
Introduction to Evolutionary Bioinformatics David H. Ardell,Forskarassistent.
77
Introduction to Evolutionary Bioinformatics David H. Ardell,Forskarassistent
-
date post
18-Dec-2015 -
Category
Documents
-
view
218 -
download
1
Transcript of Introduction to Evolutionary Bioinformatics David H. Ardell,Forskarassistent.

Introduction to Evolutionary Bioinformatics
David H. Ardell,Forskarassistent

Outline, v. 4
Wed. Jan. 26 Sequences and substitution matrices.
Thurs. Jan. 27 Alignments: basic theory and practice.
Fri. Jan. 28 Trees: basic theory and practice.
Mon. Jan. 31 Population sequence data: theory and practice.

Lecture Outline: Intro. to Sequence Evolution and Substitution Matrices
Part I: TheoryHomology, paralogy and orthology
Molecular clock
Divergence, saturation and evolutionary distance
Poisson correction
PAM and other substitution matrices
Markov and other assumptions of bioinformatics
Sequence compositions
Part II: PracticeEvolving sequences on a computer
Calculating evolutionary distances
Exploring Substitution matrices
Calculating evolutionary distance with substitution matrices

Richard Owen (1804-1892)
HOMOLOGY: descent from a common ancestor(Darwin, 1859)
Original definition: "the same organ in different animals under every variety of form and function." (Owen, 1843).
Homology need not imply similarity of form nor function because of divergence.
Similarity need not imply homology because of convergence.

Homology applied to DNA sequences:
GCCACTTTCGCGATCA
GCCACTTTCGCGATCA
GCCACTTTCGCGATTA
GACAGTTTCGCGATTA
GGCAGTTTTGCGATTA
GGCAGTTTCGCGATTT
GCCACTTTCGCGATCG
GCCACTTTCGTGATCG
GCCACGTTCGTGATCG
GCCACGTTCGCGATCG
GCCACGTTCGCGATCG
GCCACGTTCGCGATCG| || |||||||GGCAGTCTCGCGATTT
Ancestral sequence
Homologous sequences
GCCACGTTCGCGATCG GGCAGTTTCGCGATTT
Homologous residues

Hardison PNAS 2001 98: 1327-1329
Sequence homologs can be paralogs or orthologs.
Paralogs are members of a “gene family.” They arise by gene duplication. Ex: -hemoglobin and-hemoglobin are paralogs

Hardison PNAS 2001 98: 1327-1329
Paralogs arise by gene or chromosome duplications
they arose by tandem gene duplication - a chunks of chromosomes duplicating locally
Paralogs are members of a “gene family.” They arise by gene duplication. Ex: -hemoglobin and-hemoglobin are paralogs

Hardison PNAS 2001 98: 1327-1329
Orthologs arise by speciation
Orthologs duplicate by speciation. In practice we assume they retain the same have function.Ex: -hemoglobin in eutherians and marsupials
(pungdjur)

Hardison PNAS 2001 98: 1327-1329
Evolution of the Hemoglobin Gene Family
(pungdjur)
Speciation between marsupials and eutherians
Orthologs duplicate by speciation. In practice we assume they are the “same” gene in the family (have same function).Ex: -hemoglobin in eutherians and marsupials

Hardison PNAS 2001 98: 1327-1329
Ancient polyploidization event
Paralogs also arise through whole chromosome duplications (polyploidizations).

Hardison PNAS 2001 98: 1327-1329
Functional divergence can occur in orthologs
change in function

Hardison PNAS 2001 98: 1327-1329
Paralogs can be lost in some species

Hardison PNAS 2001 98: 1327-1329
Orthology is rarer than paralogy
ORTHOLOGY:Homology byspeciation, same function
PARALOGY:Homology by duplication

Hardison PNAS 2001 98: 1327-1329
Hemoglobins and other gene families evolve by speciation, duplication, loss and divergence
duplications
speciations
speciations
duplication
losses

