
Lab Qca Practicas Hyper
43
FACULTAD DE FARMACIA Departamento de Química Orgánica y Farmacéutica Prácticas de QUÍMICA COMPUTACIONAL APLICADA 2003-2004 Responsables Dra. María Font Arellano Dr. Miguel Romero Cuevas
-
Upload
william-ochoa-rojas -
Category
Documents
-
view
96 -
download
1
Transcript of Lab Qca Practicas Hyper

FACULTAD DE FARMACIA
Departamento de Química Orgánica y
Farmacéutica
Prácticas de
QUÍMICA COMPUTACIONAL APLICADA
2003-2004
Responsables
Dra. María Font Arellano Dr. Miguel Romero Cuevas


Prácticas de Química Computacional Aplicada
3
1. Descripción del programa Hyperchem
En estas prácticas trabajaremos principalmente con el programa Hyperchem 5.1 Standard, en
máquinas Pentium III bajo el sistema operativo Windows XP. Describiremos primero la pantalla general
y los iconos de las herramientas principales de Hyperchem junto con algunas convenciones a tener en
cuenta en este manual.
Hyperchem es un versátil paquete de modelización molecular que permite la visualización, edición y
cálculos de mecánica molecular y mecánica cuántica de diversos sistemas moleculares.
1.1. Descripción de pantalla

Prácticas de Química Computacional Aplicada
4
1.2. Herramientas
1.3. Convenio de pantalla
WORKSPACE área de trabajo
STATUS ángulo inferior izquierdo
METHOD ángulo inferior derecho
2. Construcción de un modelo tridimensional, opciones básicas
de Hyperchem: Estudio de la acetanilida
2.1. El archivo .log
Es posible guardar en un fichero de texto todos los datos acerca de la evolución de los cálculos, así
como de los resultados que se obtienen. Este fichero suele ser bastante grande dependiendo del nivel
de datos que queramos grabar en él, se activa mediante la siguiente opción:
Una vez finalizado el seguimiento de los cálculos, se puede desactivar esta opción mediante:
El fichero se puede leer e imprimir con cualquier editor de texto, como MS Word.
File / Start Log... • Guardar en: (elegir un directorio) • Nombre del archivo: • Mechanics print level: • Quantum print level: • OK
File / Stop Log

Prácticas de Química Computacional Aplicada
5
2.2. Opciones de trabajo
Antes de empezar a trabajar con cualquier molécula es conveniente establecer algunas opciones de
trabajo como el método de cálculo que vamos a utilizar, ya sea de mecánica molecular o mecánica
cuántica. Todas las opciones vienen en el menú Setup, elegiremos el campo de fuerzas MM+:
2.3. Dibujo en 2 dimensiones
Una molécula se dibuja átomo a átomo con la herramienta de dibujo , seleccionando primero el
elemento de la tabla periódica:
Con el elemento elegido se van uniendo los diferentes átomos entre si, picando en el átomo 1 y
desplazando el ratón con el botón izquierdo todavía presionado hasta la posición del nuevo átomo 2.
Los átomos de hidrógeno no es necesario dibujarlos, ya que Hyperchem los colocará en aquellas
posiciones que queden valencias libres, por lo tanto hay que especificar correctamente el tipo de enlace
entre cada par de átomos. Para dibujar la acetanilida vamos a seguir la siguiente secuencia de dibujo:
C
C
C
C C
C
C C
C
CC
C
C C
C
CC
N
C
C C
C
CC
N
C C
O
C
C C
C
CC
N
C C
O
Setup / Molecular Mechanics • MM+ • Options
• Atomic charges • OK
• OK Comprobar que en la zona METHOD aparece MM+
Build / Default Element... • C (o el elemento correspondiente: N, O, ...)
Podemos picar en Properties... para ver las propiedades de cada elemento. Es conveniente
desplazar la ventana de los elementos para dejar el WORKSPACE más despejado.

Prácticas de Química Computacional Aplicada
6
A continuación tenéis algunos consejos para guiarles en la construcción de la molécula:
Para cambiar el tip o de enlace (simple, doble, ...):
Para cambiar el tipo de átomo:
Para borrar átomos o enlaces:
Para centrar la figura en WORKSPACE:
Para desplazar la molécula por WORKSPACE:
2.4. Construcción del modelo tridimensional
Antes de seguir trabajando es conveniente cerrar la ventana de la tabla periódica. Vamos ahora a
añadir los hidrógenos que falta y a construir el modelo tridimensional a partir de los datos estándar que
Hyperchem tiene (distancias, ángulos, diedros, ...):
N
H
C
O
C
H
H
H
H H
HH
HC
C C
C
CC
N
C C
O
Add H & Model Build
Picar una vez más en el punto medio del enlace... ...o volver a dibujar el enlace sobre el anterior Si se pica dos veces seguidas sobre un ciclo este se volverá de tipo aromático
Picar con el botón derecho sobre el átomo o enlace
Pulsar la barra espaciadora del teclado
Picar la herramienta de desplazamiento
Elegir el elemento en la tabla periódica Picar dos veces con el botón izquierdo sobre el átomo
Build / Add H & Model Build

Prácticas de Química Computacional Aplicada
7
2.5. Visualización de estructuras
Para conocer las herramientas de visualización de Hyperchem vamos a utilizar la acetanilida como
ejemplo .
Activando los modos de giro en los ejes xy y giro en el eje z , a la vez que movemos el
ratón con el botón izquierdo pulsado, podemos observar la molécula final en tres dimensiones. Si
activamos el modo de zoom podemos aumentar o disminuir el tamaño de la molécula. Con el
modo de desplazamiento podemos desplazar la molécula por la ventana WORKSPACE hacia
la izquierda y derecha, arriba y abajo.
Hyperchem nos ofrece distintos modos de representación de la molécula: líneas, varillas, bolas, ....
Para cambiar de un modo a otro, por ejempo a varillas y bolas:
Dejar finalmente el modo de representación en sticks.
Las imágenes del WORKSPACE se pueden copiar con el comando:
Estas imágenes copiadas se pueden pegar tanto en el MS Word como en el MS Powerpoint.
También tenemos la posibilidad de ver distintas propiedades de los átomos, residuos, moléculas,
mediante el etiquetado de cada una de estas unidades. Para cada una de ellas, son diferentes las
propiedades que podemos conocer. Para ver, por ejemplo, el símbolo de cada átomo:
Display / Rendering... • Method
• Balls and Cylinders • Aceptar
En las distinas pestañas (Sticks, Balls, ...) hay más opciones para cada modo de representación.
Display / Labels... • Symbols • OK
Cada elemento se encuentra representado por un color diferente.
Edit / Copy Image También hace la misma función la tecla F9

Prácticas de Química Computacional Aplicada
8
2.6. Grabación
Cuando terminemos un paso, ya sea la simple construcción de una molécula o cálculos más complejos,
es conveniente guardar la estructura en archivo:
2.7. Obtención de parámetros geométricos: medidas de distancias,
ángulos y diedros
Una de las propiedades más interesantes es la propia geometría de la molécula. Además de la idea
general que nos da la visualización en 3D, explicada anteriormente, las medidas de distancias, ángulos
y diedros nos dará una idea más exacta de la geometría, aparte de unos valores concretos que luego
podrán ser comparados con los de otras moléculas. Es conveniente apuntar en papel cada uno de
estos valores.
Necesitaremos elegir dos átomos para medir distancias, tres para medir ángulos y cuatro para
medir diedros. Las opciones de selección múltiple de átomos ya estarán activadas, aunque conviene
comprobarlo. Elegimos luego los átomos implicados en nuestras mediciones:
Para dejar de seleccionar los átomos:
File / Save as... • Guarda en: elegir un directorio en vuestro saco o disco
En el apartado “Comments” podemos incluir cualquier comentario que consideremos importante.
Picar en la herramienta de selección Select / Atoms Select / Multiple selections. Para poder seleccionar más de un átomo a la vez. Esas dos últimas opciones han de estar seleccionadas (3)
Picar con el botón izquierdo del ratón sobre alguna zona libre del WORKSPACE (para elegir toda la molécula) o sobre un átomo o enlace concreto.
Picar con el botón derecho del ratón sobre alguna zona libre del WORKSPACE o sobre un átomo o enlace concreto

