m-AAA and i-AAA complexes work coordinately regulating...
25
© 2018. Published by The Company of Biologists Ltd. m-AAA and i-AAA complexes work coordinately regulating OMA1, the stress- activated supervisor of mitochondrial dynamics Francesco Consolato * 1 , Francesca Maltecca * 1 , Susanna Tulli 1 , Irene Sambri 2 and Giorgio Casari 1,2 . 1 Vita-Salute San Raffaele University & Neurogenomics Unit, Division of Genetics and Cell Biology, San Raffaele Scientific Institute, Milan, Italy 2 Telethon Institute of Genetics and Medicine (TIGEM), Pozzuoli (Naples), Italy *These authors contributed equally. Correspondence to: Prof. Giorgio Casari Telethon Institute of Genetics and Medicine (TIGEM), Viale Campi Flegrei 34, 80078, Pozzuoli (Naples), Italy Email: [email protected] Tel: +39 0819230683 Journal of Cell Science • Accepted manuscript JCS Advance Online Article. Posted on 15 March 2018
Transcript of m-AAA and i-AAA complexes work coordinately regulating...

© 2018. Published by The Company of Biologists Ltd.
m-AAA and i-AAA complexes work coordinately regulating OMA1, the stress-
activated supervisor of mitochondrial dynamics
Francesco Consolato *1, Francesca Maltecca *1, Susanna Tulli 1, Irene Sambri 2 and Giorgio
Casari 1,2.
1Vita-Salute San Raffaele University & Neurogenomics Unit, Division of Genetics and Cell Biology,
San Raffaele Scientific Institute, Milan, Italy
2Telethon Institute of Genetics and Medicine (TIGEM), Pozzuoli (Naples), Italy
*These authors contributed equally.
Correspondence to:
Prof. Giorgio Casari
Telethon Institute of Genetics and Medicine (TIGEM),
Viale Campi Flegrei 34, 80078, Pozzuoli (Naples), Italy
Email: [email protected]
Tel: +39 0819230683
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt
JCS Advance Online Article. Posted on 15 March 2018

Abstract
The proteolytic processing of dynamin like GTPase OPA1, mediated by the activity of both
YME1L1 (i-AAA protease complex) and OMA1, is a crucial step in the regulation of mitochondrial
dynamics. OMA1 is a zinc metallopeptidase of the inner mitochondrial membrane that undergoes pre-
activating proteolytic and auto-proteolytic cleavage after mitochondrial import. Here, we identify
AFG3L2 (m-AAA complex) as the major protease mediating this event by maturing the pre-pro-OMA1
of 60 kDa to the pro-OMA1 form of 40 kDa by severing the amino-terminal part without recognizing
specific consensus sequence. Therefore, m-AAA and i-AAA complexes coordinately regulate OMA1
processing and turnover, and consequently OPA1 isoforms, thus adding new information in the
comprehension of the molecular mechanisms in mitochondrial dynamics and of neurodegenerative
diseases affected by these phenomena.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Introduction
Mitochondrial network optimizes different activities by continuously changing its morphology
by the essential and antagonistic activities of fission and fusion (Pernas and Scorrano, 2016;
Westermann, 2010). These dynamic processes are regulated by the pro-fission dynamin related proteins
(DRPs), DRP1, Fis1 and MFF1; (Gandre-Babbe and van der Bliek, 2008; James et al., 2003; Smirnova
et al., 2001); and pro-fusion proteins, MFN1-MFN2 and OPA1, involved in the fusion of the OMM
and IMM, respectively (Cipolat et al., 2004; Pernas and Scorrano, 2016).
The GTPase protein OPA1 exists in different splicing isoforms that can be partially or totally
proteolytically processed at one or two distinct sites (S1 and S2) (Delettre et al., 2001). The
combination of splicing isoforms and proteolytic cleavage results in five electrophoretic distinguishable
protein bands, named L1, L2 (the long forms) and S1, S2 and S3 (the short forms), which promote
fusion and fission (L and S forms, respectively) (Anand et al., 2014). The balance between long and
short OPA1 forms is finely regulated by two mitochondrial inner membrane proteases, OMA1 (Ehses
et al., 2009a) and YME1L1, both cleaving OPA1 at different sites (Song et al., 2007).
The i-AAA protease YME1L1 exposes its catalytic domain to the inter membrane space (IMS)
(Leonhard et al., 1996) and is responsible of the generation of the S2-OPA1 form by proteolytic
cleavage, while OMA1 gives rise to the S1 and S3 forms (Anand et al., 2014; MacVicar and Langer,
2016; Quiros et al., 2012).
OMA1 harbors a M48 metallopeptidase domain and is the major actor for OPA1 processing in stress
conditions (Quiros et al., 2012). Different stress stimuli, such as dissipation of Δφ, increased ROS
production, decreased ATP level, heat shock and loss of mtDNA have been demonstrated to over-
activate OMA1 (Anand et al., 2014; Baker et al., 2014; Ehses et al., 2009a; Head et al., 2009). Very
recently it has been identified a threshold of Δφ as a determinant of mitochondrial homeostasis that is
mediated by OMA1 and DRP1, which cooperatively exert a regulation in OPA1 maintenance and
processing, and therefore fission and fusion pathways (Jones et al., 2017).
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

The absence of YME1L1, which generates organellar stress, also activates OMA1 (Stiburek et al.,
2012); in fact, YME1L1 and OMA1 are reciprocally regulated in response to specific stress stimuli.
OMA1 is degraded by YME1L after toxic insults that depolarize mitochondrial membrane and cause
ATP depletion. The balance of degradation activities of these two proteins tunes the proteolytic
processing of OPA1, which in turn affects the mitochondrial morphology state, thus profoundly
modulating mitochondrial functions (Rainbolt et al., 2016).
