
Isolation of novel tissue-specific genes from cDNA...
11
INTRODUCTION All the somatic tissues and the germ line of the mouse are derived from a single epithelial sheet, the epiblast (reviewed by Beddington, 1981; Hogan et al., 1994). This cup-shaped epithelium diversifies during gastrulation to generate three distinct tissues known as the germ layers, comprising an inner layer of ectoderm, an intermediate layer of mesoderm and an enveloping layer of endoderm (Fig. 1). Effectively each layer will give rise to distinct components of the fetal organ primordia. For example, the central nervous system and epidermis will arise from the ectoderm, whereas endoderm derived from the epiblast is the precursor of the gut epithelium and cardiac muscle is mesodermal in origin. Mesoderm and endoderm delaminate from the epiblast in a specialised region, the primitive streak, which constitutes a narrow stripe of egressing and differentiating epiblast cells running down one side of the cup. At the same time that the germ layers are dif- ferentiating, the basic body plan of the organism is established such that the definitive rostrocaudal, dorsoventral and right/left axes become explicit. Consequently, gastrulation represents a particularly dramatic and crucial phase of cytodifferentiation, morphogenesis and pattern formation, within 48 hours trans- forming an epithelial sheet into an embryo with recognizable vertebrate form. At present, we understand little about the molecular mechanisms underlying gastrulation although both the expression patterns of certain genes and the phenotypes of specific mutants indicate that, once initiated, the genetic control of gastrulation may be highly conserved amongst ver- tebrates (Beddington and Smith, 1993; Conlon and Bedding- ton, 1995). It is possible to identify genes likely to play an important role during mouse gastrulation by searching for homologues of genes shown to be instrumental in comparable developmental processes in lower vertebrates or invertebrates (e.g. Blum et al., 1992; Dush and Martin, 1992). An alternative strategy is to assess whether normal gastrulation depends on expression of members of known growth factor families and their receptors or of genes identified due to their oncogenic potential (e.g. Hebert et al., 1991; McMahon, 1992; Yamaguchi et al., 1992). However, neither of these routes can directly identify genes whose function may be peculiar to mouse development and a plausible but somewhat biased catalogue of genes expressed and required during gastrulation may emerge. In theory, a mutant screen could yield the most telling informa- tion about the molecular basis of gastrulation but in practice it is unlikely that the required saturation screen can be achieved in the mouse (Mullins et al., 1994). Consequently, one of the only potentially comprehensive methods available for examining the repertoire of genes active during mouse gastru- lation is to construct cDNA libraries reflecting the differential gene expression between the germ layers. Construction of such libraries from mouse embryos is hampered by the small size of the mammalian embryo 2479 Development 121, 2479-2489 (1995) Printed in Great Britain © The Company of Biologists Limited 1995 A total of 5 conventional, directionally cloned plasmid cDNA libraries have been constructed from the entire embryonic region of the mid-gastrulation mouse embryo and from its four principal tissue constituents (ectoderm, mesoderm, endoderm and primitive streak). These libraries have been validated with respect to the number of independent clones, insert-size and appropriate represen- tation of diagnostic marker genes. Subtractive hybridisa- tion has been used to remove clones common to the Endoderm and Mesoderm cDNA libraries resulting in an Endoderm minus Mesoderm subtracted library. Probe prepared from this subtracted library has been hybridised to a grid containing approximately 18,500 Embryonic Region library clones. Three novel clones have been recovered as well as expected genes already known to be highly expressed in the primitive endoderm lineage at this stage of development. In situ hybridisation to early postim- plantation embryos has revealed the expression patterns of these novel genes. One is highly expressed exclusively in visceral endoderm, one is expressed in ectodermal and endodermal tissues, and the third proves to be an early marker of prospective and differentiated surface ectoderm as well as being expressed in endoderm and its derivatives. Key words: mouse, gastrulation, cDNA library, ectoderm, mesoderm, endoderm, primitive streak SUMMARY Isolation of novel tissue-specific genes from cDNA libraries representing the individual tissue constituents of the gastrulating mouse embryo S. M. Harrison 1 , S. L. Dunwoodie 1 , R. M. Arkell 1 , H. Lehrach 2 and R. S. P. Beddington 1 1 National Institute for Medical Research, The Ridgeway, Mill Hill, London NW7 1AA, UK 2 Max-Planck-Institut für Molekulare Genetik, Ihnestraße 73, D-14195 Berlin, Germany
-
Upload
truongphuc -
Category
Documents
-
view
221 -
download
0
Transcript of Isolation of novel tissue-specific genes from cDNA...

INTRODUCTION
All the somatic tissues and the germ line of the mouse arederived from a single epithelial sheet, the epiblast (reviewedby Beddington, 1981; Hogan et al., 1994). This cup-shapedepithelium diversifies during gastrulation to generate threedistinct tissues known as the germ layers, comprising an innerlayer of ectoderm, an intermediate layer of mesoderm and anenveloping layer of endoderm (Fig. 1). Effectively each layerwill give rise to distinct components of the fetal organprimordia. For example, the central nervous system andepidermis will arise from the ectoderm, whereas endodermderived from the epiblast is the precursor of the gut epitheliumand cardiac muscle is mesodermal in origin. Mesoderm andendoderm delaminate from the epiblast in a specialised region,the primitive streak, which constitutes a narrow stripe ofegressing and differentiating epiblast cells running down oneside of the cup. At the same time that the germ layers are dif-ferentiating, the basic body plan of the organism is establishedsuch that the definitive rostrocaudal, dorsoventral and right/leftaxes become explicit. Consequently, gastrulation represents aparticularly dramatic and crucial phase of cytodifferentiation,morphogenesis and pattern formation, within 48 hours trans-forming an epithelial sheet into an embryo with recognizablevertebrate form. At present, we understand little about themolecular mechanisms underlying gastrulation although boththe expression patterns of certain genes and the phenotypes of
specific mutants indicate that, once initiated, the geneticcontrol of gastrulation may be highly conserved amongst ver-tebrates (Beddington and Smith, 1993; Conlon and Bedding-ton, 1995).
It is possible to identify genes likely to play an importantrole during mouse gastrulation by searching for homologues ofgenes shown to be instrumental in comparable developmentalprocesses in lower vertebrates or invertebrates (e.g. Blum etal., 1992; Dush and Martin, 1992). An alternative strategy isto assess whether normal gastrulation depends on expressionof members of known growth factor families and theirreceptors or of genes identified due to their oncogenic potential(e.g. Hebert et al., 1991; McMahon, 1992; Yamaguchi et al.,1992). However, neither of these routes can directly identifygenes whose function may be peculiar to mouse developmentand a plausible but somewhat biased catalogue of genesexpressed and required during gastrulation may emerge. Intheory, a mutant screen could yield the most telling informa-tion about the molecular basis of gastrulation but in practice itis unlikely that the required saturation screen can be achievedin the mouse (Mullins et al., 1994). Consequently, one of theonly potentially comprehensive methods available forexamining the repertoire of genes active during mouse gastru-lation is to construct cDNA libraries reflecting the differentialgene expression between the germ layers.
Construction of such libraries from mouse embryos ishampered by the small size of the mammalian embryo
2479Development 121, 2479-2489 (1995)Printed in Great Britain © The Company of Biologists Limited 1995
A total of 5 conventional, directionally cloned plasmidcDNA libraries have been constructed from the entireembryonic region of the mid-gastrulation mouse embryoand from its four principal tissue constituents (ectoderm,mesoderm, endoderm and primitive streak). Theselibraries have been validated with respect to the number ofindependent clones, insert-size and appropriate represen-tation of diagnostic marker genes. Subtractive hybridisa-tion has been used to remove clones common to theEndoderm and Mesoderm cDNA libraries resulting in anEndoderm minus Mesoderm subtracted library. Probeprepared from this subtracted library has been hybridisedto a grid containing approximately 18,500 Embryonic
Region library clones. Three novel clones have beenrecovered as well as expected genes already known to behighly expressed in the primitive endoderm lineage at thisstage of development. In situ hybridisation to early postim-plantation embryos has revealed the expression patterns ofthese novel genes. One is highly expressed exclusively invisceral endoderm, one is expressed in ectodermal andendodermal tissues, and the third proves to be an earlymarker of prospective and differentiated surface ectodermas well as being expressed in endoderm and its derivatives.