Prácticas de Química Computacional Aplicada
9
2.8. Edición de parámetros geométricos
Para la edición de las distintas propiedades geométricas se utilizará la opción correspondiente en el
menú Edit. Como ejemplo para aprender realizaremos la edición de un diedro de la molécula
acetanilida.
Es necesario tener en cuenta la estereoquímica de la molécula a la hora de construir la misma, ya que
se ha comprobado en numerosas ocasiones. Cómo el simple cambio de una conformación por otra
puede dar lugar a cambios drásticos en las propiedades de esta molécula. Podemos, en este caso,
observar la disposición CIS o TRANS del enlace amida y colocarlo finalmente en TRANS.
N
O R2
R1 H
3. Caracterización de la molécula de acetanilida por cálculos de
Mecánica Molecular
Vamos ahora a calcular diversas propiedades de la acetanilida utilizando el campo de fuerzas MM+.
3.1. Geometría molecular
La primera propiedad que podemos comprobar es la geometría de la molécula. Para ello realice las
siguientes medidas y apunte los datos en la tabla adjunta al final de la práctica:
N
O
H
H
H
H
HH
HH
H N
O
H
H
H
H
HH
HH
H N
O
H
H
H
H
HH
HH
H
Picar en los 4 átomos implicados: H, N, C y O En el STATUS aparece el valor del ángulo diedro. Ej: Torsion angle of atoms 15-6-7-9 is 0º
Edit / Set Bond Torsion... • Bond torsion: 180 (o el valor que deseemos) • OK
Comprobar que la configuración final del enlace es TRANS.

Prácticas de Química Computacional Aplicada
10
o Distancias N-O, Corto_1-O y Corto_2-O.
o Ángulos N-C-O y N-C-C.
o Diedros C-N-C-O y H-N-C-O.
3.2. Propiedades de átomos
Otra de las propiedades que vamos a observar es la carga parcial sobre cada átomo, por lo que
vamos a activar las etiquetas para esta propiedad:
La propiedad “Type” nos informa acerca de los tipos atómicos, que tendrá unas características
determinadas para cada campo de fuerza. Probar esta etiqueta y anotar los distintos tipos en la tabla .
3.3. Energía del sistema
Los cálculos en Hyperchem requieren en general dos pasos: elección del método de cálculo y sus
parámetros asociados y el cálculo en si mismo. Antes de iniciar cualquier cálculo es conveniente
asegurar que los parámetros son los correctos. En este caso hay que comprobar que tenemos activado
el campo de fuerzas MM+ y las opciones que elegimos al principio. Calcularemos las propiedades de la
conformación actual, haciendo un cálculo Single Point. Para hacer el cálculo de la conformación actual:
Apuntar estos valores en la tabla, junto con las medidas antes indicadas y guardar el archivo como
“acetanilida-mm”.
3.4. Comparación de resultados de Mecánica molecular
Realice ahora la construcción del sistema desde el paso a las tres dimensiones por los distintos
campos de fuerza que ofrece el programa Hyperchem y rellene la tabla al final.
Por último, cierre el archivo .log creado durante toda la práctica. Lo visualizaremos con un editor de
texto. A continuación, realice distintos cálculos single point grabando los resultados en archivos .log con
mechanics print level : 1, 2, 3 y compare la información obtenida.
Display / Labels... • Charge • OK
Compute / Single Point
En la ventana de STATUS aparece el valor de la energía y de gradiente.

Prácticas de Química Computacional Aplicada
11
MM+ AMBER BIO+ OPLS
Energía
Gradiente
N-O
Corto_1-O
Corto_2-O
N-C-O
N-C-C
C-N-C-O
H-N-C-O
TIPOS
ATÓMICOS
4. Caracterización de la molécula de acetanilida por cálculos de
Mecánica Cuántica
Pasamos ahora a precisar los cálculos utilizando el método semiempírico AM1. Como antes, primero
hemos de elegir el método y sus opciones, para luego hacer el cálculo.
Realizar la construcción del modelo 3D. Calcular la energía Observar y anotar los valores de carga
que aparecen sobre los átomos y que con los métodos de Mecánica Molecular no podían ser
calculados. Anote los valores obtenidos en la tabla correspondiente y guardar la estructura como
“acetanilida-am1”.
A continuación vamos a calcular alguna de las propiedades moleculares que podemos obtener
mediante este tipo de cálculos:
Setup / Semi-empirical • AM1

Prácticas de Química Computacional Aplicada
12
4.1. Momento dipolar
El momento dipolar, µ, es el vector cuyo módulo, dirección y sentido viene determinado por la
distribución de cargas en el interior de la molécula. Nos dará información acerca de la polaridad de una
molécula. Todas las moléculas no polares tienen un momento dipolar igual a cero.
4.2. Densidad electrónica
Representa el número de electrones por unidad de volumen alrededor de una molécula (e -/Å3). Esta
propiedad suele representarse como contornos de un nivel determinado de densidad.
Observar la distribución de la densidad de carga.
4.3. Potencial electrostático molecular
El potencial electrostático molecular se define como el trabajo realizado para acercar una carga positiva
desde el infinito hasta un punto determinado alrededor de la molécula. Dependerá de la distribución
electrónica y carga nuclear de esta molécula. Esta propiedad se relaciona con las interacciones a larga
distancia.
Observar la distribución de los valores de potencial electrostático.
Compute / Plot Molecular Properties... • Molecular Properties:
• Property: Total Charge Density • Representation: 3D isosurface
• Aceptar Podemos crear distintos tipos de superficie y a distintos niveles de densidad cambiando los
parámetros que aparecen al picar en las distintas pestañas. Probar con un nivel de densidad
de 1, valor que se puede cambiar con las opciones:
• Isosurface Rendering: • Contour value: 1
Compute / Plot Molecular Properties... • Molecular Properties:
• Property: Electrostatic Potential • Representation: 3D isosurface
• Aceptar
Display / Show Dipole Moment

Prácticas de Química Computacional Aplicada
13
Otra representación interesante se consigue con las siguientes opcio nes:
Observar el mapa de la distribución de zonas desde valores negativos a positivos. Comparar entre sí la
distribución de potencial electrostático, la de densidad electrónica y los valores de carga parcial sobre
los átomos.
4.4. Orbitales moleculares
Un orbital molecular es la región de la molécula en la que hay mayor probabilidad de encontrar un
electrón.
Vamos a ver primero la energía del orbital HOMO:
Para ver mejor la distribución del orbital y de los nodos, puede alinear la molécula respecto a los tres
ejes X, Y y Z. Para el eje X:
Compute / Plot Molecular Properties... • Molecular Properties: • Electrostatic potential
• Representation: 3D mapped isosurface • Aceptar
Compute / Orbitals...
Aparece una nueva ventana. En la derecha podemos ver el diagrama de niveles de energía de los
orbitales. Para elegir un orbital, picamos en el mismo o lo elegimos respecto al HOMO o el LUMO:
• Orbital: • HOMO - • Number: 0 Anote el valor que aparece en energy. Active “Labels” para ver los orbitales rellenos.
Pasamos ahora a dibujar el orbital HOMO. Conviene desplazar la ventana de orbitales
hacia una esquina de la pantalla para ver la molécula y el orbital en el WORKSPACE:
• Orbital Plotting: 3D Isosurface Comprobar que está desactivada Orbital squared
• Plot En “Options...” podemos cambiar algunas opciones de dibujo del orbital seleccionado.
• OK

