MERCK RESEARCH CLINICAL STUDY REPORT LABORATORIES I....
118
/MK-0476/CSR/RC1441.DOC APPROVED 06OCT97 MERCK RESEARCH LABORATORIES CLINICAL STUDY REPORT I. SYNOPSIS Montelukast Sodium Tablets (MK-0476) PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Crossover Study Comparing the Clinical Effect of Combination MK-0476/Loratadine to MK-0476 in Patients With Chronic Asthma #062 INVESTIGATORS/STUDY CENTERS: Nineteen investigators in 19 centers, U.S. PRIMARY THERAPY PERIOD: 23JUL96 to 19DEC96. The in- house cutoff date for case report forms was 15JAN97. CLINICAL PHASE: V DURATION OF TREATMENT: Two active-treatment periods lasting 2 weeks, each followed by a 2-week washout period. OBJECTIVES: (1) To confirm the safety and tolerability of concurrent administration of montelukast and loratadine; (2) To determine the clinical efficacy of montelukast + loratadine compared with montelukast alone over 2 weeks of treatment; (3) To determine the clinical effects after withdrawal of therapy; (4) To compare the effects of montelukast + loratadine and montelukast alone on peripheral blood eosinophils. STUDY DESIGN: Ten-week, multicenter, double-blind, placebo-controlled, 2 active-treatment period (II and IV), crossover study (with in-house blinding). PATIENT ACCOUNTING: ENTERED: 136 Male (age range) 64 (15 to 64) Female (age range) 72 (16 to 64) COMPLETED: 117 DISCONTINUED: 19 Clinical adverse experience 10 Laboratory adverse experience 0 Protocol deviation 5 Patient withdrew consent 3 Lost to follow-up 1 DOSAGE/FORMULATION NOS.: Placebo (MR-3305) once daily at bedtime during Periods I, III, and V. Montelukast 10-mg tablet (MR-3197) and loratadine two 10-mg capsules (MR-3371) or loratadine placebo capsules (MR-3379 and MR-3380) once daily at bedtime during Periods II and IV. Albuterol (6ZPA017, 5ZPA171) inhaler as needed during Periods I through V. DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking asthmatic patients with a forced expiratory volume in 1 second (FEV 1 ) ≥50% and ≤80% of predicted value, demonstrating reversibility of at least 15% with β-agonist, a minimal predefined level of daytime symptoms and β-agonist use; males or females age of 15 to 65 years. EVALUATION CRITERIA: EFFICACY: Primary End Point: FEV 1 Secondary End Points : AM and PM peak expiratory flow rate (PEFR), total daily β-agonist use, daytime symptom score. Other End Points: patient’s and investigator’s global evaluations, nocturnal symptom score, nocturnal awakenings, and peripheral blood eosinophil count. SAFETY: Number (%) of clinical and laboratory adverse experiences. Percents of patients outside the predefined limits of change for specified laboratory tests. STATISTICAL PLANNING AND ANALYSIS: EFFICACY: For most efficacy end points, the change (or percent change) from prerandomization baseline for both treatment periods was analyzed. Between-group comparisons were performed using a linear model (analysis of variance [ANOVA]) with factors for stratum (seasonal allergy status), sequence, patient within sequence-by-stratum, period and treatment. Using least square (LS) means, 95% confidence intervals (CI) were computed for the within-group changes and the between-group
Transcript of MERCK RESEARCH CLINICAL STUDY REPORT LABORATORIES I....

/MK-0476/CSR/RC1441.DOC APPROVED 06OCT97
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTI. SYNOPSIS
Montelukast Sodium Tablets(MK-0476)
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Crossover Study Comparingthe Clinical Effect of Combination MK-0476/Loratadine to MK-0476 in Patients WithChronic Asthma
#062
INVESTIGATORS/STUDY CENTERS: Nineteen investigators in 19 centers, U.S. PRIMARY THERAPY PERIOD: 23JUL96 to 19DEC96. The in-
house cutoff date for case report forms was 15JAN97.CLINICAL PHASE: V
DURATION OF TREATMENT: Two active-treatment periods lasting 2 weeks, each followed by a2-week washout period.
OBJECTIVES: (1) To confirm the safety and tolerability of concurrent administration of montelukast andloratadine; (2) To determine the clinical efficacy of montelukast + loratadine compared with montelukastalone over 2 weeks of treatment; (3) To determine the clinical effects after withdrawal of therapy; (4) Tocompare the effects of montelukast + loratadine and montelukast alone on peripheral blood eosinophils.
STUDY DESIGN: Ten-week, multicenter, double-blind, placebo-controlled, 2 active-treatment period(II and IV), crossover study (with in-house blinding).
PATIENT ACCOUNTING:
ENTERED: 136Male (age range) 64 (15 to 64)Female (age range) 72 (16 to 64)
COMPLETED: 117DISCONTINUED: 19
Clinical adverse experience 10Laboratory adverse experience 0Protocol deviation 5Patient withdrew consent 3Lost to follow-up 1
DOSAGE/FORMULATION NOS.: Placebo (MR-3305) once daily at bedtime during Periods I, III,and V. Montelukast 10-mg tablet (MR-3197) and loratadine two 10-mg capsules (MR-3371) orloratadine placebo capsules (MR-3379 and MR-3380) once daily at bedtime during Periods II and IV.Albuterol (6ZPA017, 5ZPA171) inhaler as needed during Periods I through V.
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking asthmatic patients with a forced expiratory volumein 1 second (FEV1) ≥50% and ≤80% of predicted value, demonstrating reversibility of at least 15% withβ-agonist, a minimal predefined level of daytime symptoms and β-agonist use; males or females age of15 to 65 years.
EVALUATION CRITERIA:EFFICACY: Primary End Point: FEV1 Secondary End Points: AM and PM peak expiratory flow rate
(PEFR), total daily β-agonist use, daytime symptom score. Other End Points: patient’s andinvestigator’s global evaluations, nocturnal symptom score, nocturnal awakenings, and peripheralblood eosinophil count.
SAFETY: Number (%) of clinical and laboratory adverse experiences. Percents of patients outside thepredefined limits of change for specified laboratory tests.
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: For most efficacy end points, the change (or percent change) from prerandomization
baseline for both treatment periods was analyzed. Between-group comparisons were performed usinga linear model (analysis of variance [ANOVA]) with factors for stratum (seasonal allergy status),sequence, patient within sequence-by-stratum, period and treatment. Using least square (LS) means,95% confidence intervals (CI) were computed for the within-group changes and the between-group

MERCK RESEARCHLABORATORIES
CSR Synopsis (Cont.)Protocol 062
Montelukast Sodium Tablets(MK-0476) -2-
/MK-0476/CSR/RC1441.DOC APPROVED 06OCT97
differences. The study was designed, with a total sample size of 90 patients, to have 80% power (atα=0.050 level, 2-sided test) to detect a 5.13 percentage point between-group difference in FEV1
percent change from baseline.
SAFETY: McNemar’s test was used to compare between-group incidences of adverse experiences andthe percent of patients outside the predefined limits of change for selected laboratory tests.
RESULTS:EFFICACY: Montelukast + loratadine improved asthma control end points compared with montelukast
alone. Significant (p<0.050) treatment differences in FEV1 (primary end point) and the secondary endpoints (total daily β-agonist use, AM PEFR, PM PEFR, daytime symptom scores) occurred.
Primary Clinical End Point: Mean PercentChange From Baseline
FEV1 Percent ChangeFrom Baseline
Treatment Mean LS Mean
Montelukast + Loratadine 13.62 13.86Montelukast 9.47 9.72Difference in LS mean 4.15*
95% CI for difference (1.65, 6.65)* p=0.001 compared with montelukast alone.
SAFETY: Both treatments were comparable in the percent of patients with clinical and laboratoryadverse experiences.
Adverse Experience Summary
Montelukast +Loratadine
MontelukastAlone
Clinical adverse experience (AEs) (N=130) (N=132)Number (%) of Patients: n (%) n (%)
with 1 or more AEs 53 (40.8) 64 (48.5)with no AEs 77 (59.2) 68 (51.5)
with serious AEs 0 ( 0.0) 0 ( 0.0)who died 0 ( 0.0) 0 ( 0.0)with drug-related AEs 12 ( 9.2) 5 ( 3.8)discontinued due to an AE 5 ( 3.8) 5 ( 3.8)discontinued due to a drug-related AE 1 ( 0.8) 1 ( 0.8)
Laboratory AEsNumber of patients with at least one laboratory test (N=130) (N=132)Number (%) of patients: n (%) n (%)
with 1 or more AEs 1 ( 0.8) 4 ( 3.0)with no AE 129 (99.2) 128 (97.0)
with drug-related AEs 0 ( 0.0) 0 ( 0.0)with serious AEs 0 ( 0.0) 0 ( 0.0)who died 0 ( 0.0) 0 ( 0.0)discontinued due to an AE 0 ( 0.0) 0 ( 0.0)

MERCK RESEARCHLABORATORIES
CSR Synopsis (Cont.)Protocol 062
Montelukast Sodium Tablets(MK-0476) -3-
/MK-0476/CSR/RC1441.DOC APPROVED 06OCT97
CONCLUSIONS: Treatment with montelukast (10 mg) + loratadine (2 x 10 mg) once daily at bedtimeover 2 weeks, compared with montelukast alone demonstrated: (1) Significant improvement in asthmacontrol end points. (2) A comparable incidence of clinical and laboratory adverse experiences. (3) Nodifference in the change in peripheral blood eosinophil counts. (4) No rebound worsening of asthma withthe withdrawal of either therapy.
AUTHORS: Donna Weinland, B.S., M.B.A.Associate Medical Program CoordinatorPulmonary-Immunology
Ha Nguyen, Ph.D.Senior BiometricianCBARDS
Izabella Peszek, Ph.D.BiometricianCBARDS
Rudolf Baumgartner, M.D.Associate DirectorPulmonary-Immunology

/MK-0476/CSR/RC3122.DOC VERSION 8.1 APPROVED 10-Aug-2001
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTI. SYNOPSIS
MK-0476Montelukast Sodium Film-Coated TabletExercise-Induced Bronchoconstriction
PROTOCOL TITLE/NO.: A Randomized, Double-Blind, Multicenter Study Evaluatingthe Effect of Montelukast Sodium as Compared to Salmeterol on the Inhibition ofExercise-Induced Bronchoconstriction in Adult Asthmatics
#064
INVESTIGATORS/STUDY CENTERS: Seventeen study centers in the United States.PUBLICATIONS: (1) Turpin J, Edelman J, DeLucca P, Pearlman D for the Exercise Study Group.
Chronic Administration of Montelukast (MK-0476) is Superior to Inhaled Salmeterol in the ProtectionAgainst Exercise-Induced Bronchoconstriction (EIB). Am J Respir and Crit Care Med (1998)157:A456. (2) Edelman J, Turpin J, DeLucca P, and the Exercise Study Group. Comparison of OralMontelukast, a Leukotriene Antagonist and Inhaled Salmeterol in Exercise Induced Asthma (EIA). EurRespir J (1998) 12:18s. (3) Edelman J, Turpin J, Bronsky E, Grossman J, Kemp J, Ghannam A,DeLucca P, Gormley G, Pearlman D, for the Exercise Study Group. Oral Montelukast Compared withInhaled Salmeterol to Prevent Exercise-Induced Bronchoconstriction. Ann Int Med (2000) 132:97-104.
PRIMARY THERAPY PERIOD: 03-Sep-1996 to 17-Jun-1997.Study is completed. The in-house cutoff date for case report formswas 19-Sep-1997.
CLINICAL PHASE: III
DURATION OF TREATMENT: Period I: 2-week placebo run-in period; Period II: 8-weekactive-treatment period.
OBJECTIVES:PRIMARY: To determine the effect after 8 weeks of chronic treatment with montelukast or salmeterol
on EIB in adult asthmatics as measured by the postexercise Maximum Percent Fall in forcedexpiratory volume in 1 second (FEV1) (mean change from baseline1) obtained at the end of the dosinginterval for salmeterol and montelukast.2
SECONDARY: To determine and compare between montelukast and salmeterol for: (1) Thepostexercise Maximum Percent Fall in FEV1 at Week 4 of treatment; (2) The Time to Recovery forFEV1 to within 5% of pre-exercise FEV1 (hereafter referred to as Time to Recovery) at Weeks 4 and 8of treatment; (3) The postexercise area under the FEV1 time curve during the first 60 minutes(AUC0-60 min) at Weeks 4 and 8 of treatment; (4) Persistence of Effect over 8 weeks of treatment;(5) The safety and tolerability of montelukast and salmeterol.
STUDY DESIGN: This was a randomized, double-blind, 2-period, active-control, parallel-group,multicenter study conducted under in-house blinding procedures.
PATIENT ACCOUNTING: Montelukast Salmeterol Total
ENTERED: 97 94 191Male (age range) 46 (15 to 45) 54 (15 to 45) 100 (15 to 45)Female (age range) 51 (15 to 46) 40 (15 to 44) 91 (15 to 46)
SCREENING FAILURES -- -- 263COMPLETED PERIOD II: (%) 91 (93.8) 86 (91.5) 177 (92.7)DISCONTINUED: Total (%) 6 (6.2) 8 (8.5) 14 (7.3)
Clinical adverse experience (%) 1 (1.0) 4 (4.3) 5 (2.6)Laboratory adverse experience (%) 0 (0.0) 0 (0.0) 0 (0.0)Other (%) 5 (5.2) 4 (4.3) 9 (4.7)
1 The term “baseline” in this clinical study report refers to the prerandomization values (obtained at
Visit 2) used in the statistical analyses of change from baseline. FEV1 values obtained prior toindividual exercise challenges are visit-specific and termed “pre-exercise.”
2 All exercise challenges occurred at the end of the dosing interval (approximately 20 to 24 hours afterdosing for montelukast and 8 to 12 hours after dosing for salmeterol).

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 064
MK-0476Montelukast Sodium Film-Coated TabletExercise-Induced Bronchoconstriction
-2-
/MK-0476/CSR/RC3122.DOC VERSION 8.1 APPROVED 10-Aug-2001
DOSAGE/FORMULATION NOS.: Period I: 1 montelukast matching-image placebo film-coated tablet(MR-3305) once daily at bedtime; 2 puffs salmeterol matching-image placebo inhaler (MR-3368) every12 hours. Period II: 1 montelukast 10-mg film-coated tablet (MR-3197) or 1 matching-image placebo��������� �� ����� �� � ���� � � � ����� ��� ������� ���� � �� �� �� � ������!"� � � �����matching-image placebo (MR-3368) every 12 hours.
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women (using appropriatecontraception) between and including 15 and 45 years of age, with a clinical history of asthma for atleast 1 year, with evidence of EIB (decrease in FEV1 after exercise of at least 20% compared with pre-exercise FEV1), airway reversibility ≥12% (as demonstrated by FEV1 after inhaled β-agonistadministration) within the past 6 months, and an average FEV1 ≥65% of the predicted value with�������#�� ������� ���$ ������������� ���� ������ ���� ���������� ���������� �
pre-exercise.EVALUATION CRITERIA:
EFFICACY: Primary Endpoint: Maximum Percent Fall in FEV1 after exercise at Week 8. SecondaryEndpoints: Maximum Percent Fall in FEV1 after exercise at Week 4; Time to Recovery atWeeks 4 and 8; AUC0-60 min at Weeks 4 and 8; Persistence of Effect over 8 weeks of treatment.Additional Endpoints: Percentage of Patients Requiring Rescue Medication During (or at the end of)Exercise Challenge; Maximum Percent Fall in FEV1 after exercise at Days 1 to 3;3 Time to Recoveryat Days 1 to 3; AUC0-60 min at Days 1 to 3; Percentage of patients with <20% Maximum Percent Fall inFEV1 after exercise at Week 8.
SAFETY: Frequency of clinical and laboratory adverse experiences, frequency of laboratory valuesoutside the predefined limits of change for selected laboratory safety tests and vital signs.
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: For most efficacy endpoints, the change from baseline was analyzed. Between-group
comparisons were performed using a linear model (analysis of variance [ANOVA]) with factors fortreatment group and study center. Using least-squares (LS) means, 95% confidence intervals (CIs)were computed for the within-group changes and between-group differences. The study was designedto have 160 patients complete the study, with 80 patients in each group. The study had 95% power todetect a 7-percentage-point difference between treatment groups in the mean change in MaximumPercent Fall in FEV1 after exercise, assuming a standard deviation (SD) of 12.5-percentage points.
SAFETY: Fisher’s exact test and 95% CIs were used to compare between-group incidences of adverseexperience categories: overall, drug-related, and serious adverse experiences, and adverse experiencescausing discontinuation from therapy. Ninety-five percent CIs for between-group differences inindividual adverse experiences and in the percentage of patients with values outside the predefinedlimits of change for selected laboratory parameters were based on Wilson’s score method.
RESULTS:EFFICACY: Montelukast showed significantly greater attenuation of EIB than salmeterol as
demonstrated in Maximum Percent Fall in FEV1 after exercise at Week 8 (primary endpoint) and atWeek 4 (secondary endpoint). Montelukast also showed significantly greater attenuation of EIB asdemonstrated in the secondary endpoints Time to Recovery and AUC0-60 min at Weeks 4 and8; montelukast and salmeterol provided similar attenuation of EIB at Days 1 to 3. The effect ofmontelukast was persistent over the 8-week treatment period, while the effect of salmeterol decreased.A lower percentage of patients in the montelukast treatment group (26.0%) than in the salmeteroltreatment group (39.8%) required Rescue Medication During Exercise Challenge at 1 or more visits(p=0.063). A greater percentage of patients in the montelukast treatment group (66.7%) than in thesalmeterol treatment group (45.6%) reached a commonly used threshold level of effect (defined as amaximum percent fall in FEV1 after exercise of <20%) at Week 8 (p=0.005).
3 The first postrandomization exercise challenge occurred within 1 to 3 days after taking at least 1 full day
of study medication.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 064
MK-0476Montelukast Sodium Film-Coated TabletExercise-Induced Bronchoconstriction
-3-
/MK-0476/CSR/RC3122.DOC VERSION 8.1 APPROVED 10-Aug-2001
Exercise Challenge Endpoints†—Analysis of Within-Group and Between-Group Differences at Week 8
Maximum Percent Fall in FEV1 (%) Time to Recovery (min) AUC0-60 min (%•min)
TreatmentBaseline(Mean)
Change FromBaseline (Mean)‡
Baseline(Mean)
Change FromBaseline (Mean)
Baseline(Mean)
Change FromBaseline (Mean)
Montelukast (N=93)Salmeterol (N=90)
36.5536.59
-16.53-9.34
62.3258.12
-31.19-12.49
1389.71305.4
-753.0-303.6
Difference in LS means95% CI for difference
-7.24(-11.84, -2.65)
-18.53(-29.45, -7.60)
-447.0(-698.6, -195.5)
p-Value 0.002 0.001 0.001† In the analysis of these endpoints, the lower the value, the greater the response to therapy.‡ Percentage-point difference.FEV1 = Forced expiratory volume in 1 second; AUC = Area under the curve; LS = Least-Squares; CI = Confidence Interval.
Exercise Challenge Endpoints†—Analysis of Within-Group and Between-Group Differences at Week 4
Maximum Percent Fall in FEV1 (%) Time to Recovery (min) AUC0-60 min (%•min)
TreatmentBaseline(Mean)
Change FromBaseline (Mean)‡
Baseline(Mean)
Change FromBaseline (Mean)
Baseline(Mean)
Change FromBaseline (Mean)
Montelukast (N=91)Salmeterol (N=86)
36.3436.94
-16.64-9.93
62.0257.40
-30.16-11.88
1375.31295.2
-798.7-214.9
Difference in LS means95% CI for difference
-6.89(-11.74, -2.05)
-18.25(-28.48, -8.03)
-588.8(-870.0, -307.5)
p-Value 0.006 0.001 <0.001† In the analysis of these endpoints, the lower the value, the greater the response to therapy.‡ Percentage-point difference.FEV1 = Forced expiratory volume in 1 second; AUC = Area under the curve; LS = Least-Squares; CI = Confidence Interval.
Exercise Challenge Endpoints†—Analysis of Within-Group and Between-Group Differences at Days 1 to 3
Maximum Percent Fall inFEV1 (%) Time to Recovery (min) AUC0-60 min (%•min)
TreatmentBaseline(Mean)
Change FromBaseline (Mean)‡
Baseline(Mean)
Change FromBaseline (Mean)
Baseline(Mean)
Change FromBaseline (Mean)
Montelukast (N=92)Salmeterol (N=87)
36.5436.98
-14.88-13.24
62.1056.54
-30.28-21.08
1373.51297.9
-814.0-558.1
Difference in LS means95% CI for difference
-1.79(-5.72, 2.14)
-9.59(-19.55, 0.37)
-266.0(-489.7, -42.3)
p-Value 0.369 0.059 0.020† In the analysis of these endpoints, the lower the value, the greater the response to therapy.‡ Percentage-point difference.FEV1 = Forced expiratory volume in 1 second; AUC = Area under the curve; LS = Least-Squares; CI = Confidence Interval.
SAFETY: Both montelukast and salmeterol were well tolerated and were similar in the percentage ofpatients with clinical and laboratory adverse experiences. One patient in the salmeterol treatmentgroup had a serious clinical adverse experience (death due to bronchial asthma), determined by theinvestigator to be definitely not drug related. No other serious adverse experiences were reported.Two patients in the salmeterol treatment group discontinued from therapy due to a clinical adverseexperience determined by the investigator to be drug related.
MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 064
MK-0476Montelukast Sodium Film-Coated TabletExercise-Induced Bronchoconstriction
-4-
/MK-0476/CSR/RC3122.DOC VERSION 8.1 APPROVED 10-Aug-2001
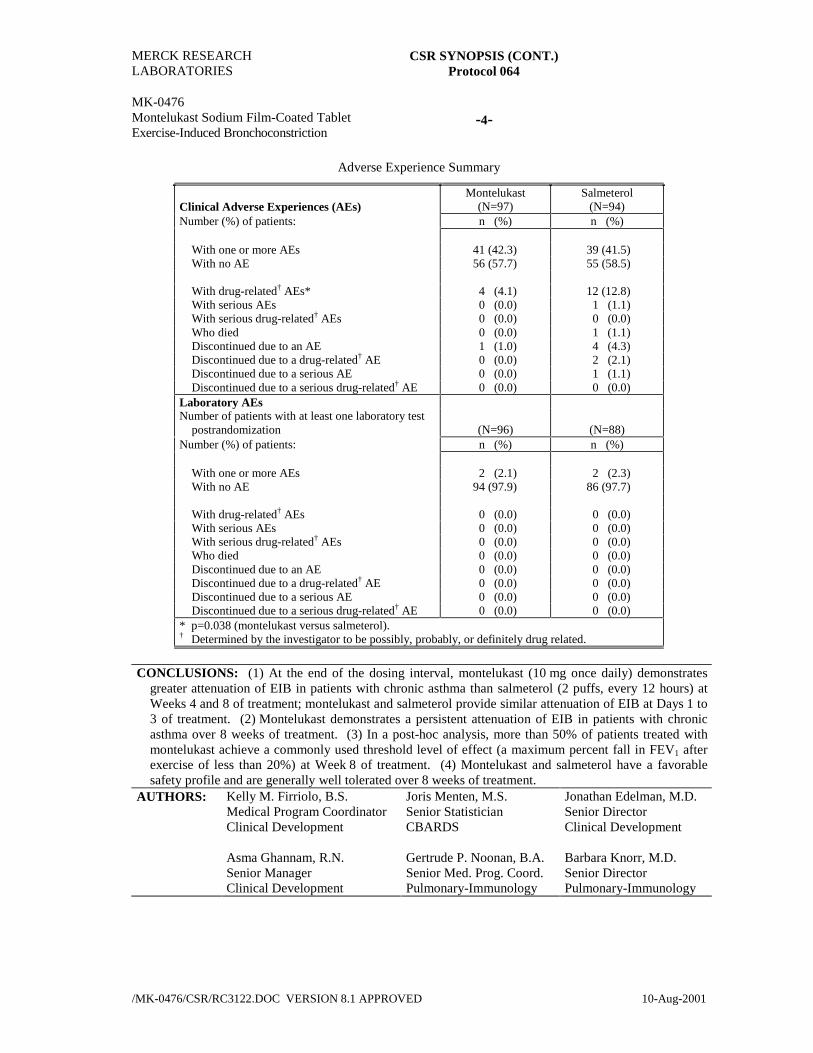
Adverse Experience Summary
Clinical Adverse Experiences (AEs)Montelukast
(N=97)Salmeterol
(N=94)Number (%) of patients: n (%) n (%)
With one or more AEs 41 (42.3) 39 (41.5)With no AE 56 (57.7) 55 (58.5)
With drug-related† AEs* 4 (4.1) 12 (12.8)With serious AEs 0 (0.0) 1 (1.1)With serious drug-related† AEs 0 (0.0) 0 (0.0)Who died 0 (0.0) 1 (1.1)Discontinued due to an AE 1 (1.0) 4 (4.3)Discontinued due to a drug-related† AE 0 (0.0) 2 (2.1)Discontinued due to a serious AE 0 (0.0) 1 (1.1)Discontinued due to a serious drug-related† AE 0 (0.0) 0 (0.0)
Laboratory AEsNumber of patients with at least one laboratory test
postrandomization (N=96) (N=88)Number (%) of patients: n (%) n (%)
With one or more AEs 2 (2.1) 2 (2.3)With no AE 94 (97.9) 86 (97.7)
With drug-related† AEs 0 (0.0) 0 (0.0)With serious AEs 0 (0.0) 0 (0.0)With serious drug-related† AEs 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0)Discontinued due to an AE 0 (0.0) 0 (0.0)Discontinued due to a drug-related† AE 0 (0.0) 0 (0.0)Discontinued due to a serious AE 0 (0.0) 0 (0.0)Discontinued due to a serious drug-related† AE 0 (0.0) 0 (0.0)
* p=0.038 (montelukast versus salmeterol).† Determined by the investigator to be possibly, probably, or definitely drug related.
CONCLUSIONS: (1) At the end of the dosing interval, montelukast (10 mg once daily) demonstratesgreater attenuation of EIB in patients with chronic asthma than salmeterol (2 puffs, every 12 hours) atWeeks 4 and 8 of treatment; montelukast and salmeterol provide similar attenuation of EIB at Days 1 to3 of treatment. (2) Montelukast demonstrates a persistent attenuation of EIB in patients with chronicasthma over 8 weeks of treatment. (3) In a post-hoc analysis, more than 50% of patients treated withmontelukast achieve a commonly used threshold level of effect (a maximum percent fall in FEV1 afterexercise of less than 20%) at Week 8 of treatment. (4) Montelukast and salmeterol have a favorablesafety profile and are generally well tolerated over 8 weeks of treatment.
AUTHORS: Kelly M. Firriolo, B.S.Medical Program CoordinatorClinical Development
Joris Menten, M.S.Senior StatisticianCBARDS
Jonathan Edelman, M.D.Senior DirectorClinical Development
Asma Ghannam, R.N.Senior ManagerClinical Development
Gertrude P. Noonan, B.A.Senior Med. Prog. Coord.Pulmonary-Immunology
Barbara Knorr, M.D.Senior DirectorPulmonary-Immunology

/MK-0476/CSR/BC1891.DOC APPROVED 17-Jan-1999
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTI. SYNOPSIS
Montelukast Sodium (MK-0476), tablets
PROTOCOL TITLE/NO.: A Randomized, Double-Blind, Placebo-Controlled,Multicenter Study Evaluating the Effect of Montelukast Sodium Compared to InhaledBeclomethasone Dipropionate in Adult Asthmatics
#070
INVESTIGATORS/STUDY CENTERS: Sixty-four centers in the United States.PRIMARY THERAPY PERIOD: Aug-1997 to Mar-1998. The
study is completed. The in-house cutoff date for case report formswas 14-Apr-1998.
CLINICAL PHASE: IIIb
DURATION OF TREATMENT: Period I, 2 weeks; Period II, 6 weeks. There was also a 1-weekprestudy screening period prior to Period I.
OBJECTIVES: (1) To determine between montelukast and beclomethasone, the overlap in thedistribution of patient response in the percent of Asthma Control Days; (2) To compare the effectsamong montelukast, beclomethasone, and placebo on the percent of Asthma Control Days; (3) Tocompare the effects between montelukast and beclomethasone on: (a) the proportion of patients withoutCorticosteroid Rescue medication, (b) the proportion of patients without Asthma Attacks, (c) theproportion of patients without Asthma Flare-Ups, (d) Patient’s and Physician’s Global Evaluations,(e) the mean change from baseline forced expiratory volume in 1 second (FEV1) and the overlap indistribution of change from baseline FEV1, (f) the percent and overlap in distribution of SustainedAsthma Control Episode Days, (g) the Average Daily β-Agonist Use, and (j) safety and tolerability.
STUDY DESIGN: Double-blind, randomized, placebo- and active-controlled, 3-arm, parallel-groupstudy.
PATIENT ACCOUNTING:
Placebo Montelukast Beclomethasone TotalENTERED: Total 111 339 332 782Male (age range) 54 (15 to 64) 162 (15 to 69) 156 (15 to 74) 372 (15 to 74)Female (age range) 57 (15 to 53) 177 (15 to 71) 176 (15 to 73) 410 (15 to 73)COMPLETED: 106 (95.5%) 328 (96.8%) 318 (95.8%) 752 (96.2%)DISCONTINUED: Total 5 (4.5%) 11 (3.2%) 14 (4.2%) 30 (3.8%)Clinical adverse experience† 3 (2.7%) 6 (1.8%) 8 (2.4%) 17 (2.2%)Laboratory adverse experience 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%)Other 2 (1.8%) 5 (1.5%) 6 (1.8%) 13 (1.6%)† One patient in the placebo group and 2 patients in the beclomethasone group discontinued due to adverse experiences that
began prior to randomization.
DOSAGE/FORMULATION NOS.: Period I: One montelukast-matching placebo tablet (MR-3305)once daily at bedtime and beclomethasone-matching placebo inhaler (MR-3472) 4 puffs twice daily.Period II: One montelukast 10-mg (MR-3459) or matching placebo tablet once daily at bedtime andbeclomethasone inhaler (50 µg per actuation) (MR-3473) or matching placebo inhaler 4 puffs twice daily.
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking male and female (using appropriatecontraception) asthmatic patients ≥15 years of age with a clinical history of asthma for ≥1 year, FEV1≥50 and ≤85% of predicted value with an increase of ≥15% after 2 puffs of β-agonist administration, anda weekly average of >2 puffs per day of β-agonist.
EVALUATION CRITERIA: EFFICACY: Primary Endpoint: Percent Asthma Control Days (a day with ≤2 puffs β-agonist use, nonighttime awakenings, and no Asthma Attacks). Secondary Endpoints: Use of Corticosteroid Rescuemedication, Asthma Attack (oral steroid rescue and/or use of unscheduled asthma care in a doctor’soffice, emergency room, or hospital setting), Asthma Flare-Up (period of at least 3 consecutive days thatdid not meet the definition of an Asthma Control Day), Patient’s and Physician’s Global Evaluations,change in FEV1 from baseline, Sustained Asthma Control Episode (period of at least3 consecutive Asthma Control Days), and Average Daily β-Agonist Use.SAFETY: Frequency of clinical and laboratory adverse experiences. Percent of patients outside thepredefined limits of change for selected laboratory tests.

MERCK RESEARCHLABORATORIES
CSR Synopsis (Cont.)Protocol 070
Montelukast Sodium (MK-0476), tablets-2-
/MK-0476/CSR/BC1891.DOC APPROVED 27-Sep-1999 * c10arfor16at
STATISTICAL PLANNING AND ANALYSIS: EFFICACY: Primary Analysis: Percent of overlap in the distribution of patient response betweentreatment groups was analyzed for Asthma Control Days using the nonparametric Mann-WhitneyU-statistic. The primary hypothesis was met if the lower bound of the 95% confidence interval of theoverlap was >70%. In order to have 95% power, the study was designed with 89, 267, and267 patients in the placebo, montelukast, and beclomethasone groups, respectively. AdditionalAnalyses: Sustained Asthma Control Episodes and FEV1 were analyzed in terms of percent overlap indistribution based on the Mann-Whitney U-statistic. Between-group comparisons were performed forAsthma Control Days, change from baseline in FEV1, change in Average Daily β-Agonist Use, andPatient’s and Physician’s Global Evaluations using analysis of variance (ANOVA). A logisticregression model was used to analyze patients with or without Asthma Attacks, Asthma Flare-Ups,Sustained Asthma Control Episodes, and Corticosteroid Rescue medication use.SAFETY: Fisher’s exact test was used to compare between-group incidences of clinical and laboratoryadverse experiences and the proportion of patients outside the predefined limits of change for selectedlaboratory tests.
RESULTS: EFFICACY: Montelukast demonstrated at least 89% overlap in percent Asthma Control Days(primary endpoint) compared with beclomethasone. In addition, there was no statistically significantdifference between montelukast and beclomethasone in the secondary endpoints of CorticosteroidRescue Medication Use, Asthma Attack, Asthma Flare-Up, Sustained Asthma Control Episode, andAverage Daily β-Agonist Use. Beclomethasone had a greater effect compared with montelukast in termsof Patient’s and Physician’s Global Evaluation and Change in FEV1.
Primary Clinical Endpoint: Percent Asthma Control Days
Treatment N Mean SD LS Mean† 95% CI for Mean
Placebo 111 26.19 30.02 26.76 (20.04, 33.49)Montelukast 337 40.32 37.59 41.39 (37.37, 45.40)Beclomethasone 329 40.09 35.29 41.14 (37.00, 45.29)Comparison Between TreatmentsPrimary Analysis—Percent Overlap U* Overlap (%) 95% CI for Overlap (%)Montelukast vs. Beclomethasone 0.51 97.73 (88.95, 100.00)
† LS Mean is the least-squared mean resulting from the ANOVA model including effects for treatment group and study center.SD = standard deviation; CI = Confidence Interval; U* = Standardized Mann-Whitney U-Statistic.
SAFETY: There were no differences in the frequency of clinical or laboratory adverse experiencesbetween the montelukast and beclomethasone groups. Adverse experiences were generally similar tothose of the placebo group. There were no serious drug-related adverse experiences. One patient in theplacebo group and 1 patient in the montelukast group discontinued the study due to a drug-relatedclinical adverse experience.

MERCK RESEARCHLABORATORIES
CSR Synopsis (Cont.)Protocol 070
Montelukast Sodium (MK-0476), tablets-3-
/MK-0476/CSR/BC1891.DOC APPROVED 27-Sep-1999 * c10arfor16at
Adverse Experience Summary
Placebo Montelukast BeclomethasoneClinical Adverse Experiences (AEs) (N=111) (N=339) (N=332)
Number (%) of patients: n (%) n (%) n (%)
With one or more AEs 57 (51.3) 161 (47.5) 152 (45.8)With no AE 54 (48.7) 178 (52.5) 180 (54.2)
With drug-related AEs† 4 (3.6) 22 (6.5) 11 (3.3)With serious AEs 0 (0.0) 2 (0.6) 1 (0.3)With serious drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to an AE‡ 3 (2.7) 6 (1.8) 8 (2.4)Discontinued due to a drug-related AE 1 (0.9) 1 (0.3) 0 (0.0)Discontinued due to a serious AE 0 (0.0) 2 (0.6) 0 (0.0)Discontinued due to a serious drug-related AE 0 (0.0) 0 (0.0) 0 (0.0)
Laboratory AEsNumber of patients with at least one laboratory testpostrandomization:
(N=110) (N=336) (N=329)
Number (%) of patients: n (%) n (%) n (%)
With one or more AEs 5 (4.5) 13 (3.9) 10 (3.0)With no AE 105 (95.5) 323 (96.1) 319 (97.0)
With drug-related AEs† 1 (0.9) 6 (1.8) 4 (1.2)With serious AEs 0 (0.0) 0 (0.0) 0 (0.0)With serious drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to an AE 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to a drug-related AE 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to a serious AE 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to a serious drug-related AE 0 (0.0) 0 (0.0) 0 (0.0)† Determined by the investigator to be possibly, probably, or definitely drug related.‡ One patient in the placebo group (AN 1842) and 2 patients in the beclomethasone group (ANs 1902 and
2068) discontinued due to adverse experiences that began prior to randomization.This table does not include those adverse experiences that occurred before randomization.
CONCLUSIONS: (1) The overlap of percent Asthma Control Days is at least 80% betweenmontelukast and beclomethasone. (2) Montelukast and beclomethasone provide effects with nodifferences in the secondary endpoints of Corticosteroid Rescue Medication Use, Asthma Attack,Asthma Flare-Up, Sustained Asthma Control Episode, and Average Daily β-Agonist Use.(3) Beclomethasone provides larger improvement than montelukast in the secondary endpoints ofPhysician’s and Patient’s Global Evaluations and change in FEV1. (4) Compared with placebo,montelukast provides improvements in all asthma control endpoints; beclomethasone, compared withplacebo, provides improvements in most study endpoints (except Corticosteroid Rescue medication andAsthma Attack). (5) Montelukast, beclomethasone, and placebo have a similar safety profile.
AUTHORS: Carol S. Skalky, B.A.Medical Program CoordinatorClinical Development
Steven Bird, M.S.Senior StatisticianClinical Development
Jonathan M. Edelman, M.D.DirectorClinical Development
Adam Polis, M.S.BiometricianClinical Development

RC3027.DOC VERSION 8.0 APPROVED 13-Jun-2001
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTI. SYNOPSIS
MK-0476Montelukast Sodium,Film-Coated TabletChronic Asthma
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized Study Comparingthe Clinical Effect of Concomitant Montelukast+Loratadine With Montelukast,Loratadine, and Inhaled Beclomethasone in Patients With Chronic Asthma
#074
INVESTIGATORS/STUDY CENTERS: Forty-two investigators in 42 centers, United States.PRIMARY THERAPY PERIOD: May-1998 to Nov-1998. The in-
house cutoff date for receipt of all case report forms was 22-Dec-1998. Patients had the option to continue an open label extension(Period V).
CLINICAL PHASE: IIb
DURATION OF TREATMENT: Two-week placebo run-in period, 2 active-treatment periods of6 weeks separated by a 2-week washout period.
OBJECTIVES: (1) To compare, over a 6-week treatment period, the clinical efficacy on primary,secondary, and other endpoints between; (a) montelukast+loratadine† and montelukast,(b) montelukast+loratadine and loratadine, (c) montelukast+loratadine and inhaled beclomethasone, and(d) montelukast and loratadine; (2) To determine the safety and tolerability of montelukast+loratadine;(3) To determine whether tolerance to concomitant administration of montelukast+loratadine develops;(4) To determine whether montelukast+loratadine causes rebound worsening upon withdrawal.______________________________
† Montelukast 10 mg + loratadine 10 mg, hereafter referred to as montelukast+loratadine.STUDY DESIGN: Seventeen-week, multicenter, double-blind, randomized, 2 active-treatment periods
(II and IV), 3-part, 2x2 crossover study (with in-house blinding).PATIENT ACCOUNTING: Total number of patients randomly allocated is summarized. Due to the
crossover design, the information presented is for the total number of patients only.
ENTERED: 406Male (age range) 195 (15 to 63)Female (age range) 211 (16 to 65)
COMPLETED: 362DISCONTINUED: 44
Clinical adverse experience 12Laboratory adverse experience 3Patient withdrew consent 10Lost to follow-up 3Protocol deviation 15Lack of efficacy 1
DOSAGE/FORMULATION NOS.: Placebo run-in period: Placebo montelukast tablets (MR 3305) andplacebo loratadine tablets (MR 3629), 1 tablet of each taken daily at bedtime and placebo inhaler(4 puffs twice daily) (MR3613). Periods I and III: Montelukast 10-mg tablets (MR 3459) and/orloratadine 10-mg tablets (MR 3651), or placebo montelukast or placebo loratadine tablets taken oncedaily at bedtime, or inhaled beclomethasone 4 puffs (50 µg/puff) (MR 3587) or placebo inhaler takentwice daily during Periods II and IV.
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking asthmatic men or women (using appropriatecontraception) ≥15 and ≤65 years of age with a forced expiratory volume in 1 second (FEV1) ≥50% and≤85% of predicted, demonstrating reversibility of at least ≥"#� $��� ��������% ��& � �������' �&�(���&������(&���������'������& ��������)��*
EVALUATION CRITERIA: EFFICACY: Primary Endpoint: FEV1; Secondary Endpoints: Morningand Evening ���+,-'� ��� � ��$���.�, �%���������� �����������%��&�������/��'����score. Other Endpoints: Patient’s and Physician’s Global Evaluations, Nocturnal Asthma score,Nocturnal Awakenings, Asthma-Specific Quality of Life, Peripheral Blood Eosinophil Counts, andAsthma Attacks (episodes of worsening asthma that resulted in the utilization of health-care resources).

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 074
MK-0476Montelukast SodiumFilm-Coated TabletChronic Asthma
-2-
RC3027.DOC VERSION 8.0 APPROVED 13-Jun-2001
SAFETY: Number (%) of clinical and laboratory adverse experiences. Percents of patients outside thepredefined limits of change for specified laboratory tests.
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: The change (or percent change) from prerandomization baseline for both treatment
periods was analyzed. Between-group comparisons were performed using a mixed model with fixedfactors for study center, period, treatment, and patient as a random effect. Least-squares (LS) meansand 95% confidence intervals (CI) were computed for the within-group changes and the between-group differences. The study was designed, with a total sample size of 360 patients, to have 90%power to detect a 3.5 percentage-point difference between montelukast+loratadine and montelukasttreatments for the primary endpoint of FEV1 percent change from baseline.
SAFETY: McNemar’s and Fisher’s tests were used to compare between-group incidences of adverseexperiences.
RESULTS:EFFICACY: Montelukast+loratadine was numerically superior to montelukast but did not show a
significant difference in the percent change from baseline for the primary endpoint of FEV1.Montelukast+loratadine compared to montelukast significantly improved the Evening PEFR(p=0.030), Nocturnal Asthma Symptom score (p=0.006), Nocturnal Awakenings (p=0.045), andAsthma-Specific Quality-of-Life (p=0.029). The differences were not significant for any of theremaining endpoints. There were no significant differences between montelukast+loratadine andbeclomethasone except for FEV1 percent change from baseline and Morning PEFR, for whichsignificant improvements in favor of beclomethasone were observed (p=0.037 and 0.023,respectively). Montelukast+loratadine produced significant treatment differences compared withloratadine for all endpoints except Daytime Symptoms score (p=0.152) and Peripheral BloodEosinophil Counts (p=0.446). Montelukast produced significant treatment differences compared withloratadine for all endpoints except Evening PEFR, for which a borderline significant improvement(p=0.053) was detected in favor of montelukast, and Asthma Attacks, which were not significantlydifferent from loratadine (p=0.227).
Primary Endpoint: Mean Percent Change From Baseline in FEV1
Treatment Comparison LS Mean Difference p-Value
Montelukast+loratadine therapy versus montelukast† 1.60 0.054Montelukast+loratadine therapy versus loratadine‡ 5.80 <0.001Montelukast+loratadine therapy versus beclomethasone§ -2.35 0.037Montelukast versus loratadine§ 5.94 <0.001Beclomethasone versus montelukast‡ 4.65 0.004† Based on pooled estimate.‡ Based on between-patient estimate.§ Based on within-patient estimate.
SAFETY: All treatments were generally comparable in the percent of patients with clinical andlaboratory adverse experiences. Due to the crossover design of the study, the total number of patientsexposed to a treatment will exceed the number of patients randomly allocated (N=406).

