
Journal of Industrial and Engineering Chemistry analysis was performed with a X-ray diffractometer...
10
Crystallization behavior of tantalum and chlorine co-substituted hydroxyapatite nanopowders Bahman Nasiri-Tabrizi a, *, Belinda Pingguan-Murphy b , Wan Jefrey Basirun c , Saeid Baradaran d a Advanced Materials Research Center, Materials Engineering Department, Najafabad Branch, Islamic Azad University, Najafabad, Iran b Department of Biomedical Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia c Department of Chemistry, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia d Department of Mechanical Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia Introduction Bioceramics are an important class of ceramics that range in biocompatibility from ceramic oxides to bioresorbable ceramic materials [1]. Among them, calcium phosphate is the most important inorganic constituent of biological tissues, and synthetic calcium phosphate has been widely used as biomaterials [2]. The two general biomedical applications of calcium phosphate can be outlined: (i) using them in powder form as filling materials to impart bioactivity to various biocomposites and hybrid biomaterials; (ii) manufacturing of either dense compacts or porous scaffolds, possessing the sufficient mechanical properties [3]. However, insertion of implants to replace the injured parts often gives discomfort for patients, due to brittleness and inadequate biological properties of the ceramic implants [4]. Most studies have shown that the incorporation of ions such as Na + , Ag + , K + , Mg 2+ , Zn 2+ , Cd 2+ , Sr 2+ , Ba 2+ , Mn 2+ , La 3+ , In 3+ , Bi 3+ , Ga 3+ , Eu 3+ , Y 3+ , Pb 4+ , V 5+ , AsO 4 3 , VO 4 3 , SO 4 3 , CO 3 2 , SiO 4 3 , F , Cl , Br , and O 2 can improve the crystal structure, crystallinity, surface charge, the solubility and other vital properties, leading to major changes in biological response upon implantation [5–7]. Accordingly, these substituted apatites can be used for repairing and replacing diseased and damaged parts of musculoskeletal systems, and also as a drug or gene delivery agent, as a bioactive coating on metallic osseous implants, biomagnetic particles and fluorescent markers [8]. In the apatite lattice, monovalent anions (F , Cl ) substitute OH ions in the anion channel without charge imbalance. Besides, the anionic complexes can substitute the phosphate group and a large number of monovalent, bivalent, trivalent, tetravalent, and pentavalent metal cations can replace for calcium, suggesting the apatite is able to incorporate almost half of the elements of the periodic system in its atomic structure [9–13]. Hence, over the past decade, many studies have been devoted to the synthesis of substituted apatites by various methods such as wet chemical precipitation [14], co-precipitation [15], emulsion method [16], sol–gel synthesis [17], hydrothermal reaction [18], mechanochem- ical method [19], and microwave irradiation technique [20]. Among them, powder synthesized using a mechanochemical route usually possesses a well-defined structure due to the perturbation of surface-bonded species as a result of pressure, which enhancing the thermodynamic and kinetic reactions between solids [21,22]. Indeed, the mechanochemical process has the advantages of simplicity and reproducibility of a solid-state procedure to perform mass production and the basic character- istics of an ordinary wet reaction to generate a powder with an appropriate microstructure [23–26]. Journal of Industrial and Engineering Chemistry 33 (2016) 316–325 A R T I C L E I N F O Article history: Received 16 August 2015 Received in revised form 19 September 2015 Accepted 5 October 2015 Available online 24 October 2015 Keywords: Crystallization Hydroxyapatite Co-substitution Nanopowders Reaction mechanism A B S T R A C T The crystallization behavior of tantalum and chlorine co-substituted hydroxyapatite nanopowders was examined. Results showed that combined processing by mechanical alloying and annealing is conducive to the structural changes and crystal growth. A series of nanocrystalline tantalum and chlorine co- substituted hydroxyapatite (Ta/Cl–HA) with different dopant contents were synthesized as a result of the progressive mechanochemical reaction. During the subsequent annealing, crystallization of the as- milled powders occurred, leading to a significant increase in the fraction of crystalline phase. TEM images revealed that the crystallized and doped nanopowders composed of crystalline nanoneedles with an average size of 61 26 nm. ß 2015 The Korean Society of Industrial and Engineering Chemistry. Published by Elsevier B.V. All rights reserved. * Corresponding author. Tel.: +98 9132275822; fax: +98 3142291016. E-mail address: [email protected] (B. Nasiri-Tabrizi). Contents lists available at ScienceDirect Journal of Industrial and Engineering Chemistry jou r n al h o mep ag e: w ww .elsevier .co m /loc ate/jiec http://dx.doi.org/10.1016/j.jiec.2015.10.019 1226-086X/ß 2015 The Korean Society of Industrial and Engineering Chemistry. Published by Elsevier B.V. All rights reserved.
Transcript of Journal of Industrial and Engineering Chemistry analysis was performed with a X-ray diffractometer...

Journal of Industrial and Engineering Chemistry 33 (2016) 316–325
Crystallization behavior of tantalum and chlorine co-substitutedhydroxyapatite nanopowders
Bahman Nasiri-Tabrizi a,*, Belinda Pingguan-Murphy b, Wan Jefrey Basirun c,Saeid Baradaran d
a Advanced Materials Research Center, Materials Engineering Department, Najafabad Branch, Islamic Azad University, Najafabad, Iranb Department of Biomedical Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysiac Department of Chemistry, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysiad Department of Mechanical Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia
A R T I C L E I N F O
Article history:
Received 16 August 2015
Received in revised form 19 September 2015
Accepted 5 October 2015
Available online 24 October 2015
Keywords:
Crystallization
Hydroxyapatite
Co-substitution
Nanopowders
Reaction mechanism
A B S T R A C T
The crystallization behavior of tantalum and chlorine co-substituted hydroxyapatite nanopowders was
examined. Results showed that combined processing by mechanical alloying and annealing is conducive
to the structural changes and crystal growth. A series of nanocrystalline tantalum and chlorine co-
substituted hydroxyapatite (Ta/Cl–HA) with different dopant contents were synthesized as a result of
the progressive mechanochemical reaction. During the subsequent annealing, crystallization of the as-
milled powders occurred, leading to a significant increase in the fraction of crystalline phase. TEM
images revealed that the crystallized and doped nanopowders composed of crystalline nanoneedles with
an average size of 61 � 26 nm.
� 2015 The Korean Society of Industrial and Engineering Chemistry. Published by Elsevier B.V. All rights
reserved.