Prácticas de Química Computacional Aplicada
14
Hacer un esquema sobre papel del orbital HOMO. Puede ver los ejes con la opción:
Dibuje sobre el papel la distribución del orbital HOMO respecto de los ejes Y y Z.
Represente ahora los orbitales LUMO, HOMO-23 y LUMO+22.
4.5. Espectro vibracional
Calculando las derivadas segundas de la energía respecto a las coordenadas espaciales de los
núcleos (matriz hesiana) es posible hallar las fuerzas con que se mueven los átomos y por tanto las
vibraciones que sufren.
4.6. Espectro electrónico
Para realizar el espectro electrónico UV-visible es necesario tener en cuenta algunos estados excitados
de la molécula. Por este motivo hay que modificar algunos parámetros relacionados con la Interacción
de Configuraciones. Utilizaremos en este caso el método semiempírico ZINDO/S, ya que se encuentra
parametrizado para esta propiedad, estimándose que se sobrestima la longitud de onda máxima en 20
nm. Analice el espectro electrónico por el método PM3.
Edit / Align Viewer... • With: X • OK
Display / Show Inertial Axes
Compute / Vibrations Compute / Vibrational Spectrum
Aparece una nueva ventana con las líneas espectrales. En la zona superior se encuentran
representadas todas las frecuencias encontradas, mientras que en la de abajo aparece la
intensidad de cada una.
• Seleccionar una frecuencia de alta intensidad • Apply Observar el modo de vibración y relacionarlo con el comportamiento esperado para este
grupo funcional en la espectroscopía de infrarrojo. Probar otras frecuencias.
• OK Cancel (para la animación)

Prácticas de Química Computacional Aplicada
15
4.7. Comparación de resultados de Mecánica Cuántica
Realice distintos cálculos con distintos métodos semiempíricos y con distintos conjuntos de funciones
base de métodos ab initio y rellene la tabla correspondiente. Compare los resultados.
A continuación, realice distintos cálculos single point grabando los resultados en archivos .log con
quantum print level : 1, 2, 3 y compare la información obtenida.
Semiempíricos Ab initio
AM1
Energía
Gradiente
Carga N
Carga O
Carga C=O
µ
∆Hº
Bandas IR
Elegimos el método semiempírico ZINDO/S • Options...:
• Configuration interaction...: • CI Method: Single excited • Orbital Criterion
• Occupied: 1 • Unoccupied: 1 Estos valores indican la cantidad de orbitales que tendremos en cuenta al crear
la combinación lineal de estados excitados.
Compute / Electronic Spectrum
En la parte inferior del gráfico aparecen las transiciones significativas a nivel espectroscópico.
Anote los valores que según este espectro serán los máximos esperables para un espectro
UV de esta molécula.

Prácticas de Química Computacional Aplicada
16
5. Algoritmos de Optimización Geométrica
Para calcular las propiedades de una manera mucho más exacta, se necesita generar una estructura
óptima. El cálculo usualmente requiere una estructura estable que representa un mínimo en la
superficie de energía potencial. Para ello se han desarrollado diversos algoritmos de optimización
geométrica. En Hyperchem todos los algoritmos de cálculo se encuentran en el menú Compute.
5.1. Importancia de la optimización geométrica
Un primer ejemplo para comprobar la importancia de obtener estructuras optimas para tener datos
correctos, es la comparación de la propiedades obtenidas para una molécula pequeña como es el
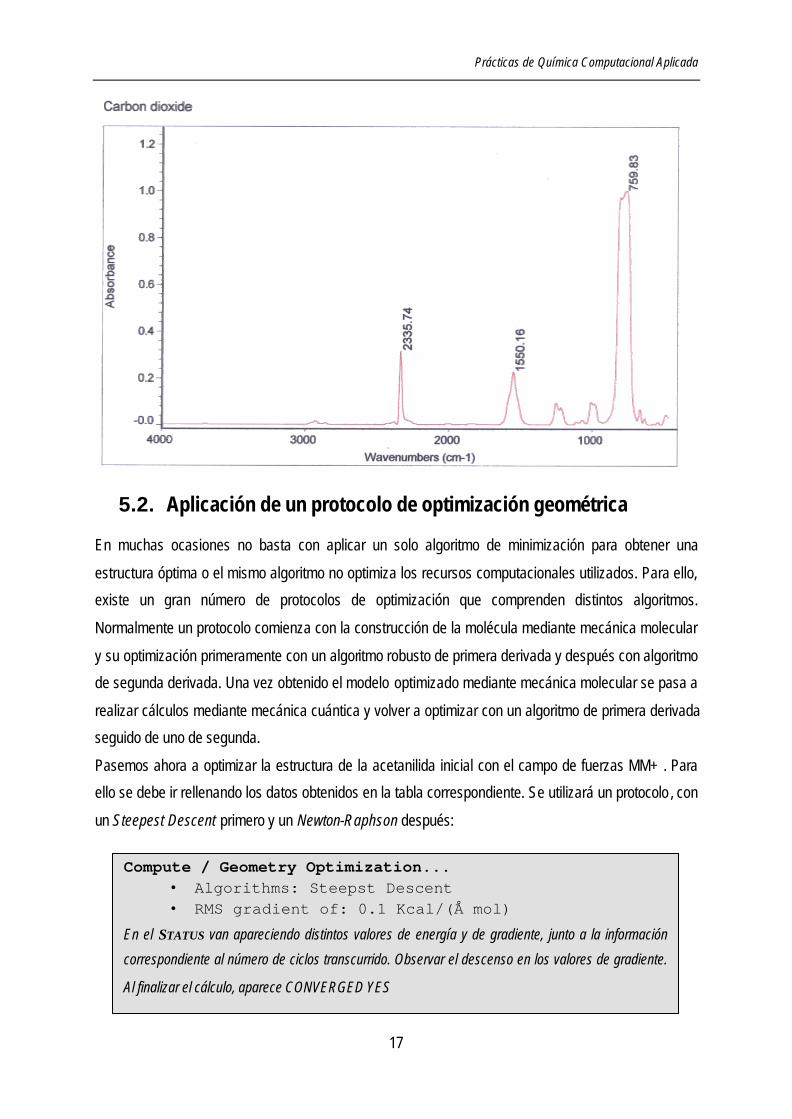
dióxido de carbono sin optimizar y optimizada. Para ello, construya ahora un modelo inicial del dióxido
de carbono con el método PM3 sin optimizar, grabe la estructura y calcule y anote las propiedades
necesarias para describir el sistema: energía, longitud de enlaces y espectro vibracional.
Con la conformación inicial construida con el método semiempírico PM3 realice ahora una optimización
geométrica utilizando el algoritmo Polak-Ribiere hasta una convergencia de 0,00001. Vuelva a realizar
las medidas anteriores y compárelas entre sí y con resultados experimentales.
Una de las características más importantes para poder tener datos correctos de bandas IR es la
necesidad de hacer una optimización geométrica bastante buena para obtener unas líneas espectrales
muy aproximadas (< 0,01 Kcal / Å mol para el gradiente del RMS) y calcular las vibraciones con el
mismo método que la optimización geométrica.
Compute / Geometry Optimization... • Algorithms: Steepst Descent • RMS gradient of: 0.1 Kcal/(Å mol)
En el STATUS van apareciendo distintos valores de energía y de gradiente, junto a la información
correspondiente al número de ciclos transcurrido. Observar el descenso en los valores de gradiente.
Al finalizar el cálculo, aparece CONVERGED YES
Prácticas de Química Computacional Aplicada
17
5.2. Aplicación de un protocolo de optimización geométrica
En muchas ocasiones no basta con aplicar un solo algoritmo de minimización para obtener una
estructura óptima o el mismo algoritmo no optimiza los recursos computacionales utilizados. Para ello,
existe un gran número de protocolos de optimización que comprenden distintos algoritmos.
Normalmente un protocolo comienza con la construcción de la molécula mediante mecánica molecular
y su optimización primeramente con un algoritmo robusto de primera derivada y después con algoritmo
de segunda derivada. Una vez obtenido el modelo optimizado mediante mecánica molecular se pasa a
realizar cálculos mediante mecánica cuántica y volver a optimizar con un algoritmo de primera derivada
seguido de uno de segunda.
Pasemos ahora a optimizar la estructura de la acetanilida inicial con el campo de fuerzas MM+ . Para
ello se debe ir rellenando los datos obtenidos en la tabla correspondiente. Se utilizará un protocolo , con
un Steepest Descent primero y un Newton-Raphson después:
Compute / Geometry Optimization... • Algorithms: Steepst Descent • RMS gradient of: 0.1 Kcal/(Å mol)
En el STATUS van apareciendo distintos valores de energía y de gradiente, junto a la información
correspondiente al número de ciclos transcurrido. Observar el descenso en los valores de gradiente.
Al finalizar el cálculo, aparece CONVERGED YES

