
Kir2.2 inward rectifier potassium channels are inhibited by an endogenous factor in Xenopus oocytes...
6
K ir 2.2 Inward Rectifier Potassium Channels Are Inhibited by an Endogenous Factor in Xenopus Oocytes Independently From the Action of a Mitochondrial Uncoupler ANTHONY COLLINS 1 * AND MAUREEN K. LARSON 2 1 Cardiovascular Biomedical Research Centre, School of Medicine and Dentistry, Queen’s University, Belfast, UK 2 Depertment of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, Corvallis, Oregon We previously showed inhibition of K ir 2 inward rectifier K þ channels expressed in Xenopus oocytes by the mitochondrial agents carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) and sodium azide. Mutagenesis studies suggested that FCCP may act via phosphatidylinositol 4,5-bisphosphate (PIP 2 ) depletion. This mechanism could be reversible in intact cells but not in excised membrane patches which preclude PIP 2 regeneration. This prediction was tested by investigating the reversibility of the inhibition of K ir 2.2 by FCCP in intact cells and excised patches. We also investigated the effect of FCCP on K ir 2.2 expressed in human embryonic kidney (HEK) cells. K ir 2.2 current, expressed in Xenopus oocytes, increased in inside-out patches from FCCP-treated and untreated oocytes. The fraction of total current that increased was 0.79 0.05 in control and 0.89 0.03 in 10 mM FCCP-treated ( P > .05). Following ‘‘run-up,’’ K ir 2.2 current was re-inhibited by ‘‘cramming’’ inside-out patches into oocytes. Therefore, run-up reflected not reversal of inhibition by FCCP, but washout of an endogenous inhibitor. K ir 2.2 current recovered in intact oocytes within 26.5 h of FCCP removal. Injection of oocytes with 0.1 U apyrase completely depleted ATP ( P < .001) but did not inhibit K ir 2.2 and inhibited K ir 2.1 by 35% ( P < .05). FCCP only partially reduced [ATP] ( P < .001), despite inhibiting K ir 2.2 by 75% ( P < .01) but not K ir 2.1. FCCP inhibited K ir 2.2 expressed in HEK cells. The recovery of K ir 2.2 from inhibition by FCCP requires intracellular components, but direct depletion of ATP does not reproduce the differential inhibitory effect of FCCP. Inhibition of K ir 2.2 by FCCP is not unique to Xenopus oocytes. J. Cell. Physiol. 219: 8–13, 2009. ß 2008 Wiley-Liss, Inc. Mitochondria are becoming increasingly recognized for their importance in cellular signaling (Hajnoczky et al., 2006; Carreras and Poderoso, 2007; Zhang and Gutterman, 2007). Several aspects of mitochondrial function are altered during the myocardial remodeling process that leads to heart failure (Das and Harris, 1990; Tokoro et al., 1995; Atlante et al., 1998; Seccia et al., 1998; Fukunaga et al., 2000; Murakami et al., 2002; Gaussin et al., 2003; Sayen et al., 2003; Benderdour et al., 2004; Nojiri et al., 2006; Prabhu et al., 2006; Lin et al., 2007). Remodeling also includes changes in cardiac electrophysiology that can promote arrhythmias. An important component of arrhythmogenic remodeling is a decrease in the cardiac inward rectifier potassium current (Pogwizd et al., 2001) that is mediated in mammalian hearts by K ir 2 channel proteins (Liu et al., 2001; Zaritsky et al., 2001; Miake et al., 2003; Schram et al., 2003). We previously provided evidence for regulation of K ir 2 inward rectifier potassium channels by changes in mitochondrial function (Collins and Larson, 2002). Our previous mutagenesis studies suggested that phosphatidylinositol 4,5-bisphosphate (PIP 2 ) may be a link in this mechanism. However, the apparent recovery in isolated membrane patches of K ir 2 channels from inhibition by the mitochondrial uncoupler carbonyl cyanide p- trifluoromethoxyphenylhydrazone (FCCP) was inconsistent with a role of PIP 2 (Collins et al., 2005). The aim of the present study was to determine how much of the apparent recovery could be attributed to spontaneous ‘‘run-up’’ of the current. We report that the inhibitory effect of FCCP on K ir 2.2 is in fact irreversible in inside-out membrane patches, and that the observed increase in current is because of washout of an endogenous inhibitor in Xenopus oocytes. Therefore the run-up in inside-out patches is independent of the effect of FCCP. Inhibition by FCCP is nevertheless reversible in intact oocytes, unlike inside-out patches, consistent with the notion that FCCP depletes an intracellular factor that is required for K ir 2 channel activity. This factor is unlikely to be ATP because direct ATP depletion did not mimic the effect of FCCP on K ir 2 channels. Our results in human embryonic kidney (HEK) cells suggest that K ir 2 channels may be modulated by changes in mitochondrial function in mammalian cells. Materials and Methods Materials Apyrase (grade VI from potato), bovine serum albumin, KCl and KOH were from Sigma (St. Louis, MO). Potassium pyrophosphate, KF and NaVO 3 were from Acros Organics (Morris Plains, NJ). Spermine, MgCl 2 and EGTA were from Fluka (Buchs, Switzerland). FCCP was from MP Biomedicals (Solon, OH). HEPES was from Contract grant sponsor: American Heart Association; Contract grant number: 0465358Z. *Correspondence to: Anthony Collins, Cardiovascular Biomedical Research Centre, School of Medicine and Dentistry, Queen’s University, 97 Lisburn Road, Belfast BT9 7BL, UK. E-mail: [email protected] Received 7 December 2007; Accepted 16 October 2008 Published online in Wiley InterScience (www.interscience.wiley.com.), 18 November 2008. DOI: 10.1002/jcp.21644 ORIGINAL ARTICLE 8 Journal of Journal of Cellular Physiology Cellular Physiology ß 2008 WILEY-LISS, INC.
-
Upload
anthony-collins -
Category
Documents
-
view
213 -
download
1
Transcript of Kir2.2 inward rectifier potassium channels are inhibited by an endogenous factor in Xenopus oocytes...

