
Materials Research Bulletin - UAntwerpenematweb.cmi.ua.ac.be/emat/pdf/1938.pdf3460 Batuk et...
7
Structure and magnetic properties of a new anion-deficient perovskite Pb 2 Ba 2 BiFe 4 ScO 13 with crystallographic shear structure Maria Batuk a, *, Oleg A. Tyablikov b , Alexander A. Tsirlin c,d , Sergey M. Kazakov b , Marina G. Rozova b , Konstantin V. Pokholok b , Dmitry S. Filimonov b , Evgeny V. Antipov b , Artem M. Abakumov a,b , Joke Hadermann a a Electron Microscopy for Materials Research (EMAT), University of Antwerp, Groenenborgerlaan 171, B-2020, Antwerp, Belgium b Department of Chemistry, Moscow State University, 119991 Moscow, Russia c National Institute of Chemical Physics and Biophysics, 12618 Tallinn, Estonia d Max Planck Institute for Chemical Physics of Solids, No ¨thnitzer Str. 40, 01187 Dresden, Germany 1. Introduction The crystallographic shear (CS) mechanism implies a cut of a given structure along a certain crystallographic plane, and a displacement of one part of the structure relative to the other part over a vector that is a fraction of the translation of the basic unit cell. The CS planes are designated as 1/m[u v w](h k l), where 1/m[u v w] is the displacement vector (fraction of the [u v w] vector of the basic structure) and (h k l) is the orientation of the CS planes [1–10]. In perovskites, periodically spaced ½[1 1 0] ð ¯ 1 0 1Þ p CS planes lead to the formation of an oxygen-deficient homologous series A n B n O 3n2 [11]. Members of this series with n = 4–6 are already known [12–18], their crystal structures are represented in Fig. 1. The structures are built of perovskite blocks comprising (n 2) layers of the BO 6 octahedra. The blocks are connected by chains of edge-sharing BO 5 distorted tetragonal pyramids. Fe 3+ is the typical B-cation in these structures, as it is able to adopt both a five-fold and six-fold coordination environment. The chains of the BO 5 pyramids and BO 6 octahedra together delimit six-sided tunnels that are occupied by two columns of A-cations with a lone electron pair (Pb 2+ ) . The lone electron pair plays an essential role in the formation of the tunnels by completing the asymmetric oxygen environment of the Pb 2+ cations. An important feature of these structures is the simultaneous presence of Pb 2+ and alkali- earth metal cations (Sr 2+ or Ba 2+ ) in the A-positions of the perovskite blocks. The alkali-earth cations promote the formation of structures with the simplest ð ¯ 101Þ p CS planes, otherwise, more complex incommensurately modulated structures occur [19]. The A n B n O 3n2 compounds feature unit-cell parameters that are related to the parameters of the basic perovskite subcell as a H2a p , b b p , c 9.7 A ˚ + (n 2)H2a p , where the nearly constant separation of 9.7 A ˚ refers to the thickness of the oxygen-deficient CS plane, and a p 4.0 A ˚ is the unit cell parameter of the cubic perovskite subcell. Similar to Pb 2+ , Bi 3+ cations have a lone electron pair and thus promote the formation of the CS planes. The partial replacement of Pb 2+ by Bi 3+ allows varying n in A n B n O 3n2 while keeping only trivalent cations in the B-position of the perovskite blocks. To Materials Research Bulletin 48 (2013) 3459–3465 A R T I C L E I N F O Article history: Received 8 February 2013 Received in revised form 12 March 2013 Accepted 9 May 2013 Available online 17 May 2013 Keywords: A. Oxides C. Electron microscopy C. X-ray diffraction D. Magnetic properties A B S T R A C T Pb 2 Ba 2 BiFe 4 ScO 13 , a new n = 5 member of the oxygen-deficient perovskite-based A n B n O 3n2 homologous series, was synthesized using a solid-state method. The crystal structure of Pb 2 Ba 2 BiFe 4 ScO 13 was investigated by a combination of synchrotron X-ray powder diffraction, electron diffraction, high-angle annular dark-field scanning transmission electron microscopy and Mo ¨ ssbauer spectroscopy. At 900 K, it crystallizes in the Ammm space group with the unit cell parameters a = 5.8459(1) A ˚ , b = 4.0426(1) A ˚ , and c = 27.3435(1) A ˚ . In the Pb 2 Ba 2 BiFe 4 ScO 13 structure, quasi-two-dimensional perovskite blocks are periodically interleaved with ½[1 1 0] ð ¯ 1 0 1Þ p crystallographic shear (CS) planes. At the CS planes, the corner-sharing FeO 6 octahedra are transformed into chains of edge-sharing FeO 5 distorted tetragonal pyramids. B-positions of the perovskite blocks between the CS planes are jointly occupied by Fe 3+ and Sc 3+ . The chains of the FeO 5 pyramids and (Fe,Sc)O 6 octahedra delimit six-sided tunnels that are occupied by double columns of cations with a lone electron pair (Pb 2+ ). The remaining A-cations (Bi 3+ , Ba 2+ ) occupy positions in the perovskite block. According to the magnetic susceptibility measurements, Pb 2 Ba 2 BiFe 4 ScO 13 is antiferromagnetically ordered below T N 350 K. ß 2013 Elsevier Ltd. All rights reserved. * Corresponding author. Tel.: +32 32653695; fax: +32 32653257. E-mail address: [email protected] (M. Batuk). Contents lists available at SciVerse ScienceDirect Materials Research Bulletin jo u rn al h om ep age: ww w.els evier.c o m/lo c ate/mat res b u 0025-5408/$ – see front matter ß 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.materresbull.2013.05.028
Transcript of Materials Research Bulletin - UAntwerpenematweb.cmi.ua.ac.be/emat/pdf/1938.pdf3460 Batuk et...

Materials Research Bulletin 48 (2013) 3459–3465
Structure and magnetic properties of a new anion-deficient perovskitePb2Ba2BiFe4ScO13 with crystallographic shear structure
Maria Batuk a,*, Oleg A. Tyablikov b, Alexander A. Tsirlin c,d, Sergey M. Kazakov b, Marina G. Rozova b,Konstantin V. Pokholok b, Dmitry S. Filimonov b, Evgeny V. Antipov b, Artem M. Abakumov a,b,Joke Hadermann a
a Electron Microscopy for Materials Research (EMAT), University of Antwerp, Groenenborgerlaan 171, B-2020, Antwerp, Belgiumb Department of Chemistry, Moscow State University, 119991 Moscow, Russiac National Institute of Chemical Physics and Biophysics, 12618 Tallinn, Estoniad Max Planck Institute for Chemical Physics of Solids, Nothnitzer Str. 40, 01187 Dresden, Germany
A R T I C L E I N F O
Article history:
Received 8 February 2013
Received in revised form 12 March 2013
Accepted 9 May 2013
Available online 17 May 2013
Keywords:
A. Oxides
C. Electron microscopy
C. X-ray diffraction
D. Magnetic properties
A B S T R A C T
Pb2Ba2BiFe4ScO13, a new n = 5 member of the oxygen-deficient perovskite-based AnBnO3n�2 homologous
series, was synthesized using a solid-state method. The crystal structure of Pb2Ba2BiFe4ScO13 was
investigated by a combination of synchrotron X-ray powder diffraction, electron diffraction, high-angle
annular dark-field scanning transmission electron microscopy and Mossbauer spectroscopy. At 900 K, it
crystallizes in the Ammm space group with the unit cell parameters a = 5.8459(1) A, b = 4.0426(1) A, and
c = 27.3435(1) A. In the Pb2Ba2BiFe4ScO13 structure, quasi-two-dimensional perovskite blocks are
periodically interleaved with ½[1 1 0] ð1 0 1Þp crystallographic shear (CS) planes. At the CS planes, the
corner-sharing FeO6 octahedra are transformed into chains of edge-sharing FeO5 distorted tetragonal
pyramids. B-positions of the perovskite blocks between the CS planes are jointly occupied by Fe3+ and
Sc3+. The chains of the FeO5 pyramids and (Fe,Sc)O6 octahedra delimit six-sided tunnels that are occupied
by double columns of cations with a lone electron pair (Pb2+). The remaining A-cations (Bi3+, Ba2+) occupy
positions in the perovskite block. According to the magnetic susceptibility measurements,
Pb2Ba2BiFe4ScO13 is antiferromagnetically ordered below TN �350 K.
