
Lie algebraic approach to multiphoton excitation of diatomic molecules in intense laser fields
7
— — < < Lie Algebraic Approach to Multiphoton Excitation of Diatomic Molecules in Intense Laser Fields YING DAI, SHI-LIANG DING Department of Chemistry, Shandong University, Jinan 250100, People’s Republic of China Received 25 October 1997; revised 15 May 1998; accepted 26 May 1998 ABSTRACT: The quadratic anharmonic oscillator Lie algebraic model is used to study the multiphoton transition of the diatomic molecule placed in intense laser fields. The multiphoton excitation of vibration and vibration-rotation of diatomic molecules in intense laser fields are discussed. In the pure vibration transition we calculate the transition probability versus the frequency of the laser fields for the CO molecule. We also investigate the roles of rotational motion in multiphoton processes and compare with pure vibration for the LiH molecule. The influences of the angular quantum number l and the molecular orientations in laser fields on the multiphoton processes are discussed. The averaged absorb energy changing with the laser field’s frequency is calculated. Q 1999 John Wiley & Sons, Inc. Int J Quant Chem 71: 201]207, 1999 Key words: Lie algebra; multiphoton; vibration-rotation; laser Introduction he phenomenon of infrared multiphoton exci- T tation and dissociation is one of the many interesting spectroscopic discoveries of the previ- ous decades. The interest of experimentalists and theoreticians alike has been aroused in recent years by the development of high-powered lasers that can be used to induce molecular processes previ- Correspondence to: Y. Dai. Project grant sponsors: National Science Foundation of China, the State Key Laboratory of Theoretical and Computa- tional Chemistry of Jilin University, and the Natural Science Foundation of Shandong Province, China. ously termed forbidden. To understand theoreti- cally how molecules absorb energy under such conditions requires a nonperturbative analysis. It is impossible to solve the time-dependent Schro- ¨ dinger equation accurately for most molecules of experimental interest, so the simplest realistic sys- tem to study is a diatomic molecule interacting with a laser. There are some theories on the study of the multiphoton processes of this system, like wx Coulter transformation theory 1 , Floquet theory w x wx 2a, b , and Lie algebraic theory 3 . Most of them use the anharmonic Morse oscillator model and solve the Schrodinger equation as an initial value ¨ problem. However, it is usually difficult and com- wx plicated. In Ref. 3 , in order to investigate the multiphoton excitation, the ‘‘simulated potential’’ ( ) International Journal of Quantum Chemistry, Vol. 71, 201 ]207 1999 Q 1999 John Wiley & Sons, Inc. CCC 0020-7608 / 99 / 020201-07
Transcript of Lie algebraic approach to multiphoton excitation of diatomic molecules in intense laser fields

— —< <
Lie Algebraic Approach to MultiphotonExcitation of Diatomic Molecules inIntense Laser Fields
YING DAI, SHI-LIANG DINGDepartment of Chemistry, Shandong University, Jinan 250100, People’s Republic of China
Received 25 October 1997; revised 15 May 1998; accepted 26 May 1998
ABSTRACT: The quadratic anharmonic oscillator Lie algebraic model is used to studythe multiphoton transition of the diatomic molecule placed in intense laser fields. Themultiphoton excitation of vibration and vibration-rotation of diatomic molecules inintense laser fields are discussed. In the pure vibration transition we calculate thetransition probability versus the frequency of the laser fields for the CO molecule. Wealso investigate the roles of rotational motion in multiphoton processes and comparewith pure vibration for the LiH molecule. The influences of the angular quantum numberl and the molecular orientations in laser fields on the multiphoton processes arediscussed. The averaged absorb energy changing with the laser field’s frequency iscalculated. Q 1999 John Wiley & Sons, Inc. Int J Quant Chem 71: 201]207, 1999
Key words: Lie algebra; multiphoton; vibration-rotation; laser
Introduction
he phenomenon of infrared multiphoton exci-T tation and dissociation is one of the manyinteresting spectroscopic discoveries of the previ-ous decades. The interest of experimentalists andtheoreticians alike has been aroused in recent yearsby the development of high-powered lasers thatcan be used to induce molecular processes previ-
Correspondence to: Y. Dai.Project grant sponsors: National Science Foundation of
China, the State Key Laboratory of Theoretical and Computa-tional Chemistry of Jilin University, and the Natural ScienceFoundation of Shandong Province, China.
