


Luisa De Stefano Business Support Leader Medical Affairs ... · E-submission e Osservatorio Il...
39
Luisa De Stefano – Business Support Leader Medical Affairs & Clinical Operations
Transcript of Luisa De Stefano Business Support Leader Medical Affairs ... · E-submission e Osservatorio Il...

Luisa De Stefano – Business Support Leader
Medical Affairs & Clinical Operations

Agenda
E-submission e Osservatorio
Il nuovo Regolamento Europeo
l’Italia e altri esempi di scenari in Europa

Agenda
E-submission e Osservatorio
Il nuovo Regolamento Europeo
l’Italia e altri esempi di scenari in Europa

Legge 189/2012 (Legge «Balduzzi»): l’art.
12 - Procedure concernenti i medicinali
«Conversione in legge, con modificazioni, del decreto-legge 13 settembre
2012, n. 158, recante disposizioni urgenti per promuovere lo sviluppo del
paese mediante un più alto livello di tutela della salute»
Comma 9: l’AIFA diventa Autorità Centrale (AC)
per tutte le sperimentazioni cliniche
Commi 10 e 11: la rete nazionale dei Comitati
Etici (CE) viene riorganizzata con riduzione
significativa del loro numero *
* Criteri per la composizione e il funzionamento dei
Comitati Etici definiti nel Decreto Ministeriale 8 Febbraio
2013

Legge 189/2012 (Legge «Balduzzi»)(2):
Art.12 comma 12 – e-submission

31.12.2012
interruzione dei servizi dell’OsSC
dal 01.01.2013
Gestione delle sottomissioni in cartaceo (procedure transitorie
07.2013
fase pilota del nuovo OsSC
da 02-2014 Censimento Richiedenti e CE
03-04.2014
corsi per gli utenti (Richiedente e Comitato Etico)
E-submission - l’implementazione
Riattivazione

E-submission: gli obiettivi del progetto
Gestione esclusivamente telematica delle sperimentazioni
Integrazione nel flusso dell’Autorità Competente - Integrazione nel flusso dell’ISS per le sperimentazione di fase I
Aggiornamento Appendici DM 21 dicembre 2007
Gestione del processo di CTA / emendamento sostanziale / dichiarazione di conclusione
Gestione dell’istruttoria (convalida) e della valutazione (CE / AIFA – ISS)
Gestione dello stato della sperimentazione /dati amministrativi per singolo centro
Scambio di informazioni tra Richiedente, AIFA e Comitato Etico (forum e messaggi circolari)

E-submission: le modalità transitorie
dopo il ripristino
Gestione tramite il nuovo OsSC
Domande di autorizzazione di nuove sperimentazioni cliniche e di emendamenti a SC inserite in
OsSC e relativi pareri Comitato Etico
Comunicazione stato sperimentazione. (Conclusione singolo centro e in toto di sperimentazioni
inserite in OsSC )
Via Forum
Comunicazione sintesi risultati di sperimentazioni inserite in OsSC
Gestione emendamenti urgenti per questioni di sicurezza (via OsSC se non presente altro
emendamento)
Per via cartacea/CD ROM
Domanda di autorizzazione di emendamenti a SC non inserite in OsSC
Notifiche alle Regioni inerenti le SC

E-submission: le criticità
Il punto di vista del Promotore
Gestione esclusivamente telematica: GCP compliance?
Sottomissione in un’unica soluzione ad AIFA e tutti i CE
Notifiche via Forum: tracciabilità
Sottomissione sequenziale degli emendamenti

E-submission: i prossimi passi
Inoltro automatico CTA a Eudra-CT
Evoluzione gestione emendamenti
Accesso Regione per visualizzazione dati di competenza territoriale
Gestione telematica delega/revoca alla CRO da parte del Promotore
Evoluzioni reportistica predefinita
Portale Ricerca Clinica – Consultazione dati pubblici

