
Kinetic Resolution of Secondary Organocatalytic...
56
ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2014 Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1191 Organocatalytic Acylation for the Kinetic Resolution of Secondary Aryl Alcohols Synthetic Applications and Mechanistic Studies LAURA MESAS SÁNCHEZ ISSN 1651-6214 ISBN 978-91-554-9075-1 urn:nbn:se:uu:diva-233734
Transcript of Kinetic Resolution of Secondary Organocatalytic...

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2014
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 1191
Organocatalytic Acylation for theKinetic Resolution of SecondaryAryl Alcohols
Synthetic Applications and Mechanistic Studies
LAURA MESAS SÁNCHEZ
ISSN 1651-6214ISBN 978-91-554-9075-1urn:nbn:se:uu:diva-233734

Dissertation presented at Uppsala University to be publicly examined in A1:111a, BMC,Husargatan 3, Uppsala, Friday, 28 November 2014 at 09:00 for the degree of Doctorof Philosophy. The examination will be conducted in English. Faculty examiner: PavelKočovský.
AbstractMesas Sánchez, L. 2014. Organocatalytic Acylation for the Kinetic Resolution of SecondaryAryl Alcohols. Synthetic Applications and Mechanistic Studies. Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty of Science and Technology 1191. 54 pp.Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-9075-1.
The research described in this thesis focuses on the catalytic acylative kinetic resolution (KR)of aromatic secondary alcohols, using a planar-chiral 4-(dimethylamino)pyridine (DMAP)organocatalyst.
In the first part of this thesis, the substrate scope of the above mentioned process wasexpanded to aromatic secondary alcohols that contain an extra functional group in the alkylmoiety, such as 1,2-azido alcohols, 2-hydroxy-2-aryl-ethylphosphonates and 2-hydroxy-2-arylesters. Thus, the preparation of highly functionalized compounds in their enantiomericallypure form with excellent enantiomeric excess (up to 99% ee) was achieved. Furthermore, thesynthetic applicability of this methodology was illustrated through the synthesis of two highvalue compounds, (R)-Pronethalol and (S)-3-hydroxy-N-methyl-3-phenylpropanamide, whichis an immediate precursor of bioactive molecules such as (S)-Fluoxetine.
The second part of this thesis deals with the mechanistic study of the acylative KR catalyzedby the planar-chiral DMAP derivative. Reaction Progress Kinetic Analysis methodology wasused in the investigation of the reaction mechanism, probing that no notable product inhibition ordecomposition of the catalyst occurs in the studied system. The reaction rate showed fractionalorder dependence on the concentration of both reactants. Furthermore, NMR spectroscopywas utilized to study the equilibrium between the different catalyst states, which explains themeasured kinetics of the reaction.
Keywords: kinetic resolution, organocatalysis, secondary alcohols, reaction progress kineticanalysis
Laura Mesas Sánchez, Department of Chemistry - BMC, Synthetical Organic Chemistry, Box576, Uppsala University, SE-75123 Uppsala, Sweden.
© Laura Mesas Sánchez 2014
ISSN 1651-6214ISBN 978-91-554-9075-1urn:nbn:se:uu:diva-233734 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-233734)

A toda mi familia
“La vida es simplemente una cuestión de química”
James Watson

Cover: 3D Plot of the in situ IR spectra corresponding to the acetylation of (R)-1-phenylethanol catalyzed by a planar-chiral DMAP catalyst.

List of Publications
This thesis is based on the following publications, which are referred to in the text by their Roman numerals I-IV.
I Mesas-Sánchez, L., Díaz-Álvarez, A. E., Dinér, P. Non-enzymatic kinetic resolution of 1,2-azidoalcohols using a planar-chiral DMAP derivative catalyst. Tetrahedron 2013, 69, 753−757.
II Mesas-Sánchez, L., Díaz-Álvarez, A. E., Koukal, P., Dinér, P. Kinet-ic resolution of 2-hydroxy-2-aryl-ethylphosphonates by a non-enzymatic acylation catalyst. Tetrahedron 2014, 70, 3807−3811.
III Díaz-Álvarez, A. E.†, Mesas-Sánchez, L.†, Dinér, P. Access to opti-cally pure β-hydroxy esters via non-enzymatic kinetic resolution by a planar-chiral DMAP catalyst. Molecules 2014, 19, 14273-14291.
IV Mesas-Sánchez, L., Dinér, P. A mechanistic investigation of the ki-netic resolution of secondary aromatic alcohols using a ferrocene-based planar chiral DMAP catalyst. Manuscript 2014.
Reprints were made with permission from the respective publishers.
Publications not included in this thesis:
V Díaz-Álvarez, A. E., Mesas-Sánchez, L., Dinér, P. Non-enzymatic dynamic kinetic resolution of secondary aryl alcohols: planar chiral ferrocene and ruthenium catalyst in cooperation. Angew. Chem. Int. Ed. 2013, 52, 502−504.
† Authors contributed equally to the publication and are presented in alpha-betical order.

The author wishes to clarify her contribution to the papers I-IV in the the-sis.
I Prepared most of the racemic substrates; planned and performed the kinetic resolutions and characterized all the final products. Partially involved in the preparation of the manuscript; dis-cussed the results and commented on the manuscript.
II Formulated the research problem. Partially involved in the syn-thesis of racemic substrates; designed and carried out all the ki-netic resolutions and characterized all the final products. Wrote the entire manuscript.
III Formulated the research problem. Partially involved in the syn-thesis of racemic substrates; performed the complete initial sub-strate screening for the determination of the selectivity factor; performed the experiments for the assignation of the optical configuration; synthesized the precursor of (S)-Fluoxetine. Wrote virtually the complete manuscript.
IV Designed, performed and analyzed the experiments for the ki-netic studies and the 1H-NMR spectroscopy studies. Partially involved in the preparation of the manuscript; discussed the re-sults and commented on the manuscript.

Contents
1. Introduction ................................................................................................. 91.1. Chirality ............................................................................................. 101.2. Preparation of optically pure compounds .......................................... 141.3. Kinetic resolution .............................................................................. 151.4. Organocatalysts for acylative kinetic resolutions of chiral secondary alcohols .................................................................................... 18
1.4.1. 4-Aminopyridine derivatives ..................................................... 191.5. The aim of this thesis ......................................................................... 22
2. Kinetic resolution of secondary aryl alcohols catalyzed by a planar- chiral DMAP derivative (Papers I-III) .......................................................... 23
2.1. The importance of chiral functionalized alcohols ............................. 232.2. Substrate scope .................................................................................. 24
2.2.1. Kinetic resolution of β-azido alcohols ....................................... 242.2.2. Kinetic resolution of β-hydroxy phosphonates .......................... 262.2.3. Kinetic resolution of β-hydroxy esters ...................................... 28
2.3. Synthetic applications ........................................................................ 302.4. Conclusions ....................................................................................... 31
2.4.1. Structure-selectivity trends ........................................................ 32
3. Mechanistic studies of the acetylation by a planar-chiral DMAP catalyst (Paper IV) ......................................................................................... 33
3.1. Introduction ....................................................................................... 333.1.1. Reaction progress kinetic analysis ............................................. 34
3.2. Results and discussion ....................................................................... 373.3. Conclusions ....................................................................................... 41
4. Concluding remarks and outlook .............................................................. 42
Populärvetenskaplig sammanfattning ........................................................... 43
Resumen divulgativo en español ................................................................... 46
Acknowledgements ....................................................................................... 49
References ..................................................................................................... 51

Abbreviations
BOX Bis(oxazoline) BTM Benzotetramisole CALB Candida Antarctica lipase B Conv. Conversion DCM Dichloromethane DIPT Diisopropyl tartrate DKR Dynamic kinetic resolution DMAP 4-(Dimethylamino)pyridine DNA Deoxyribonucleic acid DOPA 3,4-Dihydroxyphenylalanine ee Enantiomeric excess EMA European Medicines Agency Equiv Equivalents FDA Food and Drug Administration of United States of
America GABA Gamma-aminobutyric acid HBTM Homobenzotetramisole IR Infrared KR Kinetic resolution LDA Lithium diisopropylamide NMR Nuclear magnetic resonance rac Racemic RNA Ribonucleic acid RPKA Reaction progress kinetic analysis s Selectivity sec Secondary TBHP tert-Butyl hydroperoxide THF Tetrahydrofuran

9
1. Introduction
This thesis deals with the synthesis of enantiopure functionalized aromatic secondary alcohols, via kinetic resolution promoted by a planar-chiral 4-(dimethylamino)pyridine (DMAP) organocatalyst. Furthermore, the mecha-nism of this reaction has been studied in order to get a better understanding of this process.
This thesis is structured as follows: chapter 1 starts with a brief introduc-tion of the concept of chirality and its importance in life. In this chapter dif-ferent methodologies for the synthesis of optically pure chiral compounds are also introduced, emphasizing on the description of kinetic resolutions. In this context, the contributions of different families of organocatalysts to this field are presented through representative examples, including a more de-tailed description of the planar-chiral DMAP derivative used as catalyst in the research performed in this thesis.
Chapter 2 summarizes the results presented in papers I to III. The synthe-sis of enantiomerically pure secondary aryl alcohols with an additional het-eroatom-containing functional group in the alkyl moiety represents an addi-tional contribution to the substrate scope of the planar-chiral DMAP catalyst, which was previously used in the kinetic resolution of secondary aryl alkyl alcohols as model substrates.
Finally, chapter 3 discusses the mechanism of the enantioselective acyla-tion of aromatic secondary alcohols using the planar-chiral DMAP catalyst, as presented in manuscript IV. The manuscript provides both computational and experimental data, but here, the focus will be on the kinetic analysis experiments performed following the Reaction Progress Kinetic Analysis protocol and the 1H-NMR spectroscopy study of the equilibrium between two catalyst states, which represent this author’s contribution to the manu-script.

10
1.1. Chirality Chirality or “handedness” is a property that derives from the asymmetric nature of a given object, and it is a key concept in several branches of sci-ence. In chemistry, a molecule is chiral when it can exist in two distinct structural configurations, known as enantiomers, which are non-superimposable mirror images of each other.1 For instance, the amino acid alanine has two enantiomers that are mirror images of each other, and do not overlap when placed on top of one another, regardless of the rotational posi-tion of the molecules (Figure 1). An illustrative example of chiral objects is a pair of hands; they are mirror images of each other and cannot be superim-posed. Basically, two pure enantiomers have identical physical and chemical properties, unless they are in a chiral environment, except for optical rotatory dispersion, i.e. they rotate plane-polarized light in equal but opposite degree.
Figure 1. Representation of chiral objects: enantiomers of the amino acid alanine and a pair of hands.
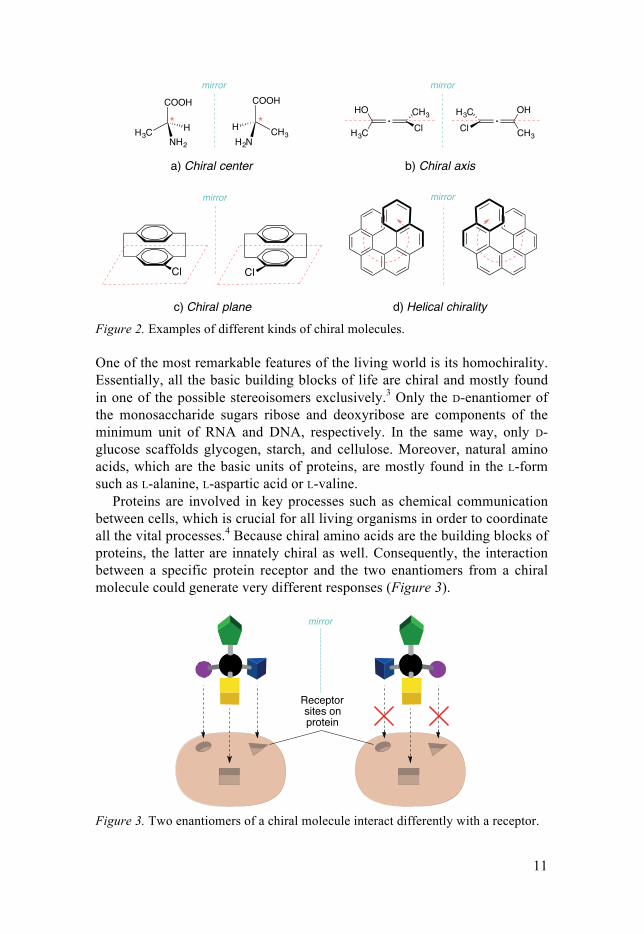
There are different types of arrangements of the atoms in a molecule from which chirality stems (Figure 2).2 The most common one is the chiral center generated by a carbon atom binding four different substituents, as in the case of alanine. Axial chirality is presented in molecules such as biphenyl, binaphthyls or certain allenes (Figure 2b). Another kind of chirality is exhib-ited in molecules possessing a chiral plane, i.e. a plane located so that placement of a substituent group in that plane destroys a perpendicular plane of symmetry. The most common molecules possessing a chiral plane are cyclophanes, such as the biphenylene structure shown in Figure 2c, and polysubstituted metallocenes. Finally, helical chirality is presented in hexa-helicene, for example, in which the chirality results from the handedness of the helicity itself (Figure 2d).
L-alanine(S)-enantiomer
D-alanine(R)-enantiomer
HOOC
H
H2NCH3H3C
NH2
H
COOH
mirror
cannot be superimposed
H3CNH2
H
COOH
HNH2
CH3
COOH
11
Figure 2. Examples of different kinds of chiral molecules.
One of the most remarkable features of the living world is its homochirality. Essentially, all the basic building blocks of life are chiral and mostly found in one of the possible stereoisomers exclusively.3 Only the D-enantiomer of the monosaccharide sugars ribose and deoxyribose are components of the minimum unit of RNA and DNA, respectively. In the same way, only D-glucose scaffolds glycogen, starch, and cellulose. Moreover, natural amino acids, which are the basic units of proteins, are mostly found in the L-form such as L-alanine, L-aspartic acid or L-valine.
Proteins are involved in key processes such as chemical communication between cells, which is crucial for all living organisms in order to coordinate all the vital processes.4 Because chiral amino acids are the building blocks of proteins, the latter are innately chiral as well. Consequently, the interaction between a specific protein receptor and the two enantiomers from a chiral molecule could generate very different responses (Figure 3).
Figure 3. Two enantiomers of a chiral molecule interact differently with a receptor.
a) Chiral center
mirror
H3CNH2
H
COOH COOH
H
H2NCH3
b) Chiral axis
mirror
H3C
HO
Cl
CH3 H3C
Cl
OH
CH3
c) Chiral plane
mirror
d) Helical chirality
mirror
* *
Cl Cl
Receptorsites onprotein
mirror

