
Interpretazione di profili STR: misture e DNA Low Template
38
Metodi statistici in genetica forense Bologna, 6-7 giugno 2016 Interpretazione di profili STR: misture e DNA Low Template
Transcript of Interpretazione di profili STR: misture e DNA Low Template

Metodi statistici
in genetica forense
Bologna, 6-7 giugno 2016
Interpretazione di profili STR: misture e DNA Low Template

Approccio Binario e Approccio Probabilistico
Modello Binario
Modello Semi-continuo
Modello Continuo
Aumenta la complessità Sempre più difficile da spiegare
Metodi statistici di base
Metodi statistici avanzati
RMNE Binary LR (uso dei soli alleli)
LRmixstudio, FST (alleli, drop in/drop out)
EuroForMix, STRmix, DNA view mix (drop in, drop out, altezza/area dei picchi)
Compatibilità Esclusione
Approccio Probabilistico: tutto è possibile

Modello Binario
Modello Binario
Metodi statistici di base
RMNE Binary LR (uso dei soli alleli)
Compatibilità Esclusione
LR= 1/freq. profilo singolo contributore Pr(E|Hp)<1 LTDNA
RMNE: profili non ambigui con alleli sopra la ST LR = Pr(E|Hp)/Pr(E|Hd)

Modelli semi-continuo e continuo
Modello Semi-continuo
Modello Continuo
Metodi statistici avanzati
LRmixstudio, FST (alleli, drop in/drop out)
EuroForMix, STRmix, DNA view mix (drop in, drop out, altezza/area dei picchi)
Approccio Probabilistico: tutto è possibile
LR = Pr(E|Hp)/Pr(E|Hd) Pr(E|Hp)<1 Low template DNA

# Step 1: identificare la presenza di una mistura sulla base di:
• Numero di alleli
- > 2 alleli per locus per più loci del profilo
(no stutters, no mutazioni somatiche e genetiche)
• Peak imbalance = profilo sbilanciato
- Heterozygote balance (Hb) < 60%
- Stutter peaks > 15%
Procedura per interpretare un profilo misto (Clayton rules and ISFG Recommendations)

# Step 2: attribuzione allelica - no aspecifici (stutters, picchi pull-up, spikes, Off Ladder) - ST e AT -no drop-out
Procedura per interpretare un profilo misto (Clayton rules and ISFG Recommendations)
Split peaks
Pull-up peaks
Spikes

• # Step 3: determinare il numero di contributori
- numero di alleli
- dati circostanziali del caso in esame
- possibili soggetti correlati
- maggior/minor contributore deconvoluzione del profilo
- misture complesse (es. numero di alleli/locus > 6) profilo non
interpretabile con LR (solo RMNE)
Procedura per interpretare un profilo misto (Clayton rules and ISFG Recommendations)

# Step 4: con due contributori posso stimare
il rapporto delle due componenti
• Calcolo Hb = φ12/φ15 dove φ = area o altezza
• Mixture proportion (MX)
• MX = φ12+ φ15/φ12+ φ13+ φ14+ φ15
• Mixture ratio (MR)
• MR= φ12+ φ15/ φ13+ φ14
MX=1097+1186/1097+253+368+1186 = 0.78 = 78%
MR= 1097+1186/253+368 = 3.67
Procedura per interpretare un profilo misto (Clayton rules and ISFG Recommendations)

# Step 5: combinazioni genotipiche - tutti i possibili genotipi unrestricted combinatorial approach
- sulla base di PHR > 60% e Mx si escludono i genotipi non possibili
Restricted combinatorial approach
Possibili genotipi
PHR
12,13 253/1097=0.23
12,14 368/1097=0.33
12,15 1097/1186=0.92 √
13,14 253/368=0.67 √
13,15 253/1186=0.21
14,15 368/1186=0.31
Procedura per interpretare un profilo misto (Clayton rules and ISFG Recommendations)

