
Limited Tryptic Digestion Near the Amino Terminus … JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 268, No....
10
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 268, No. 21, Issue of July 25, pp. 15611-15620,1993 Printed in USA. Limited Tryptic Digestion Near the Amino Terminus of Bovine Liver Rhodanese Produces Active Electrophoretic Variants with Altered Refolding* (Received for publication, December 24, 1992, and in revised form, March 20, 1993) Gerald A. MerrillSgll, Michael Butler& and Paul M. Horowitzgll)) From the $Department of Clinical Znuestigatwn, Brooke Army Medical Center, Ft. Sam Houston, Texas 78234-6200 and the §Department of Biochemistry and the VCenter for the Development and Application of Bioannlytical Methods, University of Tex& Health Science Center, Sun Antonio, Texas 78284 When the enzyme rhodanese was partially digested by immobilized trypsin, it retained greater than 50% of its original activity although less than 10% of the undigested enzyme remained. The predominant daugh- ter species were two 3 1-kDapolypeptides whose amino termini corresponded to either residue 44 or 45 of the enzyme’s sequence. Following digestion, charged spe- cies were isolated by ion exchange chromatography. Denaturing electrophoresis revealedthat a 4-kDa pep- tide remained associated with the 31-kDa fragment. This 4-kDa peptide appears to correspond to the amino-terminal 45 residues of rhodanese. Further pro- teolysis ghve a2.5-kDa peptide that dissociated under non-denaturing conditions without apparent change in migration of the 31-kDa fragment on SDSgels. Refolding of undigested, urea-denatured rhodanese restored much of its activity. Similar treatment of rhodanese following limited tryptic digestion resulted in no regain of activity. Refolding of a mixture of intact and digested rhodanese resulted in regain of activity appropriate for the amount of intact rhodanese in the sample, indicating that clipped rhodanese does not in- hibit refolding of intact rhodanese. It is concluded that portions of the amino terminus of rhodanese are im- portant in the enzyme’s folding, but are not essential for the enzyme’s sulfurtransferase activity. Rhodanese (thiosu1fate:cyanide sulfurtransferase, EC 2.8.1.1) is a ubiquitous two-domain enzyme which is being increasingly used as a model protein for studies of structure- function relationships (1-4). The enzyme, as isolated from bovine liver is a single polypeptide having a molecular mass of 33 kDa composed of 296 amino acids (5). In its active form, all four of the rhodanese sulfhydryl groups are reduced (6, 7). The structure of the amino-terminal region of rhodanese has been implicated in several aspects of the enzyme’s targeting and function (3, 8). Rhodanese has several electrophoretic variants which can be separated by non-denaturing polyacrylamide gel electro- phoresis (9, 10). Evidence suggests that theleast mobile form search Grants ES05729 and GM25177 and Welch Grant AQ723 (to * This work was supported by National Institutes of Health Re- P. M. H.), as well as Brooke Army Medical Center Grant C-18-88 (to G. A. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. 11 TO whom correspondence should be addressed:Dept. of Biochem- istry, University of Texas Health Science Center, 7703 Floyd Curl Dr., San Antonio, T X 78284. Tel.: 512-567-3737; Fax: 512-567-3719. of the enzyme corresponds to the full length 296-amino acid form of the enzyme (5). Cleavage of small peptides from the carboxyl terminus of the full-length enzyme has been reported to result inloss of positive charges associated with conversion of the least mobile forms of rhodanese to more negative species (5, 10). Other modifications to the enzyme, including phosphorylation (ll), have been proposed to account for charge modifications associated with rhodanese electropho- retic variants. Trypsinhas been previously utilized to proteolyze rho- danese for the purpose of producing fragments useful for mapping of rhodanese epitopes recognized by anti-rhodanese antibodies and for producing fragments for immunization to produce region-specific polyclonal anti-rhodanese antisera. Trypsin has also been used to define the exposed sites of proteolytic susceptibility on the folded enzyme to aid in the understanding of the three dimensional solution structures of rhodanese conformers (12). These latter studies have revealed apparent differences in the stabilities of the two catalytic forms of rhodanese: the ES form in which the active site sulfhydryl is involved in apersulfide bond with a transferable sulfur; and the E form in which the active site sulfhydryl is not sulfur-substituted. Differences in proteolytic susceptibil- ity have also differentiated the conformations of oxidized and native rhodanese (12,13) as well as conformers with perturbed structures resulting from use of chaotropes such as urea. All amino acids involved in the sulfurtransferase chemistry of rhodanese are located on the carboxyl-terminal domain of the enzyme which suggests that this domain of the enzyme could function independently of the amino-terminal domain. This notion is consistent with reports of low molecular weight species with rhodanese-like activity from procaryotes (14). Although the amino terminus of rhodanese has recently been proposed to be involved in the targeting and translocation of the enzyme, it has also been suggested that theamino-termi- nal domain of rhodanese is necessary only to aid in folding of the enzyme to itsactive conformation and to further stabilize this conformer. Specifically, it has been suggested that the terminal segments of the enzyme have a role in the folding of the polypeptide chain via unassisted and both detergent and chaperonin-assisted mechanisms (3, 15). Trypsin proteolysis of rhodanese results in the disappearance of the parental form of the enzyme with the appearance of a daughter species at approximately 31 kDa (16). Thus, limited trypsin proteolysis of rhodanese must result in the cleavage of small peptides from one or both ends of the polypeptide chain. As such, the 31-kDa daughter fragment must include all amino acids in- volved in the active site of the enzyme. It should be noted that the extent of limited trypsin digestion of rhodanese does 15611
Transcript of Limited Tryptic Digestion Near the Amino Terminus … JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 268, No....

THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 268, No. 21, Issue of July 25, pp. 15611-15620,1993 Printed in U S A .
Limited Tryptic Digestion Near the Amino Terminus of Bovine Liver Rhodanese Produces Active Electrophoretic Variants with Altered Refolding*
(Received for publication, December 24, 1992, and in revised form, March 20, 1993)
Gerald A. MerrillSgll, Michael Butler& and Paul M. Horowitzgll)) From the $Department of Clinical Znuestigatwn, Brooke Army Medical Center, Ft. Sam Houston, Texas 78234-6200 and the §Department of Biochemistry and the VCenter for the Development and Application of Bioannlytical Methods, University of Tex& Health Science Center, Sun Antonio, Texas 78284
When the enzyme rhodanese was partially digested by immobilized trypsin, it retained greater than 50% of its original activity although less than 10% of the undigested enzyme remained. The predominant daugh- ter species were two 3 1-kDa polypeptides whose amino termini corresponded to either residue 44 or 45 of the enzyme’s sequence. Following digestion, charged spe- cies were isolated by ion exchange chromatography. Denaturing electrophoresis revealed that a 4-kDa pep- tide remained associated with the 31-kDa fragment. This 4-kDa peptide appears to correspond to the amino-terminal 45 residues of rhodanese. Further pro- teolysis ghve a 2.5-kDa peptide that dissociated under non-denaturing conditions without apparent change in migration of the 31-kDa fragment on SDS gels.
Refolding of undigested, urea-denatured rhodanese restored much of its activity. Similar treatment of rhodanese following limited tryptic digestion resulted in no regain of activity. Refolding of a mixture of intact and digested rhodanese resulted in regain of activity appropriate for the amount of intact rhodanese in the sample, indicating that clipped rhodanese does not in- hibit refolding of intact rhodanese. It is concluded that portions of the amino terminus of rhodanese are im- portant in the enzyme’s folding, but are not essential for the enzyme’s sulfurtransferase activity.
Rhodanese (thiosu1fate:cyanide sulfurtransferase, EC 2.8.1.1) is a ubiquitous two-domain enzyme which is being increasingly used as a model protein for studies of structure- function relationships (1-4). The enzyme, as isolated from bovine liver is a single polypeptide having a molecular mass of 33 kDa composed of 296 amino acids (5 ) . In its active form, all four of the rhodanese sulfhydryl groups are reduced (6, 7). The structure of the amino-terminal region of rhodanese has been implicated in several aspects of the enzyme’s targeting and function (3, 8).
