
July 2021- National Hemochromatosis Awareness Month
24
July 2021- National Hemochromatosis Awareness Month A word from the author— Happy July NCP friends! Over the last two weeks, I have had several conversations with family members about health concerns. As a nurse, I want to emphasize to all the important people in my life how incredibly important it is to be your own health advocate. What does this mean? This means asking those questions… being that patient. If you go to the doctor, don’t be afraid to ask questions, why are you being prescribed that medication? What does this new diagnosis mean and how will it impact your life? What can you do to help prevent needing surgery, another medication or treatment plan, or stop the growth of something that you do not want getting bigger? If you do not understand, tell your doctor, nurse practitioner, or physician’s assistant that you do not understand. Educate yourself. And above all else, know your body. Only you know how you feel every day. Did you wake up and not feel quite right? Did that pain just start? If something is not right… do not ignore your body. Pay attention and call your doctor. And if you need someone to talk to, there are a ton of social media outlets, support groups, and medical professionals to help you navigate through any and all of your medical concerns. This month, the NCC Health Ministry awareness topic is focused on a condition that, although becoming increasingly more prevalent in society, is still very much misunderstood by the general public. Hemochromatosis (pronounced heme- o – chrome- a- tosis) is considered the most common genetic disorder in the world, yet most people dismiss it as “rare.” Why? This condition so often goes untreated that it is rarely diagnosed before symptoms manifest. However, if individuals are diagnosed with the condition early, many of the complications are reversible and the symptom severity can be significantly decreased with treatment. What is Hemochromatosis? Hemochromatosis is an iron storage disorder that can cause the body to absorb too much iron from foods and other sources, such as multivitamin supplements with iron. Iron is an essential nutrient found in many foods. Iron carries oxygen (in hemoglobin) to all parts of the body. Normally, humans absorb about 8-10% of the iron in foods that they eat. People with Hemochromatosis (too much iron) can absorb four times more iron than normal. This extra iron can gradually build up in the body’s tissues and organs, particularly in the liver cells, heart, pancreas, joints, and pituitary gland. If left untreated,
Transcript of July 2021- National Hemochromatosis Awareness Month

July 2021- National Hemochromatosis Awareness Month
A word from the author—
Happy July NCP friends! Over the last two weeks, I have had several conversations with family members about health concerns. As a nurse, I want to emphasize to all the important people in my life how incredibly important it is to be your own health advocate. What does this mean? This means asking those questions… being that patient. If you go to the doctor, don’t be afraid to ask questions, why are you being prescribed that medication? What does this new diagnosis mean and how will it impact your life? What can you do to help prevent needing surgery, another medication or treatment plan, or stop the growth of something that you do not want getting bigger? If you do not understand, tell your doctor, nurse practitioner, or physician’s assistant that you do not understand. Educate yourself. And above all else, know your body. Only you know how you feel every day. Did you wake up and not feel quite right? Did that pain just start? If something is not right… do not ignore your body. Pay attention and call your doctor. And if you need someone to talk to, there are a ton of social media outlets, support groups, and medical professionals to help you navigate through any and all of your medical concerns.
This month, the NCC Health Ministry awareness topic is focused on a condition that, although becoming increasingly more prevalent in society, is still very much misunderstood by the general public. Hemochromatosis (pronounced heme- o – chrome- a- tosis) is considered the most common genetic disorder in the world, yet most people dismiss it as “rare.” Why? This condition so often goes untreated that it is rarely diagnosed before symptoms manifest. However, if individuals are diagnosed with the condition early, many of the complications are reversible and the symptom severity can be significantly decreased with treatment.
What is Hemochromatosis? Hemochromatosis is an iron storage disorder that can cause the body to absorb too much iron from foods and other sources, such as multivitamin supplements with iron. Iron is an essential nutrient found in many foods. Iron carries oxygen (in hemoglobin) to all parts of the body. Normally, humans absorb about 8-10% of the iron in foods that they eat. People with Hemochromatosis (too much iron) can absorb four times more iron than normal. This extra iron can gradually build up in the body’s tissues and organs, particularly in the liver cells, heart, pancreas, joints, and pituitary gland. If left untreated,