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 074
MK-0476Montelukast SodiumFilm-Coated TabletChronic Asthma
-3-
RC3027.DOC VERSION 8.0 APPROVED 13-Jun-2001
Montelukast+Loratadine
Montelukast10 mg
Loratadine10 mg
Beclomethasone 400 µg
Clinical Adverse Experiences (AEs) (N= 308) (N= 258) (N= 85) (N= 128)n (%) n (%) n (%) n (%)
Number (%) of patients:With one or more AEs 145 (47.1) 128 (49.6) 43 (50.6) 65 (50.8)With no AE 163 (52.9) 130 (50.3) 42 (49.4) 63 (49.2)With drug-related AEs† 10 (3.2) 8 (3.1) 0 (0.0) 2 (1.6)With serious AEs 2 (0.6) 0 (0.0) 0 (0.0) 1 (0.8)With serious drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to an AE 2 (0.6) 4 (1.6) 3 (3.5) 3 (2.3)Discontinued due to a drug-related AE 1 (0.3) 1 (0.4) 0 (0.0) 0 (0.0)Discontinued due to a serious AE 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to a serious drug-
related AE0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)
Laboratory AEsNumber of patients with at least one
laboratory test301 255 83 127
Number (%) of patients: n (%) n (%) n (%) n (%)
With one or more AEs 7 (2.3) 4 (1.6) 2 (2.4) 3 (2.4)With no AEs 294 (97.7) 251 (98.8) 81 (97.5) 124 (97.6)With drug-related AEs 3 (1.0) 2 (0.8) 1 (1.2) 1 (0.8)With serious AEs 1 (0.3) 0 (0.0) 0 (0.0) 0 (0.0)With serious drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to an AE 1 (0.3) 1 (0.4) 0 (0.0) 0 (0.0)Discontinued due to a drug-related AE 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to a serious AE 1 (0.3) 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to a serious drug-related AE
0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)
† Adverse experiences determined by the investigator to be possibly, probably, or definitely drug related.
CONCLUSIONS: Over a 6-week treatment period, (1) Montelukast+loratadine (10 mg each) wasnumerically superior to montelukast but not significantly different for the primary endpoint of FEV1
percent change from baseline. However, montelukast+loratadine significantly improved the endpointsof Evening PEFR, Nocturnal Asthma score, and the Average of Four Domains of the Asthma-SpecificQuality-of-Life score relative to montelukast; (2) There was no significant difference betweenmontelukast+loratadine and beclomethasone in Evening PEFR, Nocturnal Asthma score, and NocturnalAwakenings, and the Average of Four Domains of the Asthma-Specific Quality-of-Life scores.Montelukast+loratadine therapy showed significant improvement relative to montelukast for theseendpoints; (3) Beclomethasone demonstrated significant improvement compared withmontelukast+loratadine therapy for the endpoints of FEV1 and Morning PEFR; (4) All treatments werewell tolerated, with no significant differences in clinical, laboratory, or other safety parameters.
AUTHORS:
Carmen Ayala, R.N., B.S.Medical Program CoordinatorPulmonary-Immunology
Janet van Adelsberg, M.D.Associate DirectorPulmonary-Immunology
Sumiko Shingo, M.S.BiometricianCBARDS
Chang Yu, Ph.D.BiometricianCBARDS

RC3100.DOC VERSION 8.1 APPROVED 08-Oct-2001
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast + Loratadine, Tablet Chronic Asthma
PROTOCOL TITLE/NO.: A Multicenter, Open-Label, Randomized Study Comparing
Montelukast + Loratadine With Inhaled Beclomethasone in Patients With Chronic Asthma (Open Extension to Protocol 074-01)
#074/074-10
INVESTIGATORS/STUDY CENTERS: Multicenter (30 study centers in the United States) PRIMARY THERAPY PERIOD: 02-Sep-1998 through
07-Dec-1999. The in-house cutoff date for receipt of all case report forms for the first and second open-label extension (Period V of Protocols 074-01 and 074-10, respectively) was 28-Feb-2000.
CLINICAL PHASE: IIb
DURATION OF TREATMENT: First extension (Period V of Protocol 074-01): 48 weeks; second extension (Protocol 074-10): 3 days.
OBJECTIVE: To collect additional long-term safety, tolerability, and exploratory efficacy data when montelukast + loratadine (concomitant montelukast 10 mg + loratadine 10 mg, hereafter referred to as montelukast + loratadine) are administered, concomitantly and then as a combination tablet, once daily at bedtime compared with beclomethasone.
STUDY DESIGN: A multicenter, open-label, randomized study. PATIENT ACCOUNTING:
Montelukast + Loratadine
Beclomethasone 400 Τg
Total
COMPLETED BASE STUDY 362 ENTERED EXTENSION 85 41 126†
Male (age range) 35 (15 to 62) 16 (15 to 63) 51 Female (age range) 50 (17 to 62) 25 (16 to 62) 75
DISCONTINUED EXTENSION 32 16 48 Clinical adverse experience 7 3 10 Laboratory adverse experience 1 0 1 Lack of efficacy 4 0 4 Lost to follow-up 2 1 3 Patient withdrew consent 15 10 25 Protocol deviation 3 2 5
COMPLETED FIRST EXTENSION‡ 53 25 78 ____________________________________
† One hundred thirty-one patients entered the extension. Data for 5 of the 131 patients were received after the in-house cutoff date for receipt of case report forms and are not included in this clinical study report.
‡ Three patients continued into a second open-label extension after completing the first extension, and their therapy was switched from concomitant montelukast + loratadine to a combination tablet containing 10 mg montelukast and 10 mg loratadine (hereafter referred to as montelukast/loratadine). The study was terminated 3 days after the initiation of the second extension.
DOSAGE/FORMULATION NOS.: Montelukast 10-mg tablets once daily (MR3459), loratadine 10-mg tablets once daily (MR3651), combination montelukast 10 mg/loratadine 10-mg tablets once daily (MR3797), beclomethasone 400 Τg (50 Τg/puff, 4 puffs twice daily) (MR3587, MR4009).
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking asthmatic men or women (using appropriate contraception) ≥15 and ≤65 years of age who successfully completed Protocol 074-01 base study (a multicenter, double-blind, randomized study comparing the clinical effect of concomitant montelukast + loratadine with montelukast, loratadine, and inhaled beclomethasone in patients with chronic asthma).
EVALUATION CRITERIA: EFFICACY: Forced Expiratory Volume in 1 second (FEV1), Daytime Symptoms score, Peak
Expiratory Flow Rate (PEFR), Total Daily β-Agonist Use, Nocturnal Asthma Symptoms score, Nocturnal Awakenings, and Peripheral Eosinophil Count.

MERCK RESEARCH LABORATORIES
CSR SYNOPSIS (CONT.) Protocol 074/074-10
MK-0476 Montelukast + Loratadine, Tablet Chronic Asthma
-2-
RC3100.DOC VERSION 8.1 APPROVED 08-Oct-2001
SAFETY: Frequency of clinical and laboratory adverse experiences. Percentages of patients outside
the predefined limits of change for selected laboratory tests. STATISTICAL PLANNING AND ANALYSIS:
EFFICACY: The change (or percent change) from baseline was summarized. The treatment effects of montelukast + loratadine and beclomethasone were estimated using an analysis of variance model with factors for treatment group and study center. The averages over the entire extension period were analyzed for each efficacy endpoint.
SAFETY: Summary statistics by visit were provided for laboratory parameters (numeric values only) and vital signs. Adverse experiences and the percentage of patients outside the predefined limits of change for selected laboratory safety tests were summarized and evaluated by clinical review.
RESULTS: EFFICACY: Both montelukast + loratadine and beclomethasone improved endpoints of asthma control.
Summary of Efficacy Endpoints
Mean of Raw Values Change (or Percent Change)
From Baseline
Endpoint Treatment N Baseline Extension
Period Least-Squares
Mean 95% CI for
Mean FEV1 (L) M+L 84 2.38 2.58 11.05† (7.28, 14.81) B 38 2.48 2.78 14.06† (8.64, 19.49) Daytime Symptoms M+L 74 2.43 1.91 -0.62 (-0.84, -0.40)
(score) B 36 2.40 1.69 -0.82 (-1.12, -0.51) Total Daily β-Agonist M+L 73 5.87 3.47 -43.29† (-53.32, -33.27)
Use (Puffs/day) B 36 4.88 2.87 -42.28† (-56.12, -28.44) Morning PEFR (L/Min) M+L 74 382.14 444.32 75.14 (52.57, 97.70) B 36 381.23 441.16 65.99 (34.77, 97.21) Evening PEFR (L/Min) M+L 74 410.98 461.10 59.36 (36.00, 82.72) B 36 403.24 453.34 52.52 (20.20, 84.84) Nocturnal Asthma
Symptoms (score) M+L
60 0.98 0.48 -0.54 (-0.65, -0.42)
(Nocturnal Asthmatics Only) B 27 0.85 0.41 -0.34 (-0.51, -0.18) Nocturnal Awakenings M+L 60 5.35 2.81 -2.68 (-3.31, -2.05)
(Nights/Week) (Nocturnal Asthmatics Only) B 27 5.25 2.50 -2.08 (-2.97, -1.19) Peripheral Blood Eosinophil M+L 83 6.18 5.28 -0.91 (-1.54, -0.28) Count (%) B 39 6.03 4.79 -1.19 (-2.10, -0.28) † Percent change. CI = Confidence interval, FEV1 = Forced expiratory volume in 1 second, M+L = Montelukast + loratadine, B = Beclomethasone, and PEFR = Peak expiratory flow rate.
SAFETY: Both treatments were comparable with respect to the percentage of patients with clinical and
laboratory adverse experiences.

MERCK RESEARCH LABORATORIES
CSR SYNOPSIS (CONT.) Protocol 074/074-10
MK-0476 Montelukast + Loratadine, Tablet Chronic Asthma
-3-
RC3100.DOC VERSION 8.1 APPROVED 08-Oct-2001
Clinical Adverse Experience Summary
Montelukast +
Loratadine Beclomethasone
400 Τg (N=85) (N=41) n (%) n (%)
Number (%) of patients:
With one or more adverse experiences 73 (85.9) 35 (85.4) With no adverse experience 12 (14.1) 6 (14.6) With drug-related† adverse experiences 2 (2.4) 3 (7.3) With serious adverse experiences 0 (0.0) 1 (2.4) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 7 (8.2) 3 (7.3) Discontinued due to drug-related adverse experiences 1 (1.2) 1 (2.4) Discontinued due to serious adverse experiences 0 (0.0) 1 (2.4) Discontinued due to serious drug-related adverse experiences 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related. Although a patient may have had 2 or more clinical adverse experiences, the patient is counted only once in a category. The same patient may appear in different categories.
Laboratory Adverse Experience Summary
Montelukast +
Loratadine Beclomethasone
400 Τg (N=85) (N=41) n (%)† n (%)†
Number (%) of patients:
With at least one laboratory test postbaseline 83 40 With one or more adverse experiences 8 (9.6) 2 (5.0) With no adverse experience 75 (90.4) 38 (95.0) With drug-related‡ adverse experiences 2 (2.4) 0 (0.0) With serious adverse experiences 0 (0.0) 1 (2.5) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 1 (1.2) 0 (0.0) Discontinued due to drug-related adverse experiences 0 (0.0) 0 (0.0) Discontinued due to serious adverse experiences 0 (0.0) 0 (0.0) Discontinued due to serious drug-related adverse experiences 0 (0.0) 0 (0.0)
† Percent of patients within the laboratory adverse experience category relative to the number of patients with one or more laboratory tests postbaseline.
‡ Determined by the investigator to be possibly, probably, or definitely drug related. Although a patient may have had 2 or more laboratory adverse experiences, the patient is counted only once in a category. The same patient may appear in different categories.

MERCK RESEARCH LABORATORIES
CSR SYNOPSIS (CONT.) Protocol 074/074-10
MK-0476 Montelukast + Loratadine, Tablet Chronic Asthma
-4-
RC3100.DOC VERSION 8.1 APPROVED 08-Oct-2001
CONCLUSIONS: (1) Montelukast + loratadine and inhaled beclomethasone are generally well tolerated
for up to 48 weeks in adult patients with chronic asthma. (2) Montelukast + loratadine and inhaled beclomethasone improve the signs and symptoms of chronic asthma.
AUTHORS: Scott C. Brancato, B.S. Sumiko Shingo, M.S. Assistant Medical Program Coordinator Senior Biometrician Pulmonary—Immunology CBARDS Janet van Adelsberg, M.D. Nancy Liu, Ph.D. Associate Director Biometrician Pulmonary—Immunology CBARDS

CSR Synopsis_0476_097_P097 VERSION 1.1 APPROVED 21-Sep-2004 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast Sodium, Chewable Tablet Asthma
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized, Parallel-Group
Study Comparing the Effect on Linear Growth of Montelukast With Placebo AND Inhaled Beclomethasone in Pediatric Patients (Prepubertal, Tanner Stage I) With Mild Asthma
#097
INVESTIGATORS/STUDY CENTERS: Multicenter (30) in Africa (3), Asia (2), Central America (3), Europe (1), North America (14), and South America (7).
PRIMARY THERAPY PERIOD: 19-Oct-2000 to 14-Dec-2003 (Period II). The frozen file date was 05-Mar-2003. The study is complete.
CLINICAL PHASE: IV
DURATION OF TREATMENT: Period I—16 weeks (placebo run in); Period II—56 weeks (treatment). OBJECTIVES: (1) To determine the average rate of linear growth over the 56-week, double-blind
treatment period for montelukast, compared with placebo. (2) To determine the average rate of linear growth over the 56-week, double-blind treatment period for beclomethasone, compared with placebo. (3) To determine the average rate of linear growth over the 56-week, double-blind treatment period for beclomethasone, compared with montelukast. (4) To evaluate the safety and tolerability of the administration of montelukast for up to 56 weeks. (5) To evaluate the effects of montelukast in comparison with placebo and beclomethasone on markers of bone turnover in an exploratory manner. (6) To evaluate the effects of montelukast in comparison with placebo and beclomethasone on the following exploratory efficacy endpoints: change from baseline in forced expiratory volume in one second (FEV1), days without β-agonist use, discontinuations from blinded study therapy (BST) due to worsening asthma, oral corticosteroid rescues for worsening asthma, total peripheral blood eosinophil counts.
STUDY DESIGN: This was a multicenter, double-blind, randomized, active- and placebo-controlled, parallel-group study. Period I was a single-blind, placebo run-in period; Period II was conducted under in-house blinding procedures with patients being randomized to receive montelukast, beclomethasone, or placebo.
PATIENT DISPOSITION: Montelukast
5 mg Beclomethasone 400 µg
Placebo
Total
SCREENING FAILURES: 215 RANDOMIZED: 120 119 121 360
Male (age range) 73 (6 to 9) 80 (6 to 9) 79 (6 to 9) 232 (6 to 9) Female (age range) 47 (6 to 8) 39 (6 to 8) 42 (6 to 8) 128 (6 to 8)
COMPLETED STUDY: 109 108 108 325 DISCONTINUED STUDY: 11 11 13 35
Clinical adverse experience† 0 0 0 0 Laboratory adverse experience 0 0 0 0 Other 11 11 13 35
_____________________________________________________
† No patient was discontinued from the study due to a clinical adverse experience. Five patients, however, discontinued from BST due to a clinical adverse experience but remained in the study (see Safety section). Four (AN 2360, montelukast; AN 2728, beclomethasone; ANs 2642 and 2548, placebo) of the patients completed the study; 1 patient (AN 2482, beclomethasone) subsequently withdrew consent (approximately 2 weeks after discontinuing from BST).
DOSAGE/FORMULATION NOS.: One montelukast 5-mg chewable tablet (CT) or matching-image placebo was administered once daily at bedtime; beclomethasone inhaler was administered twice daily as two 100-µg puffs upon arising in the morning and as two 100-µg puffs every evening (total daily dose of 400 µg). Montelukast 5-mg CT (MR-3857, MR-4485) or matching-image placebo (MR-3612, MR-3618, MR-3619, MR-3855, MR-3856, MR-4288, MR-4374, MR-4389); beclomethasone inhaler (MR-4135, MR-4136, MR-4184, MR-4265, MR-4266, MR-4596) or matching-image placebo (MR-3613, MR-3889, MR-4152, MR-4586).

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 097
MK-0476 Montelukast Sodium, Chewable Tablet Asthma
-2-
CSR Synopsis_0476_097_P097 VERSION 1.1 APPROVED 21-Sep-2004 Restricted Confidential – Limited Access
DIAGNOSIS/INCLUSION CRITERIA: Boys (aged 6 to 8 years at Visit 1) and girls (aged 6 to 7 years
at Visit 1) at Tanner Stage I (prepuberty) with at least a 6-month history of asthma with typical symptoms (including, but not limited to cough, wheeze, and shortness of breath, with periodic episodes requiring treatment with inhaled β-agonists). Patients who used more than 2 courses of inhaled corticosteroids within the 12 months prior to Visit 1 were excluded.
EVALUATION CRITERIA: GROWTH: Linear growth rate over the 56-week treatment period.
EXPLORATORY ENDPOINTS: FEV1, β-agonist use, oral corticosteroid rescues for asthma, discontinuations due to worsening asthma, peripheral blood eosinophil count, and markers of bone formation.
SAFETY: Overall frequency of clinical and laboratory adverse experiences, frequency of laboratory values outside the predefined limits of change for selected laboratory safety tests, and vital signs.
STATISTICAL PLANNING AND ANALYSIS: GROWTH: Growth rates over the 56-week treatment period for the modified intention-to-treat (MITT)
population were calculated using linear regression of height measurements over time. All patients with at least 2 scheduled visits on BST after randomization were included in the MITT population; for these patients, all height measurements collected on BST were used in the MITT analysis. For patients who discontinued from BST but remained in the study, height data collected after discontinuation of BST were not used in the MITT analysis. The growth rate was assessed using an analysis of covariance (ANCOVA) model with the factors of treatment, gender, and country, and with baseline growth rate as a covariate. The difference in growth rates between montelukast and placebo was estimated and the 95% confidence interval (CI) for the between-treatment difference in growth rates was constructed. The comparison of montelukast and placebo with beclomethasone was done using a significance test comparing mean growth velocities over the 56-week treatment period. The study was designed to provide, with 100 completed patients per treatment group, the half-length of the 95% CI for the difference in linear growth rates between the montelukast and placebo treatment groups of 0.28 cm/year and 80% power to detect 0.4 difference between the beclomethasone and placebo treatment groups, for a standard deviation of 1.0 cm/year.
EXPLORATORY ENDPOINTS: Change from baseline in FEV1 was analyzed by a mixed model. The percent of days without β-agonist use was assessed by a non-parametric ANCOVA model. The use of oral corticosteroid rescues was assessed by comparing the percentages of patients that used oral corticosteroid rescues on treatment, tabulation of the number of courses of oral corticosteroid rescues, and summaries of the percent of days with oral corticosteroid rescues. Discontinuations due to worsening asthma were tabulated. Total peripheral blood eosinophil counts were analyzed by mixed model. Markers of bone turnover (serum osteocalcin [marker for bone formation] and urine cross-linked N-telopeptides of Type I collagen (NTx) levels [marker for bone resorption]) were analyzed by an analysis of variance model on changes from baseline in log-transformed values.
SAFETY: Ninety-five percent CIs were computed for pairwise differences in overall and drug-related adverse experiences (by system organ class and specific adverse experiences), serious adverse experiences, discontinuations from therapy due to adverse experiences, and laboratory adverse experiences. Percentages of patients with values outside the predefined limits of change for selected laboratory safety tests were similarly analyzed. Changes from baseline in vital signs and laboratory safety parameters were summarized by visit.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 097
MK-0476 Montelukast Sodium, Chewable Tablet Asthma
-3-
CSR Synopsis_0476_097_P097 VERSION 1.1 APPROVED 21-Sep-2004 Restricted Confidential – Limited Access
RESULTS: GROWTH: The primary analysis (MITT population) resulted in similar estimates of the treatment period
growth rate in the montelukast and placebo treatment groups, while the growth rate in the beclomethasone treatment group was significantly smaller than that seen with either montelukast or placebo (p<0.001). The results of the analysis of per-protocol population (excluding patients or height data points following important protocol violations), sensitivity analysis, analyses of the secondary growth endpoints, and confirmatory analyses were consistent with the results of the primary analysis.
Analysis of the Linear Growth Rate (cm/year) Over 56 Weeks of Treatment
Primary Analysis
Baseline
Growth Rate Treatment Period Growth Rate
Treatment Group N Mean SD Mean SD LS
Mean 95% CI for LS Mean
Montelukast 5 mg 117 5.96 1.79 5.64 1.14 5.67 (5.46, 5.88) Beclomethasone 400 µg 116 5.74 1.43 4.81 1.15 4.86 (4.64, 5.08) Placebo 112 5.72 1.52 5.61 0.97 5.64 (5.42, 5.86)
Estimate of Difference in Growth Rates (Montelukast 5 mg - Placebo) Difference in LS Means 95% CI for Difference in LS Means 0.03 (-0.26, 0.31)
Pairwise Comparisons from ANCOVA Model
Pairwise Comparison Difference in
LS Means
95% CI for Difference in LS
Means p-Value Beclomethasone 400 µg versus Placebo -0.78 (-1.06, -0.49) <0.001 Montelukast 5 mg versus Beclomethasone 400 µg 0.81 ( 0.53, 1.09) <0.001 ANCOVA=Analysis of Covariance; CI=Confidence Interval; LS Mean=Least-Squares Mean; SD=Standard Deviation.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 097
MK-0476 Montelukast Sodium, Chewable Tablet Asthma
-4-
CSR Synopsis_0476_097_P097 VERSION 1.1 APPROVED 21-Sep-2004 Restricted Confidential – Limited Access
EXPLORATORY ENDPOINTS: Both the montelukast and the beclomethasone treatment groups were
statistically significantly different from the placebo treatment group in the percentage of days on treatment without β-agonist use (p=0.024 and <0.001, respectively). The percentage of patients that used rescue corticosteroids for worsening asthma at least once on BST during the treatment period was numerically higher with placebo (34.71%) than with montelukast or beclomethasone (25.00% and 23.53%, respectively). The percentage of patients that used rescue corticosteroids for worsening asthma more than once on BST during the treatment period was higher with placebo (15.70%) than with montelukast or beclomethasone (5.83% (p=0.021) and 5.88% (p=0.021), respectively). Both montelukast and beclomethasone were significantly different from placebo in the average change from baseline in eosinophil counts across Visits 7 and 11 (p=0.005 and 0.004, respectively). There were no statistically significant pairwise treatment differences between the treatment groups in average change from baseline in FEV1 across the 56-week treatment period. There was no difference between the placebo and montelukast treatment groups in change from baseline in markers of bone turnover. Compared with placebo, the beclomethasone treatment group, however, had a significantly smaller change from baseline in serum osteocalcin (p=0.003) and marginally significantly smaller change from baseline in urine NTx (p=0.068).
SAFETY: Asthma was the most frequently reported adverse experience in the montelukast (36.7%), beclomethasone (42.9%), and placebo (50.4%) treatment groups; the difference (95% CI) between the montelukast and placebo treatment groups was -13.7% (-25.7, -1.2). Nasopharyngitis was the second most frequent adverse experience reported clinical adverse experience in the montelukast (23.3%), beclomethasone (23.5%), and placebo (24.0%) treatment groups. Pharyngitis (13.3%) was the third most frequently reported clinical adverse experience in the montelukast treatment group; upper respiratory tract infection was the third most common clinical adverse experience reported in both the beclomethasone (17.6%) and placebo (19.0%) treatment groups.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 097
MK-0476 Montelukast Sodium, Chewable Tablet Asthma
-5-
CSR Synopsis_0476_097_P097 VERSION 1.1 APPROVED 21-Sep-2004 Restricted Confidential – Limited Access
Adverse Experience Summary
Montelukast Beclomethasone 5 mg 400 µg Placebo
Clinical Adverse Experiences (AEs) (N=120) (N=119) (N=121) Number (%) of patients: n (%) n (%) n (%) With one or more AEs 110 (91.7) 107 (89.9) 110 (90.9) With no AE 10 (8.3) 12 (10.1) 11 (9.1) With drug-related† AEs 3 (2.5) 1 (0.8) 2 (1.7) With serious AEs 7 (5.8) 5 (4.2) 5 (4.1) With serious drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) 0 (0.0) Discontinued BST due to AEs 1‡ (0.8) 2‡ (1.7) 2‡ (1.7) Discontinued BST due to drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0) Discontinued BST due to serious AEs 1 (0.8) 0 (0.0) 0 (0.0) Discontinued BST due to serious drug-related AEs
0 (0.0) 0 (0.0) 0 (0.0)
Laboratory AEs (N=120) (N=119) (N=121) Number (%) of patients: n (%)§ n (%)§ n (%)§ With at least one laboratory test postbaseline 116 117 117 With one or more AEs 2 (1.7) 4 (3.4) 4 (3.4) With no AE 114 (98.3) 113 (96.6) 113 (96.6) With drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0) With serious AEs 0 (0.0) 0 (0.0) 0 (0.0) With serious drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) 0 (0.0) Discontinued BST due to AEs 0 (0.0) 0 (0.0) 0 (0.0) Discontinued BST due to drug-related AEs 0 (0.0) 0 (0.0) 0 (0.0) Discontinued BST due to serious AEs 0 (0.0) 0 (0.0) 0 (0.0) Discontinued BST due to serious drug-related AEs
0 (0.0) 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related. ‡ These patients discontinued from BST due to a clinical AE but remained in the study. No patient was
discontinued from the study due to a clinical AE (See Patient Disposition section). § The percent = number of patients within the laboratory adverse experience category/number of patients with
one or more laboratory tests postbaseline multiplied by 100. BST=blinded study therapy.
CONCLUSIONS: Over the 56-week treatment period: (1) The observed growth rate in the montelukast
treatment group is similar to the observed growth rate in the placebo treatment group. (2) The growth rate in the beclomethasone treatment group is significantly below the growth rate in the placebo treatment group. (3) The growth rate in the beclomethasone treatment group is significantly below the growth rate in the montelukast treatment group. (4) Montelukast and beclomethasone demonstrate significant benefit, compared with placebo, in the following exploratory efficacy endpoints: percentage of days on treatment without β-agonist use, percentage of patients who used more than 1 oral corticosteroid rescue, and average change from baseline in eosinophil counts. (5) Montelukast and beclomethasone are generally well tolerated.
AUTHORS: Gertrude Noonan, B.A. Olga Kuznetsova, Ph.D. Barbara Knorr, M.D. Clinical Associate Senior Biometrician Senior Director Respiratory, Allergy, &
Urology Biostatistics and Research
Decision Sciences Respiratory, Allergy, &
Urology

RC3127.DOC VERSION 13.0 APPROVED 23-Jul-2001
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTI. SYNOPSIS
MK-0476Montelukast Sodium, Film-Coated TabletSeasonal Allergic Rhinitis
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized, Parallel-GroupStudy Investigating the Clinical Effect of Montelukast in Patients With SeasonalAllergic Rhinitis—Fall Study
#192
INVESTIGATORS/STUDY CENTERS: Multicenter (29 study sites all in the United States).PRIMARY THERAPY PERIOD: 11-Aug-2000 to 31-Oct-2000.
The in-house case report form cutoff was 15-Nov-2000. The studyis completed.
CLINICAL PHASE: III
DURATION OF TREATMENT: Period I: 3- to 5-day placebo run-in period; Period II: 2-week,double-blind treatment period.
OBJECTIVES: (1) To assess the treatment effect of montelukast 10 mg versus placebo on the primary,secondary, and other endpoints over a 2-week period in patients with seasonal allergic rhinitis. (2) Todetermine the safety and tolerability profile of montelukast in patients with seasonal allergic rhinitis.
STUDY DESIGN: This was a multicenter, double-blind, randomized, 2-period, active- and placebo-controlled, parallel-group study (3 treatment groups in Period II [montelukast 10 mg, loratadine 10 mg,and placebo]) conducted under in-house blinding procedures during the fall allergic rhinitis season.
PATIENT ACCOUNTING:
Montelukast 10 mg Loratadine 10 mg Placebo Total
ENTERED: 326 170 333 829Male (age range) 99 (15 to 75) 51 (15 to 63) 111 (15 to 75) 261 (15 to 75)Female (age range) 227 (15 to 74) 119 (15 to 66) 222 (15 to 82) 568 (15 to 82)
SCREENING FAILURES: -- -- -- 413COMPLETED STUDY: 309 163 323 795DISCONTINUED STUDY: 17 7 10 34Clinical adverse experience 8 2 4 14Lack of efficacy 4 2 3 9Other 5 3 3 11
DOSAGE/FORMULATION NOS.: Study medication was administered once daily at bedtime. Period I:Matching-image placebo tablet (Formulation Numbers: MR-4219 and MR-3974). Period II: Montelukast(10 mg) film-coated tablet (MR-3640) or matching-image placebo tablet (MR-4219), loratadine (10 mg)compressed tablet (MR-4013) or matching-image placebo tablet (MR-3974).
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women (aged 15 to 85 years), with atleast a 2-year documented clinical history of seasonal allergic rhinitis that exacerbated during the studyseason; a positive skin test (wheal ≥3 mm greater than saline control) to one of the allergens activeduring the study season, and a minimum predefined level of combined Daytime Nasal Symptoms score.
EVALUATION CRITERIA:EFFICACY: Primary Endpoint: Daytime Nasal Symptoms score (combined score of Congestion,
Rhinorrhea, Itching, and Sneezing). Secondary Endpoints: Nighttime Symptoms score (combined scoreof Nasal Congestion Upon Awakening, Difficulty Going to Sleep, and Nighttime Awakenings), DaytimeEye Symptoms score (combined score of Tearing, Itchy, Red, and Puffy Eyes), Patient’s andPhysician’s Global Evaluations of Allergic Rhinitis, and Rhinoconjunctivitis Quality-of-Life score.Other Endpoints: Composite Symptoms score (average of Daytime Nasal and Nighttime Symptomsscores), Individual Daytime Nasal Symptoms (Congestion, Rhinorrhea, Itching, and Sneezing) scores,Nasal Congestion Upon Awakening. Exploratory Endpoints: Peripheral Blood Eosinophil count, End-of-Day Nasal Symptoms score (combined score of Congestion, Rhinorrhea, Itching, and Sneezing asrated by patients using instantaneous recall at the end of the day, before taking study medication), andEnd-of-Day Eye Symptoms score (combined score of Tearing, Itchy, Red, and Puffy Eyes usinginstantaneous recall at the end of the day, before taking study medication).
SAFETY: Frequency of clinical and laboratory adverse experiences, frequency of laboratory valuesoutside the predefined limits of change for selected laboratory tests, and vital signs.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 192
MK-0476Montelukast Sodium, Film-Coated TabletSeasonal Allergic Rhinitis
-2-
RC3127.DOC VERSION 13.0 APPROVED 23-Jul-2001
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: The changes from baseline in the primary, secondary, other, and exploratory efficacy
parameters were analyzed. Between-group comparisons were mostly performed using an analysis ofcovariance model with treatment and study center as factors and the baseline value of the dependentvariable as a covariate. Using the least-squares means, 95% confidence intervals (CIs) for treatmentdifferences were obtained. The study was designed to have 750 patients complete the study: 300,150, and 300 in the montelukast, loratadine, and placebo treatment groups, respectively, in order tohave 93% power to detect a difference in Daytime Nasal Symptoms score of 0.15 (change frombaseline) between the montelukast and placebo treatment groups, assuming a standard deviation of0.53.
SAFETY: Ninety-five percent CIs were computed for pairwise differences in overall and drug-relatedadverse experiences (by body system and specific adverse experience), serious adverse experiences,and discontinuations from therapy due to adverse experiences. Percentages of patients with valuesoutside the predefined limits of change for selected laboratory safety tests were similarly analyzed.
RESULTS:EFFICACY: The difference between montelukast and placebo in the primary endpoint, Daytime Nasal
Symptoms score, was borderline significant (p=0.100) in favor of montelukast. The differencebetween the 2 treatment groups reached significance (p=0.008) for the secondary endpoint, NighttimeSymptoms score, and was borderline significant for the secondary endpoints, Daytime Eye Symptomsscore (p=0.073) and the Patient’s (p=0.099) and Physician’s (p=0.088) Global Assessment of AllergicRhinitis. No significant difference (p=0.215) was found between the 2 treatment groups for thesecondary endpoint of Rhinoconjunctivitis Quality of Life. Per-protocol results were generally in linewith the intention-to-treat results, although per-protocol results reached significance (p=0.041) for theprimary endpoint, Daytime Nasal Symptoms score. The loratadine treatment effect was significant(p≤0.001), compared with placebo, for the primary endpoint, Daytime Nasal Symptoms score; it wasborderline significant for the secondary endpoint, Nighttime Symptoms score (p=0.064), andsignificant for the secondary endpoints of Daytime Eye Symptoms score (p=0.006), the Patient’s(p=0.012) and Physician’s (p=0.029) Global Assessment of Allergic Rhinitis, and the RhinoconjunctivitisQuality of Life (p=0.019).
Daytime Nasal Symptoms Score
Change From BaselineTreatment Group N Mean LS† Mean
Montelukast 10 mg 326 -0.38 -0.38Loratadine 10 mg 168 -0.45 -0.47Placebo 331 -0.30 -0.32
Between-Group DifferenceComparison LS† Mean 95% CI‡ p-Value
Montelukast 10 mg versus Placebo -0.06 (-0.14, 0.01) 0.100Loratadine 10 mg versus Placebo -0.16 (-0.25, -0.06) ≤0.001Montelukast 10 mg versus Loratadine 10 mg 0.09 (-0.00, 0.18)† LS = Least-squares.‡ CI = Confidence interval.
MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 192
MK-0476Montelukast Sodium, Film-Coated TabletSeasonal Allergic Rhinitis
-3-
RC3127.DOC VERSION 13.0 APPROVED 23-Jul-2001
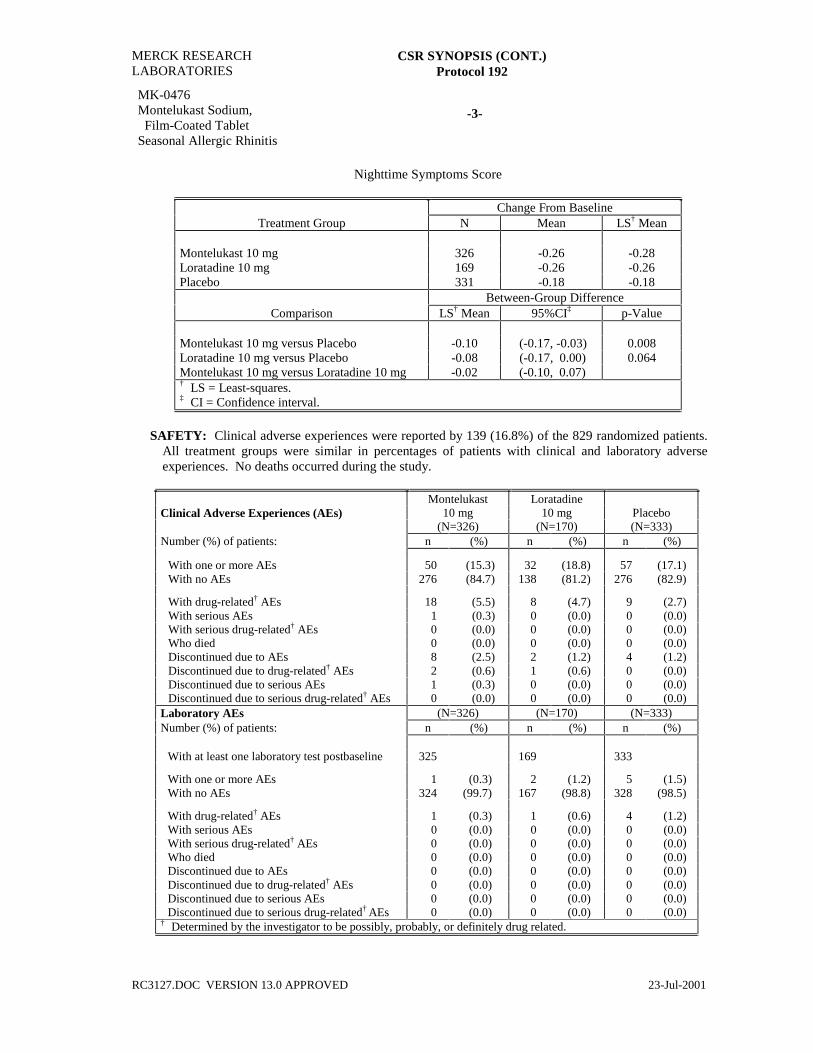
Nighttime Symptoms Score
Change From BaselineTreatment Group N Mean LS† Mean
Montelukast 10 mg 326 -0.26 -0.28Loratadine 10 mg 169 -0.26 -0.26Placebo 331 -0.18 -0.18
Between-Group DifferenceComparison LS† Mean 95%CI‡ p-Value
Montelukast 10 mg versus Placebo -0.10 (-0.17, -0.03) 0.008Loratadine 10 mg versus Placebo -0.08 (-0.17, 0.00) 0.064Montelukast 10 mg versus Loratadine 10 mg -0.02 (-0.10, 0.07)† LS = Least-squares.‡ CI = Confidence interval.
SAFETY: Clinical adverse experiences were reported by 139 (16.8%) of the 829 randomized patients.All treatment groups were similar in percentages of patients with clinical and laboratory adverseexperiences. No deaths occurred during the study.
Montelukast LoratadineClinical Adverse Experiences (AEs) 10 mg 10 mg Placebo
(N=326) (N=170) (N=333)Number (%) of patients: n (%) n (%) n (%)
With one or more AEs 50 (15.3) 32 (18.8) 57 (17.1)With no AEs 276 (84.7) 138 (81.2) 276 (82.9)
With drug-related† AEs 18 (5.5) 8 (4.7) 9 (2.7)With serious AEs 1 (0.3) 0 (0.0) 0 (0.0)With serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to AEs 8 (2.5) 2 (1.2) 4 (1.2)Discontinued due to drug-related† AEs 2 (0.6) 1 (0.6) 0 (0.0)Discontinued due to serious AEs 1 (0.3) 0 (0.0) 0 (0.0)Discontinued due to serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)
Laboratory AEs (N=326) (N=170) (N=333)Number (%) of patients: n (%) n (%) n (%)
With at least one laboratory test postbaseline 325 169 333
With one or more AEs 1 (0.3) 2 (1.2) 5 (1.5)With no AEs 324 (99.7) 167 (98.8) 328 (98.5)
With drug-related† AEs 1 (0.3) 1 (0.6) 4 (1.2)With serious AEs 0 (0.0) 0 (0.0) 0 (0.0)With serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to serious AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 192
MK-0476Montelukast Sodium, Film-Coated TabletSeasonal Allergic Rhinitis
-4-
RC3127.DOC VERSION 13.0 APPROVED 23-Jul-2001
CONCLUSIONS: In adult patients with seasonal allergic rhinitis: (1) Montelukast 10 mg, administeredonce daily, demonstrates numerically greater improvement compared with placebo in the primaryendpoint, Daytime Nasal Symptoms score. In some secondary endpoints, montelukast 10 mgdemonstrates improvement compared with placebo. (2) Loratadine 10 mg, administered once daily,compared with placebo, demonstrates improvement in the primary endpoint, Daytime Nasal Symptomsscore. In many secondary endpoints, loratadine 10 mg also demonstrates improvement compared withplacebo. (3) Montelukast 10 mg is generally well tolerated with a safety profile of clinical andlaboratory adverse experiences comparable with that of placebo. Loratadine 10 mg is also generallywell tolerated.
AUTHORS: Joan McComas, M.S.Assoc. Med. Prog. Coord.
France Vrijens, M.Sc.Senior Statistician
Barbara Knorr, M.D.Senior Director
Pulmonary-Immunology CBARDS-Brussels Pulmonary-Immunology

RC3265.doc VERSION 7.0 APPROVED 18-Dec-2001 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast, Chewable Tablet Seasonal Allergic Rhinitis
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized, Parallel-Group
Study Comparing Montelukast With Placebo in Pediatric Patients Aged 2 Through 14 Years With Seasonal Allergic Rhinitis
#219
INVESTIGATORS/STUDY CENTERS: Multicenter (31 study sites in the United States) PRIMARY THERAPY PERIOD: 12-Mar-2001 to 20-Jun-2001.
The in-house case report form cutoff was 09-Jul-2001. The study is complete.
CLINICAL PHASE: III
DURATION OF TREATMENT: Period I: 3- to 5-day placebo run-in period; Period II: 2-week, double-blind treatment period.
OBJECTIVES: (1) To determine the adverse experience profile of montelukast compared with placebo in pediatric patients with allergic rhinitis over the 2-week, double-blind treatment period. (2) To determine the measurement characteristics of new allergic rhinitis symptom scales and select scales with adequate measurement properties. (3) To explore the efficacy of montelukast, compared with placebo, as assessed by global evaluations of change in allergic rhinitis, validated quality-of-life questionnaires, and peripheral blood eosinophil counts; as well as the selected allergic rhinitis symptom scales.
STUDY DESIGN: This was a multicenter, double-blind, randomized, 2-period, placebo-controlled, parallel-group study (2 treatment groups in Period II [montelukast 4 mg or matching-image placebo in patients aged 2 to 5 years and 5 mg or matching-image placebo in patients aged 6 to 14 years]) conducted under in-house blinding procedures during the Spring allergic rhinitis season.
PATIENT ACCOUNTING:
Montelukast 4 mg and 5 mg
Placebo
Total
ENTERED: Total 280 133 413
Boys (age range, years) 157 (2 to 15) 77 (2 to 15) 234 (2 to 15) ≥2 to <5 59 24 83 ≥6 to ≤15 98 53 151
Girls (age range, years) 123 (2 to 14) 56 (2 to 13) 179 (2 to 14) ≥2 to <5 41 21 62 ≥6 to ≤15 82 35 117
Screening failures -- -- 241 COMPLETED: 269 (96.1) 127 (95.5) 396 (95.9) DISCONTINUED: Total 11 (3.9) 6 (4.5) 17 (4.1)
Clinical adverse experience 5 (1.8) 1 (0.8) 6 (1.5) Lack of efficacy 0 (0.0) 4 (3.0) 4 (1.0) Other 6 (2.1) 1 (0.8) 7 (1.7)
DOSAGE/FORMULATION NOS.: Study medication was administered once daily at bedtime. Period I: Matching-image placebo tablets (Formulation Numbers: MR-4373 and MR-4288). Period II: Montelukast (4 mg) chewable tablet (MR-4385) or matching-image placebo tablet (MR-4373), montelukast (5 mg) chewable tablet (MR-4384) or matching-image placebo tablet (MR-4288).
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking boys and girls (aged 2 to 14 years) with a clinical history of seasonal allergic rhinitis that exacerbated during the study season; a positive skin test (wheal ≥3 mm greater than saline control) to one of the allergens active during the study season, and a minimum predefined level of combined Daytime Rhinitis Symptoms score.
EVALUATION CRITERIA: SAFETY: Frequency of clinical and laboratory adverse experiences, frequency of values outside
predefined limits of change for selected laboratory safety tests and vital signs.