Rhodanese has several electrophoretic variants which can be separated by non-denaturing polyacrylamide gel electro- phoresis (9, 10). Evidence suggests that the least mobile form
search Grants ES05729 and GM25177 and Welch Grant AQ723 (to * This work was supported by National Institutes of Health Re-
P. M. H.), as well as Brooke Army Medical Center Grant C-18-88 (to G. A. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
11 TO whom correspondence should be addressed: Dept. of Biochem- istry, University of Texas Health Science Center, 7703 Floyd Curl Dr., San Antonio, TX 78284. Tel.: 512-567-3737; Fax: 512-567-3719.
of the enzyme corresponds to the full length 296-amino acid form of the enzyme (5). Cleavage of small peptides from the carboxyl terminus of the full-length enzyme has been reported to result in loss of positive charges associated with conversion of the least mobile forms of rhodanese to more negative species (5, 10). Other modifications to the enzyme, including phosphorylation (ll), have been proposed to account for charge modifications associated with rhodanese electropho- retic variants.
Trypsin has been previously utilized to proteolyze rho- danese for the purpose of producing fragments useful for mapping of rhodanese epitopes recognized by anti-rhodanese antibodies and for producing fragments for immunization to produce region-specific polyclonal anti-rhodanese antisera. Trypsin has also been used to define the exposed sites of proteolytic susceptibility on the folded enzyme to aid in the understanding of the three dimensional solution structures of rhodanese conformers (12). These latter studies have revealed apparent differences in the stabilities of the two catalytic forms of rhodanese: the ES form in which the active site sulfhydryl is involved in a persulfide bond with a transferable sulfur; and the E form in which the active site sulfhydryl is not sulfur-substituted. Differences in proteolytic susceptibil- ity have also differentiated the conformations of oxidized and native rhodanese (12,13) as well as conformers with perturbed structures resulting from use of chaotropes such as urea.
All amino acids involved in the sulfurtransferase chemistry of rhodanese are located on the carboxyl-terminal domain of the enzyme which suggests that this domain of the enzyme could function independently of the amino-terminal domain. This notion is consistent with reports of low molecular weight species with rhodanese-like activity from procaryotes (14). Although the amino terminus of rhodanese has recently been proposed to be involved in the targeting and translocation of the enzyme, it has also been suggested that the amino-termi- nal domain of rhodanese is necessary only to aid in folding of the enzyme to its active conformation and to further stabilize this conformer. Specifically, it has been suggested that the terminal segments of the enzyme have a role in the folding of the polypeptide chain via unassisted and both detergent and chaperonin-assisted mechanisms (3, 15). Trypsin proteolysis of rhodanese results in the disappearance of the parental form of the enzyme with the appearance of a daughter species at approximately 31 kDa (16). Thus, limited trypsin proteolysis of rhodanese must result in the cleavage of small peptides from one or both ends of the polypeptide chain. As such, the 31-kDa daughter fragment must include all amino acids in- volved in the active site of the enzyme. It should be noted that the extent of limited trypsin digestion of rhodanese does
15611

15612 Rhodanese NH2-terrninal Deletion Alters Refolding
not correlate with the loss of sulfurtransferase activity and, depending on the conditions of proteolysis, limited trypsin proteolysis may even cause an increase in the specific activity of the enzyme (16).
Intact rhodanese can be induced to refold in dilute solution into an active state (15, 17) without addition of refolding assistants. It seemed plausible to use rhodanese, which had been subjected to limited trypsin proteolysis as a model to investigate the necessity for the terminal segments in refold- ing of the enzyme. The objectives of this work were to deter- mine the sites where trypsin proteolysis of native rhodanese occur and to determine the consequences of limited tryptic digestion on rhodanese activity and refolding. Evidence is provided suggesting that limited trypsin proteolysis of rho- danese results in cleavage of an amino-terminal fragment which results in the loss of the ability of the clipped enzyme to refold into an active conformer.
MATERIALS AND METHODS
Bovine liver rhodanese was isolated by modification of the method of Horowitz (18) involving purification by Affi-Gel Blue (Bio-Rad) affinity chromatography (performed by batch method) and repeated ammonium sulfate fractionations. Two different enzyme preparations were utilized in the experiments discussed in this report. One prepa- ration was used in the experiments shown in Figs. 1-4, and a second preparation was used for experiments shown in the remaining figures. We have observed that different preparations of rhodanese vary in their proportions of electrophoretic variants (see below). In addition, for control experiments not shown, bovine liver rhodanese was also prepared as described by Kurzban and Horowitz (19). Recombinant enzyme was prepared as previously described (5). Purified rhodanese was stored in 1.8 M ammonium sulfate at -70 "C. The rhodanese- dependent transfer of sulfur from thiosulfate to cyanide was moni- tored spectrophotometrically a t 460 nm (Spectronics 20, Bausch and Lomb, Rochester NY) according to Sorb0 (20).
Rhodanese was subjected to limited trypsin digestion using 1-4 units of L-1-tosylamido-2-phenylethyl chloromethyl ketone-treated trypsin immobilized to agarose (Sigma, catalog no. T-4019) for each milligram of rhodanese (at 0.5 mg/ml) as detailed in the figure legends. To help clarify the interpretation of the progressive changes in charge and size which resulted from trypsin proteolysis of rho- danese, for some experiments (as noted in the results and associated figure legends), the rhodanese was purified into its variant I and variant I1 components by anion exchange HPLC' (Mono Q FPLC 5/ 5 anion exchange column, Pharmacia LKB Biotechnology Inc.) as detailed helow for isolation of digest fragments. Each pre-purified rhodanese variant was then separately subjected to limited trypsin digestion. Digestion was allowed to proceed at 30 "C with constant mixing on a tube rotator (Scientific Equipment Products, Baltimore, MD) for periods extending to 2 h. Digestion was terminated by either centrifugation (2 min at 12,000 X g ) in an Eppendorf tabletop micro- centrifuge (Brinkmann) or by filtration (2 min at 12,000 X g ) through a 0.22-pm centrifugal filter (Lida Manufacturing Corp., Kenosha WI).
Analysis of Digests-Rhodanese digests were analyzed by non- denaturing polyacrylamide gel electrophoresis (PAGE) essentially as described by Cannella et al. (9); by discontinuous SDS-PAGE (21); by SDS-PAGE (with and without 6 M urea) utilizing the greater resolving ability for small peptides achieved by the replacement of glycine in the anode buffer with Tricine as described by Schiigger and von Jagow (22); by conventional size exclusion chromatography; and by anion exchange HPLC each as briefly described below.
Non-denaturing PAGE used 7.5% polyacrylamide gels with 0.8% cross-linker with dimensions of 12 X 14 X 0.15 cm without stacker gels. The gels were pre-electrophoresed for 40 min at constant current (72 mA) with a buffer solution composed of 100 mM glycine, 12 mM Tris (pH 8.3), and 1 mM sodium thiosulfate. Samples of rhodanese or rhodanese digests were concentrated and desalted when necessary using a Centricon centrifugal concentrator (Amicon, Beverly, MA) with a M, 10,000 cut-off centrifuged in a Sorvall RC5C centrifuge
The abbreviations used are: HPLC, high performance liquid chro- matography; PAGE, polyacrylamide gel electrophoresis; FPLC, fast protein liquid chromatography; CAPS, 3-(cyclohexylamino)propane- sulfonic acid; Tricine, 2-amino-2-hydroxymethylpropane-1,3-diol.
(Sorvall Instruments, Newtown, CT) with an SS-34 fixed angle rotor (Sorvall) and dialyzed against 10 mM Tris-HC1 (pH 7.5) prior to application to the gels. Electrophoresis was at 50 mA (constant current) for 3.5 h in a fresh glycine-Tris-sodium thiosulfate buffer solution as described above for pre-electrophoresis. Gels were fixed and stained according to Fairbanks et al. (23) and destained in isopropano1:acetic acidwater (1:1:9).
Discontinuous denaturing SDS-PAGE involved electrophoresis of rhodanese and/or rhodanese digests through 4% stacking gels at pH 6.8 using 60 V (constant voltage) followed by electrophoresis through 14% resolving gels (1.5 mm thick by approximately 8 cm wide by 5 cm long) a t pH 8.8 using 100 V constant voltage (21). Electrophoresis was performed at room temperature in a model SE200 electrophoresis unit (Hoeffer Scientific, San Francisco, CA). Fixing, staining, and destaining were as described above. Calibration of the gels was achieved by plotting the log of the molecular weights of electropho- resed standard proteins (Bio-Rad low molecular weight standards) as a function of relative mobility. Gels were fixed, stained, and destained as described above for non-denaturing gels.