12
For example, it is well known that chiral discrimination is an important prin-ciple of odor perception.5 Inside of the nasal cavity, there are numerous ol-factory protein receptors that are highly specialized in differentiating mole-cules with different structure and/or configuration.6 The scent molecules interact with some of these receptors, which send signals to the brain where they are perceived as a particular smell.7 For instance, the (R)-enantiomer of the carvone is perceived as spearmint-smelling while the (S)-enantiomer smells of caraway (Figure 4).8
Figure 4. Enantiomers of carvone.
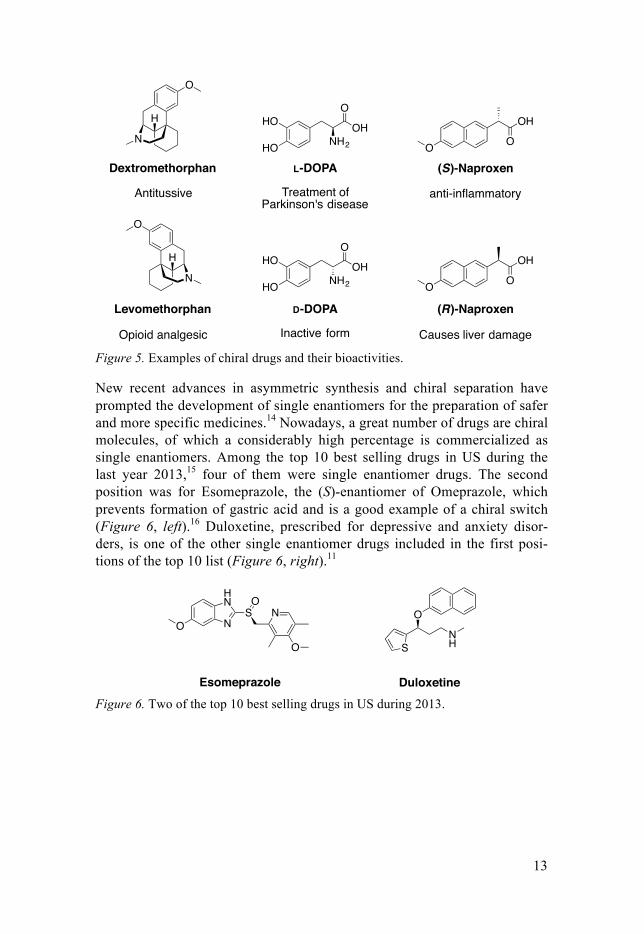
Likewise, chiral drug molecules could interact differently with protein recep-tors and as a result, two enantiomers could exhibit disparate pharmacologic or toxicological effects (Figure 5). For instance, Dextromethorphan is a cough suppressant and it is one of the active compounds in many cold and cough medicines that can be purchased without prescription, while its enan-tiomer, Levomethorphan, is an opioid analgesic.9 In other cases, one member of a pair of enantiomers is pharmacologically active and the other one is less active or even inactive. This is the case of L-DOPA, a precursor to neuro-transmitters such as dopamine used for treat Parkinson’s disease,10 and D-DOPA, which is inactive. Although inactivity of one of the enantiomers might be considered trivial, it is important when determining the dosage required for a given treatment. Furthermore, there are also examples in which toxicity has been linked to one of the enantiomers. For example, (R)-Naproxen causes liver poisoning and only the (S)-Naproxen has the benefi-cial anti-inflammatory effects.11
The regulatory authorities, such as FDA (Food and Drug Administration of US) and EMA (European Medicines Agency) provide specific guidelines on the development of chiral drugs.12 For some drugs that have been already commercialized as racemates, the authorities have allowed the single enanti-omer drugs to be patented and marketed under another name. This strategy is known as chiral switches of established racemates and mainly, try to offer enhanced therapeutic benefits and further profitability as a “line extension” of a major racemic drug with patents that are expiring.13
(R)-carvone
spearmint
(S)-carvone
caraway
O
H H
O
mirror
13
Figure 5. Examples of chiral drugs and their bioactivities.
New recent advances in asymmetric synthesis and chiral separation have prompted the development of single enantiomers for the preparation of safer and more specific medicines.14 Nowadays, a great number of drugs are chiral molecules, of which a considerably high percentage is commercialized as single enantiomers. Among the top 10 best selling drugs in US during the last year 2013,15 four of them were single enantiomer drugs. The second position was for Esomeprazole, the (S)-enantiomer of Omeprazole, which prevents formation of gastric acid and is a good example of a chiral switch (Figure 6, left).16 Duloxetine, prescribed for depressive and anxiety disor-ders, is one of the other single enantiomer drugs included in the first posi-tions of the top 10 list (Figure 6, right).11
Figure 6. Two of the top 10 best selling drugs in US during 2013.
L-DOPA
Treatment of Parkinson's disease
D-DOPA
Inactive form
HO
HO
OH
O
NH2
HO
HO
OH
O
NH2
(S)-Naproxen
anti-inflammatory
(R)-Naproxen
Causes liver damage
OH
O
OH
O
O
O
H
N
O
O
N
H
Dextromethorphan
Antitussive
Levomethorphan
Opioid analgesic
O
Esomeprazole Duloxetine
N
HN
SO
N
O S
O
NH

14
1.2. Preparation of optically pure compounds The methods employed for the preparation of enantiomerically pure com-pounds can be classified into three categories based on the type of starting material used (Figure 7).17
Figure 7. Classification of methods for the synthesis of optically pure compounds.
The Chiral pool is a collection of natural products that are readily available and cheap enough to be used as starting material for organic synthesis, such as alkaloids, monosaccharides and α-amino acids.18 The chiral pool strategy is based on the partial or complete incorporation of these compounds into the target molecule in a stereospecific manner. This way, the chiral center could be preserved in the final compound.
An alternative is Stereoselective synthesis that starts from a prochiral compound, and has been traditionally known as asymmetric synthesis. In this approach, the chirality is transferred from the chiral auxiliary or the chi-ral catalyst to the prochiral substrate to create new stereogenic centers.17
The third approach, Resolution, consists on the separation of the enantio-mers of a racemic mixture.19 Despite outstanding progress in asymmetric synthesis, the resolution of racemates is still one of the most common pro-duction method to prepare single enantiomers in industrial synthesis.14b The resolution of enantiomers, in turn, can be divided into three classes: classical resolution, chiral chromatographic methods and kinetic resolution. The latter will be further explained in the section 1.3, since it is the methodology used in this thesis.
The decision of which methodology to use for the preparation of a specif-ic compound must be made on a case-by-case basis.20 Several factors should be taken into consideration, including the efficiency of the technology for a particular substrate, access to the reagents and their cost, the substrate sensi-tivity to the reaction conditions, potential hazards, and even the scientific background of the synthetic chemist involved. Every contribution to broaden the collection of methodologies is beneficial to allow the best solution for a particular product.
O p t i c a l l y p u r e c o m p o u n d s
Chiral compound(from the Chiral pool)
Prochiral compound Racemate
Resolution
- Classical resolution- Chromatography- Kinetic resolution
Stereospecific SynthesisStereoselective Synthesis
- Chiral auxiliaries- Chiral catalysts

15
1.3. Kinetic resolution A kinetic resolution (KR) is defined as the resolution of a racemate by virtue of different rates of reaction of the enantiomers with a chiral agent.1 If the KR is efficient, one of the enantiomers of the racemate remains unreacted while the other is transformed to the product. The result is a mixture of the starting material (SM) and product (P), which then can be isolated with high enantiomeric purity (Figure 8).
Figure 8. General scheme for a catalytic KR. Main recovered starting material and product are marked in blue.
The enantiomeric purity of the chiral compounds is usually expressed as the enantiomeric excess (ee), which describes the excess of one enantiomer over the other (equation (1)).
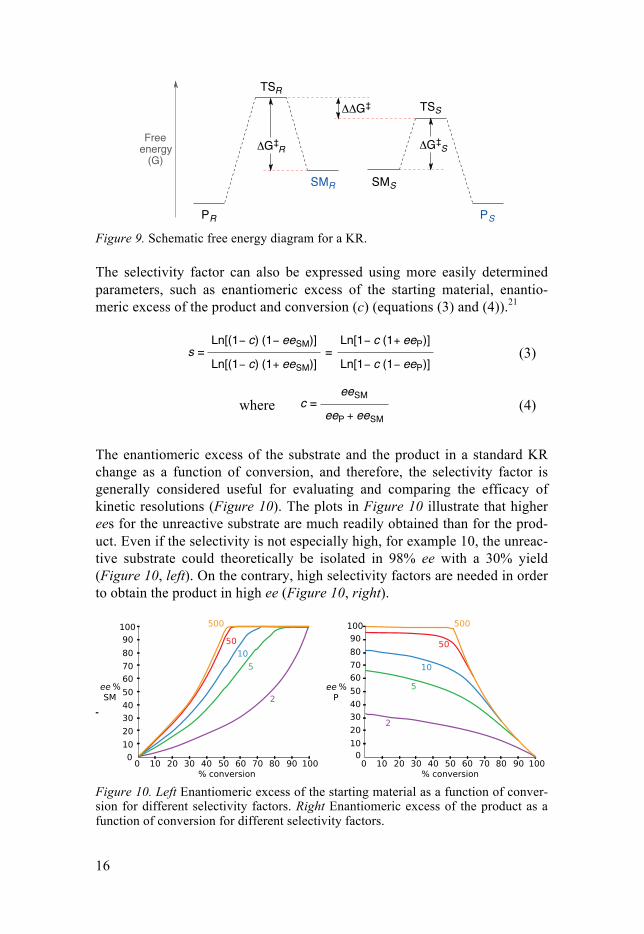
In a catalytic KR, the relation between the reaction rates of the substrate enantiomers is known as selectivity factor, s (equation (2)). The relative rates of the reactions correspond to the difference in energy between the diastere-omeric transition states (TS) in the selectivity-determining step of the reac-tion, i.e. ΔΔG‡ (Figure 9).
The larger the difference between the diastereomeric transition states (ΔΔG‡) is, or in other words, the larger difference between the reaction rates of the two enantiomers with the chiral catalyst is, the larger the selectivity factor will be.
SMS
Cat*
SMR
PS
PR
kfast
Cat*
kslow
+ +
% ee =[R] − [S]
[R] + [S]100 where [R] > [S] (1)
s =kfast
kslow
= e (2)ΔΔG‡/RT
16
Figure 9. Schematic free energy diagram for a KR.
The selectivity factor can also be expressed using more easily determined parameters, such as enantiomeric excess of the starting material, enantio-meric excess of the product and conversion (c) (equations (3) and (4)).21
The enantiomeric excess of the substrate and the product in a standard KR change as a function of conversion, and therefore, the selectivity factor is generally considered useful for evaluating and comparing the efficacy of kinetic resolutions (Figure 10). The plots in Figure 10 illustrate that higher ees for the unreactive substrate are much readily obtained than for the prod-uct. Even if the selectivity is not especially high, for example 10, the unreac-tive substrate could theoretically be isolated in 98% ee with a 30% yield (Figure 10, left). On the contrary, high selectivity factors are needed in order to obtain the product in high ee (Figure 10, right).
Figure 10. Left Enantiomeric excess of the starting material as a function of conver-sion for different selectivity factors. Right Enantiomeric excess of the product as a function of conversion for different selectivity factors.
PR PS
ΔG‡R
ΔΔG‡
ΔG‡S
SMSSMR
TSS
TSR
Free energy
(G)
s =Ln[(1− c) (1− eeSM)]
Ln[(1− c) (1+ eeSM)]=
Ln[1− c (1+ eeP)]
Ln[1− c (1− eeP)]
where c =eeSM
eeP + eeSM
(3)
(4)

17
The main drawback of the KR is the maximum theoretical yield of 50% based on the racemic substrate. Nevertheless, this procedure could be con-sidered the optimal among other preparation methods if the racemate and the catalyst are inexpensive; the resolution proceeds selectively at low catalyst loadings (s>10); and if the starting material and the product are easily sepa-rated.20 In addition, the KR step could be coupled to in situ racemization of the slow-reacting enantiomer in order to overcome the 50% yield limitation. If the enantioselectivity is sufficient and the racemization fast enough, the enantioenriched product can be isolated with a theoretical yield of 100%. This strategy is known as dynamic kinetic resolution (DKR) (Figure 11).
Figure 11. General scheme for a DKR. Main recovered specie is marked in blue.
The KR of racemic substrates can be achieved by metal catalysis, organoca-talysis, and biocatalysis. Microorganisms, such as yeast,22 or isolated en-zymes23 have been widely used in synthesis for the KR of chiral racemic substrates, affording optically pure compounds in high ee. Furthermore, the compatibility of some enzymes with metal complexes for the DKR of race-mic compounds have been reported.24 In this field, Bäckvall’s group and others have reported the combination of several ruthenium racemization catalysts with different lipases for the acylative DKR of a wide variety of alcohols and amines.25 For instance, Candida Antarctica lipase B (CALB) in combination with the Bäckvall’s catalyst 1 have been used for the DKR of many secondary alcohols, leading to the corresponding enantiopure acetates (Figure 12).26
Figure 12. DKR of sec-alcohols with CALB and Bäckvall’s racemization catalyst.
SMS
Cat*
SMR
PS
PR
kfast
Cat*
kslow
+krac
CALB, Na2CO3
R1 R2 R1 R21, t-BuOKToluene, r.t.
OH OAc
OAc+
OAc OAcOAcCN
Ph
OAcPh OAc
S
98% yield>99% ee
85% yield97% ee
98% yield>99% ee
97% yield>99% ee
98% yield>99% ee
RuCl
OCOC
Ph
PhPh
Ph
Ph
1