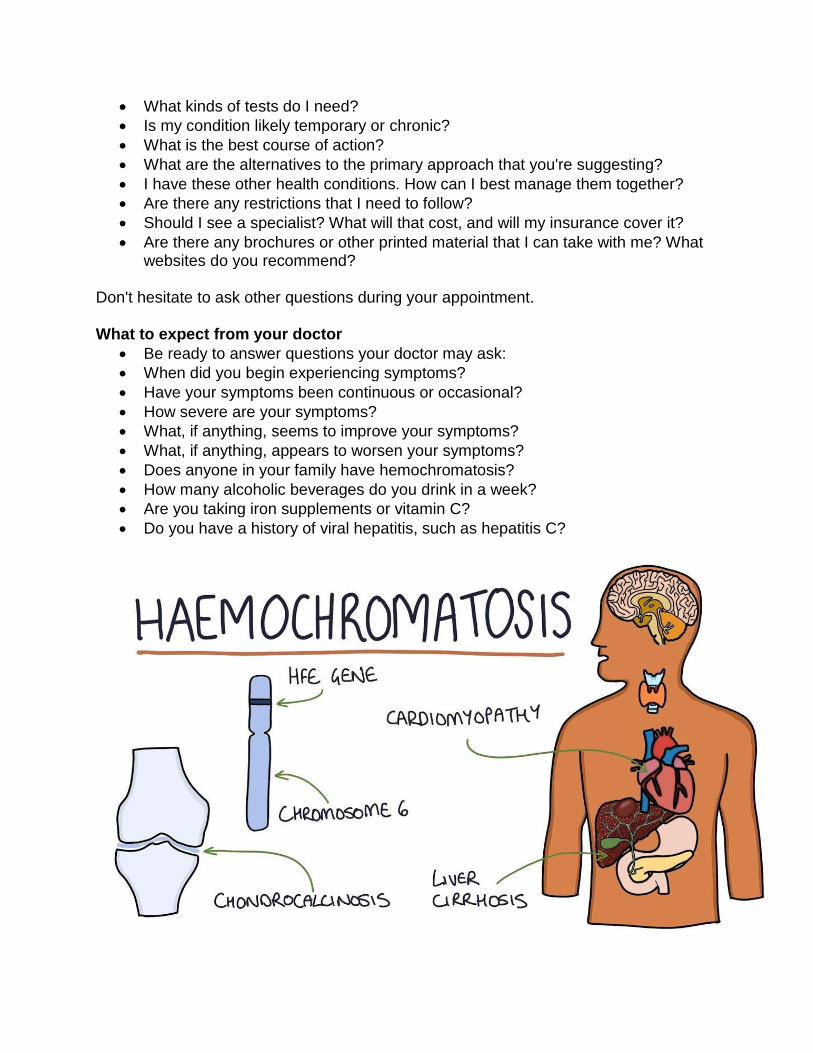
this iron buildup may lead to tissue and organ damage over time because the excess iron can poison these organs. Life-threatening conditions may result, such as cancer, heart arrhythmias, and cirrhosis of the liver. Primary hemochromatosis, also called hereditary hemochromatosis, is an inherited disorder. Secondary hemochromatosis is caused by anemia, alcoholism, and other disorders. In the United States, the most common form of hemochromatosis in adults is hereditary hemochromatosis.
Hereditary hemochromatosis is caused mainly by specific inherited alterations (mutations) in the HFE gene. When an individual inherits two altered copies of the gene—one from each parent—they are at risk of developing high iron levels which may lead to illness or organ damage over time. However, most people born with two altered copies of the HFE gene will not develop serious complications. People who inherit an altered gene from only one parent are carriers for the disorder, but are not typically affected themselves.
Juvenile hemochromatosis causes the same problems in young people that hereditary hemochromatosis causes in adults. But iron accumulation begins much earlier, and symptoms usually appear between the ages of 15 and 30. This disorder is caused by a mutation in the HJV gene. Neonatal hemochromatosis is a severe disorder during which iron builds up rapidly in the liver of the developing fetus. It is thought to be an autoimmune disease, in which the body attacks itself. Secondary hemochromatosis is the form of the disease that is not inherited. The iron deposits the same way, but are due to another disease, such as anemia, chronic liver disease or an infection. Secondary hemochromatosis is caused by disorders of

erythropoiesis and treatment of the diseases with blood transfusions. After damage of transfused erythrocytes by macrophages, iron freed from heme is accumulated in the body (liver, heart, skin). Secondary hemochromatosis is mainly induced by diseases of erythropoiesis, including thalassemia, sickle cell anemia, X-linked sideroblastic anemia, pyruvate kinase deficiency, hereditary spherocytosis, and congenital dyserythropoietic anemia (CDA).
Why Hemochromatosis is different than other health awareness topics Millions of people around the world, especially in Western countries, carry the genes for a disorder which, if not detected in time, results in a myriad of crippling and, far too often, fatal diseases; among them is cancer of the liver, which occurs 200 times more frequently in patients with this defect than in the general population. There could be someone at risk in every family, business, factory or office—depending on the genetic or ethnic background of the people concerned. In the early 1990s, one Vancouver doctor predicted that it could become the “scourge of the 21st century.” A Mayo Clinic doctor says, ”The disorder has reached epidemic proportions.”
How does hemochromatosis affect organs? Iron plays an essential role in several body functions, including helping in the formation of blood. A peptide hormone called hepcidin, secreted by the liver, plays a key role in the body's use of iron. It controls how much iron is absorbed by the intestines, how iron is used in various body processes and how it's stored in various organs. In hemochromatosis, the normal role of hepcidin is disrupted and your body absorbs more iron that it needs. This excess iron is stored in the tissues of major organs,