Contents lists available at ScienceDirect
Journal of Industrial and Engineering Chemistry
jou r n al h o mep ag e: w ww .e lsev ier . co m / loc ate / j iec
Introduction
Bioceramics are an important class of ceramics that range inbiocompatibility from ceramic oxides to bioresorbable ceramicmaterials [1]. Among them, calcium phosphate is the mostimportant inorganic constituent of biological tissues, and syntheticcalcium phosphate has been widely used as biomaterials [2]. Thetwo general biomedical applications of calcium phosphate can beoutlined: (i) using them in powder form as filling materials to impartbioactivity to various biocomposites and hybrid biomaterials; (ii)manufacturing of either dense compacts or porous scaffolds,possessing the sufficient mechanical properties [3]. However,insertion of implants to replace the injured parts often givesdiscomfort for patients, due to brittleness and inadequate biologicalproperties of the ceramic implants [4]. Most studies have shown thatthe incorporation of ions such as Na+, Ag+, K+, Mg2+, Zn2+, Cd2+, Sr2+,Ba2+, Mn2+, La3+, In3+, Bi3+, Ga3+, Eu3+, Y3+, Pb4+, V5+, AsO4
3�, VO43�,
SO43�, CO3
2�, SiO43�, F�, Cl�, Br�, and O2� can improve the crystal
structure, crystallinity, surface charge, the solubility and other vitalproperties, leading to major changes in biological response uponimplantation [5–7]. Accordingly, these substituted apatites can be
* Corresponding author. Tel.: +98 9132275822; fax: +98 3142291016.
E-mail address: [email protected] (B. Nasiri-Tabrizi).
http://dx.doi.org/10.1016/j.jiec.2015.10.019
1226-086X/� 2015 The Korean Society of Industrial and Engineering Chemistry. Publi
used for repairing and replacing diseased and damaged parts ofmusculoskeletal systems, and also as a drug or gene delivery agent,as a bioactive coating on metallic osseous implants, biomagneticparticles and fluorescent markers [8].
In the apatite lattice, monovalent anions (F�, Cl�) substituteOH� ions in the anion channel without charge imbalance. Besides,the anionic complexes can substitute the phosphate group and alarge number of monovalent, bivalent, trivalent, tetravalent, andpentavalent metal cations can replace for calcium, suggesting theapatite is able to incorporate almost half of the elements of theperiodic system in its atomic structure [9–13]. Hence, over the pastdecade, many studies have been devoted to the synthesis ofsubstituted apatites by various methods such as wet chemicalprecipitation [14], co-precipitation [15], emulsion method [16],sol–gel synthesis [17], hydrothermal reaction [18], mechanochem-ical method [19], and microwave irradiation technique[20]. Among them, powder synthesized using a mechanochemicalroute usually possesses a well-defined structure due to theperturbation of surface-bonded species as a result of pressure,which enhancing the thermodynamic and kinetic reactionsbetween solids [21,22]. Indeed, the mechanochemical processhas the advantages of simplicity and reproducibility of a solid-stateprocedure to perform mass production and the basic character-istics of an ordinary wet reaction to generate a powder with anappropriate microstructure [23–26].
shed by Elsevier B.V. All rights reserved.

B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325 317
Although the incorporation of tantalum (Ta5+) into the apatitestructure improves its biological responses [27,28], to ourknowledge, the crystallization behavior of the Ta/Cl–HA nano-powders and its effect on the crystal structure have not previouslybeen investigated. Therefore, the present study establishes for thefirst time the role of subsequent annealing on the phase transitionsand morphological changes of nanosize Ta and Cl co-substitutedHA. Besides, the structural features were assessed to figure out thepossible doping mechanism.
Experimental procedures
Raw materials
Calcium hydroxide (Ca(OH)2, �96 wt%), phosphoric acid(H3PO4, �85 wt% in H2O), and tantalum (V) chloride (TaCl5,99.99 wt%) were used as the raw materials. All chemicals werepurchased from Sigma–Aldrich Co., USA and used without furtherpurification.
Explanation
Depending on the milling conditions, the mechanical activationis classified in two categories: (i) gradual mode in which thereaction may extend to a very small volume during each collisionand (ii) mechanically induced self-sustaining reaction (MSR)wherein a self-propagating combustion reaction can be initiated,if the reaction enthalpy is sufficiently high [29]. In the second modeof reaction, which can be predicted by calculating the adiabatictemperature (Tad), the incidence of the combustion reaction resultsin a rapid rise in temperature of the milling medium, and providesappropriate conditions for a quick transformation. The reactionmechanism can be predicted by calculating the adiabatictemperature or by describing this property (�DH298/
PCp). For
self-sustaining mode reaction to take place it is necessary thatthese quantities be at least Tad > 1800 K and �DH298/P
Cp > 2000 K [30]. Here, it can be hypothesized that the modeof the reaction is progressive due to the lack of high exothermicreactions. This reaction involves the following steps: (i) thereactive absorption of atmospheric carbon dioxide during milling,and (ii) the mechanically induced reaction of the mixed powders. Itshould be noted that this assessment is provided according to thefollowing assumptions: (i) milling vial is sealed and isolated (ii) allthe reactions occur in the standard conditions (DG = DG8) (iii) theactivity coefficient is equal to 1, and (iv) the reactions occur inthe same physical conditions. During the subsequent annealing,the crystallinity degree is expected to increase significantly dueto the crystallization and removal of carbonaceous materials.
Mechanical activation
A series of Ta/Cl–HA powders with different dopant contentsand general formula Ca10–2.5xTax(PO4)6(OH)2�yCly were synthe-sized by mechanical activation of the desired amounts of the
Table 1Details of the powder components.
Sample Powder components (g) Ta/(Ca + Ta) rat
Ca(OH)2 H3PO4 TaCl5
TaCl0 2.23 1.77 – 0
TaCl25 2.06 1.68 0.26 0.025
TaCl50 1.91 1.60 0.49 0.050
TaCl100 1.65 1.46 0.89 0.100
a N Taions, number of Ta ions.b N Clions, number of Cl ions.
reagents (see Table 1), using a high-energy planetary ball mill(Retsch, PM100) with zirconia vials (50 ml) and balls (10 mmdiameter) for 3 h without using any process control agent (PCA). Inthe apatite formula, the values x and y define the deviation fromthe complete stoichiometry, within the intervals 0 � x � 0.5 and0 � y � 1.5. In all the experiments, the (Ca + Ta)/P ratio was fixed at1.67. On the other hand, the Ta/(Ca + Ta) ratio ranged from 0 to0.1. The ball-to-powder (BPR) weight ratio, total powder mass androtational speed were 8:1, 4 g, and 500 rpm, respectively. Thegrinding was suspended for 10 min after 45 min run to avoidexcess temperature increase inside the mill vials.