18
Over the last two decades, transition metal-mediated, and more recently or-ganocatalyzed, kinetic resolutions have emerged as a popular strategy due to the progress made in the development of chiral catalysts for asymmetric reactions.27 Sharpless et al. reported one of the earliest highly selective and most relevant examples of catalytic non-enzymatic KR in 1981, dealing with the titanium alkoxide tartrate epoxidation of allylic alcohols (Figure 13).28
Figure 13. Sharpless titanium alkoxide tartrate epoxidation of allylic alcohols.
Currently, many reactions based on a non-enzymatic KR strategy have been achieved with high efficiency, such as the acylation of alcohols (approach used in this thesis), oxidative KR of alcohols, ring opening reactions of epoxides, epoxidation of alkenes, oxidative transfer hydrogenation, ring-closing metathesis, hydrogenation of alkenes and imines, Pd-catalyzed al-lylic alkylation, 1,4-conjugate addition and cycloadditions.29 Moreover, im-portant advances in the application of non-enzymatic chiral catalysts for DKR has been made.30
1.4. Organocatalysts for acylative kinetic resolutions of chiral secondary alcohols While the use of enzymes for KR of racemic substrates to afford enantiopure counterparts has been commonly used among the synthetic community, the application of non-enzymatic chiral catalysts has only recently gained popu-larity.29 In particular, the use of metal-free catalysts is seen as an attractive alternative, since they are usually robust, inexpensive, readily available and non-toxic. The development of low molecular weight chiral organocatalysts for asymmetric synthesis has been the driving force of this field, over the last 20 years.31 Nowadays, a significant number of enantioselective organocata-lysts are available and are comparable to biocatalysts, in terms of enantiose-lectivity and activity. KR of racemic alcohols by the formation of esters is probably one of the most intensively studied aspect of organocatalysis.32
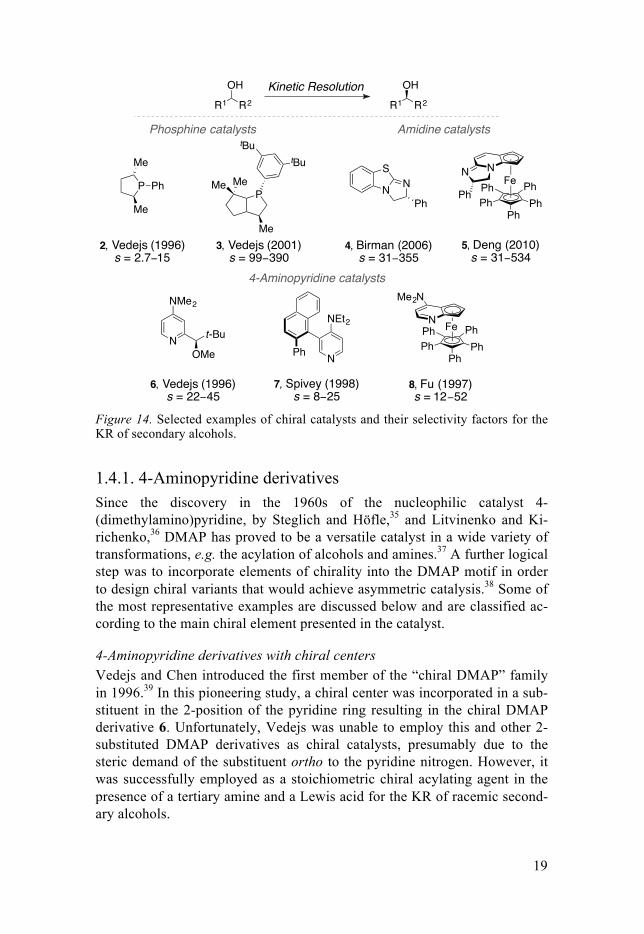
The known chiral organocatalysts for enantioselective acylation of alco-hols can be divided into different groups,32 and some of the most significant examples reported to date are phosphines,33 amidines34 and DMAP deriva-tives (Figure 14).
(+)-DIPTTi(Oi-Pr)4R R'
OH
R R'
OH+ R R'
OH
O
TBHPDCM, -20 °C
>99% ee 95-99% ee
19
Figure 14. Selected examples of chiral catalysts and their selectivity factors for the KR of secondary alcohols.
1.4.1. 4-Aminopyridine derivatives Since the discovery in the 1960s of the nucleophilic catalyst 4-(dimethylamino)pyridine, by Steglich and Höfle,35 and Litvinenko and Ki-richenko,36 DMAP has proved to be a versatile catalyst in a wide variety of transformations, e.g. the acylation of alcohols and amines.37 A further logical step was to incorporate elements of chirality into the DMAP motif in order to design chiral variants that would achieve asymmetric catalysis.38 Some of the most representative examples are discussed below and are classified ac-cording to the main chiral element presented in the catalyst.
4-Aminopyridine derivatives with chiral centers Vedejs and Chen introduced the first member of the “chiral DMAP” family in 1996.39 In this pioneering study, a chiral center was incorporated in a sub-stituent in the 2-position of the pyridine ring resulting in the chiral DMAP derivative 6. Unfortunately, Vedejs was unable to employ this and other 2-substituted DMAP derivatives as chiral catalysts, presumably due to the steric demand of the substituent ortho to the pyridine nitrogen. However, it was successfully employed as a stoichiometric chiral acylating agent in the presence of a tertiary amine and a Lewis acid for the KR of racemic second-ary alcohols.
R1 R2
OH
R1 R2
OHKinetic Resolution
N
Me2N
FePhPh
PhPhPh
N
NEt2
PhN
NMe2
t-Bu
OMe
4, Birman (2006)s = 31−355
N
SN
Ph
NFe
PhPh
PhPhPh
N
PhP
Me
Me MeP Ph
Me
Me
2, Vedejs (1996)s = 2.7−15
Phosphine catalysts Amidine catalysts
4-Aminopyridine catalysts
3, Vedejs (2001)s = 99−390
tButBu
5, Deng (2010)s = 31−534
7, Spivey (1998)s = 8−25
6, Vedejs (1996)s = 22−45
8, Fu (1997)s = 12−52

20
4-Aminopyridine derivatives with chiral axis Spivey and co-workers reported the first members of a novel family of chiral DMAP-based nucleophilic catalysts, including 7, inspired by axially chiral biaryls such as BINOL.40 The asymmetry of these derivatives relies on the restricted rotation about an aryl-aryl bond at the 3-position of the DMAP moiety. They showed moderate selectivity factors for the acylation of alco-hols. However, due to the relatively uncrowded environment of the nucleo-philic nitrogen and the efficient conjugation between the 4-amino group lone pair and the pyridine ring, these catalysts display high catalytic activity.41
4-Aminopyridine derivatives with chiral plane In 1996, Fu and Ruble presented a new subgroup of DMAP derivatives that possess a chiral plane.42 In order to design the chiral environment around the nucleophilic nitrogen atom they explored the possibility of breaking the two symmetry planes of the DMAP molecule by coupling the 4-aminopyridine structure to a metallocene (Figure 15). The resulting planar-chiral complex presents a well-pronounced chiral environment in the vicinity of the nucleo-philic nitrogen atom. Viewing along the lone pair-nitrogen atom axis, the new planar-chiral DMAP derivative now differentiates left from right (Cp vs. H) and top from bottom (vacant vs. R). Even though the C5R5 moiety is not directly bounded to the pyridine ring, its bulkiness is enough to block the bottom part of the nucleophilic nitrogen.
Figure 15. Design of a planar-chiral DMAP derivative.
In spite of the planar-chiral DMAP derivative containing an iron atom, it is mainly considered as an organocatalyst since the metal atom apparently shows no direct catalytic activity and can mainly be considered as a part of the catalyst’s framework.32 The incorporation of an electron-rich ferrocenyl moiety increases the nucleophilicity of the nitrogen atom in the pyridine ring and provides better stabilization of the acylium cation than the metal-free analogues according to calculated acetylation enthalpies by Zipse.43 Fur-
DMAP
NMe2N N HH
N H
R
planar-chiral DMAP
σ'
σ
σ'
σ
M
NMe2N
RR
R RR
Cp

21
thermore, the annulated cyclopentadiene ring in the 2,3-position is small enough to avoid the loss of catalytic activity typically suffered by DMAP derivatives with a bulky chiral group in the 2-position of the DMAP.
Fu and co-workers reported the effective KR of secondary aryl alkyl al-cohols using low loading of (−)-8 (2 mol%) and acetic anhydride as the acyl donor in ether.44 Thereafter, the role of the solvent was studied and tert-amyl alcohol was found to be the optimal solvent, both in terms of reactivity and selectivity.45 The efficiency of the planar-chiral DMAP derivative (−)-8 in the KR of secondary alcohols was also demonstrated for chiral propargylic46 and allylic47 alcohols (Figure 16).
Figure 16. KR of racemic secondary alcohols catalyzed by (−)-8.
The presence of heteroatoms or functional groups in the alkyl chain of the substrates reacted under 8 catalyzed KR conditions has been scarcely inves-tigated. In fact, only two substrates bearing a heteroatom in the alkyl chain have been studied under this conditions: 2-chloro-1-phenylethanol (s=32)44 and an allylic β-hydroxy ketone, key intermediate of Epothilone A (s=107).47
Recently, Fu et al. reported the first effective non-enzymatic DKR, cata-lyzed by 8 in the presence of the racemization ruthenium catalyst 1 (Figure 17).48 Changing the acylating agent from acetic anhydride, used in the KR, to acetyl isopropyl carbonate solved the incompatibility of the ruthenium-based catalyst with the anhydride.
Figure 17. DKR of racemic secondary alcohols catalyzed by (+)-8 and 1.
(−)-8 (1-2.5 mol%)
NEt3 (0-0.75 equiv)t-amyl alcohol, 0 °C
R1 R2
OH
O
O O+
R1 R2
OH
R1 R2
O+
O
OH
55% conv.99% ees = 43
tBu
OH
51% conv.96% ees = 95
Ph
OH
61% conv.99% ees = 22
OH
58% conv.96% ees = 20
Ph
(+)-8 (1 mol%)1 (5.2 mol%), KOtBu (4.4 mol%)
t-amyl alcohol/toluene (1:1)10 °C
R1 R2
OH
O
O
OiPr
O+
R1 R2
O
O
iPr
OAc
91% ee95% yield
OAc
Ph
OAcOAc
93% ee92% yield
90% ee94% yield
88% ee90% yield

22
1.5. The aim of this thesis In general, most of the organocatalysts are tested for the KR of model sub-strates, such as aryl alkyl alcohols. The incorporation of other functional groups in the model substrates provides new options for further derivatiza-tion, giving access to high value compounds. Therefore, study of the versa-tility of these organocatalysts for the preparation of more synthetically useful substrates containing additional functional groups is of interest.
In this context, the main objective of the research performed in this thesis has been to expand the substrate scope of organocatalytic KR methodologies to functionalized alcohols that could be synthetically useful. For this pur-pose, Fu’s catalyst 8 was chosen because its low catalyst loadings (1-2 mol%), recoverability,45 low sensitivity to oxygen, moisture, and impurities, and its compatibility with racemization catalysts for an appealing DKR. Fur-thermore, both (−)-8 and (+)-8 enantiomers are commercially available providing access to both enantiomers of the substrates. Thus, the primary aim of the first stage of this thesis has been to broaden the substrate scope of the KR catalyzed by (−)-8 to secondary aryl alcohols that contain an addi-tional heteroatom-containing functional group in the alkyl moiety, which includes β-azido alcohols (paper I), β-hydroxy phosphonates (paper II) and β-hydroxy esters (paper III) (Figure 18).
Figure 18. Substrate scope of the KR of secondary aryl alcohols catalyzed by (−)-8.
The second goal of his thesis has been to improve the understanding of the KR catalyzed by (−)-8. The mechanism of this process had been previously speculated to be similar to the mechanism presented for other DMAP deriva-tive catalysts, but had not been carefully studied. In order to get more insight into the mechanism, Reaction Progress Kinetic Analysis of the process and 1H-NMR spectroscopy study of the equilibrium between the free and acylat-ed catalyst have been performed (Paper IV).
Ar
OHFG
Work performed in this thesis
Ar
OHFG
(−)-8
Previous work
-FG = -N3 ,-P(O)(OR)2 ,-COOR
t-amyl alcohol, 0 °CAr Ralkyl
OH
Ar Ralkyl
OH
Ac2O, (NEt3)
(−)-8 (1-2 mol%)
(−)-8 (1-2.5 mol%)N
Me2N
FePhPh
PhPhPh
t-amyl alcohol, 0 °CAc2O,( NEt3)

23
2. Kinetic resolution of secondary aryl alcohols catalyzed by a planar-chiral DMAP derivative (Papers I-III)
2.1. The importance of chiral functionalized alcohols Alcohols are among the more extensively used compounds in organic syn-thesis because they can be found in nature and they can also be converted into and from many other types of compounds.49 In addition, the presence of additional functional groups provide for further derivatization options.
For instance, many bioactive molecules contain the structure 1-arylethanol with a functional group in the alkyl chain, e.g. an amine group in the 2-position (Figure 19).
Figure 19. Structures of some natural and synthetic agonists and antagonists of ad-renergic receptor. Elements identical to those found in adrenaline are in blue.
Adrenaline and noradrenaline are 2-amino-1-arylethanol neurotransmitters that act on adrenoreceptors involved in many functions regulating the body’s reaction to stress or emergency. Many adrenergic drugs are based on the structure of these ligands, sharing the chemical features of the natural ligand as is illustrated in Figure 19.
HO
HO
HN
OH
Adrenaline
HO
HO
NH2
OH
(R)-Noradrenalineβ-Agonists:
HO
HO
HN
OH
IsoprenalineHO
HN
OH
Levosalbutamol
OH
HO
HN
OH
(R,R)-Carmoterol
HN
O O
β-Antagonists:
OHN
OH
Butaxamine
NH
HN
OH
Sotalol
HN
OH
(R)-Pronethalol
O SO O

24
2.2. Substrate scope The most commonly used methods to obtain optically pure secondary aryl alcohols are: (i) the enantioselective reduction of the corresponding ketones and (ii) the KR of racemic alcohols or other derivatives. The latter are par-ticularly attractive since the racemic alcohols are often readily available and inexpensive. The KR approach involves different transformations, such as oxidative methods, hydrolysis of the corresponding racemic esters or acyla-tion reaction, which are mainly promoted by biocatalysts.
2.2.1. Kinetic resolution of β-azido alcohols The azido group is easily reduced to amines, and therefore, 1,2-azido alco-hols are often used as direct precursors of 1,2-aminoalcohols, a motif shared by many adrenergic drugs 50 and also by several chiral ligands for asymmet-ric catalysis.51 Moreover, azides can react very differently under different reaction conditions52 to give access to many nitrogen-containing heterocy-cles, such as aziridines53 and triazoles54 (Figure 20).
Figure 20. 2-Azido-1-arylethanol as important precursors.
The KR of 1,2-azido alcohols has been traditionally accomplished by enzy-matic systems.55 To the best of our knowledge, the investigation reported in paper I represents the first effective organocatalyzed KR of racemic chiral aromatic 1,2-azido alcohols.
Firstly, the racemic 1,2-azido alcohols where easily prepared in “one pot” two steps from the corresponding α-bromoacetophenone analogues, via the addition of sodium azide followed by the reduction of the ketone using sodi-um borohydride (Figure 21).
Figure 21. “One pot” two steps synthesis of racemic 1,2-azido alcohols.
ArN3
OH
ArNH
ArNH2
OHReduction
"Click chemistry"R
ArN
OH NNR
Triazoles
1,2-aminoalcoholsAziridines
1) NaN3, EtOH
Ar 2) NaBH4
O
70-95% yield
BrAr
OHN3
25
Several 1,2-azido alcohols with different substituents in the aromatic ring were resolved using 2 mol% of (−)-8 and triethylamine as auxiliary base at 0 °C in tert-amyl alcohol. After the required reaction time, the unreactive al-cohols were isolated with excellent enantiomeric excess up to 99% and with selectivity factors up to 45 (Table 1).
Table 1. Kinetic Resolution of aromatic 1,2-azido alcohols by (−)-8.a
Entry Ar Time/h eealcohol (%)b Conv. (%)c sc 1 Ph 24 99.6 73 17 2 2-naphthyl 3 99.9 60 45 3 4’-NO2-Ph 3 99.2 58 33 4 4’-CN-Ph 10 99.9 62 35 5 4’-Me-Ph 5 80.6 70 4 6 4’-MeO-Ph 13 98.6 65 14 7 4’-Benzoyl-Ph 20 92.7 58 16 8 2’,5’-(MeO)2-Ph 13 99.0 60 24 9 3’-Br-Ph 9 98.6 61 20 10 4’-Br-Ph 4 96.6 63 13
a Reaction conditions: rac-alcohol (0.25 mmol), Ac2O (0.75 equiv), triethylamine (0.75 equiv), (−)-8 (0.005 mmol, 2 mol%) in tert-amyl alcohol (1.0 mL) at 0°C. b Determined by chiral HPLC. c Calculated using equations (3) and (4) (section 1.3).
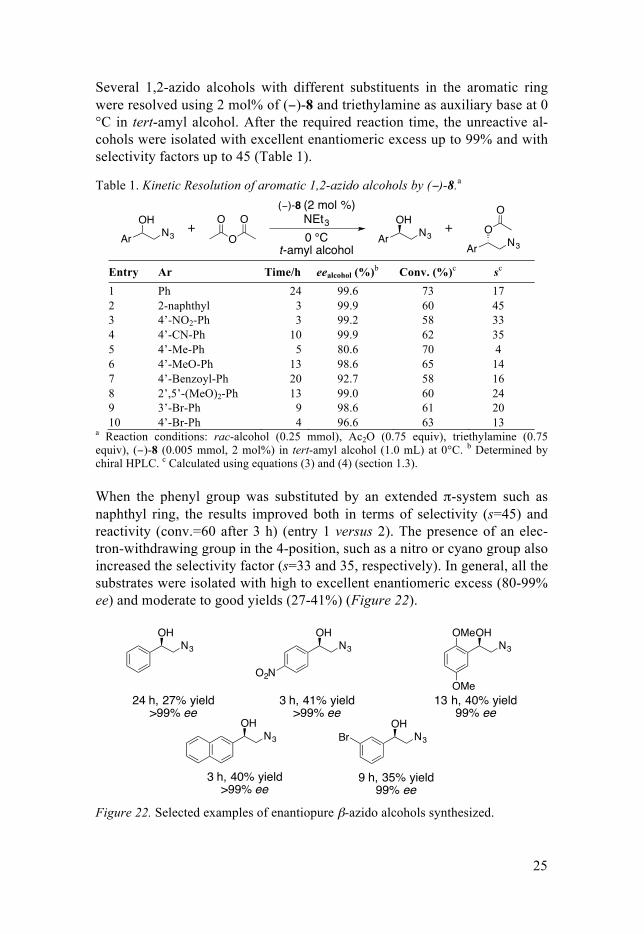
When the phenyl group was substituted by an extended π-system such as naphthyl ring, the results improved both in terms of selectivity (s=45) and reactivity (conv.=60 after 3 h) (entry 1 versus 2). The presence of an elec-tron-withdrawing group in the 4-position, such as a nitro or cyano group also increased the selectivity factor (s=33 and 35, respectively). In general, all the substrates were isolated with high to excellent enantiomeric excess (80-99% ee) and moderate to good yields (27-41%) (Figure 22).
Figure 22. Selected examples of enantiopure β-azido alcohols synthesized.
ArN3
OH+
O
O
O
ArN3
OH
ArN3
O
O
+
(−)-8 (2 mol %)NEt3
0 °Ct-amyl alcohol
3 h, 40% yield>99% ee
N3
OH
9 h, 35% yield99% ee
N3
OH
3 h, 41% yield>99% ee
N3
OH
O2N
24 h, 27% yield>99% ee
N3
OH
13 h, 40% yield99% ee
N3
OHOMe
OMe
Br