ously termed forbidden. To understand theoreti-cally how molecules absorb energy under suchconditions requires a nonperturbative analysis. Itis impossible to solve the time-dependent Schro-¨dinger equation accurately for most molecules ofexperimental interest, so the simplest realistic sys-tem to study is a diatomic molecule interactingwith a laser. There are some theories on the studyof the multiphoton processes of this system, like
w xCoulter transformation theory 1 , Floquet theoryw x w x2a, b , and Lie algebraic theory 3 . Most of themuse the anharmonic Morse oscillator model andsolve the Schrodinger equation as an initial value¨problem. However, it is usually difficult and com-
w xplicated. In Ref. 3 , in order to investigate themultiphoton excitation, the ‘‘simulated potential’’
( )International Journal of Quantum Chemistry, Vol. 71, 201]207 1999Q 1999 John Wiley & Sons, Inc. CCC 0020-7608 / 99 / 020201-07

DAI AND DING
Žusing a cubic order polynomial to simulated.Morse potential was used to present Hamiltonian
and obtained the time-evolution operator in theSchrodinger picture. However, only the lower ex-¨cited state transitions can be discussed because offewer adjustable parameters in the simulated poly-nomial.
In this work, we use Lie algebraic theory tostudy the diatomic molecular system interactionwith infrared laser fields. The Lie algebra modelused here is a quadratic anharmonic oscillatormodel that represents a nonrotating Morse oscilla-
w xtor 4 . We found a three-dimensional dynamicalLie algebra and studied the multiphoton processthrough solving the time-evolution operator in theinteraction picture. Because it is coinciding withthe Morse oscillator, the model can be used tostudy the molecule’s higher excited transitions inprinciple. We calculated the averaged absorb en-ergy as a function of time for the CO moleculedriven by continuous and shaped-wave laser fields,respectively. The time-averaged absorbed energy² Ž .:« t between the frequencies 0.9450v and01.0280v is compared. It is shown that the multi-0photon resonance appears and the oscillators maybe damping and the single photon resonance hasno signs of damping. We also studied the time-averaged energy as a function of the laser fre-quency v for the rotating LiH molecule. Becauselof the mathematical simplicity of handling aquadratic Hamiltonian, the numerical calculationbecomes relatively easy. Also using the Lie alge-braic method, the result is exact and the computa-tional time is saved relatively.
Theory
TIME-DEPENDENT HAMILTONIAN
The Lie algebraic Hamiltonian of quadratic an-harmonic oscillator for a free diatomic molecule isw x4 :
1ˆ ˆ ˆ ˆ Ž .H s "v A A q I , 1ž /1 0 q y 02
ˆ ˆwhere v is the frequency of the oscillator, A , A0 q yare creation and annihilation operators. The com-
ˆ ˆ ˆmutation relations of I , A , A are:0 q y
ˆ ˆ ˆ ˆ ˆ ˆ Ž .A , A s I , I , A s .2 x A , 2y q 0 0 " 0 "
where the anharmonicity parameter x s v r4D,0 0and D is the dissociation energy of the molecule.
ˆIn the harmonic limit x ª 0, I is an identity0 0operator.
The coupling Hamiltonian between the moleculew xand the laser field is 2 :
ˆ Ž . Ž .H s ym x E cos v tˆ2 0 l
Ž . Ž .s y m q m x E cos v tˆ0 1 0 l
Ž . Ž . Ž .s ym E cos v t y m xE cos v t , 3ˆ0 0 l 1 0 l
Ž .where the molecular dipole operator m x has beenexpanded through first-order terms in the dis-placement operator x s r y r , r is the equili-ˆ ˆ ˆ ˆe ebrant distance, E is the laser field strength that is0
Ž .1r2related to the intensity by E s 8p Irc , and v0 lis the laser frequency.
Ž .The linear approximation according to Eqs. 14Ž . w xand 17 of Ref. 4 is:
"v 10 ˆ ˆ Ž .x s A q A , 4ˆ ( ž /q y'2 D 2 a
where a is the range parameter of the Morse po-tential.