Key words: mouse, gastrulation, cDNA library, ectoderm,mesoderm, endoderm, primitive streak
SUMMARY
Isolation of novel tissue-specific genes from cDNA libraries representing the
individual tissue constituents of the gastrulating mouse embryo
S. M. Harrison1, S. L. Dunwoodie1, R. M. Arkell1, H. Lehrach2 and R. S. P. Beddington1
1National Institute for Medical Research, The Ridgeway, Mill Hill, London NW7 1AA, UK2Max-Planck-Institut für Molekulare Genetik, Ihnestraße 73, D-14195 Berlin, Germany

2480
(Rothstein et al., 1992), the volume of tissue in the embryonicregion being approximately 2×10−2 mm3 at this stage of devel-opment. As a result the amount of RNA that can be preparedfrom each microdissected germ layer is extremely small.Although PCR methods can be employed to overcome com-parable limitations in starting material (e.g. Belyavsky et al.,1989; Brady et al., 1990), the amplification steps involved canintroduce bias and generate artifacts (e.g. Smith and Gridley,1992) as well as reducing the likelihood of obtaining full-length clones for subsequent expression cloning strategies.Here we report the construction of 5 conventional, direction-ally cloned plasmid libraries prepared from the following con-stituents of mid-gastrulation mouse: (1) ectoderm, (2)mesoderm, (3) endoderm, (4) primitive streak and (5) areference cDNA library derived from the entire embryonicregion. Characterisation of these libraries has demonstratedthat they contain sufficient numbers of clones to indicate thatrelatively rare transcripts will be represented and the averagesize of inserts argues that they should contain a high propor-tion of full-length clones. Most significantly, different librariesshow appropriate enrichment for diagnostic cDNAs known tobe expressed differentially between germ layers and thereforeshould serve as enriched sources of genes expressed duringgastrulation. By subtracting common clones of one germ layerlibrary from another, we can show further enrichment forcDNA clones exhibiting restricted germ layer expressionpatterns. In particular, we have identified one abundant, novelprimitive endoderm marker, another novel gene that appears tobe expressed in epithelial tissue and one gene that is expressedthroughout the endoderm and pregastrulation epiblast butwhose transcripts become restricted during gastrulation to theprospective surface ectoderm region of the epiblast and laterin development is expressed in surface ectoderm but notneurectoderm.
MATERIALS AND METHODS
Embryo dissectionGastrulating mouse embryos were collected at 7.5 days post coitum(dpc) from C57BL6 × DBA matings. Embryos that had developedhead folds were excluded. All extraembryonic tissues were removedin M2 medium (Hogan et al., 1994) plus 10% foetal calf serum, andthe remaining embryonic region was either rinsed in PBS and frozenat −80°C in 200 µl denaturing solution (Chomczynski and Sacchi,1987) or used for recovery of isolated ectoderm, mesoderm, endodermand primitive streak fractions. These fractions were isolated using pro-teolytic enzyme digestion followed by mechanical dissection (Bed-dington, 1987) and frozen in denaturing solution.
RNA isolation and analysisEmbryonic tissue was pooled such that a single pool containedbetween 25 and 72 embryonic regions or embryonic region fractions.Embryonic tissue was thawed and denaturing solution was added toa final volume of 500 µl. In the case of the germ layer and primtivestreak fractions, 5 µg of yeast tRNA was added as carrier. Total RNAwas then isolated by scaling down the method of Chomczynski andSacchi (1987). The RNA was resuspended in 1 µl H2O per embryoand stored at −70°C. The amount of total RNA was estimated visuallyby comparing a given amount of each embryo sample to serialdilutions of liver total RNA after hybridisation to a random-primed28S/18S/5S ribosomal RNA probe (Rothstein et al., 1992) by northernblot procedure (Sambrook et al., 1989).
Library constructioncDNA plasmid libraries were constructed using the SuperScriptPlasmid System for cDNA Synthesis and Plasmid Cloning (GIBCOBRL). First strand cDNA was synthesised using a primer-adapter 44nucleotides in length containing 15 dT residues and a NotI restrictionsite. cDNA was synthesised from total RNA with SuperScript IIreverse transcriptase. Second strand cDNA was synthesised with E.coli DNA polymerase in the presence of E. coli DNA ligase and E.coli RNase H. The double-stranded cDNA was blunt ended with T4DNA polymerase and SalI adapters added using T4 DNA ligase. ThecDNA was digested with NotI and size fractionated using sephacryl500 chromatography columns. 10 ng representing the largest cDNAspecies was ligated with 50 ng of NotI-SalI-cut pSPORT1 vectorusing T4 DNA ligase. Each ligation was ethanol precipitated, washedwith 70% ethanol and resuspended in 5 µl 0.5× TE.
Bacterial electrotransformation and cDNA libraryamplificationAll electrotransformations were performed in 0.1 cm electrode gapGene Pulser/E. coli Pulser Cuvettes (BIORAD) using a Gene Pulser(BIORAD) set at 1.8 kV, 200 W, 25 µF (resulting in a field strengthof 18 kV/cm). A transformation efficiency of 5-8×109 cfu/µg wasobtained with 10 pg of pUC19. The total ligation was electroporatedin 5 separate aliquots each containing 1 µl of the ligation and 20 µlof ElectroMAX DH12S cells (GIBCO BRL). Following electropora-tion, 1 ml of prewarmed SOC medium (Hanahan et al., 1991) wasadded and the cells incubated with shaking at 250 revs/minute for 1hour at 37°C. The transformed cells were combined and aliquotsplated to determine library size (number of independent clones). Theremainder of the cells were stored on ice overnight and 5×105 clonesfrom each library were then amplified as described by Hanahan et al.(1991) and stored as aliquots (each representing approximately 1000copies of the library) at −70°C.
Gridding of Embryonic Region clones37,000 clones from the Embryonic Region library were randomlypicked directly into 384-well plates (Genetix Ltd.) using an automaticcolony picker, and 18,500 clones spotted in duplicate in a regulararray on multiple 22×22 cm Hybond-N+ membranes (Meier-Ewert etal., 1993).
Southern blot analysis500 ml Luria Broth/ampicillin cultures were grown overnight at 37°Cfollowing innoculation with bacteria representing several hundredcopies of each amplified library. DNA was isolated by alkaline lysisand density gradient ultracentrifugation (Sambrook et al., 1989).Plasmid DNA was digested with NotI and SalI and 1 µg was size frac-tionated by electrophoresis in 1% agarose/TBE and Southern blotted(Sambrook et al., 1989). 1 µg corresponds to approximately 105
copies of each individual library.
Probes and hybridisationGene-specific probes were prepared from cloned mouse cDNAfragments by polymerase chain reaction using specific oligonu-cleotide primers internal to the cDNA. The PCR product was gelpurified using the QIAEX gel extraction procedure (QIAGEN). 32P-labelled random-primed probes were made to a specific activity of~5×108cts/minute/µg DNA using the Random-Primed DNALabelling Kit (Boehringer-Mannheim).
The following primers were used for each probe: β actin cDNA: bases 865-1182 (Tokunaga et al., 1986). 5′TGCCTGACGGCCAGGTCATCAC3′ and 5′AGGGGCCG-
GACTCATCGTACTC3′T (Brachyury) cDNA: bases 1213-1695 (Herrmann et al., 1990) 5′AACGGGCTGGGAGCTCAGTTCTTT3′ and 5′GAATTCCA-
GGATTTCAAAGTCACA3′
S. M. Harrison and others

2481cDNA libraries from gastrulating mouse embryos
Oct-4 cDNA: bases 714-1140 (Scholer et al., 1990)5′TTCGAGTATGGTTCTGTAA3′ and 5′AGGCTCCTGAT-
CAACAGCAT3′AFP cDNA: bases 881-1725 (Law and Dugaiczyk, 1981)5′AAGTCATGACATATATATGTT3′ and 5′CTGTCAGTTCAG-
GCTTTTGCTTCA3′Follistatin cDNA: bases 545-960 (Albano et al., 1994)5′TTCCGAGATGGAGTTGC3′ and 5′ATCAGACCAATAAT-
GCC3′Probes of total library were prepared by random-primed labelling
approximately 300 ng of all library cDNA. Total library plasmid DNAprepared as described above was digested with NotI and SalI andcDNA was separated from vector by electrophoresis on a 1.2%agarose gel. Total cDNA was purified by QIAEX gel extraction andsubjected to a further round of digestion, electrophoresis and purifi-cation. To eliminate hybridisation of residual vector to the probe ablocking hybridisation was performed according to Baxendale et al.(1991) using 1 µg of linearised pSPORT1 vector. All hybridisationswere carried out using 1×106 cts/minute/ml of probe in 0.5 Mphosphate buffer (pH 7.2), 7% SDS, 1 mM EDTA (Church andGilbert, 1984) at 65°C for 16-18 hours. The membranes were washedin 0.5× SSC, 0.1% SDS and then 0.2× SSC, 0.1% SDS at 65°C.