Resolution of smaller peptides (less than 10,000 Da) required the use of a gel system utilizing a slower moving trailing ion than the glycine of the Laemmli discontinuous gel electrophoresis system. The Tricine gel system (22) used to resolve small peptides involved use of a 4% stacking gel (with 3% cross-linking layered above either a 16.5% resolving gel with 6% cross-linker (14 cm wide X 0.15 cm thick X approximately 11 cm high) containing 6 M urea; or above a two-part resolving gel consisting of a 2 cm high (0.075 cm thick X 14 cm wide) spacer gel (10% acrylamide with 3% cross-linker) layered above a 16.5% acrylamide gel (6% cross-linker) which was 10 cm in height without urea as specified in the figure legends. The anode electropho- resis buffer was 200 mM Tris (pH 8.9). Cathode buffer was 100 mM Tris containing 100 mM Tricine and 0.1% (w/v) SDS (pH 8.25). Gels were fixed in 50% methanol with 12% acetic acid for 1 h and stained with a colloidal suspension of Coomassie Blue G (24) or fixed in 5% glutaraldehyde in 400 mM borate at pH 8.0 for 1 h (24), stained with Coomassie Blue (25), and destained in methano1:acetic acidwater (18874) (25). Gels were calibrated with standard proteins (Bio-Rad low molecular weight standards; Rainbow Molecular Weight Stand- ards, Amersham Corp.; and/or myoglobin fragments, Gallard-Schles- inger, Carle, NY).
Conventional size exclusion chromatography of a 250-p1 rhodanese digest at 4.16 mg/ml was performed on a column (0.9 cm diameter X 90 cm) of ACA2,, (Spectrum Medical Industries, Los Angeles, CAI having an estimated exclusion limit (for globular proteins) of approx- imately 10,000 Da. The column was equilibrated and run isocratically
by the absorbance at 226 nm as determined using a LKB UV detector in 50 mM ammonium bicarbonate (pH 6.75). Elution was monitored
(LKB-Produkter AB, Bromma, Sweden). Anion exchange HPLC utilized a Mono Q FPLC 5/5 anion ex-
change column (Pharmacia) connected to a Waters HPLC system using Baseline 810 software (Millipore Corporation, Waters Chro- matography Div., Milford, MA) with UV detection at 280 nm by a Waters 486 UV detector (Millipore). The column was equilibrated with 10 mM Tris at pH 7.5 (buffer A) prior to application of intact rhodanese or trypsin digested samples (400 p1 at 0.5 mg/ml). Samples were applied at 1 ml/min. Equilibration buffer was continued for 4 min at a rate of 1 ml/min before initiation of the elution gradient. The elution gradient included an initial linear increase to 7% buffer B (10 mM Tris with 500 mM NaCl at pH 7.5) over 30 s and was continued at 7% buffer B for 3.5 min. Starting at 8 min, the percentage of buffer B was increased to 60% by a 20-min linear gradient. The column was brought to 100% buffer B by a final linear phase of the gradient over a 4-min period. Peaks were collected and concentrated by lyophilization in a Speed-Vac lyophilizing system (Savant Instru- ments, Farmingdale, NY) for further analysis. Prior to application Of additional sample, the column was reequilihrated using a minimum of 20 ml of buffer A at 1 ml/min.
Blotting and Sequencing-Rhodanese digests, resolved according to size by SDS-PAGE, were electrophoretically transferred to Im- mobilon ProBlot (Applied Biosystems, Foster City, CA) solid support membranes using a semidry transfer system (American Bionetics, Inc., Hayward, CA). The gels were briefly (15 min) soaked in 10 mM CAPS (Sigma) buffer (pH 11) which included 10% v/v methanol. The gel was placed above a stack consisting of (from bottom to top) two layers of filter paper (the lower which was soaked in 0.3 M Tris with 20% v/v methanol (pH 10.4) and the upper which was soaked in 25 mM Tris with 20% v/v methanol) and a single layer of nitrocel- lulose soaked in 10 mM CAPS (pH ll), which was immediately below
Rhodanese NH2-termiml Deletion Alters Refolding 15613 a sheet of Immobilon ProBlot which (following a rinse in 100% methanol) had been soaked in 10 mM CAPS buffer with 10% v/v methanol at pH 11. The gel was covered by two layers of filter paper soaked in 10 mM CAPS buffer with 10% v/v methanol at pH 11. Transfer was accomplished at 130 mA constant current for 20 min (to transfer from a gel having an area of 52.29 cmz X 1.5 mm thick). The membrane was stained 10 min with 0.1% w/v Amido Black 10B (Sigma), dissolved in 2% v/v acetic acid, and destained in a solution containing 45% v/v methanol and 7% v/v acetic acid. Stained bands were cut from the membrane and subjected to five cycles of sequencing in an Applied Biosystems model 477A sequenator (Foster City, CA). Sequences were compared to the known sequence of rhodanese as determined by Miller et al. (5).
Refolding-Following digestion of rhodanese with immobilized trypsin, the rhodanese sample was diluted to 50 pg/ml with a solution of 50 mM Tris at pH 7.8 containing 7.65 M urea. Intact rhodanese was similarly diluted in the urea/Tris denaturing buffer. These sam- ples were allowed to denature a t 23 "C for 45 min. Both samples were assayed to assure that denaturation was complete. Refolding of clipped rhodanese, intact rhodanese, or an equal mix of intact and clipped rhodanese was performed at 5 pg/ml in either refolding buffer (200 mM 8-mercaptoethanol, 50 mM sodium thiosulfate, and 50 mM Tris at pH 7.8) at 23 "C (15); in refolding buffer a t 4 "C (15); or in refolding buffer containing 5 mg/ml lauryl maltoside (Anatrace, Mau- mee, OH) at 23 "C (17). The extent of effective refolding in each case was monitored for 135 min by assessment of rhodanese sulfurtrans- ferase activity.