Agenda
E-submission e Osservatorio
Il nuovo Regolamento Europeo
l’Italia e altri esempi di scenari in Europa

Il nuovo Regolamento Europeo sulla
Sperimentazione Clinica (CTR)

Riduzione del numero delle sperimentazioni cliniche dal 2007 al 2011 del 25%
(dal 2007 al 2010 12%)
Aumento dei costi di conduzione delle sperimentazioni cliniche:
107% di aumento personale e relativi costi per promotori commerciali
per la gestione delle procedure di autorizzazione
98% aumento dei costi amministrativi per i promotori non commerciali
800% aumento costi per premi assicurativi per promotori commerciali
90% aumento del tempo medio d’attesa per avviare una sperimentazione clinica
(media 152gg)
Dalla Direttiva (CTD) al Regolamento
(CTR) : Il razionale
Rendere l’Europa più attrattiva e più competitiva per
la ricerca clinica

Dalla Direttiva (CTD) al Regolamento
(CTR) : l’obiettivo
La Direttiva 2001/20/CE (CTD) per l’ARMONIZZAZIONE del processo di
sviluppo dei farmaci in Europa
…ma l’obiettivo è stato raggiunto solo parzialmente.
Il recepimento della CTD a livello nazionale ha determinato
• tempistiche variabili per l’approvazione di uno studio clinico in Europa
[processo di valutazione AC e CE separato]
• specificità diverse dei singoli Stati Membri
• sottomissioni multiple e revisioni multiple dei medesimi documenti
(poca efficienza nell’uso delle risorse e dell’expertise di queste)
• varietà e molteplicità nelle richieste
Aumento di risorse, costi, tempistiche sia per il Promotore sia per le Autorità
Regolatorie
DALLA DIRETTIVA [Flessibile] al REGOLAMENTO [Vincolante].

Dal coordinamento all’armonizzazione della disciplina applicabile.
Ridotti gli ambiti di autonomia normativa di competenza degli stati membri
1 sportello unico a livello europeo
1 procedura coordinata di valutazione
1 decisione finale
Dalla Direttiva (CTD) al Regolamento (CTR)
-Riduzione costi degli studi clinici
-Assicurare ai pazienti l’accesso a
terapie innovative
-Tempistiche certe e stringenti
Dalla Direttiva (CTD) al Regolamento
(CTR) : l’implementazione

07/2012 La Commissione Europea accetta una Proposta di EU CTR (per sostituire la
Direttiva 2011/20/CE)
2012 - 2013 Fase di revisione e negoziazione, richiesta di emendamenti da parte del
Parlamento e del Consiglio EU, fase di consultazione pubblica
12/2013 Raggiunto accordo su testo definitivo a livello di Trilogo
Q2/2014 2-3 Aprile 2014 – votazione del CTR al Parlamento Europeo
19-20 Giugno 2014 – Approvazione formale (Presidenza Greca)
07/ 2014 (?) – CTR diventa effettiva 20 giorni dopo la pubblicazione sulla
Gazzetta Ufficiale (Official Journal - OJ)
Metà 2016 - CTR diventerà applicabile 2 anni dopo la pubblicazione della versione finale
sul OJ,
- Applicabilità vincolata al funzionamento del Portale
(i.e. possibili ritardi se il Portale Eu non dovesse essere attivo entro 6 mesi dalla
data di entrata in vigore del CTR)
2016-2019 3 anni di transizione: permesso utilizzo del vecchio (CTD) e/o nuovo (CTR)
processo
!
Il Regolamento (CTR):
le tempistiche di implementazione

Il Regolamento (CTR):
le principali novità
Portale unico per la domanda di autorizzazione
Procedura coordinata di valutazione e definizione dei tempi di autorizzazione
Introduzione del concetto di Sperimentazione Clinica «a basso livello di intervento»
Obbligo per gli Stati Membri di garantire l’esistenza di sistemi di risarcimento dei danni subiti da un soggetto in SC
Sistema più trasparente per quanto concerne la disponibilità di informazioni sulle sperimentazioni cliniche
Nuove norme sull’acquisizione del Consenso Informato
Possibilità di co-sponsorizzazione

