
From infection to genbank torsten seemann - lscc - 27 apr 2012

Introduction to NGS
Peter MacCallum Cancer Centre - Fri 27 July 2012
Dr Torsten Seemann

What we will cover today
● High throughput sequencing● Read sequences● Base quality values● FASTQ and FASTA files● Sequence alignment● BAM files● Visualising alignments

Sequencing

In an ideal world...
● Collect a human genomic DNA sample
● Run it through the lab sequencing machine
● Get back 46 files○ phased, haplotype chromosomes○ each one a single contiguous sequence of AGTC○ maybe some extra files if cancer sample

Reality bites
● Unfortunately, no such instrument exists○ can't read long stretches of DNA (yet)
● But we can read short pieces of DNA○ shred DNA into ~500 bp fragments○ we can read these reliably
● High-throughput sequencing○ sequence millions of different fragments in parallel○ various technologies to do this
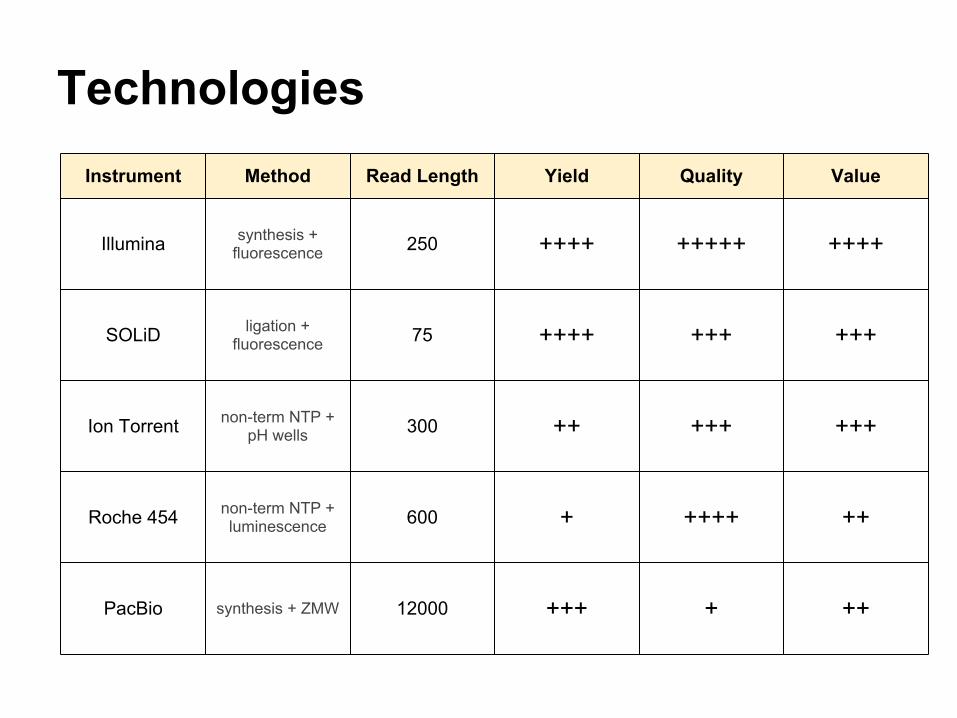
TechnologiesInstrument Method Read Length Yield Quality Value
Illumina synthesis + fluorescence 250 ++++ +++++ ++++
SOLiD ligation + fluorescence 75 ++++ +++ +++
Ion Torrent non-term NTP + pH wells 300 ++ +++ +++
Roche 454 non-term NTP + luminescence 600 + ++++ ++
PacBio synthesis + ZMW 12000 +++ + ++

Illumina
● HiSeq 2000○ 1 week run○ 300 Gb ○ 3 billion reads (100bp)○ "big jobs"
● MiSeq○ 1 day run○ 1.5 Gb○ 5 million reads (150bp)○ "benchtop sequencer"