MERCK RESEARCH LABORATORIES
CSR SYNOPSIS (CONT.) Protocol 219
MK-0476 Montelukast, Chewable Tablet Seasonal Allergic Rhinitis
-2-
RC3265.doc VERSION 7.0 APPROVED 18-Dec-2001 Restricted Confidential – Limited Access
VALIDATION: Average daily symptom scores for the Patient and Caregiver diaries at baseline
(Period I) were used to determine the grouping of symptoms into scales. Scores for the scales were computed from the average daily scores of the questions comprising the scale.
EXPLORATORY EFFICACY: Global evaluations: Caregiver’s, Patient’s and Physician’s Global Evaluations of Allergic Rhinitis. Quality of Life: Domain scores of Pediatric Rhinoconjunctivitis Quality-of-Life Questionnaire, Concept scores of Child Health Questionnaire. Peripheral Blood Eosinophil Count. Allergic Rhinitis Symptom Scales: Validated scales from Caregiver’s and Child’s Diary Symptom Scores.
STATISTICAL PLANNING AND ANALYSIS: SAFETY: Ninety-five percent confidence intervals (CIs) were computed for pairwise differences in
overall and drug-related adverse experiences (by body system and specific adverse experience), serious adverse experiences, and discontinuations from therapy due to adverse experiences. Percentages of patients with values outside the predefined limits of change for selected laboratory safety tests were analyzed similarly. Summary statistics for laboratory safety parameters, vital signs, height, and weight were provided.
VALIDATION: Standard psychometric techniques, which included exploratory Principal Components Factor Analysis, Internal Consistency using Cronbach’s Coefficient Alpha and Item-to-Item Correlations using Spearman Correlation Coefficients, were utilized in the development of scales for the Daytime and Nighttime Patient and Caregiver Diaries. The measurement characteristics for the scales were assessed using test-retest reliability as measured by intraclass correlation coefficient, construct validity comparing the questions/scales with domains of the Pediatric Rhinoconjunctivitis Quality-of-Life Questionnaire and Child Health Questionnaire, discriminant validity comparing the mean change in scales of the Patient and Caregiver Diaries between categories of global change from Caregiver, Physician, and Patient Global Questionnaires using an analysis of covariance (ANCOVA).
EXPLORATORY EFFICACY: The study was not powered to detect between-group differences in exploratory endpoints. Between-group comparisons of global evaluations were performed using an analysis of variance model with treatment and study center, and age group as factors. Between-group comparisons for changes from baseline in other exploratory efficacy endpoints were performed using an ANCOVA model with treatment, study center, and age group as factors, and the baseline value of the dependent variable as covariate. Using the least-squares means, 95% CIs for treatment differences were obtained by both variance and covariance analyses.
RESULTS: SAFETY: Clinical adverse experiences were reported by 108 (26.2%) of the 413 randomized patients.
All treatment groups were similar in percentages of patients with clinical and laboratory adverse experiences. No deaths occurred during the study.
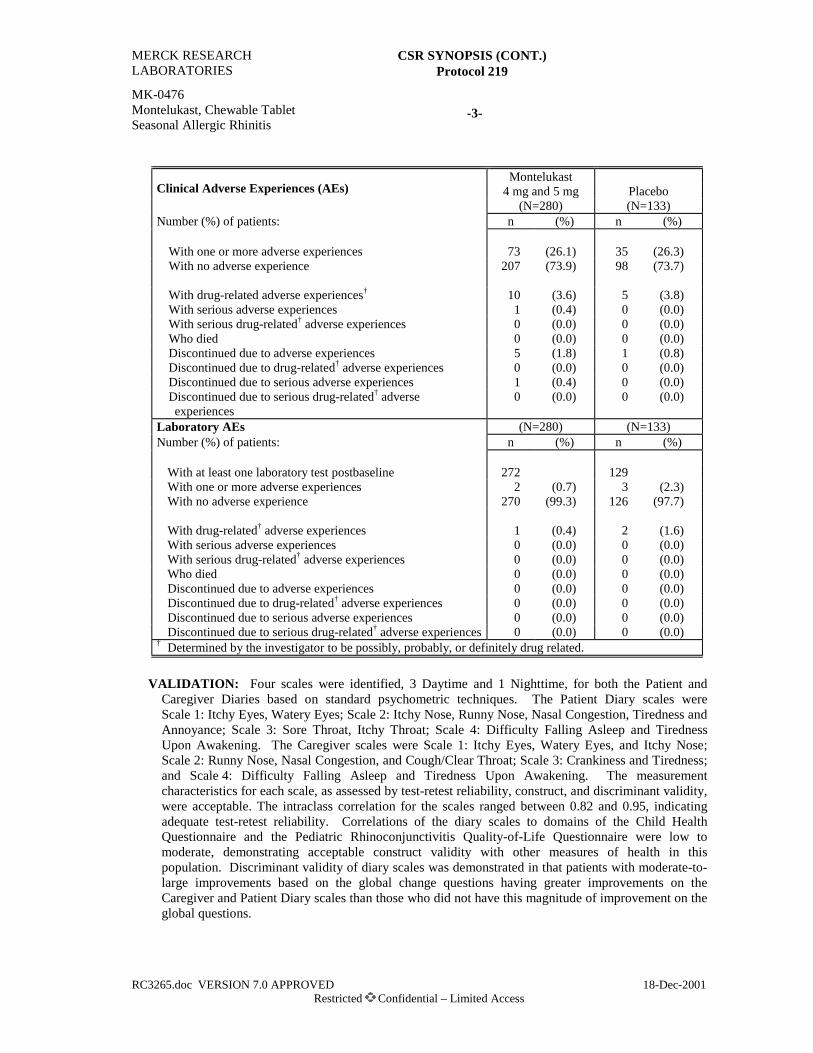
MERCK RESEARCH LABORATORIES
CSR SYNOPSIS (CONT.) Protocol 219
MK-0476 Montelukast, Chewable Tablet Seasonal Allergic Rhinitis
-3-
RC3265.doc VERSION 7.0 APPROVED 18-Dec-2001 Restricted Confidential – Limited Access
Montelukast
Clinical Adverse Experiences (AEs) 4 mg and 5 mg Placebo (N=280) (N=133) Number (%) of patients: n (%) n (%)
With one or more adverse experiences 73 (26.1) 35 (26.3) With no adverse experience 207 (73.9) 98 (73.7) With drug-related adverse experiences† 10 (3.6) 5 (3.8) With serious adverse experiences 1 (0.4) 0 (0.0) With serious drug-related† adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 5 (1.8) 1 (0.8) Discontinued due to drug-related† adverse experiences 0 (0.0) 0 (0.0) Discontinued due to serious adverse experiences 1 (0.4) 0 (0.0) Discontinued due to serious drug-related† adverse experiences
0 (0.0) 0 (0.0)
Laboratory AEs (N=280) (N=133) Number (%) of patients: n (%) n (%)
With at least one laboratory test postbaseline 272 129 With one or more adverse experiences 2 (0.7) 3 (2.3) With no adverse experience 270 (99.3) 126 (97.7)
With drug-related† adverse experiences 1 (0.4) 2 (1.6) With serious adverse experiences 0 (0.0) 0 (0.0) With serious drug-related† adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 0 (0.0) 0 (0.0) Discontinued due to drug-related† adverse experiences 0 (0.0) 0 (0.0) Discontinued due to serious adverse experiences 0 (0.0) 0 (0.0) Discontinued due to serious drug-related† adverse experiences 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related.
VALIDATION: Four scales were identified, 3 Daytime and 1 Nighttime, for both the Patient and
Caregiver Diaries based on standard psychometric techniques. The Patient Diary scales were Scale 1: Itchy Eyes, Watery Eyes; Scale 2: Itchy Nose, Runny Nose, Nasal Congestion, Tiredness and Annoyance; Scale 3: Sore Throat, Itchy Throat; Scale 4: Difficulty Falling Asleep and Tiredness Upon Awakening. The Caregiver scales were Scale 1: Itchy Eyes, Watery Eyes, and Itchy Nose; Scale 2: Runny Nose, Nasal Congestion, and Cough/Clear Throat; Scale 3: Crankiness and Tiredness; and Scale 4: Difficulty Falling Asleep and Tiredness Upon Awakening. The measurement characteristics for each scale, as assessed by test-retest reliability, construct, and discriminant validity, were acceptable. The intraclass correlation for the scales ranged between 0.82 and 0.95, indicating adequate test-retest reliability. Correlations of the diary scales to domains of the Child Health Questionnaire and the Pediatric Rhinoconjunctivitis Quality-of-Life Questionnaire were low to moderate, demonstrating acceptable construct validity with other measures of health in this population. Discriminant validity of diary scales was demonstrated in that patients with moderate-to-large improvements based on the global change questions having greater improvements on the Caregiver and Patient Diary scales than those who did not have this magnitude of improvement on the global questions.

MERCK RESEARCH LABORATORIES
CSR SYNOPSIS (CONT.) Protocol 219
MK-0476 Montelukast, Chewable Tablet Seasonal Allergic Rhinitis
-4-
RC3265.doc VERSION 7.0 APPROVED 18-Dec-2001 Restricted Confidential – Limited Access
EXPLORATORY EFFICACY: The differences between montelukast and placebo favored
montelukast for the global evaluations, some domains of the Pediatric Rhinoconjunctivitis Quality-of-Life Questionnaire, some concepts of the Child Health Questionnaire, Total Peripheral Blood Eosinophil Counts, and some Diary Domains. There was a notable decrease in Total Peripheral Blood Eosinophil Count between montelukast and placebo (p<0.05).
CONCLUSIONS: In pediatric patients aged 2 to 14 years with seasonal allergic rhinitis: (1) Montelukast 4 mg and 5 mg is generally well tolerated, with a safety profile (both clinical and laboratory adverse experiences) comparable with that of placebo. (2) The Caregiver and Patient Diaries show acceptable internal consistency, test-retest reliability, and construct and discriminant validity. The responsiveness of the Caregiver and Patient Diaries could not be established in this study. (3) Montelukast 4 mg and 5 mg, administered once daily, compared with placebo, demonstrates improvement in Peripheral Blood Eosinophil Count. (4) Montelukast 4 mg and 5 mg, administered once daily, compared with placebo, does not demonstrate improvement in several exploratory endpoints (Caregiver, Physician, and Patient [aged 10 to 14 years] Global Evaluations of Allergic Rhinitis, Pediatric Rhinoconjunctivitis Quality-of-Life Questionnaire, Child Health Questionnaire, and selected Allergic Rhinitis Symptom Scales).
AUTHORS: Ms. Joan McComas, M.S., M.M.H. Jean-Louis Marchal, Ph.D. Assoc. Med. Prog. Coordinator Contract Biometrician Pulmonary-Immunology CBARDS-Brussels Barbara Knorr, M.D. Nancy Santanello, M.D. Senior Director Senior Director Pulmonary-Immunology Epidemiology Marie-Pierre Malice, Ph.D. Jean Marie Arduino, Sc.D. Associate Director Senior Epidemiologist CBARDS-Brussels Epidemiology

RC3828.DOC VERSION 3.0 APPROVED 19-Nov-2003 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast Sodium, Oral Granules Asthma
PROTOCOL TITLE/NO.: A Multicenter, Open-Label, Controlled, Extended Safety
Study of Montelukast in Infants and Young Children With Chronic Asthma #232
INVESTIGATORS/STUDY CENTERS: Multicenter (27) in 12 countries (Africa, Europe, North America, and South America).
PUBLICATION: Van Adelsberg J, Montalvo J, Liu N, Knorr B, Wei L.X, Reiss TF. Safety and tolerability of montelukast, a leukotriene receptor antagonist in >6 to <31-month-old asthmatic patients: Evaluation at 3 months. Am J Respir Crit Care Med 2003;167(7):A271 [1.1.8].
PRIMARY THERAPY PERIOD: 01-May-2001 to 04-Mar-2003. The frozen file date was 16-Jun-2003. The study was terminated due to an administrative decision by Merck Research Laboratories.
CLINICAL PHASE: III
DURATION OF TREATMENT: Fifty-two weeks. OBJECTIVES: (1) To collect safety and tolerability data when montelukast sprinkles are administered
to infants and young children, once daily, in the evening. (2) To evaluate the effects of montelukast in comparison with usual care in infants and young children on exploratory efficacy endpoints over the 52-week study period.
STUDY DESIGN: This was an open-label, controlled extended safety study. Patients were treated with either montelukast 4-mg sprinkles or usual care. Patients who completed Protocol 176-01 had the option to enroll in this study. Additionally, patients with asthma who were 6 to 11 months of age and who had not participated in Protocol 176-01, could also enroll.
PATIENT DISPOSITION:
Montelukast 4-mg Sprinkles
Usual Care
Total
RANDOMIZED: 158 32 190
Male (age range in months)† 113 (6 to 31) 21 (6 to 28) 134 (6 to 31) Female (age range in months)† 45 (6 to 31) 11 (6 to 29) 56 (6 to 31)
COMPLETED: 86 17 103 SCREENING FAILURES: -- -- 20 DISCONTINUED: 72 15 87
Clinical adverse experience 6 0 6 Lost to follow-up 4 0 4 Other 10 4 14 Site terminated‡ 52 11 63
_____________________ † The age range was 6 months to <2 years at the Prestudy Visit (Visit 1) of Protocol 176.
Those patients who completed Protocol 176-01 were eligible to enter this study. Patients who were not previously enrolled in Protocol 176, were between the ages of ≥6 months and ≤11 months. The age range of patients at Visit 1 of Protocol 232 was 6 months to 31 months.
‡ The study was terminated due to an administrative decision by Merck Research Laboratories.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 232
MK-0476 Montelukast Sodium Asthma
-2-
RC3828.DOC VERSION 3.0 APPROVED 19-Nov-2003 Restricted Confidential – Limited Access
DOSAGE/FORMULATION NOS.: One pouch of montelukast 4-mg granules (hereafter referred to as
sprinkles) (MR-4491 and MR-4284) was mixed with 1 tablespoon of soft food and administered once daily in the evening. Usual care (defined as inhaled/nebulized cromolyn or nedocromil or inhaled/nebulized corticosteroids) was administered according to the investigator’s usual clinical practice.
DIAGNOSIS/INCLUSION CRITERIA: All patients with asthma who successfully completed Visit 5 of the Multicenter, Double-Blind, Randomized, Parallel-Group Study Comparing Montelukast With Placebo in Pediatric Patients Aged 6 to 24 Months With Asthma (Protocol 176-01) could enroll in this 52-week, open-label, controlled, extended safety study. Patients aged 6 to 11 months with asthma, who had not participated in Protocol 176-01, could also enroll.
EVALUATION CRITERIA: SAFETY: Frequency of clinical and laboratory adverse experiences, frequency of abnormal laboratory
values outside predefined limits of change for selected laboratory safety tests, and vital signs. EXPLORATORY EFFICACY: Days Without β-Agonist Use, Number of β-Agonist Treatment
Episodes Per Day (hereafter referred to as β-Agonist Treatments Per Day), Discontinuations from the Study Due to Worsening Asthma Symptoms (hereafter referred to as Discontinuations Due to Worsening Asthma), Oral Corticosteroid Rescues for Worsening Asthma Symptoms (hereafter referred to as Oral Corticosteroid Rescues), Number of Unscheduled Physician Visits or Emergency Room Visits or Hospital Visits Due to Worsening Asthma Symptoms (hereafter referred to as Patients With Unscheduled Visits for Asthma), Asthma Attacks, and Total Peripheral Blood Eosinophil Counts.
STATISTICAL PLANNING AND ANALYSIS: SAFETY: The percentages of patients with one or more adverse experiences (overall and drug related
by category and body system) and the percentages of patients outside predefined limits of change for selected laboratory safety tests were compared between montelukast and usual care using Fisher’s exact test; 95% confidence intervals (CIs) for the between-treatment-group difference were also determined. Frequencies of individual adverse experiences were summarized by treatment group and 95% CIs for the differences. Summary statistics were tabulated (by treatment group at each time point during the treatment period) for all laboratory safety parameters, vital signs, height, and weight using change from baseline values.
EXPLORATORY EFFICACY: The study was not powered to detect between-group differences in
efficacy endpoints. The efficacy analyses were performed using all the data collected during the study and received by the in-house cutoff date. Continuous exploratory efficacy endpoints were analyzed using an analysis of variance model for those endpoints without baseline values or an analysis of covariance model for those endpoints with baseline values. Models included factors for treatment, study country, age group (<1 year, ≥1 year), and stratum defined by concomitant medication (inhaled/nebulized corticosteroid use, inhaled/nebulized cromolyn use, nedocromil, or none). Fisher’s exact test was used to analyze categorical data.
RESULTS: SAFETY: Clinical adverse experiences were reported by 187 patients (98.4% of the 190 randomized
patients). Overall, there were no clinically meaningful differences between treatment groups in the incidences of clinical and laboratory adverse experiences. No deaths occurred during this study.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 232
MK-0476 Montelukast Sodium Asthma
-3-
RC3828.DOC VERSION 3.0 APPROVED 19-Nov-2003 Restricted Confidential – Limited Access
Clinical Adverse Experience Summary
Montelukast 4-mg Sprinkles Usual Care
(N=158) (N=32) n (%) n (%) Number (%) of patients:
With one or more adverse experiences 156 (98.7) 31 (96.9) With no adverse experience 2 (1.3) 1 (3.1) With drug-related adverse experiences† 8 (5.1) 0 (0.0) With serious adverse experiences 15 (9.5) 1 (3.1) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 6 (3.8) 0 (0.0) Discontinued due to drug-related adverse experiences 1 (0.6) 0 (0.0) Discontinued due to serious adverse experiences 1 (0.6) 0 (0.0) Discontinued due to serious drug-related adverse
experiences 0 (0.0) 0 (0.0)
Laboratory Adverse Experience Summary (N=158) (N=32)
n (%)‡ n (%)‡ Number (%) of patients:
With at least one laboratory test postbaseline 155 31 With one or more adverse experiences 7 (4.5) 2 (6.5) With no adverse experience 148 (95.5) 29 (93.5) With drug-related adverse experiences† 0 (0.0) 0 (0.0) With serious adverse experiences 0 (0.0) 0 (0.0) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 0 (0.0) 0 (0.0) Discontinued due to drug-related adverse experiences 0 (0.0) 0 (0.0) Discontinued due to serious adverse experiences 0 (0.0) 0 (0.0) Discontinued due to serious drug-related adverse experiences 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related. ‡ The percent=number of patients within the laboratory adverse experience category/number of
patients with one or more laboratory tests postbaseline.
CONCLUSIONS: (1) Montelukast, administered to patients aged 6 to 31 months with asthma, once
daily in the evening, is generally well tolerated during long-term treatment for ≥12 months. (2) The safety and tolerability profile is consistent with data previously reported in adults and older pediatric patients. (3) There are no significant differences between the montelukast and usual care treatment groups in the exploratory efficacy endpoints.
AUTHORS: Joan McComas, M.S.; M.M.H. Nancy Liu, Ph.D. Barbara Knorr, M.D. Med. Prog. Clin. Specialist Biometrician Senior Director Respiratory & Allergy Clinical Biostatistics Respiratory & Allergy

RC3256.DOC VERSION 4.1 APPROVED 02-Jan-2002Restricted Confidential – Limited Access
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTI. SYNOPSIS
MK-0476Montelukast SodiumFilm-Coated TabletSeasonal Allergic Rhinitis
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized, Parallel-GroupStudy Investigating the Clinical Effect of Montelukast in Patients With SeasonalAllergic Rhinitis—Spring 2001 Study
#235
INVESTIGATORS/STUDY CENTERS: Multicenter (32 Study Sites in Canada and the United States).PRIMARY THERAPY PERIOD: 05-Apr-2001 to 08-Jul-2001.
The frozen file date was 25-Sep-2001. The study is completed.CLINICAL PHASE: III
DURATION OF TREATMENT: Period I: 3- to 5-day, single-blind, placebo run-in period; Period II:2-week, double-blind, treatment period.
OBJECTIVES: (1) To assess the treatment effect of montelukast 10 mg versus placebo on the primary,secondary, and other endpoints over a 2-week period in patients with seasonal allergic rhinitis. (2) Todetermine the safety and tolerability profile of montelukast in patients with seasonal allergic rhinitis.
STUDY DESIGN: This was a multicenter, double-blind, 2-period, active- and placebo-controlled,parallel-group study (3 treatment groups in Period II [montelukast 10 mg, loratadine 10 mg, andplacebo] ) conducted under in-house blinding procedures during the spring allergic rhinitis season.
PATIENT ACCOUNTING:
Montelukast 10 mg Loratadine 10 mg Placebo Total
ENTERED: Total 522 171 521 1214Male (age range) 196 (15 to 67) 71 (15 to 70) 183 (15 to 77) 450 (15 to 77)Female (age range) 326 (15 to 82) 100 (15 to 72) 338 (15 to 73) 764 (15 to 82)
SCREENING FAILURES: 564COMPLETED: 501 165 492 1158DISCONTINUED: Total 21 6 29 56
Clinical adverse experience 7 1 8† 16Laboratory adverse experience 0 0 0 0Lack of efficacy 5 1 11 17Other 9 4 10 23
_____________________________
† This table summarizes the status of patients who participated in the study. Two patients discontinued afterrandomization due to an adverse experience that began before randomization. Therefore, a total of 6 patientsdiscontinued from placebo therapy due to a postrandomization clinical adverse experience.
DOSAGE/FORMULATION NOS.: Study medication was administered once daily at bedtime. Period I:Matching-image placebo tablet (Formulation Numbers: MR-4219 and MR-3974). Period II:Montelukast (10 mg) film-coated tablet (MR-4137) or matching-image placebo tablet (MR-4219),loratadine (10 mg) compressed tablet (C0476 OCT001P001) or matching-image placebo tablet(MR-3974).
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women (aged 15 to 85 years), with atleast a 2-year documented clinical history of seasonal allergic rhinitis that exacerbated during the studyseason; a positive skin test (wheal ≥3 mm greater than saline control) to one of the allergens activeduring the study season, and a minimum predefined level of Daytime Nasal Symptoms score.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 235
MK-0476Montelukast SodiumFilm-Coated TabletSeasonal Allergic Rhinitis
-2-
RC3256.DOC VERSION 4.1 APPROVED 02-Jan-2002Restricted Confidential – Limited Access
EVALUATION CRITERIA:EFFICACY: Primary Endpoint: Daytime Nasal Symptoms score (combined score of Congestion,
Rhinorrhea, Itching, and Sneezing). Secondary Endpoints: Nighttime Symptoms score (combined scoreof Nasal Congestion Upon Awakening, Difficulty Going to Sleep, and Nighttime Awakenings), DaytimeEye Symptoms score (combined score of Tearing, Itchy, Red, and Puffy Eyes), Patient’s andPhysician’s Global Evaluations of Allergic Rhinitis, and Rhinoconjunctivitis Quality-of-Life score.Other Endpoints: Composite Symptoms score (average of Daytime Nasal and Nighttime Symptomsscores), Individual Daytime Nasal Symptoms (Congestion, Rhinorrhea, Itching, and Sneezing) scores, andNasal Congestion Upon Awakening score. Exploratory Endpoints: End-of-Day Nasal Symptoms score(combined score of Congestion, Rhinorrhea, Itching, and Sneezing as rated by patients usinginstantaneous recall at the end of the day, before taking study medication); endpoints prespecified inthe Data Analysis Plan were Peripheral Blood Eosinophil count, and End-of-Day Eye Symptoms score(combined score of Tearing, Itchy, Red, and Puffy Eyes using instantaneous recall at the end of theday, before taking study medication).
SAFETY: Frequency of clinical and laboratory adverse experiences, frequency of laboratory valuesoutside the predefined limits of change for selected laboratory tests, and vital signs.
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: Changes from baseline in the primary, secondary, other, and exploratory efficacyparameters were analyzed. In general, between-group comparisons were performed using an analysisof covariance model with treatment and study center as factors and the baseline value of the dependentvariable as a covariate. Ninety-five percent confidence intervals (CIs) for treatment differences wereobtained using the least-squares means. The study was designed to have 700 patients complete thestudy: 300, 100, and 300 in the montelukast, loratadine, and placebo treatment groups, respectively, inorder to have 93% power to detect a difference in Daytime Nasal Symptoms score of 0.15 (changefrom baseline) between the montelukast and placebo treatment groups, assuming a standard deviationof 0.53.
SAFETY: Ninety-five percent CIs were computed for pairwise differences in overall and drug-relatedadverse experiences (by body system and specific adverse experience), serious adverse experiences,and discontinuations from therapy due to adverse experiences. Percentages of patients with valuesoutside the predefined limits of change for selected laboratory safety tests were similarly analyzed.
RESULTS:EFFICACY: Over the 2-week treatment period, montelukast showed significant improvement
compared with placebo for the primary endpoint (p=0.003), Daytime Nasal Symptoms score, and allsecondary endpoints (p≤0.050). The loratadine effect was significant, compared with placebo, for theprimary (p≤0.001) and most secondary endpoints. In addition, montelukast was significantly superiorto placebo for the following predefined other and exploratory endpoints: Composite score, DaytimeCongestion score, Daytime Rhinorrhea score, Daytime Sneezing score, Nasal Congestion UponAwakening score, and Peripheral Blood Eosinophil count. A borderline significant (p=0.056)treatment effect of montelukast was seen for End-of-Day Nasal Symptoms score; no significantmontelukast effect was seen on Daytime Itching score and End-of-Day Eye Symptoms score.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 235
MK-0476Montelukast SodiumFilm-Coated TabletSeasonal Allergic Rhinitis
-3-
RC3256.DOC VERSION 4.1 APPROVED 02-Jan-2002Restricted Confidential – Limited Access
Daytime Nasal Symptoms Score
Change From BaselineTreatment Group N Mean LS Mean
Montelukast 10 mg 519 -0.39 -0.38Loratadine 10 mg 170 -0.50 -0.47Placebo 521 -0.31 -0.29
Between-Group DifferenceComparison LS Mean 95% CI p-Value
Montelukast 10 mg versus Placebo -0.09 (-0.16, -0.03) 0.003Loratadine 10 mg versus Placebo -0.19 (-0.28, -0.10) ≤0.001Montelukast 10 mg versus Loratadine 10 mg 0.09 ( 0.00, 0.18)LS = Least-squares.CI = Confidence interval.
Nighttime Symptoms Score
Change From BaselineTreatment Group N Mean LS Mean
Montelukast 10 mg 519 -0.29 -0.28Loratadine 10 mg 170 -0.29 -0.28Placebo 521 -0.20 -0.20
Between-Group DifferenceComparison LS Mean 95% CI p-Value
Montelukast 10 mg versus Placebo -0.08 (-0.13, -0.02) 0.009Loratadine 10 mg versus Placebo -0.07 (-0.15, 0.01) 0.073Montelukast 10 mg versus Loratadine 10 mg 0.00 (-0.08, 0.08)LS = Least-squares.CI = Confidence interval.
SAFETY: Clinical adverse experiences were reported by 198 (16.3%) of the 1214 randomized patients.All treatment groups were similar in percentages of patients with clinical and laboratory adverseexperiences. There were no serious clinical or laboratory experiences. No deaths occurred during thestudy.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 235
MK-0476Montelukast SodiumFilm-Coated TabletSeasonal Allergic Rhinitis
-4-
RC3256.DOC VERSION 4.1 APPROVED 02-Jan-2002Restricted Confidential – Limited Access
Montelukast LoratadineClinical Adverse Experiences (AEs) 10 mg 10 mg Placebo
(N=522) (N=171) (N=521)Number (%) of patients: n (%) n (%) n (%)
With one or more AEs 89 (17.0) 26 (15.2) 83 (15.9)With no AEs 433 (83.0) 145 (84.8) 438 (84.1)
With drug-related† AEs 12 (2.3) 7 (4.1) 18 (3.5)With serious AEs 0 (0.0) 0 (0.0) 0 (0.0)With serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to AEs 7 (1.3) 1 (0.6) 6‡ (1.2)Discontinued due to drug-related† AEs 0 (0.0) 0 (0.0) 1 (0.2)Discontinued due to serious AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)
Laboratory AEs (N=522) (N=171) (N=521)Number (%) of patients: n (%) n (%) n (%)
With at least one laboratory test postbaseline 516 171 515
With one or more AEs 5 (1.0) 3 (1.8) 2 (0.4)With no AEs 511 (99.0) 168 (98.2) 513 (99.6)
With drug-related† AEs 5 (1.0) 2 (1.2) 1 (0.2)With serious AEs 0 (0.0) 0 (0.0) 0 (0.0)With serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to serious AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related.‡ This table includes information about AEs that began after randomization; it does not include information
about AEs that began before randomization. Although 6 patients discontinued from placebo therapy due toan AE that began after randomization, 2 patients discontinued from placebo therapy after randomization dueto an AE that began before randomization. (See Patient Accounting section on Page 1.)
CONCLUSIONS: In adult patients with seasonal allergic rhinitis: (1) Montelukast 10 mg, administeredonce daily, compared with placebo, demonstrates improvement in the primary endpoint, Daytime NasalSymptoms score. In all secondary endpoints, montelukast also demonstrates improvement comparedwith placebo. (2) Loratadine 10 mg, administered once daily, compared with placebo, demonstratesimprovement in the primary endpoint: Daytime Nasal Symptoms score. In most secondary endpoints,loratadine also demonstrates improvement compared with placebo. (3) Montelukast 10 mg is generallywell tolerated with a safety profile of clinical and laboratory adverse experiences comparable with thatof placebo. Loratadine 10 mg is also generally well tolerated.
AUTHORS:
Joan R. Lord, B.A.Assoc. Med. Prog. CoordinatorRespiratory and Allergy
Joris Menten, M.S.Senior StatisticianCBARDS
Janet van Adelsberg, M.D.Associate DirectorRespiratory and Allergy

RC3364.DOC VERSION 6.0 APPROVED 10-May-2002Restricted Confidential – Limited Access
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTI. SYNOPSIS
MK-0476Montelukast Sodium,Film-Coated TabletSeasonal Allergic Rhinitis
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized, Parallel-GroupStudy Investigating the Clinical Effect of Montelukast in Patients With SeasonalAllergic Rhinitis Over a 4-Week Treatment Period—Fall 2001
#240
INVESTIGATORS/STUDY CENTERS: Multicenter (47 Study Sites in the United States) PRIMARY THERAPY PERIOD: 03-Aug-2001 to 05-Nov-2001.
The frozen file date was 08-Mar-2002. The study is complete.CLINICAL PHASE: III
DURATION OF TREATMENT: Period I: 3- to 5-day, single-blind, placebo run-in period; Period II:4-week, double-blind treatment period.
OBJECTIVES: Primary: (1) To assess the treatment effect of montelukast 10 mg, administered in themorning, versus placebo, on the primary, secondary, other, and exploratory endpoints over the first2 weeks of a 4-week treatment period, in patients with seasonal allergic rhinitis. (2) To determine thesafety and tolerability profile of montelukast in patients with seasonal allergic rhinitis. Secondary:(1) To assess the treatment effect of montelukast 10 mg, administered in the morning, versus placebo,on the primary, secondary, other, and exploratory endpoints over the entire 4-week treatment period.(2) To assess the treatment effect of loratadine 10 mg, administered in the morning, versus placebo, onthe primary, secondary, other, and exploratory endpoints over the first 2 weeks and over the entire4-week treatment period. (3) To estimate the efficacy of montelukast 10 mg versus loratadine 10 mg,both administered in the morning, on the primary, secondary, other, and exploratory endpoints over thefirst 2 weeks and over the entire 4-week treatment period.
STUDY DESIGN: This was a multicenter, double-blind, randomized, parallel-group study (3 treatmentgroups in Period II [montelukast 10 mg, loratadine 10 mg, and placebo]) conducted under in-houseblinding procedures during the fall allergic rhinitis season. Study medication was administered in themorning.
PATIENT ACCOUNTING:
Montelukast 10 mg Loratadine 10 mg Placebo Total
ENTERED: Total 448 180 451 1079Male (age range) 147 (15 to 82) 61 (15 to 70) 150 (15 to 67) 358 (15 to 82)Female (age range) 301 (15 to 71) 119 (17 to 79) 301 (15 to 82) 721 (15 to 82)
COMPLETED: 420 170 410 1000DISCONTINUED: Total 28 10 41 79
Clinical adverse experience 13† 1 15† 29†
Laboratory adverse experience 0 0 1 1Lack of efficacy 7 2 10 19Other
____________________________________8 7 15 30
† This table summarizes the status of patients who participated in the study. Three patients (2montelukast,1placebo) discontinued after randomization due to an adverse experience that began before randomization.Therefore, 11 and 14 patients in the montelukast and placebo treatment groups, respectively, discontinued due toa clinical adverse experience that began after randomization.
DOSAGE/FORMULATION NOS.: Study medication was administered once daily in the morning.Period I: Matching-image placebo tablet (Formulation Numbers: MR-4219 and MR-3974). Period II:Montelukast (10 mg) film-coated tablet (MR-4380) or matching-image placebo tablet (MR-4219),loratadine (10 mg) compressed tablet (4471) or matching-image placebo tablet (MR-3974).
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women (aged 15 to 82 years), with atleast a 2-year documented clinical history of seasonal allergic rhinitis that exacerbated during the studyseason; a positive skin test (wheal ≥3 mm greater than saline control) to one of the allergens activeduring the study season, and a minimum predefined level of Daytime Nasal Symptoms score.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 240
MK-0476Montelukast Sodium,Film-Coated TabletSeasonal Allergic Rhinitis
-2-
RC3364.DOC VERSION 6.0 APPROVED 10-May-2002Restricted Confidential – Limited Access
EVALUATION CRITERIA:EFFICACY: Primary Endpoint: Daytime Nasal Symptoms score (averaged score of Congestion,
Rhinorrhea, Itching, and Sneezing). Secondary Endpoints: Nighttime Symptoms score (averagedscore of Nasal Congestion Upon Awakening, Difficulty Going to Sleep, and Nighttime Awakenings),Composite Symptoms score (averaged score of Daytime Nasal and Nighttime Symptoms), DaytimeEye Symptoms score (averaged score of Tearing, Itchy, Red, and Puffy Eyes), Patient’s andPhysician’s Global Evaluations of Allergic Rhinitis, and Rhinoconjunctivitis Quality-of-Life overallscore. Other Endpoints: Individual Daytime Nasal Symptom scores (Congestion, Rhinorrhea,Itching, and Sneezing), Individual Nighttime Nasal Symptom scores (Congestion Upon Awakening,Difficulty Going to Sleep, and Nighttime Awakenings), scores for individual Domains of theRhinoconjunctivitis Quality-of-Life Questionnaire (Activity, Sleep, Nasal Symptoms, Eye Symptoms,Non-Nose/Non-Eye Symptoms, Practical Problems, and Emotions), and Total Peripheral BloodEosinophils. Exploratory Endpoints: Morning (End-of-Dosing Interval) Nasal Symptoms score(averaged score of Congestion, Rhinorrhea, Itching, and Sneezing), Morning (End-of-Dosing Interval)Eye Symptoms score (averaged score of Tearing, Itchy, Red, and Puffy Eyes), and IndividualMorning (End-of-Dosing Interval) Nasal Symptom scores. All Morning (End-of-Dosing Interval)symptoms were rated by patients using instantaneous recall in the morning at the end of the dosinginterval, before the next dose of study medication was administered.
SAFETY: Frequency of clinical and laboratory adverse experiences, frequency of laboratory valuesoutside the predefined limits of change for selected laboratory safety tests, and vital signs.
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: Changes from baseline over the first 2 weeks and over the entire 4 weeks (includes both
the first and last 2 weeks) of treatment in the primary, secondary, other, and exploratory efficacyparameters were analyzed; the primary analysis was prespecified to examine the first 2 weeks oftreatment. Statistical tests were performed using the analysis of covariance (ANCOVA) model withtreatment (montelukast, loratadine, and placebo), study site as factors, and baseline value of thedependent variable as covariate. Treatment comparisons were based on testing specific contrastsusing the ANCOVA model, the primary comparison being between the montelukast 10-mg andplacebo treatment groups.
SAFETY: Ninety-five percent confidence intervals were computed for pairwise differences in overalland drug-related adverse experiences (by body system and specific adverse experience), seriousadverse experiences, and discontinuations from therapy due to adverse experiences. Percentages ofpatients with values outside the predefined limits of change for selected laboratory safety tests weresimilarly analyzed.
RESULTS:EFFICACY: Over the first 2 weeks of the active treatment period (primary analysis time point),
montelukast showed significant improvement compared with placebo for the primary endpoint,Daytime Nasal Symptoms score (p=0.003), and all secondary endpoints (p≤0.050). In addition,montelukast showed significant improvements compared with placebo for most of the predefinedother and exploratory endpoints. The loratadine treatment effect was significant, compared withplacebo, for the primary (p≤0.001) and most secondary endpoints.
Over the entire 4-week active treatment period, montelukast showed significant improvementcompared with placebo, for the primary endpoint, Daytime Nasal Symptoms score (p=0.003), and allsecondary endpoints (p≤0.050). In addition, montelukast showed significant improvements comparedwith placebo for most of the predefined other and exploratory endpoints. The loratadine effect wassignificant, compared with placebo, for the primary (p≤0.001) and most secondary endpoints.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 240
MK-0476Montelukast Sodium,Film-Coated TabletSeasonal Allergic Rhinitis
-3-
RC3364.DOC VERSION 6.0 APPROVED 10-May-2002Restricted Confidential – Limited Access
Daytime Nasal Symptoms Score
First 2 Weeks of the ActiveTreatment Period
(Primary Analysis Time Point)
Entire 4-Week ActiveTreatment Period
(Secondary Analysis Time Point)Change From Baseline Change From Baseline
Treatment Group N Mean LS Mean N Mean LS Mean
Montelukast 10 mg 445 -0.32 -0.33 445 -0.42 -0.43Loratadine 10 mg 180 -0.45 -0.45 180 -0.51 -0.50Placebo 448 -0.20 -0.23 448 -0.30 -0.33
Between-Group Difference Between-Group DifferenceComparison LS Mean 95% CI p-Value LS Mean 95% CI p-Value
Montelukast 10 mg versus Placebo -0.10 (-0.16, -0.03) 0.003 -0.10 (-0.17, -0.04) 0.003Loratadine 10 mg versus Placebo -0.22 (-0.31, -0.13) ≤0.001 -0.17 (-0.26, -0.08) ≤0.001Montelukast 10 mg versus Loratadine 10 mg 0.12 (0.03, 0.21) 0.07 (-0.02, 0.16)CI = Confidence interval; LS = Least-squares.
Nighttime Symptoms Score
First 2 Weeks of the ActiveTreatment Period
(Primary Analysis Time Point)
Entire 4-Week ActiveTreatment Period
(Secondary Analysis Time Point)Change From Baseline Change From Baseline
Treatment Group N Mean LS Mean N Mean LS Mean
Montelukast 10 mg 445 -0.27 -0.28 445 -0.35 -0.36Loratadine 10 mg 180 -0.27 -0.25 180 -0.34 -0.32Placebo 448 -0.16 -0.18 448 -0.23 -0.26
Between-Group Difference Between-Group DifferenceComparison LS Mean 95% CI p-Value LS Mean 95% CI p-Value
Montelukast 10 mg versus Placebo -0.10 (-0.16, -0.04) 0.002 -0.09 (-0.15, -0.03) 0.002Loratadine 10 mg versus Placebo -0.07 (-0.15, 0.01) 0.080 -0.06 (-0.14, 0.02) 0.165Montelukast 10 mg versus Loratadine 10 mg -0.03 (-0.10, 0.05) -0.04 (-0.12, 0.04)CI = Confidence interval; LS = Least-squares.
SAFETY:Clinical adverse experiences were reported by 289 (26.8%) of the 1079 randomized patients. Alltreatment groups were similar in percentages of patients with clinical and laboratory adverseexperiences. There were 2 patients with serious clinical adverse experiences (pregnancy andappendicitis), both in the montelukast treatment group. No serious laboratory adverse experiencesand no deaths occurred during the study.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 240
MK-0476Montelukast Sodium,Film-Coated TabletSeasonal Allergic Rhinitis
-4-
RC3364.DOC VERSION 6.0 APPROVED 10-May-2002Restricted Confidential – Limited Access
Montelukast LoratadineClinical Adverse Experiences (AEs) 10 mg 10 mg Placebo
(N=448) (N=180) (N=451)Number (%) of patients: n (%) n (%) n (%)
With one or more AEs 133 (29.7) 45 (25.0) 111 (24.6)With no AEs 315 (70.3) 135 (75.0) 340 (75.4)
With drug-related† AEs 15 (3.3) 10 (5.6) 21 (4.7)With serious AEs 2 (0.4) 0 (0.0) 0 (0.0)With serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to AEs 11‡ (2.5) 1 (0.6) 14‡ (3.1)Discontinued due to drug-related† AEs 0 (0.0) 0 (0.0) 1 (0.2)Discontinued due to serious AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)
Laboratory AEs (N=448) (N=180) (N=451)Number (%) of patients: n (%) n (%) n (%)
With at least one laboratory test postbaseline 446 179 450
With one or more AEs 6 (1.3) 1 (0.6) 5 (1.1)With no AEs 440 (98.7) 178 (99.4) 445 (98.9)
With drug-related† AEs 2 (0.4) 0 (0.0) 3 (0.7)With serious AEs 0 (0.0) 0 (0.0) 0 (0.0)With serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to AEs 0 (0.0) 0 (0.0) 1 (0.2)Discontinued due to drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to serious AEs 0 (0.0) 0 (0.0) 0 (0.0)Discontinued due to serious drug-related† AEs 0 (0.0) 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related.‡ This table includes information about AEs that began after randomization: it does not include
information about AEs that began before randomization. The 3 patients (2montelukast, 1placebo)that discontinued because of a clinical adverse experience that began before randomization are notshown in this table.
CONCLUSIONS: In adult patients with seasonal allergic rhinitis: (1) Montelukast 10 mg, administeredonce daily in the morning, compared with placebo, demonstrates improvement in the primary endpoint,Daytime Nasal Symptoms score, over both 2 and 4 weeks of treatment. In all secondary endpoints andthe majority of other endpoints, montelukast also demonstrates improvement compared with placebo.(2) Montelukast 10 mg, relative to placebo, improves allergic rhinitis symptoms over 24 hours,including nasal symptoms assessed at the end of the dosing interval. (3) Loratadine 10 mg, administeredonce daily, compared with placebo, demonstrates improvement in the primary endpoint, Daytime NasalSymptoms score, over 2 and 4 weeks of treatment. In most secondary endpoints, loratadine alsodemonstrates improvement compared with placebo. (4) Montelukast 10 mg is generally well tolerated,with a safety profile comparable with that of placebo. Loratadine 10 mg is also generally well tolerated.
AUTHORS: Joan R. Lord, B.A. Joris Menten, M.S. Janet van Adelsberg, M.D.Assoc. Med. Prog. Coord. Senior Statistician Associate DirectorRespiratory and Allergy CBARDS Respiratory and Allergy
BC3384.DOC VERSION 10.1 APPROVED 25-Feb-2003 Restricted Confidential – Limited Access R 19-Sep-2003 R 14-May-2004
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast Sodium, Film-Coated Tablet Perennial Allergic Rhinitis
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized, Placebo-
Controlled, Parallel-Group Study Investigating the Clinical Effects of Montelukast in Patients With Perennial Allergic Rhinitis
#246
INVESTIGATORS/STUDY CENTERS: Multicenter (74 study sites in the United States). PRIMARY THERAPY PERIOD: 27-Nov-2001 to 05-May-2002.
The frozen file date was 13-Aug-2002. The study is completed. CLINICAL PHASE: III
DURATION OF TREATMENT: Period I: 5- to 7-day, placebo period; Period II: 6-week treatment period. OBJECTIVES: (1) To assess the treatment effect of montelukast 10 mg versus placebo on the primary,
secondary, and other/exploratory endpoints, over the first 4 weeks of a 6-week treatment period, in patients with perennial allergic rhinitis. (2) To determine the tolerability profile of montelukast 10 mg in patients with perennial allergic rhinitis.
STUDY DESIGN: This was a multicenter, double-blind, randomized, 2-period, active- and placebo-controlled, parallel-group study. Period I was a single-blind, placebo run-in period; in Period II, patients were treated either with montelukast 10 mg, cetirizine 10 mg, or placebo. The study was conducted under in-house blinding procedures in patients with perennial allergic rhinitis.
PATIENT ACCOUNTING:
Montelukast 10 mg
Cetirizine 10 mg
Placebo
Total
ENTERED: Total 630 122 613 1365 Male (age range) 211 (15 to 74) 37 (16 to 67) 195 (15 to 77) 443 (15 to 77) Female (age range) 419 (15 to 76) 85 (15 to 75) 418 (15 to 82) 922 (15 to 82)
Screening Failures -- -- -- 1198
COMPLETED: 562 (89.2) 106 (86.9) 530 (86.5) 1198 (87.8) DISCONTINUED: Total 68 (10.8) 16 (13.1) 83 (13.5) 167 (12.2)
Clinical adverse experience 29† (4.6) 4 (3.3) 24 (3.9) 57 (4.1) Laboratory adverse experience 2 (0.3) 0 (0.0) 3 (0.5) 5 (0.4) Lack of efficacy 3 (0.5) 2 (1.6) 9 (1.5) 14 (1.0) Other 34 (5.4) 10 (8.2) 47 (7.7) 91 (6.7)
† Includes one patient in the montelukast treatment group (AN 5654) who discontinued after randomization due to an adverse experience that began prior to randomization.
DOSAGE/FORMULATION NOS.: Study drug was administered once daily at bedtime. Period I: Matching image placebo tablet (formulation numbers: MR-4309 and MR-4568). Period II: Montelukast (10 mg) film-coated tablet (MR-4380) or matching-image placebo tablet (MR-4309), cetirizine (10 mg) film-coated tablet (MR-4584) or matching-image placebo tablet (MR-4568).
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking male and female patients (aged 15 to 85 years), with at least a 2-year documented clinical history of perennial allergic rhinitis symptoms, a positive skin test (wheal ≥3 mm greater than saline control) to 1 of the relevant perennial allergens, and a minimum predefined level of Daytime Nasal Symptoms score.
EVALUATION CRITERIA: Efficacy: Primary Endpoint: Daytime Nasal Symptoms score (average of Nasal Congestion, Rhinorrhea, Nasal Itching, and Sneezing). Secondary Endpoints: Composite Symptoms score (average of Daytime Nasal Symptoms and Nighttime Symptoms), Nighttime Symptoms score (average of Nasal Congestion Upon Awakening, Difficulty Going to Sleep, and Nighttime Awakenings), and Patient’s and Physician’s Global Evaluations of Allergic Rhinitis. Other Endpoints: Daytime Eye Symptoms score (average of Tearing and Itchy Eyes), Individual Daytime Nasal Symptoms scores (Nasal Congestion, Rhinorrhea, Nasal Itching, and Sneezing), Overall and Individual Daytime Throat