# Step 5: confronto con il campione di riferimento
- in caso di match si valuta il peso dell’evidenza mediante calcolo statistico idoneo
- testare le ipotesi formulate, nel caso considerare ipotesi alternative
- la deconvoluzione del profilo risulta più complessa in caso di alleli condivisi e in posizione stutter
Procedura per interpretare un profilo misto (Clayton rules and ISFG Recommendations)

Metodi statistici: profili misti e DNA Low template
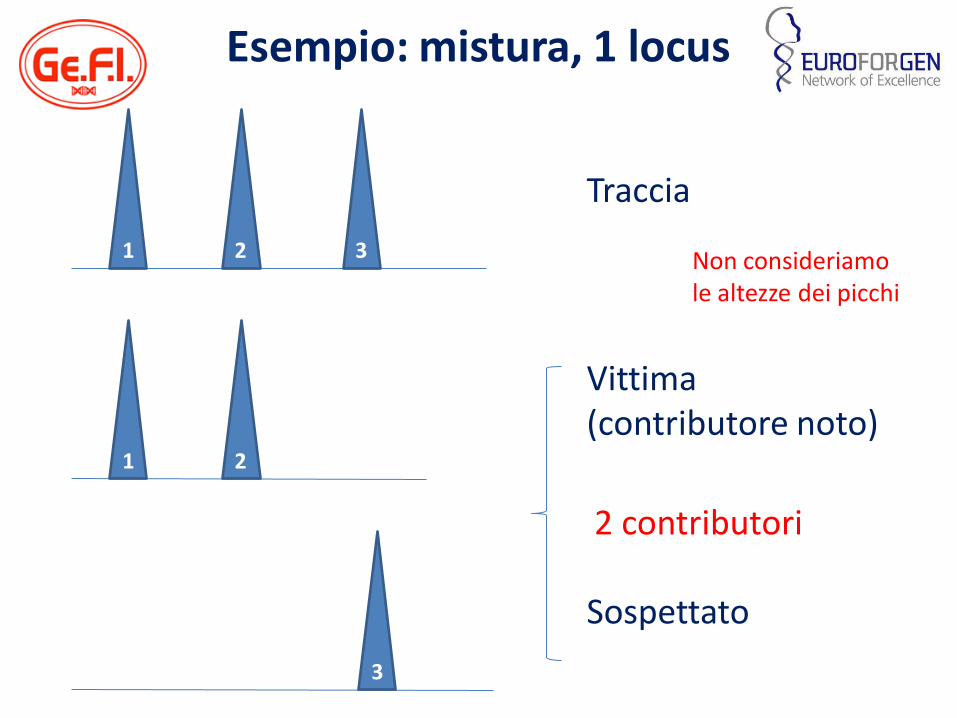
Esempio: mistura, 1 locus
Traccia
Sospettato
Vittima (contributore noto)
Non consideriamo le altezze dei picchi
1 2 3
1 2
3
2 contributori

1 2 3
1 2
3
RMNE: probabilità che un uomo a caso non possa essere escluso come contributore della mistura
RMNE = (p1+p2+p3)2 =
= p12+p2
2+p32+ 2p1p2+2p2p3+2p1p3= 0.36
pi=0.2
Nessuna ipotesi No numero di contributori No correlazione tra contributori No effetti stocastici (se sotto ST il locus non viene incluso)

Si formulano le ipotesi :
Hp= Vittima (V)+Sospettato(S)
Hd= Vittima (V)+sconosciuto(U)
LR = P(E|Hp)
P(E|Hd)
Rapporto tra due probabilità:
probabilità dell’evidenza (E) data l’ipotesi dell’accusa (Hp)
probabilità dell’evidenza (E) data l’ipotesi della difesa (Hd)
LIKELIHOOD RATIO
1 2 3
1 2
3
LR unrestricted: considera solo gli alleli (1, 2, 3) LR restricted: utilizza anche l’altezza dei picchi per determinare le combinazioni possibili (Hb e Mx)