Thermally induced crystallization
To appraise the effect of the subsequent annealing on thecrystallization behavior and structural evolution, the mechan-osynthesized powders were separately filled in a quartz boat andannealed under atmospheric pressure at 800 8C for 1 h. The heatingrate from room temperature up to the desired temperature wasfixed at 10 8C min�1. Fig. 1 shows a schematic illustration of thestrategy for the fabrication and crystallization of Ta/Cl–HAnanopowders.
Characterization techniques
Phase analysis was performed with a X-ray diffractometer(XRD, PANalytical Empyrean, Netherlands) using a Cu–Ka
radiation at 30 kV and 35 mA over a 2u range from 108 to 708.To examine the XRD profiles, Rietveld refinement and ‘‘PANalytical
X’Pert HighScore’’ software were also employed and the patternswere compared to standards compiled by the Joint Committee onPowder Diffraction and Standards (JCPDS), which involved card#24-0033 for HA, #27-0074 for chlorapatite (CA), #01-072-1651for CaCO3 (CC), and #01-072-0010 for Ca2PO4Cl (CCP). The phasepercentages (WP) in the synthesized powders were determinedfrom the relative intensity ratio of the representing major phases,using Eq. (I) [27].
WP ¼ðIÞPPni¼1ðIÞi
�100 (I)
where (I)P and (I)i are the intensity of the major peak of the phaseand intensity of major peaks of all phases.
Considering that the phase constitution and transformationcharacteristics appear to be critically dependent on crystallite size(D) and lattice strain (h), the measurement of these features in themechanically alloyed powders is very essential. Hence, thecrystallite size and lattice strain of the samples were calculatedusing the well-known equation [31]:
B cosu ¼ 0:9lDþ h sinu (II)
where l, D, h and u are the wavelength of the X-ray used(0.154056 nm), crystallite size, internal micro-strain and the Braggangle (8), respectively. Note that B in the above equation is the peakwidth (in radians) after subtracting the peak width due to
io (Ca + Ta)/P ratio aN Taions� 1023 bN Clions� 1023
1.67 0 0
1.67 0.004 0.02
1.67 0.008 0.04
1.67 0.01 0.07

Fig. 1. A schematic view of the sample specifications and the thermally induced crystallization of Ta/Cl–HA.
B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325318
instrumental broadening from the experimentally recordedprofile. When B cos u was plotted against sin u, straight lines wereobtained with the slope as h and the intercept as 0.9 l/D. It shouldbe noted that LaB6 powder standard is usually used to determineinstrumental effect on the peak broadening. Besides, if we assumethat a crystallite is a sphere of diameter D surrounded by a shell ofgrain boundary with thickness t, the volume fraction of the grainboundary (f) is approximately [32]:
f ¼ 1� D
ðD þ tÞ
� �3
(III)
The values of f were calculated by substituting the experimentalcrystallite size obtained by XRD into D under the assumption oft = 1 nm.
The crystallinity degree (XC) of the samples was determined bytaking the sum total of relative intensities of the characteristicpeaks of apatite phases for the samples ((I1/In)Apatite) and standard((I1/In)Standard), using Eq. (IV) [33]:
XC ¼PðI1 : InÞApatitePðI1 : InÞStandard
�100 (IV)
The hexagonal lattice constants ‘‘a’’ and ‘‘c’’ of the samples werecalculated using Eq. (V), which is based on Bragg’s equation [34]:
1
d2¼ 4
3
h2 þ hk þ k2
a2þ l2
c2(V)
where h, k, l are the Miller indices of the reflection planes. Thehexagonal unit cell volume of (V) of the as-prepared materials werecalculated using Eq. (VI) [35]:
V ¼ ða2cÞsin60� (VI)
Attenuated total reflection (ATR) analysis was executed on aTensor 27 (Bruker FT-IR spectrophotometer, Germany) with afrequency range of 4000–400 cm�1 to recognize the functionalgroups. The size and morphology of the synthesized nanopowderswere observed on a TEM (HT-7700, Hitachi, Japan) that operated atthe accelerating voltages of 60–100 kV. To verify the structuralchanges, the HRTEM images in addition to the TEM observationsand the selected-area electron diffraction (SAED) were employed.Energy-dispersive X-ray spectrometry (EDS) and elementalmapping analysis equipped in a field emission scanning electronmicroscopy (FESEM, SU8000, Hitachi, Japan) were operated toexamine the chemical constituents and the spatial distribution ofelements. A full particle size distribution can be obtained based on
a count of many particles using automated counting techniques.These experimental data are typically represented as a numberaverage [36]:
d¯n¼P
inidiPini
(VII)
where ni symbolizes the number of particles with a diameter, di,which in practice is typically a mean value within a specified size.However, of greater use is the surface-weighted average diameter:
d¯s¼P
inid3iP
inid2i
(VIII)
which allows comparison to crystallite sizes determined bychemisorption methods, and the volume-weighted averagediameter:
d¯v¼P
inid4iP
inid3i
(XI)
which allows comparison to crystallite sizes obtained from XRDanalysis or magnetic measurements. A narrow crystallite sizedistribution will result in similar values for Eqs. (VIII) and (XI), andthese are frequently in good agreement with the XRD measure-ments. The broader the distribution causes, the greater thedisparities among the different d
¯values. A bimodal crystallite
size distribution can also cause large discrepancies among thesedifferently weighted d
¯values, regardless of the method used for
characterization.