ORIGINAL ARTICLE 8J o u r n a l o fJ o u r n a l o f
CellularPhysiologyCellularPhysiology
Kir2.2 Inward Rectifier PotassiumChannels Are Inhibited by anEndogenous Factor in XenopusOocytes Independently From theAction of a MitochondrialUncoupler
ANTHONY COLLINS1* AND MAUREEN K. LARSON21Cardiovascular Biomedical Research Centre, School of Medicine and Dentistry, Queen’s University, Belfast, UK2Depertment of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, Corvallis, Oregon
We previously showed inhibition of Kir2 inward rectifier Kþ channels expressed in Xenopus oocytes by the mitochondrial agents carbonylcyanide p-trifluoromethoxyphenylhydrazone (FCCP) and sodium azide. Mutagenesis studies suggested that FCCP may act viaphosphatidylinositol 4,5-bisphosphate (PIP2) depletion. This mechanism could be reversible in intact cells but not in excised membranepatches which preclude PIP2 regeneration. This prediction was tested by investigating the reversibility of the inhibition of Kir2.2 by FCCP inintact cells and excised patches. We also investigated the effect of FCCP on Kir2.2 expressed in human embryonic kidney (HEK)cells. Kir2.2 current, expressed in Xenopus oocytes, increased in inside-out patches from FCCP-treated and untreated oocytes. Thefraction of total current that increased was 0.79� 0.05 in control and 0.89� 0.03 in 10 mM FCCP-treated (P> .05). Following‘‘run-up,’’ Kir2.2 current was re-inhibited by ‘‘cramming’’ inside-out patches into oocytes. Therefore, run-up reflected not reversal ofinhibition by FCCP, but washout of an endogenous inhibitor. Kir2.2 current recovered in intact oocytes within 26.5 h of FCCP removal.Injection of oocytes with 0.1 U apyrase completely depleted ATP (P< .001) but did not inhibit Kir2.2 and inhibited Kir2.1 by 35% (P< .05).FCCP only partially reduced [ATP] (P< .001), despite inhibiting Kir2.2 by 75% (P< .01) but not Kir2.1. FCCP inhibited Kir2.2 expressed inHEK cells. The recovery of Kir2.2 from inhibition by FCCP requires intracellular components, but direct depletion of ATP does notreproduce the differential inhibitory effect of FCCP. Inhibition of Kir2.2 by FCCP is not unique to Xenopus oocytes.
J. Cell. Physiol. 219: 8–13, 2009. � 2008 Wiley-Liss, Inc.
Contract grant sponsor: American Heart Association;Contract grant number: 0465358Z.
*Correspondence to: Anthony Collins, Cardiovascular BiomedicalResearch Centre, School of Medicine and Dentistry, Queen’sUniversity, 97 Lisburn Road, Belfast BT9 7BL, UK.E-mail: [email protected]
Received 7 December 2007; Accepted 16 October 2008
Published online in Wiley InterScience(www.interscience.wiley.com.), 18 November 2008.DOI: 10.1002/jcp.21644
Mitochondria are becoming increasingly recognized for theirimportance in cellular signaling (Hajnoczky et al., 2006;Carreras and Poderoso, 2007; Zhang and Gutterman, 2007).Several aspects of mitochondrial function are altered during themyocardial remodeling process that leads to heart failure (Dasand Harris, 1990; Tokoro et al., 1995; Atlante et al., 1998; Secciaet al., 1998; Fukunaga et al., 2000; Murakami et al., 2002; Gaussinet al., 2003; Sayen et al., 2003; Benderdour et al., 2004; Nojiriet al., 2006; Prabhu et al., 2006; Lin et al., 2007). Remodeling alsoincludes changes in cardiac electrophysiology that can promotearrhythmias. An important component of arrhythmogenicremodeling is a decrease in the cardiac inward rectifierpotassium current (Pogwizd et al., 2001) that is mediated inmammalian hearts by Kir2 channel proteins (Liu et al., 2001;Zaritsky et al., 2001; Miake et al., 2003; Schram et al., 2003).
We previously provided evidence for regulation of Kir2inward rectifier potassium channels by changes inmitochondrial function (Collins and Larson, 2002). Ourprevious mutagenesis studies suggested thatphosphatidylinositol 4,5-bisphosphate (PIP2) may be a link inthis mechanism. However, the apparent recovery in isolatedmembrane patches of Kir2 channels from inhibition by themitochondrial uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) was inconsistentwith a role of PIP2 (Collins et al., 2005). The aim of the presentstudy was to determine how much of the apparent recoverycould be attributed to spontaneous ‘‘run-up’’ of the current.We report that the inhibitory effect of FCCP on Kir2.2 is in factirreversible in inside-out membrane patches, and that theobserved increase in current is because of washout of anendogenous inhibitor in Xenopus oocytes. Therefore the
� 2 0 0 8 W I L E Y - L I S S , I N C .
run-up in inside-out patches is independent of the effect ofFCCP. Inhibition by FCCP is nevertheless reversible in intactoocytes, unlike inside-out patches, consistent with the notionthat FCCP depletes an intracellular factor that is requiredfor Kir2 channel activity. This factor is unlikely to be ATPbecause direct ATP depletion did not mimic the effect of FCCPon Kir2 channels. Our results in human embryonic kidney(HEK) cells suggest that Kir2 channels may be modulated bychanges in mitochondrial function in mammalian cells.