RESULTS
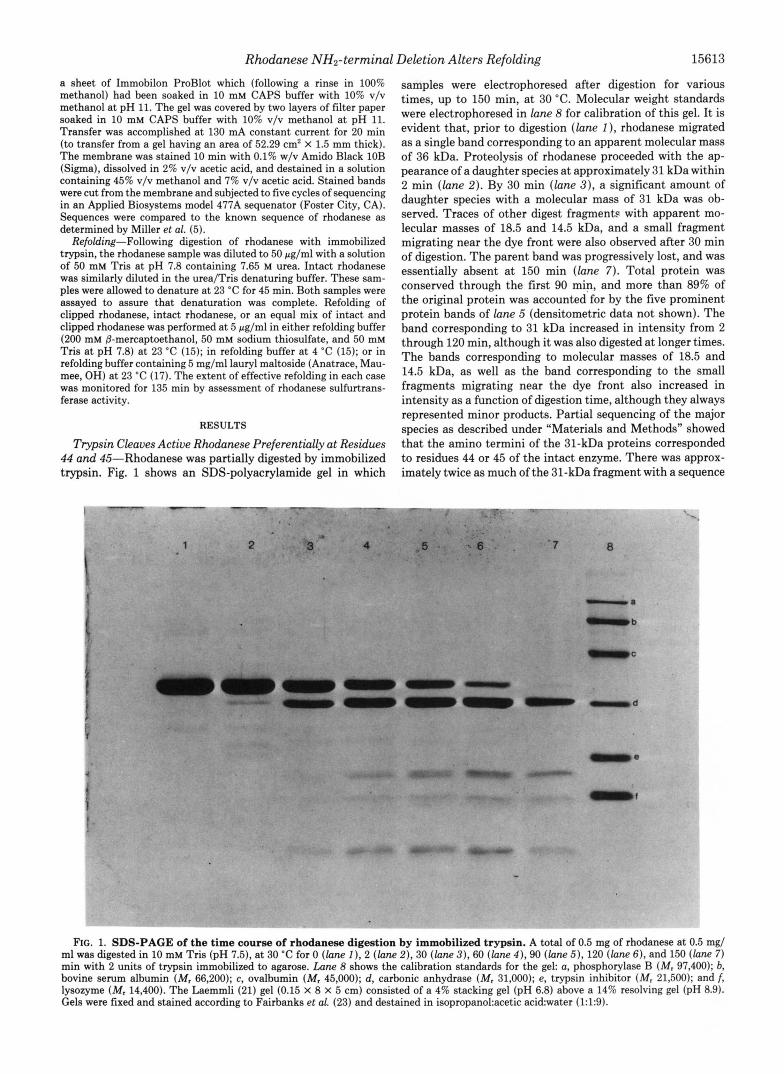
Trypsin Cleaves Active Rhodanese Preferentially a t Residues 44 and 45-Rhodanese was partially digested by immobilized trypsin. Fig. 1 shows an SDS-polyacrylamide gel in which
samples were electrophoresed after digestion for various times, up to 150 min, at 30 "C. Molecular weight standards were electrophoresed in lane 8 for calibration of this gel. It is evident that, prior to digestion ( l a n e 1 ), rhodanese migrated as a single band corresponding to an apparent molecular mass of 36 kDa. Proteolysis of rhodanese proceeded with the ap- pearance of a daughter species at approximately 31 kDa within 2 min ( l a n e 2). By 30 min ( l a n e 3), a significant amount of daughter species with a molecular mass of 31 kDa was ob- served. Traces of other digest fragment9 with apparent mo- lecular masses of 18.5 and 14.5 kDa, and a small fragment migrating near the dye front were also observed after 30 min of digestion. The parent band was progressively lost, and was essentially absent at 150 min ( l a n e 7). Total protein was conserved through the first 90 min, and more than 89% of the original protein was accounted for by the five prominent protein bands of hne 5 (densitometric data not shown). The band corresponding to 31 kDa increased in intensity from 2 through 120 min, although it was also digested at longer times. The bands corresponding to molecular masses of 18.5 and 14.5 kDa, as well as the band corresponding to the small fragments migrating near the dye front also increased in intensity as a function of digestion time, although they always represented minor products. Partial sequencing of the major species as described under "Materials and Methods" showed that the amino termini of the 31-kDa proteins corresponded to residues 44 or 45 of the intact enzyme. There was approx- imately twice as much of the 31-kDa fragment with a sequence
FIG. 1. SDS-PAGE of the time course of rhodanese digestion by immobilized trypsin. A total of 0.5 mg of rhodanese at 0.5 mg/ ml was digested in 10 mM Tris (pH 7.5), at 30 "C for 0 ( l a n e I ) , 2 ( l a n e 2), 30 ( l a n e 3 ) , 60 ( l a n e 4 ) , 90 ( l a n e 5), 120 ( l a n e 61, and 150 ( l a n e 7) min with 2 units of trypsin immobilized to agarose. Lane 8 shows the calibration standards for the gel: a, phosphorylase B (M, 97,400); b, bovine serum albumin (M, 66,200); c, ovalbumin (M, 45,000); d, carbonic anhydrase (Mr 31,000); e, trypsin inhibitor (M, 21,500); and /, lysozyme (M, 14,400). The Laemmli (21) gel (0.15 X 8 X 5 cm) consisted of a 4% stacking gel (pH 6.8) above a 14% resolving gel (pH 8.9). Gels were fixed and stained according to Fairbanks et al. (23) and destained in isopropano1:acetic acidwater (1:1:9).
15614 Rhodanese NH2-terminal Deletion Alters Refolding
starting at lysine 44 as there was with a sequence starting with glutamic acid 45 (sequence data not shown). The frag- ment(s) having an apparent molecular mass of about 18.5 kDa also had amino termini corresponding to residues 44 or 45 of the intact enzyme, thus representing the amino-terminal domain of rhodanese. The daughter fragment with an appar- ent molecular mass of 14.5 kDa had an amino terminus corresponding to residue 187 of the full length protein, thus representing the carboxyl-terminal domain of rhodanese.
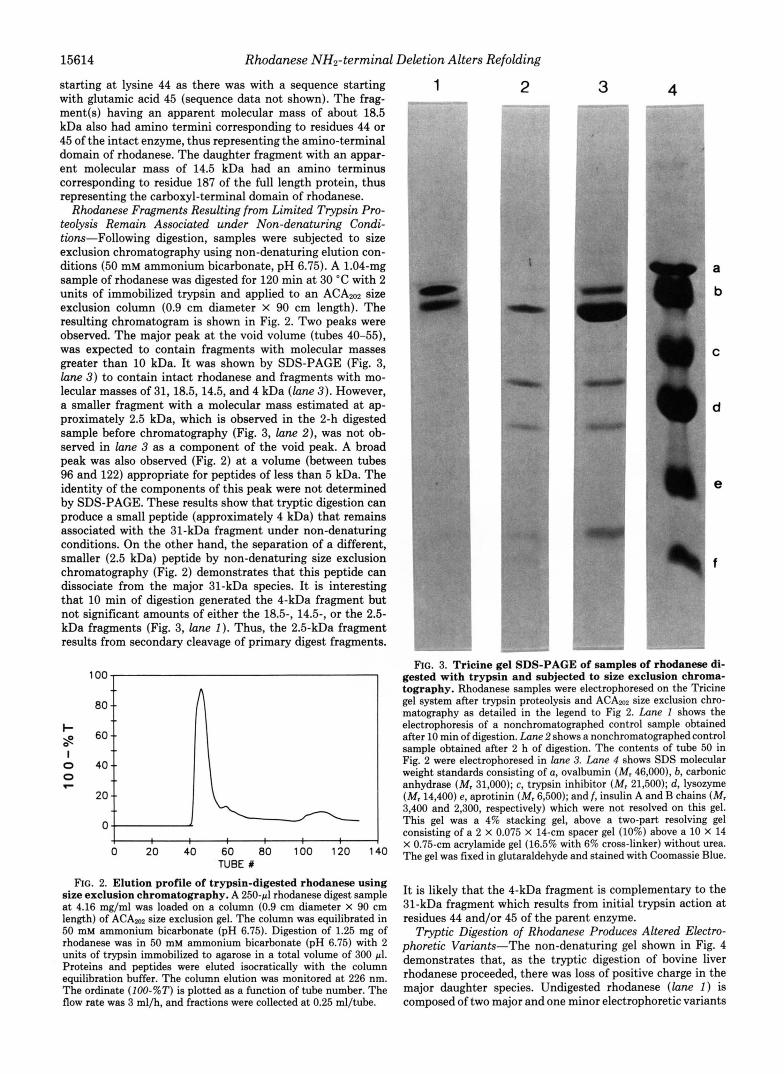
Rhodanese Fragments Resulting from Limited Trypsin Pro- teolysis Remain Associated under Non-denaturing Condi- tions-Following digestion, samples were subjected to size exclusion chromatography using non-denaturing elution con- ditions (50 mM ammonium bicarbonate, pH 6.75). A 1.04-mg sample of rhodanese was digested for 120 min at 30 "C with 2 units of immobilized trypsin and applied to an ACAm2 size exclusion column (0.9 cm diameter x 90 cm length). The resulting chromatogram is shown in Fig. 2. Two peaks were observed. The major peak at the void volume (tubes 40-55), was expected to contain fragments with molecular masses greater than 10 kDa. It was shown by SDS-PAGE (Fig. 3, lane 3) to contain intact rhodanese and fragments with mo- lecular masses of 31, 18.5,14.5, and 4 kDa (lane 3). However, a smaller fragment with a molecular mass estimated at ap- proximately 2.5 kDa, which is observed in the 2-h digested sample before chromatography (Fig. 3, lune Z ) , was not ob- served in lane 3 as a component of the void peak. A broad peak was also observed (Fig. 2) at a volume (between tubes 96 and 122) appropriate for peptides of less than 5 kDa. The identity of the components of this peak were not determined by SDS-PAGE. These results show that tryptic digestion can produce a small peptide (approximately 4 kDa) that remains associated with the 31-kDa fragment under non-denaturing conditions. On the other hand, the separation of a different, smaller (2.5 kDa) peptide by non-denaturing size exclusion chromatography (Fig. 2) demonstrates that this peptide can dissociate from the major 31-kDa species. It is interesting that 10 min of digestion generated the 4-kDa fragment but not significant amounts of either the 185, 14.5-, or the 2.5- kDa fragments (Fig. 3, lane I). Thus, the 2.5-kDa fragment results from secondary cleavage of primary digest fragments.