� 2013 Elsevier Ltd. All rights reserved.
Contents lists available at SciVerse ScienceDirect
Materials Research Bulletin
jo u rn al h om ep age: ww w.els evier .c o m/lo c ate /mat res b u
1. Introduction
The crystallographic shear (CS) mechanism implies a cut of agiven structure along a certain crystallographic plane, and adisplacement of one part of the structure relative to the otherpart over a vector that is a fraction of the translation of the basicunit cell. The CS planes are designated as 1/m[u v w](h k l), where1/m[u v w] is the displacement vector (fraction of the [u v w]vector of the basic structure) and (h k l) is the orientation of the CSplanes [1–10]. In perovskites, periodically spaced ½[1 1 0] ð1 0 1ÞpCS planes lead to the formation of an oxygen-deficient homologousseries AnBnO3n�2 [11]. Members of this series with n = 4–6 arealready known [12–18], their crystal structures are represented inFig. 1. The structures are built of perovskite blocks comprising(n � 2) layers of the BO6 octahedra. The blocks are connected bychains of edge-sharing BO5 distorted tetragonal pyramids. Fe3+ isthe typical B-cation in these structures, as it is able to adopt both a
* Corresponding author. Tel.: +32 32653695; fax: +32 32653257.
E-mail address: [email protected] (M. Batuk).
0025-5408/$ – see front matter � 2013 Elsevier Ltd. All rights reserved.
http://dx.doi.org/10.1016/j.materresbull.2013.05.028
five-fold and six-fold coordination environment. The chains of theBO5 pyramids and BO6 octahedra together delimit six-sidedtunnels that are occupied by two columns of A-cations with alone electron pair (Pb2+). The lone electron pair plays an essentialrole in the formation of the tunnels by completing the asymmetricoxygen environment of the Pb2+ cations. An important feature ofthese structures is the simultaneous presence of Pb2+ and alkali-earth metal cations (Sr2+ or Ba2+) in the A-positions of theperovskite blocks. The alkali-earth cations promote the formationof structures with the simplest ð101Þp CS planes, otherwise, morecomplex incommensurately modulated structures occur [19]. TheAnBnO3n�2 compounds feature unit-cell parameters that arerelated to the parameters of the basic perovskite subcell as a
�H2ap, b �bp, c �9.7 A + (n � 2)H2ap, where the nearly constantseparation of 9.7 A refers to the thickness of the oxygen-deficientCS plane, and ap �4.0 A is the unit cell parameter of the cubicperovskite subcell.
Similar to Pb2+, Bi3+ cations have a lone electron pair and thuspromote the formation of the CS planes. The partial replacement ofPb2+ by Bi3+ allows varying n in AnBnO3n�2 while keeping onlytrivalent cations in the B-position of the perovskite blocks. To
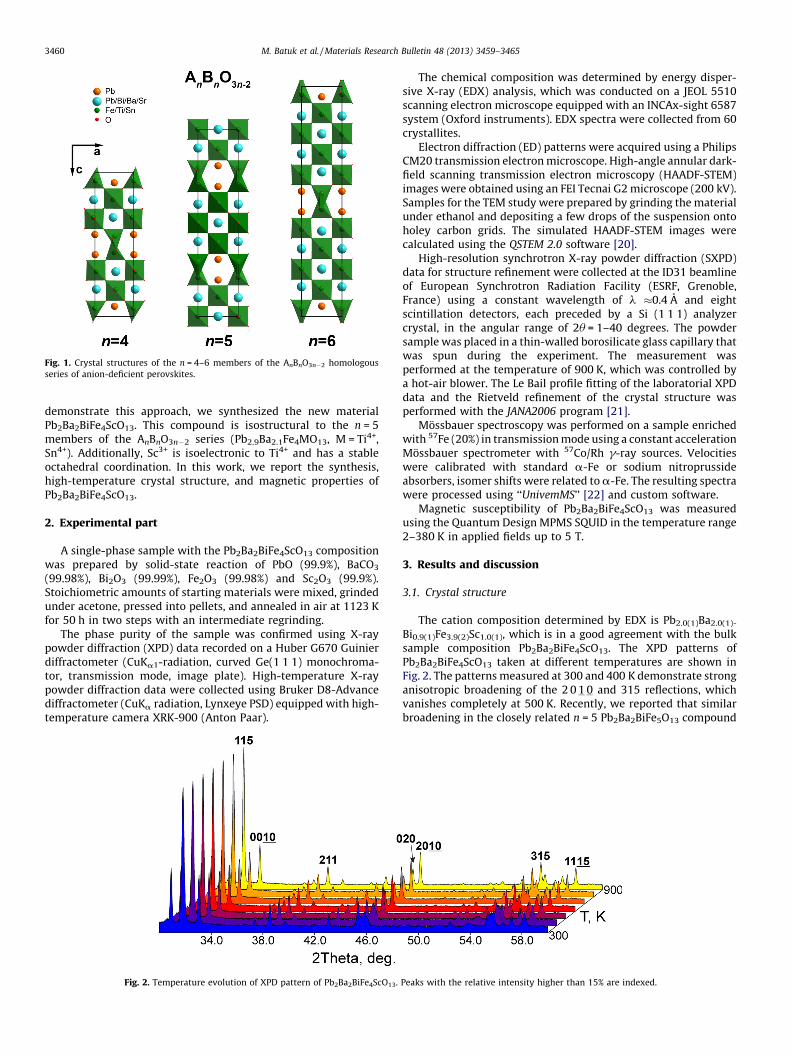
Fig. 1. Crystal structures of the n = 4–6 members of the AnBnO3n�2 homologous
series of anion-deficient perovskites.
M. Batuk et al. / Materials Research Bulletin 48 (2013) 3459–34653460
demonstrate this approach, we synthesized the new materialPb2Ba2BiFe4ScO13. This compound is isostructural to the n = 5members of the AnBnO3n�2 series (Pb2.9Ba2.1Fe4MO13, M = Ti4+,Sn4+). Additionally, Sc3+ is isoelectronic to Ti4+ and has a stableoctahedral coordination. In this work, we report the synthesis,high-temperature crystal structure, and magnetic properties ofPb2Ba2BiFe4ScO13.