SequencingClones from each pSPORT1 library were sequenced using the 373ADNA Sequencing System (Applied Biosystems). The DNA templatewas isolated by use of the Wizard Minipreps DNA PurificationSystem (Promega), and sequence generated from the 3′ end of theclone using the Ready Reaction DyeDeoxy Terminator CycleSequencing Kit (Applied Biosystems) from the −21M13 forwardprimer. 200 bp of 3′ sequence was compared to sequences present inthe EMBL and GenBank data bases using the FASTA commands ofthe GCG sequence analysis program (Devereux et al., 1984).
Generation of pSPORT2-cDNA driver librariesIn order to avoid polylinker homology during subtractive hybridisa-tion, all libraries were subcloned into pSPORT2 which has the NotIand SalI restriction enzyme sites inverted with respect to the rest ofthe polylinker. Total cDNA from each pSPORT1 library was purifiedby electrophoresis and gel extraction as described for total librarypobe preparation. Approximately 10 ng of cDNA was ligated into 50ng of NotI/SalI-digested pSPORT2 vector using T4 DNA ligase(GIBCO BRL). Ligations were ethanol precipitated and electrotrans-formed into DH12S electroMAX cells as previously described.
Generation of subtracted cDNA librariesSubtracted cDNA libraries were generated according to the protocolssupplied with the pSPORT2 vector (GIBCO BRL), using single-stranded tracer DNA made from the pSPORT1 Endoderm library andbiotinylated RNA driver generated from the pSPORT2 Endoderm andMesoderm libraries. Modifications to the generation of RNA driverand DNA tracer are described below.
The T7 MEGAscript kit (Ambion) was used for RNA driversynthesis according to the manufacturers instructions with thefollowing modifications. The total reaction volume was scaled-up to80 µl and contained 5 µg of SalI linearised Endoderm or MesodermpSPORT2 library plasmid DNA; 20 µl 10 mM biotin-21-UTP(Clontech) and 5 µl 75 mM UTP. The reaction was allowed to proceedat 37°C for 5 hours, followed by 2 rounds of LiCl precipitation. TheRNA was resuspended in DEPC treated water at 5 µg/µl.
To generate single-stranded phagemid DNA an aliquot of amplifiedEndoderm pSPORT1 library was used to innoculate 50 ml superbroth(35 g tryptone, 20 g yeast extract, 5 g NaCl/litre, pH7.5) to an OD600of 0.05. The culture was grown to an OD600 of 0.2 and then infectedwith M13K07 helper phage at a multiplicity of infection of 10. Theculture was grown with shaking for a further 6 hours and single-stranded DNA isolated by the M13 rescue method described by Vieira
and Messing (1987). Double-stranded DNA contamination wasremoved by two successive cycles of purification with theDYNABEADS lacZ Vector Purification Kit (Dynal) according to themanufacturers instructions.
Following subtractive hybridisation the remaining single-strandedDNA was converted to double-stranded DNA and used to transformDH12S electroMAX cells. Transformed cells were used to innoculateovernight cultures from which plasmid DNA was isolated using theQIAGEN plasmid kit (QIAGEN). To remove vector lacking a cDNAinsert, total cDNA from the subtracted libraries was recloned intopSPORT1 exactly as described for the generation of pSPORT2 cDNAlibraries.
Whole-mount in situ hybridisationThe expression patterns of genes represented by cDNAs identified inthe libraries were studied by whole-mount in situ hybridisation.pSPORT1-cDNA plasmid DNA was linearised with SalI, andantisense digoxygenin-labelled riboprobes prepared and used to probemouse embryos as previously described (Wilkinson, 1992). Embryosfrom 5.5 to 9.5 dpc were recovered from the uterus. Following reflec-tion of Reichert’s membrane, some egg cylinders were bisected intoanterior and posterior halves to avoid ambiguities due to trapping ofprobe in internal cavities. In addition, the endoderm was enzymati-cally removed from some embryos to allow better assessment ofinternal hybridisation patterns.
RESULTS
Preliminary characterisation of the cDNA librariesTotal RNA was isolated from 399 embryonic regions and upto a further 265 embryos were used to generate isolatedectoderm, mesoderm, endoderm and primitive streak fractions(Fig. 1; Table 1). Libraries containing 0.58-1.8×106 indepen-dent clones were generated in each case from 10 ng cDNAderived from the largest cDNA fractions (Table 1). It isestimated with a probability of >99% that rare transcripts (lessthan 10 copies per cell) are present in a cDNA library con-taining 5×105 clones (Sambrook et al., 1989; Ausubel et al.,1994). Therefore the probability of finding cDNA clones rep-resentative of rare transcripts in these cDNA libraries is high.The range of insert sizes is about 0.5-3 kb for each libraryexcept Ectoderm where they are slightly smaller. Since theaverage transcript length in the preimplantation mouse embryois estimated to be about 2 kb (Clegg and Piko, 1983), thelibraries are expected to have a proportion of full-length tran-scripts. This has been confirmed by Southern blot analysis (seebelow). More than 200 clones have been sampled from thelibraries and sequenced from the 3′ end. In all cases the cDNAhad a 3′ poly(A) tail. A fifth of these have also been sequencedfrom the 5′ end and all known sequences confirm the absenceof chimeric clones (data not shown).
Characterisation of the Embryonic Region cDNAlibraryThe gridded Embryonic Region library was further tested forthe presence of cDNA clones representing genes known to beexpressed in the 7.5 dpc embryonic region. As expected, usingspecific cDNA probes, we identified cytoskeletal actin(Tokunaga et al., 1986), Brachyury (T, Wilkinson et al., 1990),Oct-4 (Rosner et al., 1990; Scholer et al., 1990), alphafeto-protein (AFP; Dziadek and Adamson, 1978) and follistatin(Albano et al., 1994) clones. In the case of cytoskeletal actin

2482
cDNA clones, the β actin cDNA probe (which also hybridisesto γ actin cDNA; data not shown) hybridised with 0.23%(86/37,000) of colonies. Therefore the frequency of actincDNA clones falls within the range of previous estimations ofthe prevalence of actin mRNA (Ausubel et al., 1994; Rothsteinet al., 1992; Weng et al., 1989). In contrast to β actin, the Tprobe hybridised to 5 colonies (0.014%), Oct-4 to 6 (0.016%),AFP to 9 (0.024%) and follistatin to 2 (0.0054%). Since theEmbryonic Region library cDNA was synthesised from totalRNA, it is possible that it may contain ribosomal RNA (rRNA)sequences. However no positively hybridising colonies wereobserved using a 28S/18S/5S rRNA probe (Rothstein et al.,1992). cDNA sequence was obtained from 51 randomlyselected clones from the Embryonic Region library. Homologysearches revealed that, of these 51 clones, 17 (33%) hadhomology to known genes in mouse, rat or human, while 31clones (60%) are of unknown identity including 5 that showsignificant homology (77-88%) to randomly sequenced humancDNAs. Finally, 3 clones contained no cDNA insert, indicat-ing that vector background in the Embryonic Region library isapproximately 6%. As expected, these results indicate thatearly embryos are a rich source of previously unidentifiedexpressed sequences (Rothstein et al., 1992).
