
Cystic Fibrosis (CF) - gene location. Cystic Fibrosis (CF): Molecular defect.

Cystic Fibrosis-Update
Manu Jain MD MS
Professor of Medicine and Pediatrics
Director of Adult CF
Feinberg School of Medicine
Lecture Outline
• Epidemiology of CF
• Pathophysiology and genetics
• Diagnostics
• Targeting the genetic defect

CF TimelineYear Journal Article / Event
1938 CF of the pancreas and its relation to celiac disease.
1951 Heat prostration in fibrocystic disease of the pancreas and other conditions.
1953 Abnormal electrolyte composition of sweat in CF
1955 Creation of CFF – spearheaded by families of children with CF
1959 A test for concentration of electrolytes in sweat in CF utilizing pilocarpine by iontophoresis.
1964 A therapeutic regimen for patients with CF.
1966 CFF patient registry formed
1979 Dried-blood spot screening for CF in the newborn.
1983 Chloride impermeability in CF.
1988 Effect of nutrition on survival is confirmed
1989 Identification of the CF gene: genetic analysis.
2003 Evidence for benefit leads to recommendation for universal CF NBS
2011 A CFTR potentiator in patients with CF and the G551D mutation.
Epidemiology of CF
• Prevalence1
• United States ~ 30,000 (1 of every 3,500 babies born)
• Worldwide ~ 70,000
• Change in patient demographics
• Increased life expectancy1
• Improved overall lung function1,2
1) Cystic Fibrosis Foundation, 2015. 2) VanDevanter et al. Pediatr Pulmunol. 2008;43:739-44.

Newborn Screening for CF
• Mandated nationwide• 66% of new diagnoses made in the first year of life• Step-wise process:
– Measurement of IRT– If elevated, then repeat IRT or CFTR genotyping– If still elevated or mutation detected, then sweat chloride test
• Sweat chloride test = best initial diagnostic test
Age ≤ 6 mo Age > 6 mo
CF is unlikely < 30 mmol/L < 40 mmol/L
Intermediate 30-59 mmol/L 40-59 mmol/L
Consistent with CF ≥ 60 mmol/L ≥ 60 mmol/L
Cystic Fibrosis Foundation, 2015.
Diagnostic Complexity
• Patients not identified via NBS– Normal or equivocal sweat chloride test results
• 6.8% patients diagnosed ≥ 16 years old• Clinical presentation
– Often diagnosed in adolescence or adulthood– Genetic mutation – less common defect– Mild disease – respiratory, GI, pancreatic– Atypical symptoms – infertility, sinusitis– Infection with typical CF respiratory pathogens
Cystic Fibrosis Foundation, 2015. Knowles, et al. N Engl J Med. 2002;347(6):439-42.

Patients with CFCFF Patient Registry, 2013
Num
ber
of P
atie
nts
Year
49.7%
29.2%
Median Predicted Age of SurvivalCFF Patient Registry, 2013
Pat
ient
Age
Year
33.4
40.7

40
50
60
70
80
90
100
6 8 10 12 14 16 18 20 22 24 26 28 30
CF Pulmonary Function Decline
Age
Med
ian
% P
redi
cted
F
EV
1
Parallel lines
Cystic Fibrosis Foundation Patient Registry 2007.
20071990
}
Gender Gap
Improved Patient Survival:Evidence-Based Strategies
NBS program and step-wise
diagnostics
Complete nutritional
management
Pancreatic enzyme
replacement
Enhanced chest
physiotherapy
Specialized CF care centers
Aggressive antibiotic regimens
Parkins, et al. Ther Adv Respir Dis. 2011;5(2):105-19.

Pathophysiology of CF
• Heterogeneous, autosomal recessive disorder
• Over 2,000 mutations in the CFTR gene have been described
• CFTR protein is a chloride channel
• Mutated CFTR protein leads to lower or absent chloride transport
Cystic Fibrosis Foundation, 2015.
CFTR Channel
• CF is due to genetic mutations that affect the CFTR protein Impacts synthesis and transfer of the CFTR
protein to the apical membrane of epithelial cells Influences gating or conductance of chloride and
bicarbonate ions through the channel
• CFTR dysfunction results in: Imbalance of ion transport and epithelial
secretions in the lungs, GI tract, pancreas, and liver
Derichs, et al. Eur Respir Rev. 2013;22:58-65.