Il Regolamento (CTR):
le principali novità (1)
Creazione di un fascicolo di autorizzazione armonizzato
Realizzazione di un portale Unico gestito da EMA in virtù dell’esperienza
maturata con EudraCT
La commissione EU ha precisato che EudraCT diventerà parte del / verrà
assorbita dal portale unico Europeo
Portale unico per la domanda di autorizzazione

Il Regolamento (CTR):
le principali novità (2)
Procedura coordinata di valutazione e definizione dei tempi di autorizzazione
«Stato Membro Relatore» proposto dal
promotore
Tutti gli Stati Membri in cui lo sponsor
intende condurre una SC partecipano
alla valutazione
Termini precisi basati sul principio di
approvazione tacita

One Dossier
• Therapeutic & public health
benefit aspects
• Risks & inconveniences for the
subject
• Manufacturing/importation of
IMPs/AMPs
• Labelling
• Investigator’s brochure
• Informed consent
• Compensation/ rewarding
arrangements
• Recruitment arrangements
• Data protection aspects
• Suitability of
- individuals
- trial sites
• Damage compensation
• Biological samples
Part I – «General» Part II – «National»
EU Portal
Il Regolamento (CTR):
le principali novità (3)
Procedura coordinata di valutazione e definizione dei tempi di autorizzazione

Il Regolamento (CTR):
le principali novità (4)
Procedura coordinata di valutazione e definizione dei tempi di autorizzazione: il ruolo del Comitato etico
libera scelta agli Stati Membri in merito al coinvolgimento dei CE ma nel
rispetto dei tempi definiti;
la Commissione si aspetta il coinvolgimento del CE esclusivamente per la
valutazione sulla “fattibilità locale”, visto che l’autorizzazione viene
espressa centralmente;
Qualora uno Stato Membro intenda coinvolgere il CE per le valutazioni
etico-scientifiche, e non si vede come sia possibile evitarlo, il CE dovrà
interagire anch’esso con il Portale unico.
N.B. la Commissione ha chiaramente indicato che, qualora coinvolti, I CE dovranno prevedere frequenze di riunione
almeno settimanali
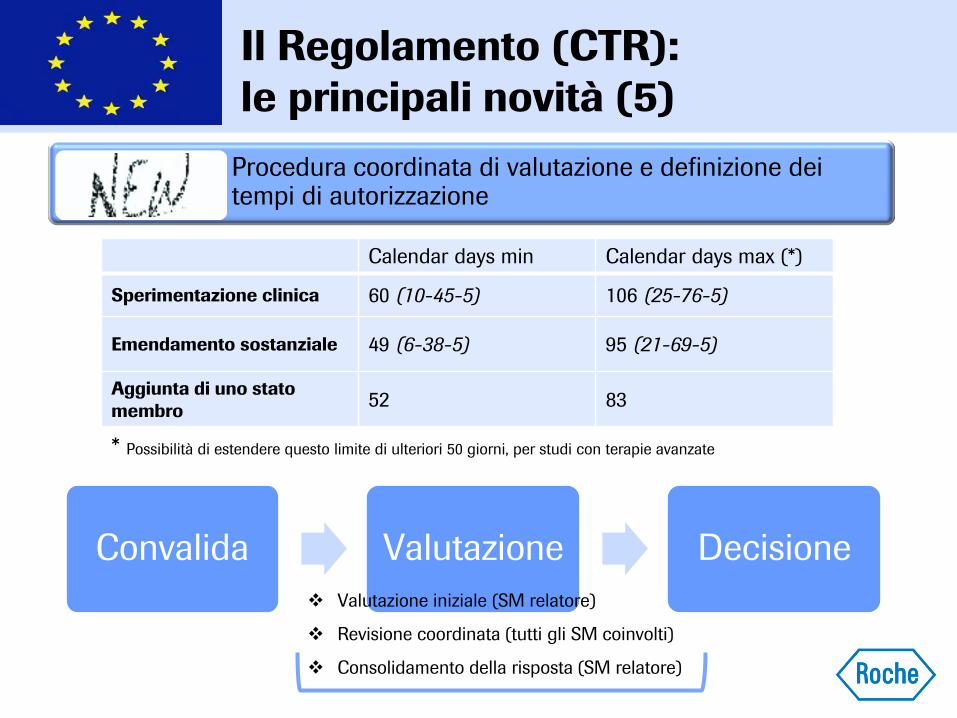
Calendar days min Calendar days max (*)
Sperimentazione clinica 60 (10-45-5) 106 (25-76-5)
Emendamento sostanziale 49 (6-38-5) 95 (21-69-5)
Aggiunta di uno stato
membro 52 83
* Possibilità di estendere questo limite di ulteriori 50 giorni, per studi con terapie avanzate
Procedura coordinata di valutazione e definizione dei tempi di autorizzazione
Convalida Valutazione Decisione
Valutazione iniziale (SM relatore)
Revisione coordinata (tutti gli SM coinvolti)
Consolidamento della risposta (SM relatore)
Il Regolamento (CTR):
le principali novità (5)