2. Experimental part
A single-phase sample with the Pb2Ba2BiFe4ScO13 compositionwas prepared by solid-state reaction of PbO (99.9%), BaCO3
(99.98%), Bi2O3 (99.99%), Fe2O3 (99.98%) and Sc2O3 (99.9%).Stoichiometric amounts of starting materials were mixed, grindedunder acetone, pressed into pellets, and annealed in air at 1123 Kfor 50 h in two steps with an intermediate regrinding.
The phase purity of the sample was confirmed using X-raypowder diffraction (XPD) data recorded on a Huber G670 Guinierdiffractometer (CuKa1-radiation, curved Ge(1 1 1) monochroma-tor, transmission mode, image plate). High-temperature X-raypowder diffraction data were collected using Bruker D8-Advancediffractometer (CuKa radiation, Lynxeye PSD) equipped with high-temperature camera XRK-900 (Anton Paar).
Fig. 2. Temperature evolution of XPD pattern of Pb2Ba2BiFe4ScO13.
The chemical composition was determined by energy disper-sive X-ray (EDX) analysis, which was conducted on a JEOL 5510scanning electron microscope equipped with an INCAx-sight 6587system (Oxford instruments). EDX spectra were collected from 60crystallites.
Electron diffraction (ED) patterns were acquired using a PhilipsCM20 transmission electron microscope. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM)images were obtained using an FEI Tecnai G2 microscope (200 kV).Samples for the TEM study were prepared by grinding the materialunder ethanol and depositing a few drops of the suspension ontoholey carbon grids. The simulated HAADF-STEM images werecalculated using the QSTEM 2.0 software [20].
High-resolution synchrotron X-ray powder diffraction (SXPD)data for structure refinement were collected at the ID31 beamlineof European Synchrotron Radiation Facility (ESRF, Grenoble,France) using a constant wavelength of l �0.4 A and eightscintillation detectors, each preceded by a Si (1 1 1) analyzercrystal, in the angular range of 2u = 1–40 degrees. The powdersample was placed in a thin-walled borosilicate glass capillary thatwas spun during the experiment. The measurement wasperformed at the temperature of 900 K, which was controlled bya hot-air blower. The Le Bail profile fitting of the laboratorial XPDdata and the Rietveld refinement of the crystal structure wasperformed with the JANA2006 program [21].
Mossbauer spectroscopy was performed on a sample enrichedwith 57Fe (20%) in transmission mode using a constant accelerationMossbauer spectrometer with 57Co/Rh g-ray sources. Velocitieswere calibrated with standard a-Fe or sodium nitroprussideabsorbers, isomer shifts were related to a-Fe. The resulting spectrawere processed using ‘‘UnivemMS’’ [22] and custom software.
Magnetic susceptibility of Pb2Ba2BiFe4ScO13 was measuredusing the Quantum Design MPMS SQUID in the temperature range2–380 K in applied fields up to 5 T.
3. Results and discussion
3.1. Crystal structure
The cation composition determined by EDX is Pb2.0(1)Ba2.0(1)-
Bi0.9(1)Fe3.9(2)Sc1.0(1), which is in a good agreement with the bulksample composition Pb2Ba2BiFe4ScO13. The XPD patterns ofPb2Ba2BiFe4ScO13 taken at different temperatures are shown inFig. 2. The patterns measured at 300 and 400 K demonstrate stronganisotropic broadening of the 2 0 1 0 and 315 reflections, whichvanishes completely at 500 K. Recently, we reported that similarbroadening in the closely related n = 5 Pb2Ba2BiFe5O13 compound
Peaks with the relative intensity higher than 15% are indexed.
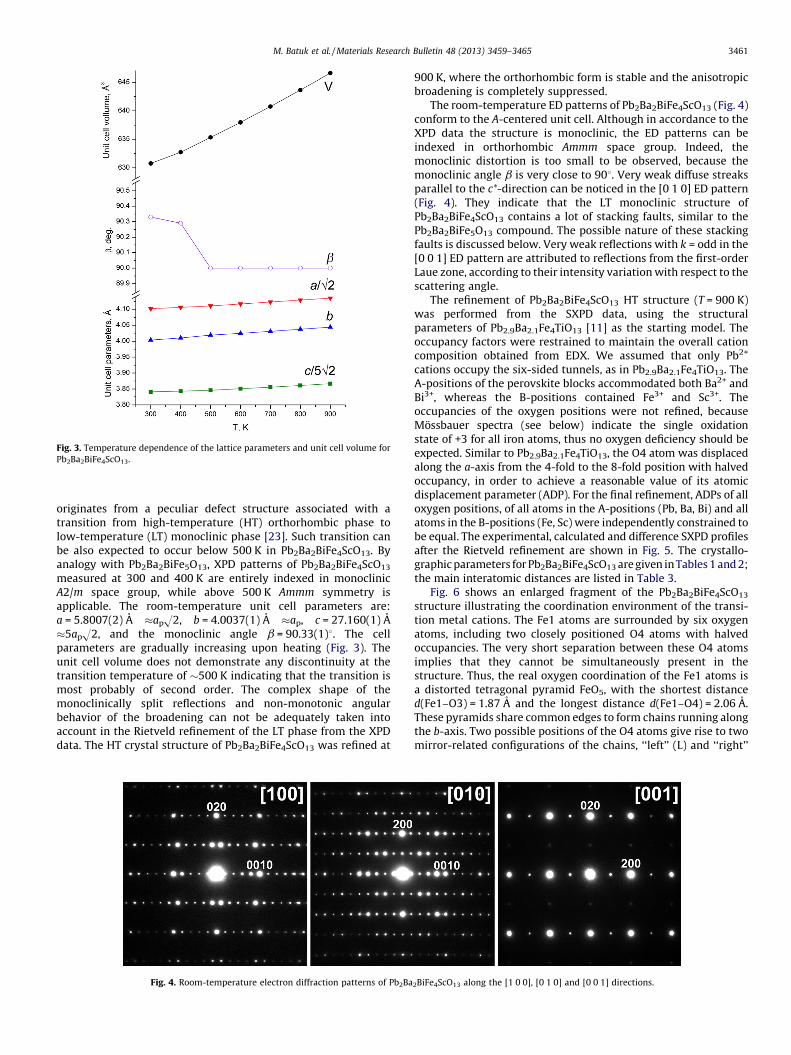
Fig. 3. Temperature dependence of the lattice parameters and unit cell volume for
Pb2Ba2BiFe4ScO13.