What you get back
Millions to billions of reads (big files):
ATGCTTCTCCGCCTTTAATTAAAATTCCATTTCGTGCACCAACACCCGTTCCTACCATAATAGCTGTTGGAGTCGCTAAACCTAATGCACATGGACACGC <- 1st read
CTAAGATACTGCCATCTTCTTCCAACGTAAATTGTACGTGATTTTCGATCCATTTTCTTCGAGGTTCTACTTTGTCACCCATTAGTGTGGTTACTCGACG <- 2nd read
GAATATGCGTGGACAGATGACGAATTGGCAGCAATGATTAAAAAAGTCGGCAAAGGATATATGCTACAGCGATATAAAGGACTTGGAGAGATGAATGCGG
ATCAATGCAAATACAAGATGTGACAATGCGCGCAATGCAATGATAACTGGTGTTGTCAAAAAGAAACCGAATGTCGTACCTAGTGCAACAGCCACTGCAA
GGAAAAAATGAGAAAAAATTCAGTTCGAAAACTAACGATTTCTGCTTTATTGATTGGGATGGGGGTCATTATCCCAATGGTTATGCCTAAAATCATGATC
GATGAAACAATCCAACAAATACCATTCAATAATTTCACAGGGGAAAATGAGACNCTAAGTTTCCCCGTATCAGAAGCAACAGAAAGAATGGTGTTTCGCT
...
... <--- 100 bp --->
...
AGGCATCTTGAAAAAACAAGTGTGTGCCTCTGCGATAATCAATGCCACAGAGGTGCATAAAATTAGTTGTCGAAAAAATAATCGCTACCGTTGAGACTTC
AAAGGAGCATTCTTCGCACGCGGCAAAAAAAGAATACAAACGCATGTCTATAAAAGAGACAACCCAAATTACCAGACAGTTAAACGCGATTTATAAGGCT
GTGACAAAAATCGTGTCACAGCTTCTTTTATATCCTGTCTTTTTTTAGTTATTTATTTTTCAACCTTATCAATATGACTTGATAGCCTTTTCTTTTTCGA
AACTTGTTAAAAAAGACGTCAATGCCTTAACTGTACGTGATTCTTCTGCAGTTAGGGGATGACCTTTGACTACTAAAACAGATGCCATATGCTTACCTTC
ACAAAGCATATTTGTAGGAACGATTGAAAGCATCACTCAAGTAGAAGCGGAAGAAGAAACGATTCAACTGAAACTCGTCGATGTCATGGCCAAAGAAGAT
AATTGGACTTTGTCACCGATTTTCAGTTCATCTATGTCCACGCTTATTTTTTCAGCAGTAGCATTCAAAATCACTCCGTCATTGCTGAATGATGTCCCCA
CTCCTGTTTCTTTATCTATAATTGAACTGTAAACATGAGGAATCACTTTTTTTACACCTGCATCGATTGCAATTTTCAGAATTTCTTCAAAGTTTGAAAG
AAACTGCCATTCAAATGCTGCAAGACATGGGAGGTACTTCAATCAAGTATTTCCCGATGAAAGGCTTAGCACATAGGGAAGAATTTAAAGCAGTTGCGGA
ATCATTCCTACGCCAGTCATTTCGCGTAGTTCTTTTACCATTTTAGCTGTAACGTCTGCCATGTTTAACTCCTCCTGTGTGTGTTCTTTTTAAAAAAAGC <- last read

● garbage reads○ instrument wierdness
● duplicate reads○ low complexity library, PCR duplicate
● adaptor read-through○ fragment too short
● indel errors○ skipping bases, inserting extra bases
● uncalled base○ couldn't reliably estimate, replace with "N"
● substitution errors○ reading wrong base
Sequences have errors
More common
Less common

Illumina reads
GATGAAACAATCCAACAAATACCATTCAATAATTTCACAGNGGGAAAATGA
● Usually 100 bp, will be 150-250 bp soon● Indel errors rare● Substitution errors < 1%● Error rate higher at 3' end● Adaptor issues
○ rare in HiSeq (TruSeq prep)○ more common in MiSeq (Nextera prep)
● Very high quality (more details later)

Applications
● If you can transform your assay in to sequencing many short pieces of DNA, then NGS can help!
● Not just genomic DNA○ Exome (targeted subsets of genomic DNA)○ RNA-Seq (transcripts via cDNA)○ ChIP-Seq (protein:DNA binding sites)○ HITS-CLIP (protein:RNA binding sites)○ Methylation (bisulphite treatment of CpG)○ ... ○ even methods to sequence peptides now!

FASTA files

FASTA
>dnaA chromosomal replication initiator protein DnaAMSLSLWQQCLARLQDELPATEFSMWIRPLQAELSDNTLALYAPNRFVLDWVRDKYLEALRDLLALQEKLVTIDNIQKTVAEYYKIKVADLLSKRRSRSVARPRQMAMALAKELLHAVGNGIMARKPNAKVVYMHSERFVQDMVKALQNNAIEEFKRYYRSVDALLIDDFSLPEIGDAFGGRDHTTVLHACRKIEQLREESHDIKEDFSNLIRTLSS
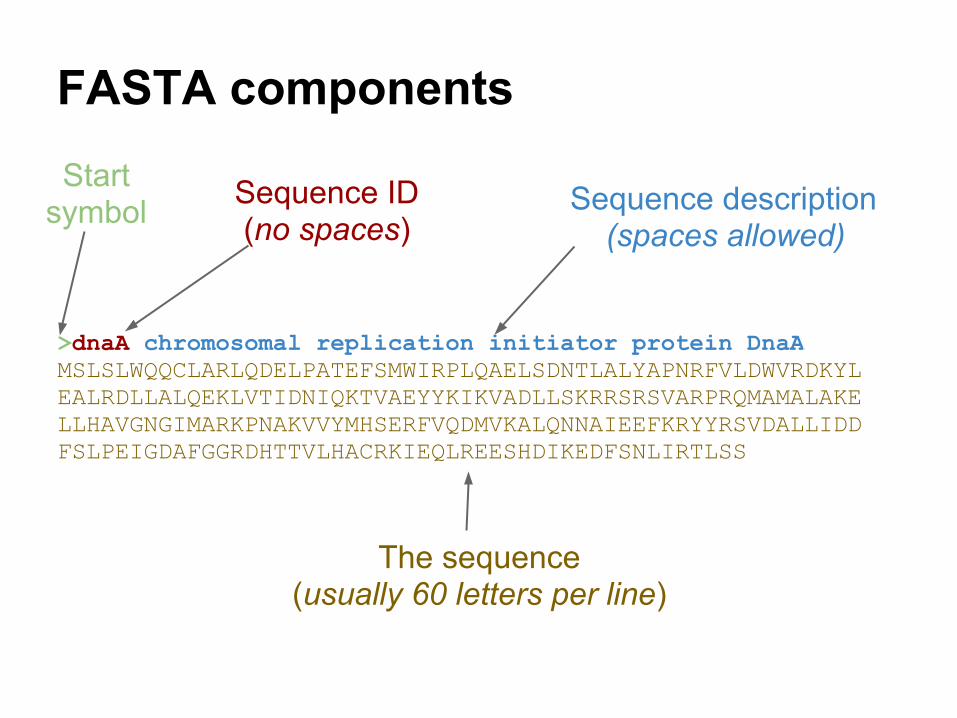
FASTA components
>dnaA chromosomal replication initiator protein DnaAMSLSLWQQCLARLQDELPATEFSMWIRPLQAELSDNTLALYAPNRFVLDWVRDKYLEALRDLLALQEKLVTIDNIQKTVAEYYKIKVADLLSKRRSRSVARPRQMAMALAKELLHAVGNGIMARKPNAKVVYMHSERFVQDMVKALQNNAIEEFKRYYRSVDALLIDDFSLPEIGDAFGGRDHTTVLHACRKIEQLREESHDIKEDFSNLIRTLSS
Start symbol Sequence ID
(no spaces)
The sequence(usually 60 letters per line)
Sequence description(spaces allowed)