26
2.2.2. Kinetic resolution of β-hydroxy phosphonates Chiral 2-amino-2-aryl-ethylphosphonic acids have been reported to be po-tential GABAB receptor antagonists,56 as well as strong fungicides in the agriculture industry.57 They are traditionally prepared from their 2-hydroxy phosphonates counterparts by Mitsunobu reaction and in situ reduction of the azide to the amine (Figure 23). Previous KR strategies for the enantiose-lective preparation of aromatic 2-hydroxy phosphonates are mainly based on the hydrolysis of the corresponding esters catalyzed by lipases.58 In addition, Onomura reported the first acylative KR, catalyzed by a copper (II) tri-flate/(R,R)-Ph-BOX system.59
Figure 23. 2-Hydroxy-2-aryl-ethylphosphonates as precursors of 2-amino-2-aryl-ethylphosphonates.
We have investigated the KR of 2-hydroxy-2-aryl-ethylphosphonates via an acylation, catalyzed by the planar-chiral DMAP catalyst (−)-8, which repre-sent the second non-enzymatic method and the first organocatalyzed KR of these substrates.
First of all, the racemate of the desired substrates were synthesized by the Arbuzov reaction of the α-bromoacetophenones with trialkyl phosphite, followed by reduction of the ketone (Figure 24).
Figure 24. Synthesis of racemic 2-hydroxy-2-aryl-ethylphosphonates.
Previous studies reported that the used of triethylamine as auxiliary base in the KR of more activated secondary alcohols, such as propargylic alcohols,46 accelerated significantly the background reaction, decreasing the selectivity factor of the process. Therefore, we initially investigate the effect of triethyl-amine on selectivity and reactivity in the KR of the parent compound, dieth-yl (2-hydroxy-2-phenylethyl)phosphonate. When the reaction was performed in the absence of base, the selectivity improved significantly but the reaction rate decreased. We decided to sacrifice the speed of the reaction in favor of selectivity, excluding triethylamine in the successive experiments. Thus, the optimal conditions found for this process were using racemic alcohol (0.25
ArP(OR)2
OH
ArP(OR)2
NH2
2-amino-2-aryl-ethylphosphonates
O OMitsunobuconditions
ArBr
O
ArP(OR)2
OP(OR)3 (1.1 equiv)
90 °C, 5 min
O
EtOH or MeOHr.t., 1 h
NaBH4
ArP(OR)2
OH O
73-99% yield26-78% yield
27
mmol), acetic anhydride (0.75 equiv), and (−)-8 (0.0025 mmol, 1 mol%) in tert-amyl alcohol (1.0 mL) at 0 °C.
Enantiomeric excess and conversion were determined by 31P-NMR spec-troscopy using quinine as a chiral solvating agent. 31P-NMR spectroscopy is a convenient tool for the determination of the enantiomeric excess of phos-phorus-containing compounds due to the large chemical shift dispersion and the simplicity of the 31P-NMR spectra.60 In order to obtain more accurate integration and selectivity factors for the comparison of the efficiency of the synthetic method of the different substrates, in the first stage of the study the selectivity factor of the KR of several 2-hydroxy-2-arylethylphosphonates was determined after 4 hours (Table 2).
Table 2. Selectivity factor of KR of 2-hydroxy-2-arylethylphosphonates by (−)-8.a
Entry Ar R eealcohol (%)b Conv. (%)b sc
1 Ph Me 38 31 24 2 4-NO2-Ph Me 23 16 5 3 3-MeO-Ph Me 36 31 16 4 Ph Et 32 25 43 5 4-NO2-Ph Et 31 25 62 6 3-MeO-Ph Et 22 20 19 7 2-Naphthyl Et 55 37 68 8 2,4-Cl2-Ph Et 35 28 35 9 4-Me-Ph Et 15 14 25 10 4-F-Ph Et 43 38 11
a Reaction conditions: rac-alcohol (0.25 mmol), Ac2O (0.75 equiv), (−)-8 (0.0025 mmol, 1 mol%) in tert-amyl alcohol (1.0 mL) at 0 °C, 4 h. b Determined by 31P-NMR spectroscopy using quinine as the chiral solvating agent. The reported values are an average of two experi-ments. c The best selectivity of two experiments. Calculated using equations (3) and (4) (sec-tion 1.3).
Initially, the effect of the size of the phosphonate ester substituents was evaluated using either the dimethyl or diethyl hydroxy phosphonates. The substrates bearing the larger ethyl group in the phosphonate ester (entries 4-6) show significantly higher selectivity than the dimethyl hydroxy phospho-nates (entries 1-3). With these results at hand, we continued with the study of the effect of different substituents in the aromatic ring among diethyl phos-phonates. The larger 2-naphthyl derivate gave better results in terms of both selectivity and reactivity (s=68, 37% conv.), followed by substrates having electron-withdrawing substituents, which showed higher selectivity factors than the substrates having electron-donating substituents (e.g. entry 5 versus 9).
In the second stage of the study, the KR of the substrates where the selec-tivity was s>10 were performed with longer reaction times in order to isolate
ArP(OR)2
OH+
O
O
O+
(−)-8 (1 mol%)
0 °C, 4 ht-amyl alcohol
O
ArP(OR)2
OH O
ArP(OR)2
OAcO

28
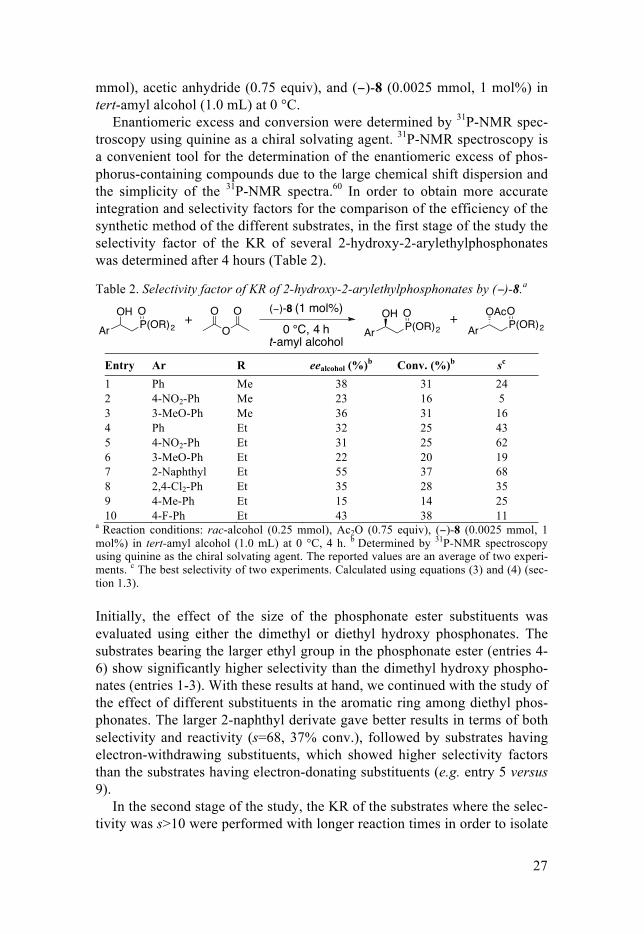
the enantiomerically pure 2-hydroxy-2-aryl-ethylphosphonates and to demonstrated the practical use of this methodology (Figure 25). In general, all the substrates were obtained with enantiomeric excess higher than 95% (a single peak in the 31P-NMR spectra using quinine as the chiral solvating agent was judged as >95% ee) and good yields (29-49%). For instance, the 2-naphthyl derivative was isolated enantiomerically pure (>95% ee) in 32% yield after 24 h. For the less reactive parent compound, longer reaction times were necessary (96 h) in order to obtain the products in high ee (>95%).
Figure 25. Selected examples of enantiopure hydroxyarylphosphonates synthesized.
2.2.3. Kinetic resolution of β-hydroxy esters Optically pure 2-hydroxy-2-aryl esters are another example of useful highly functionalized chiral precursors to important chemicals, such as flavane de-rivatives61 and 1,3-aminoalcohols (Figure 26). The aryloxypropanamine is a basic scaffold shared by many pharmaceuticals used for the treatment of depression, anxiety or eating disorders.62 For instance, Fluoxetine (Prozac), which is commercialized as a racemate, is one of the most widely prescribed antidepressant. It has been shown that the different enantiomers display dif-ferent pharmaceutical properties63 and therefore the enantioselective synthe-sis of the optically pure forms is of interest.64
Figure 26. 2-Hydroxy-2-aryl esters as important precursors of value compounds.
24 h, 32% yield>95% ee
P(OEt)2
OH O
48 h, 44% yield>95% ee
P(OEt)2
OH O
F
45 h, 33% yield>95% ee
P(OEt)2
OH O
O2N96 h, 41% yield
>95% ee
P(OEt)2
OH O
70 h, 31% yield>95% ee
P(OEt)2
OH OMeO
Ar
OH
Ar
OH
1,3-aminoalcoholsFlavane derivatives
O
OR
OAr
R' NHR'
NH
O
F3C
(S)-Fluoxetine
NH
O
(S)-Tomoxetine
NH
O
(S)-Nisoxetine
O
29
The synthesis of optically pure hydroxy esters by means of KR has been widely investigated for different kind of transformations promoted mainly by biocatalysts.65 Two non-enzymatic kinetic resolutions have been reported to date, an oxidative KR, which takes place in the presence of a palladium (−)-spartein complex;64a and a dehydrative KR, in which a chiral amino alcohol in the presence of a zinc complex dehydrates the chiral alcohol.61 In addition, Chen et al. reported the acylative KR of 2,2-difluoro-3-hydroxy-3-aryl-propionates catalyzed by the amidine-organocatalyst 4, and selectivity fac-tors up to 47 were obtained.66
We have studied the synthesis of optically pure 2-hydroxy-2-aryl esters by means of acylative KR catalyzed by the planar-chiral DMAP catalyst (−)-8. The starting racemic substrates were easily prepared by simple aldol addi-tion between the lithium enolate of ethyl acetate or tert-butyl acetate and the desired aromatic aldehyde (Figure 27).
Figure 27. Synthesis of racemic 2-hydroxy-2-aryl esters.
In this study we first investigated the influence of the ester on the selectivity factor of the KR after 3 hours of reaction (Table 3). Substrates containing either ethyl or the bulkier tert-butyl carboxylate group were examined (en-tries 1-6). In general, the substrates bearing the bulkier tert-butyl group showed slightly higher selectivity factors than the substrates with the ethyl group (e.g. entry 2 versus 5), which is in accordance to previously reported studies.
Table 3. Substrate screening for the KR of 2-hydroxy-2-aryl esters by (−)-8.a
Entry R Ar eealcohol (%)b eeacetate (%)b Conv. (%)c sc 1 Et Ph 32.4 95.4 25.4 58 2 Et 4-NO2-Ph 84.2 93.8 44.6 71 3 Et 4-MeO-Ph 39.8 94.6 29.6 53 4 tert-Bu Ph 28.8 95.2 23.2 54 5 tert-Bu 4-NO2-Ph 88.4 93.6 48.6 89 6 tert-Bu 4-MeO-Ph 39.4 96.0 29.1 72 7 tert-Bu 2-Naphthyl 83.4 95.2 46.7 107 8 tert-Bu 4-Cl-Ph 75.0 95.2 44.1 92 9 tert-Bu 2,6-Cl2-Ph 40.4 96.6 29.6 86 a Reaction conditions: rac-alcohol (0.25 mmol), Ac2O (0.75 equiv), (−)-8 (0.005 mmol, 2 mol%) in 1.0 mL of tert-amyl alcohol at 0 °C, 3 h. b Determined by chiral HPLC. c Calculated using equations (3) and (4) (section 1.3).
LDA, THF
OR -78 °C
O
80-98% yield
Ar
OH
OR
OLi
-78 °C
ArCHO O
OR
ArCOOR
OH+
O
O
O+
(−)-8 (2 mol%)
0 °C, 3 ht-amyl alcohol
ArCOOR
OH
ArCOOR
OAc