1001 1
a0 -- r 60 --
40 - -
20"
0- L
4 I 0 20 40 60 80 100 120 140
TUBE #
FIG. 2. Elution profile of trypsin-digested rhodanese using size exclusion chromatography. A 250-pl rhodanese digest sample at 4.16 mg/ml was loaded on a column (0.9 cm diameter X 90 cm length) of ACAm size exclusion gel. The column was equilibrated in 50 mM ammonium bicarbonate (pH 6.75). Digestion of 1.25 mg of rhodanese was in 50 mM ammonium bicarbonate (pH 6.75) with 2 units of trypsin immobilized to agarose in a total volume of 300 81. Proteins and peptides were eluted isocratically with the column equilibration buffer. The column elution was monitored at 226 nm. The ordinate (100-%T) is plotted as a function of tube number. The flow rate was 3 ml/h, and fractions were collected at 0.25 ml/tube.
1 2 3 4 "
I
e
f
FIG. 3. Tricine gel SDS-PAGE of samples of rhodanese di- gested with trypsin and subjected to size exclusion chroma- tography. Rhodanese samples were electrophoresed on the Tricine gel system after trypsin proteolysis and ACAm size exclusion chro- matography as detailed in the legend to Fig 2. Lane 1 shows the electrophoresis of a nonchromatographed control sample obtained after 10 min of digestion. Lane 2 shows a nonchromatographedcontrol sample obtained after 2 h of digestion. The contents of tube 50 in Fig. 2 were electrophoresed in lone 3. Lane 4 shows SDS molecular weight standards consisting of a, ovalbumin (Mr 46,000), b, carbonic anhydrase (M, 31,000); c, trypsin inhibitor (Mr 21,500); d, lysozyme (M, 14,400) e, aprotinin (Mr 6,500); and f, insulin A and B chains (M, 3,400 and 2,300, respectively) which were not resolved on this gel. This gel was a 4% stacking gel, above a two-part resolving gel consisting of a 2 X 0.075 X 14-cm spacer gel (10%) above a 10 X 14 X 0.75-cm acrylamide gel (16.5% with 6% cross-linker) without urea. The gel was fixed in glutaraldehyde and stained with Coomassie Blue.
It is likely that the 4-kDa fragment is complementary to the 31-kDa fragment which results from initial trypsin action at residues 44 and/or 45 of the parent enzyme.
Tryptic Digestion of Rhodanese Produces Altered Electro- phoretic Variants-The non-denaturing gel shown in Fig. 4 demonstrates that, as the tryptic digestion of bovine liver rhodanese proceeded, there was loss of positive charge in the major daughter species. Undigested rhodanese ( l a n e I ) is composed of two major and one minor electrophoretic variants

Rhodanese NH2-terminul Deletion Alters Refolding 15615 - " . -,..
FIG. 4. Non-denaturing gel electrophoresis showing the time course of proteolysis of rhodanese with trypsin. Non-denaturing gel electrophoresis of rhodanese samples obtained, prior to initiation of trypsin digestion (hne I ) , and at 30 ([ane Z), 60 ( l a n e 31, and 150 min (hne 4 ) after initiation of digestion. Digestion was performed at 30 "C, for the indicated times, in a total volume of 354 p1 of 50 mM sodium phosphate, pH 7.5, with 0.5 mg/ml rhodanese and 2 units of immobilized trypsin. Gels were fixed and stained according to Fairbanks et al. (23) and destained in isopropano1:acetic acidwater (1:1:9).
designated in order of increasing mobility as variants I, 11, and 111. Rhodanese, electrophoresed after 30 min of trypsin digestion (lane 2) , showed a loss of variant I and an increase in variant 111. There was no apparent loss of variant I1 after 30 min of digestion, but a new variant (designated as variant I V ) appears below the variant I11 band in lane 2. By 90 min (lane 3 ) , there was further loss of variant I as well as variant I1 with obvious increase in the proportion of variants I11 and IV. By 150 min of digestion (lane 4 ) , there was little if any remaining rhodanese in variants I or 11, with the predominant species being a more negatively charged variant IV. There did not appear to be complete conservation of protein as a func- tion of digestion time.
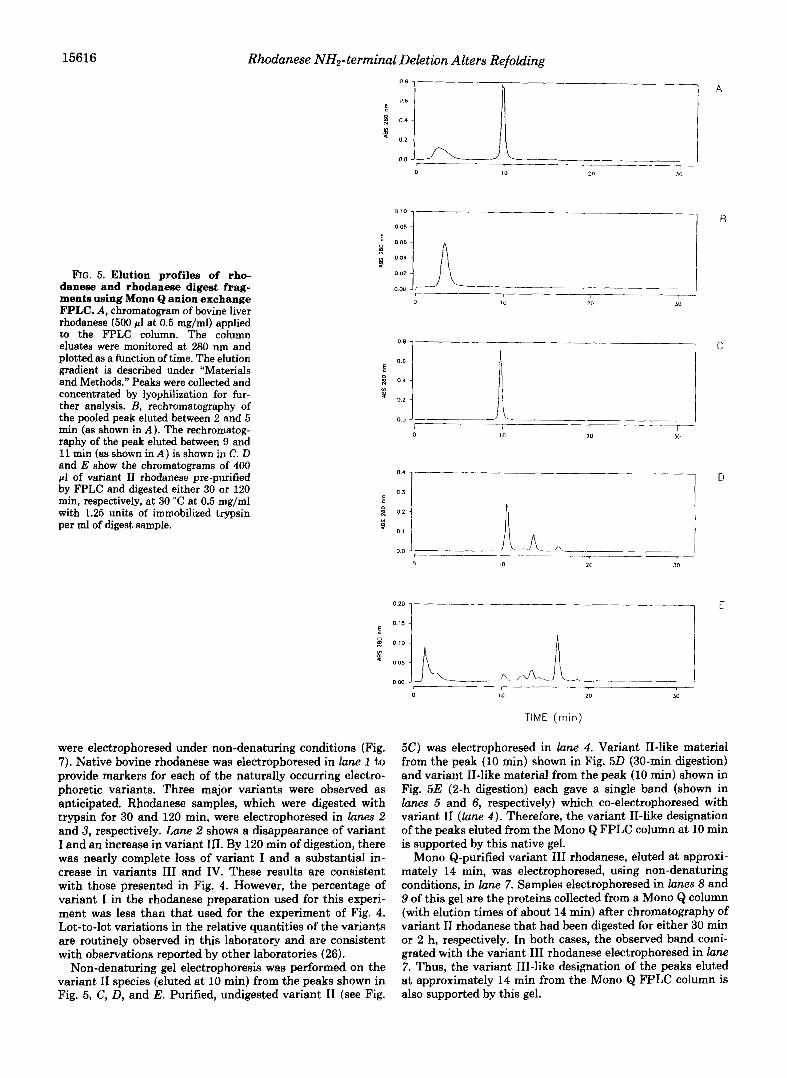
Variant Z and Variant ZZ Forms of Rhodanese Are Isolated by Anion Exchange Chromatography-Rhodanese electropho- retic variants were isolated using non-denaturing Mono Q anion exchange FPLC. A typical Mono Q FPLC chromato- gram of rhodanese is shown in Fig. 5A. The earliest eluted variant of the enzyme (2-5 min) proved to be the variant I form (see below). The variant that eluted a t approximately 10 min was shown to be variant I1 (see below). The Mono Q isolated variant I (Fig. 5B) and variant I1 (Fig. 5C) rechro- matographed as single peaks with retention times similar to those seen with the original mixture (Fig. 5A). Both FPLC- purified variant I and variant I1 showed approximately the same specific activity as the starting mixture (within 5%). Fig. 6, lane 2, shows that SDS-PAGE of the fraction collected from the peak shown in Fig. 5C gave a single band correspond- ing to an apparent molecular mass of 36 kDa. FPLC-purified variant I or variant I1 rhodanese was utilized in all subsequent experiments.
