
Late Transition Metal and Aluminium Complexes for the Polymerization of Ethene and Acrylates
59
Late Transition Metal and Aluminum Complexes for the Polymerization of Ethene and Acrylates Katariina Yliheikkilä Laboratory of Inorganic Chemistry Department of Chemistry Faculty of Science University of Helsinki Finland Academic Dissertation To be presented, with the permission of the Faculty of Science of the University of Helsinki, for public criticism in Auditorium A129 of the Department of Chemistry, A. I. Virtasen Aukio 1, on November 25 th 2006 at 12 o’clock noon. Helsinki 2006
Transcript of Late Transition Metal and Aluminium Complexes for the Polymerization of Ethene and Acrylates

Late Transition Metal and Aluminum Complexes for the Polymerization of Ethene and Acrylates
Katariina Yliheikkilä
Laboratory of Inorganic Chemistry
Department of Chemistry Faculty of Science
University of Helsinki Finland
Academic Dissertation
To be presented, with the permission of the Faculty of Science of the University of Helsinki, for public criticism in Auditorium A129 of the Department of Chemistry, A. I. Virtasen Aukio 1, on November 25th 2006 at 12 o’clock noon. Helsinki 2006

2
Supervisors Professor Markku Leskelä and Docent Timo Repo Laboratory of Inorganic Chemistry Department of Chemistry University of Helsinki Finland Reviewers Professor George J. P. Britovsek Department of Chemistry Imperial College of Science, Technology& Medicine London Professor Tuula Pakkanen Department of Chemistry University of Joensuu Finland Opponent Professor Jouni Pursiainen Laboratory of Physical Chemistry Department of Chemistry University of Oulu Finland ©Katariina Yliheikkilä 2006 ISBN 952-91-1335-2 (paperback) ISBN 952-10-3551-X (PDF) http://ethesis.helsinki.fi Helsinki University Printing House Helsinki 2006

3
Abstract Polyethene, polyacrylates and polymethyl acrylates are versatile materials that
find wide variety of applications in several areas. Therefore, polymerization of ethene,
acrylates and methacrylates has achieved a lot attention during past years. Numbers of
metal catalysts have been introduced in order to control the polymerization and to
produce tailored polymer structures. Herein an overview on the possible polymerization
pathways for ethene, acrylates and methacrylates is presented.
In this thesis iron(II) and cobalt(II) complexes bearing tri- and tetradentate
nitrogen ligands were synthesized and studied in the polymerization of tertbutyl acrylate
(tBA) and methyl methacrylate (MMA). Complexes are activated with
methylaluminoxane (MAO) before they form active combinations for polymerization
reactions. The effect of reaction conditions, i.e. monomer concentration, reaction time,
temperature, MAO to metal ratio, on activity and polymer properties were investigated.
The described polymerization system enables mild reaction conditions, the possibility to
tailor molar mass of the produced polymers and provides good control over the
polymerization. Moreover, the polymerization of MMA in the presence of iron(II)
complex with tetradentate nitrogen ligands under conditions of atom transfer radical
polymerization (ATRP) was studied.
Several manganese(II) complexes were studied in the ethene polymerization with
combinatorial methods and new active catalysts were found. These complexes were also
studied in acrylate and methacrylate polymerizations after MAO activation and converted
into the corresponding alkyl (methyl or benzyl) derivatives.
Combinatorial methods were introduced to discover aluminum alkyl complexes
for the polymerization of acrylates and methacrylates. Various combinations of
aluminum alkyls and ligands, including phosphines, salicylaldimines and nitrogen donor
ligands, were prepared in situ and utilized to initiate the polymerization of tBA.
Phosphine ligands were found to be the most active and the polymerization MMA was
studied with these active combinations. In addition, a plausible polymerization
mechanism for MMA based on ESI-MS, 1H and 13C NMR is proposed.

4
Preface The experimental part of this thesis was carried out between 2002 and 2006 in the
Laboratory of Inorganic Chemistry, University of Helsinki.
I am most grateful to my supervisors Professor Markku Leskelä and Docent Timo
Repo for their guidance and continuous support during the course of this work. I would
like also to acknowledge Professor Adnan Abu-Surrah, Hashemite University, Jordan, for
his advice.
The Laboratory of Inorganic Chemistry has proved to be an inspiring working
atmosphere and, especially, I want to thank my colleagues at the CatLab for their
supportive attitude. Special thanks to Dr. Kristian Lappalainen, Mikko Lankinen, Dr.
Kirill Axenov, and Minna Räisänen, it has been inspiring and rewarding working with
you!
Docent Martti Klinga and Dr. Mika Polamo are thanked for their help in the
determination of the crystal structures, Eeva Kaija for taking care of the DSC and GPC
measurements, and Sirpa Pirttiperä for her assistance in printing the thesis.
I want to thank my sisters’ in law and their families for friendship and enjoyable
holidays. Special thanks to Paula and Stu for their expertise in revising the language. My
warmest thanks to my parents Rauha and Kalevi, and my parents-in-law Kyllikki and Esa
for their encouragement and continuous support. I would like to express my deepest
gratitude to my husband Janne, and to our daughters Liina and Kerttu for their love,
patience and endless understanding. Without your support this would not have been
possible.
Financial support from the Academy of Finland (Centre of Excellence, Bio- and
Nanopolymer Research Group, 77317), the Finnish Technology Development Agency
(Tekes) and Gust. Komppa foundation is gratefully acknowledged.
Mäntsälä, September 2006
Katariina Yliheikkilä

5
List of Original Publications This thesis is based on the following original publications, which are referred to in the
text by Roman numerals I-V.
I Khalid Ibrahim, Katariina Yliheikkilä, Adnan Abu-Surrah, Barbro Löfgren, Kristian
Lappalainen, Markku Leskelä, Timo Repo, Jukka Seppälä: ‘Polymerization of
methyl methacrylate in the presence of iron(II) complex with tetradentate nitrogen
ligands under conditions of atom transfer radical polymerization’ Eur. Polym. J.
2004, 40, 1095-1104.
II Kristian Lappalainen, Katariina Yliheikkilä, Adnan Abu-Surrah, Mika Polamo,
Markku Leskelä, Timo Repo: ’Nitrogen Ligands Bearing Chiral Bulky Aliphatic
and Aromatic Substituents: Crystal Structure of [CoCl2{2,6-bis[R-(+)-
bornylimino)methyl]pyridine}]’ Z. Anorg. Allg. Chem. 2005, 631, 763-768.
III Katariina Yliheikkilä, Kristian Lappalainen, Pascal M. Castro, Khalid Ibrahim,
Adnan Abu-Surrah, Markku Leskelä, Timo Repo: ‘Polymerization of acrylate
monomers with MAO activated iron(II) and cobalt(II) complexes bearing tri- and
tetradentate nitrogen ligands’ Eur. Polym. J. 2006, 42, 92-100.
IV Katariina Yliheikkilä, Kirill V. Axenov, Minna T. Räisänen, Martti Klinga, Mikko
P. Lankinen, Mika Kettunen, Markku Leskelä, Timo Repo: ‘Manganese(II)
complexes in Ethene Polymerization ’ accepted to Organometallics.
V Katariina Yliheikkilä, Mikko P. Lankinen, Minna T. Räisänen, Antti Pärssinen,
Martti Klinga, Markku Leskelä, Timo Repo: ’Aluminum Alkyl - Phosphine
Catalysts for the Polymerization of Acrylates and Methacrylates’ submitted to
Organometallics.

6
Table of Contents Abstract ............................................................................................................................... 3 Preface................................................................................................................................. 4 List of Original Publications............................................................................................... 5 Abbreviations and Symbols ................................................................................................ 7 1 Introduction............................................................................................................. 8 2 Scope of the Thesis ................................................................................................. 8 3 Background............................................................................................................. 9
3.1 Polyethene and Polyacrylates and Their Applications............................................ 9 3.2 Polymerization of Ethene...................................................................................... 10
3.2.1 Homogeneous Late Transition Metal Complexes for the Polymerization of
Ethene ................................................................................................................... 10
3.2.2 Polymerization Mechanism ......................................................................... 12
3.3 Polymerization of Acrylates and Methacrylates .................................................. 14 3.3.1 Radical Polymerization................................................................................ 14
3.3.2 Anionic Polymerization ............................................................................... 17
3.3.3 Group Transfer Polymerization ................................................................... 19
3.3.4 Lanthanide and Early Transition Metal Complexes .................................... 21
3.3.5 Late Transition Metal Complexes................................................................ 22
3.4 Aluminum Complexes as Polymerization Catalysts............................................. 23 3.5 Combinatiorial Approach in the Development of Polymerization Catalyst ......... 25
4 Results and Discussion ......................................................................................... 26 4.1 Experimental in General ....................................................................................... 26 4.2 Synthesis and characterization of Manganese(II), Iron(II) and Cobalt(II) Complexes................................................................................................................... 27 4.3 MAO Activated Manganese(II) Complexes in the Polymerization of Ethene ..... 33 4.4 Manganese(II), Iron(II) and Cobalt(II) Complexes in the Polymerization of Acrylates ..................................................................................................................... 35
4.4.1 Tert-butyl Acrylate....................................................................................... 35
4.4.2 Other Acrylates and Methacrylates.............................................................. 41
4.5 Parallel screening of Aluminum Complexes ........................................................ 42 4.6.1 Tert-butyl Acrylate....................................................................................... 43
4.6.2 Methyl Methacrylate.................................................................................... 48
4.6.3 Proposed Mechanism for Polymerization of MMA..................................... 49
5 Conclusion ............................................................................................................ 52 References and Notes........................................................................................................ 53

7
Abbreviations and Symbols acac acetylacetonate AIBN azobisisobutyronitrile ATRP atom transfer radical polymerization Bz benzyl CCT catalytic chain transfer (polymerization) Cp cyclopentadienyl EI electron impact ionization EPR electron paramagnetic reconance ESI electrospray ionization EtOH ethanol Et2O diethyl ether GC gas chromatography GTP group transfer polymerization HDPE high density polyethene HR high resolution IR infrared LDPE low density polyethene m meso dyad contains two adjacent stereocenters of the same configuration MAO methylaluminoxane MA methylacrylate Me methyl MeOH methanol MMA methylmethacrylate MMAO modified methylaluminumoxane MS mass spectrometry MTS 1-methoxy-1-(trimethylsiloxy)-2-methylprop-1-ene MWD molecular weight distribution n-BuLi n-butyllithium NMR nuclear macnetic reconance PE polyethene Ph phenyl r racemic dyad contains two centers of opposing stereochemistries RAFT reversible addition-fragmentation chain transfer RT room temperature (S)-BINAP (S)- (-)-2,2'-Bis(diphenylphosphino)-1'1-binaphthyl SFRP stable free radical mediated polymerization tBA tertbutyl acrylate TEA triethylaluminum TEMPO 2,2,6,6-tetramethyl-1-piperidine N-oxyl (radical scavenger) Tg glass transition temperature THF tetrahydrofuran TIBA triisobutylaluminum Tm melting point temperature TMA trimethylaluminum UV-Vis UV-Visible spectroscopy

8
1 Introduction Polymers are important materials of today’s society in many areas and the
demand for polymers has grown enormously over the last sixty years. The success of
polymers is to be found in the unique combination of hardness, lightness, resistance of
corrosion, flame retardant properties, weather resistance, rigidity and toughness. Today
many polymers are produced by reactions which are catalyzed by organometallic
compounds.
Catalytic production of polyolefins started in the mid 1950’s when titanium based
trialkylaluminum activated catalyst were introduced by Karl Ziegler and Giulio Natta.1
The discovery of metallocenes with methylaluminoxane (MAO) as an activator in the
beginning of 1980 is a milestone on the research field of polymerization catalysts.2
Polymerization activity was increased drastically and uniform, tailored polymers were
produced. However, metallocenes based on early transition metals have one drawback
due to their sensitivity against polar functionalities. The search of more tolerant catalyst
system led to an introduction of late transition metal catalysts in 1995.3 Since then the
development in the late transition metal catalyzed polymerization of ethene and polar
monomers has continued and these recent developments are reviewed in Chapter 3.
2 Scope of the Thesis In the beginning of this work it was known that the homopolymerization of
olefins,4 methacrylates and acrylates5 could be carried out with aluminum alkyl activated
late transition metal complexes. In the polymerization of olefins it was observed that
besides the metal, the ligand also has a significant effect on the activity, and the structure
and properties of polymer material produced. This was a starting point for a study of
various nitrogen donor ligands with already known active metals in the polymerization of
acrylates.
Only a little was known about manganese complexes as polymerization catalyst6
though manganese complexes are widely used as catalysts in many other reactions.7 The
question was raised; could manganese complexes work in polymerization if an
appropriate ligand was found? Since there is a limited amount of literature on this topic,
combinatorial chemistry tools were employed to speed up the finding of possible
catalysts.