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 246
MK-0476 Montelukast Sodium Perennial Allergic Rhinitis
-2-
BC3384.DOC VERSION 10.1 APPROVED 25-Feb-2003 Restricted Confidential – Limited Access
Symptoms scores (Mucus Dripping Down Throat/Postnasal Drip and Clearing of Throat), Individual Nighttime Symptoms scores (Nasal Congestion Upon Awakening, Difficulty Going to Sleep, and Nighttime Awakenings), Overall and Individual Domains of the Rhinoconjunctivitis Quality-of-Life score (Activity, Sleep, Nasal Symptoms, Eye Symptoms, Non-Nasal/Non-Eye Symptoms, Practical Problems, and Emotions), Overall and Individual Perennial Allergic Rhinitis Questionnaire scores, and Total Peripheral Blood Eosinophil Counts. Exploratory Endpoint: End-of-Day Nasal Symptoms score (average of Nasal Congestion, Rhinorrhea, Nasal Itching, and Sneezing).
Safety: Overall frequency of clinical and laboratory adverse experiences and incidences of common adverse experiences reported by the patients.
STATISTICAL PLANNING AND ANALYSIS: Efficacy: Changes from baseline over the first 4 weeks and over the entire 6 weeks of treatment in the primary, secondary, and other/exploratory efficacy parameters were analyzed; the primary analyses focused on the first 4 weeks of treatment. Statistical tests were performed using the analysis of covariance (ANCOVA) model with treatment (montelukast, cetirizine, and placebo) and study center as factors and baseline value of the dependent variable as covariate. Treatment comparisons were based on testing specific contrasts using the ANCOVA model; the primary comparison was between the montelukast 10-mg and placebo treatment groups. For the primary endpoint at 4 weeks, with 500 patients in the montelukast and 500 patients in the placebo treatment groups, the test had 88% power to detect a treatment difference of 0.10 score; α=0.050, 2-sided test assuming a standard deviation of 0.51.
Safety: Ninety-five percent confidence limits for pairwise differences in overall and drug-related adverse experiences (by body system and specific adverse experiences), serious adverse experiences, and discontinuations from therapy due to adverse experiences were calculated. Summary statistics were tabulated for all vital signs and laboratory changes from baseline values.
RESULTS: Efficacy: Over the first 4 weeks of the active treatment period (primary analysis time point), montelukast showed numerical but not statistically significant (p=0.150) improvement compared with placebo for the primary endpoint, Daytime Nasal Symptoms score. The difference between the montelukast and placebo treatment groups reached significance (p=0.010) for the secondary endpoint of Patient’s Global Evaluation of Allergic Rhinitis. No significant differences were found between the 2 treatment groups for the secondary endpoints of Nighttime Symptoms score (p=0.255), Composite Symptoms score (p=0.139), and Physician’s Global Evaluation of Allergic Rhinitis (p=0.289). Per-protocol results were generally in line with the modified intention-to-treat results, although per-protocol results were borderline significant (primary endpoint, p=0.083) for the Daytime Nasal Symptoms score, and for the Nighttime Symptoms score (secondary endpoint, p=0.064), and reached significance for the Composite Symptoms score (secondary endpoint, p=0.042). Over the first 4 weeks of the active treatment period (primary analysis time point), the cetirizine treatment effect was significant (p=0.038), compared with placebo, for the primary endpoint, Daytime Nasal Symptoms score. The difference was significant for the secondary endpoint of Patient’s Global Evaluation of Allergic Rhinitis (p=0.050) and borderline significant for the secondary endpoint of Composite Symptoms score (p=0.062); no significant differences were found between the 2 treatment groups for the secondary endpoints of Nighttime Symptoms score (p=0.266) and Physician’s Global Evaluation of Allergic Rhinitis (p=0.293).
Results of over the entire 6-week active treatment period were generally similar to those for the first 4 weeks of treatment.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 246
MK-0476 Montelukast Sodium Perennial Allergic Rhinitis
-3-
BC3384.DOC VERSION 10.1 APPROVED 25-Feb-2003 Restricted Confidential – Limited Access
Daytime Nasal Symptoms Score
First 4 Weeks of the Active Treatment Period
(Primary Analysis Period)
Entire 6-Week Active Treatment Period
(Secondary Analysis Period)
Change From Baseline Change From Baseline Treatment Group N Mean LS Mean N Mean LS Mean
Montelukast 10 mg 626 -0.39 -0.39 626 -0.45 -0.46 Cetirizine 10 mg 120 -0.47 -0.45 120 -0.49 -0.48 Placebo 609 -0.35 -0.36 609 -0.40 -0.41 Between-Group Difference Between-Group Difference
Comparison LS
Mean 95% CI p-Value LS
Mean 95% CI p-Value Montelukast 10 mg verus Placebo -0.04 (-0.09, 0.01) 0.150 -0.05 (-0.10, 0.01) 0.086 Cetirizine 10 mg versus Placebo -0.10 (-0.19,-0.01) 0.038 -0.07 (-0.17, 0.02) 0.133 Montelukast 10 mg versus Cetirizine 10 mg 0.06 (-0.03, 0.15) 0.03 (-0.07, 0.12) CI = Confidence Interval; LS = Least-Square.
Global Evaluation of Allergic Rhinitis at the End of the First 4 Weeks of the Active Treatment Period
Patient’s Global Evaluation
of Allergic Rhinitis Physician’s Global Evaluation of
Allergic Rhinitis Global Evaluation Score Global Evaluation Score
Treatment Group N Mean LS Mean N Mean LS Mean Montelukast 10 mg 617 2.20 2.22 603 2.24 2.26 Cetirizine 10 mg 117 2.15 2.15 115 2.20 2.20 Placebo 602 2.40 2.41 583 2.32 2.33 Between-Group Difference Between-Group Difference
Comparison LS
Mean 95% CI p-Value LS
Mean 95% CI p-Value Montelukast 10 mg verus Placebo -0.19 (-0.33, -0.05) 0.010 -0.07 (-0.21, 0.06) 0.289 Cetirizine 10 mg versus Placebo -0.26 (-0.51, -0.00) 0.050 -0.13 (-0.37, 0.11) 0.293 Montelukast 10 mg versus Cetirizine 10 mg 0.07 (-0.19, 0.32) 0.05 (-0.18, 0.29) A lower global evaluation score indicates a better global evaluation. Scores are on a scale from 0 (“very much better)” to 6 (“very much worse”). CI = Confidence Interval; LS = Least-Square.
Safety: Clinical and other adverse experiences were reported by 516 (37.8%) of the 1365 randomized patients. All treatment groups were similar in percentages of patients with clinical and laboratory adverse experiences with the exception of laboratory adverse experiences determined by the investigators to be drug related. No deaths occurred during this study.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 246
MK-0476 Montelukast Sodium Perennial Allergic Rhinitis
-4-
BC3384.DOC VERSION 10.1 APPROVED 25-Feb-2003 Restricted Confidential – Limited Access R 19-Sep-2003
Clinical Adverse Experience Summary Montelukast Cetirizine 10 mg 10 mg Placebo (N=630) (N=122) (N=613) Number (%) of patients: n (%) n (%) n (%) With one or more adverse experiences 242 (38.4) 43 (35.2) 231 (37.7) With no adverse experience 388 (61.6) 79 (64.8) 382 (62.3) With drug-related† adverse experiences 38 (6.0) 12 (9.8) 35 (5.7) With serious adverse experiences 2 (0.3) 0 (0.0) 3 (0.5) With serious drug-related† adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 28‡ (4.4) 4 (3.3) 24 (3.9) Discontinued due to drug-related† adverse experiences 2 (0.3) 1 (0.8) 3 (0.5) Discontinued due to serious adverse experiences 1 (0.2) 0 (0.0) 1 (0.2) Discontinued due to serious drug-related† adverse experiences 0 (0.0) 0 (0.0) 0 (0.0)
Laboratory Adverse Experience Summary (N=630) (N=122) (N=613) n (%) n (%) n (%)
Number (%) of patients: With at least one laboratory test postbaseline 620 (98.4) 119 (97.5) 604 (98.5) With one or more adverse experiences 16 (2.6) 4 (3.4) 10 (1.7) With no laboratory adverse experience 604 (97.4) 115 (96.6) 594 (98.3) With drug-related† adverse experiences 6 (1.0)§ 3 (2.5)§ 0 (0.0) With serious adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) With serious drug-related† adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 2 (0.3) 0 (0.0) 3 (0.5) Discontinued due to drug-related† adverse experiences 1 (0.2) 0 (0.0) 0 (0.0) Discontinued due to serious adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) Discontinued due to serious drug-related† adverse experiences 0 (0.0) 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related. ‡ One patient in the montelukast treatment group (AN 5654) discontinued after randomization due to an adverse
experience that began prior to randomization. § Notable between-group differences existed for montelukast versus placebo (1.0% difference with a 95%
confidence interval [0.1, 2.1]) and for cetirizine versus placebo (2.5% difference with a 95% confidence interval [0.7, 7.2]).
CONCLUSIONS: In adult patients with perennial allergic rhinitis: (1) Montelukast 10 mg, administered
once daily, demonstrates numeric improvement, compared with placebo, in the primary endpoint, Daytime Nasal Symptoms score, over 4 and 6 weeks of treatment. In some secondary endpoints, montelukast 10 mg demonstrates improvement compared with placebo. (2) Compared with placebo, cetirizine 10 mg, administered once daily, demonstrates improvement in the primary endpoint, Daytime Nasal Symptoms score, over 4 weeks and shows numeric improvement over 6 weeks. In some secondary endpoints, cetirizine 10 mg demonstrates improvement compared with placebo. (3) Montelukast 10 mg is generally well tolerated. Cetirizine 10 mg is also generally well tolerated.
AUTHORS: Joan McComas, M.S., M.M.H. Joris Menten, M.S. Debora Williams-Herman, M.D. Medical Program Clinical Specialist Biometrician Associate Director Respiratory & Allergy Clinical Biostatistics Respiratory & Allergy

CSR Synopsis_0476_254_P254 VERSION 2.0 APPROVED 16-Dec-2004 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast, 5-mg Chewable Tablet Indication
PROTOCOL TITLE/NO.: A Double-Blind, Double-Dummy, Randomized, Placebo-
Controlled, 2-Arm, 2x2 Crossover Study Comparing the Effects of Montelukast, Inhaled Budesonide, and Placebo on Lower Leg Growth in Children (Prepubertal, Tanner Stage I) With Mild Asthma
#254
INVESTIGATORS/STUDY CENTERS: Multicenter (2) study centers in Denmark PRIMARY THERAPY PERIOD: 01-Oct-2002 to 09-Jun-2004 CLINICAL PHASE: IV DURATION OF TREATMENT: Period I: One-week (7 days) placebo run in; Period II: 3-week
active treatment period; Period III: 3-week placebo washout period; Period IV: 3-week crossover treatment period.
OBJECTIVES: (1) To determine the lower leg length (LLL) growth rate for children treated with montelukast, compared with placebo, over the 3-week, double-blind treatment period. (2) To determine the LLL growth rate for children treated with budesonide, compared with placebo, over the 3-week, double-blind treatment period. (3) To evaluate the tolerability of montelukast when administered for up to 3 weeks in prepubertal children with mild asthma.
STUDY DESIGN: This was a 10-week, double-blind, double-dummy, randomized, placebo-controlled, 2 active-treatment period study with 2 crossover arms (and a total of 4 treatment sequences). Eligible patients were randomly allocated to 1 of the 2 crossover arms (montelukast/placebo or budesonide/placebo), and within each arm, to 1 of 2 treatment sequences (active treatment/placebo and placebo/active treatment).
PATIENT DISPOSITION: The patient disposition by treatment sequence:
MNT 5 mg/ Placebo/ BUD 400 µg/ Placebo/ Placebo MNT 5 mg Placebo BUD 400 µg Total
SCREENING FAILURES: 4 RANDOMIZED: 18 19 17 17 71
Male (age range) 9 (6 to 11) 13 (6 to 11) 9 (6 to 11) 11 (6 to 11) 42 Female (age range) 9 (6 to 10) 6 (6 to 10) 8 (6 to 10) 6 (6 to 10) 29
COMPLETED: 18 17 16 16 67 DISCONTINUED: 0 2 1 1 4
Clinical adverse experience 0 1 1 1 3 Other 0 1 0 0 1
______________________________________
MNT=montelukast; BUD=budesonide. DOSAGE/FORMULATION NOS.: Periods I and III: Placebo montelukast chewable tablets (MR-4389
and MR-4374), 1 tablet taken daily at bedtime or placebo budesonide inhaler (1 puff twice daily) (MR-4607, MR-4789, and MR-4945). Periods II and IV: Montelukast 5-mg chewable tablets (MR-4485 and MR-4863) or placebo montelukast chewable tablets (MR-4389 and MR-4374), 1 tablet taken daily at bedtime, or inhaled budesonide (MR-4599, MR-4788, and MR-4944) or placebo budesonide inhaler (MR-4607, MR-4789, and MR-4945), 1 puff taken twice daily.
DIAGNOSIS/INCLUSION CRITERIA: Prepubescent (Tanner Stage I) boys between the ages of 6 and 11 years and girls between the ages of 6 and 10 years who were within the 3rd and 97th percentile for height range and weight range for age at Visit 1 and had at least a 6-month history of asthma with typical symptoms (including, but not limited to cough, wheeze, and shortness of breath, with periodic episodes requiring treatment with inhaled β-agonists). Patients who had an asthma exacerbation that required medical treatment other than a maximum of 8 puffs of rescue short-acting β-agonist in 1 day during the 3 weeks prior to Visit 1 were excluded.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 254
MK-0476 Montelukast, 5-mg Chewable Tablet Indication
-2-
CSR Synopsis_0476_254_P254 VERSION 2.0 APPROVED 16-Dec-2004 Restricted Confidential – Limited Access
EVALUATION CRITERIA: Primary Endpoint: The growth rate of the LLL over the 3-week, double-
blind treatment period. Other Endpoints: Total Daily short-acting β-Agonist Use; Forced Expiratory Volume in 1 Second (FEV1); Morning and Evening Peak Expiratory Flow Rate (PEFR).
SAFETY: Clinical review of adverse experiences, vital signs, and laboratory safety parameters. STATISTICAL PLANNING AND ANALYSIS:
LLL GROWTH RATE: The LLL growth rate over the 3-week treatment period were calculated as the difference in LLL measurements taken at the end and at the beginning of the treatment period divided by the time between these measurements (in days). The LLL growth rate was converted to mm/week units as is the convention is knemometry studies. The growth rate of LLL was analyzed using an Analysis of Variance (ANOVA) model appropriate for a 2-period, crossover design applied to subjects in one crossover arm. The ANOVA model contained the factors: treatment sequence, subject within sequence, period, and treatment. The estimation part of the primary hypothesis was addressed by constructing a 95% confidence interval (CI) for the difference in LLL growth rate during montelukast and placebo treatments based on the model that applied to the subjects in the montelukast/placebo crossover arm. The study was designed to provide, with 26 patients per crossover arm completing the study, the expected half-length of the 95% CI for the difference in LLL growth rate during montelukast versus placebo treatment was 0.12 mm/week. The comparison of LLL growth rate during budesonide versus placebo treatment was carried out by a significance test that used the ANOVA model above as applied to the subjects in the budesonide/placebo crossover arm. The study had 90% power to detect a difference of 0.2 mm/week in LLL growth rate during budesonide versus placebo treatment.
EXPLORATORY ENDPOINTS: Total daily short-acting β-agonist used, FEV1 measurements, and morning and evening PEFR were summarized as exploratory endpoints.
SAFETY: Frequency of the clinical and laboratory adverse experiences (by category and body system) and the change from baseline in vital signs and laboratory safety parameters were summarized for each treatment received.
RESULTS: LLL GROWTH RATE: The primary analysis (modified intention-to-treat approach) resulted in similar
estimates of the treatment period growth rate for montelukast and placebo, while the growth rate for budesonide was significantly smaller than that seen with placebo (p=0.002). The results of the analysis of the per-protocol population (excluding patients or height data points following important protocol violations) were consistent with the results of the primary analysis.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 254
MK-0476 Montelukast, 5-mg Chewable Tablet Indication
-3-
CSR Synopsis_0476_254_P254 VERSION 2.0 APPROVED 16-Dec-2004 Restricted Confidential – Limited Access
Analysis of the Lower Leg Length Growth Rate in Montelukast/Placebo Arm Modified Intention-to-Treat
Lower Leg Length Growth Rate (mm/week) Treatment N Mean SD Median LS Mean 95% CI for LS Mean Montelukast 35 0.42 0.27 0.38 0.42 (0.33 , 0.50) Placebo 35 0.43 0.22 0.41 0.43 (0.35 , 0.52)
Estimate of Montelukast-Placebo Difference in LLL Growth Rate (mm/week) Montelukast – Placebo Difference in LS Means
95% CI for Difference in LS Means
-0.02 (-0.14 , 0.11)
Effect p-value Pooled SD Period 0.070 0.25 Sequence 0.557 CI=Confidence Interval, LLL=Lower Leg Length, LS Mean=Least Squares Mean, SD=Standard Deviation.
Analysis of the Lower Leg Length Growth Rate in Budesonide/Placebo Arm
Modified Intention-to-Treat
Lower Leg Length Growth Rate (mm/week) Treatment N Mean SD Median LS Mean 95% CI for LS Mean Budesonide 32 0.28 0.24 0.26 0.28 (0.22 , 0.35) Placebo 32 0.44 0.19 0.43 0.44 (0.37 , 0.51)
Estimate of Budesonide-Placebo Difference in LLL Growth Rate (mm/week) Budesonide - Placebo Difference in LS Means
95% CI for Difference in LS Means
p-value
-0.16 (-0.25 , -0.06) 0.002
Effect p-value Pooled SD Period 0.051 0.19 Sequence 0.272 CI=Confidence Interval, LLL=Lower Leg Length, LS Mean=Least Squares Mean, SD=Standard Deviation.
SAFETY: Of the 71 randomized patients, 21 patients had a total of 30 clinical adverse experiences; there
were no laboratory adverse experiences. There were no clinically relevant differences between the treatments in the incidence of adverse experiences. Three patients each had 1 adverse experience determined by the investigator to be drug related; 1 adverse experience (asthma) occurred while the patient was receiving budesonide and 2 adverse experiences (asthma and oral candidiasis) occurred while the patients were receiving placebo for budesonide. Both drug-related adverse experiences of asthma resulted in discontinuation of study therapy. One additional patient discontinued from therapy due to an adverse experience (asthma, determined not to be drug related) while receiving placebo for montelukast. There were no serious adverse experiences.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 254
MK-0476 Montelukast, 5-mg Chewable Tablet Indication
-4-
CSR Synopsis_0476_254_P254 VERSION 2.0 APPROVED 16-Dec-2004 Restricted Confidential – Limited Access
CONCLUSIONS: Over the 3-week treatment period: (1) The LLL growth rate observed with
montelukast treatment is similar to the LLL growth rate observed with placebo treatment. (2) The LLL growth rate observed with budesonide treatment is significantly below the LLL growth rate observed with placebo treatment. (3) Montelukast and budesonide are generally well tolerated.
AUTHORS: Sima Gaile, RN, BSN Nancy Liu, Ph.D. Assoc. Med. Prog. Clin. Spec. Senior Biometrician Respiratory, Allergy & Urology Biostatistics and Research Decision Sciences Barbara Knorr, M.D. Senior Director Respiratory, Allergy & Urology

CSR Synopsis_0476_256_P256 VERSION 1.1 APPROVED 15-Mar-2004 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast Sodium, Film-Coated Tablet Allergic Rhinitis
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized, Crossover Study Investigating the Clinical Effect of Montelukast in Patients With Concomitant Asthma and Allergic Rhinitis Upon Controlled Exposure to Cat Allergen
256
INVESTIGATORS/STUDY CENTERS: Multicenter (2)PRIMARY THERAPY PERIOD: 30-Apr-2002 to 28-Jan-2003. The
study was completed and the frozen file date was 03-Mar-2003. CLINICAL PHASE: III
DURATION OF TREATMENT: In Period I, 2 weeks of double-blinded study therapy. In Period II,1 week of washout period. In Period III, 2 weeks of double-blinded study therapy.
OBJECTIVES: (1) To assess the protective effect of montelukast 10 mg, compared with placebo, on the lower and upper airways of patients with concomitant asthma and allergic rhinitis upon exposure to cat allergen, as demonstrated by protection against a fall in forced expiratory volume in 1 second (FEV1)and against an increase in Nasal Symptoms score (mean of scores for Nasal Congestion, Rhinorrhea, and Sneezing); (2) To assess the protective effect of montelukast 10 mg, compared with placebo, on the lower airways of patients exposed to allergen, as demonstrated by the secondary endpoints including maximum fall from prechallenge FEV1, Chest Symptoms score (mean of scores for Wheezing and Chest Tightness), and the additional endpoints including each individual symptom of the Chest Symptoms score, the symptom score of Coughing, and the Number of Coughs; (3) To assess the protective effect of montelukast 10 mg, compared with placebo, on the upper airways of patients exposed to allergen, as demonstrated by the secondary endpoint of the Nasal Symptoms score collected postchallenge, and the additional endpoints including each individual symptom of the Nasal Symptoms score, the symptom score of Pruritus, and the Number of Sneezes; (4) To determine the safety and tolerability profile of montelukast in patients with concomitant asthma and allergic rhinitis.
STUDY DESIGN: Randomized, double-blind, placebo-controlled, crossover study with 2 treatment sequences. Study Periods: 2-week, double-blind treatment (Period I); 1-week washout (Period II); 2-week, double-blind treatment (Period III). Before randomization (Visit 2) and at the end of each treatment period (Visits 4 and 6), patients underwent allergen challenge (controlled exposure to cat allergen for ≤60 minutes).
PATIENT DISPOSITION: Montelukast 10 mg - Placebo
Placebo - Montelukast 10 mg Total
SCREENING FAILURES: --- --- 72 RANDOMIZED: 29 29 58
Male (age range) 12 (22 to 53) 6 (18 to 52) 18 (18 to 53) Female (age range) 17 (18 to 50) 23 (21 to 45) 40 (18 to 50)
COMPLETED: 25 27 52 DISCONTINUED: 4 2 6
Clinical adverse experience 1 0 1 Laboratory adverse experience 0 0 0 Other 3 2 5
DOSAGE/FORMULATION NOS.: All patients received montelukast 10 mg (MR-4533) or matching-image placebo (MR-4490) once daily upon awakening, irrespective of food intake during Periods I and III. No study therapy was administered during Period II.
DIAGNOSIS/INCLUSION CRITERIA: Patients, aged 15 to 55 years, who demonstrated skin test sensitivity to cat allergen and demonstrated both decreased pulmonary function (fall in FEV1 ≥15% from prechallenge) and increased symptoms of allergic rhinitis (Nasal Symptoms score increased ≥1.0 from prechallenge) upon controlled baseline (Visit 2) exposure to cat allergen for ≤60 minutes.
14

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 256
MK-0476 Montelukast Sodium Allergic Rhinitis
-2-
CSR Synopsis_0476_256_P256 VERSION 1.1 APPROVED 15-Mar-2004 Restricted Confidential – Limited Access
EVALUATION CRITERIA:EFFICACY: Co-primary efficacy endpoints: The area under the curve computed over the 60-minute
challenge (AUC0-60 min) for the percent fall from the absolute prechallenge FEV1 value and the AUC0-60 min for the increase from prechallenge Nasal Symptoms score (mean of scores for Nasal Congestion, Rhinorrhea, and Sneezing). Key secondary efficacy endpoints: The maximum percent fall from the prechallenge FEV1 value computed over the 60-minute challenge; the AUC0-60 min for the increase from prechallenge Chest Symptoms score (mean of scores for Wheezing and Chest Tightness), and the AUC computed over 30 minutes after terminating the challenge (AUC60-90 min) for the increase from prechallenge Nasal Symptoms score. Key additional efficacy endpoints: Lower airways: The absolute decrease in FEV1 from the prechallenge value to the end-of-challenge value; the AUC for the increase from prechallenge Chest Symptoms score (mean of scores for Wheezing and Chest Tightness) computed over 30 minutes after terminating the challenge. Upper airways: The AUC for the increase from prechallenge in each individual symptom of the Nasal Symptoms score (Nasal Congestion, Rhinorrhea, and Sneezing) and the AUC for the increase from prechallenge in Nasal Symptoms score computed over the 60-minute challenge and for the initial 30-minute period after terminating the challenge combined.
SAFETY: Clinical evaluations (including physical examination), adverse experience monitoring, and laboratory safety tests.
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: Primary analysis of study data was carried out using a modified intention-to-treat
approach including all patients with data in Periods I and III. For all endpoints except the Duration of Allergen Exposure, statistical analyses were performed using a mixed model with fixed effects of center, sequence, period, and treatment; and random effect of patient (nested within center and sequence). The between-treatment comparison of duration of allergen exposure was performed by the Wei, Lin, and Weissfeld method of regression for multivariate-censored data. With 50 patients total, the study had 90% power to detect a difference in AUC for the increase from prechallenge Nasal Symptoms score between the 2 treatments of 0.15 score•hr with an assumed within-patient standard deviation (SD) of 0.32 score•hr. The study had 98% power to detect a difference in AUC for the percent fall from the prechallenge FEV1 between the 2 treatments of 9 percentage-point•hr with an assumed within-patient SD of 15 percentage point•hr.
SAFETY: Ninety-five percent confidence interval (CI) limits for treatment group differences in percentages of patients having any adverse experience, having any adverse experience within a body system, and for any specific adverse experience were calculated. The CIs were computed on the basis of Wilson’s score interval used for paired differences.
RESULTS: EFFICACY: The main primary and secondary endpoint results are summarized below. Montelukast,
compared with placebo, demonstrated a significant protection against allergen challenge in the first co-primary endpoint and the first and third secondary endpoints.
15

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 256
MK-0476 Montelukast Sodium Allergic Rhinitis
-3-
CSR Synopsis_0476_256_P256 VERSION 1.1 APPROVED 15-Mar-2004 Restricted Confidential – Limited Access
Montelukast Versus Placebo Comparisons LS Mean
Difference 95% CI for Difference p-Value
Co-Primary Endpoints
1. AUC0-60 min-%Fall-FEV1 (Percentage•hr) -4.3 (-6.7, -1.9) ≤0.001 2. AUC0-60 min-Increase in Nasal Symptoms score (Score•hr) -0.11 (-0.25, 0.03) 0.115
Secondary Endpoints
1. Maximum %Fall-FEV1 (Percent) -7.2 (-10.9, -3.5) ≤0.001 2. AUC0-60 min-Increase in Chest Symptom score (Score•hr) -0.10 (-0.26, 0.06) 0.229 3. AUC60-90 min-Increase in Nasal Symptoms score (Score•hr) -0.07 (-0.15, -0.00) 0.044A negative LS Mean difference favors the montelukast group. LS Mean = least-squares mean; CI = confidence interval. AUC0-60 min = Area under the curve computed over the 60-minute challenge. AUC60-90 min = Area under the curve computed over the 30 minutes after terminating the challenge.
Additional: The benefit of montelukast on the lower airways was measured by the endpoints of AUC for the increase from prechallenge in Chest Symptoms score computed over 30 minutes after terminating the challenge and in the individual Chest Symptoms score. These scores were numerically larger than the placebo effect; none of the differences was significant. The benefit of montelukast on the upper airways was measured by the endpoint of AUC for the increase from prechallenge in the individual Nasal Symptoms. This benefit was numerically larger than the placebo effect; none of the differences was significant, except in nasal congestion; the benefit of montelukast on the AUC for the increase from prechallenge in Nasal Symptoms score over 90 minutes was borderline significant.
SAFETY: Clinical adverse experiences were reported in 15 (25.9%) of the 58 randomized patients. There were no clinically relevant differences between the treatments in the incidence of adverse experiences. No death occurred during the study. The following table shows a total of 56 patients because 4 patients each completed only one arm of the crossover.
16

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 256
MK-0476 Montelukast Sodium Allergic Rhinitis
-4-
CSR Synopsis_0476_256_P256 VERSION 1.1 APPROVED 15-Mar-2004 Restricted Confidential – Limited Access
Montelukast 10 mg Placebo (N=56) (N=56)
Clinical Adverse Experiences n (%) n (%) Number (%) of patients:
With one or more adverse experiences 8 (14.3) 9 (16.1) With no adverse experience 48 (85.7) 47 (83.9) With drug-related adverse experiences 0 (0.0) 0 (0.0) With serious adverse experiences 0 (0.0) 0 (0.0) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 1 (1.8) 0 (0.0) Discontinued due to drug-related adverse experiences
0 (0.0) 0 (0.0)
Discontinued due to serious adverse experiences
0 (0.0) 0 (0.0)
Discontinued due to serious drug-related adverse experiences
0 (0.0) 0 (0.0)
Montelukast 10 mg Placebo
(N=56) (N=56) Laboratory Adverse Experiences (AEs) n (%) n (%) Number (%) of patients:
With at least one laboratory test post baseline 30 26 With one or more adverse experiences 1 (3.3) 1 (3.8) With no adverse experience 29 (96.7) 25 (96.2) With drug-related adverse experiences 0 (0.0) 0 (0.0) With serious adverse experiences 0 (0.0) 0 (0.0) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 0 (0.0) 0 (0.0) Discontinued due to drug-related adverse
experiences 0 (0.0) 0 (0.0)
Discontinued due to serious adverse experiences
0 (0.0) 0 (0.0)
Discontinued due to serious drug-related adverse experiences
0 (0.0) 0 (0.0)
The percent = number of patients within the laboratory adverse experience category/number of patients with one or more laboratory tests post baseline. Determined by the investigator to be possibly, probably, or definitely drug related. Although a patient may have had 2 or more clinical adverse experiences, the patient is counted only once in a category. The same patient may appear in different categories.
17

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 256
MK-0476 Montelukast Sodium Allergic Rhinitis
-5-
CSR Synopsis_0476_256_P256 VERSION 1.1 APPROVED 15-Mar-2004 Restricted Confidential – Limited Access
CONCLUSIONS: In patients with concomitant asthma and allergic rhinitis on exposure to cat allergen:(1) Montelukast 10 mg administered once daily, does not demonstrates protection against allergen challenge, compared with placebo, in both co-primary endpoint, each measured during a 60-minute controlled exposure to cat allergen: montelukast shows protective effect on the lower-airway co-primary endpoint of Area Under Curve (AUC) for the percent fall from prechallenge FEV1 and shows a numeric improvement in the upper-airway co-primary endpoint of AUC for the increase from prechallenge Nasal Symptoms score. (2) Montelukast 10 mg administered once daily, demonstrates protection against allergen challenge, compared with placebo, in the lower-airway secondary endpoint of maximum fall from prechallenge FEV1, and shows a numeric improvement in the secondary endpoint of Chest Symptoms score, as measured during the 60-minute controlled exposure to cat allergen. (3) Montelukast 10 mg is generally well tolerated with an adverse experience profile similar to that observed for placebo.
AUTHORS: Eric H. Kim, MPH Rachid Massaad, M.S. Medical Program Clinical Specialist Senior Statistician Respiratory and Allergy Clinical Biostatistics
Marie-Pierre Malice, Ph.D. George Philip, M.D. Associate Director Director Clinical Biostatistics Respiratory and Allergy
18

CSR Synopsis_0476_265-00_P265 VERSION 2.0 APPROVED 03-Aug-2004 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast Sodium, Film-Coated Tablet Perennial Allergic Rhinitis
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized, Placebo-Controlled, Parallel-Group Study Investigating the Clinical Effects of Montelukast in Patients With Perennial Allergic Rhinitis
#265
INVESTIGATORS/STUDY CENTERS: Multicenter (117 study sites) in 12 countries including the United States, Canada and several countries in Europe.
PRIMARY THERAPY PERIOD: 27-Oct-2003 to 03-May-2004. The study is completed.
CLINICAL PHASE: III
DURATION OF TREATMENT: Period I: 5- to 7-day placebo period; Period II: 6-week treatment period.
OBJECTIVES: Primary: (1) To assess the treatment effect of montelukast 10 mg versus placebo on the primary endpoint of Daytime Nasal Symptoms score (average of scores for Congestion, Rhinorrhea, and Sneezing) over a 6-week treatment period, in patients with perennial allergic rhinitis (PAR). (2) To determine the tolerability profile of montelukast 10 mg in patients with PAR. Secondary: To assess the treatment effect of montelukast 10 mg versus placebo, over a 6-week treatment period in patients with PAR on the secondary endpoints of: (1) Global Evaluation of Allergic Rhinitis by Patient and (2) Overall Rhinoconjunctivitis Quality-of-Life score (average of scores for the 7 domains of Activity, Sleep, Non-Nose/Non-Eye Symptoms, Practical Problems, Nasal Symptoms, Eye Symptoms, and Emotions).
STUDY DESIGN: This was a multicenter, double-blind, randomized, 2-period, placebo-controlled, parallel-group study. Period I was a single-blind, placebo run-in period; in Period II, patients were treated either with montelukast 10 mg or placebo. The study was conducted under in-house blinding procedures in patients with (PAR).
PATIENT DISPOSITION: Montelukast 10 mg Placebo Total
SCREENING FAILURES: 1409 RANDOMIZED: 1002 990 1992
Male (age range) 358 (15 to 79) 358 (15 to 74) 716 (15 to 79) Female (age range) 644 (15 to 81) 632 (15 to 79) 1276 (15 to 81)
COMPLETED: 913 906 1819 DISCONTINUED: 89 84 173
Clinical adverse experience† 32 35 67 Laboratory adverse experience 0 0 0 Other 57 49 106
___________________________________________
† Includes 3 patients (montelukast—ANs 1308 and 1904; placebo—AN 2443) who discontinued after randomization due to a clinical adverse experience that began prior to randomization.
DOSAGE/FORMULATION NOS.: Study drug was administered once daily at bedtime. Period I:matching-image placebo tablet (formulation numbers: MR-4309). Period II: Montelukast (10 mg) film-coated tablet (MR-4847) or matching-image placebo tablet (MR-4309).
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women patients (aged 15 to 85 years), with at least a 2-year documented clinical history of PAR symptoms, a positive skin test (wheal 3 mm greater than saline control) to ≥2 of the relevant perennial allergens, and a minimum predefined level of Daytime Nasal Symptoms.
14

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 265
MK-0476 Montelukast Sodium, Film-Coated Tablet Perennial Allergic Rhinitis
-2-
CSR Synopsis_0476_265-00_P265 VERSION 2.0 APPROVED 03-Aug-2004 Restricted Confidential – Limited Access
EVALUATION CRITERIA:EFFICACY: Primary Endpoint: Daytime Nasal Symptoms score (average of Congestion, Rhinorrhea
and Sneezing). Secondary Endpoints: Global Evaluation of Allergic Rhinitis by Patient and Rhinoconjunctivitis Quality-of-Life score (average of Activity, Sleep, Non-Nose/Non-Eye Symptoms, Practical Problems, Nasal Symptoms, Eye Symptoms, and Emotions.). Other Endpoints: End-of-Day Nasal Symptoms score (average of Congestion, Rhinorrhea and Sneezing using instantaneous recall), Nighttime Symptoms score (average of Congestion Upon Awakening, Difficulty going to Sleep, and Nighttime Awakenings), Daily Rhinitis Symptoms score (average of Daytime Nasal Symptoms score and Nighttime Symptoms score), Daytime Nasal Symptoms + Itching score, End-of-Day Nasal Symptoms + Itching score, Individual Daytime Nasal Symptoms scores, Individual End-of-Day Nasal Symptoms scores (Congestion, Rhinorrhea, Sneezing, and Itching rated using instantaneous recall), Individual Nighttime Symptoms scores, and Individual domains of the Rhinoconjunctivitis Quality-of-Life Questionnaire.
SAFETY: Overall frequency of clinical adverse experiences and incidences of common adverse experiences reported by patient.
STATISTICAL PLANNING AND ANALYSIS: EFFICACY: The primary endpoint and all other diary-based endpoints were analyzed as the mean
change from baseline over the 6-week treatment period. Statistical tests were performed using an analysis of covariance model with factors for treatment group and study center, and the corresponding value at baseline as a covariate. Treatment comparisons were based on 2-sided F-tests from this model. For the primary endpoint, with 800 patients in each treatment group, this test had 90% power to detect a difference of -0.075 score between the treatment groups (α=0.05; SD=0.46). Endpoints based on the Rhinoconjunctivitis Quality-of-Life questionnaire were analyzed as the change from baseline after 6 weeks of study therapy using a similar model as for the primary endpoint. The Global Evaluation of Allergic Rhinitis by Patient was analyzed at Week 6 using an analysis of variance model with factors for treatment group and study center.
SAFETY: Ninety-five percent confidence limits were calculated for differences in overall and drug-related adverse experiences (as defined by MedDRA standard system organ class and broader terms), serious adverse experiences, and discontinuations from therapy due to adverse experiences.
RESULTS:EFFICACY: Main efficacy results are summarized below:
Daytime Nasal Symptoms Score + Individual Scores
Change From Baseline Montelukast 10 mg Placebo Difference in Treatments
Score N Mean (SD) N Mean (SD) LS Mean 95% CI Daytime Nasal Symptoms 1000 -0.42 (0.51) 980 -0.35 (0.48) -0.08 (-0.12; -0.04)***Congestion 1000 -0.38 (0.58) 980 -0.34 (0.53) -0.05 (-0.09; -0.00)* Rhinorrhea 1000 -0.44 (0.63) 980 -0.37 (0.60) -0.08 (-0.13; -0.03)***Sneezing 1000 -0.44 (0.63) 980 -0.33 (0.62) -0.10 (-0.15; -0.05)***Treatment Effect: * p≤0.050; *** p≤0.001.CI = Confidence Interval; LS Mean = Least-squares Means.
15

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 265
MK-0476 Montelukast Sodium, Film-Coated Tablet Perennial Allergic Rhinitis
-3-
CSR Synopsis_0476_265-00_P265 VERSION 2.0 APPROVED 03-Aug-2004 Restricted Confidential – Limited Access
Global Evaluation of Allergic Rhinitis by Patient
Montelukast Placebo Difference in Treatments Score N Mean (SD) N Mean (SD) LS Mean 95% CI
Global Evaluation Score 977 2.28 (1.29) 969 2.44 (1.29) -0.15 (-0.27; -0.04)** Treatment Effect: ** p≤0.010. CI = Confidence Interval; LS Mean = Least-squares Means.
Rhinoconjunctivitis Quality-of-Life Questionnaire
Change from Baseline Montelukast Placebo Difference in Treatments
Score N Mean (SD) N Mean (SD) LS Mean 95% CI Overall Score 977 -0.81 (1.14) 969 -0.68 (1.14) -0.15 (-0.24; -0.06)***Treatment Effect: *** p≤0.001. CI = Confidence Interval; LS Mean = Least-squares Means.
SAFETY: Clinical and other adverse experiences were reported by 557 (28.0%) of the 1992 randomized patients. Both treatment groups were similar in percentages of patients with adverse experiences. No deaths occurred during this study.
Montelukast Placebo (N=1002) (N=990) Clinical Adverse Experience Summary n (%) n (%) Number (%) of patients: With one or more adverse experiences 269 (26.8) 288 (29.1) With no adverse experience 733 (73.2) 702 (70.9) With drug-related adverse experiences† 37 (3.7) 44 (4.4) With serious adverse experiences 4 (0.4) 1 (0.1) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 30‡ (3.0) 34‡ (3.4) Discontinued due to drug-related adverse experiences 2 (0.2) 6 (0.6) Discontinued due to serious adverse experiences 1 (0.1) 1 (0.1) Discontinued due to serious drug-related adverse experiences 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related. ‡ Three patients (montelukast—ANs 1308 and 1904; placebo—AN 2443) discontinued after
randomization due to an adverse experience that began prior to randomization.
16