Multi-FASTA
>dnaA Chromosomal replication initiator protein DnaAMSLSLWQQCLARLQDELPATEFSMWIRPLQAELSDNTLALYAPNRFVLDWVRDKYLEALRDLLALQEKLVTIDNIQKTVAEYYKIKVADLLSKRRSRSVARPRQMAMALAKELLHAVGNGIMARKPNAKVVYMHSERFVQDMVKALQNNAIEEFKRYYRSVDALLIDDFSLPEIGDAFGGRDHTTVLHACRKIEQLREESHDIKEDFSNLIRTLSS>TetX tetanus toxin coding sequenceATGGCGCGCCGGGACAGAATGCCCTGCAGGAACTTCTTCTGGAAGACCTTCTCCTCCTGCAAATAAAACCTCACCCATGAATGCCTCACGCAAGTTTAATTACAGACCTGAA>CHP_3431 hypothetical proteinMTVLACRKIEQLREESHDIKEDFSNLIRTLSSMSLSLWQQCLARQDELSLPEIGDAFGGRDHTLQAELSDNTLALYAPNRFVLDWVRDKYLNNINELVNAKVVY
Simple concatenation of individual FASTAUses ">" as an entry separator
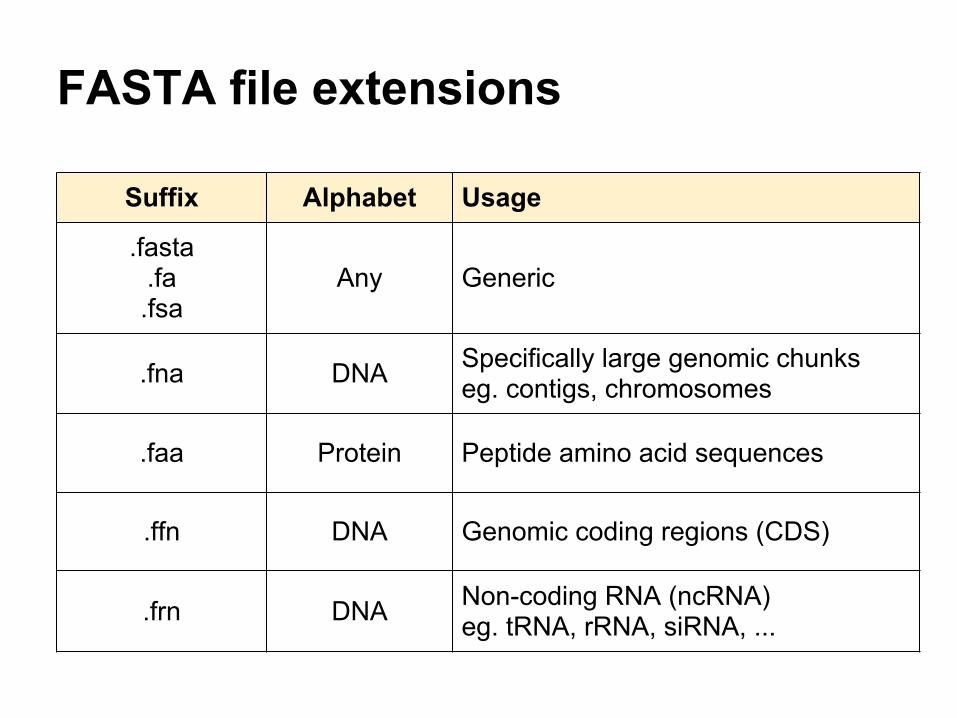
FASTA file extensions
Suffix Alphabet Usage
.fasta .fa .fsa
Any Generic
.fna DNA Specifically large genomic chunkseg. contigs, chromosomes
.faa Protein Peptide amino acid sequences
.ffn DNA Genomic coding regions (CDS)
.frn DNA Non-coding RNA (ncRNA)eg. tRNA, rRNA, siRNA, ...