E’ definita SC a basso livello d’intervento una SC che soddisfa tutte le seguenti
condizioni
• I medicinali sperimentali sono autorizzati
• In base al protocollo, i medicinali sono utilizzati in accordo all’AIC
• Le procedure diagnostiche o di monitoraggio aggiuntive pongono solo
rischi o oneri aggiuntivi minimi per la sicurezza dei soggetti rispetto alla
normale pratica clinica in qualsiasi SM interessato
Il Regolamento (CTR):
le principali novità (6)
Introduzione del concetto di Sperimentazione Clinica (SC) «a basso livello di intervento»

La Direttiva 2001/20/CE aveva introdotto un sistema di assicurazione obbligatoria /
indennizzo
Ciò ha determinato un sostanziale incremento dei costi e di oneri
amministrativo-burocratici connessi alla conduzione delle
sperimentazioni cliniche
Il nuovo Regolamento adotta un approccio proporzionato al rischio
L’assicurazione è necessaria solo per le Sperimentazioni cliniche diverse da
quelle «a basso livello di intervento»
Gli stati membri devono assicurare che vengano adeguatamente garantite
eventuali pretese risarcitorie
NB: non c’è condivisione sui parametri degli SM negli indennizzi. In Italia va prevista la costituzione di questo
meccanismo «ex-novo»
Il Regolamento (CTR):
le principali novità (7)
Obbligo per gli Stati Membri di garantire l’esistenza di sistemi di risarcimento dei danni subiti da un soggetto in SC

La banca dati europea dovrà essere accessibile al pubblico, a meno che una parte o
tutti i dati e le informazioni in essa contenute ne giustifichino la riservatezza, sulla base
di una delle seguenti motivazioni:
• Protezione dei dati personali
• Protezione di informazioni commerciali a carattere riservato
• Garanzia di una vigilanza efficace degli stati membri sulla conduzione della
sperimentazione clinica
Il nuovo approccio di EMA: dall’accesso su richiesta alla divulgazione proattiva
L’EMA ha affermato che i dati riportati negli studi clinici possono essere divulgati
senza restrizioni non contenendo informazioni commercialmente confidenziali.
Il Regolamento (CTR):
le principali novità (8)
Sistema più trasparente per quanto concerne la disponibilità di informazioni sulle sperimentazioni cliniche