Results and discussions
Mechanically alloyed Ta/Cl–HA
Fig. 2a shows the XRD patterns of the 3 h milled specimens inthe absence (TaCl0) and presence of dopant (TaCl100). In theabsence of dopant, poorly crystalline HA was formed after 3 h ofmilling, mainly as a result of the reactive absorption ofatmospheric carbon dioxide. Besides, new diffraction peakscorresponding to calcite (CC) became apparent due to theincomplete reaction of the raw materials. With the addition ofdopant, all the representing peaks corresponding to the rawmaterials and calcite disappeared completely and the XRD profileof TaCl100 indicated a major TCA (Ca10–2.5xTax(PO4)6Cl2) phase andminor concentration of Ca2PO4Cl (CCP). The main characteristicpeaks of TCA are as follows: (0 0 2) plane at 2u = 26.558, (2 1 1)
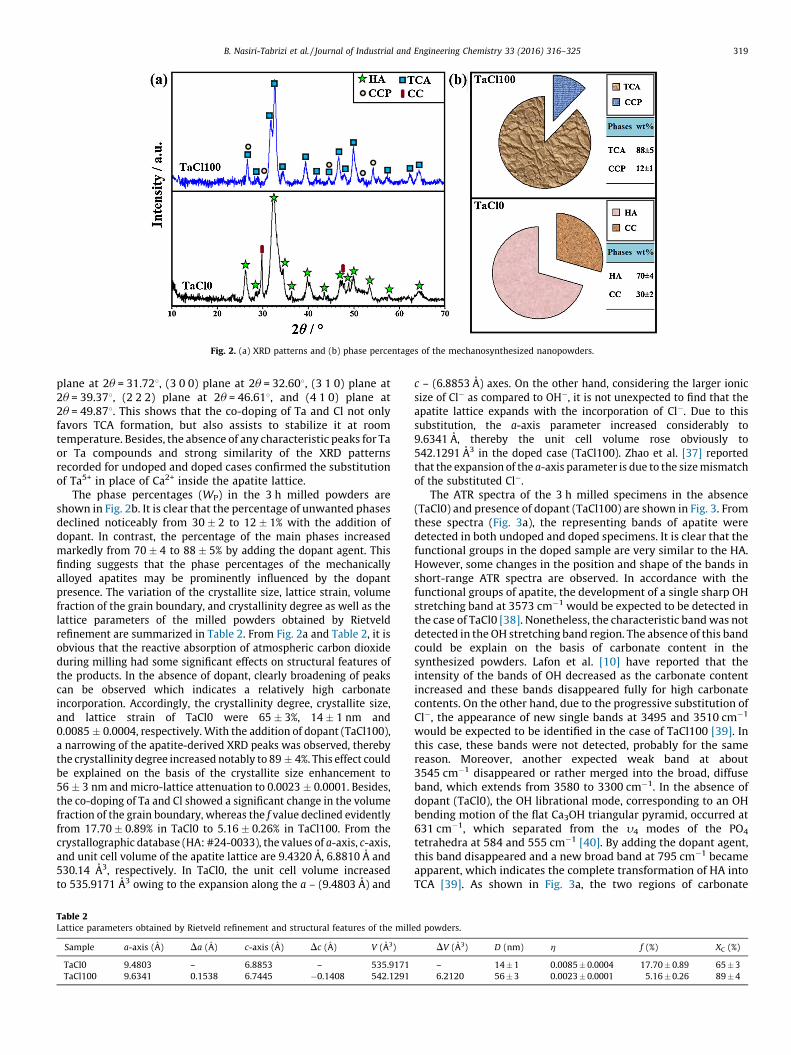
Fig. 2. (a) XRD patterns and (b) phase percentages of the mechanosynthesized nanopowders.
B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325 319
plane at 2u = 31.728, (3 0 0) plane at 2u = 32.608, (3 1 0) plane at2u = 39.378, (2 2 2) plane at 2u = 46.618, and (4 1 0) plane at2u = 49.878. This shows that the co-doping of Ta and Cl not onlyfavors TCA formation, but also assists to stabilize it at roomtemperature. Besides, the absence of any characteristic peaks for Taor Ta compounds and strong similarity of the XRD patternsrecorded for undoped and doped cases confirmed the substitutionof Ta5+ in place of Ca2+ inside the apatite lattice.
The phase percentages (WP) in the 3 h milled powders areshown in Fig. 2b. It is clear that the percentage of unwanted phasesdeclined noticeably from 30 � 2 to 12 � 1% with the addition ofdopant. In contrast, the percentage of the main phases increasedmarkedly from 70 � 4 to 88 � 5% by adding the dopant agent. Thisfinding suggests that the phase percentages of the mechanicallyalloyed apatites may be prominently influenced by the dopantpresence. The variation of the crystallite size, lattice strain, volumefraction of the grain boundary, and crystallinity degree as well as thelattice parameters of the milled powders obtained by Rietveldrefinement are summarized in Table 2. From Fig. 2a and Table 2, it isobvious that the reactive absorption of atmospheric carbon dioxideduring milling had some significant effects on structural features ofthe products. In the absence of dopant, clearly broadening of peakscan be observed which indicates a relatively high carbonateincorporation. Accordingly, the crystallinity degree, crystallite size,and lattice strain of TaCl0 were 65 � 3%, 14 � 1 nm and0.0085 � 0.0004, respectively. With the addition of dopant (TaCl100),a narrowing of the apatite-derived XRD peaks was observed, therebythe crystallinity degree increased notably to 89 � 4%. This effect couldbe explained on the basis of the crystallite size enhancement to56 � 3 nm and micro-lattice attenuation to 0.0023 � 0.0001. Besides,the co-doping of Ta and Cl showed a significant change in the volumefraction of the grain boundary, whereas the f value declined evidentlyfrom 17.70 � 0.89% in TaCl0 to 5.16 � 0.26% in TaCl100. From thecrystallographic database (HA: #24-0033), the values of a-axis, c-axis,and unit cell volume of the apatite lattice are 9.4320 A, 6.8810 A and530.14 A3, respectively. In TaCl0, the unit cell volume increasedto 535.9171 A3 owing to the expansion along the a – (9.4803 A) and
Table 2Lattice parameters obtained by Rietveld refinement and structural features of the mill
Sample a-axis (A) Da (A) c-axis (A) Dc (A) V (A3)
TaCl0 9.4803 – 6.8853 – 535.9171
TaCl100 9.6341 0.1538 6.7445 �0.1408 542.1291
c – (6.8853 A) axes. On the other hand, considering the larger ionicsize of Cl� as compared to OH�, it is not unexpected to find that theapatite lattice expands with the incorporation of Cl�. Due to thissubstitution, the a-axis parameter increased considerably to9.6341 A, thereby the unit cell volume rose obviously to542.1291 A3 in the doped case (TaCl100). Zhao et al. [37] reportedthat the expansion of the a-axis parameter is due to the size mismatchof the substituted Cl�.
The ATR spectra of the 3 h milled specimens in the absence(TaCl0) and presence of dopant (TaCl100) are shown in Fig. 3. Fromthese spectra (Fig. 3a), the representing bands of apatite weredetected in both undoped and doped specimens. It is clear that thefunctional groups in the doped sample are very similar to the HA.However, some changes in the position and shape of the bands inshort-range ATR spectra are observed. In accordance with thefunctional groups of apatite, the development of a single sharp OHstretching band at 3573 cm�1 would be expected to be detected inthe case of TaCl0 [38]. Nonetheless, the characteristic band was notdetected in the OH stretching band region. The absence of this bandcould be explain on the basis of carbonate content in thesynthesized powders. Lafon et al. [10] have reported that theintensity of the bands of OH decreased as the carbonate contentincreased and these bands disappeared fully for high carbonatecontents. On the other hand, due to the progressive substitution ofCl�, the appearance of new single bands at 3495 and 3510 cm�1
would be expected to be identified in the case of TaCl100 [39]. Inthis case, these bands were not detected, probably for the samereason. Moreover, another expected weak band at about3545 cm�1 disappeared or rather merged into the broad, diffuseband, which extends from 3580 to 3300 cm�1. In the absence ofdopant (TaCl0), the OH librational mode, corresponding to an OHbending motion of the flat Ca3OH triangular pyramid, occurred at631 cm�1, which separated from the y4 modes of the PO4
tetrahedra at 584 and 555 cm�1 [40]. By adding the dopant agent,this band disappeared and a new broad band at 795 cm�1 becameapparent, which indicates the complete transformation of HA intoTCA [39]. As shown in Fig. 3a, the two regions of carbonate
ed powders.