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 265
MK-0476 Montelukast Sodium, Film-Coated Tablet Perennial Allergic Rhinitis
-4-
CSR Synopsis_0476_265-00_P265 VERSION 2.0 APPROVED 03-Aug-2004 Restricted Confidential – Limited Access
CONCLUSIONS: In adult patients with PAR: (1) Montelukast 10 mg administered once daily over a 6-week treatment period, compared with placebo, demonstrates improvement in the primary endpoint, Daytime Nasal Symptoms score (average of scores for Congestion, Rhinorrhea, and Sneezing).(2) Montelukast 10 mg administered once daily over a 6-week treatment period, compared with placebo, demonstrates patient-perceived improvement of allergic rhinitis as assessed by the secondary endpoints of Global Evaluation of Allergic Rhinitis by Patient and Overall Rhinoconjunctivitis Quality-of-Life score. (3) Montelukast 10 mg administered once daily over a 6-week treatment period is generally well tolerated, with a safety profile comparable with that of placebo.
AUTHORS: Joan McComas, M.S., MMH Leen Gilles, M.Sc. George Philip, M.D. MPCS Senior Statistician Director Respiratory & Allergy CBARDS Respiratory & Allergy
17

CSR Synopsis_0476_269_P269 VERSION 1.1 APPROVED 23-Mar-2004 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast Sodium, 10-mg Film-Coated Tablet Seasonal Allergic Rhinitis and Asthma
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Randomized Study Investigating the Clinical Effect of Montelukast on Allergic Rhinitis in Patients With Seasonal Allergic Rhinitis and Chronic Asthma
#269
INVESTIGATORS/STUDY CENTERS: Multicenter (51 study sites in France, Italy, and the United States)
PRIMARY THERAPY PERIOD: 26-Mar to 20-Oct-2003. The frozen file was 24-Nov-2003. The study is completed.
CLINICAL PHASE: III
DURATION OF TREATMENT: Period I: 3- to 5-day, placebo period; Period II: 2-week treatment period.
OBJECTIVES: Primary: (1) To assess the treatment effect of montelukast 10 mg versus placebo on the primary endpoint of Daily Rhinitis Symptoms score, over a 2-week period, in patients with seasonal allergic rhinitis and chronic asthma. (2) To determine the safety and tolerability profile of montelukast in patients with seasonal allergic rhinitis and chronic asthma. Secondary: To assess the treatment effect of montelukast versus placebo on the secondary endpoints (Rhinoconjunctivitis Quality-of-Life Questionnaire overall score, Global Evaluation of Allergic Rhinitis by Patient, and Global Evaluation of Allergic Rhinitis by Physician), over a 2-week period, in patients with seasonal allergic rhinitis and chronic asthma. Other: To assess the treatment effect of montelukast versus placebo on the other endpoints, over a 2-week period, in patients with seasonal allergic rhinitis and chronic asthma.
STUDY DESIGN: This was a multicenter, double-blind, randomized, 2-period, placebo-controlled study. Period I was a single-blind, placebo run-in period; in Period II, patients were treated with either montelukast 10 mg or placebo. The study was conducted under in-house blinding procedures in patients with seasonal allergic rhinitis and chronic asthma.
PATIENT DISPOSITION: Montelukast
10 mg Placebo Total
RANDOMIZED: Total 415 416 831 Male (age range) 150 (15 to 78) 147 (15 to 69) 297 (15 to 78) Female (age range) 265 (15 to 73) 269 (15 to 80) 534 (15 to 80)
SCREENING FAILURES: -- -- 424 COMPLETED: 400 399 799 DISCONTINUED: Total 15 17 32
Clinical adverse experience 2 4 6 Lack of efficacy 3 3 6 Lost to follow-up 1 2 3 Patient withdrew consent 3 1 4 Protocol deviation 4 6 10 Patient discontinued for other 2 1 3
DOSAGE/FORMULATION NOS.: Study drug was administered orally once daily at bedtime. Period I: matching-image placebo tablet (formulation numbers: MR-4309 and MR-4490). Period II: Montelukast (10 mg) film-coated tablet (MR-4533) or matching-image placebo tablet (MR-4490).
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women patients (aged 15 to 85 years), with at least a 2-year documented clinical history of seasonal allergic rhinitis symptoms which exacerbate (flare up) during the study season, a positive skin test (wheal ≥3 mm greater than saline control) to ≥3allergens active during the study season, a minimal predefined level of Daily Rhinitis Symptoms score, and a documented clinical history for at least 1 year of chronic asthma (meeting at least 1 of 4 prespecified criteria for active asthma). Patients using a low and stable dose of inhaled corticosteroids were eligible.
17

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 269
MK-0476 Montelukast Sodium, 10-mg Film-Coated Tablet Seasonal Allergic Rhinitis and Asthma
-2-
CSR Synopsis_0476_269_P269 VERSION 1.1 APPROVED 23-Mar-2 Restricted Confidential – Limited Access
EVALUATION CRITERIA: EFFICACY: Primary Endpoint: Daily Rhinitis Symptoms score (average of the Daytime Nasal Symptoms
score [defined as the average of scores for Congestion, Rhinorrhea, Sneezing, and Itching] and Nighttime Symptoms score [defined as the average of scores for Nasal Congestion Upon Awakening, Difficulty Going to Sleep, and Nighttime Awakenings]). The Daytime Nasal Symptoms score and the Nighttime Symptoms score were evaluated separately as components of the primary endpoint. Secondary Endpoints: Rhinoconjunctivitis Quality-of-Life overall score and Global Evaluations of Allergic Rhinitis by Patient and by Physician. Other Endpoints: Individual components of the Daytime Nasal Symptoms score and Nighttime Symptoms score, the combined and individual components (Tearing, Itchy, Red, and Puffy Eyes) of the Daytime Eye Symptoms score, the Rhinoconjunctivitis Quality-of-Life Questionnaire individual domain scores and the Global Evaluations of Asthma by Patient and by Physician. Exploratory Endpoints: Average number of puffs per day of as-needed short acting β-agonist, and percentage of days without use of as-needed short-acting β-agonist.
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: Changes from baseline were analyzed for the diary-based and Rhinoconjunctivitis
Quality-of-Life endpoints. Data under treatment were analyzed for the Global Evaluation endpoints. Between-group comparisons were performed using an analysis of covariance (ANCOVA) model with treatment, study center, baseline use of inhaled corticosteroids, and season as factors; and (except for the Global Evaluation endpoints) the baseline value of the dependent variable as a covariate. Estimates of the treatment differences with 95% confidence intervals (CIs) were obtained from the same model using the least-squares means (LS means). The modified intention-to-treat study population was the primary population. The study was designed to have 380 patients per group complete the study, in order to have 90% power to detect a between-group difference in Daily Rhinitis Symptoms score equal to -0.11 (SD=0.46).
SAFETY: Ninety-five percent CIs were computed for treatment differences in overall and drug-related adverse experiences (by system organ class and for specific adverse experiences occurring for at least 2% of the patients in any treatment group), serious adverse experiences, and discontinuations from therapy due to adverse experiences.
RESULTS:EFFICACY: Over the 2-week treatment period, montelukast, compared with placebo, showed a significant
improvement for the primary endpoint, the Daily Rhinitis Symptoms score (p≤0.001), and for its 2 main components, the Daytime Nasal Symptoms score (p≤0.001) and the Nighttime Symptoms score (p≤0.001). Montelukast also showed a significant improvement on the 3 secondary endpoints: the overall score of Rhinoconjunctivitis Quality of Life (p=0.002), the Global Evaluation of Allergic Rhinitis by Patient (p≤0.001) and the Global Evaluation of Allergic Rhinitis by Physician (p≤0.001). Montelukast also had a significant effect compared with placebo on a majority of the other endpoints, and specifically on the Global Evaluation of Asthma by Patient (p=0.008) and on the Global Evaluation of Asthma by Physician (p=0.037). The subgroup of patients on a stable dose of inhaled corticosteroids at study entry showed a significant improvement (LS mean treatment effect and 95% CI: -0.16 [-0.26, -0.05]) in their daily rhinitis symptoms on montelukast, compared with placebo.
18

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 269
MK-0476 Montelukast Sodium, 10-mg Film-Coated Tablet Seasonal Allergic Rhinitis and Asthma
-3-
CSR Synopsis_0476_269_P269 VERSION 1.1 APPROVED 23-Mar-2 Restricted Confidential – Limited Access
Mean Change From Baseline Over the 2-Week Treatment Period, Treatment Differences and 95% CI With p-Values
(Modified Intention-to-Treat Approach)
Treatment Comparison Montelukast Placebo Montelukast versus Placebo N Mean N Mean LS Mean 95% CI p-Value
Primary Endpoint and 2 Main Components
Daily Rhinitis Symptoms 412 -0.35 413 -0.24 -0.12 (-0.18, -0.06) ≤0.001 Daytime Nasal Symptoms 412 -0.42 413 -0.29 -0.14 (-0.21, -0.07) ≤0.001 Nighttime Symptoms 412 -0.28 413 -0.20 -0.10 (-0.16, -0.04) ≤0.001
Secondary Endpoints
RQLQ Overall 403 -0.73 398 -0.55 -0.22 (-0.36, -0.08) 0.002 GE of AR by Patient† 412 2.39 406 2.77 -0.36 (-0.57, -0.15) ≤0.001 GE of AR by Physician† 413 2.41 411 2.76 -0.33 (-0.53, -0.14) ≤0.001
Other Endpoints (Selection)
Daytime Eye Symptoms 412 -0.29 413 -0.24 -0.08 (-0.15, -0.01) 0.024 GE of Asthma by Patient† 412 2.28 408 2.52 -0.24 (-0.41, -0.06) 0.008 GE of Asthma by Physician† 411 2.34 411 2.52 -0.17 (-0.33, -0.01) 0.037 † Results presented are data on treatment (no baseline data for GE endpoints). AR=Allergic Rhinitis; CI=Confidence interval; GE=Global Evaluation, LS Mean=Least-squares mean; RQLQ=Rhinoconjunctivitis Quality-of-Life.
SAFETY:
Montelukast 10 mg Placebo (N=415) (N=416)
Clinical Adverse Experience Summary† n (%) n (%) Number (%) of patients:
With one or more adverse experiences 49 (11.8) 54 (13.0) With no adverse experience 366 (88.2) 362 (87.0)
With drug-related adverse experiences‡ 5 (1.2) 13 (3.1) With serious adverse experiences 0 (0.0) 0 (0.0) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 2 (0.5) 4 (1.0) Discontinued due to drug-related adverse experiences 1 (0.2) 0 (0.0) Discontinued due to serious adverse experiences 0 (0.0) 0 (0.0) Discontinued due to serious drug-related adverse experiences 0 (0.0) 0 (0.0)
† There were no laboratory adverse experiences reported during this study. ‡ Determined by the investigator to be possibly, probably, or definitely drug related.
19

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 269
MK-0476 Montelukast Sodium, 10-mg Film-Coated Tablet Seasonal Allergic Rhinitis and Asthma
-4-
CSR Synopsis_0476_269_P269 VERSION 1.1 APPROVED 23-Mar-2 Restricted Confidential – Limited Access
CONCLUSIONS: In adult patients with seasonal allergic rhinitis and chronic asthma: (1) Montelukast 10 mg demonstrates improvement, compared to placebo, in the primary endpoint of Daily Rhinitis Symptoms score and in the 2 main components of the primary endpoint, the Daytime Nasal Symptoms score and the Nighttime Symptoms score. (2) Montelukast 10 mg demonstrates improvement, compared with placebo, in the 3 secondary endpoints: the overall score of Rhinoconjunctivitis Quality-of-Life, the Global Evaluation of Allergic Rhinitis by Patient and the Global Evaluation of Allergic Rhinitis by Physician. (3) Montelukast 10 mg demonstrates improvement, compared with placebo, in other endpoints, including the Global Evaluation of Asthma by Patient and the Global Evaluation of Asthma by Physician. (4) Montelukast 10 mg is generally well tolerated.
AUTHORS: Irena Dabrowski, M.A. France Vrijens, M.S. George Philip, M.D. Medical Program Clinical Specialist Senior Statistician Director Respiratory & Allergy CBARDS Respiratory & Allergy
20

CSR Synopsis_0476_270_P270 VERSION 1.1 APPROVED 16-Aug-2004 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast, 10-mg Film-Coated Tablet Exercise-Induced Bronchospasm
PROTOCOL TITLE/NO.: A Double-Blind, Randomized, Placebo-Controlled,
Multicenter, 2-Period, Crossover Study to Evaluate the Effects of a Single Dose of Montelukast on Exercise-Induced Bronchospasm
#270
INVESTIGATORS/STUDY CENTERS: Multicenter (9) PUBLICATION(S): PRIMARY THERAPY PERIOD: 10-Jul-2003 to 24-Nov-2003. The
frozen file date was 12-Dec-2003. The study is completed. CLINICAL PHASE: III
DURATION OF TREATMENT: Two periods, each of 2 days duration. A single witnessed dose of study medication (montelukast or matching image placebo) administered on the first day of each study period, with a minimum of 3 days between doses.
OBJECTIVES: (1) To determine the effect of montelukast on exercise-induced bronchospasm (EIB) as measured by the postexercise fall in FEV1 (change from preexercise baseline) throughout the 24 hours after a single oral dose. (2) To determine the effect of montelukast on the extent and severity of EIB as measured by the area under the percent change from baseline FEV1 time curve (AUC0-60 min), time to recovery of FEV1 to within 5% of baseline (time to recovery), and need for rescue medication following exercise challenges throughout the 24 hours after a single oral dose. (3) To determine the safety and tolerability of single doses of montelukast in patients with EIB.
STUDY DESIGN: This was a multicenter, double-blind, randomized, placebo-controlled, 2-period, crossover study.
PATIENT DISPOSITION: The patient disposition by treatment sequence:
Montelukast 10 mg Then Placebo
Placebo Then Montelukast 10 mg
Total
RANDOMIZED: 25 26 51
Male (age range) 14 (19 to 32) 10 (16 to 37) 24 (16 to 37) Female (age range) 11 (16 to 28) 16 (16 to 39) 27 (16 to 39)
COMPLETED: 24 25 49 DISCONTINUED: 1 1 2
Clinical adverse experience 1 1 2 DOSAGE/FORMULATION NOS.: Montelukast 10-mg film-coated tablet (FCT) (MR-4533) or
matching-image placebo (MR-4309), administered orally as a witnessed dose before exercise challenges in Periods I and II.
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women (using appropriate contraception) between and including the ages of 15 and 45 years of age, with stable asthma, with evidence of EIB (decrease in FEV1 after exercise of at least 20% compared with preexercise FEV1), and with preexercise FEV1 of ≥70% of the predicted value at both prestudy visits, while withholding short-acting β-agonists for ≥8 hours and long-acting β-agonists for ≥48 hours. Up to 25% of patients were allowed in the study if they had been using a low-dose inhaled corticosteroid (defined as beclomethasone dipropionate ≤400 µg/day, or equivalent1) at a stable dose for ≥4 weeks before the first visit.
EVALUATION CRITERIA: EFFICACY: Primary Endpoint: Maximum percent fall in FEV1 from preexercise baseline occurring
within the first 60 minutes after exercise challenge at 2 hours postdose. Other Endpoints: Maximum percent fall in FEV1 from preexercise baseline occurring within the first 60 minutes after exercise challenge at 12 and 24 hours postdose; area under the curve for FEV1 percent change from preexercise FEV1 over
1 Other equivalent regimens of inhaled corticosteroid therapy include: fluticasone ≤220 µg/day, budesonide
≤400 µg/day, triamcinolone ≤800 µg/day, flunisolide ≤800 µg/day.
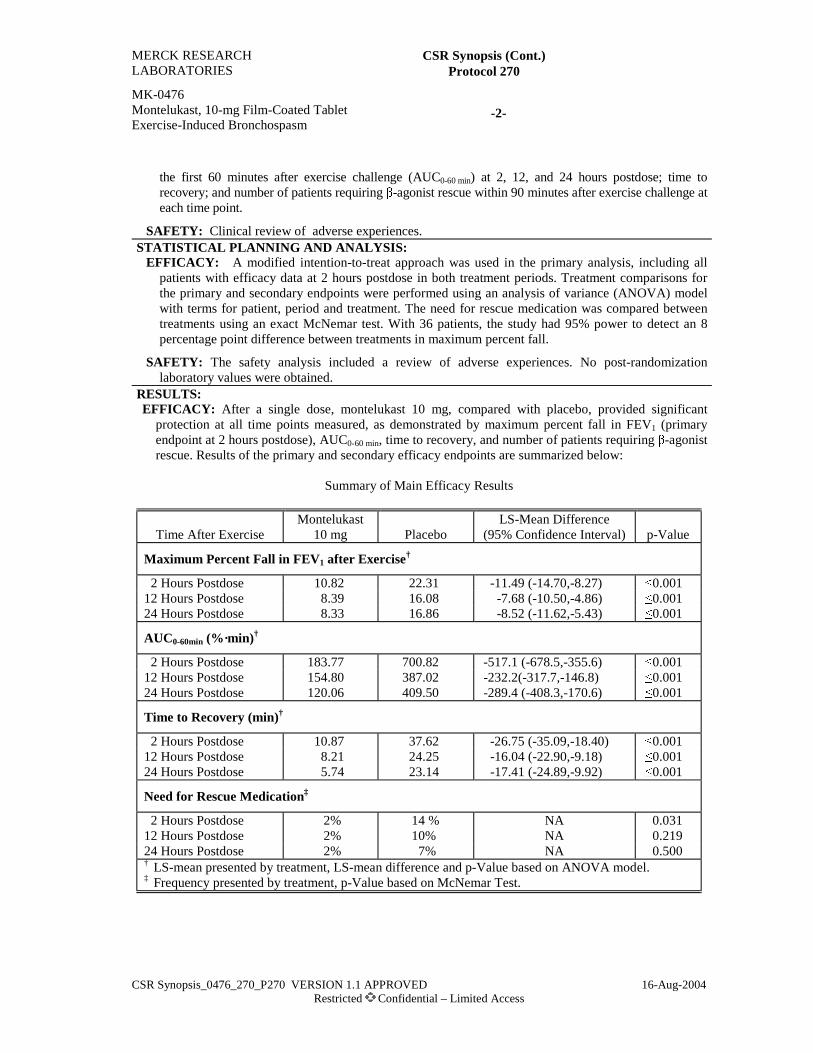
MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 270
MK-0476 Montelukast, 10-mg Film-Coated Tablet Exercise-Induced Bronchospasm
-2-
CSR Synopsis_0476_270_P270 VERSION 1.1 APPROVED 16-Aug-2004 Restricted Confidential – Limited Access
the first 60 minutes after exercise challenge (AUC0-60 min) at 2, 12, and 24 hours postdose; time to
recovery; and number of patients requiring β-agonist rescue within 90 minutes after exercise challenge at each time point.
SAFETY: Clinical review of adverse experiences. STATISTICAL PLANNING AND ANALYSIS:
EFFICACY: A modified intention-to-treat approach was used in the primary analysis, including all patients with efficacy data at 2 hours postdose in both treatment periods. Treatment comparisons for the primary and secondary endpoints were performed using an analysis of variance (ANOVA) model with terms for patient, period and treatment. The need for rescue medication was compared between treatments using an exact McNemar test. With 36 patients, the study had 95% power to detect an 8 percentage point difference between treatments in maximum percent fall.
SAFETY: The safety analysis included a review of adverse experiences. No post-randomization laboratory values were obtained.
RESULTS: EFFICACY: After a single dose, montelukast 10 mg, compared with placebo, provided significant
protection at all time points measured, as demonstrated by maximum percent fall in FEV1 (primary endpoint at 2 hours postdose), AUC0-60 min, time to recovery, and number of patients requiring β-agonist rescue. Results of the primary and secondary efficacy endpoints are summarized below:
Summary of Main Efficacy Results
Time After Exercise Montelukast
10 mg Placebo LS-Mean Difference
(95% Confidence Interval) p-Value
Maximum Percent Fall in FEV1 after Exercise†
2 Hours Postdose 10.82 22.31 -11.49 (-14.70,-8.27) ≤0.001 12 Hours Postdose 8.39 16.08 -7.68 (-10.50,-4.86) ≤0.001 24 Hours Postdose 8.33 16.86 -8.52 (-11.62,-5.43) ≤0.001
AUC0-60min (%·min)†
2 Hours Postdose 183.77 700.82 -517.1 (-678.5,-355.6) ≤0.001 12 Hours Postdose 154.80 387.02 -232.2(-317.7,-146.8) ≤0.001 24 Hours Postdose 120.06 409.50 -289.4 (-408.3,-170.6) ≤0.001
Time to Recovery (min)†
2 Hours Postdose 10.87 37.62 -26.75 (-35.09,-18.40) ≤0.001 12 Hours Postdose 8.21 24.25 -16.04 (-22.90,-9.18) ≤0.001 24 Hours Postdose 5.74 23.14 -17.41 (-24.89,-9.92) ≤0.001
Need for Rescue Medication‡
2 Hours Postdose 2% 14 % NA 0.031 12 Hours Postdose 2% 10% NA 0.219 24 Hours Postdose 2% 7% NA 0.500 † LS-mean presented by treatment, LS-mean difference and p-Value based on ANOVA model. ‡ Frequency presented by treatment, p-Value based on McNemar Test.

MERCK RESEARCH LABORATORIES
CSR Synopsis (Cont.) Protocol 270
MK-0476 Montelukast, 10-mg Film-Coated Tablet Exercise-Induced Bronchospasm
-3-
CSR Synopsis_0476_270_P270 VERSION 1.1 APPROVED 16-Aug-2004 Restricted Confidential – Limited Access
SAFETY: Clinical adverse experiences were reported in 9 of the 51 randomized patients. There were
no clinically relevant differences between the treatments in the incidence of adverse experiences. There were no post-randomization safety labs.
Montelukast
10 mg Placebo (N=50) (N=51)
Clinical Adverse Experience Summary n (%) n (%) Number (%) of patients:
With one or more adverse experiences 6 (12) 4 (8) With no adverse experience 44 (88) 47 (92) With drug-related adverse experiences† 2 (4) 0 (0) With serious adverse experiences 0 (0) 0 (0) With serious drug-related adverse experiences 0 (0) 0 (0) Who died 0 (0) 0 (0) Discontinued due to adverse experiences 2 (4) 0 (0) Discontinued due to drug-related adverse experiences
0 (0) 0 (0)
Discontinued due to serious adverse experiences
0 (0) 0 (0)
Discontinued due to serious drug-related adverse experiences
0 (0) 0 (0)
† Determined by the investigator to be possibly, probably, or definitely drug related. CONCLUSIONS: (1) Montelukast is superior to placebo in reducing the exercise-induced maximum
percent fall in FEV1 after exercise challenge 2 hours after a single oral dose in adult and adolescent patients ≥15 years old with mild-to-moderate asthma and EIB. (2) Montelukast is safe and well tolerated in patients with EIB. While there were no further specific hypotheses in this study, the results suggest the following additional conclusions: (3) Montelukast is superior to placebo in reducing the exercise-induced maximum percent fall in FEV1 after exercise challenge 12 and 24 hours after a single oral dose. (4) Montelukast is superior to placebo in reducing the area under the curve for FEV1 percent change within the first 60 minutes after exercise challenge at 2, 12, and 24 hours after a single oral dose. (5) Montelukast is superior to placebo in reducing the time to recovery from maximum percent fall in FEV1 after exercise challenges 2, 12, and 24 hours after a single oral dose.
AUTHORS: Otis Johnson, MPA Tom Loeys, Ph.D. Medical Program Clinical Specialist Biometrician Respiratory, Allergy & Urology BARDS, MSD (Europe), Inc. Barbara Knorr, M.D. Senior Director Respiratory, Allergy & Urology

MK-0476 Prot. No. 272 RSV Bronchiolitis Study
0476_P272_02_Synopsis VERSION 4.2 APPROVED 02-Mar-2007 Restricted Confidential – Limited Access
2. Synopsis
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules RSV Bronchiolitis
PROTOCOL TITLE/NO.: A Randomized, 2-Period, Multicenter, Double-Blind, Parallel-Group Study Comparing the Effects of 2 Doses of Montelukast Granules and Placebo in the Treatment of Respiratory Symptoms Associated With Respiratory Syncytial Virus-Induced Bronchiolitis in Children Aged 3 to 24 Months
#272
INVESTIGATORS/STUDY CENTERS: Multicenter (118) in Africa (8), Asia (13), Australia (3), Europe (40), North America (43), and South America (11).
PRIMARY THERAPY PERIOD: 30-Jul-2003 to 11-Oct-2006 CLINICAL PHASE: III DURATION OF TREATMENT: Period I—4 weeks (pivotal, dose ranging); Period II—20 weeks
(extended treatment). OBJECTIVES: (1) To demonstrate that montelukast, compared with placebo, results in dose-related
improvement of respiratory symptoms (as measured by percentage of symptom-free days1) over a 4-week treatment period in pediatric patients aged 3 to 24 months with RSV-induced bronchiolitis. (2) To demonstrate that montelukast, compared with placebo, results in dose-related improvement of respiratory symptoms as measured by the percentage of bronchiolitis-free days2 over a 4-week treatment period and the percentage of patients with exacerbations3 over a 24-week treatment period in pediatric patients aged 3 to 24 months with RSV-induced bronchiolitis. (3) To demonstrate that montelukast, compared with placebo, results in dose-related improvement of respiratory symptoms as measured by other endpoints over a 4-week treatment period and over a 24-week treatment period. (4) To compare the safety profile of montelukast with placebo over the 4-week treatment period in pediatric patients aged 3 to 24 months with RSV-induced bronchiolitis. (5) To explore the safety profile of montelukast over 24 weeks in patients aged 3 to 24 months with RSV-induced bronchiolitis.
STUDY DESIGN: This was a multicenter, double-blind, randomized, placebo-controlled, 2-period, parallel-group study conducted under in-house blinding procedures. After a Prestudy Visit (Visit 1), eligible patients were randomized, in a double-blind manner, to receive 1 of 3 treatments: 4-mg montelukast oral granules, 8-mg montelukast oral granules, or placebo once daily in the evening for 4 weeks (Period I). After completion of Period I, patients entered a 20-week, double-blind extended treatment period (Period II) and continued to receive the same treatment they received in Period I.
1 A day with no daytime cough, wheeze, and shortness of breath; and no nighttime cough. 2 A day with no daytime symptoms; no nighttime cough; no β-agonist use; no HRU (no systemic or
inhaled corticosteroid use, unscheduled visit to a doctor or emergency department, or hospitalization) for lung problems.
3 An exacerbation day is defined as a day with lung problems requiring systemic corticosteroid use, an unscheduled visit to the emergency department, or hospitalization.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules RSV Bronchiolitis
-2-
0476_P272_02_Synopsis VERSION 4.2 APPROVED 02-Mar-2007 Restricted Confidential – Limited Access
SUBJECT/PATIENT DISPOSITION:
Placebo Montelukast 4 mg Montelukast 8 mg Total SCREENING FAILURES: 203 RANDOMIZED (Total) 328 327 324 979 RANDOMIZED (No study drug
received 10 12 5 27
RANDOMIZED (Study drug
received): 318 315 319 952
Male (age range, months) 189 (2† to 23) 185 (3 to 22) 184 (3 to 23) 558 (2 to 23) Female (age range, months) 129 (3 to 23) 130 (3 to 23) 135 (2† to 23) 394 (2 to 23)
COMPLETED: 261 248 236 745 DISCONTINUED: 57 67 83 207
Clinical adverse experience 10 6 9 25 Laboratory adverse experience 1 2 5 8 Other 46 59 69 174‡
† Due to a database convention used to calculate age in months, 2 patients who were 90 days old at randomization are listed as 2 months old in the database; these patients are considered 3 months old in this clinical study report.
‡ Includes lack of efficacy (4), lost to follow-up (67), patient withdrew consent (49), protocol deviation (36), patient moved (6), and other (12).
DOSAGE/FORMULATION NOS.: Patients were randomized to receive 1 of 3 treatments: 4-mg montelukast oral granules, 8-mg montelukast oral granules, or placebo once daily in the evening, irrespective of food, for 24 weeks. Oral granules supplies were packaged in aluminum foil sachets; all sachets were packaged in 7-day supply blister cards. Each blister card contained 7 blister cavities (Days 1 through 7) containing 2 sachets each (Sachet A and Sachet B). Caregivers were to administer 2 sachets per day; the sachets were to be given sequentially (Sachet A followed by Sachet B) with no time interval between them. Each sachet was to be mixed with 1 tablespoon of applesauce or with 5 mL of formula and administered within 15 minutes; Sachet B was not to be opened until Sachet A had been administered.
Treatment Group
Sachet A
Sachet B Formulation Numbers for
Blister Cards Montelukast 4 mg Montelukast 4 mg Placebo MR-4897, MR-4924, MR-4927,
MR-4950, MR-5234 Montelukast 8 mg Montelukast 4 mg Montelukast 4 mg MR-4898, MR-4925, MR-4928,
MR-4951, MR-5235 Placebo Placebo Placebo MR-4899, MR-4926, MR-4929,
MR-4952, MR-5236 In addition to the above, montelukast sodium 4-mg oral granules (MR-4524) and placebo oral granules for montelukast sodium (MR-4290) were supplied as country reference samples that did not require configuration in a blister card.
DIAGNOSIS/INCLUSION CRITERIA: Males and females aged ≥3 months and ≤24 months at Visit 1
who were hospitalized (defined as a stay of at least 24 hours in a hospital or in an urgent/emergent-care setting) with his/her first or second episode of bronchiolitis, physician-diagnosed based on an acute lower respiratory tract infection with at least 2 of the following: respiratory rate >40 breaths per minute; cough; wheezing; audible rales, crackles, and/or rhonchi; and paradoxical chest movements (retractions).

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules RSV Bronchiolitis
-3-
0476_P272_02_Synopsis VERSION 4.2 APPROVED 02-Mar-2007 Restricted Confidential – Limited Access
Approximately 20% of patients were to be allowed in the study with a stay of <24 hours in a hospital or
in an urgent/emergent-care setting and a minimum predefined Respiratory Severity Score of 5 (based on the signs and symptoms listed above and oxygen saturation level). Patient had a positive test for RSV for the current episode of bronchiolitis. Patients who required intubation or mechanical ventilation prior to or during the current hospitalization or urgent/emergent-care stay were excluded as were patients who had more than one previous episode of bronchiolitis, other wheezing disorder, or asthma.
EVALUATION CRITERIA: EFFICACY: Primary endpoint: Percentage of symptom-free days over Period I (4-week treatment period). Secondary endpoints: Percentage of bronchiolitis-free days over Period I, percentage of patients with exacerbations in Periods I+II. Other endpoints evaluated over Period I, Periods I+II, and Period II: Percentage of cough-free days4, the percentage of patients with corticosteroid rescues, the percentage of patients with healthcare resource utilization (HRU)5, the percentage of patients without β-agonist use, the average value of daytime and nighttime symptoms, the number of exacerbation episodes, the time to resolution6 and the average of the wheeze and difficulty breathing symptom scores during the exacerbation7.
SAFETY: Overall frequency of clinical and laboratory adverse experiences (AEs), frequency of laboratory values outside predefined limits for selected laboratory safety tests, and vital signs.
STATISTICAL PLANNING AND ANALYSIS: EFFICACY: The percentage of symptom-free days, percentage of bronchiolitis-free days, percentage of cough-free days, percentage of days without β-agonist use, and average value of the daytime and nighttime symptom scores were analyzed using an analysis of variance (ANOVA) model with factors for treatment, region, and use of corticosteroids (Yes/No) prior to randomization for this episode of RSV-induced bronchiolitis. The montelukast doses (4 mg and 8 mg) were compared with placebo using a stepwise trend test. The percentage of patients with exacerbations, the percentage of patients with systemic corticosteroid rescue medications, and the percentage of patients with HRU were analyzed using a logistic regression model. The logistic regression model included treatment, region, and use of corticosteroids (Yes/No) prior to randomization as factors. The trend in response up to the 8-mg dose was tested using the contrast corresponding to the logarithmic scale.
SAFETY: The proportions of patients who: (1) experienced at least 1 AE; (2) experienced a drug-related AE; (3) experienced a serious AE (SAE); (4) discontinued treatment due to an AE, and (5) discontinued treatment due to a drug-related AE were compared across doses by use of the Cochran-Armitage trend test with the arithmetic and logarithmic scale dose scaling (smallest p-value was kept). For clinical and laboratory AEs with an incidence ≥2% in one or more treatment groups, the difference in proportion of patients with the AE in the active versus placebo groups were compared using 95% CIs based on Wilson's score method.
4 A day and night without cough 5 HRU = systemic or inhaled corticosteroid use, an unscheduled visit to a doctor or emergency
department, or hospitalization. 6 Resolution of bronchiolitis episode present at randomization : First day of 10 consecutive days with no
wheeze, no shortness of breath; no β-agonist use; no HRU for lung problems 7 Mean of the daily average symptom scores (for wheeze and difficulty breathing only) assessed 3 days
prior to, the day of, and 2 days post exacerbation.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules RSV Bronchiolitis
-4-
0476_P272_02_Synopsis VERSION 4.2 APPROVED 02-Mar-2007 Restricted Confidential – Limited Access
RESULTS:
EFFICACY: Primary endpoint — Percentage of Symptom-Free Days over Period I: The percentage of symptom-free days over Period I shows no significant trend from placebo through montelukast 8 mg (p=0.499). The percentage of symptom-free days over Period I is similar across treatment groups: the difference between montelukast 8 mg and placebo is equal to 1.6 [95% CI: -3.2, 6.5] and the difference between montelukast 4 mg and placebo is equal to 1.9 [95% CI: -2.9, 6.7].
Secondary endpoint — Percentage of Bronchiolitis-Free Days over Period I: The percentage of bronchiolitis-free days over Period I shows no significant trend from placebo through montelukast 8 mg (p=0.537). The percentage of bronchiolitis-free days over Period I is similar across treatment groups: the difference between montelukast 8 mg and placebo is equal to 1.4 [95% CI: -3.2, 6.1] and the difference between montelukast 4 mg and placebo is equal to 1.8 [95% CI: -2.8, 6.4].
Secondary endpoint — Percentage of Patients With Exacerbations over Periods I+II: The percentage of patients with exacerbations in Periods I+II shows no significant trend from placebo through montelukast 8 mg (p=0.565). The percentage of patients with exacerbations in Periods I+II is similar across treatment groups: 33% of the patients had at least 1 exacerbation in the placebo group, 31% in the montelukast 4 mg group and 32% in the montelukast 8 mg group.
Post-hoc analyses: Analyses were performed within the subgroup of patients in whom the percentage of symptom-free days was ≤30% during the first 2 weeks of the study. In this subgroup of 523 patients with persistent symptoms, the percentage of symptom-free days assessed over Week 3 to Week 4 is larger in the montelukast 8-mg group than in the placebo group (difference=7.3 [95% CI: 0.1, 14.4]) and in the montelukast 4-mg group than in the placebo group (difference=7.1 [95% CI: 0.1, 14.0]). When the percentage of symptom-free days is assessed over Week 3 to Week 24, the difference between montelukast 8 mg and placebo is equal to 5.9 [95% CI: 0.1, 11.7] and the difference between montelukast 4 mg and placebo is equal to 5.7 [95% CI: 0.0, 11.3].