30
We also wanted to explore the influence of the different substituents on the aromatic ring on the selectivity and reactivity of the process. The replace-ment of the phenyl group with naphthyl led to a higher enantioselectivity and a faster reaction rate compared to the parent compound (entry 4 versus 7). In general, substrates with additional substituents in the aromatic ring led to higher selectivity factors than the parent compound. For example, the com-pounds with electron withdrawing groups in the phenyl ring (entry 5) and substrates with an electron donating methoxy group in the para position (entry 6) gave higher selectivity factors compared to the parent compound (entry 4).
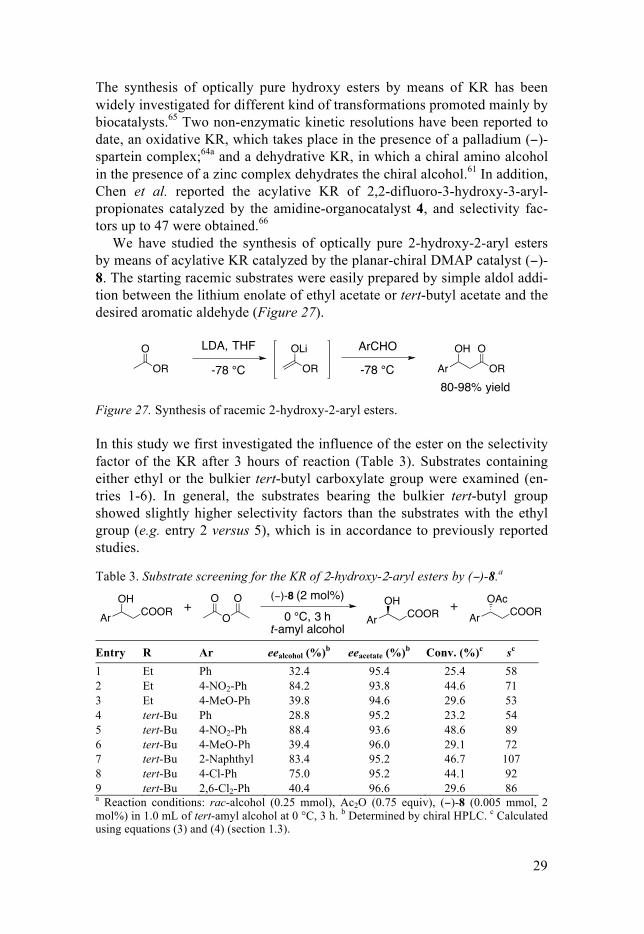
Finally, the kinetic resolutions of the different substrates were performed with longer reaction times (24 or 48 hours) in order to isolate the enantio-merically pure 2-hydroxy-2-aryl esters. All the substrates were obtained with high enantiomeric excess (95-99% ee) and moderate to good yields (23-45%) (Figure 28).
Figure 28. Selected examples of enantiopure β-hydroxy-β-aryl esters synthesized.
2.3. Synthetic applications In order to demonstrate the synthetic applicability of the methodology de-scribed herein, we have utilized catalyst (−)-8 for the KR of secondary alco-hols involved in synthetic routes of high value compounds.
(R)-Pronethalol is a β-blockers that could be synthesized from the corre-sponding 2-azido-1-(naphthalene-2-yl)ethanol (Figure 29). The KR of the 1,2-azido alcohol was scaled up to 1 mmol and the (R)-enantiomer was iso-lated in 43% yield with high enantiomeric purity (>99% ee). Thereafter, the (R)- 2-azido-1-(naphthalene-2-yl)ethanol was converted to (R)-Pronethalol in 83% yield via the reduction of the azide and subsequent in situ reductive alkylation with acetone using hydrogen/platinum (IV) oxide in ethanol at room temperature.67
24 h, 45% yield98% ee
COOtBuOH
24 h, 34% yield99% ee
COOtBuOH
Cl
24 h, 33% yield99% ee
COOtBuOH
O2N
24 h, 41% yield99% ee
COOEtOH
24 h, 41% yield99% ee
COOtBuOH
MeO

31
Figure 29. Synthesis of (R)-Pronethanol. Reagents and conditions: (i) rac-2-azido-1-(naphthalene-2-yl)ethanol (1.0 mmol), Ac2O (0.75 equiv), triethylamine (0.75 equiv), (−)-8 (2 mol%) in tert-amyl alcohol (4.0 mL) at 0 °C. (ii) (R)- 2-azido-1-(naphthalene-2-yl)ethanol (0.43 mmol), PtO2 (0.5 equiv), acetone (1.5 equiv), H2 (5 atm) in EtOH (5 mL) at r.t., 4 h.
Additionally, the synthesis of the enantiomerically pure intermediate (S)-3-hydroxy-N-methyl-3-phenylpropanamide for the synthesis of many antide-pressants, such as Fluoxetine in its enantiomerically pure form, was carried out (Figure 30). First, the KR of ethyl 3-hydroxy-3-phenylpropanoate was scaled up to 2.8 mmol, yielding 32% of the (S)-enantiomer in high enantio-meric excess (99% ee). The subsequent treatment of the β-hydroxy ester with aqueous methylamine gave access to the (S)-β-hydroxy amide in 85% yield and with retention of the configuration (99% ee).64
Figure 30. Synthesis of (S)-3-hydroxy-N-methyl-3-phenylpropanamide, an immedi-ate precursor to (S)-enantiomers of Tomoxetine, Fluoxetine and Nisoxetine. Rea-gents and conditions: (i) rac-ethyl 3-hydroxy-3-phenylpropanoate (2.8 mmol), Ac2O (0.75 equiv.), (−)-8 (0.005 mmol, 2 mol%) in tert-amyl alcohol (1.0 mL) at 0 °C, 24 h. (ii) MeNH2 (40% aq.), r.t., 3 h. (iii) See references.64
2.4. Conclusions Papers I to III illustrate the versatility of the planar-chiral DMAP derivate catalyst (−)-8 for the KR of secondary aryl alcohols that contain an addition-al heteroatom-containing functional group in the alkyl moiety, including β-azido alcohols, β-hydroxy phosphonates and β-hydroxy esters.
The strategy used herein for the preparation of optically pure secondary alcohols represents one of the first examples of organocatalyzed acylative KR for the families of substrates investigated.
iiiN3
OHN3
OH HN
OH
43% yield, >99% ee 83% yield(R)-Pronethalol
i
NH
O R= 4-CF3 (S)-FluoxetineR= 2-Me (S)-TomoxetineR= 2-OMe (S)-Nisoxetine
Ph
OH
OEt
O
Ph
OH
OEt
O
Ph
OH
NHMe
Oii
R
iii
32% yield, 99% ee 85% yield, 99% ee
32
Finally, the reaction was scale up more than ten times (from 0.25 mmol to 2.8 mmol) obtaining the product in high enantiomeric excess and good yield, and the applicability of this methodology has been further demonstrated through the synthesis of chiral compounds of high synthetic value, such as (R)-Pronethalol and (S)-3-hydroxy-N-methyl-3-phenylpropanamide.
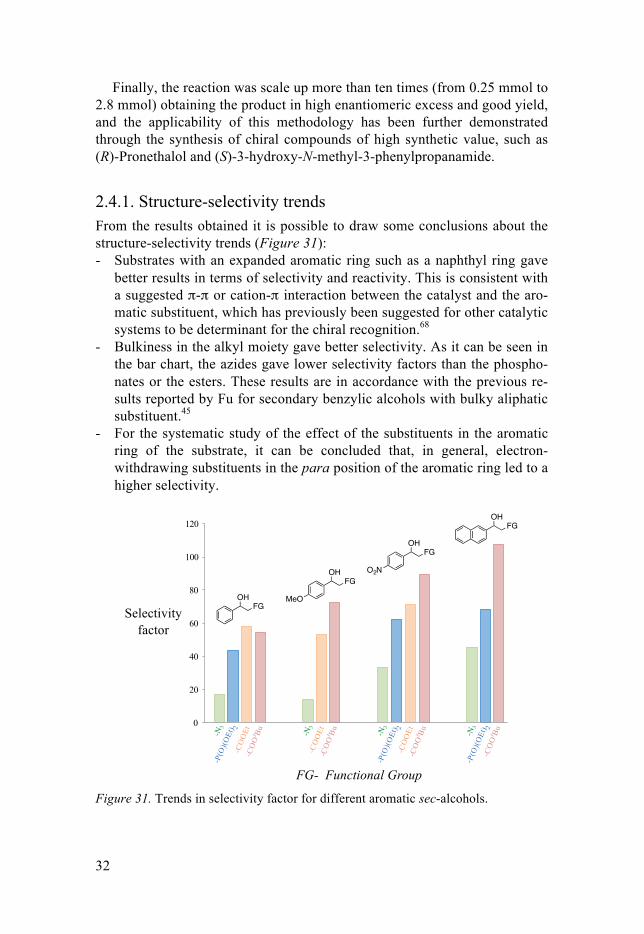
2.4.1. Structure-selectivity trends From the results obtained it is possible to draw some conclusions about the structure-selectivity trends (Figure 31): - Substrates with an expanded aromatic ring such as a naphthyl ring gave
better results in terms of selectivity and reactivity. This is consistent with a suggested π-π or cation-π interaction between the catalyst and the aro-matic substituent, which has previously been suggested for other catalytic systems to be determinant for the chiral recognition.68
- Bulkiness in the alkyl moiety gave better selectivity. As it can be seen in the bar chart, the azides gave lower selectivity factors than the phospho-nates or the esters. These results are in accordance with the previous re-sults reported by Fu for secondary benzylic alcohols with bulky aliphatic substituent.45
- For the systematic study of the effect of the substituents in the aromatic ring of the substrate, it can be concluded that, in general, electron-withdrawing substituents in the para position of the aromatic ring led to a higher selectivity.
Figure 31. Trends in selectivity factor for different aromatic sec-alcohols.
OHFG
OHFG
O2NOHFG
MeOOHFG

33
3. Mechanistic studies of the acetylation by a planar-chiral DMAP catalyst (Paper IV)
3.1. Introduction The progresses in understanding and applying the archetypical nucleophilic catalyst DMAP have been enormous since its discovery.37 A complete kinet-ic analysis of an alcohol acylation catalyzed by DMAP was reported.69 The currently accepted mechanism (Figure 32) involves the attack of the acyl donor from the nucleophilic catalyst to form an acylium cation with the cor-responding counteranion (acetate in the case of acetic anhydride) in a pre-equilibrium. The alcohol then reacts with the acylated catalyst in the turno-ver-limiting step to form the ester and the protonated catalyst. The latter is presumable regenerated via deprotonation through the auxiliary base.
Figure 32. Schematic representation of the DMAP catalyzed acylation of an alcohol.
Theoretical studies of the KR using the axially chiral DMAP catalyst 7 have been recently published.70 Kinetic studies of other related chiral catalysts, such as the amidine-based (S)-HBTM were recently described.71 However, the mechanistic studies of acylation reactions catalyzed by the planar-chiral DMAP derivative 8 are scarce.
In 1999, Fu et al.46 reported the first X-ray structural characterization of the acylated catalyst 8 with SbF6
- as a counter ion, which reveals a confor-
O
O
O
N
N
N
N
N
N
ROHO
OR
NEt3
HNEt3OAc
O
O O
HOO

34
mation in which the acetyl group is co-planar with the pyridine ring and orientated towards ring-fusion. A Nuclear Overhauser Effect between the acetyl methyl group and the pyridyl H in 2-position showed that this is also the major conformation in solution and Fu suggested that effective blocking of one face of this carbonyl by the pentaphenylcyclopentadienyl group is probably critical for chirality transfer from the chiral-planar acyl pyridinium form to the substrate. In the same communication, his group also performed NMR spectroscopy studies in CD2Cl2 of the equilibrium between the free and the acylated catalyst, which suggested that the free species are strongly favored.
In 2012, Fu and co-workers48 reported a mechanistic study based on ini-tial rates methodology for the DKR of racemic 1-phenylethanol, using the planar-chiral DMAP derivative 8 and ruthenium complex 1 as the acylation and racemization catalysts, respectively, and acetyl isopropyl carbonate as the acyl donor in a 1:1 mixture of tert-amyl alcohol/toluene. The rate law obtained was first order in 1-phenylethanol and in the acylation catalyst 8, zeroth order in the racemization catalyst 1, and “fractional” order in acetyl isopropyl carbonate, which is rationalized based on the NMR studies.46
At this stage of the thesis, we were interested to improve the understand-ing of the mechanism for the KR of aromatic secondary alcohols catalyzed by the planar-chiral DMAP derivative (−)-8 and acetic anhydride as the acyl donor. With this purpose in mind, a detailed kinetic analysis of the catalyst (−)-8 with the matched enantiopure secondary alcohol (R)-1-phenylethanol at synthetically relevant concentrations was performed using in situ IR spec-troscopy. This kinetic study is based on the Reaction Progress Kinetic Anal-ysis (RPKA).
3.1.1. Reaction progress kinetic analysis The RPKA methodology was formalized by Blackmond in the late 1990s.72 This analysis method relies on the accurate monitoring of the reaction pro-gress, for example by reaction calorimetry, NMR spectroscopy or in situ IR spectroscopy, which allows for the study of the complete reaction, and there-fore, provides further clues about side reactions, catalyst deactivation or product inhibition. This information is difficult to extract from classical ki-netic approaches. Additionally, RPKA requires, in general, fewer experi-ments than classical kinetic techniques.
The RPKA experiments are performed under synthetically relevant condi-tions and as a result, the obtained data are more representative of the “true” reaction conditions compared to pseudo-first-order analysis that contains one substrate in a large excess in order to maintain its concentration virtually constant through the reaction progress. In contrast, in RPKA the concentra-tions of all the substrates change over the course of the reaction. The RPKA makes use of the stoichiometry of the reaction and assumes that one mole-

35
cule of substrate A consumes one molecule of substrate B as it reacts to form the product P. Thus, the parameter [“excess”] can be defined as the differ-ence in the initial concentration of two substrates (equation (5)). Since [“ex-cess”] does not change during the reaction progress, it provides an extra equation to determine the concentration of one of the substrates as a function of the other (equation (6)) and, therefore, monitoring the concentration of only one substrate as the reaction progresses allows for the evaluation of the kinetics of the reaction.
The reaction system in Figure 33 represents a simple case of catalytic reac-tion mechanism with two substrates and one intermediate species, which applies to several cases including the acylation of alcohols catalyzed by DMAP derivatives.
Figure 33. Catalytic cycle for a simple reaction with two substrates and one inter-mediate specie.
The steady-state rate law expression for this case is given by equation (7). In equation (8), both the numerator and denominator of (7) are divided by the constant k-1 to give the rate equation in what is called the “one-plus” form.
These equations reveal that for a reaction with two substrates the idea of an integer reaction order in either substrates will be at best an approximation, as [A] and [B] appear in additive terms in the denominator as well as being multiplied together in the numerator.72 On the other hand, the order in cata-lyst should be one because it only appears in the numerator.
["Excess"] = [A]0 − [B]0 = [A]t − [B]t (5)
[A]t = ["Excess"] + [B]t (6)
Cat A
B
P A
Cat k1k-1
k2
.
rate = v = (7)k1 k2 [A] [B] [Cat]total
k-1 + k1 [A] + k2 [B]
v = (8)K1 k2 [A] [B] [Cat]total
1 + K1 [A] + [B]k2
k-1