DV (A3) D (nm) h f (%) XC (%)
– 14 � 1 0.0085 � 0.0004 17.70 � 0.89 65 � 3
6.2120 56 � 3 0.0023 � 0.0001 5.16 � 0.26 89 � 4

Fig. 3. (a) ATR spectra of the 3 h milled samples in the absence and presence of dopant, and (b) deconvoluted spectra in the carbonate region.
B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325320
vibrations in apatite are: (i) 1450–1400 cm�1 attributed to n3
(asymmetric stretch vibration) and (ii) 890–860 cm�1 attributed ton2 (out-of-plane bend vibration) vibrations of the carbonate groups[10]. In the absence of dopant (TaCl0), the low resolution of thePO4-derived bands and poorly hydroxylation can be explained by adecrease of crystallinity due to the increased carbonate substitu-tion in the apatite lattice [7]. It is obvious that the level ofcarbonate replacement became less in the spectrum of TaCl100.This shows that the co-doping of Ta and Cl reduces the relativeincorporation of CO3
2�, which makes the preliminary crystalliza-tion of the apatite lattice. This result is in good agreement with theXRD data, showing that the co-doping of Ta and Cl promotes themechanically induced crystallization. To evaluate more preciselythe type of carbonate substitution, deconvolution of the carbonatebands in the region between 1800 and 1200 cm�1 was carried outas shown in Fig. 3b. The presence of the carbonate bands at 1556,1464, 1404, and 889 is consistent with B- and A-type carbonatesubstituted HA, where carbonate ions replace phosphate and alsohydroxyl positions in the crystal lattice, respectively [7]. Based on
Fig. 4. TEM/HR-TEM images of (a) TaCl0 and (b) TaCl100 as well as
the ATR spectra, the position and shape of the representing bandsof apatite may be clearly influenced by the dopant presence.
Since the size, morphology, chemical composition and elemen-tal distribution of nanostructured biomaterials have significanteffects on the biomedical functionality, the morphological featuresand chemical constituents of the product were appraised by TEM/HRTEM and EDS imaging techniques, respectively. From the TEMimages in Fig. 4, the milled nanoparticles have a high tendency toagglomerate due to their large surface to volume ratio. Thisphenomenon occurs in three certain stages as follows [23]: (i) theRittinger stage, in which the interaction of particles can beneglected and the energy input is approximately proportional tothe new surface area formation, (ii) the aggregation stage, in whichthe formed new surface area is not proportional to the energy inputdue to the particle interaction (aggregation). Nevertheless, thedispersion degree is still increasing considerably. The particlesadhere on each other without structural changes in consequence ofvan der Waals forces. These aggregates can be dissolved by slightmechanical intervention, (iii) the agglomeration stage, in which an
(c) EDS spectrum and elemental mapping images of TaCl100.

B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325 321
increase in dispersion first drops to a negligible value then stopsaltogether; it may even give way to a decrease of surface area dueto the particle interaction (agglomeration). In this stage, theparticles are grown together by chemical bonds and separationbecomes impossible. It is most likely that the mechanochemicalreactions and structural changes mainly occur at this stage. FromFig. 4a, the 3 h milled sample in the absence of dopant (TaCl0) iscomposed of randomly-shaped particles with widths of 10 � 3 nmand lengths of 19 � 7 nm. The HRTEM image also shows that thedistance between the adjacent lattice fringe (0.84 nm) is in goodagreement with the d(100) spacing from the standard value(JCPDS#024-0033). As shown in Fig. 4b, the synthesized nanopow-ders in the presence of dopant (TaCl100) entailed crystallinenanoneedles with an average size of around 60 � 20 nm in lengthand 14 � 4 nm in width. Besides, the HRTEM image reveals that thecontrol of the growth occurs along the (1 0 0) surface. Regarding themorphological changes, Wang et al. [41] demonstrated that theprocessing parameters had a significant effect on the morphologicalfeatures of hydroxyapatite nanoparticles, wherein with more acidicpH values nanowires or needle-shaped particles were obtainedcompared to sphere-shaped particles in case more alkaline pH. In thepresent case, Ca(OH)2/H3PO4 molar ratio and pH decrease by addingthe dopant agent. Moreover, the addition of a solvent medium(H3PO4) in ball milling process can generate local zones of hightemperatures (up to 700 8C) and high pressures due to friction andadiabatic heating of gas bubbles, although the overall temperaturecan reach room temperature [8]. Therefore, size and morphologychanges were recorded possibly due to the combined effects of pHchanges and generation of local zones of high temperature andpressure. This shows that the morphological features of themechanochemically alloyed apatite nanopowders may be signifi-cantly influenced by the dopant content and milling conditions.Fig. 4c displays the EDS spectra and elemental mapping images of the3 h milled specimens in the presence of dopant (TaCl100). It isobvious that the main elements in the doped nanopowders aretantalum, chlorine, calcium, phosphate, oxygen and carbon. From theEDS results, the (Ca + Mg)/P molar ratio of the doped powders wasmore than 1.67 as a result of a decrease in the Ca and P concentration.This consequential increase in (Ca + Mg)/P molar ratio is evidence forthe B-type substitution. Besides, no chemically stable impurity wasfound in consequence of the excessive wearing of the vials and balls.Concerning the elemental mapping images, an appropriate spatialdistribution of elements in the nanopowders was identified,demonstrating a formation of homogenous microstructure duringthe mechanical activation.