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules RSV Bronchiolitis
-5-
0476_P272_02_Synopsis VERSION 4.2 APPROVED 02-Mar-2007 Restricted Confidential – Limited Access
Summary of Efficacy Results Assessed Over Period I
LS-Mean Difference (95% CI)
N Mean Montelukast
versus Placebo Montelukast 8 mg versus
Montelukast 4 mg p-Value†
Primary Endpoint — Percentage of Symptom-Free Days (p=0.499)‡
Placebo 302 37.0 --- --- --- Montelukast 4 mg 300 38.6 1.9 (-2.9, 6.7) --- 0.445 Montelukast 8 mg 297 38.5 1.6 (-3.2, 6.5) -0.2 (-5.1, 4.6) 0.506
Secondary Endpoint — Percentage of Bronchiolitis-Free Days (p=0.537)‡ Placebo 301 32.5 --- --- --- Montelukast 4 mg 301 33.8 1.8 (-2.8, 6.4) --- 0.446 Montelukast 8 mg 298 33.8 1.4 (-3.2, 6.1) -0.4 (-5.0, 4.3) 0.549
Percentage of Days Without β-Agonist Use (p=0.925)‡ Placebo 301 66.2 --- --- --- Montelukast 4 mg 300 63.6 -1.5 (-6.1, 3.1) --- 0.526 Montelukast 8 mg 295 66.2 -0.1 (-4.7, 4.6) 1.4 (-3.2, 6.1) 0.978
Percentage of Cough-Free Days (p=0.520)‡ Placebo 302 40.0 --- --- --- Montelukast 4 mg 300 40.8 1.1 (-3.7, 6.0) --- 0.646 Montelukast 8 mg 297 41.8 1.8 (-3.1, 6.7) 0.6 (-4.3, 5.5) 0.480
Average Symptom Score (p=0.343)‡ Placebo 302 0.63 --- --- --- Montelukast 4 mg 300 0.59 -0.04 (-0.12, 0.03) --- 0.262 Montelukast 8 mg 297 0.60 -0.03 (-0.11, 0.04) 0.01 (-0.07, 0.09) 0.376 † Comparison between placebo and montelukast dose. ‡ Adjusted Tukey's trend test from placebo through montelukast 8 mg. CI = Confidence interval.
Summary of Efficacy Results Assessed Over Periods I+II
Odds ratio (95% CI)
N Percent Montelukast
versus Placebo Montelukast 8 mg
versus Montelukast 4 mg p-Value†
Secondary Endpoint — Percentage of Patients With Exacerbations (p=0.565)‡
Placebo 318 33.3 --- --- --- Montelukast 4 mg 315 31.4 0.91 (0.65, 1.27) --- 0.562 Montelukast 8 mg 319 31.7 0.92 (0.65, 1.28) 1.01 (0.72, 1.42) 0.608
Percentage of Patients With Systemic Corticosteroid Use (p=0.814)‡ Placebo 318 19.2 --- --- --- Montelukast 4 mg 315 22.5 1.22 (0.83, 1.80) --- 0.316 Montelukast 8 mg 319 19.4 1.00 (0.67, 1.50) 0.82 (0.56, 1.21) 0.983
Percentage of Patients with Healthcare Resource Utilization (HRU) (p=0.427)‡ Placebo 318 54.1 --- --- --- Montelukast 4 mg 315 55.9 1.06 (0.77, 1.48) --- 0.708 Montelukast 8 mg 319 50.5 0.85 (0.61, 1.17) 0.80 (0.57, 1.10) 0.318 † Comparison between placebo and montelukast dose. ‡ Adjusted Tukey's trend test from placebo through montelukast 8 mg. HRU = systemic or inhaled corticosteroid use, unscheduled visit to a doctor or emergency department, or hospitalization. CI = Confidence interval.
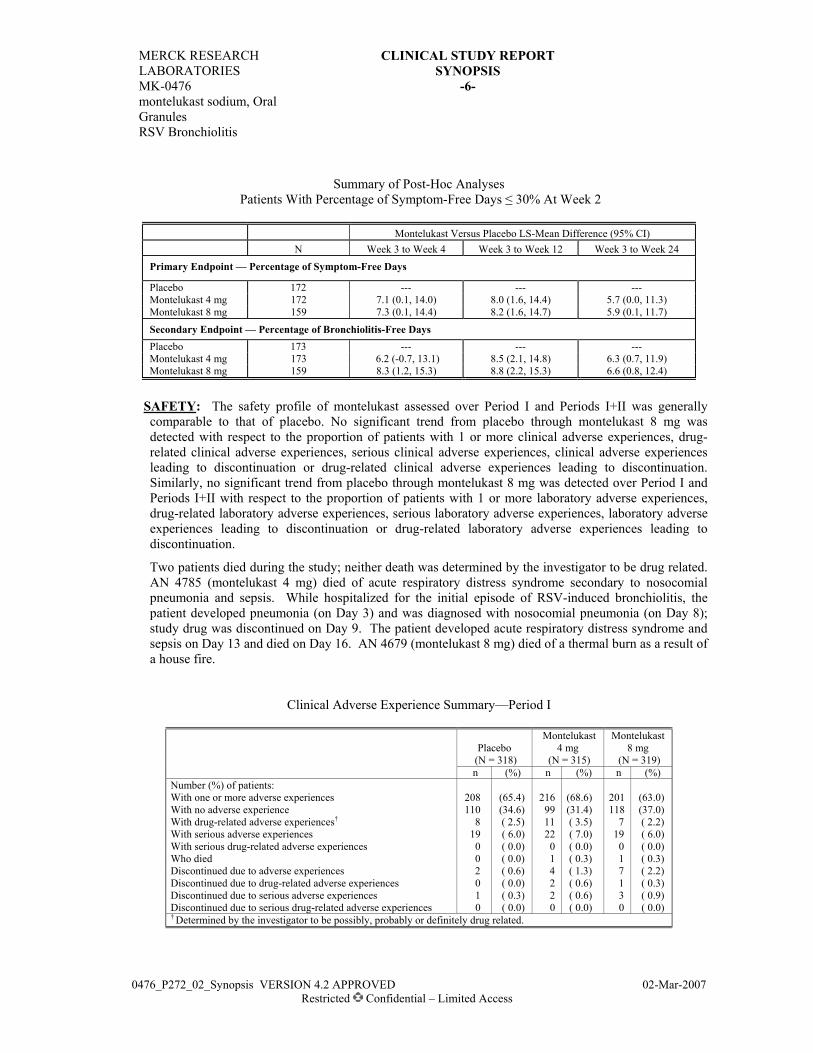
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules RSV Bronchiolitis
-6-
0476_P272_02_Synopsis VERSION 4.2 APPROVED 02-Mar-2007 Restricted Confidential – Limited Access
Summary of Post-Hoc Analyses
Patients With Percentage of Symptom-Free Days ≤ 30% At Week 2
Montelukast Versus Placebo LS-Mean Difference (95% CI) N Week 3 to Week 4 Week 3 to Week 12 Week 3 to Week 24
Primary Endpoint — Percentage of Symptom-Free Days
Placebo 172 --- --- --- Montelukast 4 mg 172 7.1 (0.1, 14.0) 8.0 (1.6, 14.4) 5.7 (0.0, 11.3) Montelukast 8 mg 159 7.3 (0.1, 14.4) 8.2 (1.6, 14.7) 5.9 (0.1, 11.7)
Secondary Endpoint — Percentage of Bronchiolitis-Free Days Placebo 173 --- --- --- Montelukast 4 mg 173 6.2 (-0.7, 13.1) 8.5 (2.1, 14.8) 6.3 (0.7, 11.9) Montelukast 8 mg 159 8.3 (1.2, 15.3) 8.8 (2.2, 15.3) 6.6 (0.8, 12.4)
SAFETY: The safety profile of montelukast assessed over Period I and Periods I+II was generally comparable to that of placebo. No significant trend from placebo through montelukast 8 mg was detected with respect to the proportion of patients with 1 or more clinical adverse experiences, drug-related clinical adverse experiences, serious clinical adverse experiences, clinical adverse experiences leading to discontinuation or drug-related clinical adverse experiences leading to discontinuation. Similarly, no significant trend from placebo through montelukast 8 mg was detected over Period I and Periods I+II with respect to the proportion of patients with 1 or more laboratory adverse experiences, drug-related laboratory adverse experiences, serious laboratory adverse experiences, laboratory adverse experiences leading to discontinuation or drug-related laboratory adverse experiences leading to discontinuation.
Two patients died during the study; neither death was determined by the investigator to be drug related. AN 4785 (montelukast 4 mg) died of acute respiratory distress syndrome secondary to nosocomial pneumonia and sepsis. While hospitalized for the initial episode of RSV-induced bronchiolitis, the patient developed pneumonia (on Day 3) and was diagnosed with nosocomial pneumonia (on Day 8); study drug was discontinued on Day 9. The patient developed acute respiratory distress syndrome and sepsis on Day 13 and died on Day 16. AN 4679 (montelukast 8 mg) died of a thermal burn as a result of a house fire.
Clinical Adverse Experience Summary—Period I
Placebo
Montelukast 4 mg
Montelukast 8 mg
(N = 318) (N = 315) (N = 319) n (%) n (%) n (%) Number (%) of patients: With one or more adverse experiences 208 (65.4) 216 (68.6) 201 (63.0) With no adverse experience 110 (34.6) 99 (31.4) 118 (37.0) With drug-related adverse experiences† 8 ( 2.5) 11 ( 3.5) 7 ( 2.2) With serious adverse experiences 19 ( 6.0) 22 ( 7.0) 19 ( 6.0) With serious drug-related adverse experiences 0 ( 0.0) 0 ( 0.0) 0 ( 0.0) Who died 0 ( 0.0) 1 ( 0.3) 1 ( 0.3) Discontinued due to adverse experiences 2 ( 0.6) 4 ( 1.3) 7 ( 2.2) Discontinued due to drug-related adverse experiences 0 ( 0.0) 2 ( 0.6) 1 ( 0.3) Discontinued due to serious adverse experiences 1 ( 0.3) 2 ( 0.6) 3 ( 0.9) Discontinued due to serious drug-related adverse experiences 0 ( 0.0) 0 ( 0.0) 0 ( 0.0) † Determined by the investigator to be possibly, probably or definitely drug related.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules RSV Bronchiolitis
-7-
0476_P272_02_Synopsis VERSION 4.2 APPROVED 02-Mar-2007 Restricted Confidential – Limited Access
Laboratory Adverse Experience Summary—Period I
Placebo
Montelukast 4 mg
Montelukast 8 mg
(N = 318) (N = 315) (N = 319) n (%)‡ n (%)‡ n (%)‡ With at least one lab test postbaseline 310 310 316 With one or more adverse experiences 12 (3.9) 15 (4.8) 13 (4.1) With no adverse experience 298 (96.1) 295 (95.2) 303 (95.9) With drug-related adverse experiences† 7 (2.3) 7 (2.3) 6 (1.9) With serious adverse experiences 1 (0.3) 0 (0.0) 0 (0.0) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 0 (0.0) 1 (0.3) 1 (0.3) Discontinued due to drug-related adverse experiences 0 (0.0) 1 (0.3) 0 (0.0) Discontinued due to serious adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) Discontinued due to serious drug-related adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) † Determined by the investigator to be possibly, probably or definitely drug related. ‡ The percent = number of patients within the laboratory adverse experience category / number of patients with one
or more laboratory tests postbaseline.
Clinical Adverse Experience Summary—Periods I+II
Placebo
Montelukast 4 mg
Montelukast 8 mg
(N = 318) (N = 315) (N = 319) n (%) n (%) n (%) Number (%) of patients: With one or more adverse experiences 259 (81.4) 265 (84.1) 262 (82.1) With no adverse experience 59 (18.6) 50 (15.9) 57 (17.9) With drug-related adverse experiences† 13 ( 4.1) 14 ( 4.4) 9 ( 2.8) With serious adverse experiences 51 (16.0) 41 (13.0) 53 (16.6) With serious drug-related adverse experiences 2 ( 0.6) 1 ( 0.3) 1 ( 0.3) Who died 0 ( 0.0) 1 ( 0.3) 1 ( 0.3) Discontinued due to adverse experiences 9 ( 2.8) 6 ( 1.9) 9 ( 2.8) Discontinued due to drug-related adverse experiences 2 ( 0.6) 3 ( 1.0) 2 ( 0.6) Discontinued due to serious adverse experiences 4 ( 1.3) 3 ( 1.0) 5 ( 1.6) Discontinued due to serious drug-related adverse experiences 1 ( 0.3) 1 ( 0.3) 1 ( 0.3) † Determined by the investigator to be possibly, probably or definitely drug related.
Laboratory Adverse Experience Summary—Periods I+II
Placebo Montelukast
4 mg Montelukast
8 mg (N = 318) (N = 315) (N = 319) n (%)‡ n (%)‡ n (%)‡ With at least one lab test postbaseline 310 310 316 With one or more adverse experiences 26 (8.4) 28 (9.0) 26 (8.2) With no adverse experience 284 (91.6) 282 (91.0) 290 (91.8) With drug-related adverse experiences† 14 (4.5) 16 (5.2) 14 (4.4) With serious adverse experiences 1 (0.3) 0 (0.0) 0 (0.0) With serious drug-related adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) 0 (0.0) Discontinued due to adverse experiences 1 (0.3) 2 (0.6) 4 (1.3) Discontinued due to drug-related adverse experiences 0 (0.0) 1 (0.3) 2 (0.6) Discontinued due to serious adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) Discontinued due to serious drug-related adverse experiences 0 (0.0) 0 (0.0) 0 (0.0) † Determined by the investigator to be possibly, probably or definitely drug related. ‡ The percent = number of patients within the laboratory adverse experience category / number of patients with one
or more laboratory tests postbaseline.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules RSV Bronchiolitis
-8-
0476_P272_02_Synopsis VERSION 4.2 APPROVED 02-Mar-2007 Restricted Confidential – Limited Access
CONCLUSIONS: In patients aged ≥3 to ≤24 months with RSV-induced bronchiolitis: (1) Montelukast,
compared with placebo, does not result in dose-related improvement of respiratory symptoms as measured by the primary endpoint of Percentage of Symptom-Free Days over Period I. (2) Montelukast, compared with placebo, does not result in dose-related improvement in respiratory symptoms as measured by secondary and other endpoints over Period I, Period II, and Periods I+II. (3) Montelukast is generally well tolerated at both dosage levels (4 mg and 8 mg).
AUTHORS: Gertrude Noonan, BA Sr. Clinical Research Specialist, Manager Clinical & Quantitative Sciences
Jean-Louis Marchal, Ph.D. Biometrician BARDS
Rachid Massaad, Ph.D.
Senior Statistician BARDS
Barbara Knorr, MD Executive Director, Clinical Research Respiratory & Allergy

CSR Synopsis_0476_275_P275 VERSION 3.0 APPROVED 16-Dec-2004 Restricted Confidential – Limited Access
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT I. SYNOPSIS
MK-0476 Montelukast, 10-mg Film-Coated Tablet Exercise-Induced Bronchospasm
PROTOCOL TITLE/NO.: A Double-Blind, Randomized, Placebo-Controlled,
Multicenter, 2-Period, Crossover Study to Evaluate the Effects of a Single Dose of Montelukast on Exercise-Induced Bronchospasm
#275
INVESTIGATORS/STUDY CENTERS: Multicenter, including the United States (7) and Peru (1) PRIMARY THERAPY PERIOD: 10-May-2004 to 04-Oct-2004.
The frozen file date was 13-Oct-2004. The study is completed. CLINICAL PHASE: III
DURATION OF TREATMENT: Two periods, each of 2 days duration. A single, witnessed dose of study medication (montelukast or matching-image placebo) administered on the first day of each study period, with a minimum of 3 days between doses.
OBJECTIVE(S): (1) To determine the effect of montelukast on EIB as measured by the maximal percent fall in FEV1 (change from pre-exercise baseline) throughout the 24 hours after a single oral dose. (2) To determine the effect of montelukast on the extent and severity of EIB as measured by the area under the curve for percent change from baseline FEV1 over time (AUC0-60 min), time to recovery of FEV1 to within 5% of baseline, and need for rescue medication following exercise challenges throughout the 24 hours after a single oral dose. (3) To determine the safety and tolerability of single doses of montelukast in patients with EIB.
STUDY DESIGN: This was a double-blind, randomized, placebo-controlled, multicenter, 2-period, crossover study.
SUBJECT/PATIENT DISPOSITION: Patient disposition by treatment sequence:
Montelukast 10 mg
Then Placebo Placebo Then
Montelukast 10 mg Total RANDOMIZED: 31 31 62
Male (age range) 17 (16-39) 16 (15-41) 33 (15-41) Female (age range) 14 (17-39) 15 (17-28) 29 (17-39)
COMPLETED: 25 (81%) 28 (90%) 53 (85%) DISCONTINUED: 6 (19%) 3 (10%) 9 (15%)
Clinical adverse experience 0 0 0 DOSAGE/FORMULATION NOS.: Montelukast 10-mg film-coated tablet (FCT) (MR-4847) or
matching-image placebo (MR-4490), administered orally as a witnessed dose before exercise challenges in Periods I and II.
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women (using appropriate contraception) between and including the ages of 15 and 45 years, with stable asthma, with evidence of EIB (decrease in FEV1 after exercise of at least 20% compared with pre-exercise FEV1), and with pre-exercise FEV1 of ≥70% of the predicted value at both prestudy visits, while withholding short-acting β-agonists for ≥8 hours and long-acting β-agonists for ≥48 hours. Up to 25% of patients were allowed in the study if they had been using low-dose inhaled corticosteroids (defined as beclomethasone dipropionate ≤400 µg/day, or equivalent) at a stable dose for ≥4 weeks before the first visit.
EVALUATION CRITERIA: Primary Endpoint: Maximum percent fall in FEV1 from pre-exercise baseline occurring within the first 60 minutes after exercise challenge at 2 hours postdose. Other Endpoints: Maximum percent fall in FEV1 from pre-exercise baseline occurring within the first 60 minutes after exercise challenge at 12 and 24 hours postdose; categorized (<10%, ≥10% to ≤20%, >20%) maximum percent fall in FEV1 at 2, 12 and 24 hours postdose, area under the curve for FEV1 percent change from pre-exercise FEV1 over the first 60 minutes after exercise challenge (AUC0-60 min) at 2, 12, and 24 hours postdose; time to recovery of FEV1 to within 5% of pre-exercise FEV1; and number of patients requiring β-agonist rescue medication within 90 minutes after exercise challenge at 2, 12, and 24 hours postdose.
SAFETY: Clinical review of adverse experiences.

MERCK RESEARCH LABORATORIES
CSR SYNOPSIS (CONT.) PROTOCOL 275
MK-0476 Montelukast, 10-mg Film-Coated Tablet Exercise-Induced Bronchospasm
-2-
CSR Synopsis_0476_275_P275 VERSION 3.0 APPROVED 16-Dec-2004 Restricted Confidential – Limited Access
STATISTICAL PLANNING AND ANALYSIS:
EFFICACY: A modified intention-to-treat approach was used in the primary analysis, including all patients with efficacy data at 2 hours postdose in both treatment periods. Treatment comparisons for the primary and secondary endpoints were performed using an analysis of variance (ANOVA) model with terms for patient, period and treatment. The categorized maximum percent fall in FEV1 was analyzed using Cochran’s Q test. The number of patients requiring β-agonist rescue medication was compared between treatments using an exact McNemar test. To adjust for multiplicity between the secondary variables and also between the secondary time points (12 and 24 hours postdose), a step down testing procedure was applied. With 36 patients, the study had 95% power to detect an 8 percentage point difference between treatments in maximum percent fall.
SAFETY: The safety analysis included a review of adverse experiences. No postrandomization laboratory values were obtained.
RESULTS: EFFICACY: After a single dose, montelukast 10 mg, compared with placebo, provided significant
protection at 2 hours postdose, as demonstrated by the maximum percent fall in FEV1 (primary endpoint at 2 hours postdose), categorized maximum percent fall in FEV1, AUC0-60 min, and time to recovery.
Summary of Main Efficacy Results
Time After Exercise Montelukast 10
mg Placebo LS-Mean Difference
(95% Confidence Interval) p-Value
Maximum Percent Fall in FEV1 after Exercise†
2 Hours Postdose 11.71 17.49 -5.78 (-8.85, -2.70) ≤0.001 12 Hours Postdose 9.88 11.72 -1.84 (-4.56, 0.88) 0.180 24 Hours Postdose 7.99 10.65 -2.66 (-5.20, -0.12) 0.041 ‡
Categorized Maximum Percent Fall §
Montelukast 10 mg Placebo <10% 10–20% >20% <10% 10–20% >20%
2 Hours Postdose 56% 28% 17% 39% 30% 31% 0.015 12 Hours Postdose 69% 19% 12% 48% 31% 21% 0.018 ‡ 24 Hours Postdose 67% 25% 8% 62% 21% 17% 0.082
AUC0-60min (%·min)†
2 Hours Postdose 283.2 584.2 -300.9 (-493.6,-108.3) 0.003 12 Hours Postdose 242.5 313.1 -70.6 (-218.6, 77.4) 0.343 24 Hours Postdose 224.3 268.4 -44.2 (-186.4, 98.1) 0.536
Time to Recovery (min)†
2 Hours Postdose 15.91 34.19 -18.29 (-27.17, -9.41) ≤0.001
12 Hours Postdose 9.40 23.43 -14.03 (-22.72, -5.34) 0.002 ‡ 24 Hours Postdose 9.47 13.81 -4.34 (-10.27, 1.59) 0.148
Patients Requiring β agonist Rescue Medication#
2 Hours Postdose 4% 13% --- 0.125 12 Hours Postdose 4% 8% --- 0.688 24 Hours Postdose 4% 4% --- 1.000 † LS-mean presented by treatment, LS-mean difference and p-Value based on ANOVA model. ‡ Significance of the effect cannot be assessed due to the step down multiplicity adjustment procedure. § Percentage of patients with maximum percent fall <10%, ≤10 to ≥20%, or >20%; p-Value based on Cochran’s Q. # Percentage of patients requiring β agonist rescue medication, presented by treatment, p-Value based on McNemar Test.

MERCK RESEARCH LABORATORIES
CSR SYNOPSIS (CONT.) PROTOCOL 275
MK-0476 Montelukast, 10-mg Film-Coated Tablet Exercise-Induced Bronchospasm
-3-
CSR Synopsis_0476_275_P275 VERSION 3.0 APPROVED 16-Dec-2004 Restricted Confidential – Limited Access
SAFETY: Clinical adverse experiences are reported by treatment received (montelukast or placebo)
regardless of study period. Four of the 58 patients who received montelukast had clinical adverse experiences while 5 of the 57 patients who received placebo had clinical adverse experiences; one patient (in the sequence Montelukast then Placebo) experienced adverse experiences in each treatment period, while a second patient (in the sequence Placebo then Montelukast) had 2 contemporaneous adverse experiences in each treatment period. There were no serious adverse experiences and no discontinuations due to adverse experiences. There were no clinically relevant differences between the treatment received and the incidence of adverse experiences. No postrandomization laboratory safety tests were performed.
CONCLUSIONS: (1) Montelukast is superior to placebo in reducing the exercise-induced maximum percent fall in FEV1 2 hours after a single oral dose in adult and adolescent patients ≥15 years old with mild-to-moderate asthma and EIB. (2) Montelukast is safe and well tolerated in patients with EIB.
AUTHORS:
Susan H. Cupo, M.S. Assoc. Medical Program Clinical Specialist Respiratory, Allergy & Urology Joan McComas M.S.; M.M.H. Medical Program Clinical Specialist Respiratory, Allergy & Urology
France Vrijens, M.S. Senior Statistician CBARDS George Philip, M.D. Director Respiratory, Allergy & Urology

MK-0476 Prot. No. 297 PK Study in 1- to 3-Month-Old Children
0476_P297_02_Synopsis VERSION 5.0 APPROVED 31-Jan-2007 Restricted Confidential – Limited Access
2. Synopsis
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 Montelukast sodium, Oral GranulesRespiratory Syncytial Virus-Induced Bronchiolitis
PROTOCOL TITLE/NO.: A Randomized, 3-Period, Multiple-Dose, Multicenter Study to Evaluate the Safety, Tolerability, and Plasma Concentration Profile of Montelukast Administered Once Daily as Oral Granules in Children Aged 1 to 3 Months
#297
INVESTIGATOR(S)/STUDY CENTER(S): Multicenter (2 sites in Chile, 1 site in Colombia, and 1 site in the United States).
PRIMARY THERAPY PERIOD: 01-Dec-2004 to 31-Oct-2005 CLINICAL PHASE: IIa DURATION OF TREATMENT: Total duration of treatment was 14 days: 1 day with montelukast 4 mg
(Period I), 6 days with montelukast 4 mg or placebo (Period II), and 7 days with montelukast 8 mg or placebo (Period III).
OBJECTIVES: Primary: To evaluate the safety and tolerability of montelukast oral granules (once-daily doses of 4 mg and 8 mg) in children aged 1 to 3 months. Secondary: To estimate the single-dose population pharmacokinetics (population area under the concentration-time curve [AUCpop], maximum plasma concentration [Cmax], time to Cmax [Tmax], and apparent elimination half-life [t½]) of montelukast 4-mg oral granules in children aged 1 to 3 months. Exploratory: To compare the pharmacokinetic parameters (AUCpop, Cmax, Tmax, and apparent elimination t½) of montelukast 4-mg oral granules in children aged 1 to 3 months with those from historical pediatric data in children aged 3 to 24 months (Protocols 136/138 and 268).
STUDY DESIGN: This was a randomized, 3-period, multiple-dose, multicenter study to evaluate the safety, tolerability, and plasma concentration-time profile of montelukast (oral granules formulation) in children aged 1 to 3 months with active viral-induced bronchiolitis or a history of viral-induced bronchiolitis and asthma-like symptoms. Period I—1-day, open-label treatment period to evaluate the population pharmacokinetics after administration of a single 4-mg dose of montelukast. All patients received montelukast 4 mg during this period and blood samples for pharmacokinetic analysis were obtained up to 24 hours after administration of the dose according to 1 of 2 possible fixed, 3 postdose timepoint plasma sampling schedules (Schedule 1 or 2). Period II—6-day, double-blind, placebo-controlled treatment period to evaluate the safety and tolerability after administration of multiple doses of montelukast (once-daily dose of 4 mg). Period III—7-day, double-blind, placebo-controlled treatment period to evaluate the safety and tolerability after administration of multiple doses of montelukast (once-daily dose of 8 mg).
PATIENT DISPOSITION: Montelukast† Placebo† Total SCREENING FAILURES: -- -- 8 RANDOMIZED: 5 7 12
Male (age range) 2 (2 months) 5 (1‡ to 2 months) 7 (1‡ to 2 months) Female (age range) 3 (1 to 2 months) 2 (1 to 2 months) 5 (1 to 2 months)
COMPLETED§: 5 6 11 DISCONTINUED: 0 1 1
Clinical adverse experience 0 0 0 Laboratory adverse experience 0 0 0 Other 0 1 1
____________________ † Patients were allocated to montelukast (Period II: 4 mg, Period III: 8 mg) or placebo after the open-label
pharmacokinetic period (Period I) during which all patients received a single dose of montelukast 4 mg. ‡ Due to a database convention used to calculate age in months, one patient (AN 1007) who was 31 days old at
randomization is listed as 0 months old in the database; this patient is considered 1 month old in this clinical study report.
§ All patients completed Period I. AN 1007 was discontinued from the study due to noncompliance with study drug during Period II.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules Respiratory Syncytial Virus-Induced Bronchiolitis
-2-
0476_P297_02_Synopsis VERSION 5.0 APPROVED 31-Jan-2007 Restricted Confidential – Limited Access
DOSAGE/FORMULATION NOS.: Period I—A single, 4-mg dose of montelukast oral granules
(MR-4910, MR-4911); Period II—montelukast 4-mg oral granules (MR-4910, MR-4911) or matching-image placebo (MR-4848); Period III—montelukast 8 mg (2 doses of 4-mg oral granules; MR-4910, MR-4911) or matching-image placebo (MR-4848).
DIAGNOSIS/INCLUSION CRITERIA: Boys and girls aged ≥1 month and <3 months old who were within the 5th and 95th percentile of height and weight for age (minimum weight, 3.5 kg) with active bronchiolitis or a history of bronchiolitis and asthma-like symptoms and who, on the basis of medical history and physical examination, were judged to be in otherwise good health were included for participation in the study. Patients must have been fed formula at least twice prior to their participation in Period I.
EVALUATION CRITERIA: PHARMACOKINETICS: Pharmacokinetic parameters of montelukast evaluated in this study were
determined by population pharmacokinetic analysis and included the following estimates: AUCpop, clearance after oral administration (CL/F, where F is oral bioavailability), Cmax, Tmax, and apparent elimination t½. All parameters were estimated using a nonlinear mixed-effects model. All parameter estimates were determined after administration of a single 4-mg dose of montelukast oral granules. As an exploratory comparison, the pharmacokinetic parameters after a single 4-mg oral granules dose in patients aged 1 to 3 month were compared with those for historical data in patients aged 3 to 24 months (Protocols 136/138 and 268).
SAFETY: Safety and tolerability were evaluated by summarizing adverse experiences, assessment of the clinical importance of laboratory safety tests that were out of the normal range, and review of vital sign measurements and physical examinations.
STATISTICAL PLANNING AND ANALYSIS: In this study, each patient was randomized to 1 of 2 possible fixed, 3 postdose timepoint plasma sampling schedules (Schedule 1 or 2) according to his/her allocation number. The pharmacokinetic analysis of the concentration-time data was performed using a population analysis approach. A 1-compartment model with first-order absorption and elimination was used to fit the concentration-time data, with the log clearance parameter and log elimination rate constant constraints assumed to be randomly distributed around a population mean. The AUCpop was computed based on the population means of the above parameters and was compared with those for patients aged 3 to 6 months (Protocol 268) and 3 to 24 months (Protocols 136/138 and 268). The 95% confidence interval (CI) for the AUCpop ratio (patients aged 1 to 3 months/3 to 6 months and patients aged 1 to 3 months/3 to 24 months) was estimated. Safety and tolerability were evaluated by tabulating adverse experiences and by clinical assessment of laboratory data.
RESULTS: PHARMACOKINETICS: The AUCpop estimates after administration of a single 4-mg oral granules
dose of montelukast to patients aged ≥1 to <3 months and after administration of a single 4-mg oral granules dose of montelukast to patients aged ≥3 to <24 months (historical data from Protocols 136/138 and 268) are in the following table. The AUCpop ratios (95% CI) were 3.62 (2.26, 5.80) and 3.64 (2.49, 5.30) for patients aged ≥1 to <3 months/≥3 to <6 months and patients aged ≥1 to <3 months/≥3 to <24 months, respectively.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Oral Granules Respiratory Syncytial Virus-Induced Bronchiolitis
-3-
0476_P297_02_Synopsis VERSION 5.0 APPROVED 31-Jan-2007 Restricted Confidential – Limited Access
Patient Population (Age Group) N AUCpop
(ng•hr/mL) Standard
Error Population Mean Ratio
95% CI† for Population Mean Ratio
≥1 to <3 months (Protocol 297) 12 13195.7 2309.8 ≥3 to <6 months (Protocol 268) 14 3644.3 481.5 3.62‡ (2.26, 5.80) ≥3 to <24 months (Protocols 136/138 and 268) 42 3629.2 243.1 3.64§ (2.49, 5.30) † Back-transformed from the log scale. ‡ Patients aged ≥1 to <3 months/≥3 to <6 months. § Patients aged ≥1 to <3 months/≥3 to <24 months. AUCpop = population area under the concentration-time curve; CI = confidence interval.
The population estimates (95% CI) for CL/F, Cmax, Tmax, and apparent elimination t½ of montelukast in patients aged ≥1 to <3 months and patients aged ≥3 to <24 months (historical data from Protocols 136/ 138 and 268) are in the following table.
Patient Population (Age Group) N
Population CL/F (mL/min) (95% CI)
Population Cmax (ng/mL)(95% CI)
Population Tmax (Hour)
(95% CI)
Population t½ (Hour)
(95% CI) ≥1 to <3 months (Protocol 297) 12 5.1 (2.9, 7.2) 1234.6 (754.1, 1715.1) 3.2 (1.7, 4.6) 1.2 (0.02, 3.3)≥3 to <6 months (Protocol 268) 14 18.3 (12.7, 23.9) 561.1 (381.3, 741.0) 2.1 (1.1, 3.1) 0.9 (0.0, 1.8) ≥3 to <24 months (Protocols
136/138 and 268) 42 18.4 (15.9, 20.9) 582.3 (486.2, 678.4) 1.9 (1.3, 2.5) 0.8 (0.3, 1.2)
CI = confidence interval; CL/F = clearance after oral administration, where F is oral bioavailability; Cmax = maximum plasma concentration; Tmax = time to Cmax; t½ = half-life.
SAFETY: There were no serious clinical or laboratory adverse experiences reported, and no patients
discontinued from the study due to an adverse experience.
CLINICAL: Six patients reported a total of 10 clinical adverse experiences; diarrhea was the only adverse experience reported more than once. None of the adverse experiences was determined by the investigator to be drug related and all patients recovered.
LABORATORY: Three patients experienced elevated serum aspartate aminotransferase (AST) levels, all determined to be drug related by the investigator. Two of the patients had received the 8-mg dose of montelukast oral granules and 1 patient had received placebo at the time of the adverse experiences.
CONCLUSIONS: (1) Multiple doses of 4 mg and 8 mg of montelukast oral granules are generally well tolerated in patients aged ≥1 to <3 months. (2) The plasma concentration-time profile of montelukast in patients aged ≥1 to <3 months is greater than that of patients aged ≥3 to <24 months after a single 4-mg oral granules dose as measured by population pharmacokinetic estimates (approximately 3.6-fold higher for AUC and approximately 2.1-fold higher for Cmax).
AUTHORS: Elizabeth Beckford, B.S.
Med. Prog. Clinical Specialist Respiratory, Allergy & Urologic Diseases
Lata Maganti, Ph.D. Biometrician Biostatistics and Research Decision Sciences
Susan Lu, Pharm.D. Clinical Associate Respiratory, Allergy & Urologic Diseases
Susie Li, Ph.D. Senior Research Pharmacokineticist Clinical Drug Metabolism

MK-0476 Prot. No. 316 Montelukast for Exercise-Induced Bronchoconstriction
0476_P316_02_Synopsis VERSION 2.0 APPROVED 27-Sep-2006 Restricted Confidential – Limited Access
2. Synopsis
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Film-Coated Tablet Exercise-Induced Bronchoconstriction
PROTOCOL TITLE/NO.: A Double-Blind, Randomized, Multicenter, 3-Period, Crossover Study to Evaluate the Effects of a Single Dose of Montelukast Compared With Placebo and Salmeterol on Exercise-Induced Bronchoconstriction
#316
INVESTIGATORS/STUDY CENTERS: Multicenter, including the United States (4) and Peru (1) PRIMARY THERAPY PERIOD: 13-Dec-2005 to 24-Jul-2006 CLINICAL PHASE: III DURATION OF TREATMENT: Period I: this period included 2 visits: during the first visit, patients
received no medication; during the second visit, patients received a single administration of oral and inhaled placebo medications in a double-dummy single-blind manner. Periods II, III, and IV: during each period, there was a single administration of oral and inhaled medications in a double-dummy double-blind manner.
OBJECTIVES: (1) To determine the effect of a single oral dose of montelukast, compared with placebo, on exercise-induced bronchoconstriction (EIB) as measured by the maximum percent-fall in FEV1 (post-exercise change from pre-exercise baseline) after exercise challenge. (2) To determine the effect of a single oral dose of montelukast, compared with placebo, on the categorized maximum percent-fall in FEV1, the time to recovery of FEV1 to within 5% of pre-exercise baseline, the area under the curve for percent-fall from pre-exercise FEV1 over time (AUC0-60min), and the need for rescue medication following challenge. (3) To estimate the effect of a single oral dose of montelukast, compared with a single inhaled dose of salmeterol, on EIB as measured by the maximum percent-fall in FEV1. (4) To determine the safety and tolerability of single doses of montelukast in patients with EIB.
STUDY DESIGN: This was a double-blind, randomized, 3-period crossover study. SUBJECT/PATIENT DISPOSITION:
SCREENING FAILURES (Total): 113 RANDOMIZED: 47
Male (age range in years) 23 (15 to 38) Female (age range in years) 24 (15 to 44)
COMPLETED: 46 DISCONTINUED: 1
Clinical adverse experience 1†
_________________________________________
† AN 1018 discontinued during the second washout period (montelukast-placebo-salmeterol sequence) due to hypersensitivity (increased allergies) (see Section 12.2.5).
DOSAGE/FORMULATION NOS.: Period I—matching-image placebo for montelukast 10-mg FCT (FL00001390 [MR-5247]) and the matching-image placebo for salmeterol 50-µg inhalation powder (FL00001370); Periods II, III and IV: montelukast 10-mg FCT (FL00000908 [MR-5252]) or matching-image placebo (FL00001390 [MR-5247]) AND inhaled salmeterol 50-µg inhalation powder (FL00001645) or matching-image placebo (FL00001370).
DIAGNOSIS/INCLUSION CRITERIA: Male and female patients, 15 to 45 years of age, with mild-to-moderate asthma and with pre-exercise FEV1 ≥70% of predicted. Patients must have demonstrated EIB with maximum percent-fall in post-exercise FEV1 of ≥20% relative to the FEV1 (in liters [L]) measured immediately before exercise.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Film-Coated Tablet Exercise-Induced Bronchoconstriction
-2-
0476_P316_02_Synopsis VERSION 2.0 APPROVED 27-Sep-2006 Restricted Confidential – Limited Access
EVALUATION CRITERIA: Spirometry measurements 5 minutes prior to dose, 5 minutes prior to
exercise challenge, and immediately, 5, 10, 15, 30, 45 and 60 minutes after exercise challenge. Exercise challenges were performed at 2, 8.5 and 24 hours postdose. Primary endpoint: Maximum percent-fall in FEV1, from pre-exercise baseline, occurring within the first 60 minutes after exercise challenge. Pre-exercise baseline FEV1 is the value (in liters) measured 5 minutes before the exercise challenge. Other endpoints: Categorized maximum percent-fall in FEV1, area under the curve for percent-fall in FEV1 from pre-exercise baseline over the first hour after exercise challenge (AUC0-60min); time to recovery from maximum percent-fall in FEV1 to within 5% of pre-exercise baseline; the proportion of patients needing short-acting β-agonist rescue within the 90 minutes after exercise challenge.
SAFETY MEASUREMENTS: Physical examinations, vital signs, blood and urine for laboratory safety measurements, and electrocardiograms were performed before allocation. No laboratory safety examinations or electrocardiograms were to be performed after the first prestudy visit, except as requested by the investigator or clinical monitor to clarify an adverse experience.
STATISTICAL PLANNING AND ANALYSIS: The analysis of the study data was carried out using a modified intention-to-treat approach. The maximum percent-fall in FEV1 after exercise, AUC0-60min, and time to recovery were analyzed at each time point separately using a crossover ANOVA model with factors for patient, treatment, and period effects. The distribution of categorized maximum percent-fall in FEV1 was compared between montelukast and placebo using Cochran’s Q. The proportions of patients who needed rescue with short-acting β-agonist within the 90 minutes following exercise were compared between montelukast and placebo using McNemar’s test. For the primary endpoint of maximum percent-fall in FEV1, the study had 95% power with a sample size of 36 patients to detect a treatment difference of 8 percentage points between montelukast and placebo (with α=0.050, two-sided test, and assumed within-patient standard deviation of 9 percentage points).
RESULTS: EFFICACY: After a single dose, montelukast 10 mg, compared with placebo, provided significant
protection at 2 hours postdose, as demonstrated by the maximum percent-fall in FEV1 (primary endpoint), categorized maximum percent-fall in FEV1, time to recovery, AUC0-60min, and need for rescue medication. This protection was sustained up to 24 hours postdose, as demonstrated by the effect of montelukast 10 mg over placebo on maximum percent-fall in FEV1, time to recovery, and AUC0-60min at 8.5 and 24 hours postdose.
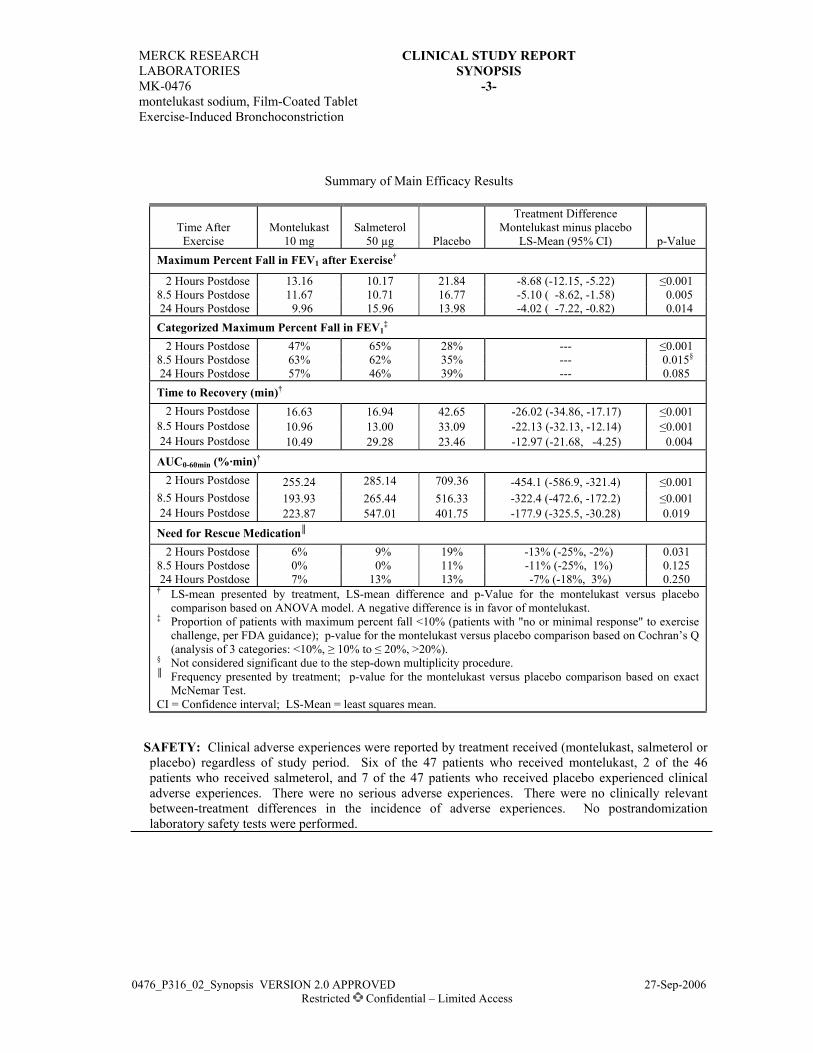
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Film-Coated Tablet Exercise-Induced Bronchoconstriction
-3-
0476_P316_02_Synopsis VERSION 2.0 APPROVED 27-Sep-2006 Restricted Confidential – Limited Access
Summary of Main Efficacy Results
Time After Exercise
Montelukast 10 mg
Salmeterol 50 µg Placebo
Treatment Difference Montelukast minus placebo
LS-Mean (95% CI) p-Value Maximum Percent Fall in FEV1 after Exercise†
2 Hours Postdose 13.16 10.17 21.84 -8.68 (-12.15, -5.22) ≤0.001 8.5 Hours Postdose 11.67 10.71 16.77 -5.10 ( -8.62, -1.58) 0.005 24 Hours Postdose 9.96 15.96 13.98 -4.02 ( -7.22, -0.82) 0.014 Categorized Maximum Percent Fall in FEV1
‡ 2 Hours Postdose 47% 65% 28% --- ≤0.001 8.5 Hours Postdose 63% 62% 35% --- 0.015§ 24 Hours Postdose 57% 46% 39% --- 0.085 Time to Recovery (min)† 2 Hours Postdose 16.63 16.94 42.65 -26.02 (-34.86, -17.17) ≤0.001 8.5 Hours Postdose 10.96 13.00 33.09 -22.13 (-32.13, -12.14) ≤0.001 24 Hours Postdose 10.49 29.28 23.46 -12.97 (-21.68, -4.25) 0.004
AUC0-60min (%·min)† 2 Hours Postdose 255.24 285.14 709.36 -454.1 (-586.9, -321.4) ≤0.001 8.5 Hours Postdose 193.93 265.44 516.33 -322.4 (-472.6, -172.2) ≤0.001 24 Hours Postdose 223.87 547.01 401.75 -177.9 (-325.5, -30.28) 0.019
Need for Rescue Medication║ 2 Hours Postdose 6% 9% 19% -13% (-25%, -2%) 0.031 8.5 Hours Postdose 0% 0% 11% -11% (-25%, 1%) 0.125 24 Hours Postdose 7% 13% 13% -7% (-18%, 3%) 0.250 † LS-mean presented by treatment, LS-mean difference and p-Value for the montelukast versus placebo
comparison based on ANOVA model. A negative difference is in favor of montelukast. ‡ Proportion of patients with maximum percent fall <10% (patients with "no or minimal response" to exercise
challenge, per FDA guidance); p-value for the montelukast versus placebo comparison based on Cochran’s Q (analysis of 3 categories: <10%, ≥ 10% to ≤ 20%, >20%).
§ Not considered significant due to the step-down multiplicity procedure. ║ Frequency presented by treatment; p-value for the montelukast versus placebo comparison based on exact
McNemar Test. CI = Confidence interval; LS-Mean = least squares mean.
SAFETY: Clinical adverse experiences were reported by treatment received (montelukast, salmeterol or placebo) regardless of study period. Six of the 47 patients who received montelukast, 2 of the 46 patients who received salmeterol, and 7 of the 47 patients who received placebo experienced clinical adverse experiences. There were no serious adverse experiences. There were no clinically relevant between-treatment differences in the incidence of adverse experiences. No postrandomization laboratory safety tests were performed.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, Film-Coated Tablet Exercise-Induced Bronchoconstriction
-4-
0476_P316_02_Synopsis VERSION 2.0 APPROVED 27-Sep-2006 Restricted Confidential – Limited Access
CONCLUSIONS: (1) Montelukast is superior to placebo in reducing the exercise-induced maximum
percent-fall in FEV1 2 hours after a single oral dose in adult and adolescent patients 15 to 45 years of age, with mild-to-moderate asthma and EIB. (2) Montelukast demonstrates an onset of action for acute prevention of EIB as early as 2 hours postdose, and a duration of action confirmed – through multiple time points – up to 24 hours postdose. (3) Using the inhaled long-acting β-agonist salmeterol as a benchmark therapy, montelukast demonstrates efficacy for acute prevention of EIB that is clinically meaningful. (4) Montelukast is well tolerated in patients with mild-to-moderate asthma and EIB.
AUTHORS: Joan McComas, M.S., M.M.H. Catherine Legrand George Philip, M.D. Clinical Research Specialist Biometrician Senior Director Respiratory, Allergy & Urology CBARDS Respiratory, Allergy & Urology
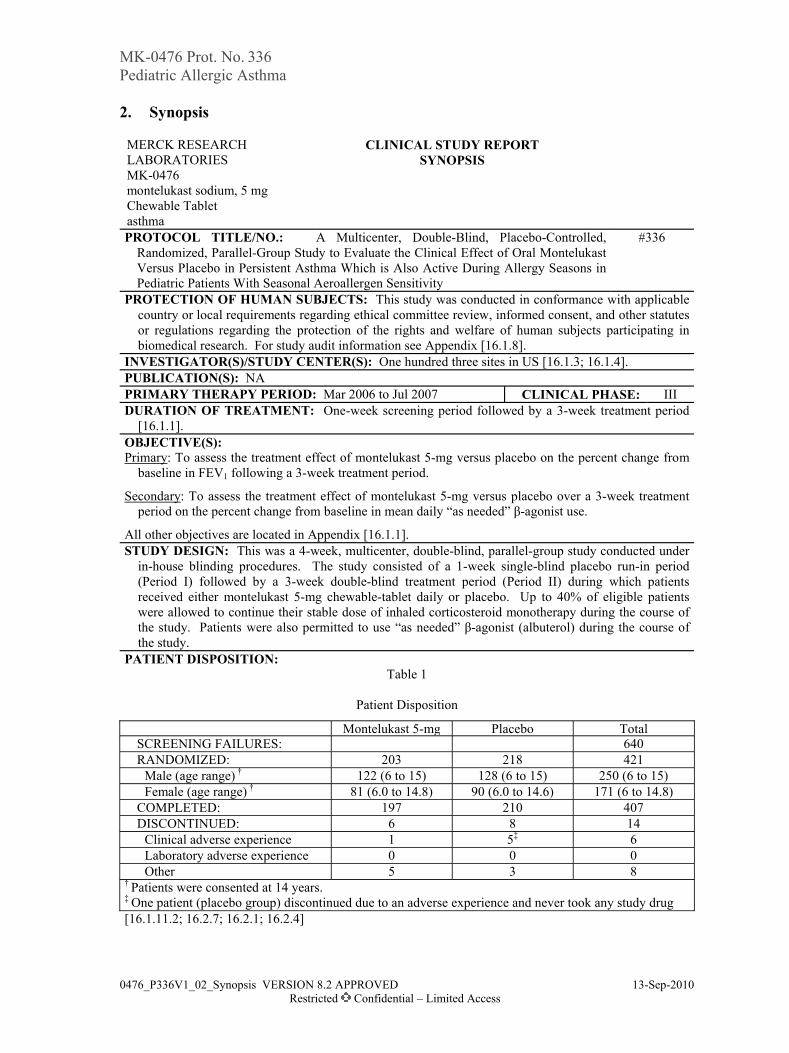
MK-0476 Prot. No. 336Pediatric Allergic Asthma
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
2. Synopsis
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable TabletasthmaPROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Placebo-Controlled,
Randomized, Parallel-Group Study to Evaluate the Clinical Effect of Oral Montelukast Versus Placebo in Persistent Asthma Which is Also Active During Allergy Seasons in Pediatric Patients With Seasonal Aeroallergen Sensitivity
#336
PROTECTION OF HUMAN SUBJECTS: This study was conducted in conformance with applicable country or local requirements regarding ethical committee review, informed consent, and other statutes or regulations regarding the protection of the rights and welfare of human subjects participating in biomedical research. For study audit information see Appendix [16.1.8].
INVESTIGATOR(S)/STUDY CENTER(S): One hundred three sites in US [16.1.3; 16.1.4].PUBLICATION(S): NAPRIMARY THERAPY PERIOD: Mar 2006 to Jul 2007 CLINICAL PHASE: IIIDURATION OF TREATMENT: One-week screening period followed by a 3-week treatment period
[16.1.1].OBJECTIVE(S):Primary: To assess the treatment effect of montelukast 5-mg versus placebo on the percent change from
baseline in FEV1 following a 3-week treatment period.
Secondary: To assess the treatment effect of montelukast 5-mg versus placebo over a 3-week treatment period on the percent change from baseline in mean daily “as needed” β-agonist use.
All other objectives are located in Appendix [16.1.1].STUDY DESIGN: This was a 4-week, multicenter, double-blind, parallel-group study conducted under
in-house blinding procedures. The study consisted of a 1-week single-blind placebo run-in period (Period I) followed by a 3-week double-blind treatment period (Period II) during which patients received either montelukast 5-mg chewable-tablet daily or placebo. Up to 40% of eligible patients were allowed to continue their stable dose of inhaled corticosteroid monotherapy during the course of the study. Patients were also permitted to use “as needed” β-agonist (albuterol) during the course of the study.
PATIENT DISPOSITION:Table 1
Patient Disposition
Montelukast 5-mg Placebo TotalSCREENING FAILURES: 640RANDOMIZED: 203 218 421
Male (age range) † 122 (6 to 15) 128 (6 to 15) 250 (6 to 15)Female (age range) † 81 (6.0 to 14.8) 90 (6.0 to 14.6) 171 (6 to 14.8)
COMPLETED: 197 210 407DISCONTINUED: 6 8 14
Clinical adverse experience 1 5‡ 6Laboratory adverse experience 0 0 0Other 5 3 8
† Patients were consented at 14 years.‡ One patient (placebo group) discontinued due to an adverse experience and never took any study drug[16.1.11.2; 16.2.7; 16.2.1; 16.2.4]

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-2-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
DOSAGE/FORMULATION NOS.: Montelukast 5-mg chewable tablet or matching-image placebo once daily. Control numbers for the single-blind placebo tablets were: CA-B567, CA-B567B and FL00004731. Control numbers for the double-blind montelukast 5-mg chewable tablet or matching-image placebo tablets were: CA-B571, CAB571B and FL00004738. Randomization details are located in Appendix [16.1.7]. A listing of compliance and drug concentration data is available in the Appendix [16.2.5].
DIAGNOSIS/INCLUSION CRITERIA: Eligible patients were male and female outpatients, ≥6 and ≤14 years of age, had at least a 1 year history of persistent asthma which is also active during allergy season, a baseline FEV1 value of ≥60% and ≤85% of predicted at randomization (Visit 2), evidence of reversible airway obstruction (≥12%) at screening (Visit 1) or within the preceding 1 year, demonstrated skin test sensitivity to at least 2 seasonal relevant aeroallergens at Visit 1 or within the preceding 1 year, at least a weekly sum of 11 for daytime symptoms, and a β-agonist daytime and nighttime weekly average of at least one puff/day during Period I (between screening and randomization).
Patients were not eligible if, in addition to asthma they: had any active, acute or chronic pulmonary disorder documented by history or physical examination; had been intubated for asthma previously; had used prohibited asthma medications prior to the Prescreen Visit; had required acute asthma treatment in an emergency room/urgent care facility/office setting within one month or had been hospitalized for asthma within one month of Visit 1; had unresolved symptoms and signs of an upper respiratory tract infection within 3 weeks of Visit 1 or between Visit 1 and Visit 2, or had evidence of active, clinically significant sinus infection within 1 week of Visit 1 or between Visit 1 and Visit 2..
EVALUATION CRITERIA: The primary endpoint was FEV1. The secondary endpoint was “as needed” β-agonist use. Additional endpoints included investigator and parent/guardian global assessment of asthma and prespecified subgroup analyses of: Gender, age, race, asthma severity, pollen levels, inhaled steroid monotherapy user at entry, season. All other efficacy and safety criteria are located in the protocol (Appendix [16.1.1]), or case report forms (Appendix [16.1.2]).
STATISTICAL PLANNING AND ANALYSIS: The primary efficacy analysis was based on the full analysis set population (i.e., includes all patients who had baseline and on-treatment measurements); a patient’s treatment group was determined by his/her randomized treatment group. A per-protocol (secondary) approach excluded patients with important protocol violations from the full analysis set. The percentage change from baseline in FEV1 measured at Week 3 was analyzed using an Analysis of Variance (ANOVA) model which will include factors for treatment, season and inhaled steroid use at entry (yes/no). Due to a non-normal distribution of the secondary endpoint, percent change from baseline in mean daily “as needed” β-agonist use, this variable was analyzed using a nonparametric method of an ANOVA model on the Tukey’s normalized ranks. The tertiary endpoints of global assessments of asthma were analyzed using the same model as the primary endpoint. The following subgroups were prespecified: Gender, age, race, asthma severity (≤ / > median baseline FEV1), pollen levels (≤ / > median pollen levels observed by season), inhaled steroid monotherapy user at entry (stratum (yes/no)), season (spring ’06, fall ’06, spring ‘07). Similar ANOVA model as used for the primary analysis was applied to each subgroup separately. The analysis plan for the other endpoints are located in Appendix [16.1.9].
All randomized patients who received double-blind study drug were included in the safety analyses. For this analysis, patients were counted in the treatment group for the treatment they actually received (All Patients as Treated (APaT) analysis). The analysis of safety followed a tiered approach. Tier I safety parameters included the overall incidence of adverse experiences (AEs), serious AEs, and drug-related AEs as well as discontinuation due to an AE. Additional planned analyses can be found in Appendix[16.1.9].