Materials and MethodsMaterials
Apyrase (grade VI from potato), bovine serum albumin, KCl andKOH were from Sigma (St. Louis, MO). Potassium pyrophosphate,KF and NaVO3 were from Acros Organics (Morris Plains, NJ).Spermine, MgCl2 and EGTA were from Fluka (Buchs, Switzerland).FCCP was from MP Biomedicals (Solon, OH). HEPES was from

I N H I B I T I O N O F K i r 2 P O T A S S I U M C H A N N E L S 9
EMD Chemicals (Cincinnati, OH). Potassium gluconate was fromTCI America (Portland, OR). Perchloric acid was from Alfa Aesar(Ward Hill, MA). Other reagents were from Fisher Scientific(Hampton, NH).
cDNA constructs and cRNA transcription
Complementary DNAs encoding Kir2.1 (IRK1) (Kubo et al., 1993)and Kir2.2 (MB-IRK2) (Takahashi et al., 1994) were subcloned intothe Xenopus expression vector pGEMHE (Liman et al., 1992).Plasmids were linearized with NheI, and cRNA was transcribedin vitro with T7 RNA polymerase (mMessage mMachine,Ambion, Austin, TX). RNA yield and integrity was assessed byagarose-ethidium bromide gel electrophoresis.
Oocyte isolation
All procedures involving animals were approved by the InstitutionalAnimal Care and Use Committee (IACUC) at Oregon StateUniversity and were in compliance with the current NIH standardsfor animal use. Ten Xenopus laevis frogs (Nasco, Fort Atkinson, WI)were used. Animals were anaesthetised by immersion in 0.03%benzocaine for 10–15 min. Animals were humanely killed bysevering the cervical spinal cord under anaesthesia. Stage V–VIoocytes were surgically removed under anaesthesia and incubatedwith 1 mg/ml collagenase (type CLS3; Worthington, Lakewood, NJ)for 2 h at 228C in 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mMHEPES, pH 7.4, with agitation, to remove connective tissue.Oocytes were then washed several times with 96 mM NaCl, 2 mMKCl, 1 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES, pH 7.4 (ND-96) andstored in the same solution at 188C. Both solutions also contained100 mg/ml streptomycin and 60 mg/ml ampicillin. Oocytes wereinjected up to 24 h later (Nanoliter; World Precision Instruments,Sarasota, FL) with 0.4 (Kir2.1) or 10 (Kir2.2) ng/oocyte cRNA in50 nl nuclease-free water (Ambion). Kir2.1 was expressed moreefficiently than Kir2.2, possibly because the Kir2.2 constructincluded original 50 and 30 untranslated regions, which were absentin the Kir2.1 construct. One day after injection oocytes weretransferred into ND-96 with FCCP added as stated, or left inND-96 as control. FCCP was added from a 100 mM stock solutionin DMSO.
Human embryonic kidney (HEK) cells
HEK293T cells were cultured in Dulbecco’s Modified EagleMedium (D-MEM) containing 4,500 mg/L glucose, 4 mML-glutamine and 110 mg/L sodium pyruvate, 10% (v/v)heat-inactivated foetal calf serum, 100 U/ml penicillin and 100 mg/ml streptomycin (all from Invitrogen, Paisley, UK) at 378C in 95%air, 5% CO2. Cells were seeded onto poly-l-lysine treated coverslips and transfected with hKir2.2/pCXN2 (Kaibara et al., 2002) andpEGFP-C1 (Clontech, Saint-Germain-en-Laye, France) at a ratio of�10:1 using JetPEI transfection reagent, according to themanufacturer’s instructions (PolyPlus, Illkirch, France).
Two-electrode voltage clamp
Inward rectifier currents were recorded using a TEC-03 amplifier(npi, Tamm, Germany) controlled by Pulse 8.4 software (Heka,Southboro, MA) via an ITC-16 computer interface (Instrutech,Long Island, NY). Currents were filtered at 500 Hz and digitizedat 1 kHz. Data were analyzed using Pulse 8.4 and Prism 3.02(GraphPad, San Diego, CA). Microelectrode pipettes wereprepared from thin-walled, 1.5 mm outer diameter borosilicateglass capillaries (TW150F-3; World Precision Instruments) on amicroprocessor-controlled puller (PUL-100; World PrecisionInstruments), and had resistances of 0.5–1.5 MOhm when filledwith 3 M KCl. Oocytes were placed in a 100 ml volume recordingchamber that was continuously perfused at a rate of approximately1.2 ml/min with 90 mM KCl/KOH, 3 mM MgCl2, 1 mM EGTA, 5 mMHEPES, pH 7.4.
JOURNAL OF CELLULAR PHYSIOLOGY
Patch-clamp
The patch-clamp amplifier was an Axopatch 200B (AxonInstruments/Molecular Devices, Union City, CA). Data wereacquired with a Digidata 1320A computer interface and pClamp8or pClamp10 software (Axon Instruments/Molecular Devices).Data were analyzed using pClamp8 or pClamp10 and Prism 4(GraphPad).
Inward rectifier currents were recorded in ‘‘giant’’ cell-attachedand inside-out patches from Xenopus oocytes (Hilgemann, 1995),2–3 days after injection with Kir2 cRNA. Membrane potential washeld at 0 mV, and every 5 sec was stepped to þ50 mV for 75 msec,then to �50 mV for 75 msec. Currents were filtered at 1 kHz, anddata were acquired at 5 kHz. Data were analyzed using pClamp 8and Prism 3.02. Patch pipettes had inner tip diameters of 20–25mm;resistance <500 kOhm. The same ‘‘FVPP’’ type solution was usedin the recording chamber and in patch pipettes, and for perfusinginside-out patches: (mM) 40 KCl, 75 potassium gluconate, 5 KF,0.1 NaVO3, 10 potassium pyrophosphate, 1 EGTA, 0.1 spermine,10 HEPES, pH 7.4.
Whole-cell inward rectifier currents were recorded intransfected HEK cells 1–2 days after transfection. Transfected cellswere identified as those expressing green fluorescent protein asdetected by fluorescence microscopy. Currents were filtered at10 kHz. Patch pipettes had resistances of 2–4 MOhm after firepolishing. The extracellular solution was (mM) 132 NaCl, 1.8CaCl2, 5.4 KCl, 10 MgCl2, 10 glucose, 10 HEPES, pH 7.4. Theintracellular pipette solution was (mM) 5 NaCl, 40 KCl, 100 KF,5 EGTA, 3 EDTA, 5 glucose, 10 HEPES, pH 7.4. Data are presentedas current density (pA/pF) to adjust for differences in cell size.