36
The RPKA methodology consists of two sets of experiments, called “dif-ferent excess” and “same excess” experiments. Firstly, the steady-state law can be streamlined by approximating to the simple power law form (equation (9)).
The “different excess” protocol provides the concentration dependence of the different reactants, i.e. the reaction order in the different substrates. Equation (9) can be normalized by the concentration of substrate A, for ex-ample, to obtain the relationship shown in equation (10).
The plot of the “normalized” rate versus concentration of the other substrate (B, in this case) for at least two reactions carried out with different values of [“excess”] will provide the following information:73
- When the correct value of “x” is inserted in the equation, the curves overlay, meaning that the reaction display x-order in the concentra-tion of the “normalized” substrate.
- When the correct value of “y” is inserted in the equation, the shape of the curves is a straight line, which reveals a y-order in the concen-tration of the substrate plotted as the x-axis.
The “Same excess” protocol addresses the question of catalyst stability and process robustness. Most of the catalytic reactions exhibit first-order de-pendence in the catalyst concentration. In order to confirm the relationship, a graphical rate equation (11) can be developed by dividing the general rate expression (7) by the total catalyst concentration [Cat]total. This expression for the rate normalized by catalyst concentration is also known as the turno-ver frequency (TOF).
If the resulting plots TOF versus [A] of two experiments carried out at dif-ferent catalyst concentration but same [“excess”] overlay, the reaction is first order in the concentration of the catalyst. Because the catalyst concentration is constant over the course of the reaction, the resulting plots are normalized by an unchanging value. In this case, the shape of this plot is not relevant and gives no further information about the reaction orders in the substrates.72
v = kobs [A]x [B]y [Cat]total (9)
= (kobs [Cat]total) [B]y (10)v
[A]x
= (11)v
[Cat]total
k1 k2 [A] [B]
k-1 + k1 [A] + k2 [B]TOF =

37
Process robustness can be investigated constructing two experiments with the same [“excess”] but different initial concentrations, which allows for the comparison of the working system, after the catalyst has completed several catalytic cycles, with the “fresh” system. If the plots of the rate, normalized by the catalyst concentration, overlay then the catalyst is doing the same work in the beginning of the reaction as when it has been working for sever-al cycles. The information extracted from these experiments is critical to probe a number of mechanistic possibilities including catalyst deactivation and/or product inhibition.
3.2. Results and discussion We have investigated the reaction between the matched enantiopure (R)-1-phenylethanol and acetic anhydride catalyzed by (−)-8 in tert-amyl alcohol at 17 °C (Figure 34). The reagents stoichiometry used was the same as in the catalytic KR, but the slow-reacting enantiomer is excluded. These conditions were chosen for the “standard conditions” experiment, which corresponds to (R)-1-phenylethanol (0.25 M), acetic anhydride (0.39 M), auxiliary base (0.38 M) and catalyst (−)-8 (0.005 M). The different experiments that were performed during the RPKA involved altering one or more of these concen-trations.
Figure 34. (−)-8 catalyzed acylation of (R)-1-phenylethanol.
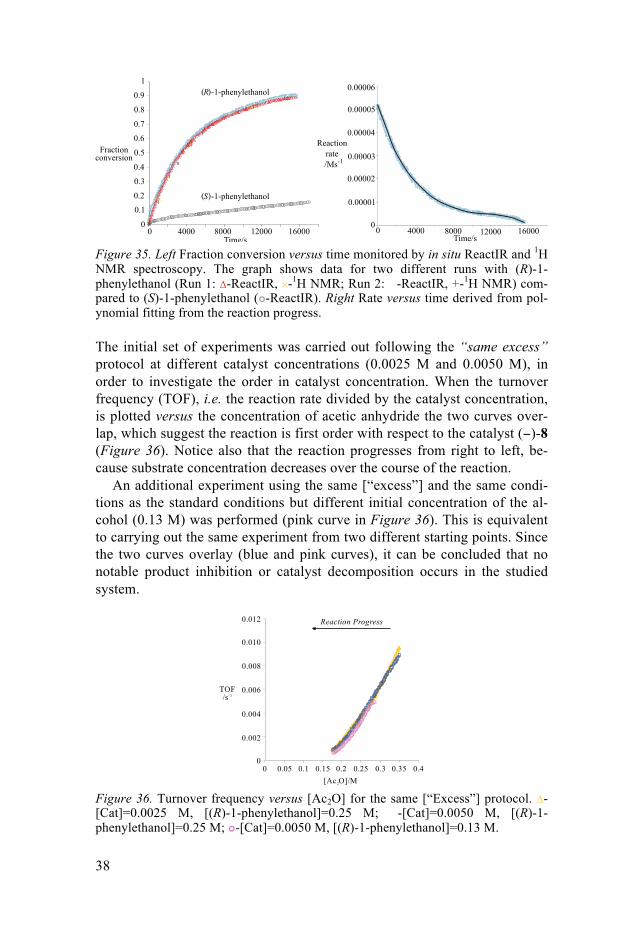
The reaction was monitored by in situ IR spectroscopy following the for-mation of product. The IR spectroscopic data were calibrated with the con-version determined from 1H-NMR analysis of periodic discrete samples. The left plot of Figure 35 shows a comparison of conversion versus time from in situ IR spectroscopy and from 1H-NMR spectroscopy of two different exper-iments, both under the “standard conditions”. The graph confirms that the data obtained is reproducible and the veracity of the in situ technique. The acetylation of the mismatched enantiomer (S)-1-phenylethanol catalyzed by (−)-8 under the same standard reaction conditions proceeds, as expected, slower compared to the (R)-1-phenylethanol. The rate of the reaction was obtained from the acquired data set (concentration versus time) by fitting a 6th order polynomial and differentiating this function (Figure 35, right).
Ph
OH+
O
O
O
Ph
OAc(−)-8, NEt3
t-amyl alcohol17 °C
38
Figure 35. Left Fraction conversion versus time monitored by in situ ReactIR and 1H NMR spectroscopy. The graph shows data for two different runs with (R)-1-phenylethanol (Run 1: Δ-ReactIR, -1H NMR; Run 2: �-ReactIR, +-1H NMR) com-pared to (S)-1-phenylethanol ( -ReactIR). Right Rate versus time derived from pol-ynomial fitting from the reaction progress.
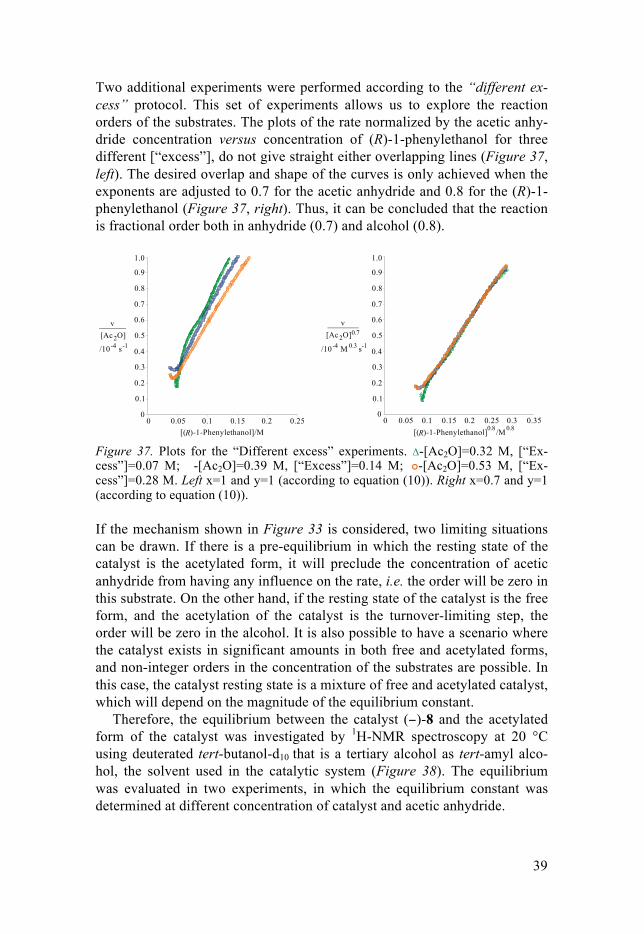
The initial set of experiments was carried out following the “same excess” protocol at different catalyst concentrations (0.0025 M and 0.0050 M), in order to investigate the order in catalyst concentration. When the turnover frequency (TOF), i.e. the reaction rate divided by the catalyst concentration, is plotted versus the concentration of acetic anhydride the two curves over-lap, which suggest the reaction is first order with respect to the catalyst (−)-8 (Figure 36). Notice also that the reaction progresses from right to left, be-cause substrate concentration decreases over the course of the reaction.
An additional experiment using the same [“excess”] and the same condi-tions as the standard conditions but different initial concentration of the al-cohol (0.13 M) was performed (pink curve in Figure 36). This is equivalent to carrying out the same experiment from two different starting points. Since the two curves overlay (blue and pink curves), it can be concluded that no notable product inhibition or catalyst decomposition occurs in the studied system.
Figure 36. Turnover frequency versus [Ac2O] for the same [“Excess”] protocol. Δ-[Cat]=0.0025 M, [(R)-1-phenylethanol]=0.25 M; �-[Cat]=0.0050 M, [(R)-1-phenylethanol]=0.25 M; -[Cat]=0.0050 M, [(R)-1-phenylethanol]=0.13 M.
39
Two additional experiments were performed according to the “different ex-cess” protocol. This set of experiments allows us to explore the reaction orders of the substrates. The plots of the rate normalized by the acetic anhy-dride concentration versus concentration of (R)-1-phenylethanol for three different [“excess”], do not give straight either overlapping lines (Figure 37, left). The desired overlap and shape of the curves is only achieved when the exponents are adjusted to 0.7 for the acetic anhydride and 0.8 for the (R)-1-phenylethanol (Figure 37, right). Thus, it can be concluded that the reaction is fractional order both in anhydride (0.7) and alcohol (0.8).
Figure 37. Plots for the “Different excess” experiments. Δ-[Ac2O]=0.32 M, [“Ex-cess”]=0.07 M; �-[Ac2O]=0.39 M, [“Excess”]=0.14 M; -[Ac2O]=0.53 M, [“Ex-cess”]=0.28 M. Left x=1 and y=1 (according to equation (10)). Right x=0.7 and y=1 (according to equation (10)).
If the mechanism shown in Figure 33 is considered, two limiting situations can be drawn. If there is a pre-equilibrium in which the resting state of the catalyst is the acetylated form, it will preclude the concentration of acetic anhydride from having any influence on the rate, i.e. the order will be zero in this substrate. On the other hand, if the resting state of the catalyst is the free form, and the acetylation of the catalyst is the turnover-limiting step, the order will be zero in the alcohol. It is also possible to have a scenario where the catalyst exists in significant amounts in both free and acetylated forms, and non-integer orders in the concentration of the substrates are possible. In this case, the catalyst resting state is a mixture of free and acetylated catalyst, which will depend on the magnitude of the equilibrium constant.
Therefore, the equilibrium between the catalyst (−)-8 and the acetylated form of the catalyst was investigated by 1H-NMR spectroscopy at 20 °C using deuterated tert-butanol-d10 that is a tertiary alcohol as tert-amyl alco-hol, the solvent used in the catalytic system (Figure 38). The equilibrium was evaluated in two experiments, in which the equilibrium constant was determined at different concentration of catalyst and acetic anhydride.

40
Figure 38. Equilibrium between the free and acetylated forms of the catalyst (−)-8.
In the first experiment, the initial concentration of the catalyst was similar to the one used in the “true” reaction conditions (0.004 M) and the sample was analyzed by 1H-NMR spectroscopy after sequential additions of different amounts of acetic anhydride, allowing the system to equilibrate for 10 minutes. The region of the 1H-NMR spectra where the proton signals from the two methyl-groups is shown in Figure 39. The spectrum of the catalyst shows a peak appearing as a singlet at 2.98 ppm that corresponds to the 6 protons of the methyl groups. After the addition of 20 μL of acetic anhydride (0.21 M), two new signals appear at 3.48 and 3.57 ppm that belong to the protons of the two methyl groups of the acetylated catalyst and represent 30% of the mixture of the total catalyst concentration. The two new signals are the result of the restricted rotation around the carbon-nitrogen bond due to the conjugation across the 4-aminopyridyl system of the acetylated cata-lyst. After a second addition of 20 μL of acetic anhydride (0.41 M), the acet-ylated specie is formed in a 38%. These conditions are close to the condi-tions used in the “true” reaction conditions, and give an idea of the catalyst state.
Figure 39. Expansion of the methyl region of the 1H-NMR spectra in tert-butanol-d10, [Cat]0=0.004 M.
+N
Me2N
FeC5Ph5 O
O ON
Me2N
FeC5Ph5O
O
O
k1
k-1
t-butanol-d10

41
Similar values of equilibrium constant were obtained in the second experi-ment, with a catalyst concentration of 0.009 M and proceeding in the same manner as before. The estimated averaged equilibrium constant is 1.74, which means that the free and acyl pyridinium states of the catalyst are in equilibrium and both species exist in significant amounts. These results are consistent with the non-integer orders, obtained for the dependence of the substrate concentrations in the kinetics expression.
On the contrary, when the experiment was performed in CDCl3, the free catalyst is strongly favored, which is in agreement with the outcome of the NMR spectroscopy studies of the equilibrium between the free and the acetylated catalyst performed in CD2Cl2 by Fu et al.46
3.3. Conclusions The results obtained with the RPKA methodology has increased our under-standing of the process studied during this thesis. The analysis reveals that the reaction is first order in catalyst (−)-8, and fractional order in acetic an-hydride (0.7) and secondary alcohol (0.8). Furthermore, the RPKA per-formed here has added valuable information about robustness of the catalytic system. This allows us to put forward the hypothesis that it does not suffer from notable catalyst deactivation or product inhibition.
The fractional order in the substrates could be explained in a situation where both free and acetylated catalyst exit in significant amounts, which have been proved by the NMR spectroscopy studies. This result differs from the first order dependence found for DMAP catalyst69 and the amidine-based catalyst (S)-HBTM.71 By contrast, it is in agreement with the fractional order of the acylative agent used for the DKR using the same chiral DMAP deriva-tive (−)-8, studied using initial rates method.48 In this case, the RPKA exper-iments provide a good representation of the “true” conditions of the KR, and suggest a fractional order in the alcohol concentration as well.
Finally, the strong influence of the solvent in the equilibrium could be demonstrated from the equilibrium experiments performed in different sol-vents, which is in accordance with the fact that polar solvents such as tert-butanol or tert-amyl alcohol favor the formation of the acylpyridinium salt, and therefore, give better results in terms of reactivity.
(13)K1 =k1
k-1
[Catacylated]=
[Cat] [Ac2O]= 1.74 ± 0.35

42
4. Concluding remarks and outlook
Nowadays, the synthesis of optically pure compounds is very important for the development of more effective and selective drugs and fungicides for agriculture, new materials, perfumes, etc. Several factors should be taken into consideration when choosing the appropriate synthetic methodology for their preparation, and therefore, the availability of a broad library of different methodologies is fundamental. In this context, the work presented herein, based on an effective organocatalyzed KR, represents the first effective non-enzymatic KR reported to date for some of the substrate studied.
The research presented in this thesis has extended the substrate scope of the KR promoted by organocatalyst (−)-8, to molecules containing an extra functional group in the alkyl moiety, including β-azido alcohols (s up to 45), β-hydroxy phosphonates (s up to 68) and β-hydroxy esters (s up to 107).
Furthermore, the kinetic analysis of the catalytic system using the novel RPKA methodology and NMR spectroscopy has increased the understanding of the mechanism and added valuable information about robustness of the catalytic system.
A goal that has always been in our mind during this thesis is to couple the effective KR to a racemization step, in order to achieve a DKR and over-come the handicap of the maximum 50% yield. During our investigation, we unsuccessfully surveyed several commercially available anhydrides, eager to find one compatible with the most common ruthenium-based racemization catalysts. Meanwhile, Fu and co-workers reported the first non-enzymatic DKR using acetyl isopropyl carbonate as acylating agent, which does not inactivate the ruthenium catalyst. This represents a starting point to further develop this methodology.
The systematic study of the selectivity factors obtained for all the sub-strates investigated revealed some selectivity trends, which could be used for the design of new catalysts or to find new interesting substrate targets. For example, the different outcomes obtained for substrates bearing groups with different electronic properties in the aromatic ring could be exploited for the KR of diaryl methanols. Preliminary studies in our group indicated that the catalyst (−)-8 shows some selectivity for the KR of 2,3,4,5,6-pentafluorobenzydrol in tert-amyl alcohol at 0 °C (s=5.5).
Future studies to increase the understanding of the π-π and π-cation stack-ing interaction between the catalyst and the substrate could help to bring about a full comprehension of enantioselective recognition step.