Prácticas de Química Computacional Aplicada
18
Si no se alcanzara la convergencia es necesario repetir el mismo cálculo partiendo de la estructura
obtenida. Podemos aumentar también el número de ciclos.
Repetir las medidas de ángulos y diedros anteriormente realizadas y anotar en la tabla. Salvar la
estructura en un archivo “acetanilida-mm-sd”.
Repetir el cálculo si no se alcanza la convergencia, anotar los datos obtenidos y guardar como
“acetanilida-mm-nr”.
A continuación optimizamos la estructura de la acetanilida obtenida en el cálculo anterior, aplicando
ahora el método AM1 mediante un protocolo, con un Steepest Descent primero y un Polak-ribiere
después:
Anotar los datos necesarios y guardar la estructura como “acetanilida-am1-first”.
Anotar los datos necesarios y guardar la estructura como “acetanilida-am1-second”.
Repetimos el procedimiento utilizando un método ab initio, guardando las estructuras optimizadas y las
cargas obtenidas.
Compare los resultados teóricos obtenidos para el espectro de IR de la molécula optimizada con el espectro experimental.
Minimizar con el algoritmo Block Diagonal Newton-Raphson hasta un gradiente de RMS de 0.01 Kcal/(Å mol).
Utilizar un algoritmo de primera derivada para llegar a un gradiente de RMS de 0.1 kcal/(Å mol)
Utilizar un algoritmo de segunda derivada para llegar a un gradiente de RMS de 0.01 kcal/(Å mol).

Tabla de mediciones de la optimización de la acetanilida
N
H
C
O
C
H
H
H
H H
HH
H
MM+ AM1 ab initio Inicial Steepest
Descent Newton-Raphson
Single Point First derivative
Second derivative
First derivative
Second derivative
Energía
Gradiente
N-O
Corto_1-O
Corto_2-O
N-C-O
N-C-C
C-N-C-O
H-N-C-O

Prácticas de Química Computacional Aplicada
20
6. Exploración del Espacio conformacional. Análisis del
Butadieno
Los métodos sistemáticos de exploración del espacio conformacional alteran la geometría de una
molécula y calculan la energía del sistema para cada uno de los puntos. Dentro del conocimiento de la
superficie de energía potencial de una molécula existen puntos en los cuales la energía no varía con
respecto de infinitesimales cambios de la geometría. Esto significa que la derivada de la energía con
respecto de las coordenadas moleculares, llamada gradiente, se acerca a cero, y se conocen como
puntos estacionarios dentro de la curva de energía potencial. Estos puntos estacionarios pueden
corresponder a máximos energéticos conocidos como puntos de ensilladura o a estados estables o de
mínima energía.
En la siguiente práctica vamos a intentar conocer mediante variación sistemática manual y mediante
dinámica molecular los puntos críticos para la molécula de butadieno estudiada posteriormente en las
reacciones de Diels-Alder.
6.1. Búsqueda sistemática
Para ello, se construirá un modelo inicial del butadieno al que se realizará una primera optimización
geométrica para liberar tensiones. A continuación se realizarán diferentes cálculos energéticos
restringiendo el valor del diedro entre los carbonos 2 y 3. Para restringir el valor del ángulo se utilizará
las siguientes opciones:
A continuación realizamos una optimización geométrica mediante el método semiempírico AM1 y
anotamos los valores de Energía y de calor de formación de la conformación obtenida.
Build / Constrain Bond Torsion... • Other: VALOR ÁNGULO • OK
Select / Name Selection... • Other: Diedro • OK
Setup / Restraints... • Add: Diedro • Other: VALOR ÁNGULO • OK
Prácticas de Química Computacional Aplicada
21
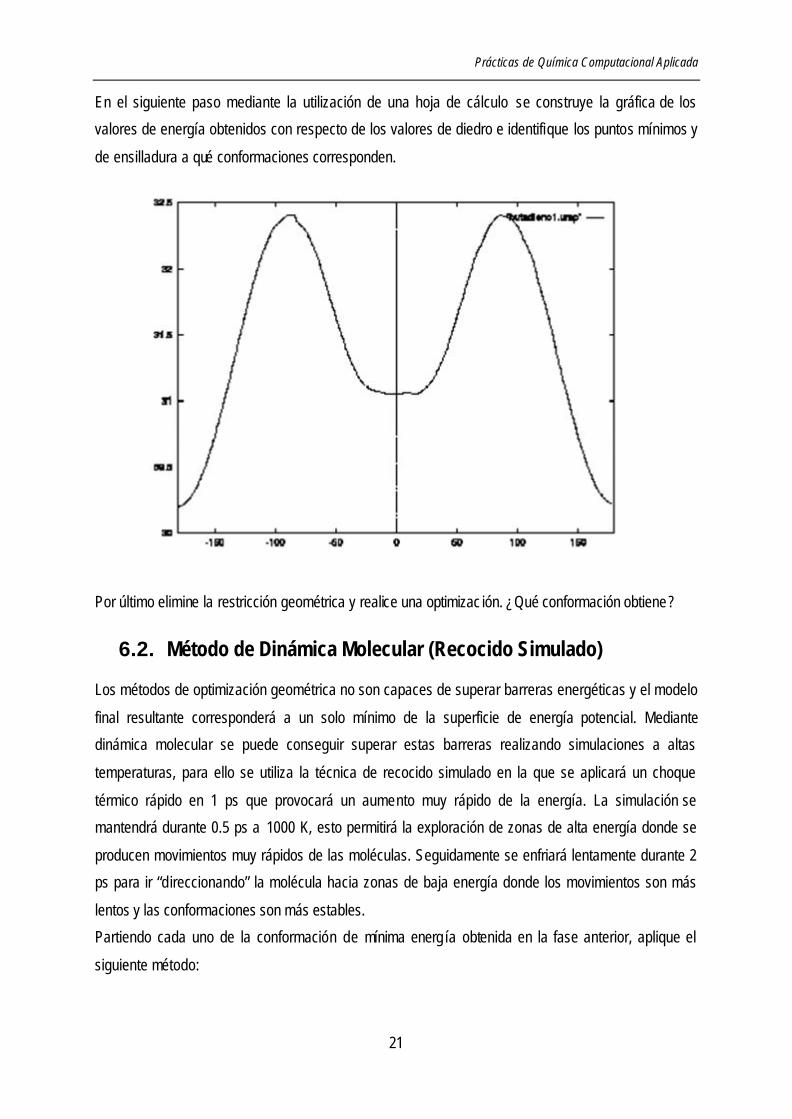
En el siguiente paso mediante la utilización de una hoja de cálculo se construye la gráfica de los
valores de energía obtenidos con respecto de los valores de diedro e identifique los puntos mínimos y
de ensilladura a qué conformaciones corresponden.
Por último elimine la restricción geométrica y realice una optimizac ión. ¿Qué conformación obtiene ?
6.2. Método de Dinámica Molecular (Recocido Simulado)
Los métodos de optimización geométrica no son capaces de superar barreras energéticas y el modelo
final resultante corresponderá a un solo mínimo de la superficie de energía potencial. Mediante
dinámica molecular se puede conseguir superar estas barreras realizando simulaciones a altas
temperaturas, para ello se utiliza la técnica de recocido simulado en la que se aplicará un choque
térmico rápido en 1 ps que provocará un aumento muy rápido de la energía. La simulación se
mantendrá durante 0.5 ps a 1000 K, esto permitirá la exploración de zonas de alta energía donde se
producen movimientos muy rápidos de las moléculas. Seguidamente se enfriará lentamente durante 2
ps para ir “direccionando” la molécula hacia zonas de baja energía donde los movimientos son más
lentos y las conformaciones son más estables.
Partiendo cada uno de la conformación de mínima energ ía obtenida en la fase anterior, aplique el
siguiente método:

Prácticas de Química Computacional Aplicada
22
7. Estudio de tautomerías
7.1. Equilibrio ceto-enólico
Los aldehídos y cetonas pueden existir como una mezcla en equilibrio de dos formas, llamadas la
forma cetónica y la forma enólica. Estas dos formas difieren en la localización de un protón y de un
doble enlace. Esta clase de isomería estructural se denomina tautomería y las distintas formas
tautómeros.
Para que un compuesto carbonílico tenga la capacidad de presentarse en la forma enólica es
indispensable que contenga un átomo de hidrógeno unido a uno de los átomos de cabono adyacentes
al grupo carbonilo. Este tipo de hidrógeno recibe el nombre de hidrógeno α.
Compute / Molecular dynamics.. • Times
• Heat time: 1 ps • Run time: 0.1 ps • Cool time: 2 ps • Step size: 0.0005 ps
• Temperature • Starting temperature: 200 K • Simulation temperature: 1000 K • Final temperature: 100 K • Temperature step: 30 K
• Options • In vacuo • Constant temperature: 0.1 ps
• Data collection period 20 time steps • Screen refresh period: 1 data steps. • Restart • Snapshots
• 10 data steps • velocities
• Averages (Graphics) • TEMP • Diedro • EPOT
• Proceed

Prácticas de Química Computacional Aplicada
23
La mayoría de los aldehídos y cetonas sencillas se presentan principalmente en la forma cetónica. El
desplazamiento del equilibrio depende de la estructura de la cetona o el aldehído, del solvente, de la
temperatura y otros factores como la conjugación o la formación de enlaces de hidrógeno.
En la siguiente práctica se evaluará el equilibrio para distintas molé culas carbonílicas: acetona,
dicetona 2,4-pentadiona y fenol. Para ello se obtendrá un modelo para cada molécula y se calculará su
calor de formación mediante el método AM1. Aproximando el calor de formación al valor de la energía
libre de Gibbs podemos calcular la constante de equilibrio para cada una de las reacciones y conocer el
desplazamiento del mismo hacia cualquiera de las formas (R=1.987 cal K-1 mol-1).
[ ][ ]RP
K
KRTHSTHG
eq
eq
=
−=∆∆−∆=∆
ln
FORMAS DE LA ACETONA
CH3 CH3
O
CH3 CH2
OH
FORMAS DE LA 2,4 PENTADIONA
CH3 CH3
O O
CH3 CH3
O OH
CH3 CH3
O O
CH3 CH3
O OH
FORMAS DEL FENOL
OH O
H
H

Prácticas de Química Computacional Aplicada
24
7.2. Aromaticidad del tiofeno
El benceno posee una estabilidad termodinámica relativa que no se corresponde a lo esperado para el
1,3,5-ciclohexatrieno. La energía de estabilización aromática del benceno se puede calcular
comparando la entalpía de la reacción de hidrogenación de un doble enlace en el benceno con una
media de las entalpías de reacción del segundo y tercer doble enlace como se muestra a continuación:
H2+
H2+
H2+
La diferencia en las entalpías de hidrogenación del benceno y de la media de los ciclohexadienos
corresponde a la estabilización aromática del benceno.
Calcula la energía de estabilización aromática del benceno y aplica el mismo procedimiento para
calcular la energía de estabilización aromática del tiofeno (el calor de formación del hidrógeno
molecular es de -5,2 Kcal/mol). ¿Cuál de los compuestos es más aromático?.
8. Reactividad: Reacciones de Diels-Alder
Una de las diversas aplicaciones de la química computacional es el estudio de la reactividad de
moléculas y de sus grupos funcionales. Uno de los métodos es el uso de la Teoría de los Orbitales
Frontera Moleculares. Se pueden usar los valores de energía, coeficientes y las propiedades nodales
obtenidas mediante cálculos sencillos de mecánica cuántica para investigar la reactividad relativa de
distintos sustituyentes, la regioselectividad de reacciones, y la especificidad de sustitución de
nucleófilos y electrófilos.
Se denominan reacciones de Diels-Alder a todas aquellas reacciones en las que se forma un
compuesto cíclico por adición 1,4 de un compuesto de doble o triple enlace (dienófilo) a un dieno
conjugado. Se les conoce también como cicloadiciones [4+2], ya que en el proceso intervienen 6
electrones π, 4 correspondiente al dieno y 2 al dienófilo. Estas reacciones están favorecidas cuando el
dienófilo posee grupos electroatrayentes como -CN, -COOH, -CHO y el dieno tiene sustituyentes
electrodonantes como -R,-OH, -OR.

Prácticas de Química Computacional Aplicada
25
Las reacciones de Diels-Alder transcurren con facilidad, incluso espontáneamente, a través de un
mecanismo concertado. Esto significa que la formación de los nuevos enlaces σ y la destrucción de los
enlaces π tiene lugar simultáneamente.
Estas reacciones son estereoespecíficas, lo que significa que a partir de un isómero geométrico del
dienófilo se obtiene sólo uno de los posibles isómeros del producto final.
De acuerdo con la teoría de los orbitales frontera, en el caso de una reacción de cicloadición, como la
reacción de Diels-Alder, es necesario establecer que pareja de orbitales frontera (HOMO de un rectivo
y LUMO del otro) debe ser considerada.
Es decir, debemos establecer si la velocidad de reacción será mayor por interacción del HOMO del
dieno y el LUMO del dienófilo o al revés (LUMO del dieno y HOMO del dienófilo). El proceso será más
rápido, cuanto menor sea el valor de ∆E = ELUMO – EHOMO de la pareja de orbitales implicados.
8.1. Flujo electrónico
Según los valores energéticos, explique entre qué orbitales se produce el flujo de electrones en la
reacción entre el 1,3-butadieno y el etileno.
+
Observe los ejemplos de un diagrama de salto energético y proponga el de la reacción.
Dienófilo Dieno Dienófilo Dieno Dienófilo Dieno

Prácticas de Química Computacional Aplicada
26
8.2. Efecto de sustituyentes
Calcule ahora los diagramas de saltos energéticos para el 1,3-butadieno y el etileno sustituidos y
analice como influyen en la reactividad distintos sustituyentes electrodonantes (D) y electroatrayentes
(A).
Por ejemplo D = -CH3, -OCH3, -NH2. A: -COOMe, -CN, -CHO, -NO2.
DA
D
A
D
A
++
8.3. Estereoselectividad
Además de los valores energ éticos, para que se lleve a cabo la reorganización electrónica es necesario
que se dé la simetría de orbitales que van a interaccionar, tal como se aprecia en la imagen del
acercamiento del 1,3-butanieno al etileno.
Explique por qué en la reacción del ciclopentadieno con él mismo se produce la forma endo y no la exo
(regla endo de Alder).
Aproximación endo Aproximación exo
8.4. Regioselectividad
Un dieno no simétrico como el piperileno reacciona con metil acrilato para dar dos aductos isoméricos
que difieren solamente en la orientación relativa de los dos sustituyentes, metilo y carbometoxilo
respectivamente.

Prácticas de Química Computacional Aplicada
27
COOMe COOMe
COOMe
++
95:5 La reacción es regioselectiva, ya que está desplazada mayoritariamente hacia uno de los isómeros.
Una explicación aceptada de la regioselectividad es valor de coeficiente de contribución al orbital
molecular de los átomos que van a formar el nuevo enlace, que puede comprobarse en el archivo .log
(como ya hemos aprendido) y que se corresponde con el tamaño de la porción de orbital que aparece
sobre él en la pantalla del programa (y que puede comprobarse aumentando hasta valores 0.1, o más
el valor de contorno de orbital en el cuadro Options del menú principal de visualización de orbitales), la
imagen se puede ver así:
COOMe COOMe
HOMO LUMO
+ Preferente
COOMe COOMe
HOMO LUMO
+No favorecido
9. Seguimiento de reacciones SN2
En esta práctica vamos a estudiar el seguimiento de reacciones con un ejemplo concreto, las
reacciones SN2. Estudiaremos la sustitución de un cloro en un metilo por un reactivo nucleofílico (Br, Cl,
I). Se trata de una reacción concertada que se produce en un solo paso:
La coordenada de reacción será la distancia entre
el nucléofilo que entra y el carbono. A medida que
esta disminuye, aumenta la distancia el carbono y el
cloro, y se produce la inversión de configuración.
Midiendo la energía a diferentes distancia Nu--C es
posible dibujar la curva de energía potencial.