Thermally induced crystallization of Ta/Cl–HA
Fig. 5 shows the XRD patterns and phase percentages of thecrystallized powders at 800 8C for 1 h. From Fig. 5a, a substantialincrease in the peaks intensity and a marked decline in breadth ofthe characteristic peaks are visible. In fact, heat treatment of theas-milled Ta/Cl–HA powders caused narrowing their apatite-derived XRD peaks. This effect was due to an increase in thecrystallite size and a reduction in the lattice strain. In the absenceof dopant, highly crystalline HA was formed, possibly as a result ofthe elimination of carbonate ions during the thermal annealing.The low intensity of the strongest peak attributed to b-TCP[(0 2 10) plane at 2u = 31.038] in the heat treated TaCl0 nano-powders serves as proof that the crystallized HA is almoststoichiometric with some minor calcium deficiency (Ca/P � 1.67) [42]. It is also obvious that calcite remained stable pastthe thermally induced crystallization. In the case of TaCl25, highlycrystalline Ta-doped hydroxychlorapatite (THCA) was formed as aresult of thermally induced crystallization. The new diffractionpeaks corresponding to calcium chlorophosphate (CCP) was also
detected. By an increase in the dopant content (TaCl50), a furtherincrease in the degree of substitution was observed. The unwantedCCP phase was also detected, signifying that the as-millednanopowders contains a secondary impurity phase, which stayedstable during thermal treatment. The substitution of CO3
2� forPO4
3� (B-type substitution) caused excess of phosphate andcalcium, which promotes the formation of unwanted phases, e.g.CC. It should be noted that the deficit in the negative charge causedby the replacement of PO4
3� by CO32�, can be compensated by the
loss of a positive charge through removal of Ca2+ from the lattice[8]. Further increasing the dopant content (TaCl100) resulted in acomplete co-substitution due to the progressive mechanochemicalreaction. Accordingly, the heat treated TaCl100 showed TCA andCCP phases in addition to narrowing the apatite-derived XRDpeaks. It is clear that the intensity of the unwanted peaks increasedwith the degree of substitution, suggesting that the phase fractionof the unwanted phases influenced by the dopant content andsynthesis condition (Fig. 5b). In the absence of dopant (TaCl0), thephase percentages of HA, b-TCP and CC were 64 � 3, 21 � 1 and15 � 1%, respectively. With the addition of dopant, the percentages ofapatite phases declined from 93 � 5% in TaCl25 to 85 � 5% in TaCl100.This shows that the subsequent annealing can significantly affectedthe phase percentages of the substituted apatite nanopowders.
The lattice parameters obtained by Rietveld refinement andstructural features of the crystallized powders are summarized inTable 3. The results show that the structural features of thecrystallized powders are strongly influenced by the subsequentannealing. In the case of the crystallized TaCl0 at 800 8C for 1 h, thecrystallite size, lattice strain and the volume fraction of the grainboundary reached 36 � 2 nm, 0.0032 � 0.0001 and 7.80 � 0.39%,respectively. With the addition of dopant, the crystallite size rosefrom 50 � 3 nm in TaCl25 to 71 � 4 nm in TaCl100. Conversely, thelattice strain of the crystallized doped powders declined clearly from0.0027 � 0.0001 to 0.0022 � 0.0001% in the case of TaCl25 andTaCl100, respectively. In a similar trend, the volume fraction of thegrain boundary ranged from 5.72 � 0.29% in TaCl25 to 4.08 � 0.21% inTaCl100. On the other hand, the crystallinity degree of the crystallizedTaCl0 was 89 � 4%, and rose gradually to 97 � 4% in the case ofcrystallized TaCl100. Therefore, it can be conclude that the startingmaterials at first reacted into a co-substituted HA, and a thermallyinduced crystallization occurred in the second stage (subsequentannealing). From the crystallographic data, the a-axis direction andunit cell volume show rising trends with increasing dopant content,which is due to the larger ionic size of Cl� as compared toOH�. Furthermore, some differences between the lattice parametersof the annealed samples and the as-milled nanopowders wereidentified, which can be explained by the surface disorder in thenanoparticles [43]. Indeed, the surface features of the nanoparticlesplay a key role in determining their physical properties due to largesurface to volume ratio. The surface disorder in substituted apatitenanoparticles may be due to the ionic exchange reaction and theformation of ionic vacancies at different sub-lattices on the surface,which becomes more pronounced with decreasing nanoparticle size.However, for the annealed nanoparticles, the higher atomic mobilityleads to a reduction in the surface stresses/strains. Hence, an increasein the lattice constants for the annealed specimens is due to thereduced surface disorder.
Fig. 6 shows the TEM micrographs together with HRTEM andSEAD patterns for the crystallized powders at 800 8C for 1 h. Well-defined nano-crystallites of HA with an average size of 34 � 5 nmcan be seen in the TEM image of TaCl0 in Fig. 6a. According to theHRTEM image (Fig. 6b), the regular spacing of the observed latticeplanes was about 0.81 nm, which was consistent with the (1 0 0)lattice spacing of HA. A similar trend was observed by SAED in Fig. 6cwhen compared with XRD analysis, both of which revealed theincreased crystallinity after annealing at 800 8C for 1 h. From the TEM

Fig. 5. (a) XRD patterns and (b) phase percentages of the crystallized powders at 800 8C for 1 h.
B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325322
image in Fig. 6d, the doped sample represents a bimodal grain sizedistribution characterized by the presence of polygonal-shapedparticles along with coarse nanoneedles (61 � 26 nm). The HRTEMimage in Fig. 6e also showed that the (1 0 0) lattice spacing of the
Table 3Lattice parameters obtained by Rietveld refinement and structural features of the crys
Sample a-axis (A) Da (A) c-axis (A) Dc (A) V
TaCl0 9.5503 – 6.8389 – 54
TaCl25 9.5078 �0.0425 6.8366 �0.0023 53
TaCl50 9.6234 0.0731 6.7928 �0.0461 54
TaCl100 9.6337 0.0834 6.7861 �0.0528 54
crystallized TaCl100 was uniformly 0.82 nm, which was similar to the(1 0 0) lattice spacing of CA. In addition, the SAED pattern in Fig. 6findicated that the crystallinity of the crystallized doped powderincreased clearly, which is in good agreement with the XRD results.
tallized powders.
(A3) DV (A3) D (nm) h f (%)
0.1893 – 36 � 2 0.0032 � 0.0001 7.80 � 0.39
5.2194 �4.9699 50 � 3 0.0027 � 0.0001 5.72 � 0.29
4.7938 4.6045 51 � 3 0.0027 � 0.0001 5.66 � 0.28
5.4327 5.2434 71 � 4 0.0022 � 0.0001 4.08 � 0.21

Fig. 6. TEM micrographs together with HRTEM and SEAD patterns for the crystallized powders at 800 8C for 1 h; (a–c) TaCl0 and (d–f) TaCl100.
B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325 323
Comparison of average crystallite diameters for the crystallizedpowders is summarized in Table 4. Based on the obtained values, thecrystallized TaCl0 showed a narrow crystallite size distribution owingto the similar values for Eqs. (VIII) and (XI). Consequently, the resultsare in reasonably good agreement with the XRD data. On the contrary,a bimodal crystallite size distribution was observed in the case of thecrystallized TaCl100, which was mainly due to the large discrepanciesamong these differently weighted d
¯values. Accordingly, the obtained
values are noticeably different from those obtained by XRD.