9
A disadvantage of the late transition metal complexes is that the complexes must
be activated with aluminum alkyls to form an active species. This introduced another
interesting question; could it be possible to discover aluminumalkyl complexes that
operate as such without activation in the polymerization of acrylates and methacrylates?
In addition, the purpose of this work was to develop new catalytic systems based
on Mn, Fe, Co and Al complexes in order to afford tailor-made polymers and finally to
achieve a better understanding of the polymerization mechanism of acrylates with
aluminum alkyl complexes bearing phosphine ligands.
3 Background
3.1 Polyethene and Polyacrylates and Their Applications Polyethene (PE) was discovered over 70 years ago and to date it is the most
widely used plastic. The commercial production of PE was over 23 Mtons in 2004, and
the annual growth from the previous year was 5.6%.8 A significant issue in the increased
consumption of polyethene products has been the continuing development in polyethene
properties and polymerization processes. Polyethene is versatile and it possesses
properties such as flexibility, strength, lightness, stability, impermeability and
processability. The list of PE products is extensive, and their applications include, for
example, food and house ware packing and films, gas and plumbing pipes, electrical
cable insulation, toys, car parts and petrol tanks, bullet proof vests, joint and bone
replacements, and waterproof coatings.9
Polyethene is classified based on its density, branching and molecular weight.
These factors strongly influence on the properties of polyethene. In today’s industrial
market low density (LD) PE is produced in high-temperature and high-pressure processes
through free radical polymerization, but in the production of high density (HD) PE the
control over polymerization, i.e. chain branching and length, is unattainable with this
method. Thus, HDPE is produced by chromium/silica catalysts, Ziegler-Natta catalysts
or metallocene catalysts.10 Besides tuning the structure of the product the use of a catalyst
enables mild reaction conditions, hence lowering the production costs.
Acrylate based polymers are utilized in a variety of applications that include
adhesives in coatings and paints,11 contact lenses,12 dental restorations and prostheses.13
Polymethylmethacrylate (PMMA) is used in applications where impact strength and high

10
transmittance is needed and it is employed as construction material and in biomedical
application, such as artificial intraocular lenses, dentures and bone cement in the fixation
of prosthetic joints.14 In cosmetic surgery, tiny PMMA microspheres suspended in
collagen solution are injected under the skin to reduce wrinkles or scars permanently.15
The PMMA market is predicted to grow in the future in applications requiring high
optical performance, such as LCD (liquid crystal display) plates. Commercial trade
names of PMMA products are for example commercial Plexiglas®, Perspex®,
Acrylite®, Acrylplast®, and Lucite®.16
The free radical polymerization process is widely used in industry to convert
acrylate monomers rapidly to solid polymer.17 In the initiation radical species attack the
monomer’s vinylic double bond. Radicals for the initiation can be formed by various
methods. Thermal initiators, such as peroxides and azo compounds, decompose to
radicals when heated. Other ways to produce radicals include redox initiators, light
decomposition of the initiator (photo polymerization) or initiation by ionizing radiation.17
The control over molar mass and molar weight distribution is poor in free radical
polymerization due to chain transfer and termination reactions.17 A number of methods to
gain control have emerged in polymerization of acrylates, including controlled radical,
anionic, group transfer and late transition metal mediated polymerization. These methods
are referred in Chapter 3.3.
Hence, the discovery of new catalysts is important in order to continuously
develop and produce tailor made polymer structures for various applications.18 With well-
defined catalytic systems it is possible to understand and control the effects of catalyst
structure on polymer structure and composition, and thus new polymer materials can be
obtained in mild reaction conditions.
The following literature review is focused on the polymerization of ethene and
acrylates and limited to literature relevant to the present study.
3.2 Polymerization of Ethene
3.2.1 Homogeneous Late Transition Metal Complexes for the Polymerization of Ethene In the mid nineties the invention of methylaluminoxane (MAO) activated
homogeneous α-diimine nickel(II) and palladium(II) complexes (Scheme 1) that

11
polymerized ethene or higher α-olefins with high activity initiated the intensive research
on the late transition metal complexes in the field of polymerization catalysis.3 By simple
ligand and reaction condition modifications various products were obtained from ethene
with these catalysts.19 Furthermore these catalysts were reported to catalyze
copolymerization of alkenes with alkyl acrylates.3(b),20 Some years later salicylaldiminato
nickel(II) catalysts for the ethene polymerization were reported.21 These catalyst
precursors are activated for example by tris(pentafluorophenyl)borane [B(C6F5)3] and
depending on the catalyst’s structure they are yielding moderately branched or linear
polyethene.21
In 1998, were discovered highly active 2,6-bis(imino)pyridine based iron(II) and
cobalt(II) catalysts (Scheme 1) that produced linear high molar weight polyethene after
activation with MAO.22 By tuning the ligand structure it is possible to produce
polyethene materials from linear α-olefins23 to high molar mass polymers,24 the
microstructure of which can range from linear24 to highly branched.3(a),25 Within this
catalyst family the cobalt complexes are reported to be less active than their iron
counterparts.24,25(b) and aldimine-derivated catalysts are less active as compared to
ketimine analogues.22(b),23(b),24 In addition, ortho substituents of imine aryl groups have
significant effect on the polymerization. The steric bulk of the ortho aryl substituents
retard chain transfer and experiments have confirmed that decreasing the size of these
ortho substituents is resulting in increased activity and decreased molar mass yielding
catalysts for ethene oligomerization.4(b),23,10 Moreover, bis(imino)pyridine iron(II) and
cobalt(II) complexes bearing single ortho substituent on the aryl rings form selective
catalysts for ethene oligomerization to α-olefins.4(b),23,10 With high MAO loadings
bimodal molar weight distributions are observed with these catalysts due to β-hydrogen
transfer and chain transfer to aluminium (Scheme 3).24
Since the early studies of 2,6-bis(imino)pyridine complexes numerous new
complexes and their use in ethene polymerization and oligomerization have been
reported.4(b),10 This type of iron(II) catalysts was also reported to polymerize various
vinylic polar monomers.26 The advantages of these catalysts over other type of catalysts
(e.g., metallocenes and early transition metal catalysts) are facile preparation and
handling, simple tuning of their polymerization activity by ligand architecture
modification and ability to tolerate polar functionalities.4(b),10 Besides the structure of the

12
catalyst, MAO concentration, ethene pressure and polymerization temperature affect the
activity and material propreties.
M
R2
N N
R2
R1 R1
(X)n
NR4
N N
R4
R3 R3M(X)n
A B Scheme 1. General structure of α-diimine (A) and 2,6-bis(imino)pyridine (B) complexes.
Despite extensive development in the field of polymerization catalysts, not highly
active polymerization catalysts with manganese metal centre has been described so
far.4(b),6 Only a few recent reports describe the ethylene and propylene polymerization
activity of complexes such as bis(imino)pyridine,27 bis(imino)carbazolide28 and
bis(cyclopentadienyl) manganese (Cp2Mn)6(a), tris(acetylacetonate) manganese
[Mn(acac)3],6(a) (salen)MnCl6(a) in combination with MAO, but no or only trace amounts
of polymer were isolated. In general, considerable lower activities are observed for
manganese complexes with ligands that provide very active catalysts with other late or
early transition metals. One report did introduce manganese catalyst with scorpionate
ligand that provided good activity for ethene polymerization when activated with
TIBA/[Ph3C][B(C6F5)4].29 In the study modified methylaluminoxane (MMAO) was also
employed as an activator resulting in lower activity. The produced polymers exhibit
moderate molar mass and unimodal narrow molar weight distribution. In addition, it was
proposed on the basis of results that coordinated anions (Cl-, Br- and NO3-) had influence
on polymerization reactions.
3.2.2 Polymerization Mechanism The conversion of the monomer into polymer depends on chain propagation and
termination rates, and therefore these rates determine molecular weight and molecular
weight distribution of the formed polymer. If the rate of propagation is slightly greater
than the rate of elimination, oligomeric products are formed. High molecular weight
polymers are produced when chain growth by insertion dominates the termination step. In

13
this chapter mechanistical aspects of complexes bearing 2,6-bis(imino)pyridine ligands
catalyzed ethene polymerization are discussed.
The active species of α-diimine nickel and palladium catalysts is known to be a
cationic alkyl metal complex,4(b),30 but the nature of the active species of
bis(imino)pyridine complexes is still under investigation and discussion. In the case of
iron complexes, the active species is proposed to be a neutral31 or cationic4(b),24,32 alkyl
complex. Neutral cationic species are formed when, trialkylaluminums, including MAO,
bind Fe2+ centers, yielding neutral catalytically active species. Ethylene coordination
mechanism apparently involves releasing of a ligating group from iron to accept the
incoming monomer.10 Cationic monoalkyl Fe2+ species are supposed to form via methyl
abstraction from dialkyl complexes, but it has been shown with Mössbauer and electron
paramagnetic resonance (EPR) studies that Fe2+ centers of precatalysts are oxidized by
excess of MAO to Fe3+ species.33 The reduction Fe3+ species back to Fe2+ species by
MAO is also proved by 1H NMR and EPR studies.31(b) In addition, it is proposed that
different active metal centers may be the reason to the broad molar weight distribution
obtained with these catalysts.
Most studies report that the activation of Co2+ precursors involves the reduction of
Co2+ species to Co+ and their further transformation into cationic methyl derivatives with
MAO-based counterion.4(b),10 This assumption is based on the color change that
bis(imino)pyridine cobalt2+ complexes go through after addition of MAO excess. This
color change is similar to the color of bis(imino)pyridine cobalt+ alkyl complexes. These
alkyl complexes are reported to polymerize ethene after addition of MAO as dichloro
complexes. However, various ways for initiation are possible for bis(imino)pyridine
cobalt complexes.34
Although the initiating species is unknown the propagation is proposed to proceed
via Cossee-Arlman mechanism by sequential monomer coordination and insertion via a
transition state as presented in Scheme 2. Propagation continues until chain transfer takes
place. These chain transfer reactions include β-H transfer to metal (a), β-H transfer to
monomer (b) and chain transfer to monomer (c) (Scheme 3).10

14
LMP
P
LM
LMP+ +
+
Scheme 2. Cossee-Arlman mechanism of chain propagation.
LMP
P
LMP
H
LMH
LM
P
R
AlR2
R2AlP
LMR
H
LM
P
LM
P
LM HP
P
LM
+
(a
(b)
(c
+
+
+
fast
slow
slow
fast
+
+
+ +
+ +
Scheme 3. Proposed chain transfer reactions in the polymerization of ethene with 2,6-
bis(imino)pyridine complexes of iron(II) and cobalt(II).10
3.3 Polymerization of Acrylates and Methacrylates
3.3.1 Radical Polymerization Acrylates are traditionally polymerized by free-radical methods.17 Radical species
for the initiation of polymerization can be formed for example by thermal decomposition
of such compound as benzoylperoxide or azobisisobutyronitrile (AIBN). Free radical
polymerization offers a little control over polymerization process and polymer properties.
To overcome the uncontrollability of the polymerization process, controlled radical
polymerization techniques have been developed. These include catalytic chain transfer
agent (CCT),35 stable free radical mediated polymerization (SFRP),36 reversible addition-
fragmentation chain transfer (RAFT)37 polymerization, and atom transfer radical

15
polymerization (ATRP).38 The main idea in these techniques is to control the amount of
free radicals in order to ensure the consistent propagation. In ATRP and CCT metal
complexes are used to gain the control while in RAFT thio-compounds and in SFRP
nitroxides are utilized.39 CCT is generally used to synthesize macromonomers and the
polymerization is initiated with free radical initiators or haloesters and the technique uses
cobalt tetraporfyrines or glyoximes to remove a hydrogen atom from a growing chain end
and transfer it to the monomer to start new chains as shown in Scheme 4.35
CH3P
R
CH2P
RH
H CH2
CH3
RCH3
CH3
R
. + [Co(II)] [Co(III)]+
[Co(III)] + . + [Co(II)]
P = polymer chainR = esther group
Scheme 4. Mechanism for CCT of methacrylates.35
ATRP was reported independently by Matyjaszewski and Sawamoto in
1995.38(a),(b) The ATRP system needs a transition metal complex and an initiator with a
transferable halogen. Many types of halogenated compounds are potential initiators but in
general initiation needs to be faster than propagation and side reactions should be
minimized. The amount of the initiator determines the final molar mass of the polymer at
the full monomer conversion. A number of transition metal complexes have been applied
in ATRP. Complexes of late and middle transition metals are most efficient ones and
within those the copper complexes are the most extensively employed for the metal
catalyzed or controlled radical polymerization.38(a),40 Copper complexes bearing pyridine
and amine based ligands form the majority of complexes used in ATPR,38(a),(b) but some
iron based systems with nitrogen and phosphine ligands have been also efficiently
employed in ATRP.41 They are fascinating due to the low price and the non toxic nature
of iron. Also ruthenium, palladium, rhodium, rhenium, molybdenum, chromium and
nickel systems have been utilized as ATRP catalysts.38(a),(b)

16
The polymerization mechanism for ATRP is based on transition metal catalyzed
reversible activation of carbon-halogen bond in a polymer chain by a transition metal
complex. Sawamoto has proposed a mechanism for ATRP and it is presented in Scheme
5.38(d) In well designed ATRP conditions chain growth is regulated and molar mass of a
polymer is increasing linearly as a function of time, which leads to narrow molar weight
distribution. Termination occurs via recombination of radical species or
disproportionation, but it is rather slow and in the ideal case polymerization ends when
all the monomer is consumed.
R X
CH2
R1
R2
R
R1
R2
H
H
CH2
R1
R2
R
R1
R2
X
H
H
R1
R2
PR
H
H
R1
R2
PR X
H
H
+ MnXnLm R. + Mn+1Xn+1Lm
. + Mn+1Xn+1LmMnXnLm
+
Mn+1Xn+1Lm+.
P = polymer chainR1 = H in acrylates and CH3 in methacrylatesR2 = esther grour
MnXnLm+
Scheme 5. Schematic presentation of ATRP by Sawamoto et al.38(b)
On the other hand, catalytic activity and selectivity is strongly ligand dependent in
ATRP.38(c) The ligand makes the transition metal salt soluble in the organic media and it
regulates the redox potential of the metal center for the appropriate reactivity and
dynamics for the atom transfer. The electronic and steric effects of the ligands are
important and the activity of N-based ligands in ATRP decreases with the number of
nitrogens coordinated to metal N4>N3>N2>>N1.38(b) Nitrogen ligands are most
commonly used and a wide variety of nitrogen ligands have been searched and utilized in
ATRP. The most common ones are a neutral chelating amine, imine or pyridine based