especially your liver. Too much iron is toxic to your body, and over a period of years, the stored iron can severely damage many organs, leading to organ failure and chronic diseases such as cirrhosis, diabetes and heart failure. Adults preserve a constant level of body iron by efficient conservation, maintaining rigorous control over absorption to balance losses. An adult man loses approximately 1 mg of iron daily, mostly in desquamated epithelium and secretions from the gut and skin. During the childbearing years, healthy women lose an average of an additional milligram of iron daily from menstrual bleeding (40 mL blood loss) and approximately 500 mg with each pregnancy. In addition, normal daily fecal loss of approximately 0.7 mL of blood (0.3 mg of iron) occurs. Only a small quantity of iron is excreted in urine (< 0.1 mg/d). In healthy adults, losses are balanced by absorption of sufficient dietary iron (1-2 mg) to maintain a relatively constant amount of body iron throughout life. Although excretion is quantitatively as important as absorption in the maintenance of iron balance, absorption usually plays the more active regulatory role. In hereditary hemochromatosis, dysregulation of intestinal iron absorption occurs, wherein iron continues to be efficiently absorbed even in the face of substantial elevation of body iron stores. Though many people have faulty genes that cause hemochromatosis, only about 10 percent of them have iron overload to the degree that causes tissue and organ damage.
Hemochromatosis- the “rare” common genetic disorder According to the CDC, hemochromatosis is misdiagnosed an average of 67% of the time. Although it is the most common genetic disorder (declared thus by the Centers for Disease Control and Prevention), many physicians, having been led to believe that it is so rare, are not on the alert for signs of the disease—because they simply do not expect

to find them.It is the only genetic disorder of which all the dreadful complications are preventable. We all probably know someone who has the disease, even if they don’t know it themselves. Are you the one among the possible four to six “carriers” on a bus, the 50+ on a ferry—or can it be your child who is among the three or more in an average class of school children? The Facts about Hemochromatosis It is transmitted as an autosomal, recessive disorder, and every child will be a carrier. According to the World Health Organization report of the meeting on the prevention and control of hemochromatosis on April 9, 1993, liver cancer was found 200 times more frequently among persons with hemochromatosis than in the general population. It is a condition that only becomes a disease when sufficient excess iron has been accumulated in the body to cause complications. It is thus the only inherited disorder of which all the complications are preventable, and the only one which is treatable with the right intervention. It takes time for iron overload to reach a level that will cause organ damage and failure. Men typically develop disease between 40 and 60 years of age, and women after menopause. Diet, vitamin pills with iron, and alcohol consumption all can have an effect. “The Celtic Curse” In some parts of the world, Hemochromatosis is known as the “celtic curse.” The best description I have found, in laymen’s terms, for Hemochromatosis was written by Stephen Cobb. I have included a large excerpt from his article below to help reinforce understanding. The article can be found here at the following URL as well http://celticcurse.org/july-is-hemochromatosis-awareness-month/

July is Hemochromatosis Awareness Month by STEPHEN COBB “Did you know that July is National Hemochromatosis Awareness Month? That simple sentence is all you need to start a conversation about the Celtic Curse. Some people may respond with “Hemo-what-osis?” and that’s when you hook them with “You know, Celtic Curse?” We’ve struck up conversations like that with lots of different people and so far nobody has objected. Most people are fascinated to learn more about a potentially fatal condition, one that might run in the family, might be killing relatives, and might explain why Uncle Fred had cirrhosis of the liver even though he swore he never touched a drop. (Here’s a hint: Uncle Fred might have been telling the truth–while drinking to excess is never a good idea, hemochromatosis can damage your liver in ways that mimic the effects of alcohol consumption.)” What causes genetic hemochromatosis? Hereditary hemochromatosis is caused by a mutation in a gene that controls the amount of iron your body absorbs from the food you eat. The mutations that cause hereditary hemochromatosis are passed from parents to children. The gene that is mutated most often in people with hereditary hemochromatosis is called HFE. You inherit one HFE gene from each of your parents. The HFE gene has two common mutations, C282Y and H63D. One of these mutations is found in about 85 percent of people who have hereditary hemochromatosis. Genetic testing can reveal whether you have these mutations in your HFE gene.
If you inherit 2 abnormal genes, you may develop hemochromatosis. About 70 percent of people who inherit two genes develop evidence of iron overload of hemochromatosis. You can also pass the mutation on to your children. If you inherit 1 abnormal gene, you won't develop hemochromatosis. You are considered a gene mutation carrier and can pass the mutation on to your children. They