1 2 3
1 2
3
Pi=0.2
Espressione corretta:
data l’evidenza, è 5 volte più probabile SE i contributori sono V+S piuttosto che V+U, non correlato con il sospettato.
Regola del prodotto se i loci sono indipendenti tra di loro es. LR=LR1*LR2*LR=5*10*2=100
LR =
P(E|Hp)
P(E|Hd)
= 1
p32+2p1p3+2p2p3
= 1
0.2
= 5
LIKELIHOOD RATIO
Hp= V+S
Hd= V+U

Vittima
1 2 3
1 2
3 Sospettato
Mistura
LR dipende dalle frequenze alleliche del sospettato
E se gli alleli del sospettato non sono frequenti?
p1= 0.3; p2= 0.25; p3= 0.05
RMNE = (p1+p2+p3)2 = (0.3+0.25+0.05)2 = 0.36
LR = P(E|Hp)
P(E|Hd)
= 1
P32+2p1p3+2p2p3
= 1
0.0575
= 17
RMNE non tiene conto dei profili di riferimento e del numero di contributori
LR tiene conto delle frequenze alleliche uso migliore dei dati
Hp= V+S
Hd= V+U

RMP: Random Match Probability
RMP = p32+2p1p3+2p2p3
Vittima
1 2 3
1 2
3 Sospettato
Mistura
Assumendo due contributori, la vittima spiega gli alleli 1 e 2, il secondo contributore deve spiegare il 3
LR = 1/RMP
Maggiore e minore contributore e altezze dei picchi diverse
Hp= V+S
Hd= V+U

RMNE e LR: pro e contro
RMNE + Facile da calcolare + Più facile da spiegare alla corte + Non richiede di stabilire il numero di contributori -Non tiene conto di tutte i dati disponibili - non applicabile a LTDNA
LR + Utilizza tutte le informazioni + Stima migliore del peso dell’evidenza + Possibilità di modellare fenomeni quali drop-out e drop-in - Più difficile da spiegare alla corte
ISFG DNA commission raccomanda l’uso della LR per le misture

Raccomandazioni ISFG DNA Commission
• Recommendation 2: Even if the legal system does not implicitly appear to support the use of the likelihood ratio, it is recommended that the scientist is trained in the methodology and routinely uses it in case notes, advising the court in the preferred method before reporting the evidence in line with the court requirements. The scientific community has a responsibility to support improvement of standards of scientific reasoning in the court-room.
• Recommendation 1: The likelihood ratio is the preferred approach to mixture interpretation. The RMNE approach is restricted to DNA profiles where the profiles are unambiguous. If the DNA crime stain profile is low level and some minor alleles are the same size as stutters of major alleles, and/or if drop-out is possible, then the RMNE method may not be conservative.

Raccomandazioni ISFG DNA Commission
• Recommendation 3: The methods to calculate likelihood ratios of mixtures (not considering peak area) described by Evett et al. [13] and Weir et al. [14] are recommended.
• Recommendation 4: If peak height or area information is used to
eliminate various genotypes from the unrestricted combinatorial method, this can be carried out by following a sequence of guidelines based on Clayton et al. [17].

Raccomandazioni ISFG DNA Commission
• Recommendation 5: The probability of the evidence under Hp is the province of the prosecution and the probability of the evidence under Hd is the province of the defence. The prosecution and defence both seek to maximise their respective probabilities of the evidence profile. To do this both Hp and Hd require propositions. There is no reason why multiple pairs of propositions may not be evaluated.

Raccomandazioni ISFG DNA Commission
• Recommendation 6: If the crime profile is a major/minor mixture, where minor alleles are the same size (height or area) as stutters of major alleles, then stutters and minor alleles are indistinguishable. Under these circumstances alleles in stutter positions that do not support Hp should be included in the assessment.
• Recommendation 7: If drop-out of an allele is required to explain
the evidence under Hp: (S = ab; E = a), then the allele should be small enough (height/area) to justify this.
(allele deve essere sotto la soglia stocastica)

Raccomandazioni ISFG DNA Commission
• Recommendation 8: If the alleles of certain loci in the DNA profile are at a level that is dominated by background noise, then a biostatistical interpretation for these alleles should not be attempted.
• Recommendation 9: In relation to low copy number, stochastic
effects limit the usefulness of heterozygous balance and mixture proportion estimates. In addition, allelic drop-out and allelic drop-in (contamination) should be taken into consideration of any assessment.