M. Batuk et al. / Materials Research Bulletin 48 (2013) 3459–3465 3461
originates from a peculiar defect structure associated with atransition from high-temperature (HT) orthorhombic phase tolow-temperature (LT) monoclinic phase [23]. Such transition canbe also expected to occur below 500 K in Pb2Ba2BiFe4ScO13. Byanalogy with Pb2Ba2BiFe5O13, XPD patterns of Pb2Ba2BiFe4ScO13
measured at 300 and 400 K are entirely indexed in monoclinicA2/m space group, while above 500 K Ammm symmetry isapplicable. The room-temperature unit cell parameters are:a = 5.8007(2) A �apH2, b = 4.0037(1) A �ap, c = 27.160(1) A�5apH2, and the monoclinic angle b = 90.33(1)8. The cellparameters are gradually increasing upon heating (Fig. 3). Theunit cell volume does not demonstrate any discontinuity at thetransition temperature of �500 K indicating that the transition ismost probably of second order. The complex shape of themonoclinically split reflections and non-monotonic angularbehavior of the broadening can not be adequately taken intoaccount in the Rietveld refinement of the LT phase from the XPDdata. The HT crystal structure of Pb2Ba2BiFe4ScO13 was refined at
Fig. 4. Room-temperature electron diffraction patterns of Pb2Ba
900 K, where the orthorhombic form is stable and the anisotropicbroadening is completely suppressed.
The room-temperature ED patterns of Pb2Ba2BiFe4ScO13 (Fig. 4)conform to the A-centered unit cell. Although in accordance to theXPD data the structure is monoclinic, the ED patterns can beindexed in orthorhombic Ammm space group. Indeed, themonoclinic distortion is too small to be observed, because themonoclinic angle b is very close to 908. Very weak diffuse streaksparallel to the c*-direction can be noticed in the [0 1 0] ED pattern(Fig. 4). They indicate that the LT monoclinic structure ofPb2Ba2BiFe4ScO13 contains a lot of stacking faults, similar to thePb2Ba2BiFe5O13 compound. The possible nature of these stackingfaults is discussed below. Very weak reflections with k = odd in the[0 0 1] ED pattern are attributed to reflections from the first-orderLaue zone, according to their intensity variation with respect to thescattering angle.
The refinement of Pb2Ba2BiFe4ScO13 HT structure (T = 900 K)was performed from the SXPD data, using the structuralparameters of Pb2.9Ba2.1Fe4TiO13 [11] as the starting model. Theoccupancy factors were restrained to maintain the overall cationcomposition obtained from EDX. We assumed that only Pb2+
cations occupy the six-sided tunnels, as in Pb2.9Ba2.1Fe4TiO13. TheA-positions of the perovskite blocks accommodated both Ba2+ andBi3+, whereas the B-positions contained Fe3+ and Sc3+. Theoccupancies of the oxygen positions were not refined, becauseMossbauer spectra (see below) indicate the single oxidationstate of +3 for all iron atoms, thus no oxygen deficiency should beexpected. Similar to Pb2.9Ba2.1Fe4TiO13, the O4 atom was displacedalong the a-axis from the 4-fold to the 8-fold position with halvedoccupancy, in order to achieve a reasonable value of its atomicdisplacement parameter (ADP). For the final refinement, ADPs of alloxygen positions, of all atoms in the A-positions (Pb, Ba, Bi) and allatoms in the B-positions (Fe, Sc) were independently constrained tobe equal. The experimental, calculated and difference SXPD profilesafter the Rietveld refinement are shown in Fig. 5. The crystallo-graphic parameters for Pb2Ba2BiFe4ScO13 are given in Tables 1 and 2;the main interatomic distances are listed in Table 3.
Fig. 6 shows an enlarged fragment of the Pb2Ba2BiFe4ScO13
structure illustrating the coordination environment of the transi-tion metal cations. The Fe1 atoms are surrounded by six oxygenatoms, including two closely positioned O4 atoms with halvedoccupancies. The very short separation between these O4 atomsimplies that they cannot be simultaneously present in thestructure. Thus, the real oxygen coordination of the Fe1 atoms isa distorted tetragonal pyramid FeO5, with the shortest distanced(Fe1–O3) = 1.87 A and the longest distance d(Fe1–O4) = 2.06 A.These pyramids share common edges to form chains running alongthe b-axis. Two possible positions of the O4 atoms give rise to twomirror-related configurations of the chains, ‘‘left’’ (L) and ‘‘right’’
2BiFe4ScO13 along the [1 0 0], [0 1 0] and [0 0 1] directions.
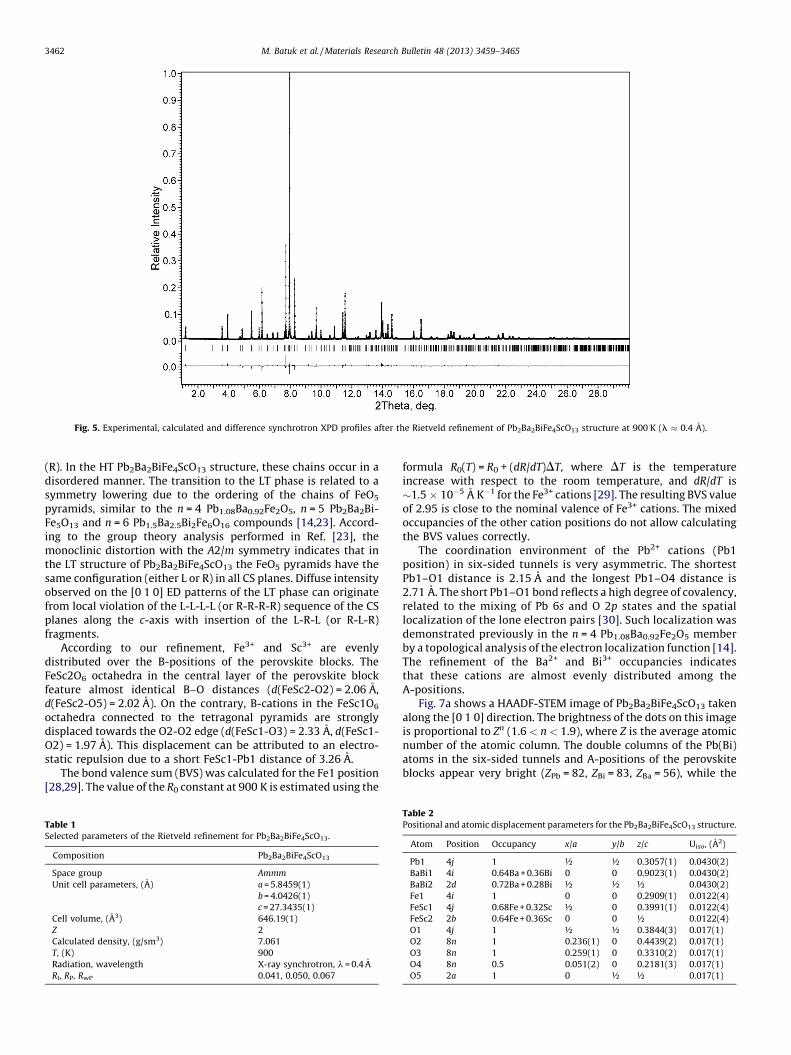
Fig. 5. Experimental, calculated and difference synchrotron XPD profiles after the Rietveld refinement of Pb2Ba2BiFe4ScO13 structure at 900 K (l � 0.4 A).