OMA1 is synthesized as a pre-pro-protein of 60 kDa that undergoes proteolytic processing upon import
into mitochondria to generate a mature form of 40 kDa (Baker et al., 2014; Head et al., 2009). It has
been proposed that OMA1 40 kDa is a stress-sensitive pro-protein that undergoes autocatalytic
cleavage of a C-terminal subunit peptide upon stress stimuli to generate the active form of OMA1 that
is responsible finally for OPA1 processing (Baker et al., 2014). All these findings point out at OMA1
as a key sensor for a plethora of mitochondrial stress.
Nevertheless, clear information on the generation of pro-OMA1 and the proteases involved in this
mechanism are still missing. Here, we add an additional tile to this complex system of interlaced and
regulated proteases by showing that (i) AFG3L2, the essential component of m-AAA, maturates
OMA1 by mediating its conversion from pre-pro-OMA1 to pro-OMA1, while (ii) YME1L1 is tightly
involved in pro-OMA1 catabolism as recently suggested (Rainbolt et al., 2016).
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Results and Discussion
AFG3L2 mediates OMA1 initial processing
With the aim of identifying the peptidase involved in OMA1 processing, HeLa cells were transfected
with OMA1-HA, and added inhibitors of different classes of peptidases to monitor the accumulation of
pre-pro-OMA1 form: PMSF to inhibit serine proteases, E64 for cysteine proteases and O-phe to block
metallopeptidases (see Material & Methods). Only O-phe treatment induced the accumulation of the
pre-pro-OMA1 form, indicating that the class of metallopeptidase is the one involved in the initial
processing of the protein (Fig. 1A).
380 putative mitochondrial proteases were selected from four different protein databases: UniProt
(UniProtConsortium), MitoCarta (Pagliarini et al., 2008), MitoMiner (Smith and Robinson, 2009) and
Gene Ontology (Ashburner et al., 2000). Combining O-phe sensitivity with the presence of the protease
in at least two of the databases, we reduced to 27 candidate proteins. Among them, all the proteins
inhibited by metal ions or recognizing specific consensus sequence not present in OMA1 or showing
deacetylase/hydrolase activity were excluded. Since OMA1 is localized in the IMM (Baker et al., 2014)
and presumably its cleavage occurs in this site four proteins localized in the IMM were prioritized:
paraplegin, AFG3L2, YME1L1 and OMA1 (Fig. 1B).
We excluded OMA1 itself from this initial step since the expression of the proteolytic inactive mutant
(OMA1-Q-HA) does not affect the pre-pro OMA1 to pro-OMA1 cleavage (Fig. 1C). However, we
confirmed that OMA1 has an active role on its own processing, in particular in the conversion from the
pro-OMA1 to the fragment of approximately 30 kDa, as previously reported (Baker et al., 2014).
Indeed, we observed that the expression of OMA1-Q-HA prevents the processing to 30 kDa OMA1
fragment, demonstrating that it is generated by auto-processing (Fig. 1C).
By transfecting OMA1-HA in HSP patient fibroblasts lacking paraplegin (Atorino et al., 2003), we
didn’t find any alteration of OMA1 processing (Fig. 1D), thus excluding paraplegin from candidates
and confirming previous findings (Ehses et al., 2009a).
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

We then evaluated OMA1 processing in the absence of AFG3L2 in MEFs (Fig. 1E). Interestingly, we
found a significant accumulation of pre-pro-OMA1 and a corresponding reduction of the pro-OMA1
form. Pre-pro-OMA1 accumulation is not due to alterations in the mitochondrial import machinery
since we previously demonstrated that the absence of AFG3L2 does not affect the import of nuclear
encoded proteins into mitochondria (Maltecca et al., 2008). Importantly, similar results were also
obtained on endogenous OMA1 (Fig.1F and Fig.1G) in both Hela and MEFs cells. In Fig.1F AFG3L2
knockdown Hela cells show accumulation of the pre-pro-OMA1 form and reduction of the pro-OMA1
form. Afg3l2 -/- MEFs (Fig. 1G) show a decreased pro-OMA1 form, indicating an active role of
AFG3L2 in pre-pro-OMA1 processing.
AFG3L2 physically interacts and processes pre-pro-OMA1 by a trimming mechanism
The diminished conversion of pre-pro-OMA1 into pro-OMA1 form in the absence of AFG3L2 can be
explained by a direct cleavage of OMA1 operated by AFG3L2 or by an indirect cleavage mediated by
another protease. To discriminate between these alternative possibilities, we co-transfected Afg3l2 -/-
MEFs with OMA1-HA and AFG3L2-myc or its proteolytic inactive mutant (AFG3L2-Q1-myc) then
monitoring the processing of both OMA1 and OPA1. In the absence of AFG3L2, OPA1 is processed to
short forms (Ehses et al., 2009b; Maltecca et al., 2012) and, coordinately, pre-pro-OMA1 accumulates
(Fig. 2A).
The re-expression of AFG3L2 induces a slight increase of OPA1 L1 band, so rescueing the
physiological processing of OPA1 and, at the same time, inducing an increase of pro-OMA1. On the
contrary, the expression of the proteolytic inactive form of AFG3L2 cannot restore OMA1 processing
(Fig. 2A). We therefore explored the possible physical interaction. To stabilize the interaction we
decided to use AFG3L2-Q1-Myc, which is still capable of substrates binding but lacks proteolytic
activity (Atorino et al., 2003) as a trap for interacting proteins. As hypothesized, AFG3L2 co-
immunoprecipitates with OMA1 (Fig. 2B).
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

To gain further evidence of the role of AFG3L2 in OMA1 processing in human cells, which differently
from murine cells do not express the AFG3L2 homologous AFG3L1, we tested the accumulation of the
pre-pro-OMA1 also in HeLa cells silenced for AFG3L2 and demonstrated that behave similarly to
Afg3l2 -/- MEFs. In fact, upon silencing of AFG3L2, we detected the accumulation of the pre-pro-
OMA1 and the presence of intermediate bands between pre-pro-OMA1and pro-OMA1 (Fig 2C, first
two lanes on the left). Moreover, the expression of proteolytic siRNA-insensitive AFG3L2 mutants
(AFG3L2-ins-Q1-Myc, AFG3L2-ins-Q2-Myc) failed to reduce the pre-pro-OMA1 form (Fig. 2C, right
panel).