ATP assay
Relative oocyte ATP content was measured by an assay reportedby Gribble et al. (1997). Batches of five oocytes were homogenizedin 200 ml of phosphate-buffered saline (PBS; 10 mM phosphate,137 mM NaCl; pH 7.4). One hundred microliters of 10% perchloricacid was added and cell debris pelleted by centrifugation. A 20 mlaliquot of the supernatant was added to 1 ml of 125 mM Tris-Cl,5 mM MgSO4, 0.5 mM EDTA, 0.5 mM dithiothreitol, 100 mg/mlbovine serum albumin, pH 7. The ATP content was quantified bythe measurement of luminescence 10 sec after the addition of 50mlreconstituted firefly luciferin-luciferase extract (Sigma), for 10 sec.
Results‘‘Run-up’’ in inside-out patches is independent ofprior inhibition by FCCP
Data from two patch-clamp experiments are shown in Figure 1.Both recordings were from Xenopus oocytes injectedwith Kir2.2 cRNA. Oocytes were pre-incubated with FCCP(part A) or in control conditions (B). Data represent inwardcurrent recorded at a membrane potential of �50 mV, initiallyin the cell-attached configuration (inverted triangles) then afterpatch excision into the inside-out configuration (continuoussolid traces). The traces were constructed from the currentrecorded at the end of 75 msec steps to �50 mV following astep to þ50 mV to monitor leak current (see inset). In bothcases the current increased upon excision and perfusion ofinside-out patches. The rate of run-up tended to be slowerin FCCP-treated cells but this tendency was not significant(P> .05 by t-test). The time to half-maximal current from thestart of patch perfusion was 3.0� 1.3 min for control (n¼ 6)and 6.6� 1.6 min (n¼ 7) for FCCP-treated.
Kir2.2 current was on average significantly less incell-attached patches on oocytes that were pre-treated withFCCP versus control oocytes. Maximum current after patchexcision had a tendency to be smaller in FCCP-treated versuscontrol but was not significant (P> .05) because of thevariability between patches (Fig. 2A). The influence of this
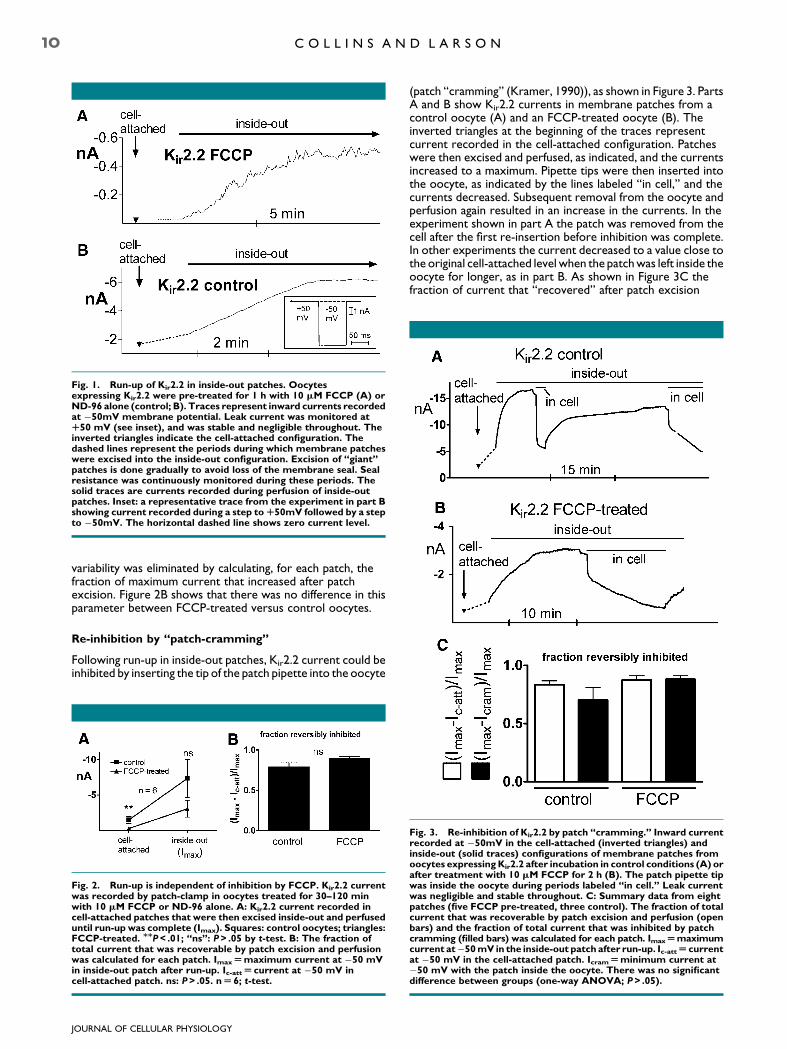
Fig. 1. Run-up of Kir2.2 in inside-out patches. Oocytesexpressing Kir2.2 were pre-treated for 1 h with 10 mM FCCP (A) orND-96 alone (control; B). Traces represent inward currents recordedat �50mV membrane potential. Leak current was monitored atR50 mV (see inset), and was stable and negligible throughout. Theinverted triangles indicate the cell-attached configuration. Thedashed lines represent the periods during which membrane patcheswere excised into the inside-out configuration. Excision of ‘‘giant’’patches is done gradually to avoid loss of the membrane seal. Sealresistance was continuously monitored during these periods. Thesolid traces are currents recorded during perfusion of inside-outpatches. Inset: a representative trace from the experiment in part Bshowing current recorded during a step to R50mV followed by a stepto �50mV. The horizontal dashed line shows zero current level.
10 C O L L I N S A N D L A R S O N
variability was eliminated by calculating, for each patch, thefraction of maximum current that increased after patchexcision. Figure 2B shows that there was no difference in thisparameter between FCCP-treated versus control oocytes.