17
ligands.38(b) A study by Sawamoto et al. showed that amine additives afford faster and
more controlled polymerization than corresponding phosphine compounds.42
The advantage in ATRP is a well defined polymer structure where the molecular
weight distribution is very narrow and the molar mass of polymer can be controlled by
regulating reaction conditions. By using multifunctional initiators, the polymer
architecture can be modified and star, branched or grafted polymers can be synthesized.
Polymer chains prepared by ATRP have halogen chain ends and thus they can be used as
macroinitiators for block copolymers. Practically, ATRP can be carried out either in bulk,
in solution, or in a heterogeneous system. The optimal temperature depends on the
monomer, the metal complex, and the desired molar mass, but typically elevated
temperatures are used. Though ATRP gives excellent control over polymer chain
structure, the major drawback in ATRP is the large amount of catalyst required leading to
residuals in the polymer.
3.3.2 Anionic Polymerization During the last decade the classical anionic polymerization of acrylates and
methacrylates has been modified towards more precisely controlled polymerization. The
main purpose has been to limit the undesired side reactions (Scheme 7). The mechanism
of anionic polymerization of acrylates and methacrylates can be described as Michael
reaction (Scheme 6). The apparent advantages of anionic polymerization of methacrylates
are the stereoselectivity of the reaction and the opportunities to modify the polymer
structures. Polymerization of acrylates with the method is more demanding because of
their higher reactivity towards nucleophiles and the presence of acidic hydrogen at the α-
position to the carbonyl group.43
R
OO
R*
IO
OR*
R
OO
R*
R
OO
R*
R
IO
OR*
RI- - -
R = H in acrylates and CH3 in methacrylatesR* = alkyl groupI- = initiator
Scheme 6. Initiation and propagation in anionic polymerization of acrylates and
methacrylates.44

18
CH2
R
OOR*
CH2
R
OOI
R
OP
H
HOR*
CH2
R
OOR*
R
OP
H
HOR*
OCH2
R*
R
OP
H
HOR*
R
OOR*
PP
R
OP
H
HOR*
O
RPP
OO
P
R
RO
OR*
R
COOR*
R* OOR*
O
RCOOR*
R
R
P
I-M++ + CH3O-MR = H in acrylates and CH3 in methacrylatesR* = alkyl groupP = polymerchainM+ = counterionI- = initiator
- M+ + + CH3O-M
- M+ + + CH3O-M
-M+ CH3O-M+
A
B
C
D
Scheme 7. Side reactions in anionic polymerization of acrylates and methacrylates; A:
initiator destruction, B: monomer carbonyl attack, C: inter-molecular polymer
termination, and D: intra-molecular back biting termination.43 In all side reactions
produced methoxy anion (CH3O-) is too weak to initiate a new chain growth, thus
polymerization is slowed.
To avoid side reactions less nucleophilic initiator, polar solvent and low
temperatures are used in the polymerization process. Depending on the polarity of the
solvent and counter ion the stereostructure of the polymer can be tailored from isotactic
to syndiotactic in methacrylate polymerization. In polar solvent, propagating carbonium
ion and counterion can be described as free ions while in non polar solvent they form
tight ion pairs. For example, the polymerization of MMA in toluene with butyllithium at
–78 °C gives virtually 100% isotactic polymer and as the polarity is increased by addition

19
of polar additives, the counter ion is solvated and the degree of isotacticity is reduced
until in the absence of a nonpolar solvent quite high syndiotacticity is observed.45
Baskaran has stated that optimal condition for the anionic polymerization of
methacrylates is the use of bulky monofunctional initiator with bulky counterion in polar
solvent at low temperature.43
Besides classical anionic initiators, such as alkali metal alkyls (e.g. BuLi), a
variety of anionic polymerization initiators and methods have been studied to gain
controlled anionic polymerization. These methods use additives, mainly inorganic salts or
Lewis acids, to stabilize the propagating centers. Improvements have aimed to a better
control over polymerization as well as to the rising of the polymerization temperature to
the room temperature or above.43,46
The disadvantage of anionic polymerization is the cooling needed to suppress the
side reactions. In addition, anionic polymerization requires inert atmosphere, pure
reagents and clean glassware since all impurities lead to termination. These facts make
the method commercially uneconomic.
3.3.3 Group Transfer Polymerization Group transfer polymerization (GTP) was developed by Webster and Sogah at
DuPont, and it first appeared in the patent literature in the eighties.47 GTP is based on a
conjugated addition of silyl ketene acetals to α,β-unsaturated carbonyl compounds, such
as acrylates and methacrylates.48 The name originates from the proposal that the silyl
group transfers to new monomer during the monomer addition. However, Webster has
recently proposed that based on the existing evidence the original trimethylsilyl transfer
process is incorrect.46(a) Nevertheless, with GTP well defined polyacrylates and
poly(methyl methacrylates) can be prepared at room temperature and well above it.48(b)
Polymerization is accomplished most successfully in polar solvents.48(a) These are
advantages when compared to the living anionic polymerization that requires low
temperature and high purity reagents. Hence, GTP is currently used by DuPont to
produce dispersing agents for pigments and for emulsion stabilizers.46(a)
GTP uses 1-methoxy-1-(trimethylsiloxy)-2-methylprop-1-ene (MTS) as initiator
and nucleophilic anions or Lewis acids as catalyst. MTS is made by addition of

20
trimethylsilane to MMA. The most active nucleophilic catalysts are fluorides and
bifluorides while carboxylates and bicarboxylates operate better above ambient
temperature. Lewis acids, such as ZnCl2 and ZnBr2, are suitable catalysts, but they are
used within rather high concentrations.46(a) When nucleophilic anions are used as catalyst,
only a little amount of catalyst relative to the initiator is needed.
The function of a nucleophilic catalyst is to ease the displacement of the
trimethylsilyl group and the Lewis acidic catalyst increases the electrophilicity of the
double bond by coordinating to the carbonyl oxygen i.e. activate the monomers. The
degree of polymerization is governed by the ratio of monomer to initiator48(a) and it is
independent of the catalyst concentration which does affect the polymerization rate49.
The similarities between GTP and anionic polymerization are remarkable.50 Even
the characteristic back biting reaction (Scheme 7) has been found to be the main
termination reaction in GTP but the rates are much lower than for propagation. 51
No universal mechanism for GTP exists due to its dependence on the nature of the
catalyst and the exact reaction conditions but two types, associative and dissociative, can
be displayed (Scheme 8 and 9).46(a) Firm evidence for a dissociative anionic process at
room temperature and above was presented by Webster.46(a) On the other hand, the
mechanism of GTP differs with the type of catalyst used for the polymerization.43
OSiMe3
OMe
OSiMe3Nu
OMe
O
OMeMe3SiNu
MMA
O
OMe
PMMAMe3SiNuOSiMe3Nu
OMe
PMMA
OSiMe3
OMe
PMMA
+ Nu-
-
+
-
Nu-+
Scheme 8. The proposed mechanism for dissociative GTP pathway.46(a)

21
OSiMe3
OMe
OSiMe3Nu
OMe
O
OMe
MMA
OSiMe3Nu
PMMA OMeOSiMe3
OMe
PMMA
OSiMe3Nu
CO2Me
OMe
+ Nu- --
Nu-+ -
Scheme 9. The proposed associative mechanism.46(a)
In the dissociative mechanism (Scheme 8), nucleophilic catalyst (Nu-) complexes
with the silyl ketene acetal end groups and reversible forms an enolate that adds
monomer. Silyl ketene ends are reformed when enolate ends react with Me3SiNu. In the
associative pathway the nucleophilic catalyst is forming a complex with the silyl acetal
group. Subsequently the silyl group transfers to the incoming monomer and remains on
the polymer chain.46(a)
Here it is worth noting that a method similar to GTP based on methylaluminum
porphyrin for polymerization of acrylic monomers was developed by Inoue.52
3.3.4 Lanthanide and Early Transition Metal Complexes Yasuda and co-workers have reported organo rare earth metal complexes in the
polymerization of acrylates and methacrylates.53 They were able to isolate a
cyclopentadienyl samarium [Cp*2Sm(μ-H)]2 complex with two MMAs and determine the
crystal structure of adduct (Scheme 10 upper right corner) which supports a metal enolate
intermediate.53(e) In Scheme 10 the initiation mechanism based on the crystal structure is
presented.54 With this catalyst high molar mass polymer with narrow molar weight
distribution was obtained at -95 °C.

22
H CH2
OSm
O
CH2
O
Sm
O
OO
CH2
OSm
O
O
O
O
Sm
O
CH2
O
O
OO
MMA
MMA
Scheme 10. Polymerization of MMA according to Yasuda et al.54 Moreover, the polymerization of MMA with group 4 metals bearing Cp*2 ligands has
been reported.55 The polymerization was supposed to proceed via similar mechanism as
proposed to samarium complexes above.56
3.3.5 Late Transition Metal Complexes As in controlled radical polymerization (Chapter 3.3.1), late transition metal
complexes have been utilized in the polymerization of acrylates and methacrylates after
activation with MAO. There are reports describing complexes of nickel in the
polymerization of MMA.57 With nickel complexes the polymerization was suggested to
happen through monomer coordination to the metal center and subsequent insertion into
the nickel-carbon bond.57(b) The polymerization of methacrylates is also reported with
MAO activated iron,57(d), 58 cobalt,57(d) palladium59 and copper60 complexes. Palladium
diimine61 and β-diketiminato59(b) complexes are known to copolymerize ethene and
propene with acrylates. In general, these catalysts yield in branched polyethene in which
acrylate units are situated at the branch ends.
Furthermore, 2,6-bis(imino)pyridine iron(II)62 and cobalt(II)57(d) complexes and
phosphine complexes of iron26(b),62,63 and cobalt63 dichlorides were reported in the MAO
activated polymerization of tBA, MA and MMA. In the case of phosphine ligands, iron

23
catalysts were proven to be more active than their cobalt analogues and the activity, as
well as the molar mass of the produced polymer, was strongly ligand dependent.63
The polymerization mechanism for MAO activated bis(imino)pyridine iron(II)
complexes was studied by Castro.64 On the basis of ESI-MS and UV-Vis potential
initiation and polymerization mechanisms for the polymerization of tBA were stated. The
active species was proposed to be a cationic monomethyl complex and two potential
initiation pathways were suggested: iron-methyl bond homolysis and insertion of
monomer into the iron-methyl bond. The former case would lead into the radical
polymerization and the latter into the radical or coordination-insertion polymerization.
Based on end group analysis, i.e. the presence of ethyl saturated chain ends, termination
was supposed to occur via β-hydrogen transfer to the metal.
Nevertheless, distinguishing between radical, anionic, GTP and coordination-
insertion polymerization mechanism is difficult and no unequivocal mechanism for the
late transition metal mediated polymerization of acrylates and methacrylates can be
established.
3.4 Aluminum Complexes as Polymerization Catalysts There has been an interest in developing well-defined aluminum catalysts for
ethene polymerization,65 but there are only a few publications regarding definite
aluminum complexes as polymerization initiators for acrylates and methacrylates.
Aluminum alkyl catalysts would be a very interesting alternative to transition metal based
catalysts due to the low cost and simplicity of the ligands. Furthermore, these catalysts
would not require methylaluminoxane as cocatalyst. Hence, a catalyst based on
aluminum could be attractive and minor activity of aluminum catalyst compared to
transition metal catalysts can be approved. Ligand variation, a key to highly active
polymerization catalysts in transition metal chemistry, has emerged in the development of
aluminum catalysts for olefin polymerization. Ligands studied include those such as
bis(imino) pyridines,65(d) amidinates,65(a),(f) aminotroponiminates65(b) and β-
diiminates.65(c),(e) Although several groups have reported olefin polymerization with
aluminum alkyl complexes, the nature of active species remains non-specific. Moreover,
computational studies support only dimerization or oligomerization of olefins, but no
polymerization.65(h),66

24
Besides olefin polymerization, aluminium alkyl complexes could offer an
interesting alternative to the polymerization of polar monomers. Aluminum alkyls solely
are not capable conduct polymerization of acrylates and meth(acrylates)67 and they can
only be used in the purification of monomers for living anionic polymerization.68
Aluminoxanes have reported to polymerize MA63, MMA69 and tBAII though low molar
mass with broad molar weight distribution were observed indicating multi centre
behaviour.
Ikeda et al. described polymerization of acrylonitrile, MA and MMA with
aluminium alkyl complexes bearing mono- and bidentate Lewis base ligands such as
bipyridine and triphenylphosphine (PPh3).67(b)(c) They found that coordination of the
Lewis base to the central metal ion of the aluminum complex reduces the
electronegativity of the metal ion and increases the ionic character of the metal-carbon
bond which in turn induces the polymerization of polar vinyl compounds. In the nineties
Hatada et al. reported polymerization of MMA and other methacrylates with
triethylaluminum (TEA) and PPh3 or triethylphosphine (PEt3) in toluene at low
temperatures.70 The living polymerization yielded syndiotactic p(MMA) but bulky ester
group prevented syndiotactic propagation.
The polymerization of MMA with a combination of t-BuLi and aluminum alkyls
bearing bulky aryloxy substiuents (Scheme 11) at ambient temperatures and above was
reported by Ballard and co-workers.71 In their study it was found out that solvents, such
as tetrahydofuran (THF), inactivate the initiators by forming a stable complex with
aluminum alkyls.
O AlR
R = H, CH3
Scheme 11. Aluminum alkyls used with t-BuLi in the polymerization of MMA at room
temperature and above.71
The latest publications on organoaluminum complexes describe the
polymerization of tBA, MA and styrene. Okamoto et al. reported the polymerization of

25
tBA with TEA and PPh3, sparteine, (S)-BINAP, or n-BuLi at -78 °C but the study was
mainly concentrated on determining the relation between polymer tacticity and glass
transition temperature.67(d) In the study by Wu et al., bulky phenoxy aluminum
complexes, similar to aluminum alkyls described by Ballard,71 were active in initiating
the polymerization of MA and styrene at ambient and elevated temperatures.72 They
observed that electron withdrawing groups on the ligand enhanced the polymerization
activity. In the presence of catalytic amounts of radical scavengers, TEMPO and
Galvinoxyl, the polymerization of MA was inhibited, suggesting that radical species were
present in the polymerization reaction, but when the styrene was polymerized, radical
scavengers were not able to inhibit the reaction. The results made them conclude that
both radical and ionic species are involved in the polymerization depending on the
monomer used. On the other hand, it has been stated later that radical scavengers react
and deactivate metal based systems via coordination73 instead of inhibiting directly
radical reactions.74
3.5 Combinatiorial Approach in the Development of Polymerization Catalyst Combinatorial methods were originally developed for biochemical applications75
and drug discovery,76 but the methods can be applied to other fields of research as well.77
In the development of new polymerization catalyst, the traditional synthetic methods are
laborious and time-consuming tasks. High-throughput synthesis and screening
approaches to catalyst discovery and optimization and it is thoroughly changing the
techniques in which catalyst research is conducted.78 Increased rates of innovation, cost
effectiveness, reduced time and an improved probability of success are some of the
attractive features that are gained with combinatorial methods. The ability to assess large
number of ligands with sufficient steric and electronic diversity makes the method
fascinating for a rapid evaluation of various catalyst candidates of one specific metal for
polymerization reactions. After the best candidates are found, they can be fully
characterized, desired reaction conditions optimized and catalytic reactions further
studied.
Combinatorial chemistry includes both parallel synthesis and split and pool
methods. Parallel synthesis reactions are carried out in separate reaction vessels and each

