
The GFF Investment Case in Priority Countries: Why, What ...

INTRODUCTION AUX FORMATS DE FICHIERSAlban LERMINEOlivier INIZANEcole bioinformatique Roscoff18 novembre 2013

Plan
• 1. Formats de séquences brutes
• 1.1. Format fastq (base-space)
• 1.2. Format csfasta/qual (color-space)
• 1.3. Les adaptateurs
• 2. Formats d’alignements
• 2.1. Format SAM
• 2.2. Format BAM
• 3. Format de Localisation / Annotation / Visualisation
• 3.1. Format BED
• 3.2. Format GFF
• 3.3. Format GTF
• 3.4. Format WIG
• 3.5. Format BEDGRAPH
• 4. Format « Variant Calling »
• 4.1. Format Pileup
• 4.2. Format VCF
• 5. Travailler avec les données NGS

1. Formats de séquences brutes
● 2 formats de données de séquences
● Format base-space (Illumina & IonTorrent)
● Format color-space (SOLiD)
● Pour chaque base séquencée, une qualité est associée
● Qualité = probabilité que la base « appelée » soit incorrecte

1.1. Format fastq (base–space)
● 1 séquence = 4 lignes dans le fichier●
●
● 1 ère ligne = identifiant de la séquence

1.1. Format fastq (base–space)
● 4ème ligne = Qualité●
● Appelée aussi Phred quality score (Sanger format)
● Probabilité qu'une base soit incorrecte

1.1. Format fastq (base–space)
● Encodée en ASCII (allège le fichier)●
●

1.2. Format csfasta/qual (color–space)
● 2 fichiers séparés pour les séquences et les qualités
● Fichier séquence :●
● Code couleur = double lecture●

1.2. Format csfasta/qual (color–space)
● Fichier qualité :●
● Pas d'encodage ASCII (fichiers plus lourds)

1.2. Format csfasta/qual (color–space)
● Conversion color space => base-space déconseillée● Très peu d'outils gèrent le format cfasta/qual
– Bioscope/lifescope
– Bowtie (sans gestion de la qualité)
● Convertir cfasta/qual en fatsq color-space– BWA– Bfast/BWA
– Bowtie (avec gestion de la qualité)

1.3. Les adaptateurs
• Un adaptateur est un court fragment d’ADN dont la séquence est connue et qui est placé à la fin des lectures
• Rôles de ces adaptateurs:
• Permettre l’accroche à la flowcell du séquenceur
• Permet d’amplifier par PCR spécifiquement les lectures contenant l’adaptateur
• Permet le séquençage multiplexe (plusieurs échantillons en mélange)
• Plusieurs outils sont disponibles pour retirer ces adaptateurs:
• Cutadapt
• SeqPrep • RmAdapter

2. Formats d’alignements
● Plusieurs format existent– SAM et BAM (= standards)
– ELAND (spécifique Illumina)
– MAQ Map

2.1 SAM Format: introduction
● NGS => a variety of new alignment tools : Bowtie (Langmead,B. et al (2009), Maq (Li,H. et al (2008), BWA (Li and Durbin, 2009), ...
● SAM : a common alignment format that supports all sequence types and aligners
● SAM : Sequence Alignment/Map format● A well-defined interface between alignment
and downstream analyses
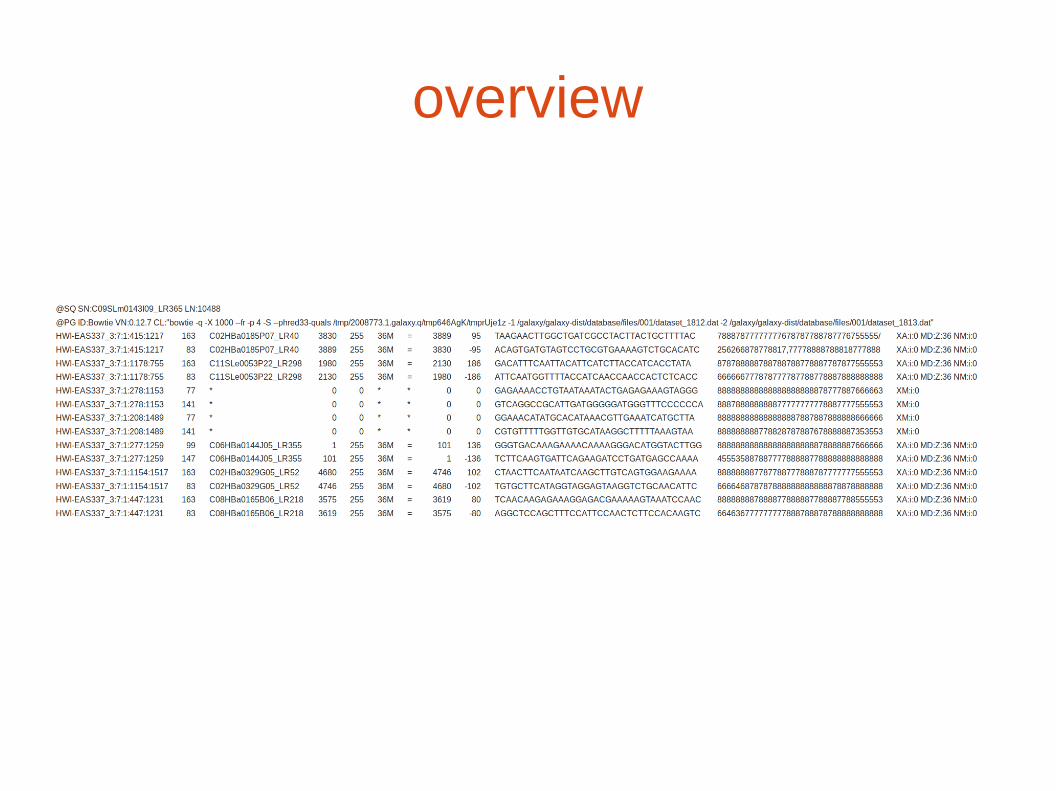
overview