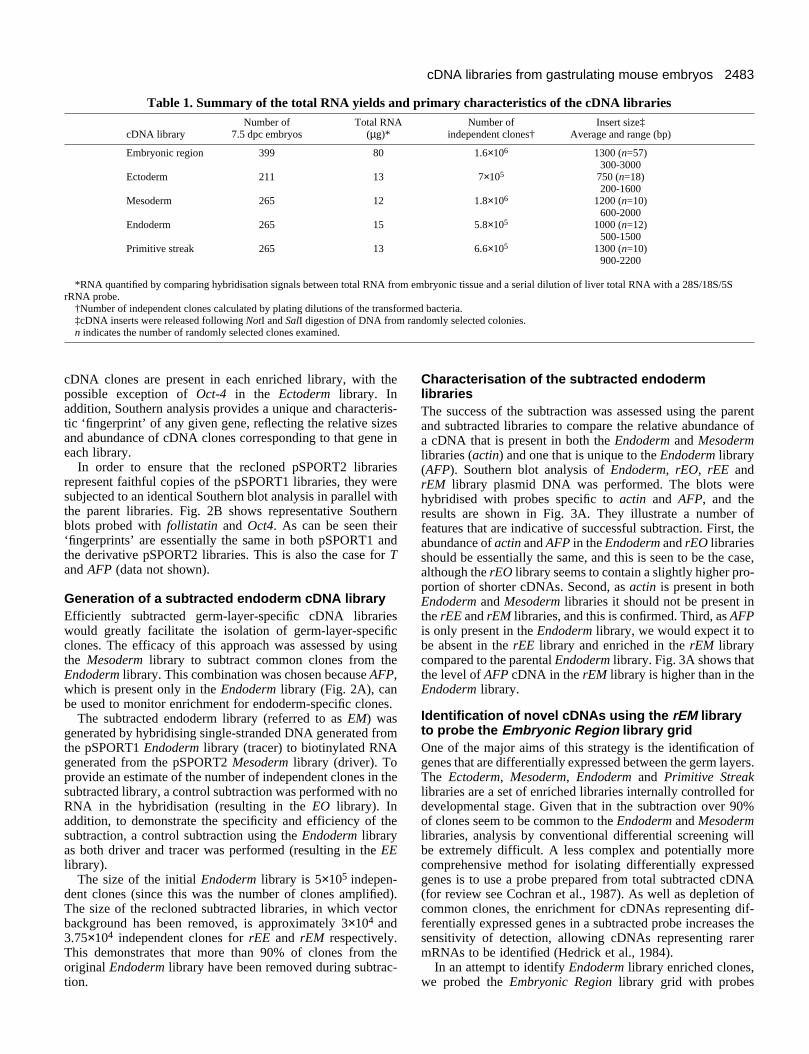
Southern blot analysis of all pSPORT1 andpSPORT2 cDNA librariesIn order to determine if the germ layer and Primitive Streaklibraries are representative of the tissues from which they werederived, a comparison was madebetween the expression pattern ofcertain marker genes at 7.5 dpc andthe distribution of their correspond-ing cDNAs between the libraries.Fig. 1 illustrates the tissue fractionsused to generate the libraries anddemonstrates that the endoderm layercontains head process/notochordprecursor, epiblast-derived definitiveendoderm and residual visceralembryonic endoderm, while theprimitive streak fraction containscontiguous ectoderm and mesoderm.Southern blot analysis, carried out ontotal cDNA insert released from eachlibrary, was used to determinewhether specific germ layer andPrimitive Streak libraries wereenriched for diagnostic cDNAs. Asshown in Fig. 2A this proved to bethe case. AFP is expressed exclu-sively in visceral embryonicendoderm at this stage of develop-ment and clones are detected only inthe Embryonic Region andEndoderm libraries, the relativeintensity of hybridisation demon-strating the predicted enrichment forAFP clones in the Endoderm library(Dziadek and Adamson, 1978). TheT gene is expressed along the lengthof the primitive streak, in the head
process and, to a lesser extent, in the mesoderm emerging fromthe primitive streak (Wilkinson et al., 1990). The hybridisationpattern is consistent with this, T being most highly representedin the Primitive Streak library, evident in the Mesoderm andbarely detectable in the Ectoderm library. The enrichment forT clones in the Endoderm library is due to this germ layer con-taining the precursor of the notochord, which expresses highlevels of T at this stage of development. Oct-4 is expressed athigh levels in the ectoderm and ectodermal component of theprimitive streak (Rosner et al., 1990). This is consistent withthe hybridisation pattern, which demonstrates enrichment ofOct-4 cDNA in the Ectoderm and Primitive Streak librariesrelative to the Embryonic Region library. The detection of Oct-4 cDNA at a low level in the Mesoderm and Endoderm librariesindicates that it is either expressed at low levels in these tissues,or that minor tissue contamination occurred during the dissec-tions. Follistatin is expressed predominantly in the primitivestreak but transcripts are detectable at lower levels in caudalectoderm and mesoderm (Albano et al., 1994). Again theSouthern analysis mimics the in vivo expression profile as fol-listatin is enriched in the Primitive Streak library and detectedin the Mesoderm and Ectoderm libraries. Follistatin cDNA isalso detected in the Endoderm library again suggesting lowlevel expression in this tissue or minor tissue contamination.
Southern blot analysis also revealed the size of the largestcDNA inserts for each of these genes in the respective libraries.The full-length transcript size for each probe is given in thelegend for Fig. 2. It is likely that for each gene full-length
S. M. Harrison and others
Fig. 1. Diagram of the germ layer separations of the embryonic region of 7.5 dpc mouse eggcylinders and illustration of the respective isolated tissues. Bar, 200 µm.
2483cDNA libraries from gastrulating mouse embryos
cDNA clones are present in each enriched library, with thepossible exception of Oct-4 in the Ectoderm library. Inaddition, Southern analysis provides a unique and characteris-tic ‘fingerprint’ of any given gene, reflecting the relative sizesand abundance of cDNA clones corresponding to that gene ineach library.
In order to ensure that the recloned pSPORT2 librariesrepresent faithful copies of the pSPORT1 libraries, they weresubjected to an identical Southern blot analysis in parallel withthe parent libraries. Fig. 2B shows representative Southernblots probed with follistatin and Oct4. As can be seen their ‘fingerprints’ are essentially the same in both pSPORT1 andthe derivative pSPORT2 libraries. This is also the case for Tand AFP (data not shown).
Generation of a subtracted endoderm cDNA libraryEfficiently subtracted germ-layer-specific cDNA librarieswould greatly facilitate the isolation of germ-layer-specificclones. The efficacy of this approach was assessed by usingthe Mesoderm library to subtract common clones from theEndoderm library. This combination was chosen because AFP,which is present only in the Endoderm library (Fig. 2A), canbe used to monitor enrichment for endoderm-specific clones.
The subtracted endoderm library (referred to as EM) wasgenerated by hybridising single-stranded DNA generated fromthe pSPORT1 Endoderm library (tracer) to biotinylated RNAgenerated from the pSPORT2 Mesoderm library (driver). Toprovide an estimate of the number of independent clones in thesubtracted library, a control subtraction was performed with noRNA in the hybridisation (resulting in the EO library). Inaddition, to demonstrate the specificity and efficiency of thesubtraction, a control subtraction using the Endoderm libraryas both driver and tracer was performed (resulting in the EElibrary).
The size of the initial Endoderm library is 5×105 indepen-dent clones (since this was the number of clones amplified).The size of the recloned subtracted libraries, in which vectorbackground has been removed, is approximately 3×104 and3.75×104 independent clones for rEE and rEM respectively.This demonstrates that more than 90% of clones from theoriginal Endoderm library have been removed during subtrac-tion.
Characterisation of the subtracted endodermlibrariesThe success of the subtraction was assessed using the parentand subtracted libraries to compare the relative abundance ofa cDNA that is present in both the Endoderm and Mesodermlibraries (actin) and one that is unique to the Endoderm library(AFP). Southern blot analysis of Endoderm, rEO, rEE andrEM library plasmid DNA was performed. The blots werehybridised with probes specific to actin and AFP, and theresults are shown in Fig. 3A. They illustrate a number offeatures that are indicative of successful subtraction. First, theabundance of actin and AFP in the Endoderm and rEO librariesshould be essentially the same, and this is seen to be the case,although the rEO library seems to contain a slightly higher pro-portion of shorter cDNAs. Second, as actin is present in bothEndoderm and Mesoderm libraries it should not be present inthe rEE and rEM libraries, and this is confirmed. Third, as AFPis only present in the Endoderm library, we would expect it tobe absent in the rEE library and enriched in the rEM librarycompared to the parental Endoderm library. Fig. 3A shows thatthe level of AFP cDNA in the rEM library is higher than in theEndoderm library.