Re-inhibition by ‘‘patch-cramming’’
Following run-up in inside-out patches, Kir2.2 current could beinhibited by inserting the tip of the patch pipette into the oocyte
Fig. 2. Run-up is independent of inhibition by FCCP. Kir2.2 currentwas recorded by patch-clamp in oocytes treated for 30–120 minwith 10 mM FCCP or ND-96 alone. A: Kir2.2 current recorded incell-attached patches that were then excised inside-out and perfuseduntil run-up was complete (Imax). Squares: control oocytes; triangles:FCCP-treated. MMP < .01; ‘‘ns’’: P > .05 by t-test. B: The fraction oftotal current that was recoverable by patch excision and perfusionwas calculated for each patch. Imax U maximum current at �50 mVin inside-out patch after run-up. Ic-att U current at �50 mV incell-attached patch. ns: P > .05. n U 6; t-test.
JOURNAL OF CELLULAR PHYSIOLOGY
(patch ‘‘cramming’’ (Kramer, 1990)), as shown in Figure 3. PartsA and B show Kir2.2 currents in membrane patches from acontrol oocyte (A) and an FCCP-treated oocyte (B). Theinverted triangles at the beginning of the traces representcurrent recorded in the cell-attached configuration. Patcheswere then excised and perfused, as indicated, and the currentsincreased to a maximum. Pipette tips were then inserted intothe oocyte, as indicated by the lines labeled ‘‘in cell,’’ and thecurrents decreased. Subsequent removal from the oocyte andperfusion again resulted in an increase in the currents. In theexperiment shown in part A the patch was removed from thecell after the first re-insertion before inhibition was complete.In other experiments the current decreased to a value close tothe original cell-attached level when the patch was left inside theoocyte for longer, as in part B. As shown in Figure 3C thefraction of current that ‘‘recovered’’ after patch excision
Fig. 3. Re-inhibition of Kir2.2 by patch ‘‘cramming.’’ Inward currentrecorded at �50mV in the cell-attached (inverted triangles) andinside-out (solid traces) configurations of membrane patches fromoocytes expressing Kir2.2 after incubation in control conditions (A) orafter treatment with 10 mM FCCP for 2 h (B). The patch pipette tipwas inside the oocyte during periods labeled ‘‘in cell.’’ Leak currentwas negligible and stable throughout. C: Summary data from eightpatches (five FCCP pre-treated, three control). The fraction of totalcurrent that was recoverable by patch excision and perfusion (openbars) and the fraction of total current that was inhibited by patchcramming (filled bars) was calculated for each patch. Imax U maximumcurrent at�50 mV in the inside-out patch after run-up. Ic-att U currentat �50 mV in the cell-attached patch. Icram U minimum current at�50 mV with the patch inside the oocyte. There was no significantdifference between groups (one-way ANOVA; P > .05).

I N H I B I T I O N O F K i r 2 P O T A S S I U M C H A N N E L S 11
(open bars) was similar to the fraction that was inhibited byinsertion of inside-out patches into the oocyte (filled bars). Thiswas true of control and FCCP-treated oocytes.
In Figure 3 the time course for inhibition by patch crammingappears slower for the FCCP-treated than for the controloocyte. There was variability in the time course of inhibitionbut on average there was no significant difference in the time tohalf-maximal inhibition between FCCP-treated (45� 8.4 sec;n¼ 5) and control (78� 41 sec; n¼ 3) oocytes (mean� SEM;P> .05 by t-test). The effect of patch cramming was clearlybiphasic in both the FCCP-treated and control cells. The rapidphase as a fraction of total inhibition was variable but there wasno significant difference between FCCP-treated (0.40� 0.04;n¼ 5) and control (0.42� 0.12; n¼ 3) oocytes (mean� SEM;P> .05 by t-test).
Reversibility of inhibition by FCCP in intact cells
Recovery of Kir2.2 current from inhibition by FCCP in intactoocytes is shown in the main part of Figure 4. Xenopus oocytesexpressing Kir2.2 were incubated with FCCP or in controlconditions. FCCP was then removed at the beginning of thetime course shown in Figure 4. Kir2.2 current was recorded ingroups of five or six oocytes at the time points indicated. Thecurrent was relatively stable in control oocytes (squares) andincreased towards the control level in FCCP-treated oocytes(triangles).
Apyrase does not mimic FCCP
The experiments shown in Figure 5 were carried out toinvestigate the effect of ATP depletion on Kir2 channels.Xenopus oocytes were treated with FCCP or injected withapyrase, an enzyme that hydrolyses ATP (Molnar and Lorand,1961). The relative ATP content of the oocytes was thenassessed using a luciferase-based assay (Gribble et al., 1997).Part A shows that FCCP decreased the ATP content to anextent (column 2 vs. column 1), but less than the higherconcentrations of apyrase (columns 4 and 5). The effect ofapyrase injection on Kir2 channels is shown in part B. The datashow that Kir2.1 (column 1 vs. column 2) was inhibited by
Fig. 4. Inhibition of Kir2.2 by FCCP is reversible in intact cells.Oocytes expressing Kir2.2 were incubated with 10mM FCCP in ND-96(triangles) or in ND-96 alone (squares) for 105 min, then transferredinto ND-96 at time U 0. Symbols are mean W SEM inward currentrecorded at �50 mV membrane potential in 90 mM extracellularKR by two-electrode voltage clamp (n U 5–6). Current-voltagerelationships were obtained as in the inset to monitor leak current.Data shown here are from one experiment in one batch of oocytes.Similar results were obtained in three other experiments. Each datapoint represents one oocyte, that is, each oocyte was used for onerecording.
JOURNAL OF CELLULAR PHYSIOLOGY
apyrase. The small inhibitory effect on Kir2.2 (column 3 vs.column 4) was not statistically significant (P> .05).Figure 5C shows the higher sensitivity of Kir2.2 to FCCPcompared to Kir2.1.