Prácticas de Química Computacional Aplicada
28
¿Qué diferencias se observan con los distintos nucleófilos? ¿Es posible localizar el estado de
transición? ¿Cómo podemos mejorar la geometría de este estado de transición y caracterizarlo?
Vamos a realizar estos estudios con dos métodos (dinámica molecular y química cuántica). Primero
construiremos el sistema:
9.1. Construcción del sistema
Construir y optimizar el clorometano. Para facilitar los cálculos y sobretodo la visualización del sistema
colocaremos esta molécula con su eje principal en el eje de las X, los hidrógenos mirando a la
izquierda, y trasladaremos el carbono al origen de coordenadas:
Dibujamos ahora el nucleófilo, por ejemplo un cloro, a la izquierda del clorometano y sin construir su
modelo (no hacer Build / Add H & Model Build) seleccionamos solamente este nuevo cloro
para trasladarlo al punto -8, 0, 0 (dinámica molecular) o -4, 0, 0 (química cuántica):
También es necesario especificar las opciones de cálculo: método AM1, carga -1 y multiplicidad 1.
Haga un Single Point y guarde el sistema en un archivo.
Edit / Align Molecules... • Major Axis
• Align: Primary • Width: X Axis
• OK Seleccionar solamente el carbono
Edit / Translate... • Translate Selection • Selection
• Origin • OK
Edit / Translate... • Translate Selection • Selection
• Other: • X: -8 (ó -4) • Y: 0.000 • Z: 0.000
• OK Deseleccionamos todo y apretamos la barra espaciadora para centrar todo el sistema.

Prácticas de Química Computacional Aplicada
29
9.2. Método 1: dinámica molecular
Con este método vamos a lanzar el nucleófilo con una velocidad inicial hacia el carbono. El sistema
evolucionará según las leyes de la dinámica molecular. Midiendo la energía en cada punto es posible
obtener la curva de superficie potencial. La curva de energía potencial se obtendrá mediante el fichero
de salida para la hoja de cálculo MSexcel que podemos realizar mediante la opción de snapchots y
collection data de dinámica molecular. Con este método es necesario dibujar enlaces extras entre el
nucleófilo que ataca y los átomos con los que puede interaccionar, el carbono y los 3 hidrógenos
(puede ayudarse de Edit / Rotate.. para ver el tercer hidrógeno).
Vamos a definir la velocidad inicial del nucleófilo:
Seleccionar solo el carbono.
Select / Name Selection... • POINT • OK
Seleccionar solo el nucleófilo.
Setup / Set Velocity.. • Magnitude: 50 • Direction: At POINT • OK
Para una mejor visualización representa el sistema con esferas.

Prácticas de Química Computacional Aplicada
30
Pasamos ahora a realizar la dinámica molecular:
Observar los desplazamientos de los diferentes átomos. Se pude volver a ver la simulación con:
9.3. Método 2: química cuántica
En este caso disminuiremos la distancia entre el nucleófilo y el. En cada ciclo de cálculo se optimizará
la geometría del sistema con la distancia restringida (fija) y después se hará un Single Point sin la
restricción. Anotaremos los valores en un documento de MSexcel.
Partimos de la molécula grabada en el paso 1, con una distancia entre el nucleófilo y el carbono de 4Å
y los mismos parámetros para el cálculo AM1. Hay que definir la restricción de la distancia:
En cada ciclo de cálculo repetiremos los siguientes pasos:
Compute / Molecular Dynamics... • Times:
• Heat time: 0 • Run time: 0.5 • Cool time: 0 • Step size: 0.0005
• Temperature: • Simulation temperature: 300
• Options: • In vacuo • Constant temperature: sin marcar
• Data collection period: 1 • Screen refresh period: 1 • Restart: marcado • Snapshots:
• Nombre de archivo: sn2md.hin • OK
• Proceed
Compute / Molecular Dynamics... • Playback: marcado • Proceed
Seleccionar el nucleófilo y el carbono.
Select / Name Selection... • Other: nu-c • OK

Prácticas de Química Computacional Aplicada
31
Por último, una vez hayamos obtenido las curvas de energía potencial mediante las dos metodologías,
podremos estudiarlas y ver las estabilidades relativas de los reactivos y de los productos. Además se
puede calcular el estado de transición de la reacción. Éste es el punto de máxima energía en el camino
de paso de reactivos a productos. Para caracterizar el estado de transición se elegirán distintas
distancias cercanas al máximo de energía de la curva y se realizará una optimización geométrica hasta
un gradiente de rms de 0.01. Al realizar el espectro de IR del sistema obtenido, se puede comprobar
que la vibración de menor energía presenta un valor negativo de frecuencia. Éste es un modo normal
de vibración imaginario que corresponde a la coordenada de reacción de la reacción relacionado con
este estado de transición.
10. Estudio ab initio del amoniaco
En esta práctica vamos a realizar crear el modelo cuántico a nivel ab initio del amoniaco al que se
realizará el análisis vib racional de su forma piramidal y del que se hallará su estado de transición por
distintos métodos.
10.1. Construcción del sistema
Primeramente, construiremos el amoniaco. A continuación elegiremos el conjunto de funciones base
para cada uno de los átomos que componen el sistema:
No tiene que estar ningún átomo seleccionado.
Setup / Restraints... • Add • Restrained Value
• Other: 4 • Force Constant
• Other: 5000 • OK
Optimizar el sistema con el algoritmo Eigenvector following hasta un gradiente de 0.01 kcal/(Åmol).
Setup / Restraints... • Remove • OK
Hacer un Single Point y apuntar el resultado

Prácticas de Química Computacional Aplicada
32
10.2. Análisis vibracional del amoniaco piramidal
Para realizar el análisis vibracional de cualquier molécula, ésta debe estar en un mínimo local estable,
por lo que debe minimizarse bien (al menos una convergencia de rms de 0.01). Anote el valor de la
energía de la conformación piramidal.
A continuación realizamos el análisis vibracional observando las frecuencias e intensidades de las
bandas más características. Para un mejor entendimiento de cada movimiento se puede visualizar los
vectores de fuerza activando la correspondiente opción:
10.3. Caracterización del estado de transición: restricciones
geométricas
La coordenada de reacción (IRC Intrinsic Reaction Coordinate) se puede definir como la trayectoria de
menor energía que conecta los reactivos con los productos a través del estado de transición. En la
Display / Labels... • Basis set • OK
Ahora elegimos un conjunto de funciones base pequeño.
Setup / Ab initio... • Small (3-21G) • OK
En un siguiente paso vamos a elegir una serie de funciones extras para definir al átomo de nitrógeno.
Para ello seleccionaremos únicamente el átomo N.
Setup / Ab initio... • Advanced Options.
• Number of d Orbitals = Six. • OK
• Extra Basis Function. • D. • Exponent = 1.0. • OK
• OK
Display / Rendering... • Sticks.
• IRvectors. • OK

Prácticas de Química Computacional Aplicada
33
práctica, los cálculos de IRC se realizan encontrando primero el estado de transición de la reacción y
minimizando después hacia uno y otro lado de este punto de ensilladura.
Los métodos matemáticos de búsqueda de estados de transición no están tan desarrollados como los
de obtención de mínimos. En general, suelen funcionar para sistemas muy cerrados y cercanos a ese
estado de transición alrededor del cual se hacen pequeñas variaciones para llegar al punto cuya
derivada primera se iguale a cero. Pero estos métodos no aseguran que sea el camino de mínima
energía entre dos mínimos y podría ser bien un máximo energético o un mínimo local de alta energía.
Por estas razones, la búsqueda de estados de transición depende todavía de factores como la intuición
química y el conocimiento de las estructuras de los reactivos de partida y de los productos resultantes,
a partir de las cuales proponer un estado intermedio que luego se optimizará y caracterizará.
A continuación crearemos una estructura plana del amoniaco la cual creemos se acerca al estado de
transición del fenómeno de inversión de esta molécula. Para ello, se utilizará una restricción geométrica
en la que se seleccionarán todos los átomos del amoniaco y se nombrará como PLANE en el menú
correspondiente1. Seguidamente se restringirá el valor de esta torsión a 180º y se realizará una
optimización geométrica hasta un gradiente de rms de 0.01.
1 El valor de este plano formado por cuatro átomos en el que uno de ellos está enlazado con los tres restantes se puede
definir mediante una torsión “impropia” y se mide como el ángulo formado por uno de los átomos con el plano formado por
los otros tres átomos.