Identification of novel cDNAs using the rEM libraryto probe the Embryonic Region library gridOne of the major aims of this strategy is the identification ofgenes that are differentially expressed between the germ layers.The Ectoderm, Mesoderm, Endoderm and Primitive Streaklibraries are a set of enriched libraries internally controlled fordevelopmental stage. Given that in the subtraction over 90%of clones seem to be common to the Endoderm and Mesodermlibraries, analysis by conventional differential screening willbe extremely difficult. A less complex and potentially morecomprehensive method for isolating differentially expressedgenes is to use a probe prepared from total subtracted cDNA(for review see Cochran et al., 1987). As well as depletion ofcommon clones, the enrichment for cDNAs representing dif-ferentially expressed genes in a subtracted probe increases thesensitivity of detection, allowing cDNAs representing rarermRNAs to be identified (Hedrick et al., 1984).
In an attempt to identify Endoderm library enriched clones,we probed the Embryonic Region library grid with probes
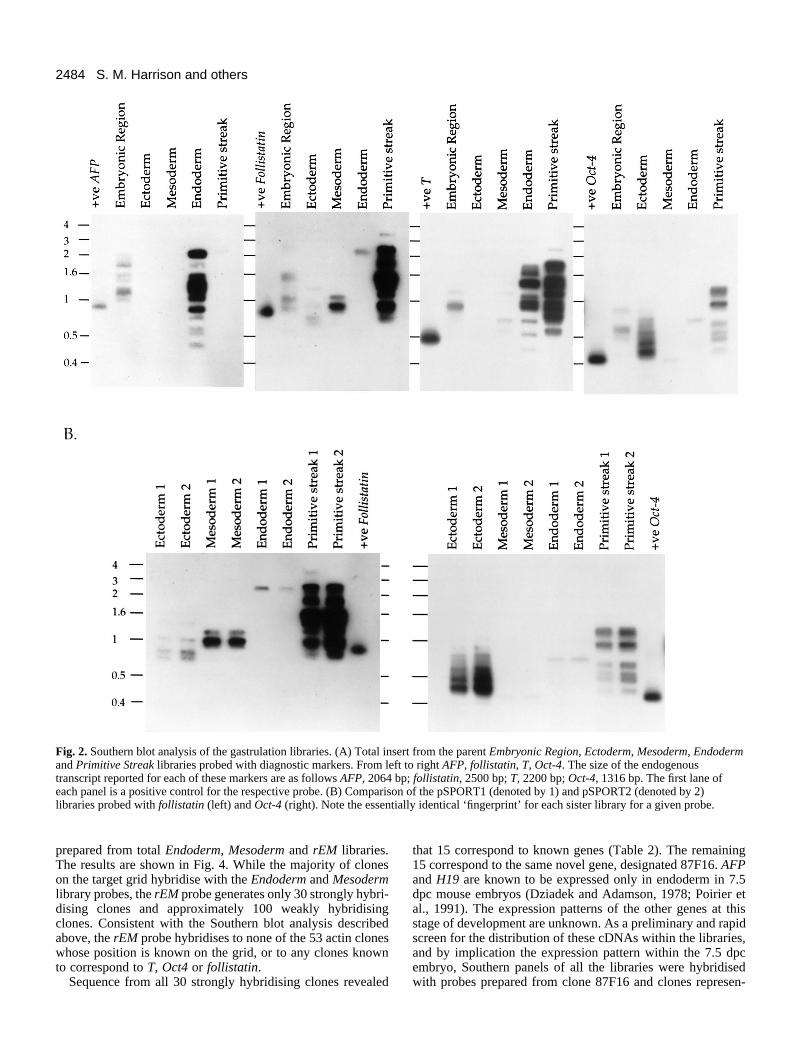
Table 1. Summary of the total RNA yields and primary characteristics of the cDNA librariesNumber of Total RNA Number of Insert size‡
cDNA library 7.5 dpc embryos (µg)* independent clones† Average and range (bp)
Embryonic region 399 80 1.6×106 1300 (n=57)300-3000
Ectoderm 211 13 7×105 750 (n=18)200-1600
Mesoderm 265 12 1.8×106 1200 (n=10)600-2000
Endoderm 265 15 5.8×105 1000 (n=12)500-1500
Primitive streak 265 13 6.6×105 1300 (n=10)900-2200
*RNA quantified by comparing hybridisation signals between total RNA from embryonic tissue and a serial dilution of liver total RNA with a 28S/18S/5SrRNA probe.
†Number of independent clones calculated by plating dilutions of the transformed bacteria.‡cDNA inserts were released following NotI and SalI digestion of DNA from randomly selected colonies. n indicates the number of randomly selected clones examined.
2484
prepared from total Endoderm, Mesoderm and rEM libraries.The results are shown in Fig. 4. While the majority of cloneson the target grid hybridise with the Endoderm and Mesodermlibrary probes, the rEM probe generates only 30 strongly hybri-dising clones and approximately 100 weakly hybridisingclones. Consistent with the Southern blot analysis describedabove, the rEM probe hybridises to none of the 53 actin cloneswhose position is known on the grid, or to any clones knownto correspond to T, Oct4 or follistatin.
Sequence from all 30 strongly hybridising clones revealed
that 15 correspond to known genes (Table 2). The remaining15 correspond to the same novel gene, designated 87F16. AFPand H19 are known to be expressed only in endoderm in 7.5dpc mouse embryos (Dziadek and Adamson, 1978; Poirier etal., 1991). The expression patterns of the other genes at thisstage of development are unknown. As a preliminary and rapidscreen for the distribution of these cDNAs within the libraries,and by implication the expression pattern within the 7.5 dpcembryo, Southern panels of all the libraries were hybridisedwith probes prepared from clone 87F16 and clones represen-
S. M. Harrison and others
Fig. 2. Southern blot analysis of the gastrulation libraries. (A) Total insert from the parent Embryonic Region, Ectoderm, Mesoderm, Endodermand Primitive Streak libraries probed with diagnostic markers. From left to right AFP, follistatin, T, Oct-4. The size of the endogenoustranscript reported for each of these markers are as follows AFP, 2064 bp; follistatin, 2500 bp; T, 2200 bp; Oct-4, 1316 bp. The first lane ofeach panel is a positive control for the respective probe. (B) Comparison of the pSPORT1 (denoted by 1) and pSPORT2 (denoted by 2)libraries probed with follistatin (left) and Oct-4 (right). Note the essentially identical ‘fingerprint’ for each sister library for a given probe.

2485cDNA libraries from gastrulating mouse embryos
tative of Apolipoprotein A, Apolipoprotein E and H19. Inaddition Southern panels were hybridised to probes preparedfrom ten of the weakly hybridising clones chosen at random.Fig. 3B shows that all four of the strongly hybridising clonesand two of the weakly hybridising clones (70C9 and 52D3) aredramatically more abundant in the Endoderm than theMesoderm library. Sequence analysis of 70C9 and 52D3revealed no matches in the database. Some of the clones appearto be confined to the Endoderm library (e.g. Apolipoprotein Aand 52D3); however, consistent with the particular subtractionfrom which the rEM probe was derived, low levels of othersare present in the Ectoderm and Primitive Streak libraries (e.g.87F16 and 70C9).
The remaining eight weakly hybridising clones were allpresent at higher levels in the Endoderm library than theMesoderm library (data not shown), but the difference in levelsis less marked. This indicates that the subtracted probe is ableto identify potentially interesting cDNAs that are enriched in
the tracer library, but which are also found at lower levels inthe driver library.