Trypsin Digestion of Variant IZ Rhodanese Produces Altered Electrophoretic Variants-A sample of variant I1 rhodanese was subjected to 30 min of digestion with trypsin, and dena- turing gel electrophoresis showed both a 31-kDa and a 4-kDa fragment (Fig. 6, lane 3) . An aliquot of this digest sample was applied to the Mono Q column, and the resulting chromato- gram is shown in Fig. 50. There was a loss of protein associ- ated with the variant I1 peak (10 min). However, two new peaks were eluted after the variant I1 peak. Urea gel SDS- PAGE of the proteins collected in the variant I1 peak (Fig. 50, lO min) demonstrated only one species with an apparent molecular mass of 36 kDa (Fig. 6, lane 4) . However, urea gel SDS-electrophoresis of the proteins associated with the FPLC
peak (Fig. 50, 14 min) demonstrated the presence of three major proteins (Fig. 6, lane 5). One of these corresponded to a molecular mass of 36 kDa indicating that variant I1 can have additional loss of positive charge without apparent de- crease in molecular mass. Fig. 6, lane 5, also shows that significant amounts of a 31-kDa fragment and a 4-kDa frag- ment were present in this FPLC peak. These results lend further support to the suggestion that limited tryptic digestion of variant I1 can produce 31- and 4-kDa fragments which remain associated under non-denaturing conditions. The small peak eluted from the FPLC column a t about 17.5 min (shown in Fig. 5 0 ) was not analyzed.
The tryptic digestion of purified variant I1 rhodanese was continued for a total of 2 h. At that time, samples were removed for non-denaturing gel electrophoresis, urea gel SDS- electrophoresis, and for anion exchange chromatography. Urea gel SDS-PAGE (Fig. 6, lane 8) shows bands correspond- ing to molecular masses of 31 and 4 kDa, in addition t o the parent band.
The results of anion exchange chromatography of the 2-h digest sample are shown in Fig. 5E. There was a substantial loss of variant I1 (10 min) and variant I11 (14 min), as compared to Fig. 5 0 . However, there was a significant increase in a species that eluted at about 17.5 min (variant IV). The proteins associated with this peak were electrophoresed in lane 10 of the SDS-urea gel shown in Fig. 6. The predominant bands correspond to proteins of molecular masses of 31 and 4 kDa, again supporting the existence of an association be- tween these fragments in trypsin cleaved rhodanese isolated under non-denaturing conditions. The components of the peak which eluted a t approximately 1 min (Fig. 5E) were electrophoresed in lane 9 of the gel shown in Fig. 6. No protein bands were observed, although a similar amount of material (based on absorbance at 280 nm) was electrophoresed. Thus, it is likely that the components of this peak were very small peptides resulting from further action of trypsin on the pro- teolytic fragments initially generated.
Similar results were obtained when rhodanese, isolated by a modified purification procedure (19), which results in recov- ery of predominantly variant I1 rhodanese, was digested by trypsin under similar conditions (data not shown).
Non-denaturing Gel Electrophoresis of Rhodanese Variants Correlates with Their Behavior on Anion Exchange Chroma- tography-Proteins and protein fragments obtained by FPLC
15616 Rhodanese NH2-terminal Deletion Alters Refolding
A
E
w I
FIG. 5. Elution profiles of rho- danese and rhodanese digest frag- ments using Mono Q anion exchange FPLC. A , chromatogram of bovine liver rhodanese (500 pl a t 0.5 mg/ml) applied to the FPLC column. The column eluates were monitored at 280 nm and plotted as a function of time. The elution gradient is described under "Materials and Methods." Peaks were collected and concentrated by lyophilization for fur- ther analysis. B, rechromatography of the pooled peak eluted between 2 and 5 min (as shown in A ). The rechromatog- raphy of the peak eluted between 9 and 11 min (as shown in A ) is shown in C. D and E show the chromatograms of 400
by FPLC and digested either 30 or 120 pl of variant I1 rhodanese pre-purified
min, respectively, a t 30 "C at 0.5 mg/ml with 1.25 units of immobilized trypsin per ml of digest sample.
B
0.6 - h 3 0 4 -
1 0 2 -
0.0 - J i- I I I I
O IO 20 30
o'20 1
were electrophoresed under non-denaturing conditions (Fig. 7). Native bovine rhodanese was electrophoresed in lane 1 to provide markers for each of the naturally occurring electro- phoretic variants. Three major variants were observed as anticipated. Rhodanese samples, which were digested with trypsin for 30 and 120 min, were electrophoresed in lanes 2 and 3, respectively. Lane 2 shows a disappearance of variant I and an increase in variant 111. By 120 min of digestion, there was nearly complete loss of variant I and a substantial in- crease in variants I11 and IV. These results are consistent with those presented in Fig. 4. However, the percentage of variant I in the rhodanese preparation used for this experi- ment was less than that used for the experiment of Fig. 4. Lot-to-lot variations in the relative quantities of the variants are routinely observed in this laboratory and are consistent with observations reported by other laboratories (26).
Non-denaturing gel electrophoresis was performed on the variant I1 species (eluted at 10 min) from the peaks shown in Fig. 5, C, D, and E. Purified, undigested variant I1 (see Fig.
0 20
TIME (rnin)
5C) was electrophoresed in lane 4. Variant 11-like material from the peak (10 min) shown in Fig. 5D (30-min digestion) and variant 11-like material from the peak (10 min) shown in Fig. 5E (2-h digestion) each gave a single band (shown in lanes 5 and 6, respectively) which co-electrophoresed with variant I1 ( l a n e 4 ) . Therefore, the variant 11-like designation of the peaks eluted from the Mono Q FPLC column at 10 min is supported by this native gel.
Mono Q-purified variant I11 rhodanese, eluted at approxi- mately 14 min, was electrophoresed, using non-denaturing conditions, in lane 7. Samples electrophoresed in lanes 8 and 9 of this gel are the proteins collected from a Mono Q column (with elution times of about 14 min) after chromatography of variant I1 rhodanese that had been digested for either 30 min or 2 h, respectively. In both cases, the observed band comi- grated with the variant I11 rhodanese electrophoresed in lane 7. Thus, the variant 111-like designation of the peaks eluted at approximately 14 min from the Mono Q FPLC column is also supported by this gel.