CFTR Channel
Mucus
Inside cell
Outside cell
Normal CFTR Defective CFTR
Normal CFTR and ENaC activity: Healthy ASL
Normal mucociliary clearance
Defective CFTR, compromised chloride transport, and ENaC
overactivity:Dehydrated ASL
Poor mucociliary clearance
ENaC
Na+
Chloride ions
CF-Related Lung Disease
AbnormalCFTR
Reduced ASL Impaired mucociliary clearance
Obstruction
Obstruction
Obstruction
Inflammation
Inflammation
Inflammation
Infection
Infection
Infection
Structural damage
Pulmonary insufficiency
Bronchiectasis
Respiratory failure
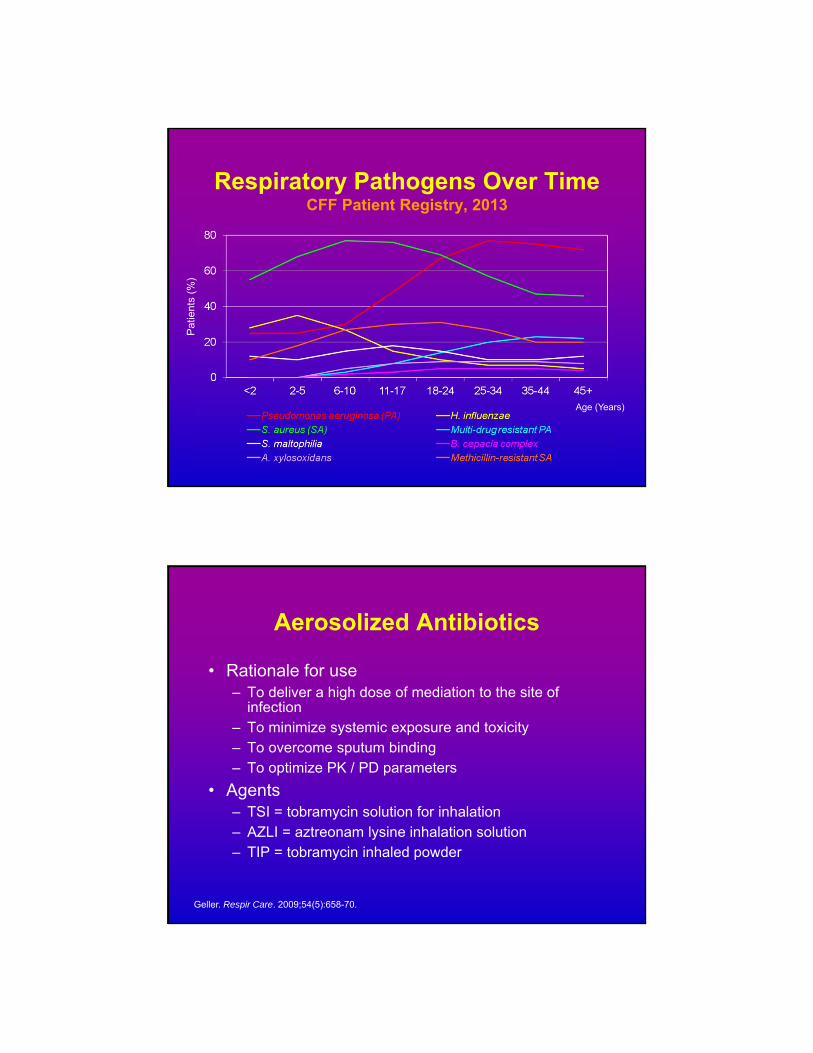
Respiratory Pathogens Over TimeCFF Patient Registry, 2013
Pat
ient
s (%
)
Age (Years)
Aerosolized Antibiotics
• Rationale for use– To deliver a high dose of mediation to the site of
infection– To minimize systemic exposure and toxicity– To overcome sputum binding– To optimize PK / PD parameters
• Agents– TSI = tobramycin solution for inhalation– AZLI = aztreonam lysine inhalation solution– TIP = tobramycin inhaled powder
Geller. Respir Care. 2009;54(5):658-70.