@SQ name LN : headerheader
ref seq name
ref seqlength
readName1 flag referenceName
readName2 flag referenceName
...
alignment
one tab-delimited line per alignment

Tab-delimited : SAM Fields

SAM Format (example)
HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0

HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0
QNAME: Query name
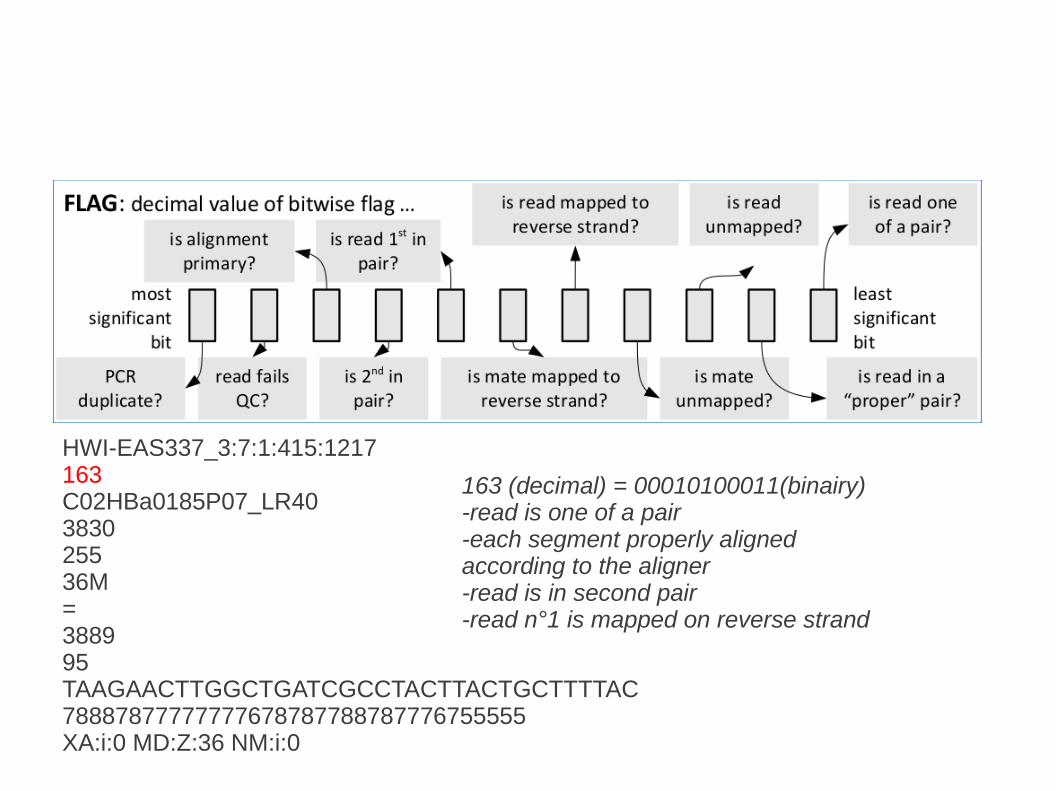
HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0
163 (decimal) = 00010100011(binairy)-read is one of a pair-each segment properly aligned according to the aligner-read is in second pair-read n°1 is mapped on reverse strand

http://picard.sourceforge.net/explain-flags.html

HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0
RNAME : reference sequence name

HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0
POS : position on reference

MAPQ : mapping quality
It equals −10 log10 Pr{mapping position is wrong}, rounded to the nearest integer.
A value 255 indicates that the mapping quality is not available.
Zero value is the lowest quality.
HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0

CIGAR
HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0
CIGAR : extended CIGAR string (Compact Idiosyncratic Gapped Alignment Report)Format: [0-9][MIDNSHP][0-9][MIDNSHP]...[0-9] : positionM = match or mismatch (?!), I/D = insertion / deletion, N = skipped bases onreference, S/H = soft / hard clip (soft means nt's still appear in sequence field), P =paddinge.g.: ”1S81M” means that the first (5'-most) nt is not part of the alignment, but thefollowing 81 nt's are either matches or mis-matches.