26
product is analyzed with an appropriate method. In split and pool methods, various
reactants are mixed together in one reactor. If one of these set turns out to be active, then
subsequently, the combinations are screened individually and studied further. In practice
where a number of reactions are carried out at the same time can be classified under the
concept of combinatorial methods. The analysis methods and automation are in the most
important position when the possible numbers of simultaneous reactions are determined.
Manual methods reduce the amount of reactions to dozens while sophisticated automatic
methods enable hundreds of reactions and analysis in a short time.78(f)
The oxidation state and electronic configuration of the metal and the ligand
system in the catalytically active species are the key issues in the catalytic performance.
The catalytic properties of a certain metal complex generally are highly dependent on the
ligand framework around the metal center and by varying the ligand substituents, the
electronic character, geometry, and steric protection of the catalytic center can be
modified. There is no doubt that mechanistic understanding is valuable in the contouring
of a reaction course, but since the screening of a large number of potential catalysts
typically leads to a discovery of an active catalyst efficiently, the combinatorial method
has its benefits. Especially when a new type of catalysts is searched or new
polymerization reactions are studied combinatorial methods offer excellent tools by
speeding up the development process.
4 Results and Discussion
4.1 Experimental in General All solvents were dried with appropriate drying agents. Other reagents used in the
syntheses were purchased from commercial sources with high purity grade, and used as
such. Monomers in publication V were distilled over calcium hydride and stored under
argon in a freezer. All syntheses and polymerization experiments were performed under
argon atmosphere in a glove box or using standard Schlenk techniques. Detailed
descriptions of the solvent purification, ligand and complex syntheses, analyses, and
polymerization procedures and polymerization activities can be found in the experimental
parts of the attached original publications.

27
4.2 Synthesis and characterization of Manganese(II), Iron(II) and Cobalt(II) Complexes The condensation of amines with aldehydes or ketones in the presence of formic
acid catalyst is an easy way to synthesize imines (Scheme 12). The reaction can be
followed by IR spectroscopy since the strong aldehyde or ketone peak disappears and
new peaks appear due to the imine bond.
NR
O
R
O
MeOHHCOOH
R'-NH2 NR
N
R
NR' R'
+ 2
Scheme 12. Synthesis of 2,6-bis(imino)pyridine ligands.III
Tertiary imine ligands were synthesized from diamine and two equivivalents of
quinoline-2-carboxaldehyde in ethanol. Tertiary amine ligands were prepared from
secondary diamines and 2-bromomethylquinoline (Scheme 13). Isolated ligands were
identified by 1H NMR, 13C NMR, IR, mass spectroscopy, and elemental analysis.
NH2
NH2 NH
O
EtOH NN
NN
N
N
R
R
H
H NBr
N
N
R
R
N
N
EtOHK2CO3
+ 2
+ 2
R R
a: R =
b: R =
c: R =
d:R =
e: R =
Scheme 13. Preparation of tertiary amine and imine ligands.I,II,III

28
It should be mentioned that in the synthesis of N,N’-di(quinoline-2-methylene)-
1,2-phenylenediimine formation of two different products is possible as was observed in
this work.79,III As can be seen from the crystal structure (Figure 1); that instead of
diimine, a five member ring can be formed. Specific reason for that is unclear but proton
transfer is a competitive mechanism to the formation of imine leading to the formation of
the ring structure. The analyses of the ligand isomers revealed that 13C NMR spectra and
determination of the crystal structure are necessary since there is only a minor distinction
in 1H NMR spectra of these two compounds. In 1H NMR spectra the signals of two
protons of the five member ring species locate at δ = 6.30 (-N2-CH2-) and in the case of
imine ligand at δ = 8.80 (-N=CH-). While in 13C NMR 13 signals appeared for diimine
ligand and 17 for its ring species. Other analyses, i.e. MS and elemental analysis, yielded
similar results for these two forms. When this five member ring structure was allowed to
react with iron dichloride, a complex presented in Figure 1, was obtained.81 Though no
polymerization was carried out with this complex, it is interesting that iron is coordinated
with only to two nitrogen atoms of the ligand.
Figure 1. ORTEP drawing of the ligand with an unexpected structure and structure of
corresponding iron dichloro complex.80,81
All complexes prepared here (Schemes 14 and 15) are relatively stable and
straightforward to synthesize when compared to the very air and moisture sensitive early

29
transition metal complexes. The straightforward preparation of the complexes from the
corresponding ligand and metal salt in THF, CH2Cl2 or EtOH, was carried out at room
temperature. The complexes were characterized by IR, mass spectroscopy and elemental
analysis. In the IR spectra, imine complexes of iron and cobalt show a shift of υ(C=N)
stretching by ca. 30-60 cm-1 as compared to the free ligand. This reflects the coordination
of the imine nitrogen atoms to the metal. In the IR spectra manganese and tertiarty amine
complexes show only slight shifting. This indicates a weaker bond between the metal and
ligand and it is also seen in the elongated bond lengths of manganese complexes.IV It is
worth mentioning that manganese complexes (Scheme 14) active in polymerization were
found with combinatorial methods and this will be discussed later (Chapter 4.3). In the
procedure catalytic amounts of complexes were prepared in separate small glass reactors.
The ligand and metal salt were allowed to react together in THF and consecutively the
reaction solvent was replaced by toluene and used in the polymerization after activation
with MAO. The active manganese catalysts (Scheme14) were synthesized in a large scale
as described above.
N
N
N
N
MCl
Cl
N
N
N
N
MCl
Cl
N
N
N
N
MCl
Cl
N
N
R
R N
N
MCl
Cl
MnN
N
N
N
Cl
Cl
3: M = Fe4: M = Co5: M = Mn
1: M = Fe 2: M= Co
7: M = Fe, R = phenyl 8: M = Fe, R = benzyl9: M = Co, R = benzyl
6: M = Mn10
Scheme 14. Complexes bearing tertiary nitrogen ligands in this work.I,III,IV

30
MCl Cl
NR
N N
R
R' R'
11: M = Fe, R = CH3, R' =
12: M = Fe, R = H, R' =
13: M = Fe, R = CH3, R' =
14: M = Fe, R = CH3, R' =
15: M = Co, R = CH3, R' =
16: M = Fe, R = CH3, R' =
17: M = Fe, R = H, R' =
18: M = Co, R = H, R' =
19: M = Fe, R = CH3, R' =
20: M = Fe, R = H, R' =
21: M = Co, R = H, R' =
22: M = Fe, R = H, R' =
23: M = Co, R = H, R' =
Scheme 15. Complexes bearing tridentate nitrogen ligands. II,III,81
The single crystal X-ray structure of the cobalt complex 23 (Figure 2.) and
manganese complexes 5, 6 (Figure 3) were determined. Suitable crystals were obtained
by slow evaporation of the solvent. The coordination of Co(II) can be described roughly
as a distorted trigonal bipyramide in 23, while distorted octahedral coordination is
observed in Mn complexes.
In complex 23, imino nitrogen atoms occupy the axial positions and chlorines and
a nitrogen atom at pyridine form the trigonal plane. The imino nitrogens are shifted to the
opposite sides of the plane formed by the pyridine and cobalt, while bornyl-sybstituents
are positioning above and below the N3Co plane. Bornyl-substituents were unable to
displace CoCl2 from the N3Co plane to the square pyramidal arrangement as happened
with earlier reported cobalt(II) complex bearing 2,6-diacetylpyridinebis(2,6-
diisopropylanil) ligand.24

31
Figure 2. Solid state structure of cobalt complex 23.II H-atoms, the second cobalt
complex and CHCl3 solvent molecules were omitted for clarity.
In complexes 5 and 6 the chiral bridging twists the N4-Mn basal plane and thus
causes a substantial rhombic distortion in octahedral geometry of Mn(II). Twisting bends
chlorine atoms from the apical position towards the cis-configuration. In complex 5, for
the two independent molecules at the unit cell, the Cl-Mn-Cl angles were 132° and 135°
and the distorted N4-Mn basal planes were practically perpendicular to the plane of
MnCl2. In 6 the pyridine nitrogen atoms are shifted away from the N4-Mn basal plane and
planes defined by N1-Mn-N2 and N3-Mn-N4 are bisecting each others with an angle of
49.0(2)º. The N2-Mn-N3 plane is perpendicular to the plane of MnCl2.
The NPy-Mn bonding with the bis(imino)pyridine ligand is remarkably shorter
(2.20 Å)82 than quinoline N-Mn bonding in 5 (2.49-2.51 Å) and 6 (2.32 -2.34 Å). This
noticeable difference in the coordination strength of the pyridyl lone pairs, together with
the elongated Mn-Cl bonds, might have significance when it comes to catalytic properties
of these Mn-complexes. Similar elongation of the N-Mn bond lengths, as in 5 and 6, can
be found, although to a lesser extent, from the previously published solid state structures
of [di(pyridine-2-methylene)diiminocyclohexane]MnCl283 and bis(bipyridine)MnCl2.84
Furthermore, the elongated NPy-Mn bonds lead to reduced electron donation from the
ligand and increase Lewis acidity of Mn. This might explain the observed ethene
polymerization activity of 5 and 6 after MAO activation.

32
Figure 3. Crystal structures of manganese(II) complexes 5 and 6.IV H-atoms and CH2Cl2
solvent molecules were omitted for clarity as well as the second manganese complex in
asymmetric unit of 5.
In order to activate transition metal complexes for olefin polymerization, they are
often converted to corresponding alkyl derivatives by the treatment with aluminum
alkyls, most commonly in situ with MAO. Such alkyl complexes are considered as
prerequisite for the formation of metal alkyl cations, generally considered as catalytic
active centers for olefin polymerization. Thus transition metal alkyl complexes,85
including manganese alkyl complexes, 86 are of significant interest.
Hence, complexes 5 and 6 were allowed to react with benzyl magnesium chloride
(BzMgCl) in diethyl ether (Et2O). Both complexes gave only monosubstituted alkyl
complexes (Scheme 16). Later attempts to introduce a second benzyl group by treatment
with excess of BzMgCl were unsuccessful and only monosubstituted species were
isolated. Both of the monobenzyl substituted complexes were adequately stable allowing
the characterization by EI-MS, HR ESI-MS and IR.IV
The methylation of complexes 5 and 6 with methyl magnesium bromide
(MeMgBr) yielded extremely air and moisture sensitive products, which were identified
by HR ESI-MS methods as alkyl derivatives. Due to air sensitivity and rapid
decomposition IR and EI-MS gave inaccurate results in analyses.IV

33
N
N
N
N
MnCl
Cl
N
N
N
N
MnCl
Cl
MgBr N
N
N
N
MnCl
MgBr N
N
N
N
MnCl
MgBr
MgBr
LMnMe2
LMnMe2
5
6
Scheme 16. Preparation of manganese alkyl complexes.IV Reaction conditions: -78 °C in
Et2O in an argon glovebox.
4.3 MAO Activated Manganese(II) Complexes in the Polymerization of Ethene Due to the rarity of manganese complexes in the ethene polymerization, the
combinatorial approach was chosen as a method to study manganese complexes in MAO
activated ethene polymerization. Since MAO and MMAO are the most active and used
co-catalysts in late transition metal mediated ethene polymerization,10 MAO was
employed.
In separate reactors in situ prepared complexes were activated with MAO.
Subsequently, the activated complexes were sealed in a 1 L steel reactor and the reactor
was charged with ethene pressure. In the primary screening three active manganese
catalysts (5, 6 and 10 in Scheme 14) were found and these complexes were then
synthesized as described in the previous chapter.
When the ethene polymerizations were carried out separately in large scale, a
rather sensitive activation of the complexes was observed. If the complexes were first
treated at room temperature with MAO and then heated to the desired polymerization
temperature, reproducible activity levels were achieved. By contrast, catalysts exhibited

34
no or very poor ethene polymerization activities when MAO treatment was done at
elevated temperatures.
The highest activity [67 kgPE/(molcat x h)] among these manganese catalysts was
reached with the MAO activated complex 5 at 80 °C and under 5 bar of ethene pressure.
Polymers produced at 60 °C exhibited very high molar masses (1200-1700 kg/mol) with
relative narrow molar weight distribution, though slight tailing towards the low molar
mass region was observed. The rise in the polymerization temperature from 60 °C to 80
°C led to PE with clear bimodal molar weight distribution and a decrease in molar mass.
Despite of variations in polymerization conditions, activity of the biphenyl-bridged 6 and
the unbridged complex 10 remained low. Both of them produced high molar mass
polyethene with bimodal molar weight distribution.
The bimodality of the polymer strongly suggests the presence of two different,
structurally defined catalytic active centers. Probably, high polymerization temperature
causes intramolecular fluctuation of the coordination sphere of the activated manganese
complexes. The existence of structural isomerization of similar manganese complexes
has been verified by crystal structure determination,87 and the three possible
conformational isomers for tetradentate ligands in an octahedral coordination sphere have
been illustrated.88 In addition, similar fluctuation of the complex geometry during the
activation has been reported for Fe(II) derivatives of 6 by Brintzinger et al.89
Alkyl complexes presented in Scheme 16 were also studied in ethene
polymerization. Complexes were activated with TIBA/B(C6F5)3, but the activated alkyl
complexes were extremely unstable and their transfer into the polymerization reactor
without a minor air contact was very complicated. In general, MAO activated dichloro
complexes were more active than corresponding TIBA/B(C6F5)3 activated alkyl
complexes. The instability of the activated alkyl complexes might induce the poor
polymerization behavior.
To conclude, with combinatorial methods new active manganese catalysts for
ethene polymerization were found and three essential requirements for the active
manganese(II) catalyst precursors can be summarized: 1) they are six-coordinated, 17-
electron species 2) there is the presence of ligands with imine pyridine/quinoline moieties
separated by two carbon atoms and 3) distortion of octahedral configuration of the
manganese by the ligand framework.IV