would not develop disease unless they also inherit another abnormal gene from another parent. What are the risk factors for hemochromatosis? Having 2 copies of a mutated HFE gene. This is the greatest risk factor for hereditary hemochromatosis. Family history. If you have a first-degree relative — a parent or sibling — with hemochromatosis, you're more likely to develop the disease. If you have a family history of alcoholism, heart attacks, diabetes, liver disease, arthritis or impotence, your risk of hemochromatosis is greater. Ethnicity. People of Northern European descent are more prone to hereditary hemochromatosis than are people of other ethnic backgrounds. Hemochromatosis is less common in African-Americans, Hispanics and Asian-Americans. Being a man. Men are more likely to develop signs and symptoms of hemochromatosis at an earlier age. Because women lose iron through menstruation and pregnancy, they tend to store less of the mineral than men do. After menopause or a hysterectomy, the risk for women increases.
Who can develop hemochromatosis? Many people inherit the faulty genes that cause hemochromatosis — it is the most common genetic disease in Caucasians. But only a minority of those with the gene develop serious problems.

Caucasians are the people most at risk for the classic type of hemochromatosis. More than one million Americans have the genes for this type. However, there are other gene combinations that result in hemochromatosis regardless of a person’s ethnicity. It is estimated that as much or more than 16 Million Americans have some degree of elevated iron and as such as at risk for the same diseases that occur in people with untreated classic type: bone and joint disease, cirrhosis, liver cancer, diabetes, hypothyroidism, hypogonadism, infertility, impotence, depression or premature death due to liver or heart failure. Hemochromatosis is more likely to be serious in men. Men and women have the same chance of inheriting two copies of the altered HFE gene. However, men are more likely than women to develop complications and at an earlier age because women lose excess iron in the blood naturally during menstruation and pregnancy. Family history is an important risk factor for hereditary hemochromatosis. People who have a close biological relative with hereditary hemochromatosis due to two altered HFE genes have a higher chance of having the altered HFE gene themselves. For example, if you have hereditary hemochromatosis due to two altered HFE genes, then your siblings have a 1 in 4 chance of also having two altered HFEgenes.

What is the prevalence of Hemochromatosis? Hereditary hemochromatosis is one of the most common genetic disorders in the United States. People of Northern European descent have a higher chance of having an altered HFE gene. Hereditary hemochromatosis is more common among U.S. non-Hispanic whites, and is less common among African Americans, Asian Americans, Hispanics/Latinos, and American Indians. What are the symptoms of Hemochromatosis? Chronic fatigue and joint pain are the most common complaints of people with hemochromatosis. For this reason, the complete diagnosis is often delayed because these two symptoms are commonly seen in other diseases. Pain in the knuckles of the pointer and middle finger “The Iron Fist” is the only sign or symptom specific to hemochromatosis, but not everyone with HHC experiences the Iron Fist.
Some complain of the following symptoms with Hemochromatosis. However these symptoms are not specific to hemochromatosis: -Lack of energy -Abdominal pain -Memory fog -Loss of sex drive -Heart flutters -Irregular heart beat

Early signs and symptoms often are nonspecific, mimicking those of other common conditions. Common symptoms of hereditary hemochromatosis include: -Joint pain -Fatigue -Weakness First signs and symptoms of the disease in men are often from organ damage. They include: -Joint pain -Diabetes -Loss of sex drive (libido) -Impotence -Heart failure When signs and symptoms typically appear When symptoms are associated with hemochromatosis, these usually begin in men in their late 20’s to early 30’s. In women, symptoms usually start about 10-15 years after they stop having a period due to menopause, birth control pills or hysterectomy. Signs and symptoms of hereditary hemochromatosis usually appear in midlife, when iron levels tend to accumulate. Some people with hereditary hemochromatosis never have symptoms. Although hereditary hemochromatosis is present at birth, most people don't experience signs and symptoms until later in life — usually between the ages of 50 and 60 in men and after age 60 in women. Women are more likely to develop symptoms after menopause, when they no longer lose iron with menstruation and pregnancy.