Reaction mechanism
The crystal structure of HA along the [1 1 1] and [0 0 1]directions is shown in Fig. 7. It is obvious that the HA structurepresents two nonequivalent Ca2+ sites. Cations at site I, Ca (I), arecoordinated by nine oxygens belonging to six PO4
3� formingtriangles and exhibiting a columnar arrangement. Cations at site(II) are heptacoordinated by six oxygen atoms corresponding tofive PO4
3� anions and one OH� group. The smallest distancebetween a cation and a coordinated oxygen is observed at site (II),while the smallest Ca–Ca distances are found between Ca2+ ions atsite (I) [44]. Accordingly, small ions are substituted preferentiallyat Ca (I). Conversely, large cations are incorporated preferably atsite (II). Here, Ta5+ ions would first replace Ca2+ at site (I) as theionic radius of Ta5+ (0.64 A) is smaller than the one of Ca2+ (0.99 A)[27]. Moreover, the more CO3
2� incorporation may change thecrystal composition of Ta/Cl–HA and induces a loss of crystallinity.In the present case, the mechanically induced co-doping of Ta andCl decreased the relative incorporation of CO3
2�, which caused anincrease in crystallinity. These alterations on crystal structure andcomposition with mechanically induced co-substitution may
Table 4Comparison of average crystallite diameters for the crystallized powders.
Crystallized sample Average diameter (nm)
TEM XRD
d¯s
(Eq. (VIII)) d¯v
(Eq. (XI))
TaCl0 33 � 5 34 � 5 36 � 2
TaCl100 54 � 21 61 � 26 71 � 4
change the morphological features, which was further confirmedby the increasing size of the doped case (TaCl100).
In general, mechanothermal process is a process that combinesshort milling duration with a low-temperature isothermalannealing. The combination of these two steps has been foundto be effective in producing different advanced materials such asnanostructured bioceramics [7,23,26,29,31]. Fig. 8 shows aschematic illustration of the strategy for the fabrication andcrystallization of Ta/Cl–HA. According to the results of the presentand previous studies [23,31,45], the formation, rupture andseverance of surface bonds that are characteristic for particleinteractions in high-energy milling can be summarized as follows.The first stage of compaction begins with the rearrangement andrestacking of particles (Fig. 8a). Particles slide past one anotherwith a minimum of deformation and fracture, producing some fine,irregularly shaped particles. In fact, after a relatively short period ofmechanical activation and at moderately low fineness theadherence of particles starts due to the van der Waals adhesionforces. In such polydisperse system, the presence of very fineparticles together with relative coarse particles will greatlypromote the formation of aggregates, and this accounts for themarked tendency for aggregation in milled products that have abroad range of particle sizes. The second stage of compactioninvolves elastic and plastic deformation of particles. During thisstage, plastic deformation at contact points increases intenselyboth the area of the adhesion forces and, thereby raises thestrength, compactness, and resistance to mechanical effects ofparticle agglomerates. From the mechanochemically point of view,the chemical interactions may also proceed at the contactingsurfaces. These interactions result in a very compact, irreversibleadhesion of the particles and forming of agglomerates by variousprocesses, e.g. by welding, coalescence of crystals, by directinitiation at the contacting surfaces, or by means of ‘‘melt-bridges’’.In fact, the particles are grown together by chemical bonds and it ismost probably that the mechanochemical reaction and structuralchanges mostly occur at this level as shown in Fig. 8a. The thirdstage of compaction, involving particle fracture, results in furtherdeformation and/or fragmentation of the particles. The release ofagglomerates by elastic energy is a dominant effect in this stage.During the subsequent heating, highly crystalline nanopowderswere formed, possibly as a result of the elimination of carbonate

Fig. 7. The crystal structure of HA along the [1 1 1] and [0 0 1] directions.
Fig. 8. (a) The process of trapping an incremental volume of powder between two balls in a randomly agitated charge of balls and powder and (b) the crystallization behavior
of the milled powders.
B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325324
ions. As shown in Fig. 8b, the crystallization behavior of the milledpowders is generally divided in three sequential stages [46]: (i*) inthe initial stage the interparticle neck forms and grows. This stepcan occur with a light or without densification and a simple modelof spherical particles can be used to schematize the initial powdercompact and the changes that happen at the microscopic scale;(ii*) in the second stage, densification is generally assumed tooccur by the shrinking of the pores. Accordingly, pores stay openand form a continuous phase. This stage covers the major part ofsintering, and it ends when the pores pinch off to become isolated;(iii*) in the final stage, the isolated pores may disappear altogether,leaving a fully or nearly fully dense ceramic. It should bementioned that the prepared nanopowders were not submittedto any biological assessment, although they take into account for
biomedical applications and it should be a subject of the futureresearch.
Conclusions
The thermally induced crystallization of tantalum and chlorineco-substituted hydroxyapatite nanopowders with different dopantcontents was examined. The results showed that the phasepercentages and structural evolution of the mechanically alloyedco-substituted hydroxyapatite may be prominently influenced bythe dopant content. From the XRD and FTIR findings, the level ofcarbonate replacement became less and less with increasingdopant content, which indicated that the co-doping of Ta and Clmakes the preliminary crystallization of apatite. According to the

B. Nasiri-Tabrizi et al. / Journal of Industrial and Engineering Chemistry 33 (2016) 316–325 325
elemental mapping images, an appropriate spatial distribution ofelements in the nanopowders was achieved, demonstrating theformation of a homogenous microstructure. During the subse-quent annealing at 800 8C for 1 h, highly crystalline nanopowderswere formed, possibly as a result of the elimination of carbonateions. From the TEM micrographs, well-defined nano-crystallites ofHA with an average size of 34 � 5 nm was formed in the case of thecrystallized TaCl0, while the doped sample represents a bimodal grainsize distribution characterized by the presence of polygonal-shapedparticles along with coarse nanoneedles (61 � 26 nm). In a nutshell,the alterations on crystal structure and composition with mechani-cally induced co-substitution may change the structural andmorphological features of synthetic hydroxyapatite and cause thegrowth of crystal in a specific direction.
Acknowledgements
This research is supported by UM High Impact Research GrantUM-MOHE UM.C/625/1/HIR/MOHE/ENG/44 (Regenerative Biome-chanics of Human Body) from the Ministry of Higher EducationMalaysia and University of Malaya grant number RP021C-13AETand RP005B 13AET. The authors are also grateful to ResearchAffairs of Islamic Azad University, Najafabad Branch for supportingof this research.