Rhodanese NH2-terminal Deletion Alters Refolding 15617
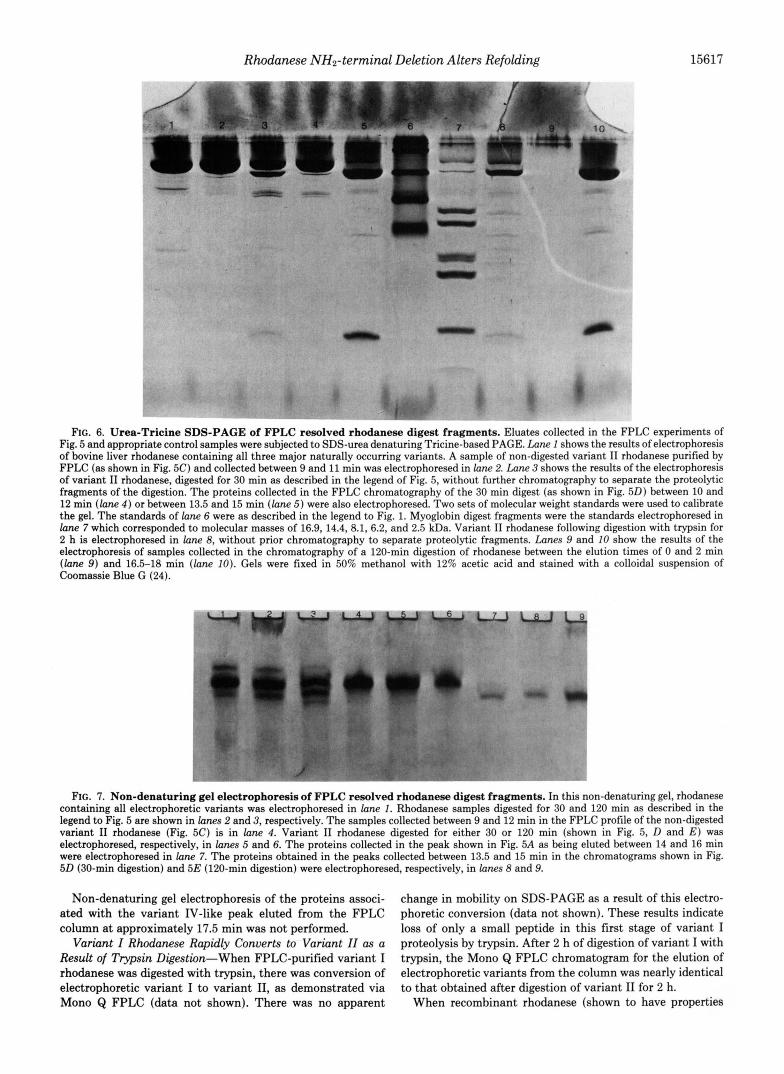
FIG. 6. Urea-Tricine SDS-PAGE of FPLC resolved rhodanese digest fragments. Eluates collected in the FPLC experiments of Fig. 5 and appropriate control samples were subjected to SDS-urea denaturing Tricine-based PAGE. Lane 1 shows the results of electrophoresis of bovine liver rhodanese containing all three major naturally occurring variants. A sample of non-digested variant I1 rhodanese purified by FPLC (as shown in Fig. 5C) and collected between 9 and 11 min was electrophoresed in lune 2. Lane 3 shows the results of the electrophoresis of variant I1 rhodanese, digested for 30 min as described in the legend of Fig. 5, without further chromatography to separate the proteolytic fragments of the digestion. The proteins collected in the FPLC chromatography of the 30 min digest (as shown in Fig. 50) between 10 and 12 min ( l a n e 4 ) or between 13.5 and 15 min ( l a n e 5) were also electrophoresed. Two sets of molecular weight standards were used to calibrate the gel. The standards of lane 6 were as described in the legend to Fig. 1. Myoglobin digest fragments were the standards electrophoresed in lane 7 which corresponded to molecular masses of 16.9, 14.4, 8.1, 6.2, and 2.5 kDa. Variant I1 rhodanese following digestion with trypsin for 2 h is electrophoresed in lane 8, without prior chromatography to separate proteolytic fragments. Lanes 9 and 10 show the results of the electrophoresis of samples collected in the chromatography of a 120-min digestion of rhodanese between the elution times of 0 and 2 min ( l a n e 9) and 16.5-18 min ( l a n e 10). Gels were fixed in 50% methanol with 12% acetic acid and stained with a colloidal suspension of Coomassie Blue G (24).
FIG. 7. Non-denaturing gel electrophoresis of FPLC resolved rhodanese digest fragments. In this non-denaturing gel, rhodanese containing all electrophoretic variants was electrophoresed in lune 1. Rhodanese samples digested for 30 and 120 min as described in the legend to Fig. 5 are shown in lanes 2 and 3, respectively. The samples collected between 9 and 12 min in the FPLC profile of the non-digested variant I1 rhodanese (Fig. 5C) is in lane 4. Variant I1 rhodanese digested for either 30 or 120 min (shown in Fig. 5, D and E ) was electrophoresed, respectively, in lanes 5 and 6. The proteins collected in the peak shown in Fig. 5A as being eluted between 14 and 16 min were electrophoresed in lane 7. The proteins obtained in the peaks collected between 13.5 and 15 min in the chromatograms shown in Fig. 50 (30-min digestion) and 5E (120-min digestion) were electrophoresed, respectively, in lanes 8 and 9.
Non-denaturing gel electrophoresis of the proteins associ- ated with the variant IV-like peak eluted from the FPLC column at approximately 17.5 min was not performed.
Variant I Rhodanese Rapidly Converts to Variant ZI as a Result of Trypsin Digestion-When FPLC-purified variant I rhodanese was digested with trypsin, there was conversion of electrophoretic variant I to variant 11, as demonstrated via Mono Q FPLC (data not shown). There was no apparent
change in mobility on SDS-PAGE as a result of this electro- phoretic conversion (data not shown). These results indicate loss of only a small peptide in this first stage of variant I proteolysis by trypsin. After 2 h of digestion of variant I with trypsin, the Mono Q FPLC chromatogram for the elution of electrophoretic variants from the column was nearly identical to that obtained after digestion of variant I1 for 2 h.
When recombinant rhodanese (shown to have properties

15618 Rhodanese NH2-terminul Deletion Alters Refolding
consistent with that of variant I (5)) was digested with trypsin in a manner similar to the digestion of Mono Q purified variant I, the results were essentially identical to those de- scribed above (data not shown).
Intact Rhodanese, but Not Tvpsinized Rhodanese, Can Be Refolded to an Active Conformution-Rhodanese, subjected to limited trypsin digestion, retained a substantial amount of thiosulfate sulfurtransferase activity, even after 2 h of diges- tion. Approximately 90% of the original activity was retained after 45 min of limited digestion. More than 50% of the activity remained after 2 h of digestion, at which time less that 10% of the protein was associated with a band corre- sponding to a molecular mass of 36 kDa (densitometric and enzymatic activity data not shown). Table I summarizes the results of refolding (as a function of time) of denatured, intact, rhodanese and/or rhodanese subjected to limited trypsin pro- teolysis. The rhodanese concentration was 5 pg/ml, and each sample (20 pl) was incubated for 10 min in the assay. Both spontaneous refolding and detergent assisted refolding were investigated. As indicated in the table, intact rhodanese re- folded spontaneously at 23 "C within 15 min to yield approx- imately 35% of the original activity. Longer incubation of the rhodanese under spontaneous refolding conditions did not increase the amount of rhodanese activity which was regained. In contrast, rhodanese which had been denatured following partial trypsin proteolysis, did not regain any rhodanese ac- tivity, even after 135 min of incubation at 23 "C. A 1:l mixture of denatured, intact, rhodanese and rhodanese subjected to limited trypsin proteolysis (to achieve a final combined rho- danese concentration of 5 pg/ml) regained a degree of activity appropriate for refolding of the intact rhodanese in the sam- ple. Similar results were obtained when the refolding was attempted at 4 "C. When refolding was attempted at 23 "C in the presence of 5 mg/ml lauryl maltoside, the denatured, nonproteolyzed rhodanese refolded progressively as a function of time to about 68% of the original activity of the sample, whereas trypsinized rhodanese did not regain any significant amount of activity. Again, a 1:1 mixture of denatured, intact, rhodanese and trypsinized rhodanese refolded to achieve an activity equivalent to that expected from refolding of the intact rhodanese in the sample. There was no apparent aggre- gation of rhodanese under any of the refolding conditions
with either intact enzyme, with trypsin digested rhodanese, or with a mixture of these rhodanese species.
DISCUSSION
Trypsin digestion of rhodanese resulted in the formation of a 31-kDa daughter species. This 31-kDa fragment was a mixture of two polypeptides with amino-terminal residues corresponding to residues 44 or 45 of intact rhodanese with an approximate 2:l predominance of the polypeptide with lysine 45 as the amino-terminal amino acid. The ease of accessibility of trypsin to these residues is apparent from the proposed structure of rhodanese as seen in Fig. 8. Results presented here indicate that the peptide corresponding to the amino-terminal 45 amino acids of intact rhodanese remains associated with the 31-kDa daughter species following the cleavage at residues 44 and 45 by trypsin. It can be seen in Fig. 8 that a portion of the amino-terminal 45 amino acids of rhodanese (residues 25-40) comprise a part of the @-sheet core of the amino-terminal domain of the enzyme. As such, these residues are both protected from proteases and involved in strong hydrophobic interactions with other core residues. Loss of residues comprising part of this hydrophobic core of the protein's amino-terminal domain would require a major disruption of the tertiary structure of rhodanese. It should be emphasized that rhodanese subjected to limited trypsin pro- teolysis does not significantly inactivate. The ability to dem- onstrate a small peptide, with an apparent molecular mass of 4 kDa, after SDS-polyacrylamide electrophoresis of rhodanese which has been subjected to limited trypsin proteolysis and thoroughly washed (during size exclusion chromatography and centrifugal ultrafiltration following ion exchange chro- matography), provides support for the continued association, under non-denaturing conditions, of the 31- and 4-kDa daugh- ter species generated as a result of trypsin action at residues 44 and 45 of rhodanese.