When to go to the doctor See your doctor if you experience any of the signs and symptoms of hereditary hemochromatosis. If you have an immediate family member who has hemochromatosis, ask your doctor about genetic tests that can determine if you have inherited the gene that increases your risk of hemochromatosis. Make an appointment with your family doctor if you have any signs or symptoms that worry you. You may be referred to a specialist in digestive diseases (gastroenterologist), or to another specialist, depending on your symptoms. Here's some information to help you get ready for your appointment, and what to expect from your doctor.
What you can do:
• Be aware of any pre-appointment restrictions. At the time you make the appointment, be sure to ask if there's anything you need to do in advance, such as restrict your diet.
• Write down any symptoms you're experiencing, including any that may seem unrelated to the reason for which you scheduled the appointment.
• Write down key personal information, including any major stresses or recent life changes. Learn if you have any liver disease in your family by asking your family members, if possible.
• Make a list of all medications, vitamins or supplements that you're taking.
• Take a family member or friend along to help you remember things.
• Write down questions to ask your doctor. Some basic questions to ask your doctor include:
• What is likely causing my symptoms or condition?
• Are there other possible causes for my symptoms or condition?
• What kinds of tests do I need?
• Is my condition likely temporary or chronic?
• What is the best course of action?
• What are the alternatives to the primary approach that you're suggesting?
• I have these other health conditions. How can I best manage them together?
• Are there any restrictions that I need to follow?
• Should I see a specialist? What will that cost, and will my insurance cover it?
• Are there any brochures or other printed material that I can take with me? What websites do you recommend?
Don't hesitate to ask other questions during your appointment. What to expect from your doctor
• Be ready to answer questions your doctor may ask:
• When did you begin experiencing symptoms?
• Have your symptoms been continuous or occasional?
• How severe are your symptoms?
• What, if anything, seems to improve your symptoms?
• What, if anything, appears to worsen your symptoms?
• Does anyone in your family have hemochromatosis?
• How many alcoholic beverages do you drink in a week?
• Are you taking iron supplements or vitamin C?
• Do you have a history of viral hepatitis, such as hepatitis C?

Early diagnosis- the importance of being tested Two blood tests can also be used to screen people who may have iron buildup due to hereditary hemochromatosis. These blood tests measure how much iron is in the body. The U.S. Preventive Services Task Force recommends against routine genetic screening for hereditary hemochromatosis in the asymptomatic general population. Knowing your family health history can help you and your doctor understand your risk for hemochromatosis. It is helpful to talk with your family members about their health history, write this information down, update it from time to time, and share it with your doctor. Family health history information can help your doctor determine which tests and screenings are recommended to help you know your health risk. To learn more about family health history, visit:http://www.cdc.gov/genomics/famhistory. Diagnosing hemochromatosis Your doctor may suggest other tests to confirm the diagnosis and to look for other problems: Liver function tests. These tests can help identify liver damage. MRI. An MRI is a fast and noninvasive way to measure the degree of iron overload in your liver. Testing for gene mutations. Testing your DNA for mutations in the HFE gene is recommended if you have high levels of iron in your blood. If you're considering genetic testing for hemochromatosis, discuss the pros and cons with your doctor or a genetic counselor. Removing a sample of liver tissue for testing. If liver damage is suspected, your doctor may have a sample of tissue from your liver removed, using a thin needle. The sample is sent to a laboratory to be checked for the presence of iron as well as for evidence of liver damage, especially scarring or cirrhosis. Risks of biopsy include bruising, bleeding and infection. Blood testing. A person can ask his or her doctor to check body iron levels with the following tests: Serum iron -- (SI) This test is best conducted after fasting for at least three hours. Also, iron or vitamin C supplements should be discontinued at least three days before taking the test. Do not discontinue other medication unless your doctor tells you to. Total iron binding capacity (TIBC) -- This test tells how well your body can bind to iron. Serum iron divided by TIBC x 100% gives you important information about the transferrin-iron saturation percentage (TS%). TS% is usually 25-35%; in some people with iron overload, the TS% is very high. There are other types of iron overload where the TS% is normal. Serum ferritin (SF) -- This measures the amount of iron contained or stored in the body.

Serum ferritin reference ranges are different for adults and children. For adults the Ideal Range is 50-150ng/mL. In the past, liver biopsy was widely used to diagnose hemochromatosis. Today, liver biopsy is not necessary to diagnose the inherited form of HHC. DNA tests are available to determine if a person has genetic hemochromatosis.
Screening healthy people for hemochromatosis. Genetic testing is recommended for all first-degree relatives (parents, siblings and children) of anyone diagnosed with hemochromatosis. If a mutation is found in only one parent, then children do not need to be tested.
Genetic testing for hemochromatosis Genetic tests can confirm a diagnosis and can help identify family members who are at risk for hemochromatosis. Type I hemochromatosis is caused by defects (mutations) in the HFE gene. HFE has many purposes, but one important role is that it helps to control the amount of iron that

is absorbed from food. There are several known mutations in the HFE gene, but presently testing for only three is available: C282Y, H63D and S65C. Everyone inherits two copies of HFE, one from Mom and one from Dad. When a person has one mutated copy, he or she is called a carrier or heterozygote. When a person has two of the same mutated copies, he or she is called a homozygote. When a person has two different, but mutated, copies, he or she is called a compound heterozygote. Genetics can be very difficult to understand at first. What is most important is that you know which gene combination causes the greatest known risk of loading iron. Most at Risk:
• C282Y homozygote and the C282Y/H63D compound heterozygote Moderate risk:
• H63D homozygote or other compound heterozygote combinations Low risk:
• C282Y heterozygote (carrier); H63D heterozygote (carrier) or S65C heterozygote (carrier)
Risk can be modified by other genes, environment or unknown factors and as such, anyone with a mutated copy of HFE should periodically ask their doctor to check iron levels: hemoglobin, fasting serum iron, TIBC and serum ferritin.