In order to identify the putative target sequence, a series of OMA1 mutants segmentally deleted
revealed that the pre-pro-OMA1 to pro-OMA1 maturation is the consequence of an N-terminal
trimming effect operated by the m-AAA, similar to what reported for MRPL32, another m-AAA
specific substrate (Bonn et al., 2011) (Fig. 2D). Moreover, since the N-term of 40 kDa OMA1 has been
mapped at amino acid 140 (Baker et al., 2014), we generated a vector expressing a truncated form of
OMA1 that mimics the pro-OMA1 by removing 53 amino acids between the mitochondrial leader
sequence and the first N terminal-amino acids of pro-OMA1. This mutant (OMA1-Δ92-144-HA)
encodes for a protein of 50 kDa and is processed as the wild type to generate the pro-OMA1 (Fig. 2E).
We also found that OMA1-Δ92-144 is proteolytically active since it is able to cleave L-OPA1 restoring
the bands S1 and S3, which both disappeared after OMA1 silencing (Fig. 2E).
Our findings demonstrate the major role of AFG3L2 in OMA1 processing, and in particular in the
conversion from the pre-pro-protein to the pro-protein. However, the presence of a residual amount of
pro-OMA1 in the absence of the m-AAA indicates that an alternative unknown salvage pathway
operated by metalloprotease is active to ensure the generation of the pro-protein.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Based on our results, we propose that the absence of AFG3L2 hampers the pre-pro-OMA1 to pro-
OMA1 conversion, leading to the accumulation of the pre-pro-protein, but at the same time the stress
caused by the absence of AFG3L2 (decreased assembling of respiratory complexes, ATP depletion,
increased ROS production, accumulation of peptides) activates OMA1 auto-catalytic cleavage, leading
to a reduction of pro-OMA1. The activated pro-OMA1 is generated by an autocatalytic processing that
involves the last C-term amino acids of pro-OMA1 (Baker et al., 2014), causing the removal of the HA
moiety from the HA-tagged OMA1 protein. In support to this, reintroducing AFG3L2 in Afg3l2 -/- cells
reduces the mitochondrial stress, hence decreasing pro-OMA1 C-term auto-cleavage and increasing
pro-OMA1 amount (Fig. 2A).
YME1L1 regulates pro-OMA1 turnover
We tested YME1L1 involvement in the generation of pro-OMA1. By silencing YME1L1 we failed to
demonstrate pre-pro-OMA1 accumulation, hence excluding a direct involvement of YME1L1 in pre-
pro-OMA1 processing (Fig.3A). On the contrary, YME1L1 depletion strongly increases the amount of
pro-OMA1 (Fig. 3A), confirming a role for YME1L1 in the regulation of pro-OMA1 turnover
(Rainbolt et al., 2016).
We excluded as possible causes of pro-OMA1 accumulation following YME1L1 knockdown the
increased amount of m-AAA or enhanced OMA1 autocatalytic activity (Fig. 3A) or the increased
protease activity of the m-AAA complex (Fig. 3B). YME1L1 is indeed the specific regulator of pro-
OMA1 since in YME1L1 knockdown cells pro-OMA1 accumulates (Fig.3C). Therefore YME1L1 is
directly involved in regulating pro-OMA1 level, thus preventing OMA1 accumulation, OPA1 over-
processing and the consequent mitochondrial network fragmentation.
Taken together, these data support for the first time that m-AAA and the i-AAA complexes actively
cooperate in controlling mitochondrial dynamics through the fine regulation of OMA1 processing and
stability. The vital necessity of accurately controlling OMA1 proteolytic activity is underlined by the
findings of a ‘tipping point’ threshold of Δφ due to both DRP1 and OMA1, which would act
coordinately to balance the fusion-fission equilibrium under Δφ stimulus (Jones et al., 2017).
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

We showed that AFG3L2 is crucial for OMA1 maturation, hence directly allowing its conversion from
pre-pro-OMA1 to pro-OMA1 by a trimming mechanism involving the solvent-exposed N-terminal part
of the protein. Possibly, another still unknown protease could be involved in the generation of pro-
OMA1, highlighting the existence of compensatory mechanism acting when AFG3L2 is absent or
mutant. We indeed demonstrated that i-AAA is involved in the regulation of pro-OMA1 turnover,
preventing its accumulation and, consequently, mitochondrial network fragmentation.
A possible model of m-AAA-i-AAA-OMA1 functional interactions is shown in Figure 4. Under
physiological condition, AFG3L2 cleaves pre-pro-OMA1 to generate abundant pro-OMA1, whose
amount is further controlled by the activity of i-AAA that regulates its accumulation and ensures the
balance between long and short OPA1 forms.
Differently, the absence of AFG3L2 induces a wide range of organellar damage including defective
assembling of respiratory complexes, ROS production, ATP depletion, and inner membrane
proteostatic stresses (Maltecca et al., 2008; Richter et al., 2015) that possibly turn on an emergency
pathway operated by other proteases that activate OMA1 and the downstream effect of mitochondrial
fragmentation.
This same effect is caused by the absence of YME1L1 and the resultant pro-OMA1 accumulation,
which enhances its autocatalytic activity and, again, induces mitochondrial fragmentation. Considering
the critical role of OMA1 in the mitochondrial fusion/fission homeostasis, it is conceivable that
multiple alternative effectors, including AFG3L2, YME1L1 and OMA1 itself, together with Δφ, are
needed to maintain the effective fine tuning of this crucial stress sensor protein.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Materials and Methods
Transfection constructs
For the generation of pcDNA3.1-Oma1-HA (in the text it appears as OMA1-HA), Oma1 mouse cDNA
clone was obtained from Thermo Scientific (Waltham, MA, USA) and an HA tag sequence introduced .
The proteolytic inactive OMA1E324Q-HA was obtained by sito-specific mutagenesis of pcDNA3.1-
Oma1-HA.