TSI for Chronic Airway Infection
Clinical Study
Study Design N Dosage Study Results
Ramsey1 RCT 520300 mg twice daily for 28 days, 3 on / off cycles
Improvement in microbial response, pulmonary function (P < 0.05)Less hospitalizations, fewer IV antibiotics used
Moss2 OL 93300 mg twice daily for 28 days, 12 on / off cycles
Improvement in pulmonary function sustained for study duration (P < 0.05)
Murphy3 OL 184300 mg twice daily for 28 days
Decrease in hospitalizations, concurrent antibiotic use (P < 0.05)
1) Ramsey, et al. NEJM. 1999;340:23-30. 2) Moss, et al. Chest. 2002;121:55-63. 3) Murphy, et al. Pediatr Pulmonol. 2004;38(4):314-20.
TSI for Chronic Airway Infection
Study design• Longitudinal,
regression analysis
• N = 12,740 patients from CFF patient registry
• Study duration = 10 years
Study results• TSI use was
associated with a 21% reduction in the odds of subsequent year patient mortality (P < 0.05)
Sawicki, et al. Pediatr Pulmonol. 2012;47:44-52.

AZLI vs TSI forChronic Airway Infection
Cha
nge
in F
EV
1(%
Pre
dict
ed)
from
Bas
elin
e
0 2 4 8 12 16 20 24 28 32 36 40 44 48
Assael, et al. J Cyst Fibros. 2013;12:130-40.
Weeks
*
Protein Repair: Mutation-Specific Treatment
Missense:AA deletion
(F508del)
VX-770VX-809
Nonsense:AA substitution
PTC124
Fajac, et al., ECFS 2011; Symposium 12 Oral
Mu
tati
on
Typ
e Missense:AA change
(G551D)
Class IV Class VClass IIClass I Class III
Reducedquantity
Altered Cl-
conductance
Processing& trafficking
defects
Proteinsynthesisdefects
Defectiveregulation
Po
ten
tial
the
rap
y
Missense:AA change
Missense:mRNA processing
VX-770
De
fec
t R
es
ult
AA = amino acid
‡

Most Common MutationsCFF Patient Registry, 2013
* Patients with one or two alleles
Homozygotes = 46.5%Heterozygotes = 39.9%{
97% of patients have had their mutations identified through
genetic testing
Patients* (%)
F508del 86.4
G542X 4.6
G551D 4.4
R117H 2.8
N1303K 2.5
W1282X 2.3
R553X 1.8
Normal
CFTR Mutations
Defect in CFTR Protein
Reduced Quantity of CFTR at Cell Surface
Reduced Function of CFTR at Cell Surface
CFTR Mutations Reduce the Amount or Function of CFTR at the Cell Surface

Approach to Restoring CFTR Function
Potentiators:Increase the flow of ions through CFTR present at the cell surfaceCorrectors:Increase the cellular processing and delivery of CFTR proteins to the cell surface ER ER
Hours
Seconds
CFTR Modulation
• Address the underlying genetic anomaly of CF
• Restore the function of the CFTR protein
• CFTR modulators:
Cystic Fibrosis Foundation, 2015.
Ivacaftor CFTR potentiator
AtalurenPremature stop codon suppressor – enables the formation of a functioning protein in patients with nonsense genetic mutations
- Lumacaftor- VX 661
CFTR correctors – move defective CFTR protein to proper place in cell membrane and improve its function as a chloride channel

Mutation Classification
Class Mutation CFTR Examples
I Nonsense;frame-shift
No functional CFTR G542X 12%
IIMissense; amino acid deletion
CFTR trafficking defect, some CFTR
F508del 86%
III MissenseAbnormal channel function, block in regulation
G551D 5%
IV MissenseAbnormal channel function, altered conductance
R117HD1152H
5%
V Missense; splicing defect
Reduced synthesis of CFTR3849+10kbC→T5TA455E
5%
VI Missense Decreased stability of CFTR 4326delTC Rare
Derichs, et al. Eur Respir Rev. 2013;22:58-65. Boyle, et al. Lancet Resp Med. 2013;1:158-63.
Ivac
afto
r
Ivacaftor:STRIVE Clinical Trial
Ramsey, et al. N Engl J Med. 2011;365:1663-72.
Day 15 8 16 24 32 40 48 Weeks
Cha
nge
in F
EV
1(%
Pre
dict
ed)
from
Bas
elin
e