However, there are numerous other cleavage sites for tryp- sin located in the first 45 amino acids of the rhodanese sequence. The x-ray crystal structure of rhodanese suggests that the amino-terminal residues are accessible to proteases. There appears to be significant heterogeneity in the extent of cleavage within the first 23 residues of the amino terminus of the enzyme. Cleavage of rhodanese at some of these residues
TABLE I Refolding of control and trypsinized rhodanese using varying refoldingprotocols
Spontaneous refolding conditionsb Spontaneous refolding conditionsb Detergent-assisted refolding conditions' Time" 23 'C 4 "C 23 "C
Control Trypsinized Mixture" Control Trypsinized Mixture Control Trypsinized Mixture min
0' 0 0 0 0 0 0 0 0 0
45 38 1 18 44 0 22 44 1 18 75 37 0 18 43 0 19 56 1 24
15 35 1 18 40 1 21 24 1 10
135 36 1 17 42 1 18 68 2 27 'Assay of a 20-rl rhodanese sample containing 0.1 pg of enzyme was assayed for 10 min in the standard assay at room temperature except
the 0 min time point as noted in e. * Refolding buffer consisted of 200 mM 8-mercaptoethanol, 50 mM sodium thiosulfate, and 50 mM tris at pH 7.8. The rhodanese concentration
during refolding was 5 pg/ml. Refolding buffer consisted of 200 mM /3-mercaptoethanol, 50 mM sodium thiosulfate, and 50 mM tris at pH 7.8 with 5 mg/ml lauryl
maltoside. The rhodanese concentration during refolding was 5 pg/ml. A 1:l mixture of the denatured control (full-length) rhodanese and trypsinized rhodanese such that the final concentration of rhodanese
of both types is 5 pglml. If only the full-length rhodanese was capable of refolding then a value equal to one-half that seen in the control column would be anticipated. Values smaller than one-half that seen in the control column would suggest an interference of the refolding of control rhodanese by the trypsinized rhodanese. Values significantly greater than the sum of one-half of the control value plue one-half of the value seen in the trypsinized column would suggest a cooperative influence of the control rhodanese on the refolding of trypsinized rhodanese.
e Asasyed at 50 pg/ml prior to dilution into each of the refolding buffers.

Rhodanese NH2-terminal Deletion Alters Refolding 15619
FIG. 8. The ribbon structure of bovine liver rhodanese. The ribbon structure shown depicts the x-ray struc- ture of bovine liver rhodanese originally presented by Ploegman et al. (6) which has been adapted from the modified ver- sion depicted by Miller et al. ( 5 ) . The view is directly into the active site pocket with the carboxyl-terminal domain on the right. The approximate locations of the residues near the ends of the poly- peptide that represent potential and/or actual trypsin cleavage sites are anno- tated. Residues 25-45, which comprise part of the hydrophobic core of the en- zyme, are shown by the shaded area.
(such as arginine 7) could result in loss of small peptides that would account for charge differences resulting in the differ- ences between electrophoretic and ion exchange rhodanese variants I1 and I11 without significant decrease in molecular weight. It has been demonstrated that processing of the car- boxyl terminus of rhodanese can account for the charge loss (due to loss of lysine 295 resulting from cleavage on the carboxyl side of lysine 292) without loss of enough mass to be detectable by SDS-PAGE (5,lO). Limited carboxyl processing has been proposed to account for the difference between variant I and I1 rhodanese (10). It is doubtful that further trypsin cleavage occurs at the carboxyl terminus, although cleavage at arginine 281 could cause a loss of charge associated with lysine 292 without a major change in apparent molecular weight. However, this residue is predicted to be resistant to trypsin proteolysis as a result of the secondary structure surrounding arginine 281. Thus, although trypsin is likely to be capable of some limited processing of the carboxyl termi- nus, the major trypsin action on rhodanese occurs at the amino terminus. It is concluded that the portion of the amino terminus peptide involved with the hydrophobic core of the enzyme (residues 25-40) acts as an anchor to hold the 4-kDa amino peptide fragment in association with the 31-kDa daughter species. Additional cleavage and dissociation from the enzyme of small peptides within the sequence consisting of residues 1-25 by the activity of trypsin is likely responsible for most of the charge differences associated with the forma- tion of electrophoretic variants (both naturally and by tryp- sinization of rhodanese). The small peptide seen in some trypsin digested rhodanese preparations with an apparent molecular mass of 2.5 kDa could result from the cleavage of small peptides from near the amino terminus such as at arginine 20 or lysine 23 with dissociation of the fragment from the enzyme. The proposed course of events which ex- plains the formation of electrophoretic variations of rho- danese based on loss of terminal peptides in no way excludes the possibility of additional variants that could result from differential phosphorylation (11) or amidation (26).
It is well documented that intact variant I and I1 rhodanese can be successfully refolded in either the presence or absence of detergents (3, 8, 15, 17) resulting in the reappearance of the sulfurtransferase activity of rhodanese. The degrees of successful refolding of intact rhodanese using each of three refolding conditions utilized in the experiments presented in this manuscript are within the ranges normally seen for each technique. However, rhodanese subjected t o trypsin proteol-
ysis prior to urea denaturation was totally resistant to SUC- cessful refolding under all three refolding protocols. This observation suggests that rhodanese refolding is significantly affected by the absence of residues 1-45 of the sequence, which are presumably dissociated from the 31-kDa fragment as a result of the urea denaturation. This result suggests a critical role for the amino terminus in the refolding of the enzyme. It is important to note that trypsinized rhodanese does not aggregate following urea denaturation and subse- quent dilution into refolding solutions. The failure of dena- tured trypsinized rhodanese to inhibit refolding of intact rhodanese co-incubated in the same refolding solutions pro- vides evidence to suggest that trypsinized rhodanese frag- ments do not associate with intact rhodanese in such a way as to inhibit normal refolding of intact rhodanese.
It is therefore concluded that there are residues within the amino-terminal rhodanese sequence which are important for effective enzyme refolding. It is possible that certain regions near the amino terminus (such as residues 25-40), which are involved in the hydrophobic core of the enzyme, act as nodes for initiation of proper refolding of rhodanese into an active enzyme. The lack of aggregation when trypsinized rhodanese is refolded indicates that the failure to regain activity is not due to extensive intermolecular associations of rhodanese monomers or digest fragments. Although limited trypsin digestion of rhodanese does not produce a carboxyl-terminal domain which can be isolated independent of all residues of the amino-terminal domain, the results indicate that the carboxyl-terminal domain of rhodanese cannot refold and function independently of the amino-terminal domain, It is further extrapolated that the amino-terminal region of rho- danese has a potential role in prevention of the misfolding of the enzyme which results in inactivation of rhodanese.
REFERENCES 1. Merrill, G. A., Miller, D., Chirgwin, J., and Horowitz, P. M. (1992) J.
2. Aird, B. A., and Horowitz, P. M. (1988) Biochim. Biophys. Acta 956 , 30- Protein Chem. 11 , 193-199
28 3. Miidoza, J. A., Lorimer, G. H., and Horowitz, P. M. (1991) J. Biol. Chem.
4. Bochkareva, E. S., Lissin, N. M., Flynn, G. C., Rothman, J. E., and
5. Miller, D., Delgado, R., Chirgwin, J., Hardies, S., and Horowitz, P. (1990)
266,16973-16976
Girshovich, A. S . (1992) J. Biol. Chem. 267, 6796-6800
J. Biol. Chem 266.4686-4691 6. Ploegman, J. H., Drent, G., Kalk, K. H., and Hal, W. G. J. (1978) J. Mol.
7. Ploegman, J. H., Drent, G., Kalk, K. H., Hol, W. G. J., Heinrikson, R. L.,
8. Mendoza, J. A., Grant, E., and Horowitz, P. M. (1993) J. Protein Chem.
Bid. 123,557-594
Keim, P., Weng, L., and Russell, J. (1978) Nature 273 , 124-129
1 2.65-69 9. Cannella, C., Costa, M., Pensa, B., Ricci, G., Pecci, L., and Cavallini, D.
- -, - - - -
(1981) Eur. J . Biochem. 119,491-495

15620 Rhodanese NH2-terminal Deletion Alters Refolding


