Within the semiclassical dipole approximation,the Hamiltonian operator for a diatomic molecularinteracting with plane-polarized monochromaticlaser radiation is:
ˆ ˆ ˆH s H q H1 2
"v0ˆ ˆ ˆ Ž .s "v A A q I y m E cos v t0 q y 0 0 0 l2d ˆ ˆ Ž .y A q A , 5ž /q y'2
where
1 "v0 Ž . Ž .d s m E cos v t . 6( 1 0 la 2 D
TRANSITION PROBABILITY
In the interaction picture, the wave function is:
< : Ž . < :N , y , t s U t , t N , y , tI 0 0 Ž .7Ž .U t , t s 1I 0
Ž .N is twice the number of bound states. U t, t isI 0the time-evolution operator; it satisfies the follow-
VOL. 71, NO. 2202

MULTIPHOTON EXCITATION OF DIATOMIC MOLECULES
ing equation:
Ž .dU t , tI 0 Ž . Ž . Ž .i" s V t U t , t . 8I I 0dt
The transition probability of the molecule fromstate y to state y is:n
Ž . <² < Ž . < : < Ž .P t s N , y U t , t N , y . 9n n I 0
The averaged energy is:
Ž . Ž . Ž .« t s P t « 10Ý n nn
Ž . Ž . Ž .« v s P v « . 11Ý n nn
TIME-EVOLUTION OPERATOR
According to the Lie algebra theory, the time-Ž . w xevolution operator U t, t can be written as 5 :I 0
Z ai iŽ . Ž .U t , t s e , 12ŁI 0i
where a is the element of dynamical Lie algebra,iand Z is the complex parameters.i
In order to find a dynamical Lie algebra a , weiŽ .divide Eq. 5 into two parts:
ˆ ˆ ˆ Ž .H s "v A A 130 0 q y
"v d0 ˆ ˆ ˆ Ž .V s I y A q A y m E cos v t .ž /0 q y 0 0 l'2 2Ž .14
Then
ˆ ˆi H tr " yi H tr "0 0Ž .V t s e VeI
"v d0 ˆi v t 2 x i v t I0 0 0 0ˆ ˆs I y e e A0 q'2 2
d ˆyi v t I0 0 ˆ Ž .y e A y m E cos v ty 0 0 l'2
"v d d0 ˆ Ž .s I y Ba y a y m E cos v tˆ ˆ0 q y 0 0 l' '2 2 23
Ž . Ž .s g a y m E cos v t , 15ˆÝ i i 0 0 lis1
where
"v0g s ,1 2
di v t 2 x0 0Ž . Ž .g s y B B s e , 162 '2
dg s y .3 '2
And a , i s 1, 2, 3 are three operatorsi
ˆ ˆi v t I yi v t I0 0 0 0ˆ ˆ ˆ Ž .I , a s e A , a s e A . 17ˆ ˆ0 q q y y
Ž .Since m E cos v t has no relation to the other0 0 lthree operators and it can be proved that it has noinfluence on the probability, the commutation rela-tions among the three time-dependent operatorsI , a , a are:ˆ ˆ0 q y
Uˆ ˆw x Ž .I , a s y2 x a , a , a s B I . 18ˆ ˆ ˆ ˆ0 " 0 " y q 0
ˆWe found the operators I , a , a are the elementsˆ ˆ0 q yof the three-dimensional dynamical Lie algebra inthe interaction picture.
In order to obtain the complex parameters Z ,iŽ . Ž .we substitute Eq. 12 into 8 . Then
"v d0 U˙ Ž .i"Z s y Z B exp y2 x Z1 2 0 1'2 2
y 2"v Z Z x BU0 2 3 0
w Ž .x1 y x y y 1 y0 Ž .= q 1 19½ 51 y 2 x y0
d˙ Ž . Ž .i"Z s y exp y2 x Z y "v 1 y 2 x y Z .3 0 1 0 0 3'2Ž .20
qŽ . y1Ž .Considering U t s U t , we obtain:I I
< < 23 Z3 Ž .Re Z s y 211 2< <6 q Z x3 0
Ž < < 2 . Ž . Ž < < 2 .6 3q Z x y i3 I Z x 6q Z x3 0 m 1 0 3 0 UZ s Z B.2 32 2Ž < < . Ž . Ž < < .y2 3q2 Z x q i3 I Z x 3q Z x3 0 m 1 0 3 0
Ž .22
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 203

DAI AND DING
Ž . Ž .Combining Eqs. 18 ] 22 , we obtain the couplednonlinear equation group about Z :i
"v0˙i"Z s1 2
< < 2d 2 x Z0 3Uy Z exp y i2 x Im Z3 0 121½ ' ž /< <2 2 q Z x3 03
Ž .1 y x y y 1 f02< <q2"v x Z y q 10 0 3 51 y 2 x y g0
Ž .23
2< <d 2 x Z0 3˙i"Z s y exp y 2 ix Im Z3 0 121' < <2 2 q Z x3 03
w Ž .x Ž .y "v Z 1 y 2 x y y 1 240 3 0
with initial condition
Z s 0, i s 1, 2, 3.i
< < 2x Z0 3 21 < <f s 1 y 1 q Z xŽ .3 0321ž /< <2 q Z x3 03
1 2< < Ž .y ix Im Z 1 y Z x , 25Ž .0 1 3 03
< < 2x Z0 3g s y q ix Im Z y 10 121ž /< <2 q Z x3 03
1 4 2< < Ž .= 1 y Z x . 26Ž .3 09
It is easy to solve the differential equation of theset group by numerical method.