FASTA alphabets● DNA
○ Standard: AGTC (4)○ Extended: adds N (unknown base)○ Full: adds RY MS WK VHDB (ambiguous bases)
■ R = A/G (puRine), Y=C/T (pYrimidine)
● Protein○ Standard: ARNDC QEGHI LKMFP SUTWY V (21)○ Extended: adds X (unknown amino acid), * (term)○ Full: adds OBZJ
■ O = pyrrolysine, B = D/N, Z =Q/E, J = I/L
● RNA○ replace T with U in DNA rules

Sequence Quality

DNA Sequence Quality
● DNA sequences often have a quality value associated with each nucleotide
● A measure of reliability for each base○ as it is derived from physical process
■ chromatogram (Sanger sequencing)■ pH reading (Ion Torrent sequencing)
● Formalised by the Phred software for the Human Genome Project
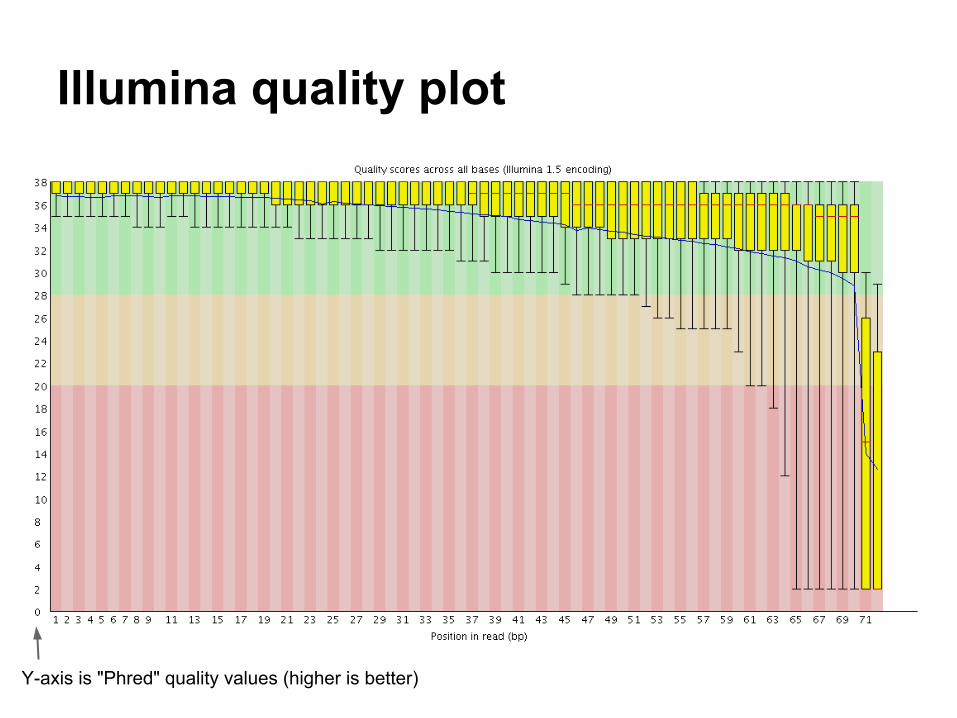
Illumina quality plot
Y-axis is "Phred" quality values (higher is better)
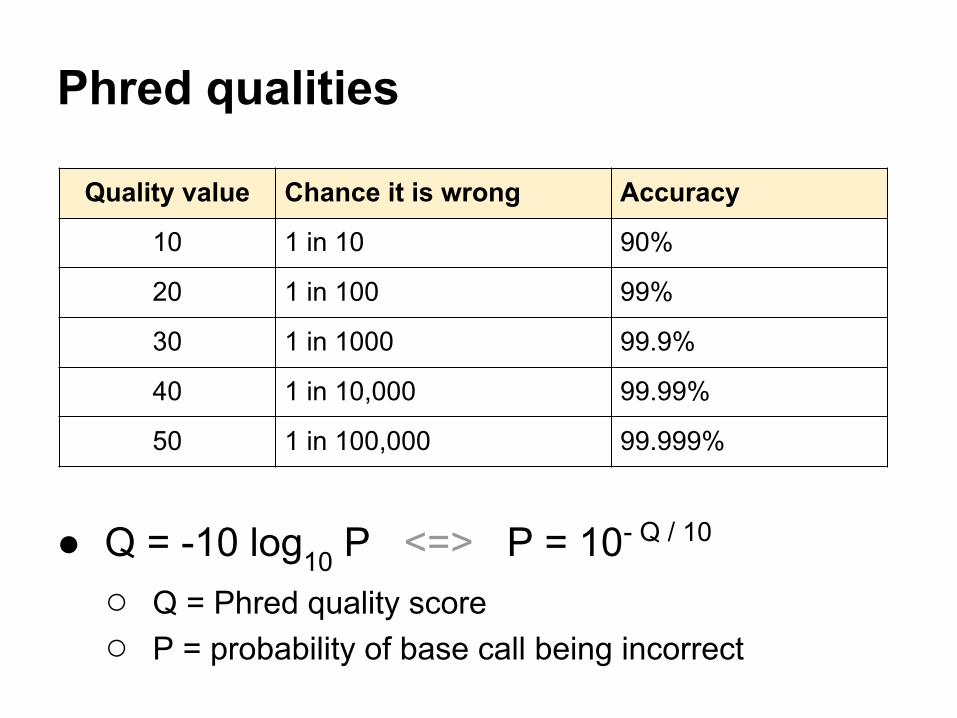
Phred qualities
● Q = -10 log10 P <=> P = 10- Q / 10
○ Q = Phred quality score○ P = probability of base call being incorrect
Quality value Chance it is wrong Accuracy
10 1 in 10 90%
20 1 in 100 99%
30 1 in 1000 99.9%
40 1 in 10,000 99.99%
50 1 in 100,000 99.999%