Emile Zuckerkandl and Linus Pauling (1965)"Evolutionary Divergence and Convergence in Proteins," in Evolving Genes and Proteins, eds. V. Bryson and H. Vogel (New York: Academic Press, 1965). pp. 97-166.
• Divergence of -, -, and -Hemoglobin are about the same regardless of which species they are in.
• Duplications preceded the divergence of mammals.
The “Molecular Clock:” orthologs evolve at typical constant rates

“There may thus exist a Molecular Evolutionary Clock”Zuckerkandl & Pauling (1965)
Divergence between and or
Divergence between , and
Approx. duplication dates (mya) from vertebrate fossil records
% a
min
o a
cid d
iffere
nce
s

PBS Evolution Library (http://www.pbs.org/wgbh/evolution/library/)
Different proteins “tick” at different rates


Also, different parts of the same gene or protein evolve at different rates
Ex: Globular proteins evolve faster at their outsides!

The molecular clock also works for DNAEx: influenza virus genes
Gojobori et al. 1990 PNAS 87 10015-10018

BUT: the Molecular Clock slows down after a long time because of SATURATION (double mutations).
Approx. duplication dates (mya) from vertebrate fossil records
% a
min
o a
cid d
iffere
nce
s

Ex: why Percent Identity (%ID) underestimates divergence
The more sequences diverge, the more substitutions we miss.
ANCESTOR

Ex: why Percent Identity (%ID) underestimates divergence
The more sequences diverge, the more substitutions we miss.
Multiple mutations hit the same site
ANCESTOR

Ex: why Percent Identity (%ID) underestimates divergence
The more sequences diverge, the more substitutions we miss.
Multiple mutations hit the same site
ANCESTOR
3 mutations,2 differences

Ex: why Percent Identity (%ID) underestimates divergence
The more sequences diverge, the more substitutions we miss.
Multiple mutations hit the same site
Back mutations undo earlier mutations
ANCESTOR
3 mutations,2 differences

Ex: why Percent Identity (%ID) underestimates divergence
The more sequences diverge, the more substitutions we miss.
Multiple mutations hit the same site
Back mutations undo earlier mutations
ANCESTOR
4 mutations,1 difference
3 mutations,2 differences

Ex: why Percent Identity (%ID) underestimates divergence
The more sequences diverge, the more substitutions we miss.
Multiple mutations hit the same site
Back mutations undo earlier mutations
Parallel mutations hide divergence
ANCESTOR
4 mutations,1 difference
3 mutations,2 differences

Ex: why Percent Identity (%ID) underestimates divergence
The more sequences diverge, the more substitutions we miss.
Multiple mutations hit the same site
Back mutations undo earlier mutations
Parallel mutations hide divergence
ANCESTOR
4 mutations,1 difference
3 mutations,2 differences
6 mutations,1 difference

The more distantly related two sequences are, the more we must correct for hidden mutations
Two strategies:
Poisson correction Quick and dirty, can be computed by hand Neglects back and parallel substitutions. These are rare at low
divergence, so works better for closer-related sequences. Includes no information about how proteins or DNA evolve. All types of
changes are equally likely.
Substitution matrices Complex to compute Accounts for back and parallel substitutions,more accurate A complete model of evolution about how sequences evolve Can be used for making alignments, database searches and trees

The Poisson Correction
Imagine mutations “raining down” on sequences:

The Poisson Correction
Imagine mutations “raining down” on sequences:

The Poisson Correction
Imagine mutations “raining down” on sequences:

The Poisson Correction
Imagine mutations “raining down” on sequences:

The Poisson Correction
1. Want to estimate avg. evolutionary distance = r t (# mutations per sequence length in sites) from %ID = 100 x (p/N).
Imagine mutations “raining down” on sequences:

The Poisson Correction
1. Want to estimate avg. evolutionary distance = r t (# mutations per sequence length in sites) from %ID = 100 x (p/N).
2. Assume mutations occur independently in space and time.
Imagine mutations “raining down” on sequences:

The Poisson Correction
1. Want to estimate avg. evolutionary distance = r t (# mutations per sequence length in sites) from %ID = 100 x (p/N).
2. Assume mutations occur independently in space and time.
3. Normalize sequence to length 1. Then each site has probability /N of mutating at distance . The average fraction of sites not mutated at this distance is then: (1 - /N)N ≈ e– ( as N ).
Imagine mutations “raining down” on sequences:

The Poisson Correction
1. Want to estimate avg. evolutionary distance = r t (# mutations per sequence length in sites) from %ID = 100 x (p/N).
2. Assume mutations occur independently in space and time.
3. Normalize sequence to length 1. Then each site has probability /N of mutating at distance . The average fraction of sites not mutated at this distance is then: (1 - /N)N ≈ e– ( as N ).
4. Therefore, if we see (p/N) sites not mutated and assume no back- or parallel mutations, we can estimate distance = – ln (p/N).
Imagine mutations “raining down” on sequences:

The Poisson Correction
1. Want to estimate avg. evolutionary distance = r t (# mutations per sequence length in sites) from %ID = 100 x (p/N).
2. Assume mutations occur independently in space and time.3. Normalize sequence to length 1. Then each site has probability
/N of mutating at distance . The average fraction of sites not mutated at this distance is then: (1 - /N)N ≈ e– ( as N ).
4. Therefore, if we see (p/N) sites not mutated and assume no back- or parallel mutations, we can estimate distance = – ln (p/N).
5. Ex: %ID of 37.8 ≈ 100 x e–1 implies = -ln( 1/e) = 1. About as many mutations as the length of the sequence have occurred.
Imagine mutations “raining down” on sequences:

Poisson-Corrected Evolutionary Distance vs. %ID
%ID
Su
bst
itu
tion
s p
er
site
61%ID = 0.5
37%ID = 1.0

Poisson-Corrected Evolutionary Distance vs. %ID
%ID
Su
bst
itu
tion
s p
er
site
61%ID = 0.5
37%ID = 1.0
Something wrong here though:Real proteins don’t evolve less than about 5% ID, and they do it much slower than this.

For most bioinformatics work we need something more sophisticated… substitution matrices.
The Poisson correction… … neglects back and parallel substitutions: %ID goes falsely to
zero at large evolutionary divergences. … uses information only from sites that are identical. Throws out
information from the mutated sites. … includes no information about which kinds of changes are more
likely to occur than other kinds of changes (Ex: hydrophobic amino acids, transition bias in DNA mutation).
…provides only a “back-of-the envelope” model of evolution.
Substitution matrices… …give a complete accounting of all possible mutational paths is
made. …use information from all sites, changed or unchanged. …provide a superior model of sequence evolution. …can be used to make alignments, search databases (GenBank)
for homologs, and make phylogenetic trees.

Q: What is a “substitution?”
A: A substitution is the fixation of a mutation in a population. It has been “accepted” by natural selection.
Population of 5 individuals at generation t = 0

Q: What is a “substitution?”
A: A substitution is the fixation of a mutation in a population. It has been “accepted” by natural selection.
Population of 5 individuals at generation t = 0
t = 2

Q: What is a “substitution?”
A: A substitution is the fixation of a mutation in a population. It has been “accepted” by natural selection.
Population of 5 individuals at generation t = 0
t = 2

Q: What is a “substitution?”
A: A substitution is the fixation of a mutation in a population. It has been “accepted” by natural selection.
Population of 5 individuals at generation t = 0
t = 2

Q: What is a “substitution?”
A: A substitution is the fixation of a mutation in a population. It has been “accepted” by natural selection.
Population of 5 individuals at generation t = 0
t = 2