35
4.4 Manganese(II), Iron(II) and Cobalt(II) Complexes in the Polymerization of Acrylates
4.4.1 Tert-butyl Acrylate It became apparent from this study that there are several factors affecting the
polymerization as well as the properties of the polymers. Firstly, the ligand’s structure
and the metal are the significant factors determining the polymerization behavior.
However, the reaction conditions, e.g. monomer concentration, temperature and reaction
time, have major influences on polymerization activity and material properties within
these complexes.
As can be seen in Figure 4, the metal has an essential effect on the monomer
conversion and molar mass. Iron complexes are more active than their cobalt analogues
and produce also polymers with remarkable higher molar mass. Similar results have been
observed in ethene polymerization with complexes of 2,6-bis(imino)pyridine
ligands.24,25(b) It is reasonable to assume that the different configuration of d-electrons or
activation processes90 of cobalt complexes are accountable for the observed phenomenon.
Manganese complex 5 is the least active (Figure 5) within the complexes bearing the
same ligand structure, but it produces relative high molar mass (Figure 6).
020406080
100120140160180
20 21 22 23Complex
Mw (k
g m
ol-1
)
FeCo
0
10
20
30
40
50
60
70
80
20 21 22 23
Complex
Con
vers
ion
(%)
FeCo
Figure 4. Comparison between iron and cobalt complexes bearing tridentate nitrogen
ligands in the polymerization of tBA.
The influence of the ligand’s structure on polymerization is diverse. However,
this versatility brings possibilities that affect the polymerization and produced materials.
Within the complexes bearing tridentate nitrogen ligands, aldimine-derivated ones are

36
more active than their ketimine analogues (Figure 5 complex 12 vs. 11 and 20 vs. 19).
This is contrary for reported to ethene polymerization activity with catalysts of this
type.22(b),23(b),24 No conclusion can be drawn regarding which structure is better on the
basis of molar masses, because aldimine complex 12 produces higher molar mass than its
ketimine analogue 11 while aldimine complex 20 produces lower molar mass than the
corresponding ketimine complex 19.
Substituents on the imino nitrogen atoms have only a minor effect on the
polymerization activity within ketimine complexes of iron, namely complexes 11, 13, 16
and 19 (Figure 5). However, complexes bearing aliphatic substituents, i.e. 13 and 19,
produced polymers with a higher molar mass than complexes 11 and 19 with aromatic
groups (Figure 6). A similar effect is not observed within aldimine complexes 12, 20 and
22. Within these three complexes, 12 with aromatic substituents on the imino nitrogen, is
the most active and produces polymer with the highest molar mass.
0
10
20
30
40
50
60
70
80
90
100
1 2 3 4 5 7 8 9 11 12 13 16 19 20 22
Complex
Con
vers
ion
(%)
Fe
Co
Mn
Figure 5. Selected examples of monomer conversion in the polymerization of tBA.III,81
Polymerization conditions: [tBA] = 3.0 M, ncat = 2.0 μmol, MAO:M = 250, RT,
tpolymerization = 24 h, solvent toluene.

37
0
50
100
150
200
250
300
350
400
450
1 2 3 4 5 7 8 9 11 12 13 16 19 20 22
Complex
Mn
(kg
mol
-1)
Fe
Co
Mn
Figure 6. Selected molar masses of p(tBA). Polymerization conditions as above in Figure
5.
For a more detailed study on the effect of the ligand and metal on tBA
polymerization activity, four complexes bearing tri- and tetradentate nitrogen ligands,
namely 3, 4, 14 and 15, were chosen. In addition, MAO only and MAO activated iron(II)
chloride was included in the study. Gas chromatography (GC) methods were utilized in
determination of monomer conversion. Samples from the polymerization vessel were
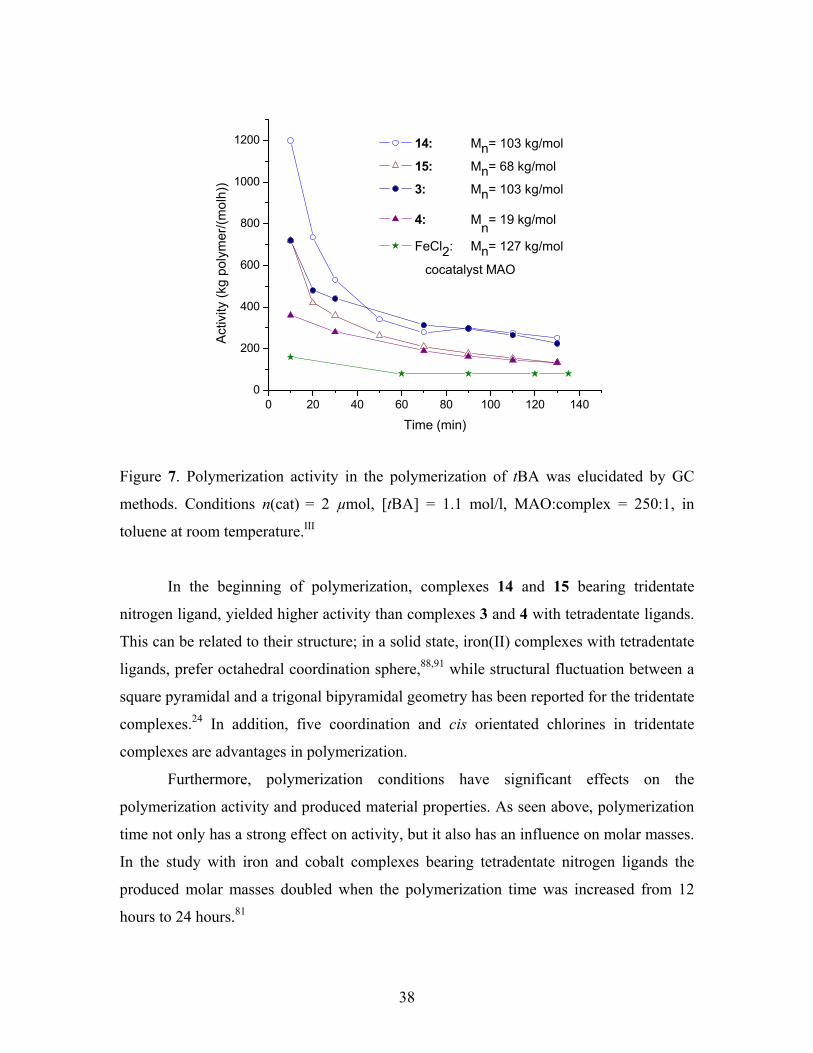
taken sequentially during the first two hours of the polymerization. Figure 7 shows that
polymerization proceeds very fast in the beginning but after a certain polymerization
time, the differences in initial activities e.g. within iron complexes are equalized.
38
0 20 40 60 80 100 120 1400
200
400
600
800
1000
1200 14: Mn= 103 kg/mol
15: Mn= 68 kg/mol
3: Mn= 103 kg/mol
4: Mn= 19 kg/mol
FeCl2: Mn= 127 kg/mol
cocatalyst MAO
Activ
ity (k
g po
lym
er/(m
olh)
)
Time (min)
Figure 7. Polymerization activity in the polymerization of tBA was elucidated by GC
methods. Conditions n(cat) = 2 µmol, [tBA] = 1.1 mol/l, MAO:complex = 250:1, in
toluene at room temperature.III
In the beginning of polymerization, complexes 14 and 15 bearing tridentate
nitrogen ligand, yielded higher activity than complexes 3 and 4 with tetradentate ligands.
This can be related to their structure; in a solid state, iron(II) complexes with tetradentate
ligands, prefer octahedral coordination sphere,88,91 while structural fluctuation between a
square pyramidal and a trigonal bipyramidal geometry has been reported for the tridentate
complexes.24 In addition, five coordination and cis orientated chlorines in tridentate
complexes are advantages in polymerization.
Furthermore, polymerization conditions have significant effects on the
polymerization activity and produced material properties. As seen above, polymerization
time not only has a strong effect on activity, but it also has an influence on molar masses.
In the study with iron and cobalt complexes bearing tetradentate nitrogen ligands the
produced molar masses doubled when the polymerization time was increased from 12
hours to 24 hours.81

39
Increase of monomer concentration increased the conversion, polymerization
activity and molar mass of the produced polymers as can be seen in Figures 8 and 9. The
linear enhancement in molar masses with the increasing monomer concentration occurred
with all studied complexes. With the highest monomer concentration 6.4 M (bulk) the
highest molar masses obtained were 1900 kg/mol while with low monomer concentration
(0.6 M) molar masses as low as 5 kg/mol were obtained.III,81
Figure 8. Selected conversions of tBA polymerization in different monomer
concentrations. Polymerization conditions: ncat = 2.0 μmol, MAO:M = 250, RT,
tpolymerization = 24 h, solvent toluene.III,81
30
40
50
60
70
80
90
100
110
0 1 2 3 4 5 6 6.5 [tBA]
3 4 11 12
Con
vers
ion
(%)

40
Figure 9. Selected molar masses of tBA-polymers in different monomer concentrations.
Polymerization conditions: ncat = 2.0 μmol, MAO:M = 250, RT, tpolymerization = 24 h,
solvent toluene.III,81
The influence of temperature has been studied with 2,6-bis(imino)pyridine
iron(II) complexes26(b) in tBA polymerization and phosphine complexes of iron(II) and
cobalt(II)63 in MA polymerization. The results on polymerization activity with iron(II)
and cobalt(II) complexes bearing tetradentate nitrogen ligands concurred with those
results and activity was increased as polymerization temperature increased.III However, a
more interesting issue is the relationship between temperature and molar mass. As the
temperature was increased from 20 °C to 70 °C, the molar mass was decreased in the
series of 2,6-bis(imino)pyridine iron(II) complexes,26(b) while the molar mass was
increased up to 40 °C within the complex group bearing tertiary nitrogen ligands.III Until
some point between 40 °C and 60 °C molar masses started to decrease. Similar behavior
was observed for phosphine complexes of iron(II) and cobalt(II).63 The possibility of
ligand dissociation from the complex was overruled by the polymerization experiments
on MAO activated metal salts.63 Thus, the decrease in molar mass originates from an
increased chain termination rate. In addition, variation in polymerization temperature had
no significant influence on the polymer microstructure when the polymerizations were
0
200
400
600
800
1000
1200
1400
1600
1800
0 1 2 3 4 5 [tBA]
3 4 11 12
Mn(
kg m
ol-1
)

41
carried out above 0 °C,III,63 but lowering the temperature to -78 °C increased the [rr]
triads.III
In earlier polymerization studies of MMA with NiCp*2 and Ni(acac)2 complexes
it was observed that large amounts of MAO are not required.92 It was found that the
optimal Ni:MAO ratio was 1:100. On the other hand, the influence of the Fe:MAO ratio
has been studied in detail with iron(II) phosphine complexes.63 There, maximum activity
was reached at a ratio of 1:125 and 1:250 after which the activity lowered and remained
at constant level. The molar mass decreased sharply until a constant level at the ratio of
1:500 is settled. With iron(II) and cobalt(II) complexes bearing a tetradentate nitrogen
ligand, similar dependence on MAO amount was observed.81
It can be concluded that in the polymerization of tBA the activity and molar
masses of polymers are influenced by the ligand structure and the choice of metal centre
and with different polymerization conditions various polymer materials were produced.
However, based on the results no mechanism for transition metal assisted
polymerizations can be conducted.
4.4.2 Other Acrylates and Methacrylates The polymerizations of methyl acrylate and methyl methacrylate were also carried
out with iron(II), cobalt(II) and manganese(II) complexes bearing tetradentate nitrogen
ligands.I,III,IV Polymerization of MMA was carried out under ATRP conditions with in
situ prepared iron dichloro complexes from ligands a-d (Scheme 13).I The
polymerizations were activated with ethyl-2-bromoisobutyrate. It was observed that the
steric effect of ligand determine the solubility of the catalyst as well as the selectivity.
The polymerization activity of the in situ formed iron dichloro complexes was in the
order of: a > c > b, while iron dichloro complex of ligand d was inactive.
The polymerization of MMA was also accomplished with MAO activated
complexes 1, 2 and 3 (dichloro complex of ligand d).III However, conversions and molar
masses remained low. Furthermore, these complexes were employed in the
polymerization of MA resulting in slightly better conversion and moderately higher
molar masses than in the polymerization of MMA. With both monomers, iron complex 3

42
gave the highest molar masses and cobalt complex 2 the lowest ones. A similar effect is
seen in the polymerization of tBA.
MAO activated manganese complex 5 was more extensively employed in the
polymerization of various acrylates and methacrylates (Table 1).93 It was observed that
acrylates were polymerized easier than methacrylates. The molar mass of the produced
polyacrylates is found to increase as the size of the ester group increased. With
methacrylates, rather low conversions were obtained though poly(ethyl methacrylate)
exhibited a substantially high molar mass. Furthermore, the molar mass of poly(methyl
methacrylates) produced with 5 (36 kg/mol) is greater than molar mass obtained with the
corresponding iron complex 3 (4.3 kg/mol). In the polymerization of tBA it was also
noticed that manganese complex 5 was able to produce a rather high molar mass though
conversion remained low (Chapter 4.4.1).
Table 1. Polymerization of acrylates and methacrylates with MAO activated 5.
Polymerization conditions: solvent toluene, RT, c(monomer) = 3.0 M, n (cat) = 2.0 µmol,
[Al]:[Mn] = 250, polymerization time 15 hours.93
Monomer Mnx10-3 Mw/Mn Activity Tg
c(ºC) Methyl acrylate 79 2.47 20.4 21
Ethyl acrylate 133 1.64 20.8 -15
Butyl acrylate 206 1.60 36.0 -
Tert-butyl acrylate 316 1.83 14.8 48
Methyl methacrylate 36 1.78 3.3 91
Ethyl methacrylate 225 1.55 8.8 -
4.5 Parallel screening of Aluminum Complexes Combinatorial screening was chosen as a method to find new aluminum based
catalysts since very scarce studies have been carried out with aluminum alkyl complexes
in the polymerization of acrylates and methacrylates. The set up for the screenings was
very simple and aluminum complexes for the polymerization were formed in situ by
stirring a ligand with aluminum alkyl in toluene. Consecutively, a sufficient amount of