FASTA Quality files - "old way"
Sequence file: sequence.fasta >contig00179agtcTGATATGCTgtacCTA TTATAATTCTAGGCGCTCATGCCCGCGGATATCGTAGCTATATgCttca
Corresponding quality file: sequence.qual >contig0017910 15 20 22 50 50 50 50 20 25 25 30 30 20 15 20 35 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 20 14 11 10 7
Sometimes lowercase is used to denote "bad" quality and
uppercase for "good" quality

FASTQ files
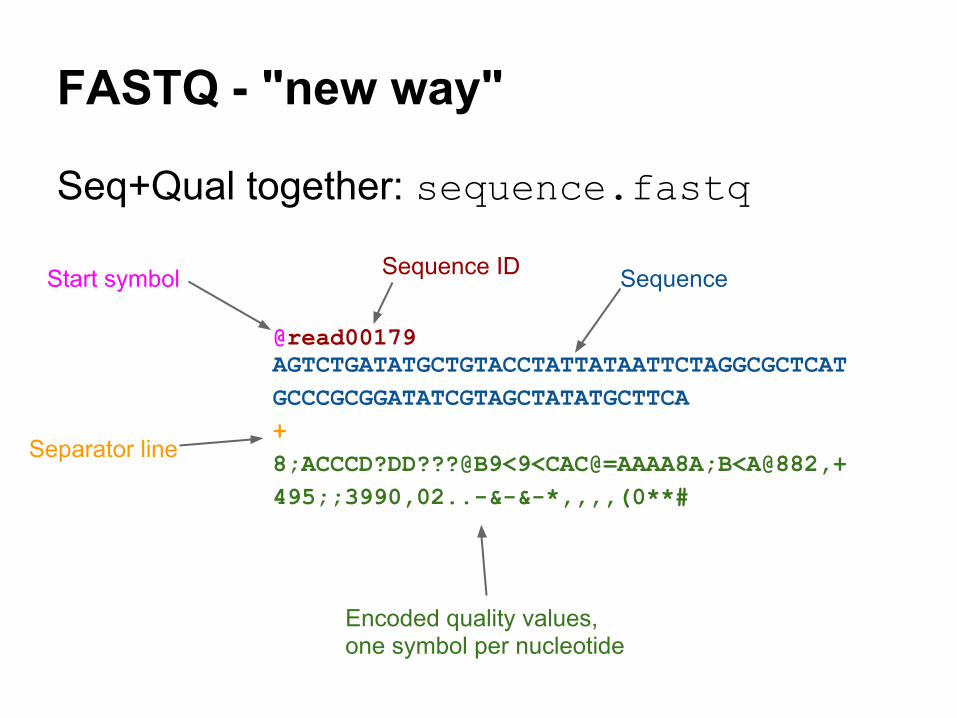
FASTQ - "new way"
Seq+Qual together: sequence.fastq
@read00179AGTCTGATATGCTGTACCTATTATAATTCTAGGCGCTCATGCCCGCGGATATCGTAGCTATATGCTTCA+8;ACCCD?DD???@B9<9<CAC@=AAAA8A;B<A@882,+495;;3990,02..-&-&-*,,,,(0**#
Start symbol Sequence ID Sequence
Separator line
Encoded quality values, one symbol per nucleotide

FASTQ quality encoding
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJ| | | | |Q0 Q10 Q20 Q30 Q40bad maybe ok good excellent
Uses letters/symbols to represent numbers:

Multi-FASTQ
Same as multi-FASTA, just concatenate:@M00267:3:000000000-A0AGE:1:1:15997:1501
CTCGTGCTCTACTTTAGAAGCTAATGATTCTGTTTGTAGAACATTTTCTACCACTACATCTTTTTCTTGCTTCGCATCTT
+
:=?DD:BDDF>FFHI>E>B9AE>4C<4CCAE+AEG3?EAGEHCGIIIIIIIIIIIIGIIIEIIIIGGIDGIID/;4C<EE
@M00267:3:000000000-A0AGE:1:1:15997:1501
GCCTATAGTAGAAGAAAAAGAAGTGGCTCAAGAAATGAGTGCACCGCAGGAAGTTCCAGCGGCTGAATTACTTCATGAAA
+
<@@FFF?DHFHGHIIIFGIIGIGICDGEGCHIIIIIIIIIGIHIIFG<DA7=BHHGGIEHDBEBA@CECDD@CC>CCCAC
@M00267:3:000000000-A0AGE:1:1:14073:1508
GTCTTGCTAAATTTAAATAATCTGAAATAATTTGTTCTGCCCGGTCCAATTCAGCTAATACGAGACGCATATAATCCTTA
+
+:?DDDDD?84CFHC><F>9EEH>B>+A4+CEH4FFEHFHIIIIIIIIIIIIIGGIIIIIIIIG>B7BBEBBB@CDDCFC
@M00267:3:000000000-A0AGE:1:1:14073:1508
ACGTACAGAGATGCAAAAGTCAGAGAAACTTAATATTGTAAGTGAGTTAGCAGCAAGTGTTGCACATGAGGTTCGAAATC
+
1@@DDDADHGDF?FBGGAFHHCHGGCGGFHIECHGIIGIGFGHGHIIHHEGCCFCB>GEDF=FCFBGGGD@HEHE9=;AD