Results and Discussion
All calculations are carried out using atomicunits. The parameters of CO and LiH are given by
w xRefs. 2b, 6 and summarized in Table I.The other parameters were calculated from pa-
rameters in this study.
MULTIPHOTON TRANSITION IN CO
Ž . w xAccording to Eq. 19 of Ref. 4 , the energyeigenvalues differences between the ground and
the excited states are « y « s 0.9983"v , « y1 0 0 2« s 1.9753"v , « y « s 5.7778"v , and « y0 0 6 0 0 11« s 10.2531"v . In Figure 1, we give the transi-0 0tion probabilities P as a function of the frequencyv
v for the continuous field strength E s 0.01 a.u.,0corresponding to an intensity of 3.5 TWrcm2. It is
Ž . 4plotted averaging over t s 1 ; 5 = 10 a.u. forevery D t s 100 a.u. The curves a, b, c, and drepresent the transitions from the ground to thefirst, second, sixth, and eleventh excited states,respectively. The maximum probabilities plottedat near v s 0.9600v , 1.0280v , v s 0.9582v ,01 0 0 02 01.002v , v s 0.9140v , 0.9450v , and v s0 06 0 0 110.8650v ; 0.8510v , respectively, correspond to0 0the one-, two-, six-, and twelve-photon resonance
Ž . w xfrequencies defined by n s « y « r"v 7 .n 0 nThis means that the higher the energy level is, thesmaller the resonance frequency is, and the smallerthe energy interval is. This is consistent with thecharacteristic of an anharmonic oscillator.
We also have given the averaged energy ab-² Ž .:sorption « t as a function of time at v sŽ .0.9450v the y s 0 to y s 6 six-photon resonance0
Žand v s 1.0280v the y s 0 to y s 1 single-pho-0
FIGURE 1. Multiphoton absorb peaks.
TABLE I
y1( ) ( ) ( ) ( ) ( ) ( )Molecule "v cm r a m D m D / a m m D eV01 e 0 0 1 0
7 1Li H 1373.3 3.0648 6.09 1.235 0.8812 2.428712 6Cl O 2169.8 1.2348 0.112 1.598 6.8562 11.09
VOL. 71, NO. 2204

MULTIPHOTON EXCITATION OF DIATOMIC MOLECULES
.ton resonance in Figure 2. The results show thatŽthe v s 0.9450v oscillator multiphoton reso-0
. 4nance vibrates with a period of about 2.8 = 10a.u. and the amplitude is damped, the vibrationalamplitude of the single-photon oscillation issmaller and no sign of damping out to 6.0 = 104
a.u.Figure 3 shows the averaged energy absorption
² Ž .:« v as a function of the frequency v for theŽ .continuous field E s 0.01 cos v t and shapedl
2Ž . Ž .field E s 0.01 sin p tr26000 cos v t a.u., re-0 lspectively. It appears to be a small power shiftingof the resonance peaks toward higher frequenciesin the latter case.
INFLUENCE OF ROTATION ONMULTIPHOTON PROCESSES IN LiH
For a rotating diatomic molecule interacting5with laser fields Z, the Hamiltonian operator is:
Ž . 2l l q 1 "ˆ ˆH s H q1 22mr
w Ž .x Ž . Ž .y m q m r y r E cos v t cos u , 27ˆ ˆ0 1 e 0 l
which is independent of the azimuthal angle w,where l is the angular moment quantum number,
u is the angle between the polarized vector andthe body-fixed molecular axis. With the initial con-
< :ditions N, y s 0 , the system has energy eigenval-ues:
10 RR Ž .Ž .« s « q « y a l l q 1 y qy l y l e 2
2w Ž .x Ž .y D l l q 1 , 28e
where the first two terms are the anharmonic oscil-lator and rigid rotor energies. The last two ener-gies are due to the inclusion of vibration-rotationinteraction and the centrifugal distortion, respec-tively.