RNEXT : mate or not mate ?
' = ' means the mate is mapped to the same reference sequence as the current read
' * ' means that the read is unpaired (has no mate)
HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0
HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0

HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0
PNEXT : mate position
' 0 ' means no info is avalaible

TLEN : insert size
read mate
insert size
HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0

SEQ and QUAL : sequence and quality c.f. FASTQ format
HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0

HWI-EAS337_3:7:1:415:1217163C02HBa0185P07_LR40383025536M=388995TAAGAACTTGGCTGATCGCCTACTTACTGCTTTTAC78887877777777678787788787776755555XA:i:0 MD:Z:36 NM:i:0
OPT : optional fieldsFollow the TAG:TYPE:VALUE format.TYPE is : [A(printable character); i(signed integer); f(floating point); z(printable string);H(hex string)]

BAM
● BAM = compressed SAM● Indexed BAM : *.bam.bai● Tools (post process, viewers) use indexed
bam to avoid all information extraction

Reference

3. Formats de Localisation/Annotation/Visualisation
● Fichiers texte tabulés● 5 formats sont le plus couramment utilisés
– BED
– GFF
– GTF (dérivé du GFF)
– WIG
– BEDGRAPH
● 1 ligne par zone

3.1. Format BED
● BED pour Browser Extensible Data● Coordonnées en base 0● 3 champs obligatoires●
●
●
●
●
●
● 9 autres champs sont optionnels et peuvent contenir :– Le brin (forward oureverse)– Le nom de l'intervalle
– De l'information sur l'intervalle (annotation)

3.2. Formats GFF
● GFF pour General Feature Format● Format utilisé pour localiser et décrire toute zone caractéristique d'un
génome (ex : un exon)● Contient 8 champs
– Nom
– Source
– Type
– Début d'intervalle
– Fin d'intervalle
– Score
– Brin
– Cadre

3.3. Format GTF
● GTF pour Gene Transfert Format● Dérivé du GFF● Contient les mêmes champs + 2 pour
l'annotation

3.4. Format WIG
● WIG pour Wiggle● Format compressé● Utilisé pour la représentation de données
continues et denses du type :– Poucentage de GC
– Probabilités
– ...

3.5. Format BEDGRAPH
● Utilité similaire au format WIG● Quelque différences :
– Non compressé
– Permet de définir des zones de longueurs différentes

4. Formats « Variant Calling »
● 2 formats sont couramment utilisés :– Format Pileup
– Format VCF
● Le format Pileup est spécifique de l'outil Samtools Mpileup
● Le format VCF est le format par défaut d'un grand nombre de SNP caller

4.1. Format Pileup
● Permet de décrire des variations de type :– Modification d'un nucléotide unique (SNP)
– Insertion/délétion
● Fichier texte tabulé– Chromosome
– Position dans la référence
– Nucléotide de référence à cette position
– Nombre de lecture alignées à cette position
– Nucléotides des lectures alignées à cette position
– Qualité d'alignement de la base
–

4.2. Format VCF
● SAM = standard for alignment● VCF = standard for storing sequence variation● SNPs, indels, large structural variants● Primary intention : to represent human genetic
variation (1000 Genome Project)● Can be used in different contexts

overview

header
meta info starting with '##'
body
Meta info : provide a standardized description of tags and annotations used in the body section.

example
#CHROM : chrom id#ID : unique identifier of variant#POS : position of the start of the variant#REF : refrence allele#ALT : comma seprated list of alternate non reference allele#QUAL : phred quality score#FILTER : site filtering information
mandatory fields


no mandatory fields
#FORMAT : describe format of #SAMPLE(s)#FORMAT : infos found in the headerSamples for this line : genotypes and read depth
#SAMPLE1: genotype '1 ' (i.e. deletion) is on each allele and read depth is 13
#SAMPLE 2: genotype '2 ' (i.e remplacement) is on each allele and read depth is 29

large struct variant

Reference

5. Travailler avec les données NGS
● Difficile sur une station locale (manque de ressources)● 1 alignement = 4 processeurs + 15 gb Ram (à multiplier par le
nombre d'échantillons)● Lecture des fichiers impossible dans les logiciels de bureautique
couramment utilisés● Espace de stockage nécessaire important● Gestion des sauvegardes● Serveur d'application connecté sur cluster de calcul et baie de
stockage– Solution commerciales (CLC Bio, NextGene, ...etc)
– Galaxy ...

Sources & Ref
● Joe Fass <[email protected]> and his « Next Generation Sequence Alignment » slides
● The Sequence Alignment/Map format and SAM tools. Li et al. 2009 Bioinformatics 25 2078-2079
● The variant call format and VCFtools. Daneck et al. 2011 Bioinformatics 27 2156-2518.


