43
monomer was added to the reaction vessels through septa. Polymerization reaction
conditions for the primary screening (temperature, monomer to initiator ratio, solvent)
were chosen on the basis of our earlier studies with acrylates.III,26(b),62,63,81
4.6 Aluminum Complexes in the Polymerization of Acrylates and
Methacrylates
4.6.1 Tert-butyl Acrylate Aluminum alkyl complexes form an interesting group of compounds for the
polymerization of acrylates and methacrylates assuming that no co-catalyst are required
with them. For the primary screening, 19 ligands (Scheme 17), including phosphines,
salicylaldimines and nitrogen ligands, were selected. The ligands for the primary
screening were selected on the basis that their transition metal complexes were already
known to be active in polymerization reactions.III,4(b),10,22,26(b),62,63 ,94

44
Scheme 17. Ligands employed in the primary screening of aluminum complexes.V
In the primary screening 57 aluminum alkyl complexes were employed in the
polymerization of tBA. In Figure 10 it can be seen that with a couple of exceptions all
complexes were able to initiate the polymerization of tBA to some extent. However, the
major finding from the primary screening was that all phosphine ligands with triethyl
aluminum (TEA) or triisopropyl aluminum (TIBA) as aluminum alkyl led to the
quantitative polymerization of tBA. Additionally these polymerizations afforded high
molar masses (from 50 to 150 kg/mol) with relatively narrow molar weight distributions
(MWD ~2). Hence, various phosphines were selected for the further studies.
NOH
F
FF
FF
NOHNOHNOH
FNOH
NOH
OMeOMe
OMe
NN N NH
F
F F
F
N N
N NH
iPr iPr
iPr
OH
N
OH
N OH
N
OH
N
OH
N
OH
N
N
N
N
NN
N NP P P
NMe2 PPh2
N N
1 2 3 4
6
5
9 107
1311
16
1514
17
iPr
18
12
8
19

45
Figure 10. Monomer converision in the primary screening of aluminum complexes.V The
reaction conditions used were room temperature, [tBA] = 1.1 M, n(complex) = 150 μmol,
monomer to initiator ratio (M:I) = 47, solvent toluene. The ligands that correspond to the
ligand numbers are shown in Scheme 17.
Scheme 18. Phosphine ligands used in the second screening.V
P P P P
PP 3
P 3
P 3
P 3
P 3
P 3
PCl
Cl Cl
P 3
PPh
Ph
P
F F
F
FF3
11
2 3
7 8 109
4 5 6
13 14 12
1
15
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 TIBA
TEA
TMA
0
20
40
60
80
100
Con
vers
ion
%
Ligand
TIBATEATMA

46
In Scheme 18, the phosphine ligands for the second screening are presented. The
selection criteria were different bulkiness and donor-acceptor properties as well as
commercial availability and affordability. Furthermore, the steric and electronic
properties of phosphines are easily modified,95 and typically a direct relation between
catalyst performance and the properties of the phosphine ligand could be established.96
Result from the secondary screening showed that, besides the phosphine ligand,
aluminum alkyl has a remarkable effect on the polymerization activity. In general,
trimethyl aluminum (TMA) yielded lower conversions than the other aluminum alkyls
(Figure 11). However, ligands 4, 10, 11, 12 and 13 (in Scheme 18) with TMA were able
to produce a high molar mass poly(tBA).
Figure 11. Monomer conversions in the secondary screening.V Polymerization conditions
as in the primary screening. The ligands that correspond to the ligand numbers on the
horizontal axis are shown in Scheme 18. Polymerization conditions as in Figure 10.
In general, phosphines with phenyl moieties (e.g. 11 and 12) formed the most
active combinations and produced the highest molar masses. While phosphines with alkyl
substituents (e.g. 2) or electron withdrawing groups (e.g. 1 and 14) yielded low
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
TMA
TEA
TIBA0
20
40
60
80
100
Con
vers
ion
%
TMATEATIBA

47
polymerization conversions and relatively low molar masses. Increasing the steric bulk in
the phenyl ring (e.g. 13 and 15) had a similar effect.
The effect of solvent, reaction temperature and monomer concentration were
studied with triphenyl phosphine (PPh3) (8) and aluminum alkyls.V Polymerization was
carried out in tertahydrofuran (THF), acetonitrile (AcN), ethyl acetate (EtOAc) and
toluene (the solvent in the primary screening). When compared to the polymerization in
toluene THF was the only solvent where the conversion was diminished. However, the
molar masses of the produced polymers in THF, AcN and EtOAc were lower than those
made in toluene with TEA and TIBA as aluminum alkyls. When TMA was used, high
conversions were reached in EtOAc and AcN. In EtOAc, a substantially high molar mass
was observed. Thus, the polymerization can be carried out in various solvents.
Here, the similar enhancement in molar masses of produced polymers with
increasing monomer concentration was observed as in the polymerization of tBA with
iron and cobalt complexes.III When the reaction temperature is elevated, molar masses are
decreased and molar weight distribution broadened, probably due to the increased chain
transfer reactions. Decreased polymerization activity at high temperatures indicates that
the aluminum species are thermally labile. At temperature under ambient molar mass
increases and molar weight distribution approaches value 1, which is indicating living
polymerization.
Figure 12 shows the comparison of produced molar masses of p(tBA) between
iron(II) and cobalt(II) complexes bearing tri- and tetradentate nitrogen ligand and
aluminum alkyl complexes. It should be mentioned that catalyst loading was greater in
the polymerizations with aluminum alkyl complexes. Due to the different catalyst
amounts, a general conclusion on behalf of either complex group can not be drawn.
Nevertheless, molar masses produced with these complexes diverge and phosphine
ligands 11 and 12 with TMA and TEA, ligand 10 with TMA, and 8 with TEA are
producing higher molar masses than the other complexes.

48
0
50
100
150
200
250
comple
x 3
comple
x 4
comple
x 14
comple
x 15
8/TMA
8/TEA
8/TIB
A
10/TMA
10/TEA
10/TIBA
11/TMA
11/TEA
11/TIBA
12/TMA
12/TEA
12/TIBA
Mn (
kgm
ol-1
)
Figure 12. Comparison between molar masses of poly(tBA) produced with selected iron
and cobalt complexes bearing tri- and tetradentate nitrogen ligandsIII and aluminum alkyl
complexes bearing phosphine ligandsV.
4.6.2 Methyl Methacrylate Subsequently, the most active phosphine ligands, i.e. 5-12, from tBA
polymerization and TIBA as aluminum alkyl, were employed in the polymerization of
MMA to gain differences between these complexes. With ligand 10 little conversions
with low molar masses were observed while conversions 60 % and over were yielded
with other combinations. Increasing monomer concentration clearly enhanced the
conversion with TIBA complexes of ligands 5 and 7, but the effect can not be seen with
other studied combinations because of the almost complete conversions obtained. Figure
13 shows that the molar masses of produced pMMAs are growing as the monomer
concentration is increased. The highest molar masses are produced with PPh3 (8), tri(m-
tolyl) phosphine (11) and tri(p-tolyl) phosphine (12). These all yielded conversions over
80 % with rather narrow molar weight distribution (under 2).

49
5 6 7 8 9 10 11 12
100
200
4000
50
100
150
200
250
300
Mn [
kg m
ol-1
]
Ligand
M:I
Figure 13. Molar masses of poly(MMA) with PPh3TIBA.V The reaction conditions: RT,
n(complex) = 150 μmol, monomer to initiator ratio (M:I) 100, 200 and 400, in toluene.
The ligands that correspond to the ligand numbers are shown in Scheme 18.
4.6.3 Proposed Mechanism for Polymerization of MMA First, to ensure the complex formation in the screening PPh3 and TEA or TIBA
were allowed to react and subsequently the formed complexes were studied in detail.
The complex of PPh3 and TEA crystallized as colorless crystals. In the complex (Figure
14) PPh3 is bound to TEA and the phenyl rings of the ligand are in a propeller shape
conformation and aluminum adopts a distorted tetrahedral geometry. Furthermore, both
complexes were identified by 1H NMR, 13C NMR, and ESI-MS.97

50
Figure 14. An ORTEP drawing of (PPh3)TEA.V Thermal ellipsoids were drawn on a 30
% probability. H-atoms and C6b atom in the disordered ethyl group (C5-C6) were
omitted for clarity.
For the mechanism studies, PPh3TIBA complex was employed. In ESI-MS, MS-
MS and NMR studies, it was confirmed that the coordination of MMA is taking place via
a carbonyl oxygen to PPh3TIBA complex (Scheme 19 left). In addition, enolization of
MMA was observed (Scheme 19).V The resonance structures of MMA·AlR3 adducts and
NMR shifts for phenolate complexes have been studied earlier,98 therefore an analogous
monomer activation is taking place. The coordination via carbonyl was also demonstrated
with methyl isobutyrate (MeiB). The shifts in NMR signals were analogous to those of
MMA except for the double bond that is absent in the MeiB, thus enolization is
impossible with MeiB. A possible pathway for the propagation is via 1,4-conjugated
(Michael) addition (Scheme 20) as proposed for GTP, but the precise mode of
propagation remains open. No evidence for an associative or dissociative pathway nor a
monometallic or bimetallic system can be provided. Furthermore, the ability for part of
the aluminum complex to act as monomer activator or propagator can not be excluded.
The role of phosphine ligands in the polymerization is to accept and donate
electron density during the polymerization process and facilitate the propagation step.
The phosphine ligand remains coordinated to aluminum throughout the process as no
changes in 13C-NMR signals of the phenyl group in PPh3 were observed during the

51
polymerization. Further, phosphine end groups were absent in NMR of polymers, though
PPh3 residues were observed.
P
AlR
R
R
P
AlR
R
RO
O
O
O
O
O
P
Al
R
R
R
R = isobutyl, C4H9 Scheme 19. The addition of first monomer (MMA) to the PPh3Al(C4H9)3 complex.
O
O
P
AlRR
R
O
OOO
O
OR
P
AlRR
OO
O
Opolymer chain
P
AlRR
R = isobutyl, C4H9
MMAMMA
Scheme 20. Representation of the proposed polymerization mechanism via a series of
conjugated additions.

52
5 Conclusion In this thesis, versatile complexes of manganese, iron, cobalt and aluminum were
synthesized and employed in the polymerization of acrylates and methacrylates. High
polymerization activity was gained and varying polymer properties were produced
depending on the complex structure and reaction conditions. The choice of catalyst and
the tuning of the polymerization conditions, enabled control of the polymer structure
from a low to high molar mass product with narrow molar weight distributions.
All preparations of ligands and complexes were straightforward to carry out. After
activation with MAO iron, cobalt and manganese complexes were efficient in the
polymerization of tBA and iron complexes with tridentate 2,6-bis(imino)pyridine ligands
turned out to be the most active ones. Moreover, the polymerization of other acrylates
and methacrylates were demonstrated with these MAO activated complexes.
Furthermore, this work confirmed the efficiency of combinatorial methods in
finding new active complexes for the polymerization reactions. Parallel screening
allowed a wide variety of ligand and metal combinations to be studied in a short time and
resulted in the discovery of new polymerization catalysts for ethene, acrylates and
methacrylates. Particularly, a series of aluminum phosphine complexes which were able
to polymerize tBA and MMA to total conversion are interesting new catalysts.
The polymerization mechanism of MMA is suggested to begin with the first
monomer coordination via a carbonyl group to aluminum alkyl complex, followed by
enolization of the monomer. Propagation is proposed to take place via 1,4-conjugated
addition as presented for GTP (Chapter 3.3.3). However, the intimate propagation mode,
i.e. monometallic vs. bimetallic and associative vs. dissociative, remains open. As the
reaction mechanism of the transition metal assisted polymerization of acrylates and
methacrylates is ambiguous, each stated mechanism interpretation will hopefully carry
the research further.