To generate pcDNA3.1-deleted-Oma1-HA mutants (in the text named as Δ1, Δ2, Δ3, Δ4, Δ5, Δ6, Δ7,
Δ8, Δ9) we introduced the indicated deletions performing site-directed mutagenesis: Δ1, pcDNA3.1-
Oma1-Δ147-149-HA; Δ2, pcDNA3.1-Oma1-Δ150-152-HA; Δ3, pcDNA3.1-Oma1-Δ153-155-HA;
Δ4, pcDNA3.1-Oma1-Δ169-172-HA; Δ5, pcDNA3.1-Oma1-Δ170-HA; Δ6, pcDNA3.1-Oma1-Δ171-
HA; Δ7, pcDNA3.1-Oma1-Δ172-HA; Δ8, pcDNA3.1-Oma1-Δ175-178-HA; Δ9, pcDNA3.1-Oma1-
Δ177-180-HA.
In pcDNA3.1-Oma1-Δ92-144-HA (in the text it appears as OMA1-Δ92-144-HA), we introduced a
deletion between amino acid 92 and 144.
pcDNA3.1-AFG3L2-Myc (in the text it appears as AFG3L2-myc), was previously described (Maltecca
et al., 2012)
pcDNA3.1-AFG3L2-E408Q-Myc and pcDNA3.1-AFG3L2-E575Q-Myc appear in the text as AFG3L2-
Q1-Myc and AFG3L2-Q2-Myc, respectively.
In order to make them insensitive to the Stealth RNAi against AFG3L2, these constructs were
mutagenized without changing the sense of encoded amino acid sequence and generating : pcDNA3.1-
AFG3L2-Ins-Myc, pcDNA3.1-AFG3L2-Ins-E575Q-Myc (in the text it appears as AFG3L2- Ins -Q1-
Myc), pcDNA3.1-AFG3L2-Ins-E408Q-Myc (in the text it appears as AFG3L2-Ins-Q2-Myc)
pMT21-YME1L1-Myc (in the text it appears as YME1L1-Myc) was previously described (Coppola et
al., 2000). pMT21-YME1L1-E543Q-Myc (in the text it appears as YME1L1-Q-Myc) was generated
by site-directed mutagenesis. To make them insensitive to the Stealth RNAi against YME1L1, these
constructs were mutagenized without changing the sense of encoded amino acid sequence.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

MrpL32 cDNA was isolated from HeLa cells, HA-tagged and cloned into pCDNA3.1 (MRPL32-HA in
the text). All primers are available on request.
We confirmed all constructs by Sanger sequencing.
Cell culture
Human immortalized fibroblasts, MEFs and HeLa cells were maintained in DMEM containing
penicillin/streptomycin 10 ug/ml, 10% fetal bovine serum, 2 mM L-glutamine and 1 mM sodium
pyruvate.
Transient transfections for the overexpression of the indicated constructs or for the silencing of the
indicated genes were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according
to the manufacturer’s instructions.
mRNAS silencing
Knockdown of AF3GL2, OMA1 and YME1L1 was performed using specific Stealth RNAi siRNAs
and scrambled Stealth RNAi siRNAs as negative control (Invitrogen, Carlsbad, CA, USA).
The sequences of the Stealth RNAi siRNAs are 5’-acgacuuccaaucugucuacuacuc-3’ for human
AFG3L2, 5’-uggacuacugcuugcugcaaaggcu-3’ for human OMA1, 5'-uccagaaacccaaucugccaucgaa-3’ for
human YME1L1. 100 pmoles of each siRNA were transfected. Down-regulation of the target genes
was monitored by immunoblot analysis on cell lysates after 72h from transfection.
Western blot analysis
Western blot was performed using standard protocols. Briefly, total cell lysate was prepared by protein
extraction using the following buffer: 50 mM Tris-HCl pH 8, 150 mM NaCl, 2% Triton, 0.5 mM
EDTA pH 8, 1X PIC (Roche Applied Science, Penzberg, Upper Bavaria, Germany). 25 µg of protein
extracts were dissolved in sample buffer (60 mM Tris-HCl pH 6.8, 5% glycerol, 1,7% SDS, 0,1 M
DTT, 0,002% bromophenol blue), were separated on SDS-PAGE and analyzed by standard
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

immunoblotting procedures. Anti-HA antibody was from Sigma-Aldrich (St. Louis, MO, USA), anti-
OPA1 antibody was from BD Transduction Laboratories (Franklin Lakes, NJ, USA), anti-Hsp60 and
anti-Hsp70 antibodies were from Stressgen (San Diego, CA, USA), anti-GAPDH was from Santa Cruz
Biotechnology (Dallas, TX, USA), anti-c-Myc antibody was from Novus biologicals (Cambrige, UK),
anti-YME1L1 antibody was from ProteinTech Group (Chicago, USA), anti-OMA1 was obtained from
Novus (Milan, Italy), and anti-AFG3L2 was previously generated in the lab (Atorino et al., 2003).
Protease inhibitors
HeLa cells were transfected with OMA1-HA for 14 hours and subsequently incubated for 10 hours
with the following protease inhibitors: 0.5 mM O-phe, 100 µM E-64d and 400 µM PMSF (Sigma-
Aldrich St. Louis, MO, USA).
Co-Immunoprecipitation
One mg of proteins was extracted from HeLa cells in lysis buffer (50 mM Tris HCl pH 8, 150 mM
NaCl, 0.5 mM EDTA pH 8, 1X PIC, 0.15% Sarkosyl, after mild sonication. After a pre-clearing step
(incubation with protein G Sepharose beads) proteins were incubated with the anti-Myc or the anti-HA
(Clone 16B12 Monoclonal Antibody, Affinity Matrix beads (Covance, Princeton, New Jersey, USA)
antibodies for an hour in constant gentle agitation. Subsequently, protein G–Sepharose was added.
Precipitates were used for Western blot analysis.