Lifestyle changes to lessen your risk for hemochromatosis symptoms You may reduce your risk of complications from hemochromatosis if you: Avoid iron supplements and multivitamins containing iron.These can increase your iron levels even more. Avoid vitamin C supplements, especially with food. Vitamin C increases absorption of iron. Try to drink vitamin C-rich juices, such as orange juice, between meals. Avoid alcohol. Alcohol increases the risk of liver damage. If you have liver disease and hereditary hemochromatosis, avoid alcohol completely. Avoid eating raw shellfish. People with hereditary hemochromatosis are susceptible to infections, especially those caused by certain bacteria in raw shellfish. Drink tea. Some evidence suggests that drinking tannin-rich tea may slow the storage of iron.
Treating hemochromatosis- the GOALS of early treatment When this condition is diagnosed early, the first step is very important--to get iron levels down to normal. Therapeutic blood removal or phlebotomy is the most common means of iron reduction. Therapeutic phlebotomy (TP) is the same as regular blood donation but TP requires a doctor’s order (prescription).

Doctors can treat hereditary hemochromatosis safely and effectively by removing blood from your body (phlebotomy) on a regular basis, just as if you were donating blood. (In fact, this blood is safe to be used by other people.) The goal is to reduce your iron levels to normal. The amount of blood drawn depends on your age, your overall health and the severity of iron overload. It may take several years to reduce the iron in your body to normal levels. Initial treatment schedule. Initially, you may have a pint (470 milliliters) of blood taken once or twice a week — usually in a hospital or your doctor's office. This process shouldn't be too uncomfortable. While you recline in a chair, a needle is inserted into a vein in your arm. The blood flows from the needle into a tube that's attached to a blood bag. Depending on the condition of your veins and the consistency of your blood, the time needed to remove a pint of blood can range from 10 to 30 minutes. Regular blood donation can be done every 8 weeks. A person with severe iron overload may need to give blood as much as 8 times in a single month! The goal is to bring blood ferritin levels to an Ideal range of 50-150ng/mL. Depending on the amount of iron overload at the time of diagnosis, reaching normal levels can require several phlebotomies. Maintenance treatment schedule. Once your iron levels have returned to normal, blood draws can be less frequent, typically every three to four months. Some people may maintain normal iron levels without any blood draws, and some may need to have blood drawn monthly. The schedule depends on how rapidly iron accumulates in your body. The TS% and serum ferritin tests can be done periodically to help determine how often blood should be removed. Treating hereditary hemochromatosis can help alleviate symptoms of tiredness, abdominal pain and skin darkening. It can help prevent serious complications such as liver disease, heart disease and diabetes. If you already have one of these conditions, phlebotomy may slow the progression of the disease, and in some cases even reverse it. If you have hemochromatosis but no complications of cirrhosis or diabetes, you have the same life expectancy as a healthy person of your same age. Phlebotomy will not reverse cirrhosis or improve joint pain. If you have cirrhosis, your doctor may recommend periodic screening for liver cancer. This usually involves an abdominal ultrasound and a blood test. When hemochromatosis is diagnosed early and treated before organs are damaged a person can live a normal life expectancy. For people who have disease at the time of diagnosis, life expectancy may be shortened depending upon the disease. If a person

is diagnosed and treated before serum ferritin is above 1,000ng/mL the risk of cirrhosis or liver cancer is less than 1%. Other treatment options- step two There are several approaches to treating hemochromatosis, all of which are dependent upon the severity of the condition and the stage at which it has been diagnosed. Treatment options, therefore, will vary dependent upon the individual patient. The treatment option discussed above is the “phlebotomy” option. This, in addition to the other treatment options listed below, are used as first and second line measures. Nowadays, surgical intervention is reserved as a last line of defense, which is discussed in the next section. Phlebotomy Treatment Phlebotomy is generally a safe and efficient method of iron removal. Encourage patients to have weekly therapeutic phlebotomy of 500 mL of whole blood (equivalent to approximately 200-250 mg of iron). Some patients can tolerate twice-weekly phlebotomy, but this regimen is tedious and often inconvenient. Therapeutic phlebotomy should be performed until iron-limited erythropoiesis develops, identified by failure of the hemoglobin level and/or hematocrit to recover before the next phlebotomy. It should be continued until transferrin saturation is less than 50% and serum ferritin levels are less than 50 ng/mL, preferably 20 ng/mL. Most patients require maintenance phlebotomy in which 1 unit of blood is removed every 2-3 months. Therapeutic phlebotomy may improve or even cure some of the manifestations and complications of the disease, such as fatigue, elevated liver enzymes, hepatomegaly, abdominal pain, arthralgias, and hyperpigmentation. Other complications usually show little or no change after phlebotomy. Avoid excessive phlebotomy and the risk of hypovolemia and dehydration.