53
References and Notes [1] Martin, H. In Ziegler Catalysts, Eds., Fink, G., Mülhaupt, R., Brintzinger, H. H., Springer Verlag, Berlin, 1995, p.15. [2](a) Andresen, A., Cordes, H.-G., Herwig, J., Kaminsky, W., Merck, A., Mottweiler, R., Pein, J., Sinn, H., Vollmer, H.-J., Angew. Chem. 1976, 88, 689-690. (b) Sinn, H., Kaminsky, W., Vollmer, H.-J., Woldt, R., Angew. Chem. Int. Ed. 1980, 19, 390-392. [3] (a) Johnson, L. K., Killian, C. M., Brookhart, M., J. Am. Chem. Soc. 1995, 117, 6414-6415. (b) Johnson, L. K., Mecking, S., Brookhart, M., J. Am. Chem. Soc. 1996, 118, 267-268. [4] (a) Gibson, V. C., Spitzmesser, S. K., Chem. Rev. 2003,103, 283-315. (b) Ittel, S. D., Johnson, L. K., Brookhart, M., Chem. Rev. 2000, 100, 1169-1203. [5] See references 51-58. [6] (a) Ban, H. T., Kase, T., Murata, M., J. Pol. Sci. Part A: Polym. Chem. 2001, 29, 3733-3738. (b) Nabika, M., Seki, Y., Miyatake, T., Ishikawa, Y., Okamoto, K., Fujisawa, K., Organometallics 2004, 23, 4335-4337. [7] (a) Murphy, M., Dubois, G., Stack T. D. P., J. Am. Chem. Soc. 2003, 125, 5250-5251. (b) McGarrigle, E. M., Gilheany, D. G., Chem. Rev. 2005, 105, 1564-1602. (c) Xia, Q.-H., Ge, H.-Q., Ye, C.-P., Liu, Z.-M., Su, K.-X., Chem. Rev. 2005, 105, 1603-1662. (c) Katsuki, T., Coord. Chem. Rev. 1995, 140, 189-214. (d) Schoumacker, S., Hamelin, O., Pécaut, J., Fontecave, M., Inorg. Chem. 2003, 42, 8110-8116. (e) Lee, N. H.; Lee, C. S.; Jung, D. S. Tetrahedron Lett. 1998, 39, 1385-1388. (f) Moreira, R. F., When, P. M., Sames, D., Angew. Chem. Int. Ed. 2000, 39, 1618-1621. (g) Canali, L., Sherrington, D. Chem. Soc. Rev. 1999, 28, 85–93. (h) Vierle, M., Zhang, Y., Herdweck, E., Bohnenpoll, M., Nuyken, O., Kühn, F., Angew. Chem. Int. Ed. 2003, 42, 1307-1310. [8] Anon., Commodity Polymers World Trade Review of 2004, International Trader Publications, Inc., April 2005, Private communication with Kari Teppola, Muoviteollisuus ry, 18.5.2006. [9]http://www.exxonmobilpe.com/public_products/polyethylene/polyethylene/Worldwide/PE_ProductFrontPage.asp and http://plastics.dow.com/plastics/ap/prod/polyethylene/ (referred 16.9.2006) [10] Bianchini, C., Giambastiani, G., Rios, I. G., Mantovani, G., Meli, A., Segarra, A. M., Coordin. Chem. Rev. 2006, 250, 1391-1418. [11] (a) Bernhard, P., Hofmann, M., Schulthess, A., Steinmann, B., Chimia 1994, 48, 427-430. (b) Decker, C., Nucl. Instrum. Methods Phys. Res. B 1999, 151, 22-28.(c) Decker, C., Elzaouk, B., J. Appl. Polym. Sci. 1997, 65, 833-844. (d) http://www.dow.com/acrylates/ (referred 16.9.2006) [12](a) Kunzler, J. F., McGee, J. A., Chem. Ind. (London) 1995, 16, 651-655. (b) Martin, S. J., O’Brien, J. E., Dowling, J., McBrierty, V. J., Eur. Polym. J. 1998, 34, 1817-1828. [13](a) Elliott, J. E., Lovell, L. G., Bowman, C. N., Dent. Mater. 2001, 17, 221-229. (b) Lovell, L. G., Berchtold, K. A., Elliot, J. E., Lu, H., Bowman,C. N., Polym. Adv. Technol. 2001, 12, 335-345. (c) Lovell, L. G., Stansbury, J. W., Syrpes, D. C., Bowman, C.N., Macromolecules 1999, 32, 3913-3921. [14] Konttinen, Y., Santavirta, S., Lääkelaitoksen julkaisusarja 6/2003, ISBN 952-5099-59-8, National Agency for Medicines, Helsinki, Finland. [15]Lemperle, G., Romano, J. J., Busso, M., Dermatol. Surg. 2003, 29, 573–587.

54
[16] http://www.cyro.com/ and http://www.perspex.co.uk/ (referred 16.9.2006) [17] Odian, G., Principles of Polymerization, 3rd ed., John Wiley & Sons, Inc., New York, USA, 1991, 198-259. [18] Arriola, D. J., Carnahan, E. M., Hustad, P. D., Kuhlman, R. L., Wenzel, T. T., Science 2006, 312, 714-719. [19](a) Killian, C. M., Johnson, L. K., Brookhart, M., Organometallics 1997, 16, 2005-2007. (b) Svejda, S. A., Brookhart, M., Organometallics 1999, 18, 65-74. [20] Mecking, s., Johnson, L. K., Wang, L., Brookhart, M., J. Am. Chem. Soc. 1998, 120, 888-899. [21] (a) Johnson, L. K., Bennet, A. M. A., Ittel, S. D., Wang, L., Parthasarathy, A., Hauptman, E., Simpson, Feldman, J., Coughlin, E. B.,1998, (DuPont) WO Patent 98/30609. (b) Wang, C., Friedrich, S., Younkin, T. R., Li, R. H., Grubbs, R. H., Bansleben, D. A., Day, M., W., Organometallics 1998, 17, 3149-3151. (c) Younkin, T. R., Connor, E. F., Henderson, J. I., Friedrich, S., Grubbs, R. H., Bansleben, D. A., Science 2000, 287, 460-462. [22]( a) Small, B. M., Brookhart, M., Bennett, A. M. A., J. Am. Chem. Soc. 1998, 120, 4049-4050. (b) Britovsek, G. J. P., Gibson, V. C., Kimberley, B. S., Maddox, P. J., McTavish, S. J., Solan, G. A., White, A. J. P., Williams, D. J., Chem. Commun.1998, 849-850. [23] (a) Small, B. L., Brookhart, M., J. Am. Chem. Soc. 1998, 120, 7143-7144. (b) Britovsek, G. J. P., Mastroianni, S., Solan, G. A., Baugh, S. P. D., Redshae, C., Gibson, V. C., White, A. J. P., Williams, D. J., Elsegoog, M. R. J., Chem. Eur. J. 2000, 6, 2221-2231. [24]Britovsek, G. J. P., Bruce, M., Gibson, V., C., Kimberley, B. S., Maddox, P. J., Mastroianni, S., McTavish, S. J., Redshaw, C., Solan, G. A., Strömberg, S., White, A. J. P., Williams, D. J., J. Am. Chem. Soc. 1999, 121, 8728-8740. [25](a)Gates, D., Svejda, S., Onate, E., Killian, C., Johnson, L., Brookhart, M., Macromolecules 2000, 33, 2320-2334. (b) Abu-Surrah, A. S., Lappalainen, K., Piiroinen, U., Lehmus, P., Repo, T., Leskelä, M., J. Organomet. Chem. 2002, 648, 55-61. (c) Leatherman, M., Svejda, S., Johnson, L., Brookhart, M., J. Am. Chem. Soc. 2003, 125, 3068-3081. [26] (a) Britovsek, G. J. P., Gibson, V. C., Spitzmesser, S. K., Tellmann, K. P., White, A. J. P., Williams, D. J. J., J. Chem. Soc. Dalton Trans. 2002, 6, 1159-1171. (b) Castro, P. M., Lappalainen, K., Ahlgren, M., Leskelä, M., Repo, T., J. Pol. Sci. Part A: Polym. Chem. 2003, 41, 1380-1389. [27](a) Gibson, B., McTavish, Redshaw, C., Solan, G., White, A., Wiliams, D., Dalton Trans. 2003, 221-226. (b) Reardon, D., Aharonian, G., Gambarotta, S., Yap, G. P. A., Organometallics 2002, 21, 786-788. [28]Gibson, V., Spitzmesser, S., White, A., Williams, D., Dalton Trans. 2003, 2718-2727. [29]Nabika, M., Seki, Y., Miyatake, T., Ishikawa, Y., Okamoto, K., Fujisawa, K., Organometallics 2004, 23, 4335-4337. [30](a) Svejda, S. A., Johnson, L. K., Brookhart, M., J. Am. Chem. Soc. 1999, 121, 10634-10635. (b) Tempel, D. J., Johnson, L. K., Huff, R. L., White, P. S., Brookhart, M., J. Am. Chem. Soc. 2000, 122, 6686-6700.

55
[31] (a) Talsi, E. P., Babushkin, D. E., Semikolenova, N. V., Zudin, V. N., Panchenko, V. N., Zakharov, V., Macromol. Chem. Phys. 2001 ,202, 2046-. (b) Bryliakov, K. P., Semikolenova, N. V., Zudin, V. N., Zakharov, V. A., Talsi, E. P., Catal. Commun. 2004, 5, 45-48. (c) Bryliakov, K. P., Semikolenova, N. V., Zakharov, V. A., Talsi, E. P., Organometallics 2004, 23, 5375-5378. [32] (a) Griffiths, E. A. H., Britovsek, G. J. P., Gibson, V. C., Gould, I. R., Chem. Commun. 1999, 1333-1334. (b) Deng, L., Margl, P. Ziegler, T., J. Am. Chem. Soc. 1999, 121, 6479-6487. (c) Small, B. L., Brookhart, M., Macromolecules, 1999, 32, 2120-2130. (d) Khoroshun, D., Musaev, D. G., Vreven, T., Morokuma, K., Organometallics 2001, 20, 2007-2026. [33] Britovsek, G. J. P., Clensmith, G. K. B., Gibson, V. C., Goodgame, D. M. L. McTavish, S. J., Pankhurst, Q. A., Catal. Commun.2002, 3, 207-211. [34] (a) Kooistra, T. M., Knijnenburg, Q., Smits, J. M. M., Horton, A. D., Budzelaar, P. H., M., Gal, A. W., Angew. Chem. Int. Ed. 2001, 40, 4719-4722. (b) Gibson, V. C., Humphries, M. J., Tellmann, K. P., Wass, D. F., White, A. J. P., Williams, D. J., Chem. Commun. 2001, 2252-2253. [35] (a) Gridnev, A., J. Polym. Chem. Part A: Polym. Chem. 2000, 38, 1753-1766. (b) Gridnev, A., Ittel, S. D., Chem. Rev. 2001, 101, 3611-3660. [36] Hawker, C. J., Bosman, A. W., Harth, E., Chem. Rev. 2001, 101, 3661-3688. [37] (a) Chiefari, J., Chong, Y. K., Ercole, F., Krstina, J., Jeffery, J., Le, T. P. T., Mayadunne, R. T. A., Meijs, G. F., Moad, C. L., Moad, G., Rizzardo, E., Thang, S., H., Macromolecules, 1998, 31, 5559-5562. (b) Hawthorne, D. G., Moad, G., Rizzardo, E., Thang, S. H., 1999, 32, 5457-5459. (c) Mayadunne, R. T. A., Rizzardo, E., Chiefari, J., Chong, Y. K., Moad, G., Thang, S. H., Macromolecules 1999, 32, 6977-6980. Favier, A., Charreyre, M.-T., Macromol. Rapid Comm., 2006, 27, 653-692. [38] (a) Wang, J.-S., Matyjaszewski, K., J. Am. Chem. Soc., 1995, 117, 5614-5615. (b)Kato, M., Kamigaito, M., Sawamoto, M., Higashimura, T., Macromolecules 1995, 28, 1721-1723. (c) Matyjaszewski, K., Xia, H., Chem. Rev., 2001, 101, 2921-. (d) Kamigaito, M., Ando, T., Sawamoto, M., Chem. Rev., 2001, 101, 3689-3746. [39] Lizotte, J. R., ”Synthesis and Characterization of Tailored Macromolecules via Stable Free Radical Polymerization Methodologies” 2003, PhD thesis, Virginia Polytevhnic Institute and State University, p. 17-53. [40] Percec, V., Barboiu, B., Macromolecules, 1995, 28, 7970-7972. [41] (a) Matyjaszewski, K., Wei, M., Xia, J., McDermott, N., Macromolecules, 1997, 30, 8161-. (b) Ando, T., Kamigaito, M., Sawamoto, M., Macromolecules, 1997, 30, 4507-. (c) Gibson, V., O’Reilly, R., Reed, W., Wass, D., White, A., Williams, D., Chem. Commun., 2002, 1850-1851. [42] Hamasaki, S., Kamigaito, M., Sawamoto, M., Macromolecules, 2002, 35, 2934-2940. [43] Baskaran, D., Prog. Polym. Sci. 2003, 28, 521-581. [44] Odian, G., Principles of Polymerization, 3rd ed., John Wiley & Sons, Inc., New York, USA, 1991, 399. [45] Davis, T., Haddleton, D., Richards, S., J. Macrom.Chem. and Physics 1994, C34, 243. [46] (a) Webster, O. W., Adv. Polym. Sci 2004, 167, 1-34. (b) Baskaran, D., Sivaram, S., Macromolecules 1997, 30, 1550-1555. Dvoránek, L., Vlcek, P., Macromolecules 1994, 27, 4881-4885. (c) Zundel, T., Teyssié, P. Jérôme, R., Macromolecules 1998, 31, 5577-