Uncoupler treatment
HeLa cells were treated with Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (Sigma
Aldrich) 10 µM for 1 hour.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Acknowledgements
This work was supported by the Italian Telethon Foundation (GGP12235), Fondazione Cariplo
(2012_0646), Ministero Italiano dell’Università e della Ricerca (MIUR, 20108WT59Y_001),
Associazione Italiana Sindromi Atassiche (AISA) and by National Ataxia Foundation (NAF),
Ministero Italiano della Salute (Giovani Ricercatori GR-2011-02351638). We are grateful to Laura
Cassina for scientific discussion, Maurizio De Fusco for technical assistance, Michela Riba and Davide
Gaudesi for bionformatic analyses. The authors declare no competing financial interests. OMA1-/- MEF
cells were kindly provided by Carlos Lopez-Otin, Universidad de Oviedo, Oviedo, Spain.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Abbreviations List
C-term, carboxy terminus
E-64, N-[N-(L-3-trans-carboxyirane-2-carbonyl)-L-leucyl]-agmatine; [1-[N-[(L-3-trans-
carboxyoxirane-2-carbonyl)-L-leucyl]amino]-4-guanidinobutane]
FCCP Carbonyl Cyanide p-Trifluoromethoxyphenylhydrazone
HSP, Hereditary spastic paraplegia
HET heterozygous
IMM, inner mitochondrial membrane
KO, knockout
MEFs, mouse embryonic fibroblasts
N-term , amino terminus
OMM, outer mitochondrial membrane
O-phe, orto-phenantroline
PMSF, Phenylmethanesulfonylfluoride
ROS, reactive oxygen species
WT, wild type
Δφ, mitochondrial membrane potential
Jo
urna
l of C
ell S
cien
ce •
Acc
epte
d m
anus
crip
t

References
Anand, R., Wai, T., Baker, M. J., Kladt, N., Schauss, A. C., Rugarli, E. and Langer, T.
(2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and
fission. J Cell Biol 204, 919-29.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A.
P., Dolinski, K., Dwight, S. S., Eppig, J. T. et al. (2000). Gene ontology: tool for the unification of
biology. The Gene Ontology Consortium. Nat Genet 25, 25-9.
Atorino, L., Silvestri, L., Koppen, M., Cassina, L., Ballabio, A., Marconi, R., Langer, T.
and Casari, G. (2003). Loss of m-AAA protease in mitochondria causes complex I deficiency and
increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol 163, 777-87.
Baker, M. J., Lampe, P. A., Stojanovski, D., Korwitz, A., Anand, R., Tatsuta, T. and
Langer, T. (2014). Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-
dependent mitochondrial dynamics. EMBO J 33, 578-93.
Bonn, F., Tatsuta, T., Petrungaro, C., Riemer, J. and Langer, T. (2011). Presequence-
dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J 30,
2545-56.
Cipolat, S., Martins de Brito, O., Dal Zilio, B. and Scorrano, L. (2004). OPA1 requires
mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A 101, 15927-32.
Coppola, M., Pizzigoni, A., Banfi, S., Bassi, M. T., Casari, G. and Incerti, B. (2000).
Identification and characterization of YME1L1, a novel paraplegin-related gene. Genomics 66, 48-54.
Delettre, C., Griffoin, J. M., Kaplan, J., Dollfus, H., Lorenz, B., Faivre, L., Lenaers, G.,
Belenguer, P. and Hamel, C. P. (2001). Mutation spectrum and splicing variants in the OPA1 gene.
Hum Genet 109, 584-91.
Ehses, S., Raschke, I., Mancuso, G., Bernacchia, A., Geimer, S., Tondera, D., Martinou, J.
C., Westermann, B., Rugarli, E. I. and Langer, T. (2009a). Regulation of OPA1 processing and
mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol 187, 1023-36.
Ehses, S., Raschke, I., Mancuso, G., Bernacchia, A., Geimer, S., Tondera, D., Martinou, J.
C., Westermann, B., Rugarli, E. I. and Langer, T. (2009b). Regulation of OPA1 processing and
mitochondrial fusion by m-AAA protease isoenzymes and OMA1. The Journal of Cell Biology 187,
1023-36.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Gandre-Babbe, S. and van der Bliek, A. M. (2008). The novel tail-anchored membrane
protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19,
2402-12.
Head, B., Griparic, L., Amiri, M., Gandre-Babbe, S. and van der Bliek, A. M. (2009).
Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell
Biol 187, 959-66.
James, D. I., Parone, P. A., Mattenberger, Y. and Martinou, J. C. (2003). hFis1, a novel
component of the mammalian mitochondrial fission machinery. J Biol Chem 278, 36373-9.
Jones, E., Gaytan, N., Garcia, I., Herrera, A., Ramos, M., Agarwala, D., Rana, M., Innis-
Whitehouse, W., Schuenzel, E. and Gilkerson, R. (2017). A threshold of transmembrane potential is
required for mitochondrial dynamic balance mediated by DRP1 and OMA1. Cell Mol Life Sci 74,
1347-1363.
Leonhard, K., Herrmann, J. M., Stuart, R. A., Mannhaupt, G., Neupert, W. and Langer,
T. (1996). AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic
system for the ATP-dependent degradation of inner membrane proteins in mitochondria. EMBO J 15,
4218-29.
MacVicar, T. and Langer, T. (2016). OPA1 processing in cell death and disease - the long and
short of it. J Cell Sci 129, 2297-306.
Maltecca, F., Aghaie, A., Schroeder, D. G., Cassina, L., Taylor, B. A., Phillips, S. J.,
Malaguti, M., Previtali, S., Guenet, J. L., Quattrini, A. et al. (2008). The mitochondrial protease
AFG3L2 is essential for axonal development. J Neurosci 28, 2827-36.
Maltecca, F., De Stefani, D., Cassina, L., Consolato, F., Wasilewski, M., Scorrano, L.,
Rizzuto, R. and Casari, G. (2012). Respiratory dysfunction by AFG3L2 deficiency causes decreased
mitochondrial calcium uptake via organellar network fragmentation. Hum Mol Genet 21, 3858-70.