Data compression
● FASTQ files are very big ○ typically > 10 gigabytes○ contains redundancy still
● Often they will be compressed○ gzip (.gz extension) ○ bzip2 (.bz2 extension)○ these are like .ZIP but different method
● Usually get to < 20% of original size○ faster transfer, less disk space○ can be slower to read and write though
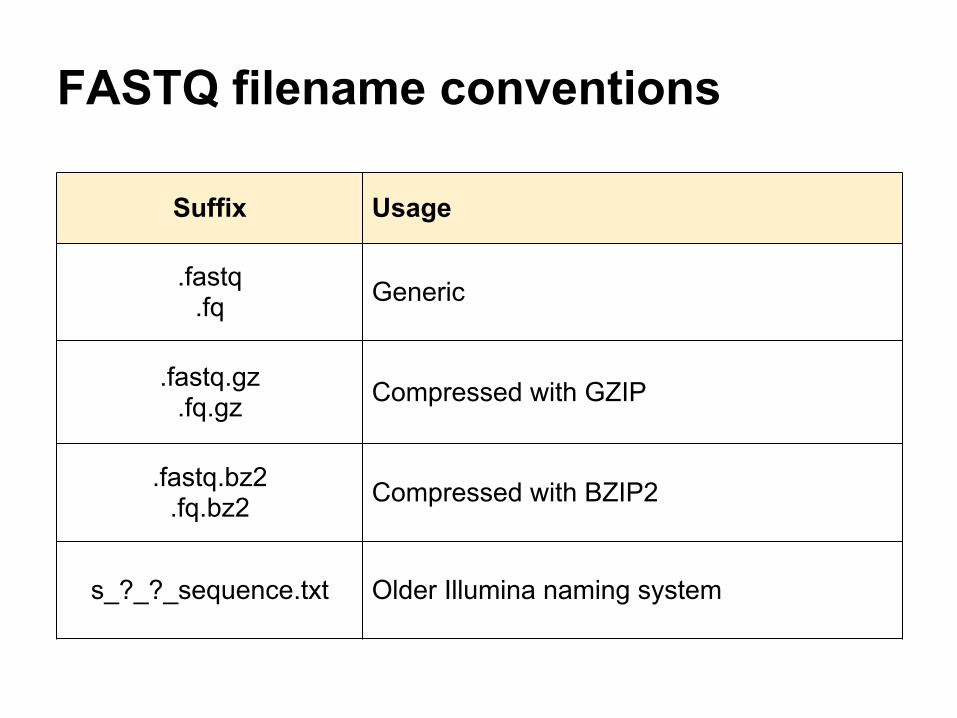
FASTQ filename conventions
Suffix Usage
.fastq.fq Generic
.fastq.gz.fq.gz Compressed with GZIP
.fastq.bz2 .fq.bz2 Compressed with BZIP2
s_?_?_sequence.txt Older Illumina naming system

Alignment

What is an alignment?
● A "best fit" to line up two or more sequences
● Use gaps symbols "-" to help things fit
● Can be considered a matrix○ Each sequence is a row○ Columns are to group corresponding letters

Sequence alignment
● Number of sequences involved (N)○ N=2 is pairwise alignment○ N>2 is multiple sequence alignment (MSA)
● Pairwise alignment○ a "query" sequence vs. a "reference" sequence○ BLAST compares 1 query to 1000s of references○ NGS read aligners do 1000s of queries to 1 ref
● Multiple sequence alignment○ jointly line up more than 2 sequences
■ infer homology, phylogenetic trees ○ ClustalW, Muscle, ....

ClustalW MSA formatCLUSTAL W (1.81) multiple sequence alignment
CYS1_DICDI -----MKVILLFVLAVFTVFVSS---------------RGIPPEEQ------------SQALEU_HORVU MAHARVLLLALAVLATAAVAVASSSSFADSNPIRPVTDRAASTLESAVLGALGRTRHALRCATH_HUMAN ------MWATLPLLCAGAWLLGV--------PVCGAAELSVNSLEK------------FH * :*.. : :. . . *. .
CYS1_DICDI FLEFQDKFNKKY-SHEEYLERFEIFKSNLGKIEELNLIAINHKADTKFGVNKFADLSSDEALEU_HORVU FARFAVRYGKSYESAAEVRRRFRIFSESLEEVRSTN----RKGLPYRLGINRFSDMSWEECATH_HUMAN FKSWMSKHRKTY-STEEYHHRLQTFASNWRKINAHN----NGNHTFKMALNQFSDMSFAE * : .. *.* * * *: * .. :: * . .:.:*.*:*:* *
CYS1_DICDI TTGNVEGQHFISQNKLVSLSEQNLVDCDHECMEYEGEEACDEGCNGGLQPNAYNYIIKNGALEU_HORVU TTGALEAAYTQATGKNISLSEQQLVDCAGGFNNF--------GCNGGLPSQAFEYIKYNGCATH_HUMAN TTGALESAIAIATGKMLSLAEQQLVDCAQDFNNY--------GCQGGLPSQAFEYILYNK *** :*. : .* :**:**:**** :: **:*** .:*::** *
CYS1_DICDI E-WQFYIGGVF-DIPCN--PNSLDHGILIVGYSAKNTIFRKNMPYWIVKNSWGADWGEQGALEU_HORVU DGFRQYKSGVYTSDHCGTTPDDVNHAVLAVGYGVENGV-----PYWLIKNSWGADWGDNGCATH_HUMAN QDFMMYRTGIYSSTSCHKTPDKVNHAVLAVGYGEKNGI-----PYWIVKNSWGPQWGMNG : : * *:: . * *:.::*.:* ***. :* : ***::*****.:** :*
CYS1_DICDI YIYLRRGKNTCGVSNFVSTSII--ALEU_HORVU YFKMEMGKNMCAIATCASYPVVAACATH_HUMAN YFLIERGKNMCGLAACASYPIPLV *: : *** *.:: .* .:

Alignment notation
* = exact match / full consensus: = conservative change. = moderate change = radical change / no consensus
CYS1_DICDI TTGNVEGQHFISQNKLVSLSEQNLVDCDHECMEYEGEEACDEGCNGGLQPNAYNYIIKNGALEU_HORVU TTGALEAAYTQATGKNISLSEQQLVDCAGGFNNF--------GCNGGLPSQAFEYIKYNGCATH_HUMAN TTGALESAIAIATGKMLSLAEQQLVDCAQDFNNY--------GCQGGLPSQAFEYILYNK *** :*. : .* :**:**:**** :: **:*** .:*::** *

Alignment type
● Global○ the whole of each sequence is included, end to end
● Local○ only the best matching parts of each sequence
● Glocal○ global in query (small), local in reference (big)
FTFTALILLAVAV F--TAL-LLA-AV
FTFTALILL-AVAV FTAL-LL
FTFTALILL-AVAV FTAL-LLAAV

Alignment complexity
● Alignment is computationally hard○ to align N sequences of length L takes LN time!○ intractable for large N or L
● Imagine L is 300aa (reasonable protein)○ to align 2
■ 300x300 = 90,000 time units○ to align 3
■ 300x300x300 = 27,000,000 time units○ to align 4
■ 300x300x300x300 = 8,100,000,000 time units
● We have a problem!

Make it go faster please
● Use (reasonable) heuristics○ usually aligning similar sequences○ don't expect lots of random gaps○ expect short, exact matches to occur ("seeds")
● Standard method○ find exact seed matches, and extend alignment
● Implications○ examines only a subset of all possible solutions○ less sensitivity - will miss some better alignments○ close to optimal for well behaved sequences○ orders of magnitude faster

NGS alignment

NGS read alignment
● Query○ Illumina reads in FASTQ format○ Huge number of them, >100M ○ Short read length, ~100bp
● Reference○ Human genome in FASTA format○ About ~27,000 contigs○ Total size 3.2 Gbp
● Lots of shorts vs. a few longs○ BLAST isn't suitable

NGS aligners
● Optimized for short reads / big reference○ will degrade disgracefully with long reads○ can fit human genome in RAM○ use all available CPUs in parallel
● Common "read mappers"○ BWA, Bowtie, Novoalign, BFAST, ....
● Trade-offs○ speed vs. sensitivity○ will miss divergent matches○ can miss indels (insertions and deletions)

Read mapping considerations
● Quality trimming○ have my reads been filtered/trimmed for quality?
● Local or Glocal alignment○ did the aligner require the whole read to map?
● Ambiguous alignment○ a read could map to several places in genome!○ ignore all, choose one, choose random, use all?
● Unaligned reads○ novel DNA, rubbish, discordant pairs?

BAM files

SAM and BAM files
● SAM○ plain text file, tab separated columns○ "a huge spreadsheet"○ inefficient to read and store
● BAM○ a compressed version of SAM (less storage)
■ ~15% original size○ can be indexed (fast access to rows)○ needs to be sorted to be useful however
● Standardized format○ readable by most software