Q: What is a “substitution?”
A: A substitution is the fixation of a mutation in a population. It has been “accepted” by natural selection.
Population of 5 individuals at generation t = 0
t = 2
t = 3
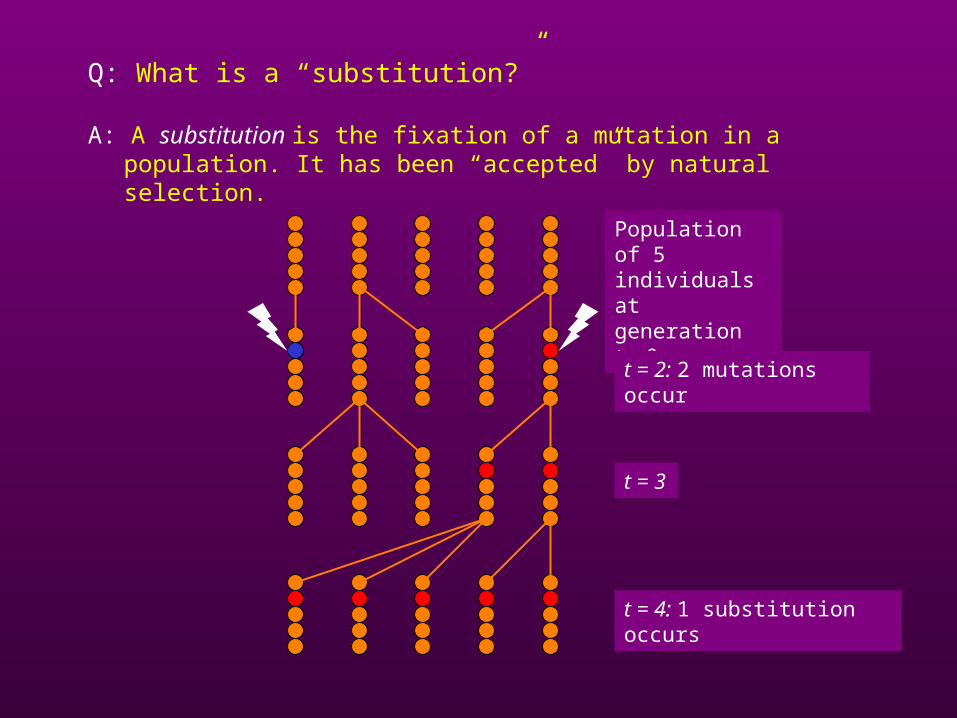
Q: What is a “substitution?”
A: A substitution is the fixation of a mutation in a population. It has been “accepted” by natural selection.
Population of 5 individuals at generation t = 0
t = 2: 2 mutations occur
t = 3
t = 4: 1 substitution occurs

HINT: Sequence differences between
species are often assumed to be
substitutions (fixed differences).
Species 2Species 1
Ancestor

Margaret Oakley Dayhoff (1925-1983) Inventor of PAM Amino Acid Substitution Matrices
Basic ideas: 1. Collect a big dataset of closely related proteins.
2. Count up amino acid changes and the total composition of amino acids in the dataset.
3. Calculate from this the transition probabilities for any amino acid to change into any other amino acid after 1% sequence divergence.
4. This defines the PAM1 matrix (“Point Accepted Mutation,” where “accepted” means “by natural selection”).
5. Assume that the transition probabilities after N% sequence divergence is given by “powering up” the PAM1 matrix.
Ex: PAM250 = PAM1250

Q: What does PAM250 – 250% change to a protein – mean?
A: just over 18%ID

Assumptions of PAM Substitution Matrices
1. Site-Independence: Probability of mutation/substitution
at a site is independent of which amino acids/bases
occupy all other sites in any protein in the organism.

Assumptions of PAM Substitution Matrices
1. Site-Independence: Probability of mutation/substitution
at a site is independent of which amino acids/bases
occupy all other sites in any protein in the organism.
2. Memorylessness: Probability of mutation/substitution at
a site depends only on its present state, not on its
history.

Assumptions of PAM Substitution Matrices
1. Site-Independence: Probability of mutation/substitution
at a site is independent of which amino acids/bases
occupy all other sites in any protein in the organism.
2. Memorylessness: Probability of mutation/substitution at
a site depends only on its present state, not on its
history.
3. Stationarity: Sequence composition is the same or will
become the same as in the alignments that were used
to make the matrix.