Dietary Restrictions Your doctor, in conjunction with another therapy (i.e. chelation or phlebotomy), may suggest dietary restrictions to aid in keeping your iron levels down. Below is more information on foods containing iron. Your doctor may limit you to a certain amount of iron, or may ask that you cut some of the following foods out of your diet completely. Humans consume two types of iron: heme and non-heme. Heme iron is the most easily absorbed form of iron. Highest in heme iron is red meat such as beef, venison, lamb, buffalo; also blue fin tuna is higher in heme iron than most other types of fish. All meat and fish also contain non-heme iron, which is found mostly in vegetables, fruits, nuts, grains and most over-the-counter iron supplements. Patients should not consume foods that contain large concentrations of bioavailable iron, such as red meats and organ meats. In addition, they should not use iron supplements, including multivitamins with iron. In addition, vitamin C supplements should be avoided. Substances in foods and drinks, including tannates (in tea), phytates, oxalates, calcium, and phosphates, can bind iron and inhibit its absorption. Alcohol abuse may accelerate disease progression. Ethanol sometimes increases iron absorption, and certain alcoholic drinks, especially red wine, contain relatively high concentrations of iron. Activity of hydroxyl free radicals is elevated by iron-containing diets combined with alcohol intake, and this is implicated in hepatocarcinogenesis. Ingestion of 30 g or more of ethanol daily potentiates hepatic injury due to iron overload and increases the relative risk for primary liver cancer in persons with cirrhosis. Patients with evidence of hepatic injury should consume little or no ethanol. Other patients should consume ethanol in moderation. Studies performed on healthy subjects living in the Spanish-Mediterranean coast showed that some genotypes (C282Y heterozygote, H63D heterozygote, and homozygote, as well as H63D/S63C compound heterozygote) together with alcohol and iron intake increased indicators of iron status; however, calcium intake decreased them.[98] These effects were not observed in S63C heterozygotes.
Vitamin C (ascorbic acid) increases intestinal absorption of inorganic iron. No reason exists to discourage patients from eating fresh fruits and vegetables containing vitamin C, but advising them to limit ingestion of vitamin C in supplements to 500 mg/d is prudent. Use mineral supplements for specific deficiencies only. Seafood from potentially contaminated waters must be cooked thoroughly. Raw or improperly cooked shellfish is sometimes contaminated with Vibrio vulnificus and can cause sepsis in patients with hemochromatosis. Chelation Therapy In patients with hemochromatosis and heart disease, anemia, or poor venous access, treatment with iron chelation agents is recommended. The therapeutic perspectives comprise compounds inhibiting intestinal absorption of iron, chelators of iron, hepcidin,

or ferroportin supplementation. In disease caused by hepcidin deficiency, protein supplementation with hepcidin is advised. If you can't undergo phlebotomy, because you have anemia, for example, or heart complications, your doctor may recommend a medication to remove excess iron. The medication can be injected into your body, or it can be taken as a pill. The medication causes your body to expel iron through your urine or stool in a process that's sometimes called chelation.
Medications- Pills for Chelation Therapy Deferasirox
Deferasirox (Exjade) is the oral iron chelator that should be taken once daily as an adjunct to phlebotomies or instead of phlebotomy in patients in whom these procedures are poorly tolerated. Deferasirox is very efficacious in liver iron removal. During treatment with deferasirox, kidney function should be controlled. In a study that evaluated the effects of deferasirox in Hjv-/- mice (knockout animals lacking hemojuvelin [HJV]; ie, an experimental model of hereditary hemochromatosis), a dose of 100 mg/kg markedly reduced the iron level in the liver and heart. However, in the pancreas, deferasirox was less effective, and the splenic iron count was not influenced. Deferasirox was administered once daily 5 times a week.