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-3-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
The observed mean between-group difference in percentage change from baseline in FEV1 for a pediatric population with asthma (Protocol 049) was of 4.6% with a standard deviation (SD) of 14%. Assuming a SD of 15%, a sample of 225 patients per treatment group provides 90% power at α-level of 0.050 (two-sided test) to detect a 5% between-group difference and 80% power to detect a 4% between-group difference in the percentage change from baseline in FEV1. Since the study faced some recruitment difficulties, the planned sample size was reduced to 200 evaluable patients per treatment group. A somewhat increased variability was expected as recruitment was extended over 3 different seasons. Assuming a SD of 16%, a sample of 200 evaluable patients per treatment group provides 80% power at α-level of 0.050 (two-sided test) to detect a 4.6% between-group difference in the percentagechange from baseline in FEV1.
Further details related to the Statistical Plan and protocol violators are located in Appendix [16.1.9] and [16.1.11.1; 16.2.2] respectively.
EFFICACY RESULTS: 1. Primary Endpoint - Percentage change from baseline in FEV1
The percentage change from baseline in FEV1 was numerically greater in the montelukast 5-mg group compared with the placebo group (Figure 1). However, the treatment difference was not statistically different (p-value=0.810). The montelukast 5-mg showed an increase of 9.53 % while the placebo group showed an increase of 9.15 % for the percentage change from baseline in FEV1.The between-group difference for the percentage change from baseline was 0.38 % (95% CI -2.74, 3.50).

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-4-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
Figure 1
Percentage Change from Baseline in FEV1 (LSmean +/- SE)(Full Analysis Set population)
Montelukast 5 mgPlacebo
Perc
enta
ge C
hang
e fro
m b
asel
ine
in F
EV1
(LSm
ean
+/- S
E)
0
5
10
15
9.53
(n=199)
*
* p=0.810 vs Placebo
9.15
(n=208)
[16.1.11.2; 16.2.3; 16.2.6]
2. Secondary Endpoint – Percentage Change from Baseline in “as needed” daily β-agonist use
Table 2 contains the percentage change from baseline in daily “as needed” β-agonist use. No significant treatment difference was observed between the montelukast 5-mg group and placebo group on the percentage change from baseline in mean daily “as needed” β-agonist use (p-value=0.173).

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-5-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
Table 2
Percentage Change from Baseline in Mean Daily “As Needed” β-Agonist UseNon-Parametric Analysis
Full Analysis Set Population
Montelukast 5-mgN=198
PlaceboN=212
Median -18.88 -12.40Standard Deviation 84.75 53.8895% CI for Median -26.28, -11.47 -20.09, -4.72
Difference in Medians (95 % CI) -5.51 (-14.3, 2.83)Treatment Effect p-value 0.173CI: Confidence Interval
[16.1.11.2; 16.2.3; 16.2.6]
Additionally, a supportive analysis of the change from baseline in the mean daily “as needed” -agonist use (puffs/day) was performed. These results can be found in Table 3 and Figure 2. The change from baseline in mean daily “as needed” -agonist use showed a between-group difference of -0.20 puffs/day (95% CI -0.44, 0.03; p-value=0.094).
Table 3
Change from Baseline in Daily β-agonist Use (puffs/day)Full-Analysis-Set Population
Montelukast 5-mgN=202
PlaceboN=216
Mean -0.55 -0.31Standard Deviation 1.26 1.25LS Mean -0.57 -0.3795% CI for LS Mean -0.76, -0.38 -0.55, -0.19
LS Mean Difference (95% CI) -0.20 (-0.44, 0.03)Treatment Effect p-value 0.094LS Mean: Least Squares MeanCI: Confidence Interval
[16.1.11.2; 16.2.3; 16.2.6]

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-6-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
Figure 2
Change from Baseline in Daily “As Needed” β-agonist Use Puffs/Day (LSmean +/- SE)Full Analysis Set population
Montelukast 5 mgPlacebo
Chan
ge fr
om B
asel
ine
in D
aily
b-a
goni
st U
se (L
Smea
n +/
- SE)
-3
-1
1
-0.57
(n=202)
*
* p=0.094 vs Placebo
-0.37
(n=216)
[16.1.11.2; 16.2.3; 16.2.6]
3. Global Evaluations for Asthma – Investigator and Parent/Guardian
Figure 3 shows the results for the investigator’s asthma global assessment after 3 weeks of treatment. There was a significant treatment difference observed in favor of the montelukast 5-mg group compared with the placebo group (p-value =0.032).
The results of the parent/guardian’s asthma global assessment after 3 weeks of treatment are shown in Figure 4. There was a significant treatment difference observed in favor of montelukast 5-mg group compared with the placebo group (p-value=0.034).

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-7-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
Figure 3
Global Evaluation by the Investigator (LSmean +/- SE)Full Analysis Set population
Montelukast 5 mgPlacebo
Glo
bal E
valu
atio
n by
the
Inve
stiga
tor (
LSm
ean
+/- S
E)
0
1
2
3
4
5
6
2.71
(n=200)
*
* p=0.032 vs Placebo
2.98
(n=210)
[16.1.11.2; 16.2.3; 16.2.6]

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-8-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
Figure 4
Global Evaluation by the Parent/Guardian (LSmean +/- SE)Full Analysis Set population
Montelukast 5 mgPlacebo
Glo
bal E
valu
atio
n by
the
Pare
nt/G
uard
ian
(LSm
ean
+/- S
E)
0
1
2
3
4
5
6
2.63
(n=201)
*
* p=0.034 vs Placebo
2.90
(n=214)
[16.1.11.2; 16.2.3; 16.2.6]
Supportive asthma global collapsed score (better, no change, worse) analyses for both the investigator and parent/guardian were also completed. The results of these analyses are located in Appendix [16.1.11.2; 16.2.3; 16.2.6].

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-9-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
4. Pre-specified Subgroup analyses on primary and secondary endpoints were: Gender, Age: <9 years versus ≥ 9 years, Race (white, black, other), Season (spring ’06, fall ’06, spring ‘07), User of stable dose of inhaled corticosteroid monotherapy at entry (stratum (yes/no), Pollen levels (≤ / > median pollen levels observed by season (spring ’06, fall ’06, spring ’07), and Asthma Severity (≤ / > median baseline FEV1).
The percentage change from baseline FEV1 for the subgroup analyses are located in Table 4. The effects between treatment groups were generally consistent among subgroups defined by gender, age, race, stratum, pollen level and asthma severity. A numerical treatment difference of 2.52 was observed in favor of montelukast for the subgroup “Spring 07”.
Table 5 contains the subgroup analyses for the change from baseline in daily “as needed” -agonist use. Subgroup analyses were performed on this endpoint (change from baseline, not percentage change from baseline) to enable appropriate parametric analyses of the subgroups. The effects between treatment groups were generally consistent among subgroups defined by gender, age, race, stratum, asthma severity and pollen level. A numerical treatment difference of -0.66 was observed in favor of montelukast for the subgroup “Fall 2006”.

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-10-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
Table 4
Analysis of Percentage Change from Baseline in FEV1by Subgroup
Montelukast 5-mg% Change from Baseline
LS Mean (CI) †
Placebo% Change from Baseline
LS Mean (CI) †
Treatment-groupDifference
LS Mean (CI) †
Gender Female 10.21 (6.72, 13.69) 11.15 (7.42, 14.89) -0.94 (-5.32, 3.43) Male 9.05 (5.58, 12.52) 8.23 (4.96, 11.51) 0.82 (-3.59, 5.23)Age (Years) < 9 years 10.16 (5.22, 15.10) 8.33 (3.34, 13.33) 1.83 (-4.78, 8.44) ≥ 9 years 9.18 (6.22, 12.13) 9.33 (6.50, 12.16) -0.15 (-3.74, 3.44)Race White 9.79 (5.99, 13.59) 10.02 (6.31, 13.74) -0.23 (-4.95, 4.49) Black 7.33 (2.73, 11.94) 4.94 (0.58, 9.31) 2.39 (-2.70, 7.48) Other 11.26 (6.12, 16.40) 12.65 (7.31, 17.98) -1.39 (-8.36, 5.59)Season of Enrollment Spring 2006 7.70 (4.10, 11.30) 7.67 (4.19, 11.15) 0.03 (-4.59, 4.64) Fall 2006 10.35 (4.66, 16.03) 12.30 (6.19, 18.41) -1.95 (-9.32, 5.41) Spring 2007 11.25 (7.10, 15.39) 8.72 (4.96, 12.49) 2.52 (-2.52, 7.56)Use of Concomitant Inhaled Corticosteroid Monotherapy No 7.31 (4.93, 9.68) 6.86 (4.48, 9.23) 0.45 (-2.88, 3.78) Yes 12.16 (6.31, 18.01) 12.02 (6.40, 17.64) 0.13 (-7.83, 8.10)Above/below median Pollen count by season ≤ median 10.92 (7.35, 14.48) 9.57 (5.91, 13.23) 1.35 (-3.35, 6.05) > median 8.25 (4.58, 11.92) 9.35 (5.84, 12.85) -1.10 (-5.50, 3.30)Above/below median Baseline FEV1
≤ median 11.39 (7.27, 15.51) 10.42 (6.61, 14.23) 0.97 (-4.07, 6.01) > median 8.01 (5.01, 11.01) 7.84 (4.77, 10.91) 0.17 (-3.59, 3.93)Above/below median Baseline Percent Predicted FEV1
≤ median 11.72 (8.09, 15.35) 11.50 (7.92, 15.08) 0.22 (-4.42, 4.87) > median 6.14 (2.94, 9.33) 6.34 (3.21, 9.47) -0.20 (-4.13, 3.72)CI = 95% Confidence Interval† LS mean, based on analysis of variance with terms for treatment, stratum, and season.SE = Standard Error LS Mean = Least Squares Mean
[16.1.11.2; 16.2.3; 16.2.6]

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-11-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
Table 5
Analysis of Change from Baseline in Daily “As Needed” β-Agonist Use (puffs/day)by Subgroup
Montelukast 5 mgChange from Baseline
LS Mean (CI) †
PlaceboChange from Baseline
LS Mean (CI) †
Treatment groupDifference
LS Mean (CI) †
Gender Female -0.36 (-0.68, -0.04) -0.22 (-0.55, 0.12) -0.14 (-0.54, 0.26) Male -0.70 (-0.93, -0.46) -0.46 (-0.68, -0.24) -0.24 (-0.54, 0.06)Age (Years) < 9 years -0.67 (-1.02, -0.32) -0.31 (-0.66, 0.05) -0.37 (-0.83, 0.10) ≥ 9 years -0.56 (-0.79, -0.33) -0.40 (-0.61, -0.18) -0.16 (-0.44, 0.12)Race White -0.56 (-0.84, -0.28) -0.30 (-0.57, -0.03) -0.27 (-0.62, 0.08) Black -0.45 (-0.92, 0.03) -0.42 (-0.86, 0.02) -0.02 (-0.54, 0.49) Other -0.69 (-0.98, -0.41) -0.50 (-0.79, -0.22) -0.19 (-0.57, 0.19)Season of Enrollment Spring 2006 -0.43 (-0.73, -0.14) -0.51 (-0.80, -0.22) 0.07 (-0.31, 0.46) Fall 2006 -0.77 (-1.10, -0.44) -0.11 (-0.45, 0.24) -0.66 (-1.09, -0.23) Spring 2007 -0.63 (-0.97, -0.29) -0.43 (-0.74, -0.13) -0.20 (-0.61, 0.22)Use of Concomitant Inhaled Corticosteroid Monotherapy No -0.49 (-0.69, -0.29) -0.24 (-0.44, -0.05) -0.25 (-0.53, 0.03) Yes -0.62 (-0.93, -0.30) -0.55 (-0.85, -0.26) -0.07 (-0.49, 0.36)Above/below median Pollen count by season ≤ median -0.72 (-0.97, -0.48) -0.46 (-0.71, -0.21) -0.26 (-0.58, 0.06) > median -0.38 (-0.69, -0.07) -0.25 (-0.54, 0.04) -0.13 (-0.50, 0.24)Above/below median Baseline FEV1
≤ median -0.70 (-0.98, -0.41) -0.43 (-0.70, -0.16) -0.27 (-0.62, 0.08) > median -0.46 (-0.72, -0.20) -0.30 (-0.56, -0.03) -0.16 (-0.48, 0.17)Above/below median Baseline Percent Predicted FEV1
≤ median -0.71 (-0.96, -0.45) -0.45 (-0.70, -0.20) -0.25 (-0.58, 0.07) > median -0.42 (-0.72, -0.13) -0.26 (-0.55, 0.02) -0.16 (-0.52, 0.20)CI = 95% Confidence Interval† LS mean, based on analysis of variance with terms for treatment, stratum, and season.SE = Standard Error LS Mean = Least Squares Mean
[16.1.11.2; 16.2.3; 16.2.6]
The results of all other study endpoints are located in Appendix [16.1.11.2; 16.2.3; 16.2.6].
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-12-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
SAFETY RESULTS: The summary of clinical adverse experiences are shown in Table 6.
Table 6
Clinical Adverse Experience Summary
Montelukast 5-mg(N=203)
Placebo(N=217)
Number (%) of Patients n (%) n (%)With one or more adverse experiences 17 (8.4) 23 (10.6)With no adverse experiences 186 (91.6) 194 (89.4)With drug-related adverse experiences 2 (1.0) 2 (0.9)With serious adverse experiences 0 (0.0) 4 (1.8)With serious drug-related adverse experiences 0 (0.0) 0 (0.0)Who died 0 (0.0) 0 (0.0)Discontinued due to adverse experiences 1 (0.5) 4 (1.8)Discontinued due to drug-related adverse experiences 0 (0.0) 0 (0.0)Discontinued due to serious adverse experiences 0 (0.0) 3 (1.4)Discontinued due to serious drug-related adverse
experiences0 (0.0) 0 (0.0)
N =Number of randomized and treated patients in each treatment group.† Determined by the investigator to be possibly, probably or definitely drug related.CI: Confidence interval.This table contains counts of patients. Although a patient may have two or more AEs, the patient is counted only once in a category or overall.
[16.1.11.2; 16.2.7]
There was no significant difference between treatment groups with respect to the percentage of patients with at least one clinical adverse experience (AE), with at least one serious clinical AE, with at least one drug-related AE, with at least one drug-related serious clinical AE, or for the percentage of patientsdiscontinued due to an AE.
The overall safety profile of the two treatment groups was similar, with about 9.5% of patients reporting at least one clinical AE, and about 1% of the patients had a least one drug-related AE in both treatment groups. A total of 4 patients reported at least one serious clinical AE; all serious clinical AEs were reported in the placebo group. Two patients reported a serious adverse experience of asthma, 1 patient reported overdose and 1 patient reported respiratory distress and femur fracture. No patient died during the treatment period. No serious drug-related AE were reported in either group.
Five patients discontinued due to an AE during the study: 1 (0.5%) patient in the montelukast 5-mg (adverse experience of asthma) and 4 (1.8%) patients in the placebo group (3 patients reported anadverse experience of asthma, 1 reported respiratory distress and femur fracture).
No laboratory AEs were reported.

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg Chewable Tabletasthma
-13-
0476_P336V1_02_Synopsis VERSION 8.2 APPROVED 13-Sep-2010Restricted Confidential – Limited Access
CONCLUSIONS: For patients 6 to 14 years old with seasonal allergen sensitivity and persistent asthma active during the allergy season: Montelukast 5-mg administered once daily over a 3-week treatment period did not show a
significant improvement in FEV1 (the primary efficacy endpoint), compared with placebo; Montelukast 5-mg was well tolerated, with incidences and overall patterns of adverse experiences
similar to those of the placebo group.
In secondary efficacy analyses (which should be interpreted with caution, since the primary efficacy analysis was not significant and secondary analyses are not adjusted for multiplicity): No significant treatment difference was observed between montelukast 5-mg and placebo on the
percentage change from baseline in mean daily “as needed” β-agonist use; A small treatment difference was observed in favor of montelukast 5-mg compared to placebo for
the investigator’s and parent/guardian’s asthma global assessment; Predefined subgroup analyses for the percentage change from baseline FEV1 were generally
consistent among subgroups, even though a numerical higher treatment difference was observed for the subgroup “Spring 07”. Likewise, the subgroup analyses for the change from baseline in daily β-agonist use were generally consistent among subgroups even though a numerical higher treatment difference was observed for the subgroup “Fall 2006”.
AUTHORS: [16.1.5] Molly Watkins, MMS Hilde GiezekClinical Research Specialist Senior Statistician
George Philip, MD Peter G. Polos, MD, PhDSenior Director Global Director

MK-0476 Prot. No. 340Back to School
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
2. Synopsis
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthmaPROTOCOL TITLE/NO.: A Multicenter, Randomized, Double-blind, Placebo-
Controlled Parallel Group 8-Week Study to Evaluate the Efficacy and Safety of Chewable Montelukast When Initiated at the Start of the School Year in Pediatric Patients With Asthma
#340
PROTECTION OF HUMAN PATIENTS: This study was conducted in conformance with applicable country or local requirements regarding ethical committee review, informed consent, and other statutes or regulations regarding the protection of the rights and welfare of human patients participating in biomedical research. For study audit information see [16.1.8].
INVESTIGATOR(S)/STUDY CENTER(S): One hundred sixty five investigators in the U.S. and Canada [16.1.3; 16.1.4].
PRIMARY THERAPY PERIOD: Jul-2006 to Nov-2006 CLINICAL PHASE: IIIDURATION OF TREATMENT: One screening period of 2-12 weeks in duration and one 8-week
treatment period [16.1.1].OBJECTIVE(S):
Primary Objective: To assess the treatment effect of montelukast 5 mg versus placebo on the primary endpoint: percentage of days of worsening asthma1, measured on daily diaries over an 8-week treatment period.
Key Secondary Objective: To assess the treatment effect of montelukast 5 mg versus placebo over an 8-week treatment period on the following individual components of the primary measure: (a) β-agonist use, (b) daytime asthma symptoms score and (c) the occurrence of health care utilization2.
All other objectives are located in appendix [16.1.1].STUDY DESIGN: This study was a randomized, double-blind, placebo-controlled, parallel-group study
performed under in-house blinding conditions. Patients had a screening period of variable duration during which they continued their current asthma controller medications. Eligible patients wererandomized 2 weeks prior to their first day of school. On the night before the patient’s first day of school, randomized patients began their 8-week double-blind treatment period while continuing to use their usual asthma treatment including “as needed” β -agonist (albuterol). Patients had a telephone contact at treatment week 4 and a clinic visit at treatment week 8.
1 A day of worsening asthma is any day with one occurrence of any one of the following events:, (a) an
increase from baseline in β-agonist use of >70% and a minimum increase of 2 puffs; (b) an increase of >50% from baseline in daytime symptoms score, (c) awake “all night” on the asthma symptom diary, (d) an increase in inhaled corticosteroid use of at least 100% from baseline or oral corticosteroid rescue for worsening asthma; or (e) unanticipated visits to the physician, emergency room or hospital for asthma.
2 Health care utilization is defined as unanticipated asthma care in an office or clinic, emergent or hospital setting.
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-2-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
PATIENT DISPOSITION: (Table 1)
Montelukast 5 mg Placebo TotalSCREENING FAILURES: 97RANDOMIZED: 580 582 1162
Male (age range)† 6 to 15 6 to 15 6 to 15Female (age range)† 6 to 15 6 to 15 6 to 15
COMPLETED: 536 545 1081DISCONTINUED: 44 37 81
Clinical adverse experience 5 5 10Laboratory adverse experience 1 0 1Other 38 32 70
† patients were consented at 14 years[16.1.11.2]DOSAGE/FORMULATION NOS.: montelukast 5 mg chewable tablet or matching-image placebo
once daily (Control Number/Lot ID = CA-B609). Randomization details are located in Appendix [16.1.7].
DIAGNOSIS/INCLUSION CRITERIA: Eligible patients were male and females ≥ 6 and ≤ 14 years of age with a history of chronic asthma, defined as symptoms including, but not limited to: dyspnea, wheezing, chest tightness, or cough, of at least one year in duration, a history of asthma exacerbations associated with apparent respiratory viral infections within the past year and a documented history of asthma that required treatment with any asthma medication within 6 months prior to Visit 1.
Patients were not eligible if: in addition to asthma, had: any active, acute or chronic pulmonary disorder documented by history or physical examination; ever had been intubated for asthma, required acute asthma therapy treated in an emergency room/urgent care facility/office setting within one month or had been hospitalized for asthma within one month of Visit 1, had been hospitalized more than three times for asthma within one year of Visit 1 or he/she had used any prohibited asthma medications prior to the randomization telephone call.
EVALUATION CRITERIA: The key efficacy parameters used to address the primary objective were:β-agonist use, daytime and nighttime asthma symptoms, health care utilization, and oral and inhaled corticosteroid use for worsening asthma, which were captured using a daily electronic diary. A copy of the case report forms, which include the diary questions, are located in Appendix [16.1.2].
STATISTICAL PLANNING AND ANALYSIS:The primary efficacy endpoint of percent days of worsening asthma during the treatment period wasanalyzed using ANOVA model with factors including treatment, school start period, investigator site type, inhaled corticosteroid use at entry, age group (6-9 vs. ≥10) and gender. The least-squares mean estimate for between treatment comparison and the corresponding 95% confidence interval from the main effect ANOVA model was used to test the primary hypothesis. The calculation of the 95% confidence limits for the differences in pre-defined adverse experience event rates were performed using Wilson’s score method.
Based on data from a prior pediatric protocol (Protocol 049) , the difference in percent of asthma exacerbation days (defined similarly as worsening asthma days in this study) between montelukast and placebo was 5.1% (placebo-montelukast) and standard deviation was 24.7% and 22.6% in the montelukast and placebo group, respectively. With a total number of 1100 patients randomized to montelukast and placebo in a 1:1 ratio (495 evaluable in each group), assuming that the placebo-adjusted treatment effect of montelukast was 5% in percent worsening asthma days and the common standard deviation was 24%, the power to demonstrate superiority of montelukast treatment compared to placebo was 90%. Further details of the Statistical Analysis Plan, including the patients not included in the analysis, can be found in appendices [16.1.9; 16.1.11.1] respectively.

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-3-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
EFFICACY RESULTS:1. Primary Endpoint - Percentage of Days with Worsening Asthma
The percentage of days with worsening asthma was numerically lower in montelukast 5 mg group compared with the placebo group (Figure 1). However, the treatment difference was not statistically significant. The least-square (LS) mean estimate of the percentage of days with worsening asthma was 24.3 (95% confidence interval [CI] = [21.52, 27.01]) for montelukast group, and the LS mean estimate for placebo group was 27.2 (95% CI = [24.45, 30]). The treatment difference in LS means was -2.96 (95% CI = [-6.21, 0.29], p=0.074).
Figure 1
Treatment Difference in Percent Worsening Asthma DayFull Analysis Set Population
-10 -5 0 5 10Treatment Difference (%)
(Montelukast - Placebo)
Favors PlaceboFavors Montelukast
-2.96%
[16.1.11.2]

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-4-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
2. Secondary Endpoints - percentage of days with increased β-agonist use, percentage of days with increased daytime asthma symptom score and healthcare utilization
The treatment comparisons between montelukast 5 mg and placebo were not statistically significant in any of the secondary endpoints (Table 2).
The results of all other study endpoints are located in Appendices [16.1.11.3; 16.1.11.4; 16.1.11.5;16.1.11.6; 16.1.11.7; 16.1.11.8].
Table 2
Key Secondary Efficacy Endpoints
Montelukast 5mg Placebo
Treatment Comparison(Montelukast 5mg-
Placebo)Percentage of Days with Increased Beta-Agonist Use N 495 491 Mean (SD) 5.59 (10.65) 6.58 (12.11) LS Means (SE) 6.04 (0.61) 7.02 (0.62) -0.98 (0.72) 95% C.I. (4.84, 7.24) (5.80, 8.23) (-2.39, 0.44) P Value 0.177 Percentage of Days with Increased Daytime Symptom ScoreN 496 491 Mean (SD) 16.09 (19.22) 17.51 (19.30) LS Means (SE) 16.03 (1.04) 17.4 (1.05) -1.36 (1.23) 95% C.I. (13.99, 18.07) (15.33, 19.47) (-3.77, 1.05) P Value 0.267 Health Care Utilizationn/N (%) 55/466 (11.8) 70/476 (14.7) Odds Ratio 0.77 95% C.I. (8.9 ,14.7) (11.5 ,17.9) (0.53, 1.13) P Value 0.182 Unscheduled Physician Visit
n/N (%) 50/466 (10.7) 65/476 (13.7) Emergency Room Visit
n/N (%) 6/463 (1.3) 8/476 (1.7) Hospitalization
n/N (%) 5/463 (1.1) 2/476 (0.4) Note:1. The LS Means and their standard errors, p-values and 95% confidence intervals were calculated using an
ANOVA model that included terms for treatment, age group (6-9 vs. 10-14), gender, use of inhaledcorticosteroid at entry, school start period and investigator type.2. The odds ratio, p-value and 95% confidence interval was calculated using a logistic regression model that
included terms for treatment, age group (6-9 vs. 10-14), gender, use of inhaled corticosteroid at entry, school start period and investigator type.
[16.1.11.2]
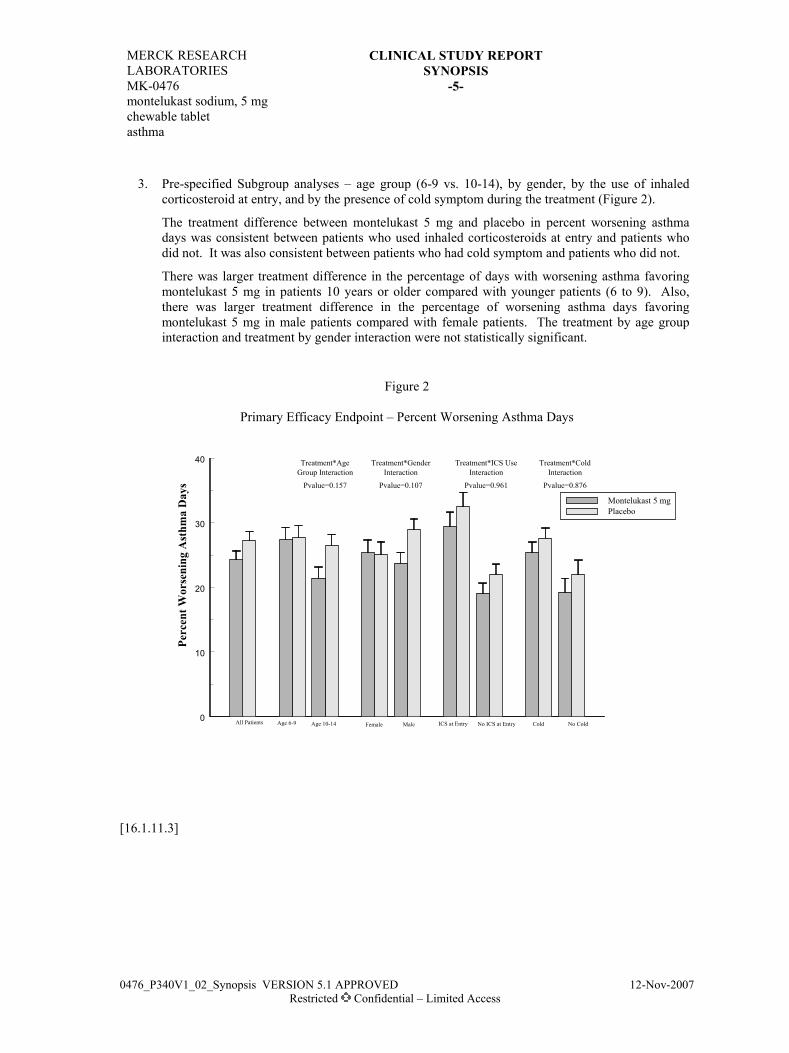
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-5-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
3. Pre-specified Subgroup analyses – age group (6-9 vs. 10-14), by gender, by the use of inhaled corticosteroid at entry, and by the presence of cold symptom during the treatment (Figure 2).
The treatment difference between montelukast 5 mg and placebo in percent worsening asthma days was consistent between patients who used inhaled corticosteroids at entry and patients who did not. It was also consistent between patients who had cold symptom and patients who did not.
There was larger treatment difference in the percentage of days with worsening asthma favoring montelukast 5 mg in patients 10 years or older compared with younger patients (6 to 9). Also,there was larger treatment difference in the percentage of worsening asthma days favoring montelukast 5 mg in male patients compared with female patients. The treatment by age group interaction and treatment by gender interaction were not statistically significant.
Figure 2
Primary Efficacy Endpoint – Percent Worsening Asthma Days
0
10
20
30
40
Pvalue=0.157 Pvalue=0.107 Pvalue=0.961 Pvalue=0.876
Montelukast 5 mgPlacebo
Age 6-9 Age 10-14 MaleFemale ICS at Entry No ICS at Entry Cold No Cold
Treatment*Age Treatment*Gender Treatment*ICS Use Treatment*ColdGroup Interaction Interaction Interaction Interaction
All Patients
Perc
ent W
orse
ning
Ast
hma
Day
s
[16.1.11.3]
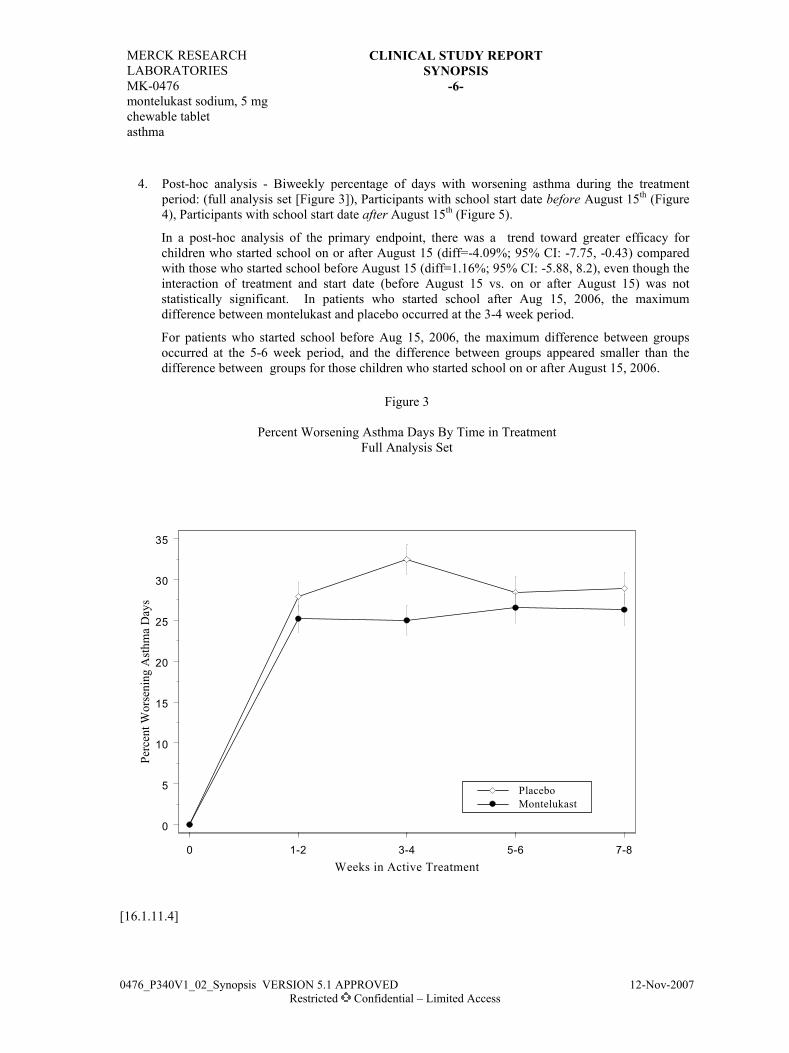
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-6-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
4. Post-hoc analysis - Biweekly percentage of days with worsening asthma during the treatment period: (full analysis set [Figure 3]), Participants with school start date before August 15th (Figure 4), Participants with school start date after August 15th (Figure 5).
In a post-hoc analysis of the primary endpoint, there was a trend toward greater efficacy for children who started school on or after August 15 (diff=-4.09%; 95% CI: -7.75, -0.43) compared with those who started school before August 15 (diff=1.16%; 95% CI: -5.88, 8.2), even though the interaction of treatment and start date (before August 15 vs. on or after August 15) was not statistically significant. In patients who started school after Aug 15, 2006, the maximum difference between montelukast and placebo occurred at the 3-4 week period.
For patients who started school before Aug 15, 2006, the maximum difference between groups occurred at the 5-6 week period, and the difference between groups appeared smaller than the difference between groups for those children who started school on or after August 15, 2006.
Figure 3
Percent Worsening Asthma Days By Time in TreatmentFull Analysis Set
0 1-2 3-4 5-6 7-8Weeks in Active Treatment
0
5
10
15
20
25
30
35
Perc
ent W
orse
ning
Ast
hma
Day
s
PlaceboMontelukast
[16.1.11.4]

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-7-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
Figure 4
Percent Worsening Asthma Days By Time in TreatmentParticipants with school start date before August 15th
0 2 4 6 8Weeks in Active Treatment
0
5
10
15
20
25
30
35
Perc
ent W
orse
ning
Ast
hma
Day
s
PlaceboMontelukast
[16.1.11.2; 16.1.11.4]

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-8-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
Figure 5
Percent Worsening Asthma Days By Time in TreatmentParticipants with school start date after August 15th
0 1-2 3-4 5-6 7-8Weeks in Active Treatment
0
5
10
15
20
25
30
35
Perc
ent W
orse
ning
Ast
hma
Day
s
PlaceboM ontelukast
[16.1.11.2; 16.1.11.4]

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-9-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
SAFETY RESULTS:
Table 3
Clinical Adverse Experience Summary
Montelukast 5 mg(N=566)
Placebo(N=566)
Number (%)of Patients n (%) n (%)With one or more adverse experiences 192 ( 33.9) 190 ( 33.6) With no adverse experience 374 ( 66.1) 376 ( 66.4) With drug-related adverse experiences† 11 ( 1.9) 13 ( 2.3) With serious adverse experiences 4 ( 0.7) 1 ( 0.2) With serious drug-related adverse experiences 0 ( 0.0) 0 ( 0.0) Who died 0 ( 0.0) 0 ( 0.0) Discontinued due to adverse experiences 4 ( 0.7) 4 ( 0.7) Discontinued due to drug-related adverse experiences 1 ( 0.2) 2 ( 0.4) Discontinued due to serious adverse experiences 2 ( 0.4) 0 ( 0.0) Discontinued due to serious drug-related adverse experiences
0 ( 0.0) 0 ( 0.0)
N=Number of randomized and treated patients in each treatment group.† Determined by the investigator to be possibly, probably or definitely drug related.Although a patient may have had two or more clinical adverse experiences the patient is counted only once in a category. The same patient may appear in different categories.
[16.1.11.2]
There was no significant difference between treatment groups with respect to the percentage of patients with at least one clinical adverse experience, with at least one serious clinical adverse experience, with at least one drug-related clinical adverse experience, with at least one drug-related serious clinical adverse experience, or for the percentage of patients discontinued due to an adverse experience.
The overall safety profiles of the two treatments were very similar, with about 34% of the patients reporting at least one clinical adverse experience in both treatment groups, and about 2% of the patients had at least one drug-related clinical adverse experience in both treatment groups. Serious adverse experiences were reported by 4 patients (0.7%) and 1 patient (0.2%) from the montelukast and placebo group, respectively. There were three patients who reported a serious AE of exacerbation of asthma, two in the montelukast treatment group and one in the placebo treatment group. The other serious AEs reported in the montelukast treatment group were pneumonia, and abdominal pain. No patient died during the treatment period. No serious drug-related AE was found in either group.
One patient in the study (montelukast group) had a laboratory AE (alanine aminotransferase increased). The AE occurred after the patient was randomized, but prior to the first dose of study medication. It was not considered to be drug-related by the investigator, and was the only laboratory AE reported for the study. There were no significant differences between treatment groups in the overall incidence of laboratory AEs, drug-related laboratory AEs, serious laboratory AEs, or discontinuations due to laboratory AEs.

MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTSYNOPSIS
MK-0476montelukast sodium, 5 mg chewable tabletasthma
-10-
0476_P340V1_02_Synopsis VERSION 5.1 APPROVED 12-Nov-2007Restricted Confidential – Limited Access
CONCLUSIONS: There is insufficient evidence to conclude that the treatment of montelukast 5 mg once daily is more effective compared with placebo in controlling worsening asthma over an 8-week period at the beginning of school year.
The sub-group analyses suggest that age and gender results may be used to guide future research in the area of asthma. Additionally, the post-hoc analyses suggest that the treatment effect appears to be larger in participants who start school later in the calendar year. Further investigation is needed to determine if there is an effect relative to season rather than school start.
There were no significant differences between treatment groups in the overall incidence of clinical or laboratory AEs, drug-related clinical or laboratory AEs, serious clinical or laboratory AEs, or discontinuations due to clinical or laboratory AEs.
AUTHORS: [16.1.5] (Clinical Research Specialist) (Statistician) (Clinical Monitor)Molly Watkins, M.M.S. Gang Jia, Ph.D. Evalyn N. Grant, M.D.