Inhibition of Kir2.2 by FCCP is not unique toXenopus oocytes
Figure 6 shows the effect of FCCP on Kir2.2 current expressedin HEK cells. Transfection with Kir2.2 cDNA resulted in theexpression of inwardly rectifying currents as shown in part A.Part B shows that current density decreased over time aftertransfer of cells from culture medium into recording solution.Current density was lower in FCCP-treated versus controlcells at all time points up to 324 min. The time-dependentdecrease was compensated for by pairing data points at similarincubation times as indicated by the dotted lines inFigure 6B. Statistical analysis of paired data showed that currentdensity was significantly lower in FCCP versus control (part C).
DiscussionMechanism of recovery from FCCP
We previously reported an increase in Kir2 channel activityin excised, inside-out membrane patches from oocytespre-treated with FCCP (Collins et al., 2005). In the presentstudy we took a closer look at this apparent recovery fromFCCP because the conditions required for recovery couldprovide important clues to the mechanism. The data in Figures 1and 2 demonstrate that the run-up of Kir2.2 in inside-outpatches occurred to the same relative degree regardless ofprior treatment of the oocyte with FCCP. The run-up ininside-out patches is independent of the effect of FCCP. Theinhibitory effect of FCCP could not be reversed by excision andperfusion of the membrane, and is inconsistent with FCCPcausing the accumulation of an inhibitory factor that can bewashed away. An alternative mechanism involves the depletionof an intracellular factor that is required for Kir2.2 channelactivity and that cannot be regenerated in an isolated patch ofmembrane, or the action of an inhibitory enzyme that maybe reversible, but not by a membrane-associated mechanism.The data in Figures 1, 2, and 4 demonstrate that reversal of theinhibition of Kir2.2 by FCCP requires at least one intracellularfactor.
Evidence for an endogenous inhibitor
The run-up of Kir2.2 current after excision and perfusion ofinside-out membrane patches (Fig. 1) suggested the wash-out ofan inhibitory factor. Further evidence for this was provided bythe re-inhibition of the current by insertion of the patch pipettetip into the oocyte and subsequent recovery after removal andreperfusion (Fig. 3). The inhibitory effect of FCCP does notappear to involve this putative inhibitor.
Inhibition of Kir2.2 by FCCP cannot be accounted forby ATP depletion
ATP is one intracellular factor that could become depletedas a result of mitochondrial impairment. We were previouslyunable to attenuate the inhibitory effect of FCCP on Kir2.2and Kir2.3 by pre-injecting the oocytes with ATP (Collins et al.,2005). However, in the absence of a positive control thisnegative result did not rule out a role for ATP depletion as themechanism of inhibition by FCCP, so we investigated thispossibility further by using apyrase to directly deplete ATP.Figure 5A shows that ATP was depleted by injection of oocyteswith apyrase or incubation with FCCP. Injection of 0.01 Uapyrase per oocyte was more effective at depleting ATP than an

Fig. 5. Inhibition by FCCP does not require ATP depletion. A: Batches of five oocytes were incubated in control conditions (column 1), or in thepresence of 10 mM FCCP for 110 min, or after injection with 0.001, 0.01 or 0.1 U reconstituted apyrase (high ATPase/ADPase ratio; Sigma) andincubation for 1 h. They were then homogenized for determination of relative ATP content by luciferase assay. Relative light units (RLU) data arebackground-subtracted and normalized to the mean control data. MMMP < .001 versus control, one-way ANOVA, n U 4–12 from two experiments.B,C: KR currents were recorded by two-electrode voltage clamp in Xenopus oocytes expressing Kir2 channels 90–220 min after injection with0.1 units apyrase (B, filled bars), after 100 min incubation with 10 mM FCCP (C, filled bars) or in control conditions (open bars). Current-voltagerelationships were obtained as in Figure 4 (inset) to monitor leak current. Current recorded at�50 mV is shown. MP < .05, MMP < .01 versus control,unpaired t-test, n U 3–6. Scale bar in (C) 10 mA (Kir2.1); 2.4 mA (Kir2.2).
12 C O L L I N S A N D L A R S O N
FCCP treatment that inhibited Kir2.2 by �75% but not Kir2.1(Fig. 5C). Figure 5B shows the effect on Kir2 channels of anapyrase treatment that depleted ATP to an undetectable level(Fig. 5A, column 5). If ATP depletion were the mechanism ofaction of FCCP then this apyrase treatment would be expectedto inhibit Kir2.2 by more than 75%, which was not the case(Fig. 5B, columns 3 and 4). Furthermore, these data cannot beexplained in terms of a submembranous compartment of ATPthat was depleted by external treatment with FCCP andpartially resistant to injection of apyrase into the oocyteinterior, because apyrase did not mimic the selectivity ofFCCP. Kir2.2 would be expected to be more sensitivethan Kir2.1 to apyrase if ATP depletion were the mechanism ofaction of FCCP on Kir2 channels. Figure 5B shows that this wasnot the case.
In a previous study we found that the sensitivity of Kir2channels to FCCP was influenced by mutations that are knownto alter PIP2 sensitivity (Collins et al., 2005). This raised thepossibility that FCCP inhibits Kir2 channels by PIP2 depletion.Such a mechanism would predict that the inhibition may bereversible in intact cells capable of regenerating PIP2, but not inisolated membrane patches. We show here that the increasein Kir2.2 current in inside-out patches was independent of priorFCCP treatment of the cell, and was apparently the result ofwashing out an endogenous inhibitor. Inhibition by FCCP istherefore irreversible in inside-out patches, although recoverycan occur in intact cells. The inability of ATP depletion to mimic
Fig. 6. Inhibition by FCCP of Kir2.2 expressed in HEK cells. A: Currents recDNA. Steps of 190 msec duration were applied from a holding potential o10 mV increments. The dashed line represents zero current. Command p�8.5 mV. B: Current density recorded at�100 mV in the presence (open ctime since transfer of cells from culture medium into recording solution. EafromtheexperimentshowninpartBandasimilarexperiment.Datafromcoas indicated by the dotted lines in part B. MP < .05 versus control, paired t-
JOURNAL OF CELLULAR PHYSIOLOGY
the effect of FCCP on Kir2 channels is difficult to reconcile witha PIP2 depletion mechanism, however, because it is expectedthat a reduction in ATP with continuing lipid phosphataseactivity would lead to a reduction in PIP2 production and thus tocurrent inhibition.