43
Populärvetenskaplig sammanfattning
Inom kemin är en molekyl kiral när den kan existera i två former som är varandras spegelbilder, men formerna kan inte läggas på varandra (Figur 1). Ett liknande exempel är våra egna händer där höger hand är den vänstra han-dens spegelbild. Dessa olika "spegelbildsmolekylerna" kallas enantiomerer. De har samma kemiska sammansättning, men en annan rumslig fördelning.
Figur 1. Representation av kirala objekt: enantiomererna av aminosyran alanin och ett par händer.
Kiralitet är ett vanligt fenomen i vår värld och naturen tenderar att föredra en enantiomer över den andra. Kroppens biomolekyler (t. ex. proteiner och receptorer) reglerar många viktiga funktioner i alla organismer och dessa biomolekyler är uppbyggda av aminosyror där endast den ena enantiomeren (spegelbildsmolekylen) förekommer. Detta innebär att proteiner och recep-torer (biomolekylerna) i våra kroppar är kirala och att olika enantiomerer (spegelbildsmolekyler) kan interagera på olika sätt i kroppen. Exempelvis interagerar de två enantiomererna av karvon på olika sätt med doftrecep-torerna, vilket gör att ena enantiomeren doftar grönmynta medan den andra doftar kummin (Figur 2).
Figur 2. Enantiomerer av karvon.
L-alanin(S)-enantiomer
D-alanin(R)-enantiomer
COOH
H
H2NCH3H3C
NH2
H
COOH
kan inte läggas på varandra
H3C NH2
H
COOH
HNH2
CH3
COOHspegel
spegel
(R)-karvongrönmynta
(S)-karvonkummin
O
H H
O

44
En mer avgörande betydelse kan detta fenomen få om den kirala substan-sen är ett läkemedel och den ena av enantiomererna har en annan effekt än den andra. I extremfallet kan en av enantiomererna ha en positiv hälsosam effekt och spegelbilden kan ha en hälsoskadlig effekt. Detta är fallet för (S)-Naproxen, som har gynnsamma antiinflammatoriska effekter, medan (R)-Naproxen orsakar leverskador. På grund av enantiomerers olika verkan i vår kropp, innehåller idag oftast läkemedel endast den aktiva enantiomeren. Detta innebär att läkemedel som är kirala måste vara enantiomert rena, det vill säga endast innehålla en av enantiomererna.
Den senaste utvecklingen inom asymmetrisk syntes och enantiomera sep-arationstekniker har främjat utvecklingen av säkrare och mer specifika läke-medel. Idag är ett av huvudområdena för syntes av kemikalier att utveckla metoder för att framställa enantiomeriskt rena föreningar som är effektiva och som är anpassade till de olika behoven i varje laboratorium. För att göra detta använder kemister en katalysator, vilket är en förening som påskyndar en kemisk reaktion utan att själv förbrukas.
Fokus i denna avhandling är på studier av en kiral katalysator som kan omvandla en av enantiomererna av en kiral molekyl, och låta den andra förbli oförändrad (Figur 3). Reaktionen kallas kinetisk resolvering och innebär att man kan erhålla den icke-reagerade enantiomeren med hög renhet om katalysatorn selektivt endast reagerar med en av enantiomererna. Denna metod används ofta inom industrin för framställning av enantiomert rena föreningar.
Figur 3. Allmänt system av en katalytisk kinetisk resolvering (substratet och produkten som erhålls vid slutet av reaktionen är markerade i blått).
Det första målet för denna forskning var att studera mångsidigheten av kata-lysatorn, för kinetisk resolvering av aromatiska alkoholer som innehåller en andra funktionell grupp (FG), och få en enantiomeriskt ren form av alkohol-er som kan användas som startmaterial för viktiga föreningar, såsom läke-medel, bekämpningsmedel, livsmedelstillsatser, etc. I detta arbete har vi utvecklat processer för tre nya typer av molekyler och visat dess tillämpning genom att framställa endast en av de två enantiomererna (spegel-bildsmolekylerna) av läkemedelsmolekyler (Figur 4).
Det andra målet med denna avhandling har varit att öka förståelsen för de katalytiska processer som har utvecklats i detta arbete. Genom att noggrannt följa hur snabbt denna process sker, det vill säga hur snabbt katalysatorn omvandlar en av enantiomererna, kan man dra slutsatser om hur processen
(S)-Substrat
KiralKatalysator
(R)-Substrat
(S)-Produkt
(R)-ProduktLångsam
+ +
Snabb
45
går till. Dessa studier har gett ny kunskap om funktionen av katalysatorn, vilket kan bidra till vidareutveckling av denna typ av katalytiska processer.
Figur 4. Katalytisk kinetisk resolvering utvecklade i denna avhandling ( = kiral katalysator; = erhållet substrat vid slutet av reaktionen).
t-amyl alkohol0 °C
Ar
OHFG
Ar
OAcFG
(−)-8
Ac2O
-FG = -N3, -P(O)(OR)2 och -COORR= -CH3, -CH2CH3 eller -C(CH3)3
(−)-8 (1-2 mol%)
N
Me2N
FePhPh
PhPhPh
+Ar
OHFG
enantiomerrena läkemedelsmolekyler

46
Resumen divulgativo en español
En química, una molécula es quiral cuando no puede ser superpuesta con su imagen especular, es decir, con su imagen reflejada en un espejo. Un ejem-plo sencillo son nuestras propias manos: la mano derecha no es superponible con su imagen especular, que es la mano izquierda (Figura 1). Así, las moléculas quirales existen en dos formas especulares, llamadas enantióme-ros, que comparten la misma composición química, pero poseen una dis-tribución espacial diferente.
Figura 1. Representación de objetos quirales: enantiómeros del aminoácido alanina y un par de manos.
La quiralidad es un fenómeno común en el universo y, además, la naturaleza suele preferir uno de los enantiómeros frente al otro. Por ejemplo, proteínas y receptores, responsables de muchas de las funciones vitales para los seres vivos, están compuestos por aminoácidos que normalmente sólo existen en una de las dos formas posibles. Esto significa que las proteínas y los recep-tores son quirales y pueden interactuar de diferentes maneras con otros enan-tiómeros. Así, cada enantiómero de carvona interactúa de manera distinta con los receptores olfativos, y percibimos el olor a menta verde de un enan-tiómero mientras que el otro huele a alcaravea (Figura 2).
Figura 2. Enantiómeros de carvona.
L-alanina(S)-enantiómero
D-alanina(R)-enantiómero
COOH
H
H2NCH3H3C
NH2
H
COOH
no pueden superponerse
H3C NH2
H
COOH
HNH2
CH3
COOHespejo
espejo
(R)-carvonamenta verde
(S)-carvonaalcaravea
O
H H
O

47
Este fenómeno es aún más crucial cuando se trata de un fármaco quiral, cu-yos enantiómeros pueden presentar diferentes efectos farmacológicos. En el caso más extremo, sólo uno de los enantiómeros tiene un efecto beneficioso, mientras que el otro es perjudicial para la salud. Éste es el caso de (S)-Naproxeno, que tiene efectos antiinflamatorios beneficiosos, mientras que el otro enantiómero, (R)-Naproxeno, causa daños en el hígado. Por este motivo, una gran parte de los fármacos contienen únicamente un enantiómero. Esto significa que las fármacos tienen que ser sintetizados y purificados de tal manera que sólo uno de los enantiómeros esté presente.
Los avances recientes en síntesis asimétrica y técnicas de separaciones de enantiómeros han fomentado la preparación de fármacos más seguros y es-pecíficos. Hoy en día, uno de los objetivos principales de los químicos sin-téticos continúa siendo el desarrollo de métodos de preparación de com-puestos enantioméricamente puros que sean eficaces y que se adapten a las diferentes necesidades de cada laboratorio. Para ello, se utilizan cataliza-dores químicos, que son compuestos que aceleran la reacción química sin ser consumidos en ella.
En esta tesis, nos hemos centrado en el estudio de un catalizador quiral que es capaz de transformar más rápidamente uno de los enantiómeros de una molécula quiral, de manera que el otro permanece prácticamente inal-terado (Figura 3). Este método, conocido como Resolución Cinética, es uti-lizado comúnmente en la industria para la preparación de compuestos enan-tioméricamente puros.
Figura 3. Esquema general de una resolución cinética catalítica (el sustrato y producto recuperados al final de la reacción están marcados en azul).
El primer objetivo de esta investigación ha sido estudiar la versatilidad del catalizador chiral utilizado para la resolución cinética de alcoholes aromáti-cos que contienen un segundo grupo funcional (FG), para así obtener un sólo enantiómero del alcohol (Figura 4). Así, se ha llevado a cabo la resolución cinética de tres familias distintas de alcoholes y se ha aplicado en la produc-ción de moléculas bioactivas (fármacos).
El segundo objetivo de esta tesis ha sido ampliar el conocimiento sobre los procesos catalíticos que se han desarrollado durante este trabajo. Gracias a la monitorización continua y completa del proceso catalítico, se ha podido estudiar los factores que influyen en la velocidad de reacción. Estos estudios han proporcionado nuevos conocimientos sobre la función del catalizador, lo
(S)-Substrato
Catalizadorquiral
(R)-Substrato
(S)-Producto
(R)-ProductoLento
+ +
Rápido
48
que puede contribuir a un mayor desarrollo de este tipo de procesos catalíti-cos.
Figura 4. Resolución cinética catalítica desarrollada en esta tesis ( = catalizador quiral; = substrato obtenido al final de la reacción).
t-amyl alcohol0 °C
Ar
OHFG
Ar
OAcFG
(−)-8
Ac2O
-FG = -N3, -P(O)(OR)2 y -COORR= -CH3, -CH2CH3 o -C(CH3)3
(−)-8 (1-2 mol%)
N
Me2N
FePhPh
PhPhPh
+Ar
OHFG
Fármacosenantiopuros

49
Acknowledgements
First of all, I would like to thank Professor Peter Dinér. I am grateful for having had the opportunity to be under your supervision. Thank you for guiding me through these years.
I would like to express my gratitude to Professor Henrik Ottosson, who encouraged me to apply for this PhD position four years ago.
Thanks to Professor Ramón Alajarín, because it was during the time that I was a scholarship holder in his group that I decided I wanted to perform a PhD. Thanks for all the good advises.
Big thanks to Huan Ma, Alban Cadu, Carmen Herrera, Supaporn Sawad-joon and the PD group for reading my thesis and giving valuable comments.
The PD group has been small, but all the members have meant a lot to me. Jia-Fei Poon, my first lab mate, always happy! Good luck with your thesis! Sebastian Kitzig, you are a live wire! I am sure you are doing very well in your PhD now in Berlin. Alban Cadu, or was it a fleeting visit to our lab? And last but not least Dr. Alba E. Díaz-Álvarez who has accompanied me through half of my PhD, to make the Spanish lab louder than any, to share complaints, successful experiences and lot of cat pictures! I also want to thank the current PD group at KTH university: Björn Blomkvist for creat-ing such a nice environment and for your patience when having a conversa-tion in Swedish with me. Good luck with your research!
I would like to thank all the persons with whom I shared the lab during all these years: Alex, Maxim, Joakin, Supaporn and Christian (although now he is enjoying his little boy). We may have different perceptions of cleanness sometimes but it has been a delight to work with you! Also special thanks to Professor Joseph Samec, even if I finally became the “intruder” in his lab, he always was concern about me, especially during this last year when my group moved to Stockholm.
These four years in this department have been unforgettable. There has been a lot of work in the lab, which would be impossible to perform without the technicians: Gunnar S., Bo F., and Tomas K.. In addition, there has also been bureaucracy and paperwork unimaginable to go through without the support of Eva O. and, especially Johanna J.. Thanks for all the help!
The lunchtime was not only the moment to fill the stomach but to ex-change ideas, problems, anecdotes and laughs. For this, I am happy to have had all the lovely people that has been in this department during this years: Aleksandra B., Aleksandra D., Andreas, Angelica, Anna L., Anon, Carina,

50
Derar, Emilien, Jiajie, Karthik, Rabia, Rikard, Xu, Åsa,… to name some of them.
Special thanks go to my favorite biochemist, Huan: thank you so much for being a “happy horse” and for being there during the good moments and more important, the bad ones. I am very glad that our friendship has grown so much these years!
Thanks to the people in KTH: Anna, Fredrik, Laure, Quentin, Robin, Bri-an, Lei, Lei… It has been a great pleasure to meet you.
After a hard working week… the best is to relax with good friends. Thanks to Pieter, Giulia, Davide, Mahsa, Ana Rosa, Carol, Fred and the rest of the Uppsala crew for all the good moments together, specially the crazy parties! My deepest gratitude also to the Spanish people: José, Alberto, Guillermo, and my girls María, Ana M., and Eva. Thanks for bringing the best things from our country (I do not mean the chorizo, now). It is warmer here with you all around. And I am not forgetting anyone, but I wanted to specially thank Carmen: five years together in Uppsala! Thank you for being my mother when I was sick and alone, my sister to share confidences, my colleague to talk about science and, of course, for being such a good friend! Gracias ratona.
I would like to thank all my friends in Spain, for make me feel I never left. Gracias Ana, Carol, Gloria y Marta por los ricos vinos tintos y los men-ta-poleo. Begoña, Eva y Sara por las noches de gossip; María, Marta, Car-los, Javier y Jorge por hacer que los años de Universidad fueran más leves.
Me gustaría agradecer a toda mi familia por su apoyo. En primer lugar, agradecer a mis padres por alentarme en los momentos difíciles, por creer siempre en mí y por valorar mi trabajo incluso sin llegar a comprenderlo muy bien. A ellos especialmente les dedico este libro. Gracias a mi hermano José Luis, por ser un ejemplo de superación. Gracias a mi hermana mayor Carolina, por hacerme sentir tan orgullosa de su arte y creer que algún día podría ser su mecenas (creo que no elegí la profesión adecuada, lo siento). Gracias a mi hermana Mónica, por los buenos momentos que hemos pasado, especialmente en la aventura de traer el pequeño Corsa desde Madrid a Upp-sala. Además, me gustaría felicitar a ella y a Miguel A., por el bebé que está en camino. Y agradecerle a él, mi sobrino Miguelín, porque sin aún haber nacido, ha sido una de las personas que más me ha animado durante este último sprint, porque sé que después de defender mi tesis podré conocerle.
Por último quiero agradecerle a Fernando, por venir a Suecia conmigo, por estar todo este tiempo junto a mí, apoyándome, por su amor, por su sen-tido del humor y por ser un sol, sobretodo en los oscuros inviernos. Gracias especialmente por éste último año lleno de emociones, ¡somos el mejor equipo! I love you. Thanks, Gracias och Tack så mycket.