Nolden, M., Ehses, S., Koppen, M., Bernacchia, A., Rugarli, E. I. and Langer, T. (2005).
The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in
mitochondria. Cell 123, 277-89.
Nunnari, J. and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148,
1145-59.
Pagliarini, D. J., Calvo, S. E., Chang, B., Sheth, S. A., Vafai, S. B., Ong, S. E., Walford, G.
A., Sugiana, C., Boneh, A., Chen, W. K. et al. (2008). A mitochondrial protein compendium
elucidates complex I disease biology. Cell 134, 112-23.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Pernas, L. and Scorrano, L. (2016). Mito-Morphosis: Mitochondrial Fusion, Fission, and
Cristae Remodeling as Key Mediators of Cellular Function. Annu Rev Physiol 78, 505-31.
Quiros, P. M., Ramsay, A. J., Sala, D., Fernandez-Vizarra, E., Rodriguez, F., Peinado, J.
R., Fernandez-Garcia, M. S., Vega, J. A., Enriquez, J. A., Zorzano, A. et al. (2012). Loss of
mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective
thermogenesis in mice. EMBO J 31, 2117-33.
Rainbolt, T. K., Lebeau, J., Puchades, C. and Wiseman, R. L. (2016). Reciprocal
Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep
14, 2041-9.
Richter, U., Lahtinen, T., Marttinen, P., Suomi, F. and Battersby, B. J. (2015). Quality
control of mitochondrial protein synthesis is required for membrane integrity and cell fitness. J Cell
Biol 211, 373-89.
Smirnova, E., Griparic, L., Shurland, D. L. and van der Bliek, A. M. (2001). Dynamin-
related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12, 2245-
56.
Smith, A. C. and Robinson, A. J. (2009). MitoMiner, an integrated database for the storage
and analysis of mitochondrial proteomics data. Mol Cell Proteomics 8, 1324-37.
Song, Z., Chen, H., Fiket, M., Alexander, C. and Chan, D. C. (2007). OPA1 processing
controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J
Cell Biol 178, 749-55.
Stiburek, L., Cesnekova, J., Kostkova, O., Fornuskova, D., Vinsova, K., Wenchich, L.,
Houstek, J. and Zeman, J. (2012). YME1L controls the accumulation of respiratory chain subunits
and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol Biol Cell 23,
1010-23.
UniProtConsortium. (2013). Activities at the Universal Protein Resource (UniProt). Nucleic
Acids Res 42, 7486.
Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nat Rev Mol
Cell Biol 11, 872-84.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt
Figures
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

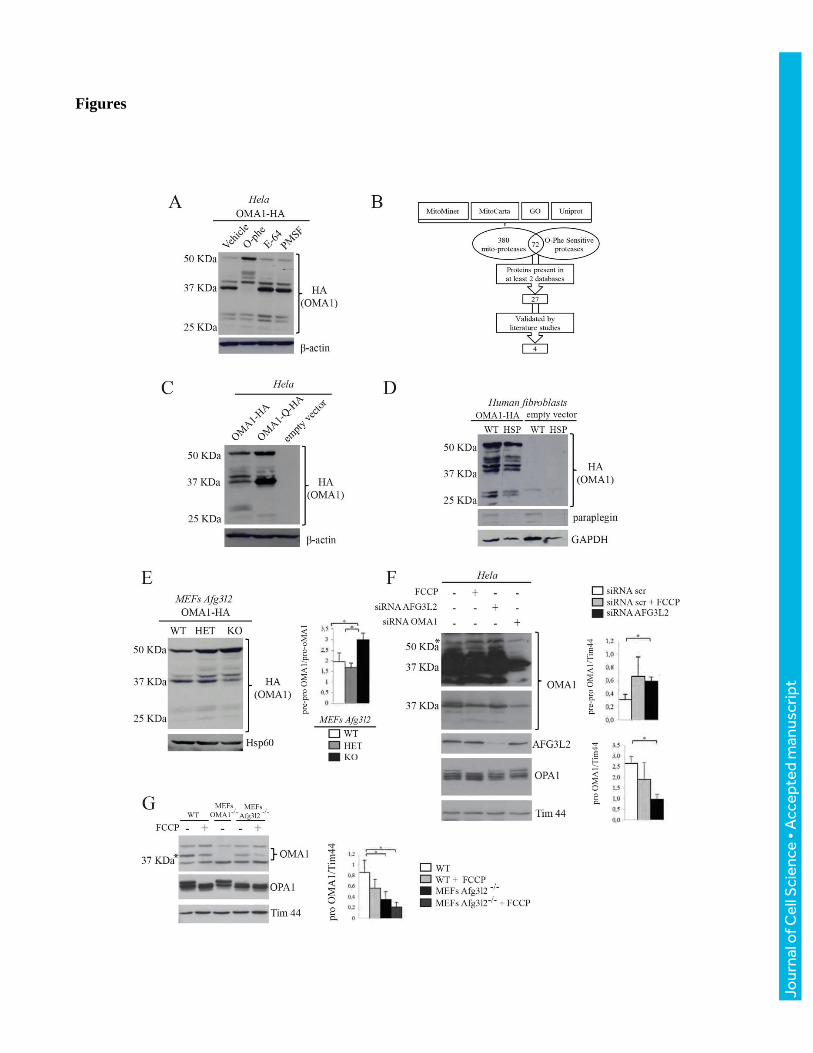
Figure 1. A metallopeptidase cleaves pre-pro-OMA1 to pro-OMA1.