Possible significance of differential sensitivity tomitochondrial agents
We have reported here and previously (Collins and Larson,2002; Collins et al., 2005) that Kir2.2 is more sensitivethan Kir2.1 to inhibition by mitochondrial agents. Kir2.1 isgenerally considered to be the dominant component of thecardiac inward rectifier potassium current (IK1), at least inmouse and rat (Nakamura et al., 1998; Zaritsky et al., 2001)but several results suggest that Kir2.2 makes a significantcontribution, despite being less abundant than Kir2.1 at themRNA level (Rose et al., 2005; Domenighetti et al., 2007).First, Kir2.2 gene knockout reduced IK1 density by 50% inmouse heart (Zaritsky et al., 2001). Second, dominant negativeconstructs of Kir2.1 and Kir2.2 were equally able to reduce IK1
density by 70% in rabbit cardiomyocytes (Zobel et al., 2003).Third, analysis of the block by extracellular Ba2þ of inwardrectifier channels indicated a significant contribution of Kir2.2to IK1 in guinea-pig (Liu et al., 2001) and human (Schram et al.,2003). Therefore it may be reasonable to speculate that theproarrhythmic decrease in IK1 in myocardial remodeling leading
corded by whole-cell voltage-clamp from a cell transfected with Kir2.2f�70 mV to test potentials between�140 mV and R40 mV inclusive inotentials as reported do not include a calculated junction potential ofircles) or absence (filled circles) of 10mM FCCP. The abscissa indicatesch data point indicates an individual cell. C: Pooled data (mean R SEM)ntrolandFCCP-treatedcellswerepairedaccordingtoincubationtimetest, n U 6.

I N H I B I T I O N O F K i r 2 P O T A S S I U M C H A N N E L S 13
to heart failure reflects a selective down-regulation of Kir2.2resulting from mitochondrial dysfunction (Marin-Garcia et al.,1995; Sharov et al., 1998; Jarreta et al., 2000; Murakami et al.,2002).
Acknowledgments
We thank Dr. Lily Jan, Dr. Yoshihisa Kurachi and Dr. MuneshigeKaibara for cDNA clones of IRK1 (Kir2.1), MB-IRK2 (Kir2.2)and hKir2.2/pCXN2 respectively.
Literature Cited
Atlante A, Seccia TM, Pierro P, Vulpis V, Marra E, Pirrelli A, Passarella S. 1998. ATP synthesisand export in heart left ventricle mitochondria from spontaneously hypertensive rat. Int JMol Med 1:709–716.
Benderdour M, Charron G, Comte B, Ayoub R, Beaudry D, Foisy S, Deblois D, Des Rosiers C.2004. Decreased cardiac mitochondrial NADPþ-isocitrate dehydrogenase activity andexpression: A marker of oxidative stress in hypertrophy development. Am J Physiol HeartCirc Physiol 287:H2122–H2131.
Carreras MC, Poderoso JJ. 2007. Mitochondrial nitric oxide in the signaling of cell integratedresponses. Am J Physiol Cell Physiol 292:C1569–C1580.
Collins A, Larson MK. 2002. Differential sensitivity of inward rectifier Kþ channels tometabolic inhibitors. J Biol Chem 277:35815–35818.
Collins A, Wang H, Larson MK. 2005. Differential sensitivity of kir2 inward-rectifierpotassium channels to a mitochondrial uncoupler: Identification of a regulatory site. MolPharmacol 67:1214–1220.
Das AM, Harris DA. 1990. Defects in regulation of mitochondrial ATP synthase incardiomyocytes from spontaneously hypertensive rats. Am J Physiol 259:H1264–H1269.
Domenighetti AA, Boixel C, Cefai D, Abriel H, Pedrazzini T. 2007. Chronic angiotensin IIstimulation in the heart produces an acquired long QT syndrome associated with IK1potassium current downregulation. J Mol Cell Cardiol 42:63–70.
Fukunaga Y, Itoh H, Hosoda K, Doi K, Matsuda J, Son C, Yamashita J, Chun TH, Tanaka T,Inoue M, Masatsugu K, Saito T, Sawada N, Nakao K. 2000. Altered gene expression ofuncoupling protein-2 and -3 in stroke-prone spontaneously hypertensive rats. J Hypertens18:1233–1238.
Gaussin V, Tomlinson JE, Depre C, Engelhardt S, Antos CL, Takagi G, Hein L, Topper JN,Liggett SB, Olson EN, Lohse MJ, Vatner SF, Vatner DE. 2003. Common genomic responsein different mouse models of beta-adrenergic-induced cardiomyopathy. Circulation108:2926–2933.
Gribble FM, Ashfield R, Ammala C, Ashcroft FM. 1997. Properties of cloned ATP-sensitiveKþ currents expressed in Xenopus oocytes. J Physiol 498:87–98.
Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. 2006.Mitochondrial calcium signalling and cell death: Approaches for assessing the role ofmitochondrial Ca2þ uptake in apoptosis. Cell Calcium 40:553–560.
Hilgemann D. 1995. The giant membrane patch. In: Sakmann B, Neher E, editors.Single-channel recording, 2nd edition. New York: Plenum Press. pp 307–327.
Jarreta D, Orus J, Barrientos A, Miro O, Roig E, Heras M, Moraes CT, Cardellach F,Casademont J. 2000. Mitochondrial function in heart muscle from patients with idiopathicdilated cardiomyopathy. Cardiovasc Res 45:860–865.