Trasparenza: comunicazione dei risultati
• Riassunto dei risultati in linguaggio facilmente comprensibile
– Entro un anno dalla fine dello studio in tutti gli Stati Membri partecipanti
– Dopo un anno solo se le motivazioni scientifiche sono riportate nel protocollo
– Secondo formato contenuto nel Regolamento stesso
• Analisi Intermedie (in accordo al protocollo): entro un anno dall’analisi
• Clinical Study Reports (CSR)
– Per studi registrativi
– Entro 30 gg (dopo l’ottenimento della MA o del ritiro della richiesta di MA)
• Patient level (raw) data:
- La Commissione deve fornire lineee guida e schemi per la pubblicazione su base
volontaria dei dati Reference: Art. 34 * Trial is not completed in all 3rd:untries is a scientific justification (Ref: Recital 25b)
Sistema più trasparente per quanto concerne la disponibilità di informazioni sulle sperimentazioni cliniche
Il Regolamento (CTR):
le principali novità (9)

27
Trasparenza: informazioni pubbliche • Il Portale EU sarà di pubblico accesso a meno che la confidenzialità sia giustificata
– Protezione di dati personali
– Protezione di informazioni commercialmente confidenziali
– Protezione delle comunicazioni tra Stati Membri in merito alle ispezioni sugli studi clinici
– Garanzia di supervisione efficace
• CTA dossier non accessibile prima della conclusione della valutazione
• In generale, le informazioni incluse bnel CSR non sono considerate commercialmente
confidenziali se
– E’ stata ottenuta l’Autorizzazione alla commercializzazione
– Il processo decisionale in merito alla MA si è concluso
– La domanda di Autorizzazione è stata ritirata
Il Regolamento (CTR):
le principali novità (10)
Sistema più trasparente per quanto concerne la disponibilità di informazioni sulle sperimentazioni cliniche

Il nuovo Regolamento non modifica sostanzialmente le norme in materia di tutela degli
individui e di consenso informato introdotte dalla direttiva 2001/20/CE
alcune disposizioni vengono riformulate e/o sintetizzate per facilitarne la
comprensione
viene omessa la norma sui CE
nuova norma sulle sperimentazioni cliniche in situazioni di emergenza
A differenza della Direttiva 2001/20/CE, il nuovo Regolamento disciplina
espressamente le fattispecie in cui, a causa delle condizioni di urgenza, non
è possibile acquisire preventivamente il consenso libero ed informato
Il Regolamento (CTR):
le principali novità (11)
Nuove norme sull’acquisizione del Consenso Informato

Una sperimentazione clinica può avere uno o più sponsor
Qualsiasi sponsor può delegare la totalità o una parte dei suoi compiti a una persona
fisica, a una società, a un’istituzione o a un organismo. Tale delega non pregiudica la
responsabilità dello sponsor.
Introdotto il concetto di co—sponsorizzazione
• I co-Sponsor possono definire in un contratto le rispettive responsabilità
• I co-Sponsor devono stabilire uno sponsor responsabile al fine del rispetto degli
obblighi individuati dall’art. 69(2)
• Ciascuno sponsor rimane civilmente e penalmente responsabile della propria
attività
Il Regolamento (CTR):
le principali novità (12)
Possibilità di co-sponsorizzazione

Agenda
E-submission e Osservatorio
Il nuovo Regolamento Europeo
l’Italia e altri esempi di scenari in Europa

Francia: quali interventi per
l’attrattività?

Francia: quali interventi per
l’attrattività? (2)

E l’Italia?

Italia: Disegno di Legge Lorenzin (2)
Decreti Legislativi inerenti le seguenti tematiche:
Requisiti dei centri autorizzati a condurre SC
Interventi a sostegno delle SC di Fase I
Semplificazione nella presentazione della domanda di
autorizzazione alla conduzione di uno SC
Valutazione dei risultati della Az. Sanitarie pubbliche
nell’ambito delle SC
Formazione del personale coinvolto nella SC
Apparato sanzionatorio
Studi senza scopo di lucro e osservazionali


Q&A

Doing now what patients need next


