51
References
1. IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins.
2. D. Nasipuri, Stereochemistry of Organic Compounds: Principles and Applications, Wiley Eastern, New Delhi, India, 1994.
3. U. Meierhenrich, Amino acids and the asymmetry of life: caught in the act of formation, Springer, Berlin, Germany, 2008.
4. M. F. Dunn, Protein-ligand interactions: general description. In eLS, John Wiley & Sons Ltd., Chichester, UK, 2010.
5. G. Ohloff, Experientia 1986, 42, 271-279. 6. J. C. Brookes, A. P. Horsfield, A. M. Stoneham, J. R. Soc. Interface 2009,
6, 75-86. 7. L. B. Buck, Angew. Chem. Int. Ed. 2005, 44, 6128-6140. 8. U. Ravid, E. Putievsky, I. Katzir, V. Weinstein, R. Ikan, Flavour Frag. J.
1992, 7, 289-292. 9. H. Morris, J. Wallach, Drug Test. Anal. 2014, 6, 614-632. 10. (a) A. Barbeau, Can. Med. Assoc. J. 1969, 101, 59-68; (b) D. N. Wade, P.
T. Mearrick, J. L. Morris, Nature 1973, 242, 463-465; (c) T. Nagatsu, M. Sawada, Parkinsonism Rel. Disord. 2009, 15, Suppl 1, S3-S8.
11. Q.-D. You, J.-F. Cheng, Chiral Drugs : Chemistry and Biological Action, John Wiley & Sons, Somerset, NJ, US, 2011.
12. FDA Guidances (Drugs). Development of New Stereoisomeric Drugs. http://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm122883.htm (access date 29/09/2014).
13. I. Agranat, H. Caner, J. Caldwell, Nat. Rev. Drug Discov. 2002, 1, 753-768. 14. (a) V. Farina, J. T. Reeves, C. H. Senanayake, J. J. Song, Chem. Rev. 2006,
106, 2734-2793; (b) A. Rouf, S. C. Taneja, Chirality 2014, 26, 63-78. 15. Top 10 best selling drugs in US during 2013.
http://www.drugs.com/stats/top100/sales (access date 31/07/2014). 16. T. Lind, L. Rydberg, A. Kylebäck, A. Jonsson, T. Andersson, G.
Hasselgren, J. Holmberg, K. Röhss, Aliment. Pharmacol. Ther. 2000, 14, 861-867.
17. P. Wyatt, S. Warren, Stereochemistry. In Organic synthesis: strategy and control, John Wiley & Sons Ltd., Chichester, UK, 2007, pp. 371-717.
18. Synthetic Methods I - Chiral Pool and Diastereoselective Methods. In Comprehensive Chirality, Vol. 2 (Eds.: E. M. Carreira, H. Yamamoto), Elsevier, Amsterdam, Netherlands, 2012.

52
19. R. Siedlecka, Tetrahedron 2013, 69, 6331-6363. 20. J. M. Keith, J. F. Larrow, E. N. Jacobsen, Adv. Synth. Catal. 2001, 343, 5-
26. 21. (a) H. B. Kagan, J. C. Fiaud, Top. Stereochem. 1988, 18, 249-330; (b) J. M.
Goodman, A.-K. Köhler, S. C. M. Alderton, Tetrahedron Lett. 1999, 40, 8715-8718.
22. S. Servi, Synthesis 1990, 1-25. 23. J. M. J. Williams, R. J. Parker, C. Neri, Enzymatic Kinetic Resolution. In
Enzyme Catalysis in Organic Synthesis (Eds.: K. Drauz, H. Waldmann), Wiley-VCH Verlag GmbH, Weinheim, Germany, 2008, pp. 287-312.
24. H. Pellissier, Use of transition metals and enzymes in tandem. In Chirality from Dynamic Kinetic Resolution (Ed.: H. Pellissier), The Royal Society of Chemistry, Cambridge, UK, 2011, pp. 191-242.
25. (a) B. Martín-Matute, J.-E. Bäckvall, Curr. Opin. Chem. Biol. 2007, 11, 226-232; (b) P. Hoyos, V. Pace, A. R. Alcántara, Adv. Synth. Catal. 2012, 354, 2585-2611.
26. B. Martín-Matute, M. Edin, K. Bogár, F. B. Kaynak, J.-E. Bäckvall, J. Am. Chem. Soc. 2005, 127, 8817-8825.
27. R. Noyori, Angew. Chem. Int. Ed. 2002, 41, 2008-2022. 28. (a) V. S. Martin, S. S. Woodard, T. Katsuki, Y. Yamada, M. Ikeda, K. B.
Sharpless, J. Am. Chem. Soc. 1981, 103, 6237-6240; (b) Y. Kitano, T. Matsumoto, F. Sato, Tetrahedron 1988, 44, 4073-4086.
29. H. Pellissier, Adv. Synth. Catal. 2011, 353, 1613-1666. 30. H. Pellissier, Chiral catalysts. In Chirality from Dynamic Kinetic
Resolution (Ed.: H. Pellissier), The Royal Society of Chemistry, Cambridge, UK, 2011, pp. 49-143.
31. A. Berkessel, H. Gröger, Kinetic resolution of racemic alcohols and amines. In Asymmetric organocatalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005, pp. 323-346.
32. C. E. Müller, P. R. Schreiner, Angew. Chem. Int. Ed. 2011, 50, 6012-6042. 33. (a) E. Vedejs, O. Daugulis, S. T. Diver, J. Org. Chem. 1996, 61, 430-431;
(b) E. Vedejs, O. Daugulis, J. A. MacKay, E. Rozners, Synlett 2001, 1499-1505; (c) E. Vedejs, O. Daugulis, J. Am. Chem. Soc. 2003, 125, 4166-4173.
34. (a) V. B. Birman, X. Li, Org. Lett. 2006, 8, 1351-1354; (b) V. B. Birman, L. Guo, Org. Lett. 2006, 8, 4859-4861; (c) B. Hu, M. Meng, Z. Wang, W. Du, J. S. Fossey, X. Hu, W.-P. Deng, J. Am. Chem. Soc. 2010, 132, 17041-17044; (d) B. Hu, M. Meng, J. S. Fossey, W. Mo, X. Hu, W.-P. Deng, Chem. Commun. 2011, 47, 10632-10634; (e) X. Li, H. Jiang, E. W. Uffman, L. Guo, Y. Zhang, X. Yang, V. B. Birman, J. Org. Chem. 2012, 77, 1722-1737.
35. (a) W. Steglich, G. Höfle, Angew. Chem. Int. Ed. 1969, 8, 981-981; (b) G. Höfle, W. Steglich, Synthesis 1972, 619-621.
36. Litvinen.L. M., Kirichen.A. I., Dokl. Akad. Nauk SSSR 1967, 176, 97-100. 37. R. Murugan, E. F. V. Scriven, Aldrichimica Acta 2003, 36, 21-27. 38. R. P. Wurz, Chem. Rev. 2007, 107, 5570-5595. 39. E. Vedejs, X. Chen, J. Am. Chem. Soc. 1996, 118, 1809-1810.

53
40. (a) A. C. Spivey, T. Fekner, H. Adams, Tetrahedron Lett. 1998, 39, 8919-8922; (b) A. C. Spivey, T. Fekner, S. E. Spey, H. Adams, J. Org. Chem. 1999, 64, 9430-9443.
41. A. C. Spivey, T. Fekner, S. E. Spey, J. Org. Chem. 2000, 65, 3154-3159. 42. J. C. Ruble, G. C. Fu, J. Org. Chem. 1996, 61, 7230-7231. 43. I. Held, A. Villinger, H. Zipse, Synthesis 2005, 1425-1430. 44. J. C. Ruble, H. A. Latham, G. C. Fu, J. Am. Chem. Soc. 1997, 119, 1492-
1493. 45. J. C. Ruble, J. Tweddell, G. C. Fu, J. Org. Chem. 1998, 63, 2794-2795. 46. B. Tao, J. C. Ruble, D. A. Hoic, G. C. Fu, J. Am. Chem. Soc. 1999, 121,
5091-5092. 47. S. Bellemin-Laponnaz, J. Tweddell, J. C. Ruble, F. M. Breitling, G. C. Fu,
Chem. Commun. 2000, 1009-1010. 48. S. Y. Lee, J. M. Murphy, A. Ukai, G. C. Fu, J. Am. Chem. Soc. 2012, 134,
15149-15153. 49. L. G. Wade, In Organic Chemistry, Pearson Education (Us), n.p., 2013,
pp. 467-512. 50. (a) D.-M. Lee, J.-C. Lee, N. Jeong, K.-I. Lee, Tetrahedron: Asymmetry
2007, 18, 2662-2667; (b) S. Connolly, L. Alcaraz, A. Bailey, E. Cadogan, J. Christie, A. R. Cook, A. J. Fisher, S. Hill, A. Humphries, A. H. Ingall, Z. Kane, S. Paine, G. Pairaudeau, M. J. Stocks, A. Young, Bioorg. Med. Chem. Lett. 2011, 21, 4612-4616.
51. D. J. Ager, I. Prakash, D. R. Schaad, Chem. Rev. 1996, 96, 835-876. 52. S. Brase, C. Gil, K. Knepper, V. Zimmermann, Angew. Chem. Int. Ed.
2005, 44, 5188-5240. 53. (a) F. M. D. Ismail, D. O. Levitsky, V. M. Dembitsky, Eur. J. Med. Chem.
2009, 44, 3373-3387; (b) H. Pellissier, Tetrahedron 2010, 66, 1509-1555. 54. H. Pellissier, Tetrahedron 2007, 63, 3235-3285. 55. (a) O. Pàmies, J.-E. Bäckvall, J. Org. Chem. 2001, 66, 4022-4025; (b) E. C.
S. Brenelli, J. L. N. Fernandes, Tetrahedron: Asymmetry 2003, 14, 1255-1259; (c) A. Kamal, A. A. Shaik, M. Sandbhor, M. S. Malik, Tetrahedron: Asymmetry 2004, 15, 935-939.
56. (a) J. Ong, D. I. B. Kerr, J. Abbenante, R. H. Prager, Eur. J. Pharmacol. 1991, 205, 319-322; (b) J. Ong, V. Marino, D. A. S. Parker, D. I. B. Kerr, Naunyn-Schmiedeberg's Arch. Pharmacol. 1998, 357, 408-412.
57. C. Xu, C. Yuan, Eur. J. Org. Chem. 2004, 4410-4415. 58. (a) Y. Zhang, Z. Li, C. Yuan, Tetrahedron Lett. 2002, 43, 3247-3249; (b)
A. Woschek, W. Lindner, F. Hammerschmidt, Adv. Synth. Catal. 2003, 345, 1287-1298.
59. A. Moriyama, S. Matsumura, M. Kuriyama, O. Onomura, Tetrahedron: Asymmetry 2010, 21, 810-824.
60. T. J. Wenzel, C. D. Chisholm, Chirality 2011, 23, 190-214. 61. E. T. Choi, M. H. Lee, Y. Kim, Y. S. Park, Tetrahedron 2008, 64, 1515-
1522. 62. J. Boot, M. Cases, B. P. Clark, J. Findlay, P. T. Gallagher, L. Hayhurst, T.
Man, C. Montalbetti, R. E. Rathmell, H. Rudyk, M. W. Walter, M. Whatton, V. Wood, Bioorg. Med. Chem. Lett. 2005, 15, 699-703.

54
63. (a) R. W. Fuller, H. D. Snoddy, Pharmacol. Biochem. Behav. 1986, 24, 281-284; (b) D. W. Robertson, J. H. Krushinski, R. W. Fuller, J. D. Leander, J. Med. Chem. 1988, 31, 1412-1417.
64. (a) I. S. Ali, A. Sudalai, Tetrahedron Lett. 2002, 43, 5435-5436; (b) H. Kakei, T. Nemoto, T. Ohshima, M. Shibasaki, Angew. Chem. Int. Ed. 2004, 43, 317-320; (c) M. Panunzio, K. Rossi, E. Tamanini, E. Campana, G. Martelli, Tetrahedron: Asymmetry 2004, 15, 3489-3493; (d) G. Wang, X. Liu, G. Zhao, Tetrahedron: Asymmetry 2005, 16, 1873-1879; (e) G. L. Khatik, V. Kumar, V. A. Nair, Org. Lett. 2012, 14, 2442-2445.
65. (a) F. F. Huerta, J.-E. Bäckvall, Org. Lett. 2001, 3, 1209-1212; (b) J. Brem, M. C. Turcu, C. Paizs, K. Lundell, M.-I. Toşa, F.-D. Irimie, L. T. Kanerva, Process Biochem. 2012, 47, 119-126.
66. H. Zhou, Q. Xu, P. Chen, Tetrahedron 2008, 64, 6494-6499. 67. J. F. Larrow, S. E. Schaus, E. N. Jacobsen, J. Am. Chem. Soc. 1996, 118,
7420-7421. 68. (a) T. Kawabata, M. Nagato, K. Takasu, K. Fuji, J. Am. Chem. Soc. 1997,
119, 3169-3170; (b) Y. Wei, I. Held, H. Zipse, Org. Biomol. Chem. 2006, 4, 4223-4230; (c) X. Li, P. Liu, K. N. Houk, V. B. Birman, J. Am. Chem. Soc. 2008, 130, 13836-13837.
69. (a) A. C. Spivey, S. Arseniyadis, Angew. Chem. Int. Ed. 2004, 43, 5436-5441; (b) S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich, H. Zipse, Chem. Eur. J. 2005, 11, 4751-4757.
70. E. Larionov, M. Mahesh, A. C. Spivey, Y. Wei, H. Zipse, J. Am. Chem. Soc. 2012, 134, 9390-9399.
71. A. J. Wagner, S. D. Rychnovsky, Org. Lett. 2013, 5504–5507. 72. D. G. Blackmond, Angew. Chem. Int. Ed. 2005, 44, 4302-4320. 73. J. S. Mathew, M. Klussmann, H. Iwamura, F. Valera, A. Futran, E. A. C.
Emanuelsson, D. G. Blackmond, J. Org. Chem. 2006, 71, 4711-4722.


Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 1191
Editor: The Dean of the Faculty of Science and Technology
A doctoral dissertation from the Faculty of Science andTechnology, Uppsala University, is usually a summary of anumber of papers. A few copies of the complete dissertationare kept at major Swedish research libraries, while thesummary alone is distributed internationally throughthe series Digital Comprehensive Summaries of UppsalaDissertations from the Faculty of Science and Technology.(Prior to January, 2005, the series was published under thetitle “Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-233734
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2014


