56
5581. (d) Schmitt, B., Stauf, W., Müller, A. H. E., Macromolecules 2001, 34, 1551-1557. (e) Ishizone, T., Yoshimura, K., Hirao, A., Macromolecules 1998, 31, 8706-8712. (f) Ishizone, T., Yoshimura, K., Yanase, E., Nakahama, S., Macromolecules 1999, 32, 955-957. (g) Reetz, M. T., Hütte, S., Goddard, R., J. Phys. Org. Chem. 1995, 8, 231-241. (h) Vlcek, P., Lochmann, L., Prog. Polym. Sci 1999, 24, 793-873. [47] Webster, O. W., Farnham, W. B., Sogah, D. Y., 1986, European Patent 0 068 887. [48] (a) Webster, O. W., Hertler, W. R., Sogah, D. Y., Farnham, W. B., Rajan Babu, T. V., J. Am. Chem. Soc. 1983, 105, 5706-5608. (b) Sogah, D. Y., Hertler, W. R., Webster, O. W., Cogen, G. M., Macromolecules 1987, 20, 1473-1488. (c) Webster, O. W., J. Polym. Sci. Part A: Polym. Chem. 2000, 38, 2855-2860. [49] (a) Mai, P., Müller, A., Macromol. Chem. Rapid Commun. 1987, 8, 99-107. (b) Brittain, W., J. Am. Chem. Soc. 1988, 110, 7440-7444. [50] Müller, A. H. E., Macromol. Chem., Macromol. Symp. 1990, 32, 87-104. [51] Brittain, W., Dicker, I., Macromolecules 1989, 22, 1054-1057. [52] (a) Adachi, T., Sugimoto, H., Aida, T., Inoue, S., Macromolecules 1993, 26, 1238-1243. (b) Inoue, S., J. Polym. Sci. Part A: Polym. Chem. 2000, 38, 2861-2871. [53] (a) Yasuda, H., Yamamoto, H., Yokota, K., Miyake, S., Nakamura, A., J. Am. Chem. Soc. 1992, 114, 4908-4910. (b) Yasuda, H., Yamamoto, H., Yamashita, M., Yokota, K., Nakamura, A., Miyake, S., Kai, Y., Kanehisa, N., Macromolecules 1993, 26, 7134-7143. (c) Yasuda, H., Prog. Polym. Sci. 1999, 25, 573-626. (d) Yasuda, H., J. Polym. Sci. Part A Polym. Chem. 2001, 39, 1955-1959. (e) Yasuda, H., J. Organomet. Chem. 2002, 647, 128-138. [54] Yasuda, H., Yamamoto, H., Yamashita, M., Yokota, K., Nakamura, A., Miyake, S., Kai, Y., Kanehisa, N., Macromolecules 1993, 26, 7134-7143. [55] (a) Collins, S., Ward, D. W., J. Am. Chem. Soc. 1992, 114, 5460-5462. (b) Collins, S., Ward, D. G., Suddaby, K. H., Macromolecules 1994, 27, 7222-7224. (c) Soga, K., Deng, H., Yano, T., Shiono, T., Macromolecules 1994, 27, 7938-7940. (d) Deng, H., Shiono, T., Soga, K., Macromolecules 1995, 28, 3067-3073. (e) Li, Y., Ward, D. G., Reddy, S. S., Collins, S., Macromolecules 1997, 30, 1875-1883. (f) Chen, Y. X. E., Metz, M. V., Li, L., Stern, C. L., Marks, T. J., J. Am. Chem. Soc. 1998, 120, 6287-6305. (g) Cameron, P. A., Gibson, V. C., Graham, A. J., Macromolecules 2000, 33, 4329-4335. [56] (a)Frauenrath, H., Keul, H., Höcker, H., Macromolecules 2001, 34, 14-19. (b) Bolig, A. D., Chen, E. Y. X., J. Am. Chem. Soc. 2004, 126, 4897-4906. (c) Rodriguez-Delgado, A., Chen, E. Y. X., Macromolecules 2005, 38, 2587-2594. [57] (a) Endo, K., Macromol. Chem. Phys. 1999, 200, 1722-1725. (b) Endo, K., Inukai, A., Polymer Int. 2000, 49, 110-114. (c) Carlini, C., Martinelli, M., Raspolli, G., Anna, M., Sbrana, G., J. Pol. Chem. Part A: Polym. Chem. 2003, 41, 1716-1724. (d) Kim, I., Hwang, J.-M., Lee, J. K., Ha, C. S., Woo, S. I., Macromol. Rapid Commun. 2003, 24, 508-511. (e) He, X., Yao, Y., Luo, X., Zhang, J., Liu, Y., Zhang, L., Wu, Q., Organometallics 2003, 22, 4952-4957. [58] Liu, J.-Y., Zheng, Y., Li, Y.-S., Polymer 2004, 45, 2297-2301. [59] (a) Ihara, E., Maeno, Y., Yasuda, H., Macromol. Chem. Phys. 2001, 202, 1518-1523. (b) Wang, L., Li, Y., Zhu, F., Wu, Q., Polymer Bulletin 2006, 57, 73-81.

57
[60] (a) Shibayama, K., Polym. J. 2003, 35, 711-713. (b) Stibrany, R. T., Schulz, D. N., Kacker, S., Patil, A. O., Baugh, L. S., Rucker, S. P., Zushma, S., Berluche, E., Sissano, J. A., Macromolecules 2003, 36, 8584-8586. [61] (a) Johnson, L. K., Mecking, S., Brookhart, M., J. Am. Chem. Soc. 1996, 118, 267-268. (b) Mecking, S., Johnson, L. K., Wang, L., Brookhart, M., J. Am. Chem. Soc. 1998, 120, 888-899. (c) Heinemann, J., Mülhaupt, R., Brinkmann, P., Luinstra, G., Macrom. Chem. Phys. 1999, 200, 384-389. [62] (a) Castro, P. M., Lankinen, M. P., Uusitalo, A.-M., Leskelä, M., Repo, T., Macromol. Symp. 2004, 213, 199-208. [63] Castro, P. M., Lankinen, M. P., Leskelä, M., Repo, T., Macromol. Chem. Phys. 2005, 206, 1090-1097. [64] Castro, P., PhD thesis, “Polymerization of Acrylates with MAO Activated Iron(II) Complexes”, Yliopistopaino, Helsinki, 2005, ISBN 952-10-2520-4 (pdf). [65] (a) Coles, M. P., Jordan, R. F., J. Am. Chem. Soc. 1997, 119, 8125-8126. (b) Ihara, E., Young, V. G., Jordan, R. F., J. Am. Chem. Soc. 1998, 120, 8277-8278. (c) Radzewich, C. E., Guzei, I. A., Jordan, R. F., J. Am. Chem. Soc. 1998, 120, 9384-9385. (d) Bruce, M., Gibson, V. C., Redshaw, C., Solan, G. A., White, A. J. P., Williams, D. J., Chem. Commun. 1998, 2523-2524. (e) Radzewich, C. E., Guzei, I. A., Jordan, R. F., J. Am. Chem. Soc. 1999, 121, 8673-8674. (f) Dagorne, S., Guzei, I. A., Coles, M. P., Jordan, R. F., J. Am. Chem. Soc. 2000, 122, 274-289. (g) Kim, J. S., Wojeinski, L. M., Liu, S., Sworen, J. C., Sen, A., J. Am. Chem. Soc. 2000, 122, 5668-5669. (h) Talarico, G., Busico, V., Organometallics 2001, 20, 4721-4726. [66] Budzelaar, P. H. M., Talarico, G., Structure and Bonding 2003, 105, 141-165. [67] (a) Yliheikkilä, K., Lankinen, M. P., Repo, T., Leskelä, M., unpublished results. (b) Ikeda,M., Hirano, T., Tsuruta, T., Macromol. Chem. 1971, 10,127-135. (c) Ikeda,M., Hirano, T., Nakayama, S., Tsuruta, T., Macromol. Chem. 1974, 175, 2775-2793. (d) Liu, W., Nakano, T., Okamoto, Y., Polymer 2000, 41, 4467-4472. [68] Baskaran, D., Prog. Polym. Sci 2003, 28, 521-581. [69] (a) Wu, B., Lenz, R. W., Hazer, B., Macromolecules 1999, 32, 6856-6859. (b) Liu, J.-Y., Zheng, Y., Li, Y.-S., Polymer 2004, 45, 2297-2301. [70] Kitayama, T., Masuda, E., Yamaguchi, M., Nishiura, T., Hatada, K., Polym. J. 1992, 24, 817-827. [71] Ballard, D., G. H., Bowles, R. J., Haddleton, D. M., Richards, S. N., Sellens, R., Twose, D. L., Macromolecules 1992, 25, 5907-5913. [72] Bi, X, Wang, D., Wu, Z., J. Mol. Cat. A 2002, 179, 53-57. [73] Albeniz, A., Espinet, P., Lopez-Fernandez, R., Sen, A., J. Am. Chem. Soc. 2002, 124, 11278-11279. [74] Nagel, M., Paxton, W., Sen, A., Macromolecules 2004, 37, 9305-9307. [75] Lowe, G., Chem. Soc. Rev. 1995, 24, 309317. [76] Drews, J., Science 2000, 287, 1960-1964. [77] (a) Hoveyda, A. H., Chem. Biol. 1998, 5, 187-191. (b) Kuntz, K. W., Snapper, M. L., Hoveyda, A. H, Curr. Opin. Chem. Biol. 1999, 3, 313-319. (c) Shimizu, K. D., Snapper, M. L., Hoveyda, A. H., Chem. Eur. J. 1998, 4, 1885-1889. [78] (a) Schlogl, R., Angew. Chem. Int. Ed. 1998, 37, 2333-2336. (b) Senkan, S. M., Ozturk, S., Angew. Chem. Int. Ed. 1999, 38, 791-795. (c) Maier, W. F., Angew. Chem.

58
Int. Ed. 1999, 38, 1216-1218. (d) Jandeleit, B., Schaefer, D. J., Powers, T. S., Turner, H. W., Weinberg, W. H., Angew. Chem. Int. Ed. 1999, 38, 2494-2532. (e) Wennemers, H., Comb. Chem. High Throughput Screening 2003, 6, 527-540. (f) Boussie, T. R., Diamond, G. M., Goh, C., Hall, K. A., LaPointe, A. M., Leclerc, M., Lund, C., Murphy, V., Shoemaker, J. A. W., Tracht, U., Turner, H., Zhang, J., Uno, T., Rosen, R. K., Stevens, J. C., J. Am. Chem. Soc. 2003, 125, 4306-4317. (g) Cho, H. Y., Hong, D. S., Jeong, D. W., Gong, Y.-D., Seong, I., Macromol. Rapid Commun. 2004, 25,302-306. (h) Adams, N., Arts, H. J., Bolton, P. D., Cowell, D., Dubberley, R. R., Friederich, N., Grant, C. M., Kranenburg, M., Sealey, A. J., Wang, B., Wilson, P. J., Cowley, A. R., Mountford, P., Schröder, M., Chem. Commun. 2004, 434-435. (i) Mason, A. F., Coates, G. W., J. Am. Chem. Soc. 2004, 126, 10798-10799. (j) Jones, D. J., Gibson, V. C., Green, S. M., Maddox, P. J., White, A. J., P., Williams, D. J., J. Am. Chem. Soc. 2005, 127, 11037-11046. [79] Abu-Surrah, A. S., Thewald, U., Rieger, B., J. Organom. Chem. 1999, 587, 58-66. [80] Ligand crystallizes in monoclinic space group Cc (No. 9) with a = 10.7056(1) Å, b = 40.0964(1) Å, c = 4.8321(1) Å, V = 1995.74 Å3 and complex in monoclinic spacegroup P21/c with a = 10.035(1) Å, b = 19.200(1) Å, c = 12.046(1) Å, V = 2320.0 Å3. Displacement ellipsoids are drawn at the 30% probability level. [81] Yliheikkilä, K., Lappalainen, K., Repo, T., Leskelä, M., unpublished results. [82] Reardon, D., Aharonian, G., Gambarotta, S., Yap, G. P. A., Organometallics 2002, 21, 786-788. [83] Schoumacker, S., Hamelin, O., Pécaut, J., Fontecave, M., Inorg. Chem. 2003, 42, 8110-8116. [84] (a) Lumme, P. O., Lindell, E., Acta Cryst. 1988, C44, 463-465. (b) McCann, S., McCann, M., Casey, R. M. T., Jackman, M., Devereux, M., McKee, V., Inorg. Chim. Acta 1998, 279, 24-29. [85] (a) Reardon, D., Conan, F., Gambarotta, S., Yap, G., Wang, Q., J. Am. Chem. Soc. 1999, 121, 9318-9325. (b) Gibson, V. C., Tellmann, K. P., Humphries, M. J., Wass, D. F., Chem. Comm. 2002, 20, 2316-2317. (c) Kitiachvili, K.D., Mindiola, D. J., Hillhouse, G. L., J. Am. Chem. Soc. 2004, 126,10554-10555. (d) Vela, J., Vaddadi, S., Cundari, T. R., Smith, J. M., Gregory, E. A., Lachicotte, R. J., Flaschenriem, C. J., Holland, P. L., Organometallics 2004, 23, 5226-5239. (e) Scott, J., Gambarotta, S., Korobkov, I., Budzelaar, H. M., J. Am. Chem. Soc. 2005,127, 13019-13029. (f) Woodman, T. J., Yann Sarazin, Y., Garratt, S., Fink, G., Bochmann, M., J. Mol. Cat. A. Chem. 2005, 235, 88-97. [86] (a) Bart, S., Hawrelak, E., Schmisseur A., Lobkovsky, E., Chirik, P., Organometallics 2004, 23, 237-246. (b) Sugiyama, H., Aharonian, G., Gambarotta, S., Yap, G. P. A., Budzelaar, P. H. M., J. Am. Chem. Soc. 2002, 124, 12268-12274. [87] (a)Li, Q.-X, Luo, Q.-H., Li, Y.-Z., Pan, Z.-Q., Shen, M.-C., Eur. J. Inorg. Chem. 2004, 22, 4447-4456. (b) Hureau, C., Blondin,G., Charlot, M. F., Philouze,C., Nierlich, M., Césario, M., Anxolabéhére-Mallart, E., Inorg. Chem., 2005, 44, 3669-3683. [88] Rieger, B., Abu-Surrah, A. S., Fawzi, R., Steinman, M., J. Organomet. Chem. 1995, 497, 73-79. [89] Vedder, C., Schaper, F., Brintzinger, H.-H., Kettunen, M., Babik, S., Fink, G., Eur. J. Inorg. Chem. 2005, 1071-1080.

59
[90] (a) Gibson, V., Humphries, M., Tellmann, K., Wass, D., White, A., Williams, D., Chem. Commun. 2001, 2252-2253. (b) Kooistra, T., Knijnenburg, Q., Smits, J., Horton, A., Budzelaar, P., Gal, A., Angew. Chem. Int. Ed. 2001, 40, 4719-4722. [91] Matouzenko, G., Bousseksou, A., Lecocq, S., Konningsbrugen, P., Perrin, M., Kahn, O., Inorg. Chem. 1997, 36,2975-2981. [92] Endo, K., Yamanaka, Y., Macromol. Rapid Commun. 2000, 21, 785-787. [93] Yliheikkilä, K., Repo, T., Leskelä, M., unpublished results. [94] (a) Bourget-Merle, L., Lappert, M. F., Severn, J. R., Chem. Rev., 2002, 102, 3031-3065. (b) Matsui, S., Mitani, M., Saito, J., Tohi, Y., Makio, H., Tanaka, H., Fujita, T., Chem. Lett. 1999, 1263-1264. (c) Matsui, S. Tohi, Y., Mitani, M., Saito, J., Makio, H., Tanaka, H., Nitabaru, M., Nakano, T., Fujita, T., Chem. Lett. 1999, 1065-1066. [95] (a) Tolman, C. A., Chem. Rev. 1977, 77, 313-348. (b) Dierkes, P., van Leeuwen, P. W. N. M., J. Chem. Soc. Dalton. Trans., 1999, 1519-1529. [96] van Leeuwen, P. W. N. M., Kamer, P. C., Reek, J. N. H., Dierkes, P., Chem. Rev., 2000, 100, 2741-2769. [97] Barron, A. R., J. Chem. Soc. Dalton Trans., 1988, 3047-3050. [98] (a) Akakura, M., Yamamoto, H., Bott, S. G., Barron, A R., Polyhedron, 1997, 16, 4389. (b) Rodriguez, A., Chen, E. Y.-X., J. Am. Chem. Soc., 2005, 127, 1253-1266.


