M. Batuk et al. / Materials Research Bulletin 48 (2013) 3459–34653462
(R). In the HT Pb2Ba2BiFe4ScO13 structure, these chains occur in adisordered manner. The transition to the LT phase is related to asymmetry lowering due to the ordering of the chains of FeO5
pyramids, similar to the n = 4 Pb1.08Ba0.92Fe2O5, n = 5 Pb2Ba2Bi-Fe5O13 and n = 6 Pb1.5Ba2.5Bi2Fe6O16 compounds [14,23]. Accord-ing to the group theory analysis performed in Ref. [23], themonoclinic distortion with the A2/m symmetry indicates that inthe LT structure of Pb2Ba2BiFe4ScO13 the FeO5 pyramids have thesame configuration (either L or R) in all CS planes. Diffuse intensityobserved on the [0 1 0] ED patterns of the LT phase can originatefrom local violation of the L-L-L-L (or R-R-R-R) sequence of the CSplanes along the c-axis with insertion of the L-R-L (or R-L-R)fragments.
According to our refinement, Fe3+ and Sc3+ are evenlydistributed over the B-positions of the perovskite blocks. TheFeSc2O6 octahedra in the central layer of the perovskite blockfeature almost identical B–O distances (d(FeSc2-O2) = 2.06 A,d(FeSc2-O5) = 2.02 A). On the contrary, B-cations in the FeSc1O6
octahedra connected to the tetragonal pyramids are stronglydisplaced towards the O2-O2 edge (d(FeSc1-O3) = 2.33 A, d(FeSc1-O2) = 1.97 A). This displacement can be attributed to an electro-static repulsion due to a short FeSc1-Pb1 distance of 3.26 A.
The bond valence sum (BVS) was calculated for the Fe1 position[28,29]. The value of the R0 constant at 900 K is estimated using the
Table 1Selected parameters of the Rietveld refinement for Pb2Ba2BiFe4ScO13.
Composition Pb2Ba2BiFe4ScO13
Space group Ammm
Unit cell parameters, (A) a = 5.8459(1)
b = 4.0426(1)
c = 27.3435(1)
Cell volume, (A3) 646.19(1)
Z 2
Calculated density, (g/sm3) 7.061
T, (K) 900
Radiation, wavelength X-ray synchrotron, l = 0.4 A
RI, RP, RwP 0.041, 0.050, 0.067
formula R0(T) = R0 + (dR/dT)DT, where DT is the temperatureincrease with respect to the room temperature, and dR/dT is�1.5 � 10�5 A K�1 for the Fe3+ cations [29]. The resulting BVS valueof 2.95 is close to the nominal valence of Fe3+ cations. The mixedoccupancies of the other cation positions do not allow calculatingthe BVS values correctly.
The coordination environment of the Pb2+ cations (Pb1position) in six-sided tunnels is very asymmetric. The shortestPb1–O1 distance is 2.15 A and the longest Pb1–O4 distance is2.71 A. The short Pb1–O1 bond reflects a high degree of covalency,related to the mixing of Pb 6s and O 2p states and the spatiallocalization of the lone electron pairs [30]. Such localization wasdemonstrated previously in the n = 4 Pb1.08Ba0.92Fe2O5 memberby a topological analysis of the electron localization function [14].The refinement of the Ba2+ and Bi3+ occupancies indicatesthat these cations are almost evenly distributed among theA-positions.
Fig. 7a shows a HAADF-STEM image of Pb2Ba2BiFe4ScO13 takenalong the [0 1 0] direction. The brightness of the dots on this imageis proportional to Zn (1.6 < n < 1.9), where Z is the average atomicnumber of the atomic column. The double columns of the Pb(Bi)atoms in the six-sided tunnels and A-positions of the perovskiteblocks appear very bright (ZPb = 82, ZBi = 83, ZBa = 56), while the
Table 2Positional and atomic displacement parameters for the Pb2Ba2BiFe4ScO13 structure.
Atom Position Occupancy x/a y/b z/c Uiso, (A2)
Pb1 4j 1 ½ ½ 0.3057(1) 0.0430(2)
BaBi1 4i 0.64Ba + 0.36Bi 0 0 0.9023(1) 0.0430(2)
BaBi2 2d 0.72Ba + 0.28Bi ½ ½ ½ 0.0430(2)
Fe1 4i 1 0 0 0.2909(1) 0.0122(4)
FeSc1 4j 0.68Fe + 0.32Sc ½ 0 0.3991(1) 0.0122(4)
FeSc2 2b 0.64Fe + 0.36Sc 0 0 ½ 0.0122(4)
O1 4j 1 ½ ½ 0.3844(3) 0.017(1)
O2 8n 1 0.236(1) 0 0.4439(2) 0.017(1)
O3 8n 1 0.259(1) 0 0.3310(2) 0.017(1)
O4 8n 0.5 0.051(2) 0 0.2181(3) 0.017(1)
O5 2a 1 0 ½ ½ 0.017(1)
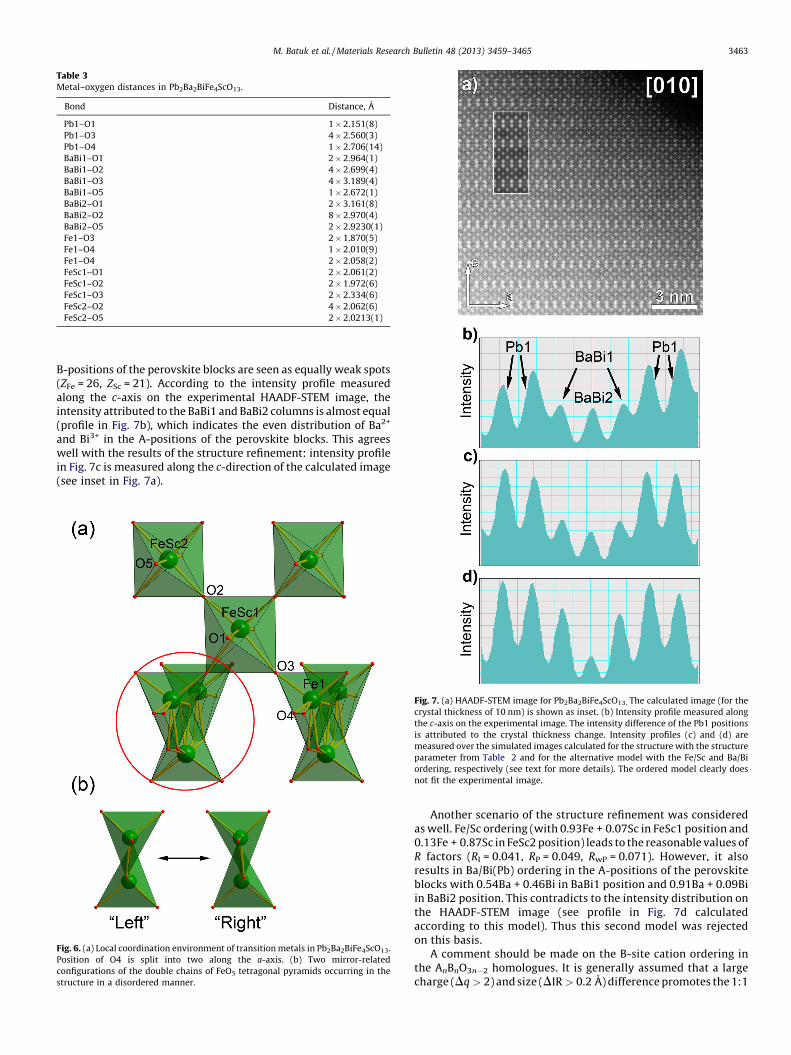
Table 3Metal–oxygen distances in Pb2Ba2BiFe4ScO13.