Assumptions of PAM Substitution Matrices
1. Site-Independence: Probability of mutation/substitution
at a site is independent of which amino acids/bases
occupy all other sites in any protein in the organism.
2. Memorylessness: Probability of mutation/substitution at
a site depends only on its present state, not on its
history.
3. Stationarity: Sequence composition is the same or will
become the same as in the alignments that were used
to make the matrix.
4. Markov Assumption: The probabilities of change remain
the same throughout history.

A G
TC
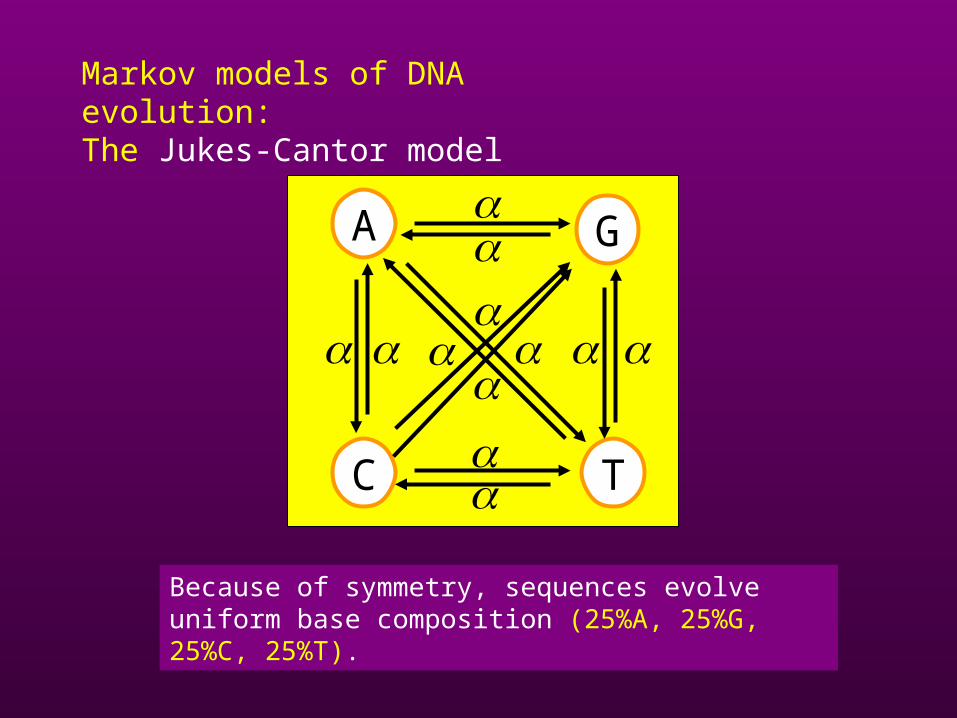
Markov models of DNA evolution: The Jukes-Cantor model

A G
TC
Markov models of DNA evolution: The Jukes-Cantor model
“Pools”

A G
TC
Markov models of DNA evolution: The Jukes-Cantor model
“Flows out”

A G
TC
Markov models of DNA evolution: The Jukes-Cantor model
“Flows in”
A G
TC
Markov models of DNA evolution: The Jukes-Cantor model
Because of symmetry, sequences evolve uniform base composition (25%A, 25%G, 25%C, 25%T).

A G
TC
€
€
Markov models of DNA evolution: The Kimura 2-parameter model
€
€

A G
TC
€
€
Markov models of DNA evolution: The Kimura 2-parameter model
€
€
Transitions
Transversions

A G
TC
€
€
Markov models of DNA evolution: The Kimura 2-parameter model
€
€

A G
TC
€
€
Markov models of DNA evolution: The Kimura 2-parameter model
€
€

Jones, Taylor, Thornton (1992) “JTT” MDM-1 score matrix
A R N D C Q E G H I L K M F P S T W Y V

JTT MDM-15 Score Matrix, 85% expected ID between proteins
A R N D C Q E G H I L K M F P S T W Y V

JTT MDM-120 Score Matrix, 36% expected ID
A R N D C Q E G H I L K M F P S T W Y V

Score Matrices vs. Substitution Matrices
To make evolutionary matrices, calculate avg. composition Ma = p(a) and transition probabilities Mab = p(b|a*)p(a*|a) that an amino acid/base mutates to b and substitutes in the population.