BC2893.DOC VERSION 9.0 APPROVED 02-Nov-2001
MERCK RESEARCHLABORATORIES
CLINICAL STUDY REPORTI. SYNOPSIS
MK-0476Montelukast Sodium, Film-Coated TabletExercise-Induced Bronchoconstriction
PROTOCOL TITLE/NO.: A Randomized, Double-Blind, Parallel-Group StudyEvaluating the Long-Term Effect of Montelukast Sodium Versus Salmeterol on theInhibition of Exercise-Induced Bronchoconstriction in Adult Patients With Asthma
#902-0B
INVESTIGATORS/STUDY CENTERS: Twenty-five study centers in Europe, South Africa, andSouth America.
PUBLICATIONS: (1) Villaran C, O’Neill S, Helbling A, van Noord J, Lee T, Chuchalin A, Langley S,Gunawardena K, Suskovic S, Laurenzi M, Jasan J, Menten J, Leff JA for the Montelukast/SalmeterolExercise Study Group. Montelukast (MK-0476) Versus Salmeterol in Patients With Asthma andExercise-Induced Bronchoconstriction (EIB). J Allergy Clin Immunol 1999;104:547-53. (2) VillaranC, O’Neill S, Helbling A, van Noord J, Lee T, Chuchalin A, Langley S, Gunawardena K, Suskovic S,Laurenzi M, Jasan J, Leff JA for the Montelukast/Salmeterol Exercise Study Group. Montelukast(MK-0476) Compared to Salmeterol in the Treatment of Exercise-Induced Asthma (EIA). Eur Respir J1998;12(suppl 28):361s.
PRIMARY THERAPY PERIOD: 27-Nov-1996 to 09-Sep-1997.Study is completed. The in-house cutoff date for case report formswas 05-Aug-1998.
CLINICAL PHASE: IV
DURATION OF TREATMENT: Period I: 2-week run-in period; Period II: 8-week treatment period.OBJECTIVES: Primary: To determine the effect of 8 weeks of chronic treatment with montelukast
sodium or salmeterol on EIB in adult asthmatics as measured by the postexercise Maximum Percent Fallin forced expiratory volume in 1 second (FEV1) (mean change from baseline1) obtained at the end of thedosing interval for montelukast and salmeterol2.
Secondary: To determine and compare between montelukast and salmeterol for: (1) The postexerciseMaximum Percent Fall in FEV1 at Week 4 of treatment; (2) The Time to Recovery for FEV1 to within5% of pre-exercise FEV1 (hereafter referred to as Time to Recovery) at Weeks 4 and 8 of treatment;(3) The postexercise area under the FEV1 time curve during the first 60 minutes (AUC0-60 min) atWeeks 4 and 8 of treatment; (4) Persistence of Effect over 8 weeks of treatment; (5) The safety andtolerability of montelukast and salmeterol.
STUDY DESIGN: This was a randomized, double-blind, double-dummy, 2-period, active-control,parallel-group, multicenter study conducted under in-house blinding procedures preceded by a placeborun-in period.
PATIENT ACCOUNTING: Montelukast Salmeterol Total
ENTERED: 102 95 197Male (age range) 53 (15 to 45) 45 (17 to 45) 98 (15 to 45)Female (age range) 49 (15 to 44) 50 (14 to 44) 99 (14 to 44)
COMPLETED PERIOD II: (%) 100 (98.0) 88 (92.6) 188 (95.4)DISCONTINUED: Total (%) 2 (2.0) 7 (7.4) 9 (4.6)
Clinical adverse experience (%) 0 (0.0) 4 (4.2) 4 (2.0)Laboratory adverse experience (%) 0 (0.0) 0 (0.0) 0 (0.0)Other (%) 2 (2.0) 3 (3.2) 5 (2.5)
1 The term “baseline” in this clinical study report refers to the prerandomization values (obtained at
Visit 2) used in the statistical analyses of change from baseline. FEV1 values obtained prior toindividual exercise challenges are visit-specific and termed “pre-exercise.”
2 All exercise challenges occurred at the end of the dosing interval (approximately 20 to 24 hours afterdosing for montelukast and 8 to 12 hours after dosing for salmeterol).

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 902-0B
MK-0476Montelukast Sodium, Film-Coated TabletExercise-Induced Bronchoconstriction
-2-
BC2893.DOC VERSION 9.0 APPROVED 02-Nov-2001
DOSAGE/FORMULATION NOS.: Period I: Montelukast matching-image placebo film-coated tablet(MR-3209) once daily at bedtime; 2 puffs salmeterol matching-image placebo inhaler (MR-3368) every12 hours. Period II: 1 montelukast 10-mg film-coated tablet (MR-3210) or 1 matching-image placebo(MR-3209) once daily at bedtime, or 2 puffs (42 µg [21 µg/puff]) salmeterol inhaler (MR-3374) or2 puffs matching-image placebo (MR-3368) every 12 hours.
DIAGNOSIS/INCLUSION CRITERIA: Nonsmoking men and women (using appropriatecontraception) with asthma between and including 15 and 45 years of age, with a clinical history ofasthma for at least 1 year, with evidence of EIB (decrease in FEV1 after exercise of at least 18%compared with pre-exercise FEV1), airway reversibility ≥12% (as demonstrated by FEV1 after inhaledβ-agonist administration) within the past 6 months, and an average FEV1 of ≥65% of the predictedvalue, with β-agonist withheld for at least 6 hours prior to spirometry testing, measured at 20 minutesand 5 minutes pre-exercise. Up to 25% of patients were allowed to continue concomitant inhaledcorticosteroids at a stable dose.
EVALUATION CRITERIA:EFFICACY: Primary Endpoint: Maximum Percent Fall in FEV1 after exercise at Week 8. Secondary
Endpoints: Time to Recovery at Week 8; AUC0-60 min at Week 8; Persistence of Effect over 8 weeksof treatment. Additional Endpoints: Maximum Percent Fall in FEV1 after exercise at Days 1 to 33
and Week 4; Time to Recovery at Days 1 to 3 and Week 4; AUC0-60 min at Days 1 to 3 and Week 4;Percentage of Patients Requiring Rescue Medication During (or at the end of) the Exercise Challenge;Percentage of patients with <20% Maximum Percent Fall in FEV1 after exercise at Week 8.
SAFETY: Frequency of clinical and laboratory adverse experiences, frequency of laboratory valuesoutside the predefined limits of change for selected laboratory safety tests, and vital signs.
STATISTICAL PLANNING AND ANALYSIS:EFFICACY: For most efficacy endpoints, the change from baseline was analyzed. Between-group
comparisons were performed using a linear model (analysis of variance [ANOVA]) with factors fortreatment group, stratum, and study center. Using least-squares (LS) means, 95% confidence intervals(CIs) were computed for the within-group changes and between-group differences. The study wasdesigned to have 160 patients complete the study, with 80 patients in each group. The study had 95%power to detect a 7-percentage point difference between treatment groups in the mean change inMaximum Percent Fall in FEV1 after exercise, assuming a standard deviation (SD) of 12.5 percentagepoints.
SAFETY: Fisher’s exact test and 95% CIs were used to compare between-group incidences of adverseexperience categories: overall, drug-related, serious adverse experiences, and adverse experiencescausing discontinuation from therapy. Ninety-five percent CIs for between-group differences inindividual adverse experiences and in the percentage of patients with values outside the predefinedlimits of change for selected laboratory parameters were based on Wilson’s score method.
3 The first postrandomization exercise challenge occurred within 1 to 3 days after taking at least 1 full day
of study medication.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 902-0B
MK-0476Montelukast Sodium, Film-Coated TabletExercise-Induced Bronchoconstriction
-3-
BC2893.DOC VERSION 9.0 APPROVED 02-Nov-2001
RESULTS:EFFICACY: Montelukast showed significantly greater attenuation of EIB than salmeterol as
demonstrated in Maximum Percent Fall in FEV1 after exercise at Week 8 (primary endpoint) and atWeek 4 (additional endpoint). Montelukast also showed significantly greater attenuation of EIB asdemonstrated in the secondary and additional endpoints, Time to Recovery and AUC0-60 min atWeeks 4 and 8; montelukast and salmeterol provided similar attenuation of EIB at Days 1 to 3 asdemonstrated in Maximum Percent Fall in FEV1 after exercise and Time to Recovery, andmontelukast provided greater attenuation of EIB at Days 1 to 3 for AUC0-60 min. The effect ofmontelukast was persistent over the 8-week treatment period, while the effect of salmeterol decreased.The percentage of patients in the montelukast and salmeterol treatment groups that required RescueMedication During Exercise Challenge at one or more visits was 31.4% and 37.2%, respectively(p=0.452). The percentage of patients in the montelukast and salmeterol treatment groups thatreached a commonly used threshold level of effect (defined as a Maximum Percent Fall in FEV1 afterexercise of <20%) at Week 8 was 64.4% and 57.8%, respectively (p=0.375).
Exercise Challenge Endpoints†—Analysis of Within-Group and Between-Group Differences at Week 8
Maximum Percent Fallin FEV1 (%) Time to Recovery (min) AUC0-60 min (%·min)
TreatmentBaseline(Mean)
Change FromBaseline (Mean)‡
Baseline(Mean)
Change FromBaseline (Mean)
Baseline(Mean)
Change FromBaseline (Mean)
Montelukast (N=101)Salmeterol (N=90)
33.5931.76
-17.08-10.61
51.1349.21
-25.56-12.09
1162.31085.8
-674.8-362.2
Difference in LS means95% CI for differencep-Value
-6.26(-10.38, -2.14)
0.003
-12.63(-21.51, -3.75)
0.006
-304.8(-518.3, -91.3)
0.005† In the analysis of these endpoints, the lower the value, the greater the response to therapy.‡ Percentage-point difference.FEV1 = Forced Expiratory Volume in 1 second; AUC = Area Under the Curve; LS = Least-Squares; CI = ConfidenceInterval.
Exercise Challenge Endpoints†—Analysis of Within-Group and Between-Group Differences at Week 4
Maximum Percent Fallin FEV1 (%) Time to Recovery (min) AUC0-60 min (%·min)
TreatmentBaseline(Mean)
Change FromBaseline (Mean)‡
Baseline(Mean)
Change FromBaseline (Mean)
Baseline(Mean)
Change FromBaseline (Mean)
Montelukast (N=101)Salmeterol (N=87)
33.5931.80
-16.00-7.40
51.1349.13
-24.69-11.33
1162.31092.7
-656.4-190.1
Difference in LS means95% CI for differencep-Value
-8.13(-12.38, -3.88)
<0.001
-12.30(-21.43, -3.17)
0.009
-443.3(-675.4, -211.2)
<0.001† In the analysis of these endpoints, the lower the value, the greater the response to therapy.‡ Percentage-point difference.FEV1 = Forced Expiratory Volume in 1 second; AUC = Area Under the Curve; LS = Least-Squares; CI = ConfidenceInterval.
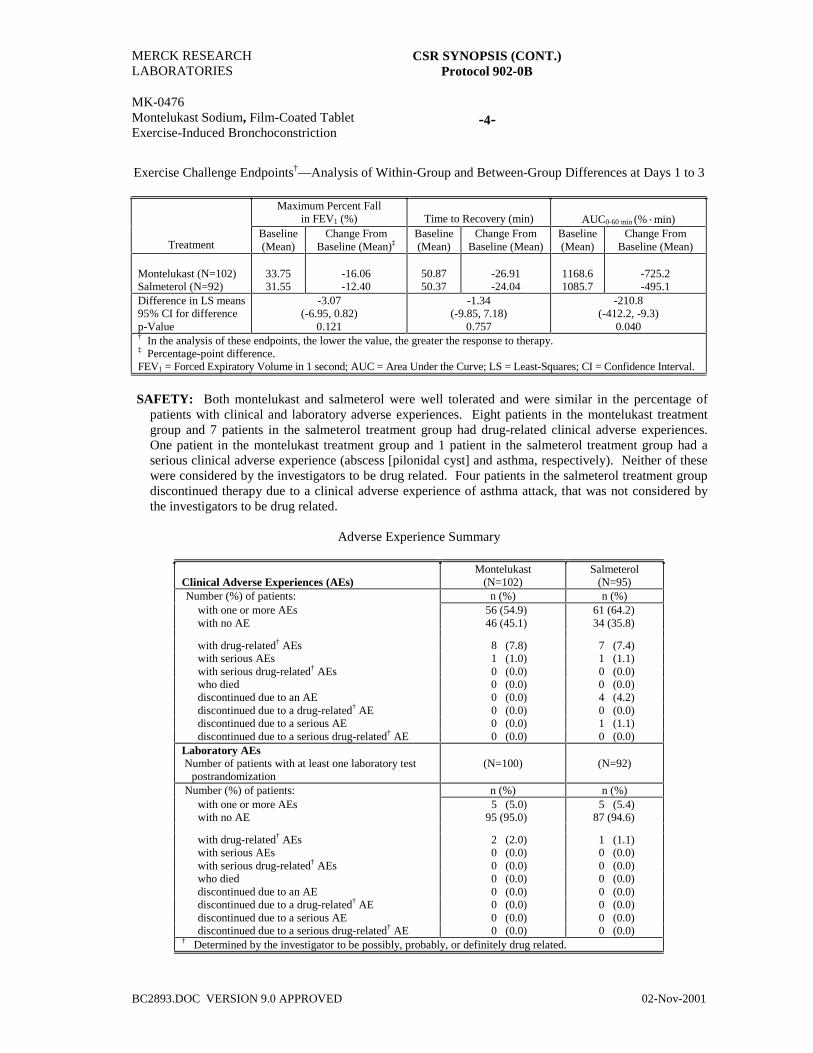
MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 902-0B
MK-0476Montelukast Sodium, Film-Coated TabletExercise-Induced Bronchoconstriction
-4-
BC2893.DOC VERSION 9.0 APPROVED 02-Nov-2001
Exercise Challenge Endpoints†—Analysis of Within-Group and Between-Group Differences at Days 1 to 3
Maximum Percent Fallin FEV1 (%) Time to Recovery (min) AUC0-60 min (%·min)
TreatmentBaseline(Mean)
Change FromBaseline (Mean)‡
Baseline(Mean)
Change FromBaseline (Mean)
Baseline(Mean)
Change FromBaseline (Mean)
Montelukast (N=102)Salmeterol (N=92)
33.7531.55
-16.06-12.40
50.8750.37
-26.91-24.04
1168.61085.7
-725.2-495.1
Difference in LS means95% CI for differencep-Value
-3.07(-6.95, 0.82)
0.121
-1.34(-9.85, 7.18)
0.757
-210.8(-412.2, -9.3)
0.040† In the analysis of these endpoints, the lower the value, the greater the response to therapy.‡ Percentage-point difference.FEV1 = Forced Expiratory Volume in 1 second; AUC = Area Under the Curve; LS = Least-Squares; CI = Confidence Interval.
SAFETY: Both montelukast and salmeterol were well tolerated and were similar in the percentage ofpatients with clinical and laboratory adverse experiences. Eight patients in the montelukast treatmentgroup and 7 patients in the salmeterol treatment group had drug-related clinical adverse experiences.One patient in the montelukast treatment group and 1 patient in the salmeterol treatment group had aserious clinical adverse experience (abscess [pilonidal cyst] and asthma, respectively). Neither of thesewere considered by the investigators to be drug related. Four patients in the salmeterol treatment groupdiscontinued therapy due to a clinical adverse experience of asthma attack, that was not considered bythe investigators to be drug related.
Adverse Experience Summary
Clinical Adverse Experiences (AEs)Montelukast
(N=102)Salmeterol
(N=95)Number (%) of patients: n (%) n (%)
with one or more AEs 56 (54.9) 61 (64.2)with no AE 46 (45.1) 34 (35.8)
with drug-related† AEs 8 (7.8) 7 (7.4)with serious AEs 1 (1.0) 1 (1.1)with serious drug-related† AEs 0 (0.0) 0 (0.0)who died 0 (0.0) 0 (0.0)discontinued due to an AE 0 (0.0) 4 (4.2)discontinued due to a drug-related† AE 0 (0.0) 0 (0.0)discontinued due to a serious AE 0 (0.0) 1 (1.1)discontinued due to a serious drug-related† AE 0 (0.0) 0 (0.0)
Laboratory AEsNumber of patients with at least one laboratory test
postrandomization(N=100) (N=92)
Number (%) of patients: n (%) n (%)with one or more AEs 5 (5.0) 5 (5.4)with no AE 95 (95.0) 87 (94.6)
with drug-related† AEs 2 (2.0) 1 (1.1)with serious AEs 0 (0.0) 0 (0.0)with serious drug-related† AEs 0 (0.0) 0 (0.0)who died 0 (0.0) 0 (0.0)discontinued due to an AE 0 (0.0) 0 (0.0)discontinued due to a drug-related† AE 0 (0.0) 0 (0.0)discontinued due to a serious AE 0 (0.0) 0 (0.0)discontinued due to a serious drug-related† AE 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related.

MERCK RESEARCHLABORATORIES
CSR SYNOPSIS (CONT.)Protocol 902-0B
MK-0476Montelukast Sodium, Film-Coated TabletExercise-Induced Bronchoconstriction
-5-
BC2893.DOC VERSION 9.0 APPROVED 02-Nov-2001
CONCLUSIONS: (1) At the end of the dosing interval, montelukast (10 mg once daily) demonstratesgreater attenuation of EIB in patients with chronic asthma compared with salmeterol (2 puffs, every12 hours) after Weeks 4 and 8 of treatment; montelukast and salmeterol provide similar attenuation ofEIB at Days 1 to 3 of treatment. (2) Montelukast demonstrates a persistent magnitude of attenuation ofEIB in patients with chronic asthma over 8 weeks of treatment. (3) In a post-hoc analysis, more than50% of patients treated with montelukast achieve a commonly used threshold level of effect (amaximum percent fall in FEV1 after exercise of less than 20%) at Week 8 of treatment. (4) Montelukastand salmeterol have a favorable safety profile and are well tolerated over 8 weeks of therapy.
AUTHORS:
Kelly M. Firriolo, B.S.Medical Program CoordinatorClinical Development
Joris Menten, M.S.Senior StatisticianCBARDS
Jonathan A. Leff, M.D.Senior DirectorClinical Development Studies Program
Jonathan M. Edelman, M.D.Senior DirectorClinical Development

MK-0476 Prot. No. 907 Montelukast Pediatric Exacerbation Study (PREVIA)
0476_P907_02_Synopsis VERSION 5.0 APPROVED 11-Aug-2005 Restricted Confidential – Limited Access
2. Synopsis
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, 4- and 5-mg Chewable Tablets Asthma
PROTOCOL TITLE/NO.: A Multicenter, Double-Blind, Placebo-Controlled, Randomized, Parallel-Group Study to Determine the Efficacy of Montelukast in the Prevention of Exacerbations in Asthmatic Patients Aged 2 to 5 Years
#907
INVESTIGATORS/STUDY CENTERS: Multicenter (68) in Africa (2), Asia (13), Europe (41), North America (4), South America (8)
PUBLICATIONS: (1) Bisgaard H, Zielen S, Garcia-Garcia ML, Johnston SL, Gilles L, Menten J, et al. Montelukast reduces asthma exacerbations in 2- to 5-year-old children with intermittent asthma. Am J Respir Crit Care Med 2005;171:315-22. (2) Online data supplement for the following publication: Bisgaard, H; Zielen, S; Garcia Garcia, ML; Johnston, S; Gilles, L; Menten, J; Tozzi, CA; Polos, P. Montelukast reduces asthma exacerbations in 2 to 5 year old children with intermittent asthma. Am J Respir Crit Care Med 2005;171:315-22.
PRIMARY THERAPY PERIOD: 06-Feb-2001 to 03-Dec-2002 CLINICAL PHASE: IV DURATION OF TREATMENT: Period I: 3 weeks (included a 2-week placebo run in).
Period II: 48 weeks (approximately 12 months). OBJECTIVES: Primary: To determine the effect of a 12-month treatment period course of montelukast,
compared to placebo, on the rate of exacerbation episodes in 2- to 5-year-old asthmatic children. Secondary: To determine the effect of a 12-month treatment course of montelukast, compared to placebo, on the: number of treatment courses of oral/inhaled steroids; total duration of exacerbation episodes; percentage of days without asthma; total severity of exacerbation episodes; blood eosinophil counts; proportion of patients with exacerbation episodes; and percentage of exacerbations caused by viral infection. Tertiary: To determine the effect of a 12-month treatment course of montelukast, compared to placebo, on the: time to first exacerbation episode; asthma-related resource utilization (as collected on the diary cards and day-care/work-loss questionnaire); and safety and tolerability profile.
STUDY DESIGN: Multicenter, double blind (with in-house blinding procedures), placebo controlled, randomized, parallel group
SUBJECT/PATIENT DISPOSITION:
Montelukast Placebo Total SCREENING FAILURES: 219 RANDOMIZED: 278 271 549
Male (age range) 173 (2 to 6) 177 (2 to 5) 350 (2 to 6) Female (age range) 105 (2 to 6) 94 (2 to 6) 199 (2 to 6)
COMPLETED: 259 241 500 DISCONTINUED: 19 30 49
Clinical adverse experience 1 10 11 Laboratory adverse experience 0 0 0 Other 18 20 38
DOSAGE/FORMULATION NOS.: One montelukast 4-mg chewable tablet (CT) (MR-4385) or matching-image placebo (MR-3778, MR-4373) was administered once daily at bedtime. Patients who turned 6 years old during the study were switched, according to their original allocation, to montelukast 5 mg CT (MR-4384) or matching-image placebo (MR-4288, MR-4374) for Visits 6 and 7.
DIAGNOSIS/INCLUSION CRITERIA: Boys and girls with a clinical history of episodic asthmatic symptoms resulting from an upper respiratory infection (common cold), consisting of, but not limited to, cough, wheeze, shortness of breath with at least 3 physician-diagnosed asthmatic episodes lasting for at

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, 4- and 5-mg Chewable Tablets Asthma
-2-
0476_P907_02_Synopsis VERSION 5.0 APPROVED 11-Aug-2005 Restricted Confidential – Limited Access
least 3 days within the past 12 months and leading to treatment with β-agonist (at least 1 of the 3
episodes must have occurred within 6 months of Visit 1). Between asthmatic episodes, patients had to have mild asthma characterized by the absence of symptoms (for example, cough, shortness of breath, chest tightness, or wheeze) and short-acting bronchodilator use in a typical week over the 3 months prior to Visit 1. To be randomized, patients were to have symptoms (defined as average score on 4 daily daytime symptom questions of >0.5) on no more than 2 consecutive days and on no more than a total of 4 days between Visits 2 and 3.
EVALUATION CRITERIA: Efficacy: (1) Number of exacerbation episodes (any 3 consecutive days with daytime symptoms [average score of 4 daily daytime symptom questions of at least 1.0/day] AND at least 2 treatments of β-agonist/day [puffs, tablets, teaspoons, or nebulization treatments] OR use of inhaled corticosteroids [during at least 3 consecutive days] or oral/rectal/intramuscular corticosteroids [during 1 or more days] OR a hospitalization for asthma). The end of an exacerbation was defined as 2 consecutive days out of the hospital in which there were no daytime symptoms (average score ≤0.25) AND <2 treatments of β-agonist/day AND no use of inhaled or oral/rectal/intramuscular corticosteroids. (2) Number of treatment courses or oral/inhaled steroids. (3) Duration of exacerbation episodes. (4) Percentage of days without asthma (a day with no daytime or overnight symptoms recorded on the diary card AND no daytime or overnight β-agonist use AND no inhaled or oral corticosteroids for asthma AND no asthma-related resource utilization [no hospitalization, no emergency room, and no unscheduled physician visit]). (5) Severity of exacerbation episodes (based on average daytime symptom score, daily β-agonist use, oral and inhaled steroid use, and hospitalization). (6) Eosinophil counts. (7) Proportion of patients with exacerbation episodes. (8) Percentage of exacerbation episodes associated with viral infection. (9) Time to first exacerbation episode. (10) Asthma-related resource utilization (as collected on the diary card and day-care/work-loss questionnaire). Safety: Vital signs and adverse experiences.
STATISTICAL PLANNING AND ANALYSIS: The primary efficacy variable, rate of exacerbation episodes, was analyzed using a Poisson regression methodology. The model for analyzing the number of episodes contained a factor for treatment group and geographic region, and an offset for number of days in study; the variance was estimated using a robust variance estimator. Safety information was evaluated by tabulating adverse experiences.
RESULTS: Efficacy: Montelukast significantly reduced the rate of exacerbation episodes (p≤0.001, primary
endpoint), the rate of inhaled corticosteroid courses (p=0.027), eosinophil counts (p=0.010), and significantly increased time to first exacerbation episode (p=0.024). Numeric and borderline significant improvements were observed for most other secondary endpoints. Treatment did not influence duration or severity of individual exacerbation episodes.
Comparison (Montelukast versus Placebo) Rate (year) Relative Rate† Rate Reduction (%)
Endpoint N Estimate (95% CI) Estimate (95% CI) Estimate (95% CI) p-Value Number of Exacerbation Episodes Montelukast 265 1.60 (1.35; 1.88) 0.68 (0.56; 0.83) 31.9 (16.9; 44.1) ≤0.001 Placebo 257 2.34 (1.97; 2.79) Number of Inhaled Corticosteroid Courses Montelukast 265 0.66 (0.46; 0.94) 0.60 (0.38; 0.94) 39.8 (5.5; 61.7) 0.027 Placebo 257 1.10 (0.83; 1.45) Number of Oral Corticosteroid Courses Montelukast 265 0.53 (0.40; 0.70) 0.82 (0.54; 1.25) 17.5 (-25.5; 45.8) 0.368 Placebo 257 0.64 (0.47; 0.88) † A relative rate <1 is in favor of montelukast.

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, 4- and 5-mg Chewable Tablets Asthma
-3-
0476_P907_02_Synopsis VERSION 5.0 APPROVED 11-Aug-2005 Restricted Confidential – Limited Access
Safety: The proportions of patients with any clinical adverse experience (p=0.193), with a drug-related
clinical adverse experience (p=0.120), or with a serious clinical adverse experience (p=0.165) were similar. Significantly more patients in the placebo treatment group discontinued due to a clinical adverse experience (p=0.005); 4 patients in the placebo group and none in the montelukast group discontinued due to asthma-related adverse experiences. Laboratory adverse experiences occurred infrequently in both treatment groups.
Montelukast Placebo
Clinical Adverse Experiences (AEs) (N=278) (N=271) Number (%) of patients: n (%) n (%) With one or more AEs 257 (92.4) 240 (88.6) With no AE 21 (7.6) 31 (11.4)
With drug-related† AEs 14 (5.0) 11 (4.1) With serious AEs 24 (8.6) 34 (12.5) With serious drug-related AEs 1 (0.4) 1 (0.4) Who died 0 (0.0) 0 (0.0) Discontinued due to AEs 1‡ (0.4) 10 (3.7) Discontinued due to drug-related AEs 0 (0.0) 3 (1.1) Discontinued due to serious AEs 1 (0.4) 3 (1.1) Discontinued due to serious drug-related AEs 0 (0.0) 1 (0.4)
Laboratory AEs (N=278) (N=271) Number (%) of patients: n (%)§ n (%)§ With at least one laboratory test postbaseline 261 251
With one or more AEs 5 (1.9) 14 (5.6) With no AE 256 (98.1) 237 (94.4)
With drug-related AEs 1 (0.4) 2 (0.8) With serious AEs 0 (0.0) 0 (0.0) With serious drug-related AEs 0 (0.0) 0 (0.0) Who died 0 (0.0) 0 (0.0) Discontinued due to AEs 0 (0.0) 0 (0.0) Discontinued due to drug-related AEs 0 (0.0) 0 (0.0) Discontinued due to serious AEs 0 (0.0) 0 (0.0) Discontinued due to serious drug-related AEs 0 (0.0) 0 (0.0)
† Determined by the investigator to be possibly, probably, or definitely drug related. ‡ Fisher Exact p-value: 0.005. § The percent = number of patients within the laboratory adverse experience category/
number of patients with one or more laboratory tests postbaseline multiplied by 100.
CONCLUSIONS: In patients aged 2 to 5 years with mild asthma with episodic exacerbations treated over
a 1-year period, it can be concluded that: (1) montelukast reduces the rate of asthma exacerbation episodes; (2) montelukast increases the time to first asthma exacerbation episode; (3) montelukast reduces rescue β-agonist and corticosteroid use; (4) montelukast does not reduce the severity or duration of asthma exacerbation episodes; and (5) montelukast is generally well tolerated.
AUTHORS: Gertrude Noonan, B.A. Leen Gilles, M.Sc. Barbara Knorr, M.D. Clinical Associate Senior Statistician Executive Director Respiratory, Allergy, & Urology CBARDS Respiratory, Allergy, & Urology

MK-0476 Prot. No. 910 Pediatric Asthma Control Study (MOSAIC)
0476_P910_02_Synopsis VERSION 5.5 APPROVED 23-Aug-2005 Restricted Confidential – Limited Access
2. Synopsis
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, 5-mg Chewable Tablets Asthma
PROTOCOL TITLE/NO.: A Double-Blind, Randomized, Double-Dummy, Multicenter Study to Evaluate and Compare Oral Montelukast and Inhaled Fluticasone in the Control of Asthma for 6- to 14-Year-Olds with Mild Persistent Asthma
#910
INVESTIGATORS/STUDY CENTERS: Multicenter (104 investigators in 24 countries ) PUBLICATIONS: (1) Polos P, Garcia MLG, Gilles L, Tozzi CA, Knorr B, Reiss TF. Montelukast
versus Fluticasone in Patients Aged 6 to 14 Years with Mild Persistent Asthma: the MOSAIC Study. Eur Respir J 2004; 24 (Suppl 48):377s. (2) Garcia-Garcia M Luz, Ulrich Wahn, Leen Gilles, Arlene Swern, Carol Tozzi, Peter Polos. Montelukast, compared with fluticasone, for Control of Asthma Among 6- to 14-Year-Old Patients With Mild Asthma: The MOSAIC Study. Pediatrics 2005;116:360-369.
PRIMARY THERAPY PERIOD: 19-Jul-2001 to 26-Jun-2003 CLINICAL PHASE: IV DURATION OF TREATMENT: Period 1—4 weeks; Period II—12 months OBJECTIVE(S): Primary objective: To compare the efficacy of montelukast with inhaled fluticasone in
the control of mild persistent asthma in patients 6 to 14 years of age, as measured by the percentage of asthma rescue-free days over 1 year of treatment. Secondary objectives: To compare the effect of 1 year of treatment with montelukast and fluticasone on the following variables: 1) the change from baseline in percent predicted forced expiratory volume in 1 second (FEV1); 2) the proportion of patients requiring additional asthma therapy; 3) the proportion of patients experiencing an asthma attack; 4) the average percentage of days with β-agonist use; 5) pediatric asthma-related quality of life; 6) peripheral blood eosinophil levels. Tertiary objectives: To compare the effect of 1 year of treatment of montelukast and fluticasone on the periodic report of asthma control and asthma-related patient school loss and parent work loss.
STUDY DESIGN: Double-blind, randomized, double-dummy, 2-arm, parallel-group study conducted under in-house blinding procedures.
SUBJECT/PATIENT DISPOSITION: Montelukast Fluticasone Total
SCREENING FAILURES: 438 RANDOMIZED: 495 499 994
Male (age range in years) 321 (6 to 14) 292 (5 to 14) 613 (5 to 14) Female (age range) 174 (6 to 14) 207 (6 to 15) 381 (6 to 15)
COMPLETED: 459 (92.7) 466 (93.4) 925 (93.1) DISCONTINUED: 36 (7.3) 33 (6.6) 69 (6.9)
Clinical adverse experience 6 (1.2) 1 (0.2) 7 (0.7) Protocol deviation 9 (1.8) 16 (3.2) 25 (2.5) Lost to follow-up 6 (1.2) 5 (1.0) 11 (1.1) Withdrew consent 11 (2.2) 4 (0.8) 15 (1.5) Lack of Efficacy 2 (0.4) 1 (0.2) 3 (0.3) Other 2 (0.4) 6 (1.2) 8 (0.8)

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, 5-mg Chewable Tablets Asthma
-2-
0476_P910_02_Synopsis VERSION 5.5 APPROVED 23-Aug-2005 Restricted Confidential – Limited Access
DOSAGE/FORMULATION NOS.: Montelukast 5-mg chewable tablet (CT) was administered once
daily at bedtime and inhaled fluticasone 100 µg (50 µg x 2 puffs) was administered twice daily. Period I: matching placebo for montelukast CT (MR-4288; MR-4374) and placebo for fluticasone metered dose inhaler (MDI) (MR-4457); Period II: montelukast 5-mg CT (MR-4384 and MR-4485) and matching placebo for fluticasone MDI (MR-4457); matching placebo for montelukast CT (MR-4288; MR-4374) and fluticasone MDI (MR-4459; MR-4655; MR-4527; MR-4594; MR-4585). Patients who turned 15 years during the study were switched to montelukast 10-mg film-coated tablets (FCTs) (MR-4380) or matching placebo for montelukast FCTs (MR-4219).
DIAGNOSIS/INCLUSION CRITERIA: Male or female patients, between 6 and 14 years of age, with a clinical history of asthma for at least 12 months and who have mild persistent asthma (at Step 2 of the Global Initiative for Asthma [GINA] guidelines). Patients must have a FEV1 or peak expiratory flow (PEF) value of ≥80% of predicted (pre-bronchodilator) at least 2 times in Period I (Visits 1, 2, or 3) and FEV1 or PEF ≥70% predicted at Visit 3, and demonstrate a reversibility of ≥12% (FEV1 or PEF). Historical reversibility within the past 12 months is acceptable. Patients may be taking short-acting β-agonist alone, or in combination with 1 controller therapy. Patients must demonstrate symptoms requiring β-agonist use on at least 2 and at most 6 days of a week for the 2 weeks prior to Visit 3.
EVALUATION CRITERIA: Diary cards collecting information on asthma rescue medication use (short-acting β-agonists, oral corticosteroids and other marketed controller therapy), and resource utilization (Emergency Department visit, Hospitalization and Visit to Physician); spirometry measurements; validated Pediatric Asthma-related Quality-of-Life Questionnaire; questionnaire based on the control domain of the validated Pediatric Asthma Therapy Assessment Questionnaire; eosinophil counts.
STATISTICAL PLANNING AND ANALYSIS: The primary efficacy variable, the percentage asthma Rescue-Free Days (RFDs), was analyzed using an analysis of covariance, with factors for treatment and region, and the baseline value (calculated from Period I) as a covariate. To address the primary hypothesis of whether montelukast is non-inferior to fluticasone, a 95% confidence interval (CI) was constructed for the difference in least squares means between treatment groups (montelukast— fluticasone). If the lower bound of the 95% CI was above the non-inferiority limit of -7%, montelukast was considered non-inferior to fluticasone.
RESULTS: Efficacy: The least squares mean percentage of RFDs during the 12-month study period was 83.6% in the montelukast treatment group and 86.4% in the fluticasone treatment group. The difference in least squares means was -2.8% in favor of fluticasone with a 95% confidence interval (-4.7%; -0.9%) entirely above the non-inferiority limit of -7%. Both montelukast and fluticasone increased FEV1, reduced the percentage of days with β-agonist use, improved the asthma-related quality of life, and decreased peripheral eosinophil counts. Fluticasone, but not montelukast, increased percent predicted FEV1. This difference between treatments may be a reflection of an underlying difference between treatments in overall growth, which was higher in the montelukast treatment group than in the fluticasone treatment group. Results for the primary and secondary endpoints are given in the table below.
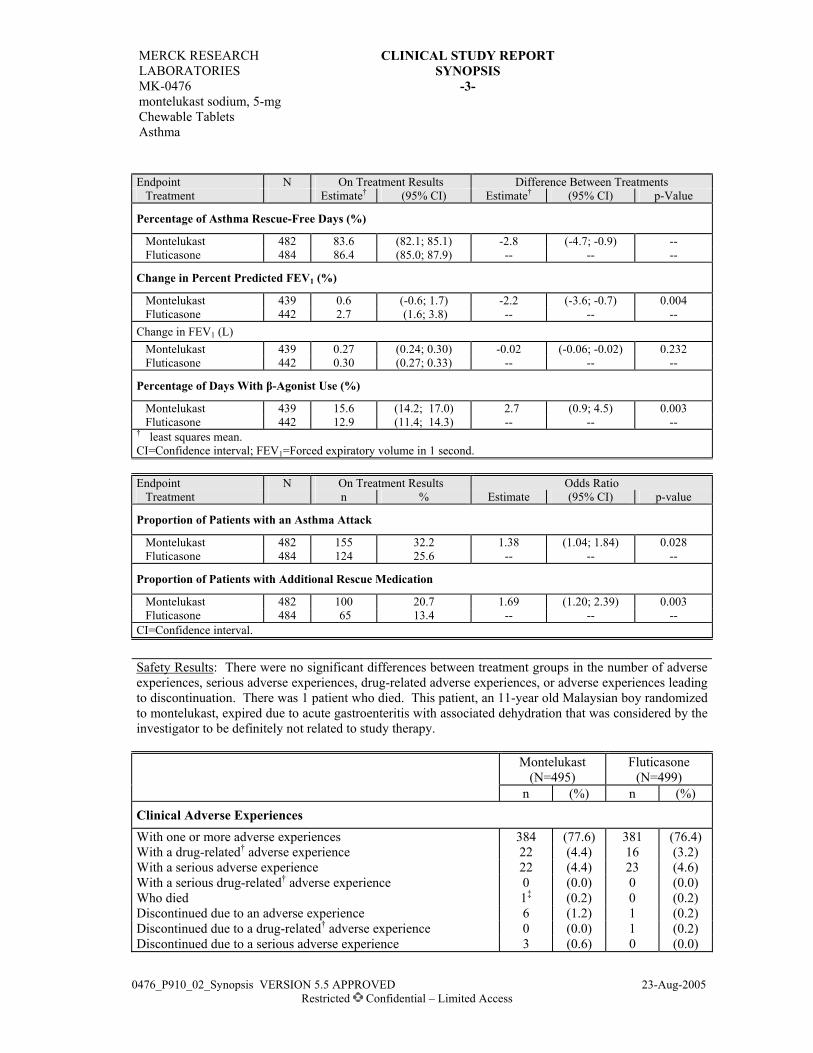
MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, 5-mg Chewable Tablets Asthma
-3-
0476_P910_02_Synopsis VERSION 5.5 APPROVED 23-Aug-2005 Restricted Confidential – Limited Access
Endpoint N On Treatment Results Difference Between Treatments Treatment Estimate† (95% CI) Estimate† (95% CI) p-Value
Percentage of Asthma Rescue-Free Days (%)
Montelukast 482 83.6 (82.1; 85.1) -2.8 (-4.7; -0.9) -- Fluticasone 484 86.4 (85.0; 87.9) -- -- --
Change in Percent Predicted FEV1 (%)
Montelukast 439 0.6 (-0.6; 1.7) -2.2 (-3.6; -0.7) 0.004 Fluticasone 442 2.7 (1.6; 3.8) -- -- -- Change in FEV1 (L) Montelukast 439 0.27 (0.24; 0.30) -0.02 (-0.06; -0.02) 0.232 Fluticasone 442 0.30 (0.27; 0.33) -- -- --
Percentage of Days With β-Agonist Use (%)
Montelukast 439 15.6 (14.2; 17.0) 2.7 (0.9; 4.5) 0.003 Fluticasone 442 12.9 (11.4; 14.3) -- -- -- † least squares mean. CI=Confidence interval; FEV1=Forced expiratory volume in 1 second.
Endpoint N On Treatment Results Odds Ratio Treatment n % Estimate (95% CI) p-value
Proportion of Patients with an Asthma Attack
Montelukast 482 155 32.2 1.38 (1.04; 1.84) 0.028 Fluticasone 484 124 25.6 -- -- --
Proportion of Patients with Additional Rescue Medication
Montelukast 482 100 20.7 1.69 (1.20; 2.39) 0.003 Fluticasone 484 65 13.4 -- -- -- CI=Confidence interval.
Safety Results: There were no significant differences between treatment groups in the number of adverse experiences, serious adverse experiences, drug-related adverse experiences, or adverse experiences leading to discontinuation. There was 1 patient who died. This patient, an 11-year old Malaysian boy randomized to montelukast, expired due to acute gastroenteritis with associated dehydration that was considered by the investigator to be definitely not related to study therapy.
Montelukast
(N=495) Fluticasone
(N=499) n (%) n (%) Clinical Adverse Experiences With one or more adverse experiences 384 (77.6) 381 (76.4) With a drug-related† adverse experience 22 (4.4) 16 (3.2) With a serious adverse experience 22 (4.4) 23 (4.6) With a serious drug-related† adverse experience 0 (0.0) 0 (0.0) Who died 1‡ (0.2) 0 (0.2) Discontinued due to an adverse experience 6 (1.2) 1 (0.2) Discontinued due to a drug-related† adverse experience 0 (0.0) 1 (0.2) Discontinued due to a serious adverse experience 3 (0.6) 0 (0.0)

MERCK RESEARCH LABORATORIES
CLINICAL STUDY REPORT SYNOPSIS
MK-0476 montelukast sodium, 5-mg Chewable Tablets Asthma
-4-
0476_P910_02_Synopsis VERSION 5.5 APPROVED 23-Aug-2005 Restricted Confidential – Limited Access
Laboratory Adverse Experiences
With at least one laboratory test postbaseline§ 434 433 With one or more adverse experiences 9/434 (2.1) 6/433 (1.4) With no adverse experiences 425 (97.9) 427 (98.6) With a drug-related† adverse experience 2/434 (0.5) 0/433 (0.0) With a serious adverse experience 0/434 (0.0) 0/433 (0.0) With a serious drug-related† adverse experience 0/434 (0.0) 0/433 (0.0) Who died 0/434 (0.0) 0/433 (0.0) Discontinued due to an adverse experience 0/434 (0.0) 0/433 (0.0) Discontinued due to drug-related adverse experiences 0/434 (0.0) 0/433 (0.0) Discontinued due to serous adverse experiences 0/434 (0.0) 0/433 (0.0) † Determined by the investigator to be possibly, probably or definitely drug related. ‡ An 11-year-old Malaysian boy died due to gastroenteritis with dehydration that was determined by the
investigator to be definitely not related to therapy with montelukast. § Laboratory safety tests, including a blood eosinophil count, were performed prerandomization and at
Visit 6 or at a discontinuation visit (blood eosinophil count only). CONCLUSIONS: In patients, aged 6 to 14 years with mild persistent asthma (Step 2 of the GINA
guidelines), and treated over a 1-year period, it can be concluded that: 1) montelukast is non-inferior to fluticasone as measured by the percentage of asthma rescue-free days; 2) montelukast and fluticasone are generally well tolerated.
AUTHORS: Joan McComas, M.S., M.M.H. Leen Gilles, M.Sc. Barbara Knorr, M.D. MPCS Senior Statistician Executive Director Respiratory, Allergy & Urology CBARDS Respiratory, Allergy & Urology


