According to the Taylor expansion:
1 1 2 1 2Ž . Ž .f y r y r s y x . 29ˆe2 2 3 2 3r r r r re e e e
Then:
ˆ ˆ ˆH s "v A A0 q y
Ž . 2"v l l q 1 " 1 20 ˆq I q y x0 2 3ž /2 2m r re e
Ž . Ž .y m q m x E cos v t cos uˆ0 1 0 l
( )FIGURE 2. a Time-dependent energy absorption for nonrotating CO with v = 0.9450v and E = 0.01 a.u.0 0( ) ( )six-photon resonance . b Time-dependent energy absorption for nonrotating CO with v = 1.0280v and E = 0.010 0
( )a.u. single-photon resonance .
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 205

DAI AND DING
( ) ( )FIGURE 3. a Time-averaged energy vs. frequency of the continuous field at E = 0.01 cos v t for the vibrational0 l( )description of CO. b Time-averaged energy vs. frequency of the continuous field at E = 0.010
( ) ( )sin p t /26000 cos v t for the vibrational description of CO.l
ˆ ˆ Ž .s "v A A y m E cos v t cos u0 q y 0 0 l
Ž . 2l l q 1 " "v0 ˆq q I02 22mre
2Ž .l l q 1 "Ž .y x E cos v t cos u qˆ 0 l 3mre
ˆ ˆ Ž .s "v A A y m E cos v t cos u0 q y 0 0 l
Ž . 2l l q 1 " "v d0 ˆ ˆ ˆq q I y A q Až /0 q y2 '22mr 2e
ˆ Ž .s H q V , 300
where
Ž . 2l l q 1 "ˆ ˆ ˆ Ž .H s "v A A q , 310 0 q y 22mre
"v d0 ˆ ˆ ˆV s I y A q Až /0 q y'2 2
Ž . Ž .y m E cos v t cos u , 320 0 l
2Ž .1 "v l l q 1 "0 Ž .d s m E cos v t cos u q .( 1 0 l 3a 2 D mre
Ž .33
Therefore the difference in calculation betweennonrotating vibration and rotating vibration is onlychanging the coefficient d.
The rotational constants of LiH molecule arew x2b :
a s 0.152 cmy1 ,e
y4 y1D s 7.633 = 10 cm ,e
B s 7.272 cmy1 .e
In order to study the influence of rotation onmultiphoton processes in LiH, we have calculatedthe time-averaged absorbed energy as a functionof the external field v with laser intensity ofl
y2 2 Ž . Ž .3.5 = 10 TWrcm . Figures 4 a and 4 b are theresults over the frequency ranges 0.700v F v F0
1.100v for nonrotating LiH and rotating LiH. In0Ž .Figure 4 a one can distinguish the multiphoton
transitions from y s 1 to y s 8 at frequencies v8
s 0.7020v , v s 0.7420v , v s 0.780v , v s0 7 0 6 0 5
0.820v , v s 0.8620v , v s 0.9060v , v s0 4 0 3 0 2Ž0.9520v , and v s 0.9920v . The energy eigen-0 1 0
value differences between the ground and the ex-cited states are, respectively: « y « s 6.8148"v ,8 0 0
« y « s 6.0926"v , « y « s 5.333"v , « y7 0 0 6 0 0 5
« s 4.537"v , « y « s 3.7037"v , « y « s0 0 4 0 0 3 0
VOL. 71, NO. 2206
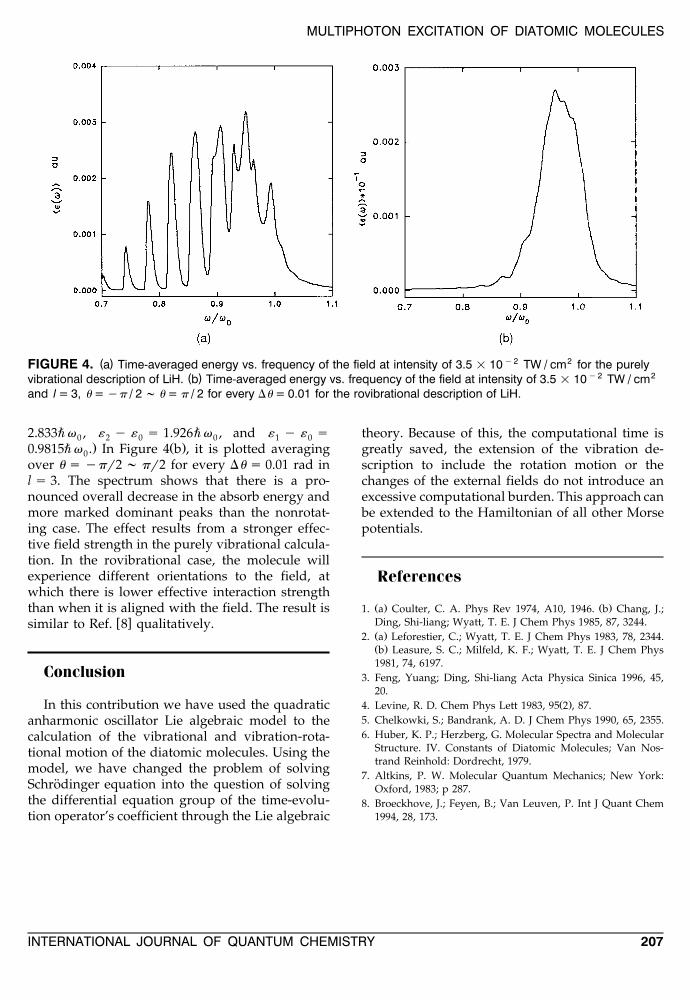
MULTIPHOTON EXCITATION OF DIATOMIC MOLECULES