Bond Distance, A
Pb1–O1 1 � 2.151(8)
Pb1–O3 4 � 2.560(3)
Pb1–O4 1 � 2.706(14)
BaBi1–O1 2 � 2.964(1)
BaBi1–O2 4 � 2.699(4)
BaBi1–O3 4 � 3.189(4)
BaBi1–O5 1 � 2.672(1)
BaBi2–O1 2 � 3.161(8)
BaBi2–O2 8 � 2.970(4)
BaBi2–O5 2 � 2.9230(1)
Fe1–O3 2 � 1.870(5)
Fe1–O4 1 � 2.010(9)
Fe1–O4 2 � 2.058(2)
FeSc1–O1 2 � 2.061(2)
FeSc1–O2 2 � 1.972(6)
FeSc1–O3 2 � 2.334(6)
FeSc2–O2 4 � 2.062(6)
FeSc2–O5 2 � 2.0213(1)
M. Batuk et al. / Materials Research Bulletin 48 (2013) 3459–3465 3463
B-positions of the perovskite blocks are seen as equally weak spots(ZFe = 26, ZSc = 21). According to the intensity profile measuredalong the c-axis on the experimental HAADF-STEM image, theintensity attributed to the BaBi1 and BaBi2 columns is almost equal(profile in Fig. 7b), which indicates the even distribution of Ba2+
and Bi3+ in the A-positions of the perovskite blocks. This agreeswell with the results of the structure refinement: intensity profilein Fig. 7c is measured along the c-direction of the calculated image(see inset in Fig. 7a).
Fig. 6. (a) Local coordination environment of transition metals in Pb2Ba2BiFe4ScO13.
Position of O4 is split into two along the a-axis. (b) Two mirror-related
configurations of the double chains of FeO5 tetragonal pyramids occurring in the
structure in a disordered manner.
Fig. 7. (a) HAADF-STEM image for Pb2Ba2BiFe4ScO13. The calculated image (for the
crystal thickness of 10 nm) is shown as inset. (b) Intensity profile measured along
the c-axis on the experimental image. The intensity difference of the Pb1 positions
is attributed to the crystal thickness change. Intensity profiles (c) and (d) are
measured over the simulated images calculated for the structure with the structure
parameter from Table 2 and for the alternative model with the Fe/Sc and Ba/Bi
ordering, respectively (see text for more details). The ordered model clearly does
not fit the experimental image.
Another scenario of the structure refinement was consideredas well. Fe/Sc ordering (with 0.93Fe + 0.07Sc in FeSc1 position and0.13Fe + 0.87Sc in FeSc2 position) leads to the reasonable values ofR factors (RI = 0.041, RP = 0.049, RwP = 0.071). However, it alsoresults in Ba/Bi(Pb) ordering in the A-positions of the perovskiteblocks with 0.54Ba + 0.46Bi in BaBi1 position and 0.91Ba + 0.09Biin BaBi2 position. This contradicts to the intensity distribution onthe HAADF-STEM image (see profile in Fig. 7d calculatedaccording to this model). Thus this second model was rejectedon this basis.
A comment should be made on the B-site cation ordering inthe AnBnO3n�2 homologues. It is generally assumed that a largecharge (Dq > 2) and size (DIR > 0.2 A) difference promotes the 1:1
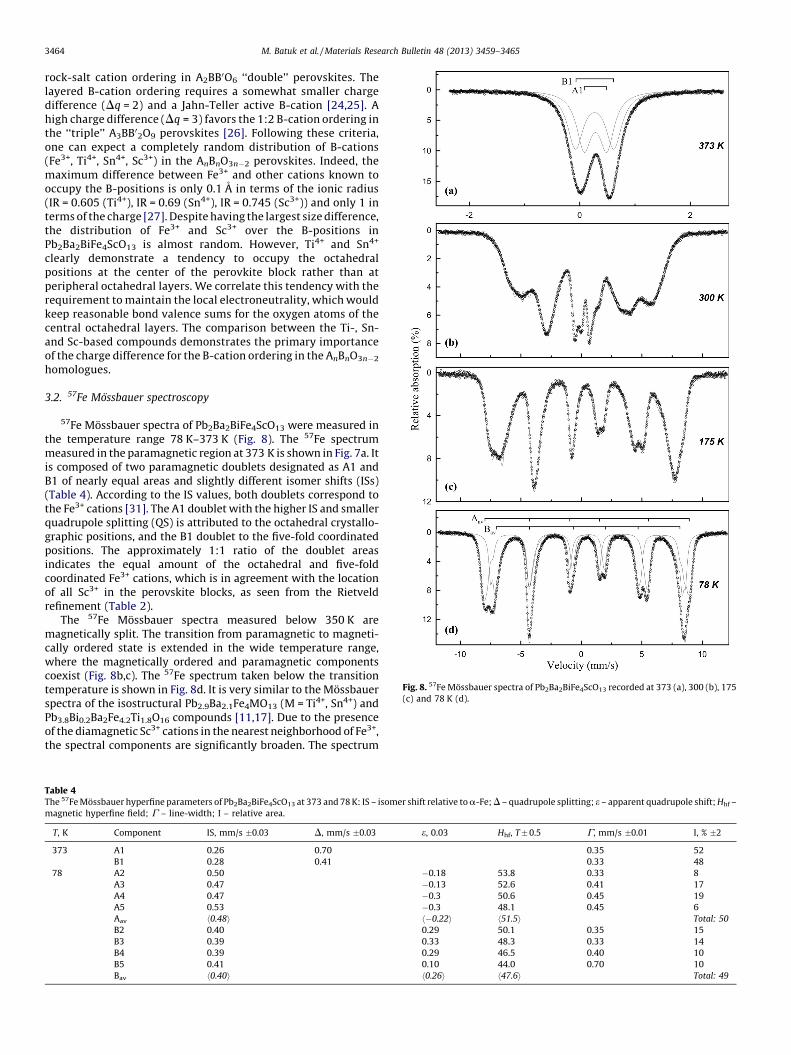
Fig. 8. 57Fe Mossbauer spectra of Pb2Ba2BiFe4ScO13 recorded at 373 (a), 300 (b), 175
(c) and 78 K (d).
