
Isothiocyanates. ХХХП. Microsynthesis of 3-Substituted ... reaction of isothiocyanate with...
5
Isothiocyanates. ХХХП. Microsynthesis of 3-Substituted Rhodanines a Ľ. DROBNICA, ьу. KNOPPOVÁ, and b E. ROMÁNOVÁ ^Department of Microbiology and Biochemistry, Slovak Technical University, Bratislava 1 ъ Department of Organic Chemistry, Slovak Technical University, Bratislava 1 Received January 27, 1971 Dedicated to Professor Dr. P. Nemec on his 60th Anniversary A simple microsynthesis of 3-substituted rhodanines starting from the isothiocyanates (alkyl, aralkyl, and aryl ITC) and thioglycolate via addition reaction and subsequent cyclization of thus formed iV-substituted thiocarb- amoylmercaptoacetates is described. Differences in u.v. spectra of prepared substances and in reactivity of aryl and aralkyl ITC with thiogly colate to give cyclization products are discussed in connection with the nature of the substituent. Also a chromatographic separation of 3-substituted rhodanines and their intermediates is described. In our previous paper [1] we reported the preparation and physicochemical properties of some 3-substituted rhodanines. Standard methods were employed, namely the reaction of JV-substituted dithiocarbamates with monochloroacetic acid, or isothiocyanates with thiogly colic acid, followed by cyclization of thus formed addition products. Yields of rhodanines prepared according to described procedures were relatively low, i.e. between 10 to 40% depending on the isothiocyanate or dithiocarbamate used. Analogous rhodani- nes synthesized by other authors were also isolated in similarly low yields longer time ago [2]. Bearing in mind our experience with the synthesis of 3-substituted rhodanines via reaction of isothiocyanate with thioglycolate and particularly the quantitative investiga- tion of this reaction with various alkyl and especially aralkyl and aryl isothiocyanates with this nucleophile [3], we worked up a more rational procedure for microsynthesis of 3-substituted rhodanines. At the same time, we elaborated also a method for chromato- graphic separation of intermediates and final products on thin layers of adsorbents. The microsynthesis is suitable for a convenient preparation of rhodanines differing markedly by the nature of the substituent at C-3, i.e. it makes it possible to start from various types of isothiocyanates. The yield is virtually 100% when calculated either on isothio- cyanate, or thiogly colic acid. Further advantage of this microsynthesis is seen in the applicability of this method to preparation of rhodanines from infrequent, or 35 S or 14 C labelled isothiocyanates [4] and in the analytical chemistry of natural and synthetic isothiocyanates. The latter will be the subject of our independent paper. 5 38 Ckfm.zpesti 26, 538-542 (1972)
Transcript of Isothiocyanates. ХХХП. Microsynthesis of 3-Substituted ... reaction of isothiocyanate with...

Isothiocyanates. ХХХП. Microsynthesis of 3-Substituted Rhodanines
a Ľ. DROBNICA, ьу. KNOPPOVÁ, and bE. ROMÁNOVÁ
^Department of Microbiology and Biochemistry, Slovak Technical University, Bratislava 1
ъDepartment of Organic Chemistry, Slovak Technical University, Bratislava 1
Received January 27, 1971
Dedicated to Professor Dr. P. Nemec on his 60th Anniversary
A simple microsynthesis of 3-substituted rhodanines starting from the isothiocyanates (alkyl, aralkyl, and aryl ITC) and thioglycolate via addition reaction and subsequent cyclization of thus formed iV-substituted thiocarb-amoylmercaptoacetates is described. Differences in u.v. spectra of prepared substances and in reactivity of aryl and aralkyl ITC with thiogly colate to give cyclization products are discussed in connection with the nature of the substituent. Also a chromatographic separation of 3-substituted rhodanines and their intermediates is described.
In our previous paper [1] we reported the preparation and physicochemical properties of some 3-substituted rhodanines. Standard methods were employed, namely the reaction of JV-substituted dithiocarbamates with monochloroacetic acid, or isothiocyanates with thiogly colic acid, followed by cyclization of thus formed addition products. Yields of rhodanines prepared according to described procedures were relatively low, i.e. between 10 to 40% depending on the isothiocyanate or dithiocarbamate used. Analogous rhodanines synthesized by other authors were also isolated in similarly low yields longer time ago [2].