What's in a SAM/BAM?
● One line per original read sequence○ where it aligned (if at all)○ how much of it aligned (soft/hard clipping)○ how well it aligned (mapping quality)○ any differences to the reference (CIGAR string)○ lots of other stuff (aligner dependent)
1:497:R:-272+13M17D24M 113 1 497 37 37M 15 100338662 0CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG 0;==-==9;>>>>>=>>>>>>>>>>>=>>>>>>>>>>XT:A:U NM:i:0 SM:i:37 AM:i:0 X0:i:1 X1:i:0 XM:i:0
19:20389:F:275+18M2D19M 99 1 17644 0 37M = 17919 314TATGACTGCTAATAATACCTACACATGTTAGAACCAT >>>>>>>>>>>>>>>>>>>><<>>><<>>4::>>:<9 RG:Z:UM0098:1 XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0
19:20389:F:275+18M2D19M 147 1 17919 0 18M2D19M = 17644 -314GTAGTACCAACTGTAAGTCCTTATCTTCATACTTTGT ;44999;499<8<8<<<8<<><<<<><7<;<<<>><< XT:A:R
NM:i:2 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 MD:Z:18^CA19
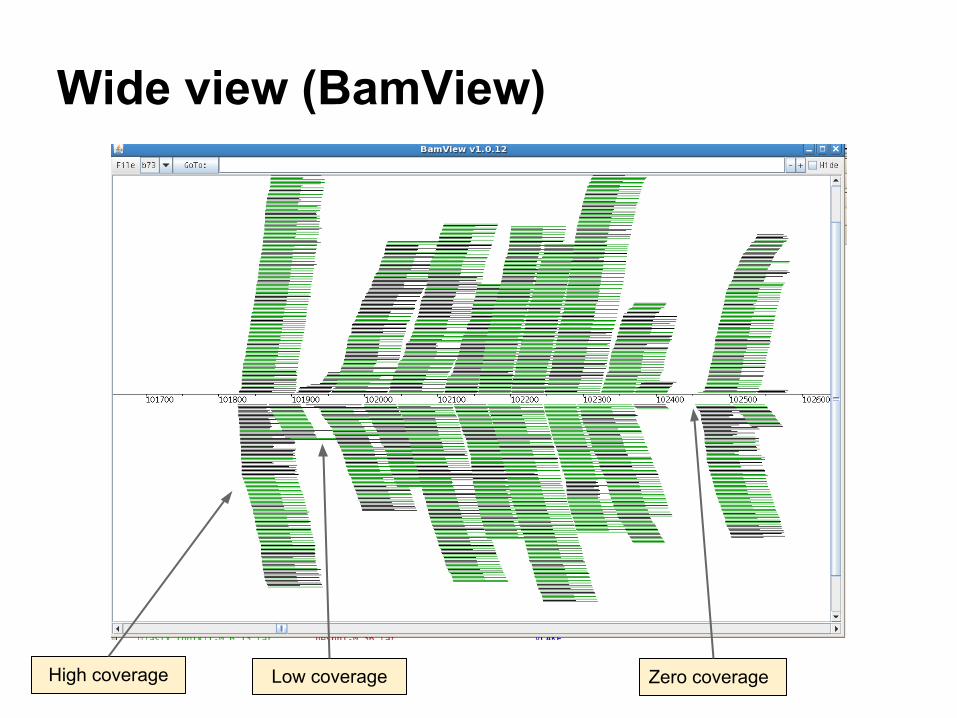
Wide view (BamView)
Zero coverageLow coverageHigh coverage

Medium view (IGV)
Reads
Reference coordinates
Ref seqA variant!

Close view (BamView)Reference
Reads
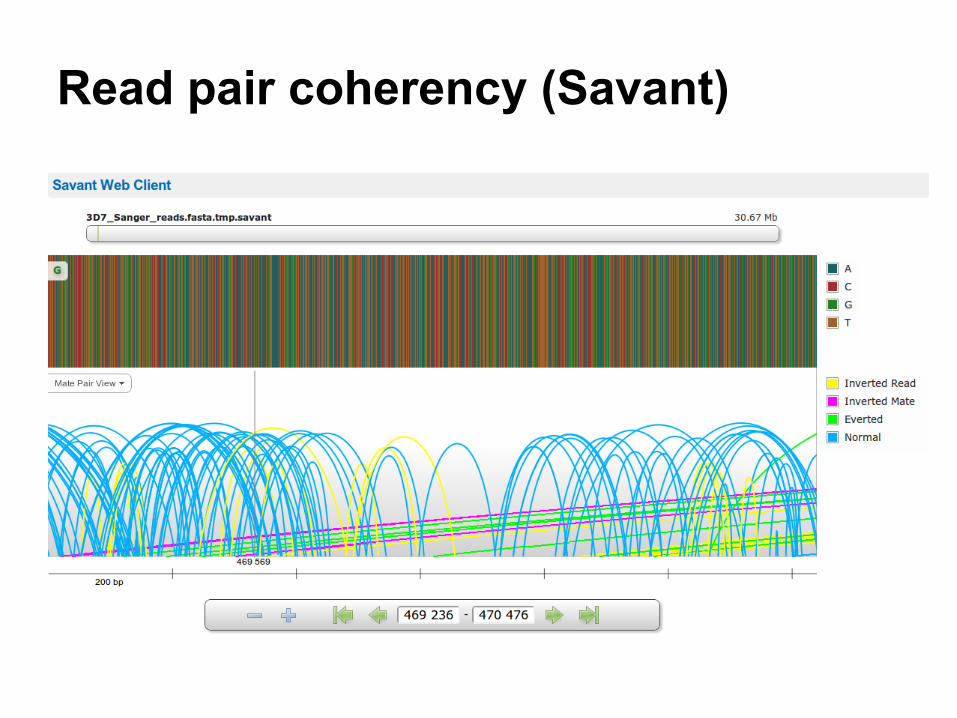
Read pair coherency (Savant)

Why should I look at BAMs?
● Pipelines help distill the data down○ gigabytes of reads to lists of variant calls etc
● But you still need to validate the results○ look at the read stack around your variant calls○ check for oddness
■ mixes of read groups - possible repeat?■ borderline mapping quality - novel DNA?
● Look at your data○ you wouldn't pipette with a blindfold on

Next week...

Next week - Fri 1 Aug @ 12:30pm
● Module 2 - variant calling○ Converting reads to "genotype"
● Methods and assumptions○ somatic/germline, cancer/normal, exome/genome
● Outcomes○ allelle frequencies, confidence metrics
● File formats○ VCF for SNPs, BED for intervals
● Objective metrics○ quality, transition/transversion, concordance
● Visualization○ examining variants in detail, BAM comparison



