(A) Immunoblot analysis of total extracts from HeLa cells transfected with OMA1-HA and treated for
10 hours with vehicle (DMSO 0,001%), O-phe, E-64 or PMSF. (B) Representative scheme of protease
selection. (C) Immunoblot analysis of total extracts from HeLa cells transfected with OMA1-HA,
OMA1-Q-HA or empty vector. (D) Immunoblot analysis of total extracts from paraplegin-null
immortalized human fibroblasts (HSP) and controls transfected with OMA1-HA or empty vector. (E)
Immunoblot analysis of total extracts from MEFs Afg3l2 wild type (WT), heterozygous (HET) and
knockout (KO) transfected with OMA1-HA and detected with the indicated antibodies. The relative
graph shows the quantification of the ratio between pre-pro-OMA1 and pro-OMA1 band after OMA1-
HA over-expression in MEFs cells by densitometric analysis (ImageJ software). Bars represent mean ±
SD, n=3, two-tailed Student’s t test: *p< 0.05.
(F) Immunoblot analysis of total extracts from Hela cells transfected with siRNAs against AFG3L2 or
OMA1 and treated or not with the uncoupler FCCP for 1h. The relative graphs show the quantification
of the ratio between pre-pro-OMA1 or pro-OMA1 band and Tim 44 by densitometric analysis (ImageJ
software).Bars represent mean ± SD, n=3, two-tailed Student’s t test: *p< 0.05.
(G) Immunoblot analysis of total extracts from MEFs wild type, OMA1-/- , AFG3L2 -/- treated or not
with the uncoupler FCCP for 1h. The * indicate the specific endogenous OMA1 band. The relative
graph shows the quantification of the ratio between pro-OMA1 band and Tim 44 by densitometric
analysis (ImageJ software). Bars represent mean ± SD, n=3, two-tailed Student’s t test: *p< 0.05.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

Figure 2. AFG3L2 is the metallopeptidase converting pre-pro-OMA1 into pro-OMA1 form.
(A) Immunoblot analysis of total extracts from MEFs cells co-transfected with OMA1-HA and empty
vector or AFG3L2-myc or AFG3L2-Q1-Myc. (B) Co-immunoprecipitation (Co-IP) of AFG3L2 with
OMA1 in HeLa cells after expression of OMA1-HA and AFG3L2-Q1-Myc or empty vector. Protein
extracts were immunoprecipitated with anti-myc or anti-HA antibodies. Precipitates were analyzed by
SDS-PAGE and immunostained using anti-AFG3L2 or anti-HA antibodies. I, input, P, pre-clearing; F,
flowthrough. We immunoprecipitated AFG3L2 and identified OMA1 in the Co-IP lane and the same
result was obtained with the reverse approach. (C) Immunoblot analysis of total extracts, from HeLa
cells silenced for AFG3L2 and co-transfected with OMA1-HA and empty vector or AFG3L2-ins-myc,
(AFG3L2-ins-Q1-myc, AFG3L2-ins-Q2-myc). (D) Immunoblot analysis of total extracts from HeLa
cells, transfected with OMA1-HA (OMA1 full length) or OMA1-HA deletion mutants (Δ1 to Δ9, see
material and methods) as indicated or empty vector. Representative scheme of the mutations sites in
OMA1 structure is shown.(E) Immunoblot analysis of total extracts from OMA1-silenced HeLa cells,
transfected with OMA1-HA or OMA1-Δ92-144-HA or empty vector.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt
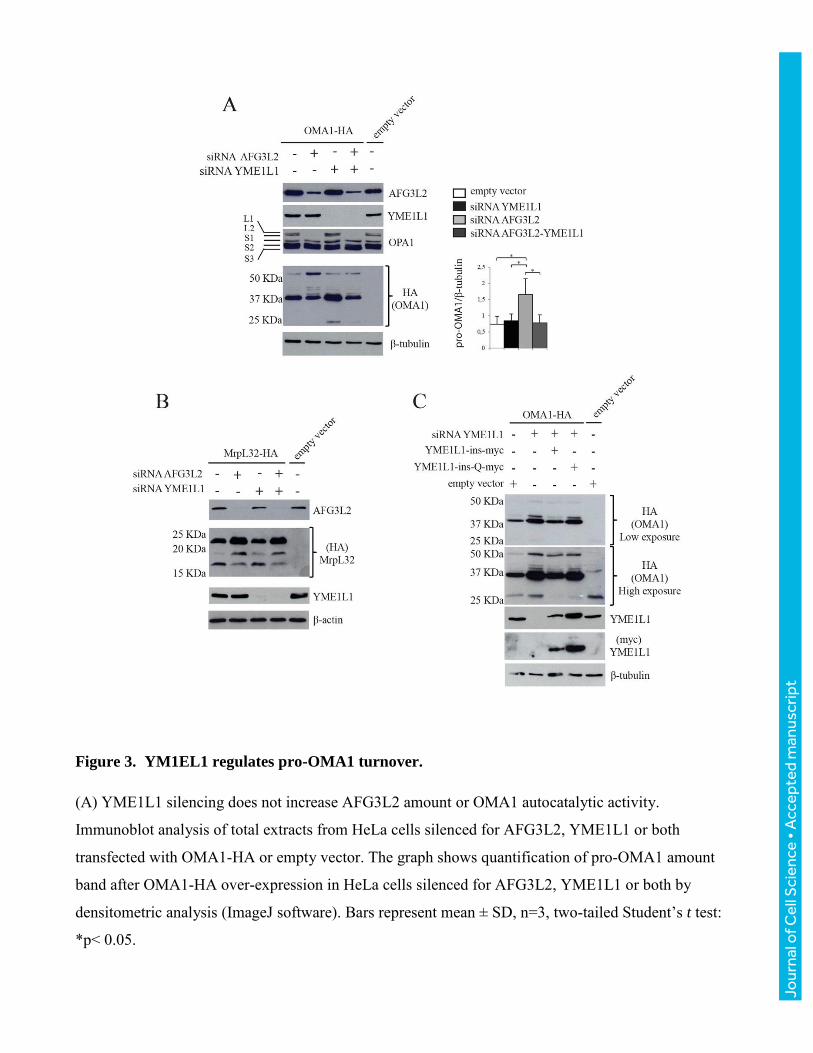
Figure 3. YM1EL1 regulates pro-OMA1 turnover.
(A) YME1L1 silencing does not increase AFG3L2 amount or OMA1 autocatalytic activity.