Dendrimers
The family of dendrimers, iron-selective chelators, have been synthesized. Dendrimers terminated with hydroxypiridinone have high affinity to iron and reduce its absorption in the rat intestine. Therefore, the application of the dendrimers in the treatment of iron overload diseases is considered. In experiments performed on rats compared the protective effect of 2 iron chelators, deferoxamine and deferiprone, on iron overload in the heart, deferiprone was found to reduce histopathologic changes in the heart of rats chronically loaded with iron. The 2 compounds were administered individually or in combination with vitamin C (vitamin C was used as the antioxidative compound aimed at preventing heart oxidative injury). Additional administration of vitamin C improved histopathologic changes and biochemical markers in the heart. Juvenile hemochromatosis
The first patient affected by juvenile hemochromatosis was successfully treated with chelation therapy. Because of severe congestive heart failure, phlebotomy was contradicted. Simultaneous administration of deferoxamine and deferiprone reduced the myocardial dysfunction and improved the clinical status of that patient. Anemia
Patients affected with anemia cannot be treated with phlebotomy. Thus, application of iron chelation agents (eg, deferoxamine, deferiprone, deferasirox) is recommended. Deferoxamine is administered intravenously or subcutaneously at doses ranging from 25 to 40 mg/kg. Intravenous infusion is usually 8-10 hours in duration and is repeated 5 nights per week. Similar effects can be obtained with subcutaneous bolus injections administered twice daily. The main adverse effects are inflammatory reactions at the sites of injection, visual and auditory disturbances, bone growth disturbances, and allergic reactions, including anaphylaxis. Deferiprone is given orally in 3 divided doses of 75 mg/kg/d. Agranulocytosis, neutropenia, arthralgia, gastrointestinal reactions, and elevation of liver enzyme levels are the main adverse effects. Cardiac iron overload is better reduced by deferiprone than deferoxamine. Deferasirox is an oral chelation agent, administered at 10-30 mg/kg. Deferasirox adverse effects can include elevation of the creatinine level, skin exanthem, diarrhea, and visual and auditory disturbances. When surgical intervention is needed - the last effort Surgical procedures are used to treat 2 important complications: end-stage liver disease and severe arthropathy.

When end-stage liver disease progresses despite iron-reduction therapy, orthotopic liver transplantation is the only therapeutic option. Another indication for liver transplantation is the development of hepatocellular carcinoma. Careful patient selection is advised for liver transplantation to treat patients with hepatocellular carcinoma. Particularly, these patients should have a single tumor of 5 cm or smaller in diameter. If multiple tumors are present, the acceptable number is 3 tumors or less, smaller than 3 cm. The 4-year survival rate can be approximately 90% if these criteria are respected. Surgical arthroplasty is considered if joint destruction becomes severe despite medical therapy.
The risk of going untreated Undiagnosed and untreated hemochromatosis (too much iron) increases the risk for diseases and conditions such as diabetes mellitus, irregular heart beat or heart attack, arthritis (osteoarthritis, osteoporosis), cirrhosis of the liver or liver cancer, gall bladder disease, depression, impotence, infertility, hypothyroidism, hypogonadism, and some cancers. Mismanaged iron in the brain has been observed in autopsies of people with neurodegenerative diseases: Alzheimer's, early onset Parkinson's, epilepsy, multiple sclerosis, and Huntington's disease. The following are the most common diseases that occur in patients with undiagnosed or untreated hemochromatosis:
• Bone and joint: (osteoarthritis, osteoporosis) knuckles, ankles and hips
• Liver: enlarged liver, cirrhosis, cancer, and liver failure diabetes
• Spleen: enlarged spleen
• Heart: irregular heart beat, enlarged heart, congestive heart failure
• Endocrine: diabetes, hypothyroidism, hypogonadism, (infertility, impotence), hormone

• imbalances
• Skin: abnormal color (bronze, reddish or ashen-gray) **This is where the term “Bronze Diabetes” comes from
Hemochromatosis can be overlooked by a doctor who is concentrating on treatment of diseases that are present in the patient. Many doctors still believe what they learned in medical school, that hemochromatosis is rare and only happens in older men. When hemochromatosis is discovered early and treated before organ damage can occur, a person can live a normal, healthy life. Any family practice physician is qualified to diagnose and order treatment for a hemochromatosis patient.
Doctors that may help to diagnose hemochromatosis If there are complications with diseased organs related to iron damage, the patient may need a specialist. Keeping in mind the diseases that may result from undiagnosed or untreated hemochromatosis (listed above), here are some of the types of specialists needed by patients with this condition:
• Cardiologist (heart specialist)
• Endocrinologist (hormone and endocrine system specialist)
• astroenterologist (gastrointestinal specialist)
• Gynecologist (female reproductive system specialist)
• Hemotologist/ Oncologist (blood or cancer specialist)
• Hepatologist (liver specialist)
• Internist (internal medicine specialist)
• Rheumatologist (immune system specialist)
• Urologist (genitourinary specialist)

Resources http://celticcurse.org/july-is-hemochromatosis-awareness-month/ http://emedicine.medscape.com/article/177216-overview#a0156 http://blogcritics.org/may-is-international-hemochromatosis-awareness-month/ http://www.hemochromatosis.org/hemochromatosis/ http://www.cdc.gov/genomics/resources/diseases/hemochromatosis.htm http://www.mayoclinic.com/health/hemochromatosis/DS00455


