Bearing in mind our experience with the synthesis of 3-substituted rhodanines via reaction of isothiocyanate with thioglycolate and particularly the quantitative investigation of this reaction with various alkyl and especially aralkyl and aryl isothiocyanates with this nucleophile [3], we worked up a more rational procedure for microsynthesis of 3-substituted rhodanines. At the same time, we elaborated also a method for chromatographic separation of intermediates and final products on thin layers of adsorbents. The microsynthesis is suitable for a convenient preparation of rhodanines differing markedly by the nature of the substituent at C-3, i.e. it makes it possible to start from various types of isothiocyanates. The yield is virtually 100% when calculated either on isothiocyanate, or thiogly colic acid. Further advantage of this microsynthesis is seen in the applicability of this method to preparation of rhodanines from infrequent, or 3 5S or 14C labelled isothiocyanates [4] and in the analytical chemistry of natural and synthetic isothiocyanates. The latter will be the subject of our independent paper.
5 38 Ckfm.zpesti 26, 538-542 (1972)
ISOTHIOCYANATES. X X X I I
Experimental
Chemicals
All chemicals were of anal, grade. Triethylamine (Kodak), was redistilled and had b.p. 83.2 —84°C, the purity of thioglycolic acid (Fluka, A. G., or Reanal) was more than 99%. Isothiocyanates were obtained from the proper amines and thiophosgene, or employing the dithiocarbamate method (c/. [5, 6]) and distilled, or crystallized before use.
Substituted rhodanines
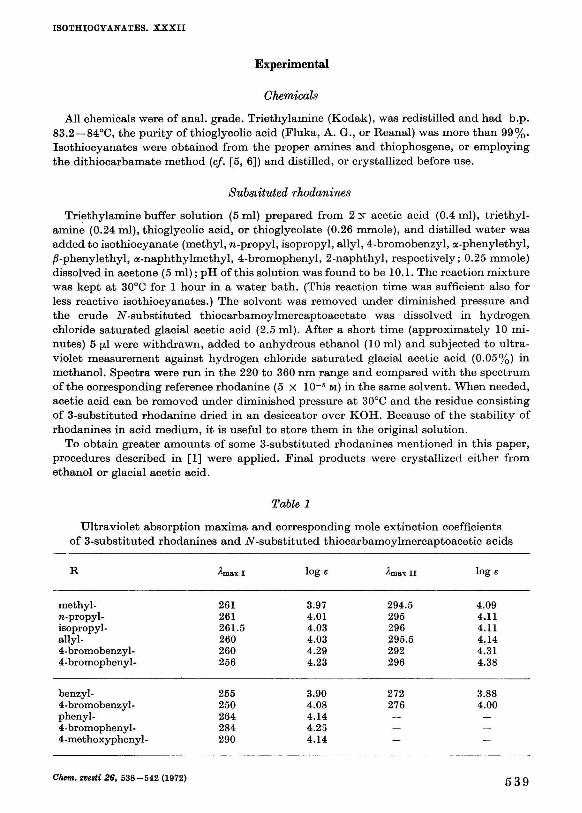
Triethylamine buffer solution (5 ml) prepared from 2 N acetic acid (0.4 ml), triethylamine (0.24 ml), thioglycolic acid, or thioglycolate (0.26 mmole), and distilled water was added to isothiocyanate (methyl, n-propyl, isopropyl, allyl, 4-bromobenzyl, a-phenylethyl, ß-phenylethyl, a-naphthylmethyl, 4-bromophenyl, 2-naphthyl, respectively; 0.25 mmole) dissolved in acetone (5 ml); pH of this solution was found to be 10.1. The reaction mixture was kept at 30°C for 1 hour in a water bath. (This reaction time was sufficient also for less reactive isothiocyanates.) The solvent was removed under diminished pressure and the crude iV-substituted thiocarbamoylmercaptoacetate was dissolved in hydrogen chloride saturated glacial acetic acid (2.5 ml). After a short time (approximately 10 minutes) 5 (JLI were withdrawn, added to anhydrous ethanol (10 ml) and subjected to ultraviolet measurement against hydrogen chloride saturated glacial acetic acid (0.05%) in methanol. Spectra were run in the 220 to 360 nm range and compared with the spectrum of the corresponding reference rhodanine (5 X 10~5 м) in the same solvent. When needed, acetic acid can be removed under diminished pressure at 30°C and the residue consisting of 3-substituted rhodanine dried in an desiccator over KOH. Because of the stability of rhodanines in acid medium, it is useful to store them in the original solution.