Day 15 Week 8 Week 16 Week 24 Week 32 Week 40 Week 48-60
-55
-50
-45
-40
-35
-30
-25
-20
-15
-10
-5
0
5
PlaceboVX-770
Ch
ang
e in
sw
eat
chlo
rid
e co
nce
ntr
atio
nm
mo
l/L
(m
ean
, 95%
CI)
Change from Baseline in Sweat Chloride
Treatment effect through Week 24– 47.9 mmol/L
P < 0.0001
Treatment effect through Week 48– 48.1 mmol/L
P < 0.0001
Ivacaftor
• Mechanism of action– Increases the time that activated CFTR channels at the
cell surface remain open; thereby, restoring CFTR function
• FDA approved for patients with the following genetic mutations:
Cystic Fibrosis Foundation, 2015. Flume, et al. Chest. 2012;142(3):718-24.
G551D R117H G178R S549N S549R
G551S G1244E S1251N S1255P G1349D

Mutation Classification
Class Mutation CFTR Examples
I Nonsense;frame-shift
No functional CFTR G542X 12%
IIMissense; amino acid deletion
CFTR trafficking defect, some CFTR
F508del 86%
III MissenseAbnormal channel function, block in regulation
G551D 5%
IV MissenseAbnormal channel function, altered conductance
R117HD1152H
5%
V Missense; splicing defect
Reduced synthesis of CFTR3849+10kbC→T5TA455E
5%
VI Missense Decreased stability of CFTR 4326delTC Rare
Derichs, et al. Eur Respir Rev. 2013;22:58-65. Boyle, et al. Lancet Resp Med. 2013;1:158-63.
CF
TR
cor
rect
ors
Combination Approach: CFTR Potentiator Ivacaftor Doubled the In Vitro Activity of VX-809
-9 -8 -7 -6 -5 -40
10
20
30
40
Log M [VX-809]
F5
08
de
l-C
FT
R A
cti
vit
y(%
wt-
CF
TR
)
VX-809+
Ivacaftor
VX-809alone
CFTR corrector
CFTR potentiator
F508del-CFTR
Van Goor et al. Pediatr Pulmonol 2009;44(S32):154absS9.4

Wainwright CE et al. N Engl J Med 2015;373:220-231.
Ivacaftor with Lumacaftor
Wainwright CE et al. N Engl J Med 2015;373:220-231.
Ivacaftor with Lumacaftor