Prácticas de Química Computacional Aplicada
34
Para calcular la energía y el espectro de vibración de la conformación plana se quitará la restricción
geométrica ya que si no se calcularía la derivada de la fuerza invertida en mantener el plano y quedaría
reflejada en la energía y el espectro.
La diferencia de energía entre la conformación plana y la conformación piramidal corresponde a la
barrera de inversión del amoniaco.
Por último, al observar cada una de las bandas del espectro se puede comprobar que la vibración de
menor energía presenta un valor negativo de frecuencia. Éste es un modo normal de vibración
imaginario que corresponde a la coordenada de reacción del amoniaco relacionado con este estado de
transición.
10.4. Caracterización del estado de transición: Eigenvector
Following
Mediante el seguimiento de los vectores propios se puede encontrar el estado de transición de una
reacción, este método de optimización sigue una coordenada que, si está bien definida, puede coincidir
con la coordenada de reacción. Por definición, si seguimos la coordenada de reacción el punto en que
la primera derivada de la trayectoria se hace cero coincidirá con el estado de transición y cumplirá que
todos los valores de los vectores propios serán positivos menos uno que será negativo.
Para trabajar con este método utilizaremos una estructura que no se encuentre en un mínimo local ni
global. Para ello construiremos el modelo ab initio del amoniaco pero no lo optimizaremos. A partir de
esta estructura utilizaremos el método eigenvector following para hallar el estado de transición:
Una vez lo hemos hallado, caracterice este estado y realizar su espectro vibracional.
Compute / Transition State... • Eigenvector following • OK
Ahora se abre la ventana con las vibraciones correspondientes a la conformación del amoniaco.
• Elegir la frecuencia de menor energía. • RMS gradient of : 0.1 • OK

Prácticas de Química Computacional Aplicada
35
11. Diseño Indirecto
Los métodos de diseño indirecto se utilizan cuando la estructura de la molécula diana (proteína,
enzima,...) no está disponible a priori. Esta metodología consiste en el análisis detallado de las
propiedades estructurales de moléculas activas y de sus análogos inactivos conocidos.
11.1. Relaciones estructura-actividad de amidas
En esta práctica vamos analizar una serie de estructuras análogas a la urea para intentar deducir las
relaciones estructura-actividad entre ellas. Para ello, se tomará como modelo el estudio realizado por
Fang y Li2 en el que se halló la relación entre los valores de energía de los orbitales HOMO y LUMO de
distintas aminas: urea, tiourea, tioacetamida y tiosemicarbazida, y su relación con la actividad
anticorrosiva.
Molécula Eficiencia anticorrosión (%)
Urea 21,8
Tiourea 70,11
Tioacetamida 70,13
Tiosemicarbazida 76,95
Para la construcción de los modelos de las diferentes moléculas se necesita conocer su estructura
molecular. Una herramienta muy útil para investigadores no familiarizados con la nomenclatura de
química orgánica es Chemfinder-Canbridgesoft (http://chemfinder.cambridgesoft.com/), con ella se
pueden realizar búsquedas de distintas moléculas según su nombre, fórmula o estructura.
Una vez conocida la estructura molecular, se realizará el modelo de cada una de esas moléculas y se
calcularán distintas propiedades moleculares: momento dipolar, valores de energía de los orbitales
HOMO y LUMO, la contribución de cada átomo a cada uno de esos orbitales (archivo .log) y la
diferencia de energía LUMO-HOMO. Mediante el análisis de los datos obtenidos se hallará la relación
estructura actividad de dichas moléculas y se podrá proponer una ecuación de actividad y elementos
estructurales importantes para la misma.
2 Quantum chemistry study on the relationschip between molecular structure and corrosion inhibition afficiency of amides.
Journal of Molecular Structure (Theochem). 2002; 583: 179-185.

Prácticas de Química Computacional Aplicada
36
Una vez hallada la relación estructura- actividad proponga distintas estructuras y calcule su valor
teórico como anticorrosivo. Para tener idea puede realizar una nueva búsqueda en Chemfinder.
11.2. Relaciones estructura-actividad de análogos de fluoxetina
En esta práctica vamos analizar una serie de estructuras análogas a la fluoxetina (antidepresivo) para
intentar deducir las relaciones estructura-actividad entre ellas. La fórmula general y parámetros
fundamentales (datos mínimos) a determinar son:
R1 = H, CF3, CH3, NO2, OCH3, Cl
R2 = H, CH3, COCH3, CSCH3
n = 0, 1, 2, 3.
O
R1
(CH2)n
HN R2
C4'
C4
qN: carga sobre N
qC4: carga sobre C4
qC4’: carga sobre C4’
d1: distancia O-N
d2: distancia O-C4
d3: distancia O-C4’
Y los datos de actividad son:
n R1 R2 IC50 (nM) 2 CF3 CH3 3 2 H CH3 6 2 H COCH3 500 2 H H 4 2 H CSCH3 300 2 NO2 CH3 3,5 0 CF3 CH3 INACTIVO 1 CF3 CH3 500 3 CF3 CH3 INACTIVO
Realice un estudio de estos compuestos para llegar a proponer un farmacóforo para la actividad
antidepresiva.
Tenga en cuenta que estas moléculas presentan un centro estereogénico. Para obtener unos
resultados correctos se debe realizar el modelo de todas las posibles formas de la molécula. Para ello
en la opción de labels, se puede activar ver Chirality. Así observaremos las propiedades
estereogénicas de los distintos carbonos de la molécula. Para cambiar la forma de R a S o viceversa de

Prácticas de Química Computacional Aplicada
37
un carbono se puede elegir la herramienta de dibujo y con la tecla SHIFT del ordenador pulsada picar
sobre el carbono estereogénico una sola vez.
12. Diseño Directo. Diseño basado en la estructura de
distintos inhibidores de la proteasa del VIH-1
Los métodos de diseño directo de nuevas moléculas con actividad biológica se apoyan en la existencia
de información experimental previa sobre la estructura tridimensional de la diana biológica y, a menudo,
del complejo diana-ligando. Para obtener esta información estructural es necesario, normalmente, aislar
y clonar el ADN responsable de la expresión de dicha diana, expresarlo, purificarlo y caracterizar
cantidades considerables de proteína, y por último conseguir una cristalización de la misma, proceso
que en ocasiones no se puede conseguir. Posteriormente, cuando se ha conseguido la cristalización,
se someten los cristales a difracción de rayos X, y a estudios de resonancia magnética nuclear, para
los cuales es necesario frecuentemente el marcaje previo con 15N y 13C.
12.1. Datos Experimentales
En esta práctica se tomará como referencia el estudio realizado por Holloway y col.3 en el que
encontraron como los datos de energía de interacción (E i) entre distintos inhibidores formando complejo
con la enzima tenían correlación con los datos de actividad in vitro hallados para distintas series de
inhibidores de la proteasa.
Una de las series estaba compuesta por moléculas con modificaciones estructurales en la posición P2’,
cuya estructura base es:
O
O
NH
OH
O
Ph
Ph
NH
R
3 Structure-Based Design of Human Immunodeficiency Virus-1 Protease Inhibitors. Chapter 3 in Computer –Aided Molecular
Design. Ed. Reynolds-Holloway-Cox. ACS Symposium Series 589. ISBN 0-8412-3160-5.