Immunoblot analysis of total extracts from HeLa cells silenced for AFG3L2, YME1L1 or both
transfected with OMA1-HA or empty vector. The graph shows quantification of pro-OMA1 amount
band after OMA1-HA over-expression in HeLa cells silenced for AFG3L2, YME1L1 or both by
densitometric analysis (ImageJ software). Bars represent mean ± SD, n=3, two-tailed Student’s t test:
*p< 0.05.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

(B) YME1L1 silencing does not increase AFG3L2 catalytic activity. AFG3L2 specifically cleaves
precursor MrpL32 to the mature form, while the absence of AFG3L2 accumulates unprocessed
MrpL32 (Nolden et al., 2005), while the absence of YME1L1 is irrelevant on MrpL32 maturation.
Immunoblot analysis of total extracts from HeLa cells AFG3L2-, YME1L1- or both silenced and
transfected with MrpL32-HA or empty vector. (C) Pro-OMA1 increases by YME1L1 depletion.
Immunoblot analysis of total extracts from HeLa cells silenced for YME1L1 and co-transfected with
OMA1-HA and YME1L1-ins-myc or YME1L1-ins-Q-myc or empty vector and detected with the
indicated antibodies.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt
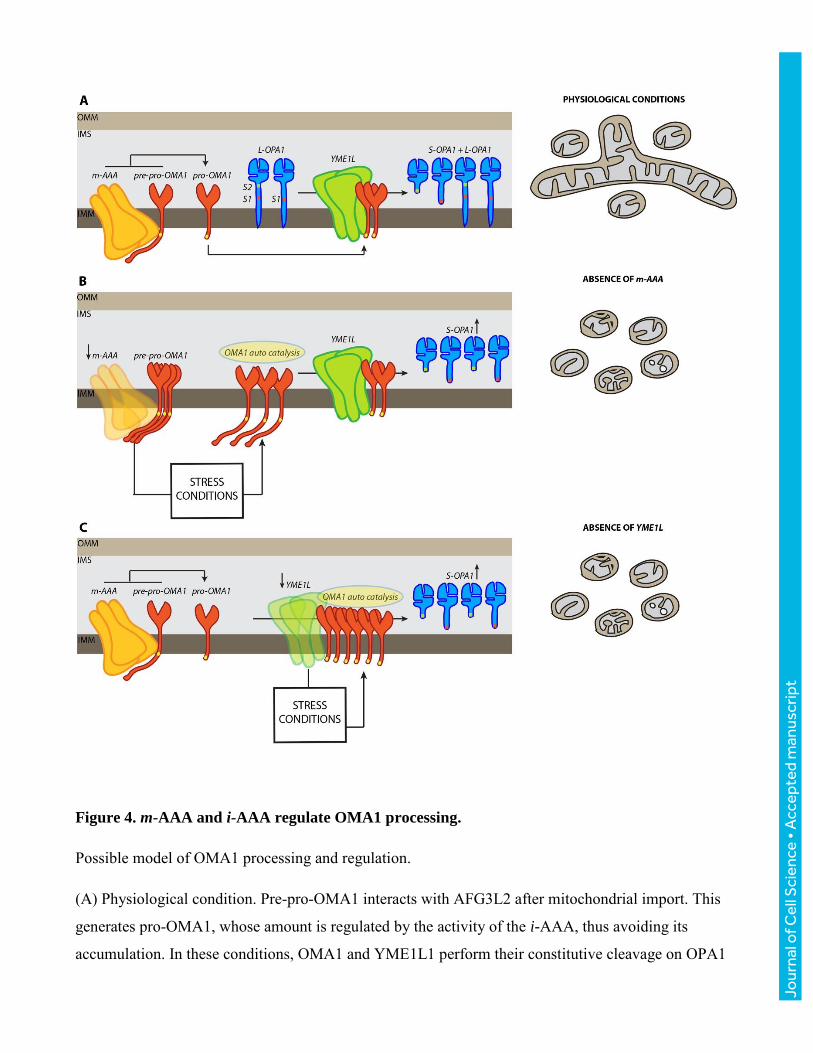
Figure 4. m-AAA and i-AAA regulate OMA1 processing.
Possible model of OMA1 processing and regulation.
(A) Physiological condition. Pre-pro-OMA1 interacts with AFG3L2 after mitochondrial import. This
generates pro-OMA1, whose amount is regulated by the activity of the i-AAA, thus avoiding its
accumulation. In these conditions, OMA1 and YME1L1 perform their constitutive cleavage on OPA1
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt

(at S1 and S2 sites, respectively) ensuring the balance between OPA1 long and short forms and hence a
physiological mitochondrial network. The yellow rectangle of OMA1 transmembrane domain
represents the leucine stretch halting the m-AAA trimming processing.
(B) Absence of AFG3L2 (m-AAA). When AFG3L2 is absent the efficiency of pre-pro-OMA1 to pro-
OMA1 conversion is minimized. The absence of m-AAA induces several stress events that are able to
activate OMA1. (C) Absence of YME1L1. When YME1L1 is absent, pro-OMA1 accumulates. This
accumulation, together with the stress caused by the absence of the i-AAA complexes, induces OMA1
activation. In both cases (B and C), the OMA1increment enhances its proteolytic activity towards
OPA1, thus causing mitochondrial network fragmentation.
Jour
nal o
f Cel
l Sci
ence
• A
ccep
ted
man
uscr
ipt



![arranged by tom wallace percussion by tony mccutchen 11 a a 10 aaa > e] aa aaa 6 aaa aaa aaa aaa aaa aaa 13 > 19 — 18 15 a a aa 16 a a 12 20 23 a > 24 aaa > 25 a > 26 aaa > 27 gÆ4k](https://static.fdocuments.in/doc/165x107/5e6c4dfc8bd84b079d5a5076/arranged-by-tom-wallace-percussion-by-tony-mccutchen-11-a-a-10-aaa-e-aa-aaa.jpg)