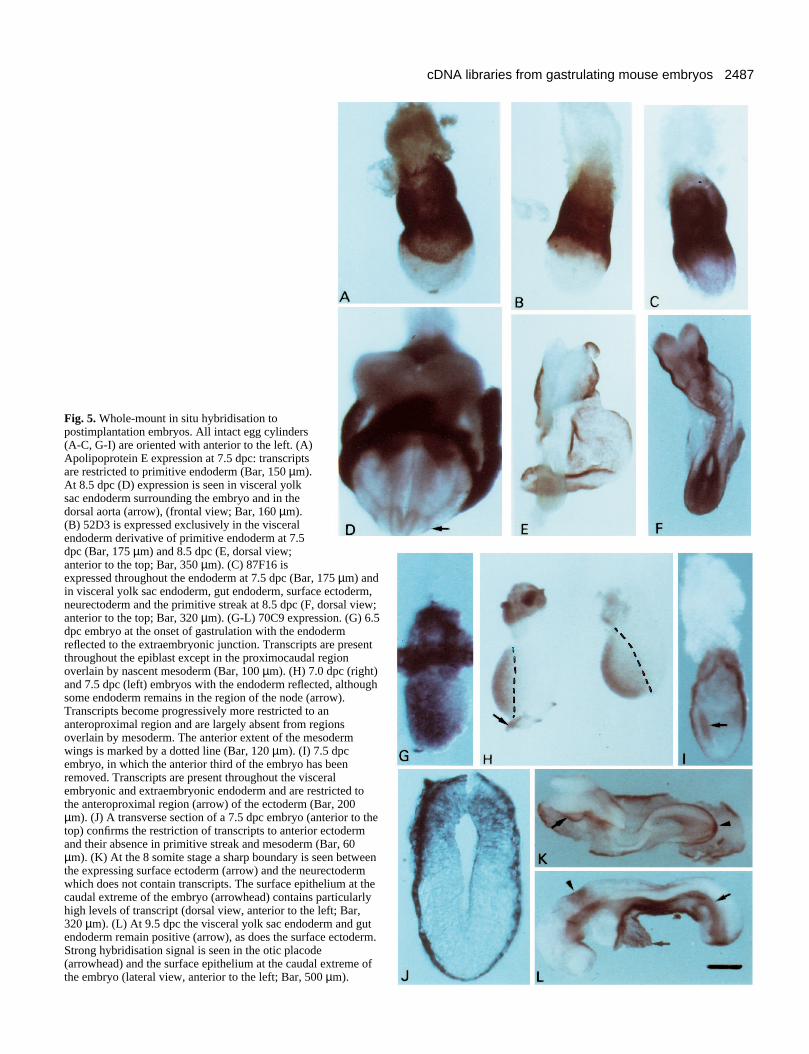
Whole-mount in situ hybridisation using novelEndoderm library enriched clonesFig. 5 illustrates the early postimplantation expression patternsof 4 genes identified by the subtracted probe. ApolipoproteinE transcripts are restricted largely to the visceral primitiveendoderm lineage although at early somite stages low levels oftranscript are seen in the dorsal aortae. Clone 52D3 isexpressed exclusively in visceral primitive endoderm exhibit-ing a pattern strongly reminiscent of AFP (Dziadek andAdamson, 1978; R. Arkell, unpublished observations). Clone87F16 is expressed throughout the visceral endoderm at 7.5dpc and transcripts are also present at reduced levels in theectoderm germ layer (data not shown). A day later hybridisa-tion is observed in visceral yolk sac endoderm, gut endodermand in both surface ectoderm and neurectoderm derivatives of
Fig. 3. (A) Southern blot analysis of the subtracted libraries probed with β actin (top) or AFP (bottom). Endoderm, parent Endoderm library;rEO, Endoderm library ‘subtracted’ in the absence of driver; rEE, Endoderm library subtracted with Endoderm library; rEM, Endoderm librarysubtracted with Mesoderm library. (B) Southern blot analysis to show the distribution within the libraries of clones from the Embryonic Regionlibrary grid which hybridise to the rEM library probe. ApoA, Apolipoprotein A; ApoE, Apolipoprotein E and H19. Clones 87F16, 70C9 and52D3 are novel.

2486
the ectoderm. Transcripts are also abundant in the region of theprimitive streak. Transcripts of clone 70C9 are abundant in thevisceral primitive endoderm from at least 5.5 dpc but are notevident in parietal endoderm (data not shown). Subsequentlyboth gut endoderm and visceral yolk sac endoderm express thisgene. Interestingly, transcripts are also present in the surfaceectoderm, but not neural epithelium, and are particularlyabundant in the otic placode and the surface epithelium at theposterior extreme of the embryo. At mid-gastrulation, region-alisation of the transcripts within the ectoderm germ layer isalready evident and they appear to be restricted to an anteriorlocation that includes the prospective origin of epidermismapped by orthotopic grafting (Beddington, 1982) and bysingle cell labelling (Lawson et al., 1991). Transcripts arepresent in pregastrulation epiblast (data not shown) but areselectively lost in posteriolateral regions underlain by nascentmesoderm indicating a repression of transcription effected byinteraction with mesoderm.
DISCUSSION
Many biological questions relate to changes in gene expressionoccurring in small populations of cells in vivo. Satisfactory invitro cell lines exist for studying some in vivo events and theseprovide almost limitless quantities of material for molecularanalysis. However, for events involving complex morphogen-esis or pattern formation, such as mammalian gastrulation, celllines are seldom suitable substitutes. In these cases, methodsare required for directly assessing the changes in gene
expression occurring in vivo. Assessing the expression ofgenes already identified due to their activity in other biologi-cal systems can be very informative but there is inherentprejudice in such a strategy and it is possible that importantpathways or responses may be overlooked. Here, we haveelected to combine precise microdissection with efficient pro-duction of conventional plasmid cDNA libraries from smallquantities of total RNA, to provide a permanent resource rep-resenting the differential gene expression of four separate cellpopulations within the gastrulating embryo.
The efficiency and precision of the microdissection, and thusthe value of the libraries, has been confirmed by the appropri-ate hybridisation profile of the marker probes to parallelSouthern blots of total insert released from each library (Fig.2). Furthermore, the Southern analysis has demonstrated thatthe desired enrichment for genes expressed in a tissue- orregion-specific manner has been achieved. Each probegenerates a characteristic banding pattern, or ‘fingerprint’, ineach library corrsponding to the prevalence of different sizedcDNA clones. Consequently, as well as validating the qualityof the libraries, this parallel Southern analysis of all librariesprovides a rapid screen for the tissue specificity of anyexpressed sequence, will recognise identical novel clones bytheir ‘fingerprint’ banding pattern and, for most genes, willgive an indication of full-length cDNA size (the largest mRNAtested being follistatin which is 2.5 kb; van den Eijnden-vanRaaij et al., 1992). The average size of inserts also makes theselibraries a suitable substrate for subtractive hybridisation (sincethe hybridisation reaction is more efficient for longersequences) and means that functional screens involvingexpression cloning may be an option (e.g. Smith and Harland,1992).
In an attempt to identify genes that are expressed differen-tially in the endoderm, we have used total cDNA prepared froma subtracted endoderm library rEM to probe a sample of theEmbryonic Region library on a grid. The simplicity of thehybridisation pattern has allowed the very rapid identificationof cDNAs representing seven different genes, three of whichare novel, whose expression is largely confined to theendoderm or to endoderm and ectoderm derivatives (Table 2;Figs 3, 5). At present, only a small proportion of the gridded
S. M. Harrison and others
Table 2. Sequence identity of the 30 clones hybridisingstrongly to the rEM probe
NumberGene of clones Reference
AFP 5 Law and Dugaiczyk, 1981Apolipoprotein A 4 Januzzi et al., 1992Apolipoprotein E 4 Horiuchi et al., 1989H19 2 Pachnis et al., 198887F16 15 Same unknown sequence
Fig. 4. Replica filters of 18,500 Embryonic Region library clones in duplicate probed with Endoderm, Mesoderm and rEM library probes.
