 Purificazione del DNA dopo PCR di sequenza
Il protocollo di purificazione del DNA dopo la PCR di sequenza prevede l’utilizzo del G50 Dye
Terminator Removal kit (RBC Bioscience, New Taipei City, Taiwan), con i seguenti passaggi:
‐ Risopendere la resina con 300 µl di acqua sterile picchiettando sulla parte superiore della
colonna fornita dal kit. Staccare il tappo rosso nella parte inferiore della provetta.

Ambra Cattani TAB3 SSMT Locarno 2012/2013
56
‐ Centrifugare le colonne a 3000 g per 2 minuti.
‐ Eliminare il tubo posto sotto la colonna, contenente l’eccesso di acqua e spostare la
colonna su una nuova Eppendorf da 1,5 ml.
‐ Caricare il prodotto della PCR al centro della colonna, senza toccare la resina con il puntale.
‐ Centrifugare i campioni a 3000 g per 3 minuti. Fare attenzione a mantenere la stessa
posizione della provetta, in modo che la resina si sposti uniformemente nella stessa
direzione.
‐ Eliminare le colonne e porre le provette nella centrifuga Speedvac (Eppendorf, Amburgo,
Germania) ad una temperatura di 30°C per un tempo di 23 minuti al buio.
‐ Risospendere i pellet in formamide e caricare il sequenziatore.


