Prácticas de Química Computacional Aplicada
38
Y donde la serie completa se presenta a continuación:
No R pIC50 No R pIC50 19
R
6,9431 27 R
OH
9,1549
20
R
OH
8,0209 28 R
O
OH
9,7447
21 R
7,4653 29 R
O
OH
7,3925
22
R
OH
Ph
6,1612 30 R
4,5229
23
R
OHO
6,7932 31 R
6,8861
24
R
OO
7,1785 32 R
6,8356
25
R
OH
CH3
6,6728 33 R
OH
OH
10,0000
26 R
OH
6,9144 34 ROH
7,4134
12.2. Bases de datos de macromoléculas
Una vez conocidos los datos experimentales, el punto de partida de cualquier estudio directo es la
recopilación de toda la información estructural que se pueda obtener sobre la diana. Para este propósito
existen diversas bases de datos de acceso libre que cuentan con herramientas eficientes de búsqueda.
La base de datos más importante de estructuras tridimensionales de proteínas es la Brookhaven
Protein DataBank (PDB; http://www.rcsb.org/pdb/). En ella, se encuentran depositados los datos de más

Prácticas de Química Computacional Aplicada
39
de 17.000 estructuras tridimensionales de proteínas y ácidos nucleicos. Los archivos de esta base de
datos presentan una estructura dividida en dos partes4. En la primera parte de toda estructura
tridimensional de la PDB se encuentra información general sobre la misma (organismo, genes,
nomenclatura, referencias bibliográficas, referencias a otras bases de datos), variables utilizadas en los
experimentos de elucidación estructural (aparato de difracción o resonancia, programas de
refinamiento, disolventes, pH, temperatura, etc, ...), y estructura de la molécula (simetría,
estereoquímica, organización y nomenclatura de los distintos dominios, regiones, cadenas o
agrupaciones moleculares o atómicas, como ligandos, cofactores, moléculas de agua, etc, ...), entre
otros. Los autores también alertan acerca de diversos aspectos con que se han encontrado en la
obtención de la estructura tridimensional, como ambigüedades en la detección de los diferentes
átomos, posiciones alternativas para un mismo átomo, ocupaciones muy bajas, o incluso ausencia de
los mismos. El estudio de la repercusión que estas cuestiones puedan tener en relación a la interacción
entre ligando y receptor es de gran importancia para la misma.
La segunda parte de los archivos es la utilizada por los programas de visualización para crear la
estructura tridimensional, ya que en ella se encuentran las coordenadas y características de cada
átomo.
En la actualidad las estructuras de la PDB sufren una rigurosa revisión antes de ser depositadas, que
en ocasiones obliga a demorar su inclusión en la misma desde meses hasta incluso años. Sin embargo,
esto no ocurría hace unos años y aquellas estructuras depositadas entonces no sufrieron estas
validaciones tan rigurosas. Esto, junto con el hecho de que las técnicas de elucidación estructural de
entonces no eran comparables a las actuales, hace que sea necesario realizar una validación de las
estructuras que se van a utilizar en cualquier estudio de modelización molecular. Errores en estas
estructuras pueden repercutir enormemente en las interacciones entre ligando y receptor bajo estudio.
Con respecto de complejos ligando-proteína, la recopilación de datos se puede realizar con la ayuda de
la REceptor-LIgand dataBASE (RELIBASE; http://relibase.ccdc.cam.ac.uk ). RELIBASE ha sido
diseñada como una herramienta rápida y flexible de obtención y visualización de información sobre
complejos ligando-proteína. Esta base de datos contiene todas las estructuras de la PDB junto a
información adicional sobre el ligando, subestructuras, similitud de secuencias de proteínas y
empaquetamiento cristalográfico. Además, se pueden llevar a cabo búsquedas basadas en la similitud
de ligandos, subestructuras, sitios de unión e interacciones ligando-proteína.
4 Información acerca de la estructura y ordenación de un fichero estándar de la PDB se puede encontrar en
http://www.rcsb.org/pdb/info.html

Prácticas de Química Computacional Aplicada
40
De todos los complejos encontrados en la PDB buscaremos uno que contenga la estructura base de
nuestros ligandos y lo bajaremos (código: 1BDR).
12.3. Visualización de estructuras macromoleculares
Una vez se ha recopilado toda la información estructural disponible y la bibliografía relacionada,
mediante la aplicación de las correspondientes técnicas computacionales, se puede llevar a cabo un
análisis estructural en el que se estudien las interacciones esenciales entre el inhibidor y la diana.
En Hyperchem existen distintas herramientas que facilitan la visualización de macromoléculas.
12.4. Construcción modelo inicial (Docking)
Una vez visualizado el complejo buscaremos sólo el inhibidor para realizar los cambios pertinentes.
Para ello pondremos en pantalla todo el complejo visualizado mediante el menú Display/Show all.
Con la herramienta de selección activada en moléculas, sele ccionamos el inhibidor y en el menú
Display hacemos que solo mantenga la selección. Veremos como nos aparece el inhibidor en pantalla.
Una vez lo tengamos solo, quitamos la selección y visualizamos todos los átomos del mismo:
Display / Rendering… • Carpeta Sticks
• Ribbons Display / Show Hydrogens
• Off Select / Select backbone A continuación en el menú display elegimos la opción de mostrar solo selección del backbone.

Prácticas de Química Computacional Aplicada
41
Ahora, con la herramienta de dibujo, borraremos todos los átomos sobrantes y dibujaremos el grupo R
que nos corresponda.
Una vez dibujados todos los átomos pesados, con la herramienta de selección en molécula, se
seleccionará el inihbidor entero y se le añadirán los átomos de Hidrógeno que le faltan.
Sólo con el inhibidor seleccionado: Build / Add Hydrogens
Quitando selección Setup / Semiempirical...
• PM3 Setup / Molecular Mechanics...
• MM+ Display / Show all
File / Save as... Lo guardamos como complejo inicial.

Prácticas de Química Computacional Aplicada
42
12.5. Optimización de la geometría del inhibidor en el complejo
Al realizar cualquier minimización hay que asegurarse de sólo tenemos seleccionado el ligando.
12.6. Cálculo de la Energía de Interacción
Una vez tenemos el complejo final, podemos visualizar sus estructuras más importantes y calcular la
energía de interacción. Para ello se utilizará la fórmula:
Einteracción=Ecomplejo-Eligando-Eproteína
La energía total del complejo se calcula deseleccionando todos los átomos y realizando un single
point.
Para calcular la energía de la proteína seleccionaremos el ligando y en el menú Edit seleccionaremos
la opción Clear. Así habremos eliminado del sistema el inhibidor.
A continuación realizamos un single point de la proteína y anotamos la energía.
Por último calculamos la energía del ligando abriendo la estructura del complejo final guardada.
Compute / Single point Anotar el valor de la energía y del gradiente.
Compute / Geometry optimization • Steepst Descent • RMS 0.1 • 32767 maximun cycles
Anotar el valor de la energía y del gradiente.
Compute / Geometry optimization • Polak-Ribiere • RMS 0.01 • 32767 maximun cycles
Anotar el valor de la energía y del gradiente.
File / Save as... Lo guardamos como complejo final.
File / Save as... Guardamos la proteína sola.
File / Open... Abrimos complejo final.

Prácticas de Química Computacional Aplicada
43
Seleccionando el inhibidor y en el menú Select eligiendo Complement selection elegimos todo lo que
no es ligando, y en el menú Edit seleccionaremos la opción Clear para borrar la proteína y las
moléculas de agua. Realizamos un single point y nos da el valor de la energía del ligando.
13. Unión de una serie de fenilimidazoles al citocromo
P450cam
Se sabe que las estructuras representadas a continuación se unen al mismo sitio activo del citocromo
P450cam, aunque no se sabe el modo en que lo hacen. Por este motivo se pretende encontrar un patrón
farmacofórico que ayude a entender el modo en que se produce esta unión. ¿Qué posibilidades de
alineamiento de las moléculas existen? ¿En qué propiedades están basadas?

![0 0-------. I:0 0-------. .-------0lent/pdf/nd/ElectronicQCA.pdf · clocked QCA cells, a QCA shift register, and power gain in QCA cells [14, IS]. At an early stage of QCA development](https://static.fdocuments.in/doc/165x107/6145851207bb162e665fbe8b/0-0-i0-0-0-lentpdfnd-clocked-qca-cells-a-qca-shift.jpg)
