Kaibara M, Ishihara K, Doi Y, Hayashi H, Ehara T, Taniyama K. 2002. Identification of humanKir2.2 (KCNJ12) gene encoding functional inward rectifier potassium channel in bothmammalian cells and Xenopus oocytes. FEBS Lett 531:250–254.
Kramer RH. 1990. Patch cramming: Monitoring intracellular messengers in intact cells withmembrane patches containing detector ion channels. Neuron 4:335–341.
JOURNAL OF CELLULAR PHYSIOLOGY
Kubo Y, Baldwin TJ, Jan YN, Jan LY. 1993. Primary structure and functional expression of amouse inward rectifier potassium channel [see comments]. Nature 362:127–133.
Liman ER, Tytgat J, Hess P. 1992. Subunit stoichiometry of a mammalian Kþ channeldetermined by construction of multimeric cDNAs. Neuron 9:861–871.
Lin L, Sharma VK, Sheu SS. 2007. Mechanisms of reduced mitochondrial Ca(2þ) accumulationin failing hamster heart. Pflugers Arch 454:395–402.
Liu GX, Derst C, Schlichthorl G, Heinen S, Seebohm G, Bruggemann A, Kummer W, VehRW, Daut J, Preisig-Muller R. 2001. Comparisonof clonedKir2 channels with native inwardrectifier Kþ channels from guinea-pig cardiomyocytes. J Physiol 532:115–126.
Marin-Garcia J, Goldenthal MJ, Pierpont ME, Ananthakrishnan R. 1995. Impairedmitochondrial function in idiopathic dilated cardiomyopathy: Biochemical and molecularanalysis. J Card Fail 1:285–291.
Miake J, Marban E, Nuss HB. 2003. Functional role of inward rectifier current in heart probedby Kir2.1 overexpression and dominant-negative suppression. J Clin Invest 111:1529–1536.
Molnar J, Lorand L. 1961. Studies on apyrases. Arch Biochem Biophys 93:353–363.Murakami K, Mizushige K, Noma T, Tsuji T, Kimura S, Kohno M. 2002. Perindopril effect on
uncoupling protein and energy metabolism in failing rat hearts. Hypertension 40:251–255.Nakamura TY, Artman M, Rudy B, Coetzee WA. 1998. Inhibition of rat ventricular IK1 with
antisense oligonucleotides targeted to Kir2.1 mRNA. Am J Physiol 274:H892–H900.Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, Ohta Y, Sami M,
Tachibana T, Ishikawa H, Kurosawa H, Kahn RC, Otsu K, Shirasawa T. 2006. Oxidativestress causes heart failure with impaired mitochondrial respiration. J Biol Chem281:33789–33801.
Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. 2001. Arrhythmogenesis and contractiledysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassiumcurrent, and residual beta-adrenergic responsiveness. Circ Res 88:1159–1167.
Prabhu S, Jainu M, Sabitha KE, Shyamala Devi CS. 2006. Effect of mangiferin on mitochondrialenergy production in experimentally induced myocardial infarcted rats. Vascul Pharmacol44:519–525.
Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, O’Rourke B, Kass DA,Marban E, Tomaselli GF. 2005. Molecular correlates of altered expression of potassiumcurrents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol 288:H2077–H2087.
Sayen MR, Gustafsson AB, Sussman MA, Molkentin JD, Gottlieb RA. 2003. Calcineurintransgenic mice have mitochondrial dysfunction and elevated superoxide production. Am JPhysiol Cell Physiol 284:C562–C570.
Schram G, Pourrier M, Wang Z, White M, Nattel S. 2003. Barium block of Kir2 and humancardiac inward rectifier currents: Evidence for subunit-heteromeric contribution to nativecurrents. Cardiovasc Res 59:328–338.
Seccia TM, Atlante A, Vulpis V, Marra E, Passarella S, Pirrelli A. 1998. Mitochondrial energymetabolism in the left ventricular tissue of spontaneously hypertensive rats: Abnormalitiesin both adeninenucleotide and phosphate translocators and enzyme adenylate-kinase andcreatine-phosphokinase activities. Clin Exp Hypertens 20:345–358.
Sharov VG, Goussev A, Lesch M, Goldstein S, Sabbah HN. 1998. Abnormal mitochondrialfunction in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol 30:1757–1762.
Takahashi N, Morishige K, Jahangir A, Yamada M, Findlay I, Koyama H, Kurachi Y. 1994.Molecular cloning and functional expression of cDNA encoding a second class of inwardrectifier potassium channels in the mouse brain. J Biol Chem 269:23274–23279.
Tokoro T, Ito H, Maenishi O, Suzuki T. 1995. Mitochondrial abnormalities in hypertrophiedmyocardium of stroke-prone spontaneously hypertensive rats. Clin Exp Pharmacol PhysiolSuppl 22:S268–S269.
Zaritsky JJ, Redell JB, Tempel BL, Schwarz TL. 2001. The consequences of disrupting cardiacinwardly rectifying K(þ) current (I(K1)) as revealed by the targeted deletion of the murineKir2.1 and Kir2.2 genes. J Physiol 533:697–710.
Zhang DX, Gutterman DD. 2007. Mitochondrial reactive oxygen species-mediated signalingin endothelial cells. Am J Physiol Heart Circ Physiol 292:H2023–H2031.
Zobel C, Cho HC, Nguyen TT, Pekhletski R, Diaz RJ, Wilson GJ, Backx PH. 2003. Moleculardissection of the inward rectifier potassium current (IK1) in rabbit cardiomyocytes:Evidence for heteromeric co-assembly of Kir2.1 and Kir2.2. J Physiol 550:365–372.







![Review Unveiling the Bmp13 Enigma: Redundant Morphogen or … · 2008-09-12 · induction in Xenopus oocytes [42,43] than the two homodimeric molecules combined. BMP13 formed heterodimers](https://static.fdocuments.in/doc/165x107/5f949e2357b1e95cc02853e4/review-unveiling-the-bmp13-enigma-redundant-morphogen-or-2008-09-12-induction.jpg)