M. Batuk et al. / Materials Research Bulletin 48 (2013) 3459–34653464
rock-salt cation ordering in A2BB0O6 ‘‘double’’ perovskites. Thelayered B-cation ordering requires a somewhat smaller chargedifference (Dq = 2) and a Jahn-Teller active B-cation [24,25]. Ahigh charge difference (Dq = 3) favors the 1:2 B-cation ordering inthe ‘‘triple’’ A3BB02O9 perovskites [26]. Following these criteria,one can expect a completely random distribution of B-cations(Fe3+, Ti4+, Sn4+, Sc3+) in the AnBnO3n�2 perovskites. Indeed, themaximum difference between Fe3+ and other cations known tooccupy the B-positions is only 0.1 A in terms of the ionic radius(IR = 0.605 (Ti4+), IR = 0.69 (Sn4+), IR = 0.745 (Sc3+)) and only 1 interms of the charge [27]. Despite having the largest size difference,the distribution of Fe3+ and Sc3+ over the B-positions inPb2Ba2BiFe4ScO13 is almost random. However, Ti4+ and Sn4+
clearly demonstrate a tendency to occupy the octahedralpositions at the center of the perovkite block rather than atperipheral octahedral layers. We correlate this tendency with therequirement to maintain the local electroneutrality, which wouldkeep reasonable bond valence sums for the oxygen atoms of thecentral octahedral layers. The comparison between the Ti-, Sn-and Sc-based compounds demonstrates the primary importanceof the charge difference for the B-cation ordering in the AnBnO3n�2
homologues.
3.2. 57Fe Mossbauer spectroscopy
57Fe Mossbauer spectra of Pb2Ba2BiFe4ScO13 were measured inthe temperature range 78 K–373 K (Fig. 8). The 57Fe spectrummeasured in the paramagnetic region at 373 K is shown in Fig. 7a. Itis composed of two paramagnetic doublets designated as A1 andB1 of nearly equal areas and slightly different isomer shifts (ISs)(Table 4). According to the IS values, both doublets correspond tothe Fe3+ cations [31]. The A1 doublet with the higher IS and smallerquadrupole splitting (QS) is attributed to the octahedral crystallo-graphic positions, and the B1 doublet to the five-fold coordinatedpositions. The approximately 1:1 ratio of the doublet areasindicates the equal amount of the octahedral and five-foldcoordinated Fe3+ cations, which is in agreement with the locationof all Sc3+ in the perovskite blocks, as seen from the Rietveldrefinement (Table 2).
The 57Fe Mossbauer spectra measured below 350 K aremagnetically split. The transition from paramagnetic to magneti-cally ordered state is extended in the wide temperature range,where the magnetically ordered and paramagnetic componentscoexist (Fig. 8b,c). The 57Fe spectrum taken below the transitiontemperature is shown in Fig. 8d. It is very similar to the Mossbauerspectra of the isostructural Pb2.9Ba2.1Fe4MO13 (M = Ti4+, Sn4+) andPb3.8Bi0.2Ba2Fe4.2Ti1.8O16 compounds [11,17]. Due to the presenceof the diamagnetic Sc3+ cations in the nearest neighborhood of Fe3+,the spectral components are significantly broaden. The spectrum
Table 4The 57Fe Mossbauer hyperfine parameters of Pb2Ba2BiFe4ScO13 at 373 and 78 K: IS – isomer shift relative to a-Fe; D – quadrupole splitting; e – apparent quadrupole shift; Hhf –
magnetic hyperfine field; G – line-width; I – relative area.
T, K Component IS, mm/s �0.03 D, mm/s �0.03 e, 0.03 Hhf, T � 0.5 G, mm/s �0.01 I, % �2
373 A1 0.26 0.70 0.35 52
B1 0.28 0.41 0.33 48
78 A2 0.50 �0.18 53.8 0.33 8
A3 0.47 �0.13 52.6 0.41 17
A4 0.47 �0.3 50.6 0.45 19
A5 0.53 �0.3 48.1 0.45 6
Aav h0.48i h�0.22i h51.5i Total: 50
B2 0.40 0.29 50.1 0.35 15
B3 0.39 0.33 48.3 0.33 14
B4 0.39 0.29 46.5 0.40 10
B5 0.41 0.10 44.0 0.70 10
Bav h0.40i h0.26i h47.6i Total: 49
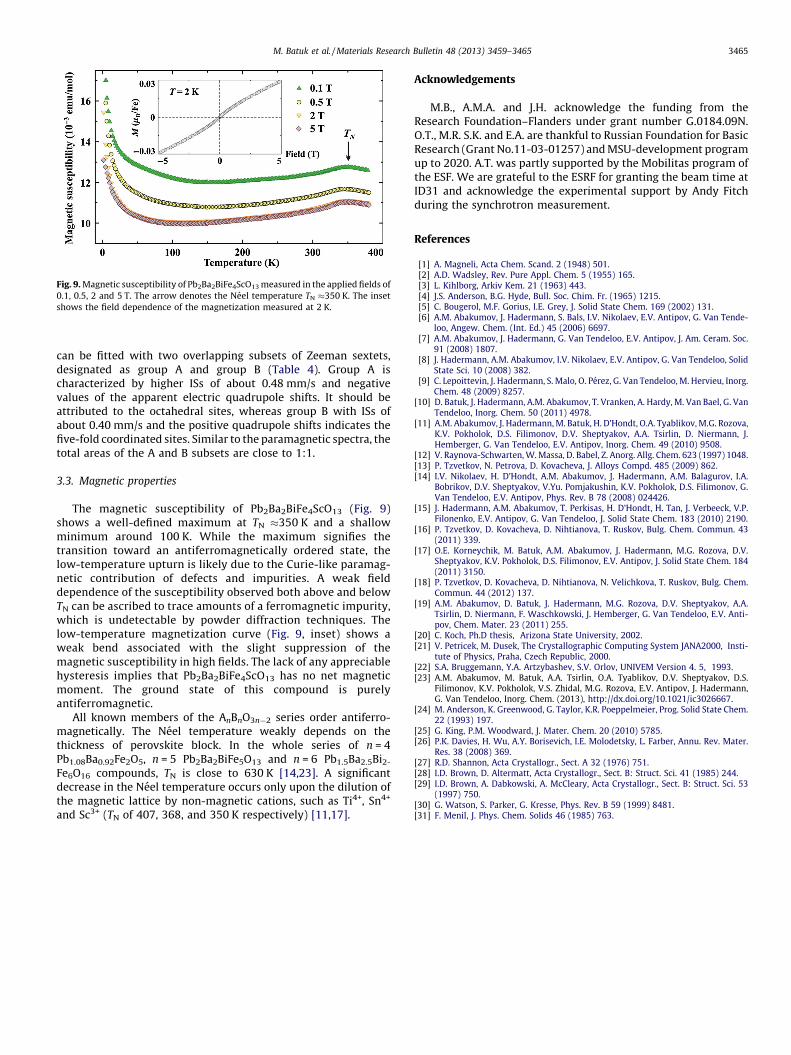
Fig. 9. Magnetic susceptibility of Pb2Ba2BiFe4ScO13 measured in the applied fields of
0.1, 0.5, 2 and 5 T. The arrow denotes the Neel temperature TN �350 K. The inset
shows the field dependence of the magnetization measured at 2 K.
M. Batuk et al. / Materials Research Bulletin 48 (2013) 3459–3465 3465
can be fitted with two overlapping subsets of Zeeman sextets,designated as group A and group B (Table 4). Group A ischaracterized by higher ISs of about 0.48 mm/s and negativevalues of the apparent electric quadrupole shifts. It should beattributed to the octahedral sites, whereas group B with ISs ofabout 0.40 mm/s and the positive quadrupole shifts indicates thefive-fold coordinated sites. Similar to the paramagnetic spectra, thetotal areas of the A and B subsets are close to 1:1.