2487cDNA libraries from gastrulating mouse embryos
Fig. 5. Whole-mount in situ hybridisation topostimplantation embryos. All intact egg cylinders(A-C, G-I) are oriented with anterior to the left. (A)Apolipoprotein E expression at 7.5 dpc: transcriptsare restricted to primitive endoderm (Bar, 150 µm).At 8.5 dpc (D) expression is seen in visceral yolksac endoderm surrounding the embryo and in thedorsal aorta (arrow), (frontal view; Bar, 160 µm).(B) 52D3 is expressed exclusively in the visceralendoderm derivative of primitive endoderm at 7.5dpc (Bar, 175 µm) and 8.5 dpc (E, dorsal view;anterior to the top; Bar, 350 µm). (C) 87F16 isexpressed throughout the endoderm at 7.5 dpc (Bar, 175 µm) andin visceral yolk sac endoderm, gut endoderm, surface ectoderm,neurectoderm and the primitive streak at 8.5 dpc (F, dorsal view;anterior to the top; Bar, 320 µm). (G-L) 70C9 expression. (G) 6.5dpc embryo at the onset of gastrulation with the endodermreflected to the extraembryonic junction. Transcripts are presentthroughout the epiblast except in the proximocaudal regionoverlain by nascent mesoderm (Bar, 100 µm). (H) 7.0 dpc (right)and 7.5 dpc (left) embryos with the endoderm reflected, althoughsome endoderm remains in the region of the node (arrow).Transcripts become progressively more restricted to ananteroproximal region and are largely absent from regionsoverlain by mesoderm. The anterior extent of the mesodermwings is marked by a dotted line (Bar, 120 µm). (I) 7.5 dpcembryo, in which the anterior third of the embryo has beenremoved. Transcripts are present throughout the visceralembryonic and extraembryonic endoderm and are restricted tothe anteroproximal region (arrow) of the ectoderm (Bar, 200µm). (J) A transverse section of a 7.5 dpc embryo (anterior to thetop) confirms the restriction of transcripts to anterior ectodermand their absence in primitive streak and mesoderm (Bar, 60µm). (K) At the 8 somite stage a sharp boundary is seen betweenthe expressing surface ectoderm (arrow) and the neurectodermwhich does not contain transcripts. The surface epithelium at thecaudal extreme of the embryo (arrowhead) contains particularlyhigh levels of transcript (dorsal view, anterior to the left; Bar,320 µm). (L) At 9.5 dpc the visceral yolk sac endoderm and gutendoderm remain positive (arrow), as does the surface ectoderm.Strong hybridisation signal is seen in the otic placode(arrowhead) and the surface epithelium at the caudal extreme ofthe embryo (lateral view, anterior to the left; Bar, 500 µm).

2488
clones that hybridise to the subtracted probe have beenanalysed, so although the efficacy of the approach has beendemonstrated its full potential, including subtractions utilisingdifferent libraries, has yet to be realised. In particular, onlyabout 25% of the 18,500 clones on a single Embryonic Regionlibrary grid will be derived from endoderm mRNA. Conse-quently, the target size is effectively quite small (i.e. about5,000 clones) and it is likely that only relatively abundantendoderm-specific genes can be identified this way. To extendthe screen either larger numbers of Embryonic Region libraryclones can be used or the Endoderm library can be substitutedas the target. However, having shown that the rEM library isselectively depleted of clones common to mesoderm, this sub-tracted library should prove the richest source of endoderm-specific cDNAs if used as a target for differential screens. Suchtargets have been shown previously to facilitate the identifica-tion of rarer differentially expressed genes (e.g. Sargent andDawid, 1983; Duguid et al., 1988).
With respect to recovering novel genes involved in mousegastrulation, it is clear that, at the very least, this strategy canrapidly generate useful tissue-specific markers. For example,52D3 is a specific marker for the primitive endodermcomponent of visceral endoderm and, given the similaritiesbetween visceral yolk sac endoderm and liver (Jollie, 1990;Thomas et al., 1990), 52D3 (like its companions AFP,Apolipoprotein A and Apolipoprotein E) may also be expressedin hepatic cells (Abelev et al., 1971). In contrast, the veryabundant clone, 87F16, appears to be an unknown gene, whichis most highly expressed in endoderm but transcripts aredetectable in most epithelial tissues, excluding nascentsomites, during gastrulation and early organogenesis. Theexpression of 70C9 in definitive ectoderm derivatives is par-ticularly interesting. In 8.5 and 9.5 dpc embryos, transcripts arerestricted to surface ectoderm and absent in neurectoderm,suggesting that this gene may mark an early change in geneexpression related to the segregation of these two principalectoderm derivatives. Intriguingly, this distribution appears tobe presaged in full-length streak embryos where expressionpredominates in the anterior region of the epiblast, coincidentwith the proposed origin of surface ectoderm (Beddington,1982; Lawson et al., 1991). Restriction of expression to thisregion appears to correlate with the expansion of mesoderm:prior to gastrulation 70C9 transcripts are present throughoutthe epiblast but as the mesoderm wings envelop the epiblast sotranscripts disappear in epiblast cells in contact withmesoderm. This is reminiscent of the expression of Otx2although this transcription factor subsequently becomesrestricted to rostral neurectoderm tissue (Simeone et al.,1992a,b) and, at late streak stages (Ang et al., 1994), appearsto have a broader domain of expression than 70C9. Recombi-nation experiments in vitro have shown that there is a likelyrepressive signal emanating from the posterior mesendodermresponsible for the restriction of Otx2 expression but that laterthis is replaced by a positive maintenance effect requiringrostral mesoderm (Ang et al., 1994). Therefore, it is not unrea-sonable to anticipate similar tissue interactions regulating theexpression of 70C9. The persistence of abundant 70C9 tran-scripts in surface epithelium at the caudal extreme of 8.5-9.5dpc embryos is also interesting since this strongly suggests thatthis population may represent the embryonic source of surfaceectoderm during axis elongation, a hypothesis that can be
tested using lineage tracers. In conclusion, from this initialscreen of the cDNA libraries, 70C9 provides an excellentexample of the kind of novel genes that can be rapidlyrecovered. Such genes reveal new details of molecular hetero-geneity within the embryo and serve as essential tools tomonitor tissue interactions involved in establishing the basicbody pattern of the embryo.
This work was supported in part by an MRC Special Project Grant(G9118913). R. B. is a Howard Hughes Medical Institute InternationalScholar and R. Arkell is supported by the Howard Hughes MedicalInstitute.
REFERENCES
Abelev, G. I. (1971). Alpha-fetoprotein in ontogenesis and its’ association withmalignant tumors. Adv. Cancer Res. 14, 295-358.
Albano, R. M., Arkell, R., Beddington, R. S. P. and Smith, J. C. (1994).Expression of inhibin subunits and follistatin during postimplantation mousedevelopment: decidual expression of activin and expression of follistatin inprimitive streak, somites and hindbrain. Development 120, 803-813.
Ang, S.-L., Conlon, R. A., Jin, O. and Rossant, J. (1994). Positive andnegative signals from mesoderm regulate the expression of mouse Otx2 inectoderm explants. Development 120, 2979-2989.
Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G.,Smith, J. A. and Struhl, K. (1994). Current Protocols in Molecular Biology.Greene Publishing and Wiley Interscience, USA.
Baxendale, S., Bates, G. P., MacDonald, M. E., Gusella, J. F. and Lehrach,H. (1991). The direct screening of cosmid libraries with YAC clones. Nucl.Acids Res. 19, 6651.
Beddington, R. S. P. (1981). The analysis of cell lineages in thepostimplantation mammalian embryo. In Genetic Approaches toDevelopmental Neurobiology. (ed. Y. Tsukada). pp. 21-44. University ofTokyo Press.
Beddington, R. S. P. (1982). An autoradiographic analysis of tissue potency indifferent regions of the embryonic ectoderm during gastrulation in themouse. J. Embryol. Exp. Morph. 69, 265-285
Beddington, R. S. P. (1987). Isolation, culture and manipulation of postimplantation mouse embryos. In Mammalian Development: A PracticalApproach. (ed. M. Monk), pp.43-69. Oxford: IRL Press.
Beddington, R. S. P. and Smith, J. C. (1993). The control of vertebrategastrulation: inducing signals and responding genes. Curr. Opin. Genet. Dev.3, 655-661.
Belyavsky, A., Vinogradova, T. and Rajewsky, K. (1989). PCR-based cDNAlibrary construction: general cDNA libraries at the level of a few cells. Nucl.Acids Res. 17, 2919-2932.
Blum, M., Gaunt, S. J., Cho, K. W. Y., Steinbeisser, H., Blumberg, B.,Bittner, D. and De Robertis, E. M. (1992). Gastrulation in the mouse: therole of the homeobox gene goosecoid. Cell 69, 1097-1106.
Brady, G., Barbara, M. and Iscove, N. N. (1990). Representative in vitrocDNA amplification from individual hemopoietic cells and colonies. Meth.Mol. Cell. Biol. 2, 17-25.
Chomczynski, P. and Sacchi, N. (1987). Single step method of RNA isolationby guanidinium thiocyanate-phenol-chloroform extraction. AnalyticalBiochemistry 162, 156-159.
Church, G. M. and Gilbert, W. (1984). Genomic Sequencing. Proc. Natl.Acad. Sci. USA 81, 1991-1995.