Score Matrices vs. Substitution Matrices
To make evolutionary matrices, calculate avg. composition Ma = p(a) and transition probabilities Mab = p(b|a*)p(a*|a) that an amino acid/base mutates to b and substitutes in the population.
Substitution matrices are made only from the transition probabilities Mab. Because Mab Mba, they are not symmetric about the diagonal.

Score Matrices vs. Substitution Matrices
To make evolutionary matrices, calculate avg. composition Ma = p(a) and transition probabilities Mab = p(b|a*)p(a*|a) that an amino acid/base mutates to b and substitutes in the population.
Substitution matrices are made only from the transition probabilities Mab. Because Mab Mba, they are not symmetric about the diagonal.
Score Matrices (or “MDMs”) are made from both Mab and Ma. They give the log-odds of two residues in a sequence being biologically homologous relative to chance.

Score Matrices vs. Substitution Matrices
To make evolutionary matrices, calculate avg. composition Ma = p(a) and transition probabilities Mab = p(b|a*)p(a*|a) that an amino acid/base mutates to b and substitutes in the population.
Substitution matrices are made only from the transition probabilities Mab. Because Mab Mba, they are not symmetric about the diagonal.
Score Matrices (or “MDMs”) are made from both Mab and Ma. They give the log-odds of two residues in a sequence being biologically homologous relative to chance.
Score matrices are symmetrical:
Sab = log (Mab / Mb) = log (Mba / Ma) = Sba.

Score Matrices vs. Substitution Matrices
To make evolutionary matrices, calculate avg. composition Ma = p(a) and transition probabilities Mab = p(b|a*)p(a*|a) that an amino acid/base mutates to b and substitutes in the population.
Substitution matrices are made only from the transition probabilities Mab. Because Mab Mba, they are not symmetric about the diagonal.
Score Matrices (or “MDMs”) are made from both Mab and Ma. They give the log-odds of two residues in a sequence being biologically homologous relative to chance.
Score matrices are symmetrical:
Sab = log (Mab / Mb) = log (Mba / Ma) = Sba.
Score matrices are used for many bioinformatic applications we will soon cover such as alignment and database searching.

Q: Score matrices are “log-odds” matrices. What are log-odds?
Odds are ratios of probabilities. Usually written like “4:1” (said like “4 to 1”) they tell you the relative chance of two events.
Score Matrices are made from the odds-ratio p(AB):p(A)p(B) that two amino acids or bases A and B are likely to be found in homologous positions in a sequence p(AB), relative to the chance of picking the pair at random p(A)p(B)
Log-odds L are made by taking the log of the odds-ratio: log p(AB):p(A)p(B) = log p(AB) – log p(A) – log p(B)
they are more convenient to compute with and understand: if L > 0, A and B more likely to occur by evolution than by chance and vice versa.


Other Amino Acid Substitution/Score Matrices
Some matrices are updates of the original Dayhoff method with more data or some technical refinementsEx: Jones, Taylor, Thornton 1992 (JTT) Gonnet, Benner and Cohen
Some matrices are for specialized kinds or parts of proteins. Ex: JTT transmembrane protein matrixGoldstein secondary structure matrices
Some matrices have different assumptionsEx: BLOSSUM: removes Markov assumption. They make a series of matrices from alignments at different %IDs. OBS: BLOSSUMs are labeled by expected %ID, so while PAM250 > PAM100, BLOSSUM30 > BLOSSUM62 !!

One last point: evolutionary distance between two sequences:
Root
Seq 1 Seq 2

One last point: evolutionary distance between two sequences:
Root
Seq 1 Seq 2
Seq 2Seq 1
Root


