To obtain greater amounts of some 3-substituted rhodanines mentioned in this paper, procedures described in [1] were applied. Final products were crystallized either from ethanol or glacial acetic acid.
Table 1
Ultraviolet absorption maxima and corresponding mole extinction coefficients of 3-substituted rhodanines and IV-substituted thiocarbamoylmercaptoacetic acids
R
methyl -n-propyl-isopropyl-allyl-4-bromobenzyl-4-bromophenyl-
benzyl-4-bromobenzyl-phenyl-4-bromophenyl-4-methoxyphenyl-
A m a x I
261 261 261.5 260 260 256
255 250 264 284 290
loge
3.97 4.01 4.03 4.03 4.29 4.23
3.90 4.08 4.14 4.23 4.14
лтах II
294.5 295 296 295.5 292 296
272 276
— —
loge
4.09 4.11 4.11 4.14 4.31 4.38
3.88 4.00
— —
Chem. zvesti 26, 538-542 (1972) 539

Ľ. DROBNICA, V. KNOPPOVÁ, E. KOMANOVÁ
Following procedure was chosen to characterize the ^-substituted thiocarbamoylacetic acids: Methanolic solution of isothiocyanate was added to a borate buffer solution of pH 9.8 (0.1 M) with thioglycolate (1 X Ю - 3 м) so as the final concentration of isothiocyanate and methanol was 5 x Ю - 5 м and 2%, respectively. Under these reaction con-dit'ons isothiocyanates react virtually at once and quantitatively to give Af-substituted thiocarbamoylmercaptoacetates, which are stable due to the high concentration of the thioglycolate. Ultraviolet spectra of the reaction products were directly determined in reaction mixtures in the 220 — 360 nm range toward the same reaction mixture without isothiocyanate. iV-Substituted thiocarbamoylmercaptoacetates are the sole products of this reaction what has been evidenced by cyclization at p H less than 2 to yield 3-substituted rhodanines.
The concentration of 3-substituted rhodanines can be determined spectrophotometri-cally either in situ, or after extraction using log e values as given in Table 1 (c/. also [1]). I t is advisable to dissolve 3-substituted rhodanines for u.v. determination in glacial acetic acid containing methanol, which avoid decomposition (decyclization) of rhodanine.
Kinetic measurements
The kinetics of reaction of 4-bromophenyl and 4-bromobenzyl isothiocyanates as representatives of aryl and aralkyl isothiocyanates with thioglycolate was investigated under such reaction conditions when this reaction had a pseudomonomolecular course and when the proper excess of thioglycolate excluded the side reaction of isothiocyanates with "OH ions: Mcllvain citrate buffer solution (pH 7.5), the initial thioglycolic acid concentration 1.25 x 10~3 м (concentration of the reactive form ~S—CH 2—COO -
0.41 x 1 0 - 5 м provided that pifsH = 10.0), isothiocyanate 3.13 X 1 0 _ 5 м ; temperature 25 ± 0.1 °C; measured in the absorption maximum range of the reaction product (Table 1). For details see [3].
After addition reaction this mixture was equally diluted with 0.4 м citrate buffer solution of pH 2.91 and measured at 296 nm and 25°C. Both addition and cyclization were characterized with rate constants h' and decomposition half life ^/2 (Table 2).
Table 2
Rate constants of the addition reaction of isothiocyanates with thioglycolic acid and cyclization of iV-substituted thiocarbamoylmercaptoacetic acids
R
4-bromobenzyl-4-bromophenyl-
k' [min - 1 ]
0.258 0.475
*1/2
[min]
2.68 1.46
fcc'yci X 10-3 [min - 1 ]
1.38 4.70
hli [min]
502 147
Chromatography
Silica gel (0.125 mm with 10% of gypsum) thin-layer chromatography was used for separation of 3-arylrhodanines from the proper isothiocyanates and ^"-substituted thiocarbamoylmercaptoacetic acids. The standard layers (0.275 mm) were stored in an desiccator and activated before use in a drying oven at 105°C for 45 minutes. All substances to be chromatographed were 0.5% solutions in chloroform. Developing time
S40 Chem. zvesti 26, 638-542 (1972)

ISOTHIOCYANATES. XXXII
approximately 30 minutes at 20°C, developing system benzene — diethyl ether (9 : 1). Developed chromatoplates were air-dried at room temperature and visualized by spraying with a 5% AgNO s solution in 10% ammonia and heating (5 minutes) at 140°C.