References
[1] L.L. Hench, J. Am. Ceram. Soc. 74 (1991) 1487.[2] S.V. Dorozhkin, Materials 2 (2009) 399.[3] S.V. Dorozhkin, Materials 2 (2009) 1975.[4] J.L. Xu, K.A. Khor, J.J. Sui, W.N. Chen, Mater. Sci. Eng. C 29 (2009) 44.[5] S. Baradaran, E. Moghaddam, B. Nasiri-Tabrizi, W.J. Basirun, M. Mehrali, M.
Sookhakian, M. Hamdi, Y. Alias, Mater. Sci. Eng. C 49 (2015) 656.[6] M. Okada, T. Furuzono, Sci. Technol. Adv. Mater. 13 (2012) 1.[7] M. Iafisco, J. Gomez Morales, M. Angeles Hernandez-Hernandez, J. Manuel Garcıa-
Ruiz, N. Roveri, Adv. Eng. Mater. 12 (2010) B218.[8] M. Supova, Ceram. Int. 41 (2015) 9203.[9] Y. Pan, M.E. Fleet, in: M.J. Kohn, J. Rakovan, J.M. Hughes (Eds.), Phosphates:
Geochemical, Geobiological and Material Importance, Reviews in Mineralogyand Geochemistry, vol. 48, Mineralogical Society of America, Washington, DC,2002, p. 13.
[10] J.P. Lafon, E. Champion, D. Bernache-Assollant, J. Eur. Ceram. Soc. 28 (2008) 139.[11] B. Wopenka, J.D. Pasteris, Mater. Sci. Eng. C 25 (2005) 131.[12] H.C.W. Skinner, Mineral. Mag. 69 (2005) 621.
[13] P.H. Schlesinger, H.C. Blair, S.L. Teitalbaum, J.C. Edwards, J. Biol. Chem. 272 (1997)18636.
[14] J.C. Merry, I.R. Gibson, S.M. Best, W. Bonfield, J. Mater. Sci. Mater. Med. 9 (1998)779.
[15] G. Ciobanu, A.M. Bargan, C. Luca, Ceram. Int. 41 (2015) 12192.[16] W.Y. Zhou, M. Wang, W.L. Cheung, B.C. Guo, D.M. Jia, J. Mater. Sci. Mater. Med. 19
(2008) 103.[17] G. Renaudin, E. Jallot, J.M. Nedelec, J. Sol–Gel Sci. Technol. 51 (2009) 287.[18] A. Aminian, M. Solati-Hashjin, A. Samadikuchaksaraei, F. Bakhshi, F. Gorjipour, A.
Farzadi, F. Moztarzadeh, M. Schmucker, Ceram. Int. 37 (2011) 1219.[19] B. Nasiri-Tabrizi, A. Fahami, Mater. Lett. 134 (2014) 42.[20] A.Z. Alshemary, Y. Goh, M. Akram, I.R. Razali, M.R.A. Kadir, R. Hussain, Mater. Res.
Bull. 48 (2013) 2106.[21] M. Sadat-Shojai, M.T. Khorasani, E. Dinpanah-Khoshdargi, A. Jamshidi, Acta
Biomater. 9 (2013) 7591.[22] B. Nasiri-Tabrizi, P. Honarmandi, R. Ebrahimi-Kahrizsangi, Mater. Lett. 63 (2009)
543.[23] P. Balaz, Mechanochemistry in Nanoscience and Minerals Engineering, 1st ed.,
Springer, Berlin Heidelberg, 2008.[24] M.H. Fathi, E.M. Zahrani, J. Alloys Compd. 475 (2009) 408.[25] A. Fahami, B. Nasiri-Tabrizi, R. Ebrahimi-Kahrizsangi, Mater. Lett. 110 (2013) 117.[26] B. Nasiri-Tabrizi, A. Fahami, R. Ebrahimi-Kahrizsangi, Ceram. Int. 40 (2014) 901.[27] J. Dhal, S. Bose, A. Bandyopadhyay, Mater. Sci. Eng. C 33 (2013) 3061.[28] S. Ligot, T. Godfroid, D. Music, E. Bousser, J.M. Schneider, R. Snyders, Acta Mater. 60
(2012) 3435.[29] C. Suryanarayana, N. Al-Aqeeli, Prog. Mater. Sci. 58 (2013) 383.[30] L. Takacs, Prog. Mater. Sci. 47 (2002) 355.[31] C. Suryanarayana, Prog. Mater. Sci. 46 (2001) 1.[32] F. Sun, F.H.S. Froes, J. Alloys Compd. 340 (2002) 220.[33] S.S. Rayalu, J.S. Udhoji, S.U. Meshram, R.R. Naidu, S. Devotta, Curr. Sci. 89 (2005)
2147.[34] B.D. Cullity, Elements of X-ray Diffraction, 2nd ed., Addison-Wesley, Boston, MA,
1978.[35] W.F. Smith, J. Hashemi, Crystal Structures and Crystal Geometry, McGraw-Hill
Science, New York, 2004p. 67.[36] M.A. Vannice, Kinetics of Catalytic Reactions, 1st ed., Springer, US, 2005.[37] J. Zhao, X. Dong, M. Bian, J. Zhao, Y. Zhang, Y. Sun, J.H. Chen, X. Wang, Appl. Surf.
Sci. 314 (2014) 1026.[38] Q. Liu, J.P. Matinlinna, Z. Chen, C. Ning, G. Ni, H. Pan, B.W. Darvell, Ceram. Int. 41
(2015) 6149.[39] G.C. Maiti, F. Freund, J. Inorg. Nucl. Chem. 43 (1981) 2633.[40] E. Garcıa-Tunon, B. Dacuna, G. Zaragoza, J. Franco, F. Guitian, Acta Cryst. B68
(2012) 467.[41] P. Wang, C. Li, H. Gong, X. Jiang, H. Wang, K. Li, Powder Technol. 203 (2010) 315.[42] W.L. Suchanek, P. Shuk, K. Byrappa, R.E. Riman, K.S. Tenhuisen, V.F. Janas,
Biomaterials 23 (2002) 699.[43] K. Nadeem, S. Rahman, M. Mumtaz, Prog. Nat. Sci. 25 (2015) 111.[44] Z.Y. Lia, W.M. Lama, C. Yangb, B. Xub, G.X. Nia, S.A. Abbaha, K.M.C. Cheunga, K.D.K.
Luka, W.W. Lua, Biomaterials 28 (2007) 1452.[45] C.L. De Castro, B.S. Mitchell, in: M.I. Baraton (Ed.), Nanoparticles from Mechanical
Attrition, American Scientific Publishers, Valencia, 2002.[46] E. Champion, Acta Biomater. 9 (2013) 5855.


