3.3. Magnetic properties
The magnetic susceptibility of Pb2Ba2BiFe4ScO13 (Fig. 9)shows a well-defined maximum at TN �350 K and a shallowminimum around 100 K. While the maximum signifies thetransition toward an antiferromagnetically ordered state, thelow-temperature upturn is likely due to the Curie-like paramag-netic contribution of defects and impurities. A weak fielddependence of the susceptibility observed both above and belowTN can be ascribed to trace amounts of a ferromagnetic impurity,which is undetectable by powder diffraction techniques. Thelow-temperature magnetization curve (Fig. 9, inset) shows aweak bend associated with the slight suppression of themagnetic susceptibility in high fields. The lack of any appreciablehysteresis implies that Pb2Ba2BiFe4ScO13 has no net magneticmoment. The ground state of this compound is purelyantiferromagnetic.
All known members of the AnBnO3n�2 series order antiferro-magnetically. The Neel temperature weakly depends on thethickness of perovskite block. In the whole series of n = 4Pb1.08Ba0.92Fe2O5, n = 5 Pb2Ba2BiFe5O13 and n = 6 Pb1.5Ba2.5Bi2-
Fe6O16 compounds, TN is close to 630 K [14,23]. A significantdecrease in the Neel temperature occurs only upon the dilution ofthe magnetic lattice by non-magnetic cations, such as Ti4+, Sn4+
and Sc3+ (TN of 407, 368, and 350 K respectively) [11,17].
Acknowledgements
M.B., A.M.A. and J.H. acknowledge the funding from theResearch Foundation–Flanders under grant number G.0184.09N.O.T., M.R. S.K. and E.A. are thankful to Russian Foundation for BasicResearch (Grant No.11-03-01257) and MSU-development programup to 2020. A.T. was partly supported by the Mobilitas program ofthe ESF. We are grateful to the ESRF for granting the beam time atID31 and acknowledge the experimental support by Andy Fitchduring the synchrotron measurement.
References
[1] A. Magneli, Acta Chem. Scand. 2 (1948) 501.[2] A.D. Wadsley, Rev. Pure Appl. Chem. 5 (1955) 165.[3] L. Kihlborg, Arkiv Kem. 21 (1963) 443.[4] J.S. Anderson, B.G. Hyde, Bull. Soc. Chim. Fr. (1965) 1215.[5] C. Bougerol, M.F. Gorius, I.E. Grey, J. Solid State Chem. 169 (2002) 131.[6] A.M. Abakumov, J. Hadermann, S. Bals, I.V. Nikolaev, E.V. Antipov, G. Van Tende-
loo, Angew. Chem. (Int. Ed.) 45 (2006) 6697.[7] A.M. Abakumov, J. Hadermann, G. Van Tendeloo, E.V. Antipov, J. Am. Ceram. Soc.
91 (2008) 1807.[8] J. Hadermann, A.M. Abakumov, I.V. Nikolaev, E.V. Antipov, G. Van Tendeloo, Solid
State Sci. 10 (2008) 382.[9] C. Lepoittevin, J. Hadermann, S. Malo, O. Perez, G. Van Tendeloo, M. Hervieu, Inorg.
Chem. 48 (2009) 8257.[10] D. Batuk, J. Hadermann, A.M. Abakumov, T. Vranken, A. Hardy, M. Van Bael, G. Van
Tendeloo, Inorg. Chem. 50 (2011) 4978.[11] A.M. Abakumov, J. Hadermann, M. Batuk, H. D’Hondt, O.A. Tyablikov, M.G. Rozova,
K.V. Pokholok, D.S. Filimonov, D.V. Sheptyakov, A.A. Tsirlin, D. Niermann, J.Hemberger, G. Van Tendeloo, E.V. Antipov, Inorg. Chem. 49 (2010) 9508.
[12] V. Raynova-Schwarten, W. Massa, D. Babel, Z. Anorg. Allg. Chem. 623 (1997) 1048.[13] P. Tzvetkov, N. Petrova, D. Kovacheva, J. Alloys Compd. 485 (2009) 862.[14] I.V. Nikolaev, H. D’Hondt, A.M. Abakumov, J. Hadermann, A.M. Balagurov, I.A.
Bobrikov, D.V. Sheptyakov, V.Yu. Pomjakushin, K.V. Pokholok, D.S. Filimonov, G.Van Tendeloo, E.V. Antipov, Phys. Rev. B 78 (2008) 024426.
[15] J. Hadermann, A.M. Abakumov, T. Perkisas, H. D’Hondt, H. Tan, J. Verbeeck, V.P.Filonenko, E.V. Antipov, G. Van Tendeloo, J. Solid State Chem. 183 (2010) 2190.
[16] P. Tzvetkov, D. Kovacheva, D. Nihtianova, T. Ruskov, Bulg. Chem. Commun. 43(2011) 339.
[17] O.E. Korneychik, M. Batuk, A.M. Abakumov, J. Hadermann, M.G. Rozova, D.V.Sheptyakov, K.V. Pokholok, D.S. Filimonov, E.V. Antipov, J. Solid State Chem. 184(2011) 3150.
[18] P. Tzvetkov, D. Kovacheva, D. Nihtianova, N. Velichkova, T. Ruskov, Bulg. Chem.Commun. 44 (2012) 137.
[19] A.M. Abakumov, D. Batuk, J. Hadermann, M.G. Rozova, D.V. Sheptyakov, A.A.Tsirlin, D. Niermann, F. Waschkowski, J. Hemberger, G. Van Tendeloo, E.V. Anti-pov, Chem. Mater. 23 (2011) 255.
[20] C. Koch, Ph.D thesis, Arizona State University, 2002.[21] V. Petricek, M. Dusek, The Crystallographic Computing System JANA2000, Insti-
tute of Physics, Praha, Czech Republic, 2000.[22] S.A. Bruggemann, Y.A. Artzybashev, S.V. Orlov, UNIVEM Version 4. 5, 1993.[23] A.M. Abakumov, M. Batuk, A.A. Tsirlin, O.A. Tyablikov, D.V. Sheptyakov, D.S.
Filimonov, K.V. Pokholok, V.S. Zhidal, M.G. Rozova, E.V. Antipov, J. Hadermann,G. Van Tendeloo, Inorg. Chem. (2013), http://dx.doi.org/10.1021/ic3026667.
[24] M. Anderson, K. Greenwood, G. Taylor, K.R. Poeppelmeier, Prog. Solid State Chem.22 (1993) 197.
[25] G. King, P.M. Woodward, J. Mater. Chem. 20 (2010) 5785.[26] P.K. Davies, H. Wu, A.Y. Borisevich, I.E. Molodetsky, L. Farber, Annu. Rev. Mater.
Res. 38 (2008) 369.[27] R.D. Shannon, Acta Crystallogr., Sect. A 32 (1976) 751.[28] I.D. Brown, D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci. 41 (1985) 244.[29] I.D. Brown, A. Dabkowski, A. McCleary, Acta Crystallogr., Sect. B: Struct. Sci. 53
(1997) 750.[30] G. Watson, S. Parker, G. Kresse, Phys. Rev. B 59 (1999) 8481.[31] F. Menil, J. Phys. Chem. Solids 46 (1985) 763.


