Results and Discussion
Ten various 3-substituted rhodanines were prepared by reaction of thioglycolic ac'd with alkyl, aralkyi and aryl isothiocyanates. Based upon data obtained from ultraviolet spectroscopy the described microsynthesis proceeded quantitatively.
3-Alkyl and also 3-arylrhodanines are characterized by two absorption maxima, namely in the 260 to 263, 294 — 296 and 250 to 266, 294 to 296 nm regions, respectively. The interpretation of the above-mentioned absorption spectra was discussed earlier [1]. The characteristic u.v. absorption regions and molar extinction coefficients of various 3-substituted rhodanines are listed in Table 1. Fig. 1 exemplifies the differences in u.v. spectra of aryl or aralkyi derivatives with 4-bromobenzyl, 4-bromophenyl isothiocyanate and corresponding 3-substituted rhodanines and ^"-substituted thiocarbamoylmercapto-acetic acids. A more significant difference than that of the appropriate 3-substituted rhodanines could be seen in the u.v. spectra of isothiocyanates and ZV-substituted thiocarbamoylmercaptoacetic acids.
^-Arylthiocarbamoylmercaptoacetic acids are markedly less stable when compared with analogous aralkyi ones and cannot be isolated; nevertheless they can be well characterized in the given medium by means of u.v. spectroscopy (c/. Table 1). I t is however, possible to isolate them in esterified form (e.g. as ethyl esters; Fig. 16, curve 4). The preparation of these substances was already reported [7]. On the other hand, iV-aralkyl substituted thiocarbamoylmercaptoacetic acid can be isolated. This difference can be
200 250 300 A [nm] 200 250 300 Л [nm]
Fig. 1. Ultraviolet absorption spectra of 4-bromobenzyl isothiocyanate (a), 4-bromophenyl isothiocyanate (b) and corresponding 3-substituted rhodanines, iV-substituted thiocarb
amoylmercaptoacetic acids and their ethyl esters. 1. isothiocyanates с = 5 X 10~5 м in methanol; 2. ^-substituted thiocarbamoylmercaptoacetic acids с = 5 X 10~5 M (in reaction mixture — see text); 3. rhodanines с = = 5 x 10~5 M in 0.05% acetic acid containing methanol; 4. ethyl N- (4-bromophenyl) -thiocarbamoylmercaptoacetate c=5x Ю - 5 м in methanol; 5. N- (4-bromobenzyl) -
thiocarbamoylmercaptoacetic acid с = 5 X 10~5 м in methanol.
Chem. zvesti 26, 538-542 (1972) 541

Ľ. DBOBNICA, V. KKOPPOVÁ, B. KOMANOVÁ
associated w i t h t h e n a t u r e of t h e s u b s t i t u e n t in posi t ion 3 of t h e r h o d a n i n e skeleton, which influences t h e e lectron dens i ty of t h e n i t rogen a t o m .