Mutation Classification
Class Mutation CFTR Examples
I Nonsense;frame-shift
No functional CFTR G542X 12%
IIMissense; amino acid deletion
CFTR trafficking defect, some CFTR
F508del 86%
III MissenseAbnormal channel function, block in regulation
G551D 5%
IV MissenseAbnormal channel function, altered conductance
R117HD1152H
5%
V Missense; splicing defect
Reduced synthesis of CFTR3849+10kbC→T5TA455E
5%
VI Missense Decreased stability of CFTR 4326delTC Rare
Derichs, et al. Eur Respir Rev. 2013;22:58-65. Boyle, et al. Lancet Resp Med. 2013;1:158-63.
Ata
lure
n
Premature Termination Codons
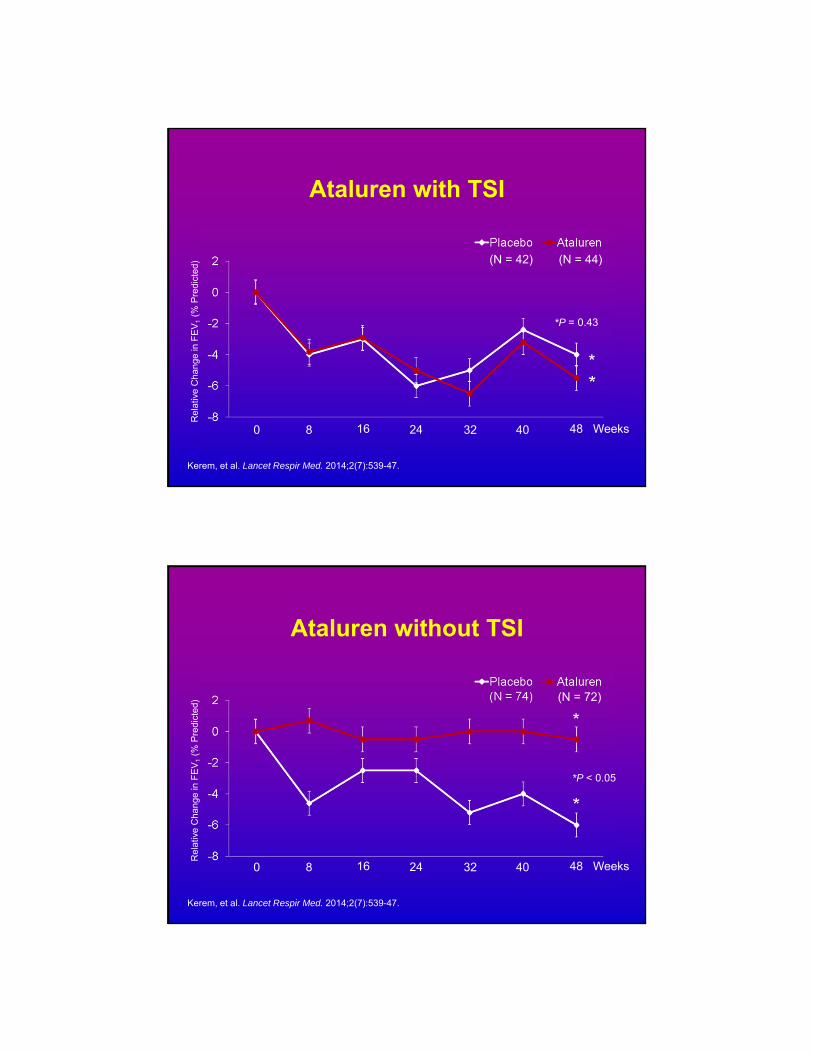
Ataluren with TSI
Kerem, et al. Lancet Respir Med. 2014;2(7):539-47.
*
*P = 0.43
Rel
ativ
e C
hang
e in
FE
V1
(% P
redi
cted
)
8 16 24 320 40 48 Weeks
(N = 44)(N = 42)
Ataluren without TSI
Kerem, et al. Lancet Respir Med. 2014;2(7):539-47.
*
*P < 0.05
Rel
ativ
e C
hang
e in
FE
V1
(% P
redi
cted
)
8 16 24 320 40 48 Weeks
(N = 72)

BMI Trend
10
12
14
16
18
20
22
24
2004 2006 2008 2010 2012 2014 2016
2004 2006 2008 2010 2012 2014 2016
24
22
20
16
14
12
10
BMI
YEAR
Before Kalydeco
After Kalydeco
24 year woman with G551D/d508F
CF Severe Pulmonary Exacerbations
2010 2011 2012 2013 2014 2015
Kalydeco StartedCFPE CFPE CFPE CFPE CFPE
42 year old man with R117H-5T/del508F

HRCT ChestPre-Ivacaftor Post-Ivacaftor
Hepatic SteatosisPre-Ivacaftor Post-Ivacaftor

Sinus DiseasePre-Ivacaftor Post-Ivacaftor
Restoring CFTR Function Decreases Rate of Decline in FEV1
*p < 0.001.
Sawicki, McKone…Konstan et al, Poster 207

Effect of Decreased Rate of Decline in FEV1
20
30
40
50
60
70
80
90
100
6 16 26 36 46 56 66 76 86
G551D With Ivacaftor
F508del/F508del
FEV
1 %
P
red
icte
d
Age in Years
Lung Transplant
FEV1 change doesn’t predict CFPE reduction-Orkambi

F508del/G542XF508del/F508del
A second corrector further enhances in vitro F508del CFTR function
Courtesy of Fred Van Goor
CFTR Modulation
Agent(s) Phase I Phase II Phase III Available
Ivacaftor
Ataluren
Ivacaftor + Lumacaftor
Ivacaftor + VX 661
Riociguat
QBW251
N91115
Cystic Fibrosis Foundation, 2015.

CF-Summary
• Genetic Disease- CFTR– Autosomal Recessive
– Defect in Ion Transport: Cl-, Na, H20
• Cycle- Infection, Inflammation, Mucus– Innate Immunity-likely primary defect
– Antibiotics, Mucolytics, Anti-inflammatory, Rehyrdrators
• CFTR Modulation– Protein Repair– Gene Therapy
– RNA editing
– ENaC Inhibition