Clegg, K. B. and Piko, L. (1983). Poly (A) length, cytoplasmic adenylation andsynthesis of Poly (A)+ RNA in early mouse embryos. Dev. Biol. 95, 331-341.
Cochran, B. H., Zumstein, P., Zullo, J., Rollins, B., Mercola, M. and Stiles,C. D. (1987). Differential colony hybridisation: molecular cloning from azero data base. Methods in Enzymology 147, 64-85.
Conlon, F. and Beddington, R. S. P. (1995). Mouse gastrulation from a frog’sperspective. Sem. Dev. Biol. (in press)
Devereux, J., Haeberli, P. and Smithies, O. (1984). A comprehensive set ofsequence analysis programs for the VAX. Nucl. Acids Res. 12, 387-395.
Duguid, J. R., Rohwer, R. G. and Seed, B. (1988). Isolation of cDNAs ofscrapie-modulated RNAs by subtractive hybridisation of a cDNA library.Proc. Nat. Acad. Sci. USA 85, 5738-5742.
S. M. Harrison and others

2489cDNA libraries from gastrulating mouse embryos
Dush, M. K. and Martin, G. R. (1992). Analysis of mouse Evx genes: Evx-1displays graded expression in the primitive streak. Dev. Biol. 151, 273-287.
Dziadek, M. and Adamson, E. D. (1978). Localisation and synthesis ofalphafetoprotein in postimplantation mouse embryos. J. Embryol. Exp.Morph. 43. 289-313.
Hanahan, D., Jesse, J. and Bloom, F. R. (1991). Plasmid transformation ofEscherichia coli and other bacteria. Methods in Enzymology 204, 63-113.
Hebert, J. M., Boyle, M. and Martin, G. R. (1991). mRNA localizationstudies suggest that murine FGF-5 plays a role in gastrulation. Development112, 407-415.
Hedrick, S. M., Cohen, D. I., Nielsen, E. A. and Davis, M. M. (1984).Isolation of cDNA clones encoding T cell-specific membrane-associatedproteins. Nature 308, 149-153.
Herrmann, B. G., Labeit, S., Poustka, A., King, T. R. and Lehrach, H.(1990). Cloning the T gene required in mesoderm formation in the mouse.Nature 343, 617-622.
Hogan, B., Beddington, R. S. P., Costantini, F. and Lacy, E. (1994).Manipulating the Mouse Embryo: A Laboratory Manual. 2nd edition. ColdSpring Harbor, New York.
Horiuchi, K., Tajima, S., Menju, M. and Yamamoto, A. (1989). Structureand expression of mouse apolipoprotein E gene. J. Biochem. 106, 98-103.
Januzzi, J. L., Azrolan, N., O’Connell, A., Aalto-Setala, K. and Breslow, J.L. (1992). Characterisation of the mouse Apolipoprotein Apoa-1/Apoc3 genelocus: genomic, mRNA, and protein sequences with comparisons to otherspecies. Genomics 14, 1081-1088.
Jollie, W. P. (1990) Development, morphology and function of the yolk sacplacenta of laboratory rodents. Teratology 41, 361-381.
Law, S. W. and Dugaiczyk, A. (1981). Homology between the primarystructure of α-fetoprotein, deduced from a complete cDNA sequence, andserum albumin. Nature 291, 201-205.
Lawson, K. A., Meneses, J. J. and Pedersen, R. A. (1991). Clonal analysis ofepiblast during germ layer formation in the mouse embryo. Development113, 891-911.
Meier-Ewert, S., Maier, E., Ahmadi, A., Curtis, J. and Lehrach, H. (1993).An automated approach to generating expressed sequence catalogues.Nature 361, 375-376.
McMahon, A. P. (1992). The Wnt family of developmental regulators. TrendsGenet. 8, 236-242.
Mullins, M. C., Hammerschmidt, M., Haffter, P. and Nusslein-Volhard, C.(1994). Large-scale mutagenesis in the zebrafish: in search of genescontrolling development in a vertebrate. Curr. Biol. 4, 189-202.
Pachnis, V., Brannan, C. I. and Tilghman, S. M. (1988). The structure andexpression of a novel gene activated in early mouse embryogenesis. EMBOJ. 7, 673-681.
Poirier, F., Chan, C.-T. J., Timmons, P. M., Robertson, E. J., Evans, M. J.and Rigby, P. W. J. (1991). The murine H19 gene is activated duringembryonic stem cell differentiation in vitro and at the time of implantation inthe developing embryo. Development 113, 1105-1114.
Rosner, M. H., Vigano, M. A., Ozato, K., Timmons, P. M., Poirier, F.,Rigby, P. W. J. and Staudt, L. M. (1990). A POU-domain transcriptionfactor in early stem cells and germ cells of the mammalian embryo. Nature345, 686-692.
Rothstein, J. L., Johnson, D., DeLoia, J. A., Skowronski, J., Solter D. andKnowles, B. (1992). Gene expression during preimplantation mousedevelopment. Genes Dev. 6, 1190-1201.
Sambrook, J., Fritsch E. F. and Maniatis, T. (1989). Molecular Cloning: ALaboratory Manual. 2nd ed. Cold Spring Harbor: Cold Spring HarborLaboratory Press.
Sargent, T. D. and Dawid, I. B. (1983). Differential gene expression in thegastrula of Xenopus laevis. Science 222, 135-139.
Scholer, H. R., Ruppert, S., Suzuki, N., Chowdhury, K. and Gruss, P.(1990). New type of POU domain in germ-line specific protein Oct-4. Nature344, 435-439.
Simeone, A., Acampora, D., Gulisano, M., Stornaiuolo, A. and Boncinelli,E. (1992a). Nested expression domains of four homeobox genes indeveloping rostral brain. Nature 358, 687-690.
Simeone, A., Gulisano, M., Acampora, D., Stornaiuolo, A., Rambaldi, M.and Boncinelli, E. (1992b). Two vertebrate homeobox genes related to theDrosophila empty spiracles gene are expressed in the embryonic cerebralcortex. EMBO J. 11, 2541-2550.
Smith, D. E. and Gridley, T. (1992). Differential screening of a PCR-generated mouse embryo cDNA library: glucose transporters aredifferentially expressed in early postimplantation mouse embryos.Development 116, 555-561.
Smith, W. C. and Harland, R. M. (1992). Expression cloning of noggin, a newdorsalising factor localised to the Spemann organiser in Xenopus embryos.Cell 70, 829-840.
Thomas, T., Southwell, B. R., Schreiber, G. and Jaworowski, A. (1990).Plasma protein synthesis and secretion in the visceral yolk sac of the fetal rat:Gene expression, protein synthesis and secretion. Placenta 11, 413-430.
Tokunaga, K., Taniguchi, H., Yoda, K., Shimizu, M. and Sakiyama, S.(1986). Nucleotide sequence of a full-length cDNA for mouse cytoskeletalβactin mRNA. Nucl. Acids Res. 14, 2829-2829.
van den Eijnden-van Raaij, A. J. M., Feijen, A., Lawson, K. A. andMummery, C. L. (1992) Differential expression of inhibin subunits andfollistatin, but not of activin receptor Type II, during early murine embryonicdevelopment. Dev. Biol. 154, 356-365.
Vieira, J. and Messing, J. (1987). Production of single-stranded plasmidDNA. Methods in Enzymol. 153, 3-11.
Weng, D. E., Morgan, R. A. and Gearhart, J. D. (1989). Estimates of mRNAabundance in the mouse blastocyst based on cDNA library analysis. Mol.Reprod. Dev. 1, 233-241.
Wilkinson, D., Bhatt, S. and Herrmann, B. G. (1990). Expression pattern ofthe mouse T gene and its role in mesoderm formation. Nature 343, 657-659.
Wilkinson, D. G. (Ed.) (1992). Whole-mount in situ hybridisation of vertebrateembryos. In In Situ Hybridisation. pp. 75-83. Oxford: IRL Press.
Yamaguchi, T. P., Conlon, R. A. and Rossant, J. (1992). Expression of thefibroblast growth factor receptor FGFR-1/flg during gastrulation andsegmentation in the mouse embryo. Dev. Biol. 152, 75-88.
(Accepted 23 May 1995)


