D a t a in Table 2 d o c u m e n t t h e fact t h a t e i ther aryl, or alkyl i sothiocyanates , d u e t o t h e electrophil i ty of t h e — NCS carbon react readi ly w i t h th iog lycolate because of t h e enormous ly high nucleophi l i ty of t h e ionized thiol g roup. U n d e r favourable condit ions, part icular ly as far as t h e suppression of t h e ionizat ion of t h e carboxyl g roup in ^ - s u b s t i t u t e d t h i o c a r b a m o y l m e r c a p t o a c e t i c acids is concerned, a cyclization t a k e s p lace a t r o o m
Table 3
RF Values of t h e s t a r t i n g c o m p o u n d s , a d d i t i o n a n d cycl izat ion p r o d u c t s
Compound
phenyl isothiocyanate ethyl iV-phenylthiocarbamoylmercaptoacetate 3-phenylrhodanine benzyl isothiocyanate ethyl Aŕ-benzylthiocaŕbamoylmercaptoacetate 3-benzylrhodanine p-to\y\ isothiocyanate ethyl N-(£>-tolyl)thiocarbamoylmercaptoacetate 3-(jo-tolyl)rhodanine £>-bromophenyl isothiocyanate ethyl N-(^-bromophenyl)thiocarbamoylmercaptoacetate 3 - (p-bromopheny 1 )rhodanine jo-dimethylaminophenyl isothiocyanate ethyl ^-(p-dimethylaminophenylJthioQarbamoylmercaptoacetate 3-(^-dimethylaminophenyl)rhodanine £>-methoxyphenyl isothiocyanate ethyl i^-(jo-methoxyphenyl)thiocarbamoylmercaptoacetate 3-(p-methoxyphenyl)rhodanine
B.p. [°C/Torr] M.p. [°C]
120/35 63
1 9 2 - 1 9 3 140/17
80 - 8 1 83 26
7 5 - 77 168.5-169
61 8 0 - 82
1 6 4 - 1 6 5 67
105-106 2 0 4 - 2 0 6 145/12 1 0 4 - 1 0 5
155.5-156
RF
0.96 0.44 0.86 0.90 0.59 0.77 0.88 0.46 0.61 0.97 0.51 0.66 0.87 0.40 0.50 0.97 0.55 0.65
t e m p e r a t u r e t o furnish 3-subst i tuted rhodanines . On microsynthe t ic scale th is can be achieved by dissolving t h e addi t ion p roduc t s in hydrogen chloride s a tu r a t ed glacial acetic acid. I t is wor th no t ing t h a t t h e cyclization r a t e in buffered solutions (e.g. p H 2 .91 ; cf. kinetic measurements) is even here de te rmined b y t h e effect of subs t i tuen t u p o n t h e n a t u r e of ni t rogen.
RF Values of t h e s t a r t ing isothiocyanates , addi t ion p roduc t s resul t ing from t h e react ion of i so th iocyanate wi th thioglycolic acid, a n d 3-subst i tuted rhodanines are l isted in Table 3.
References
1. K n o p p o v á V., Antoš К . , D r o b n i c a Ľ.. K r i s t i á n P . , Cheni. Zvesti 26, 527 (1972). 2. Brown F . С , B r a d s h e r C. K., Morgan E. C T e t e n b a u m M., Wilder P . , J r . , J. Amer.
Chem. Soc. 78, 384 (1956).
3. D r o b n i c a Ľ., A u g u s t í n J . , Collect. Czech. Chem. Commun. 30, 1618 (1965). 4. August ín J . , D r o b n i c a Ľ., Sborník prác Chemickotechnologickej fakulty SVŠT. (Collection
of Communica t ions , Section of Chemis t ry , Slovak Technical Univers i ty . ) Brat i s lava, 1966.
5. Antoš К . , Š tu l lerová A., K n o p p o v á V K r i s t i á n P . , Chem. Zvesti 19, 353 (1965). 6. D y s o n G. M., George H . J . , H u n t e r R. F . , J. Chem. Soc. 1927, 436. 7. K n o p p o v á V., D r o b n i c a Ľ., Chem. Zvesti 26, 533 (1972).
Translated by Z. Votický
5 4 2 Chem. zvesti 26, 538-542 (19 72)










![Reaction rates for mesoscopic reaction-diffusion … rates for mesoscopic reaction-diffusion kinetics ... function reaction dynamics (GFRD) algorithm [10–12]. ... REACTION RATES](https://static.fdocuments.in/doc/165x107/5b33d2bc7f8b9ae1108d85b3/reaction-rates-for-mesoscopic-reaction-diffusion-rates-for-mesoscopic-reaction-diffusion.jpg)




![Isothiocyanates. XXXVI. The synthesis of substituted 4-benzylthio … Isothiocyanates. XXXVI. The synthesis of substituted 4-benzylthio-, 4-[(phenylthio)methyl]phenyl isothiocyanates,](https://static.fdocuments.in/doc/165x107/5e62373958886e351756b249/isothiocyanates-xxxvi-the-synthesis-of-substituted-4-benzylthio-isothiocyanates.jpg)