( ) y2 2FIGURE 4. a Time-averaged energy vs. frequency of the field at intensity of 3.5 = 10 TW / cm for the purely( ) y2 2vibrational description of LiH. b Time-averaged energy vs. frequency of the field at intensity of 3.5 = 10 TW / cm
and l = 3, u = yp / 2 ; u = p / 2 for every Du = 0.01 for the rovibrational description of LiH.
2.833"v , « y « s 1.926"v , and « y « s0 2 0 0 1 0. Ž .0.9815"v . In Figure 4 b , it is plotted averaging0
over u s ypr2 ; pr2 for every Du s 0.01 rad inl s 3. The spectrum shows that there is a pro-nounced overall decrease in the absorb energy andmore marked dominant peaks than the nonrotat-ing case. The effect results from a stronger effec-tive field strength in the purely vibrational calcula-tion. In the rovibrational case, the molecule willexperience different orientations to the field, atwhich there is lower effective interaction strengththan when it is aligned with the field. The result is
w xsimilar to Ref. 8 qualitatively.
Conclusion
In this contribution we have used the quadraticanharmonic oscillator Lie algebraic model to thecalculation of the vibrational and vibration-rota-tional motion of the diatomic molecules. Using themodel, we have changed the problem of solvingSchrodinger equation into the question of solving¨the differential equation group of the time-evolu-tion operator’s coefficient through the Lie algebraic
theory. Because of this, the computational time isgreatly saved, the extension of the vibration de-scription to include the rotation motion or thechanges of the external fields do not introduce anexcessive computational burden. This approach canbe extended to the Hamiltonian of all other Morsepotentials.
References
Ž . Ž .1. a Coulter, C. A. Phys Rev 1974, A10, 1946. b Chang, J.;Ding, Shi-liang; Wyatt, T. E. J Chem Phys 1985, 87, 3244.Ž .2. a Leforestier, C.; Wyatt, T. E. J Chem Phys 1983, 78, 2344.Ž .b Leasure, S. C.; Milfeld, K. F.; Wyatt, T. E. J Chem Phys1981, 74, 6197.
3. Feng, Yuang; Ding, Shi-liang Acta Physica Sinica 1996, 45,20.
Ž .4. Levine, R. D. Chem Phys Lett 1983, 95 2 , 87.5. Chelkowki, S.; Bandrank, A. D. J Chem Phys 1990, 65, 2355.6. Huber, K. P.; Herzberg, G. Molecular Spectra and Molecular
Structure. IV. Constants of Diatomic Molecules; Van Nos-trand Reinhold: Dordrecht, 1979.
7. Altkins, P. W. Molecular Quantum Mechanics; New York:Oxford, 1983; p 287.
8. Broeckhove, J.; Feyen, B.; Van Leuven, P. Int J Quant Chem1994, 28, 173.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 207


