Misture complesse
Calcoli complessi

Drop out e Drop in
• Fenomeni stocastici che si verificano in presenza di DNA Low Template
• Drop out (no amplificazione di un allele) sotto la soglia stocastica
• Drop in (falso allele) non riproducibile
• Drop in ≠ contaminazione

Probabilità di drop out Pr(D)
• Introduce incertezza sulla presenza o meno di un allele • Non si ragiona più in termini di compatibilità /esclusione
soglia soglia
Compatibilità Esclusione
Probabilità di drop out ≈ 0
soglia soglia
Compatibilità Compatibilità?
0 < Pr(D) < 1
approccio binario classico
Approccio probabilistico

Drop out
1
1 2
Hp: drop-out dell’allele 2 no drop-out dell’allele 1 Eventi indipendenti
Evidence
Suspect
Hp: Suspect Hd: Unknown
Consideriamo per semplicità un solo contributore, ma il modello è applicabile ugualmente alle misture

Drop out
1
1 2
Hd: • sconosciuto ha un genotipo che contiene l’allele 1 • può essere un genotipo 1,1 senza drop-out, oppure essere 1,Q dove Q è un qualsiasi allele escluso 1 con drop-out di Q
Evidence
Suspect
Hp: Suspect Hd: Unknown

Drop out
1 2
1
• Evidence
• Suspect
Hp: Suspect Hd: Unknown
Binario LR = 0 p1
2
Probabilistico LR = ? p1
2
? = un numero tra 0 e 1

1 2 1
Evidence Suspect
Hp: Suspect Hd: Unknown
Definiamo:
- probabilità di drop out di un allele ad un locus eterozigote: d - probabilità di drop out di entrambi gli alleli: d2
- probabilità di drop out per gli omozigoti: d’
- probabilità di non drop out: 1-d e 1-d’
Soglia analitica
con d’≤ d2

1 1 2
Evidence Suspect Hp: Suspect Hd: Unknown
Esempio: LR considerando drop out
Affinchè sia vera Hp: l’allele 2 ha subito drop out l’allele 1 non ha subito drop out Affinchè sia vera Hd lo sconosciuto deve avere un genotipo che contenga l’allele 1:
P(E|Hp)=P(drop-out 2)*P(non drop-out 1)
= d(1-d) P(E|Hd)=p1
2(1-d’)+2p1pq(1-d)d
Genotipo Drop-out Probabilità del genotipo
1,1 1-d' p12
1,Q (1-d)d 2p1pq
Q = qualsiasi allele diverso da allele 1 Assumendo che il locus abbia 5 alleli, ciascuno con frequenza pi: Q={2,3,4,5} e pQ=p2+p3+p4+p5
LR = P(E|Hp)
P(E|Hd)
= d(1-d)
p12(1-d’)+2p1pq(1-d)d

Effetto della P(dropout) e delle frequenze alleliche su LR
1
1 2
Evidence
Suspect
p1=0.2, pQ=0.8, d=0.05, d’=0.052
LR = 0.05(1-0.05)
0.22(1- 0.052)+2*0.2*0.8(1-0.05)*0.05
= 0.86
p1=0.2, pQ=0.8, d=0.3, d’=0.32
LR = 0.3(1-0.3)
0.22(1- 0.32)+2*0.2*0.8(1-0.3)*0.3
2.03 =
p1=0.01, pQ=0.99, d=0.05, d’=0.052
LR = 0.05(1-0.05)
0.012(1- 0.052)+2*0.01*0.99(1-0.05)*0.05
45.66 =

Drop in
1
1 2
Suspect
Evidence
Hp: Suspect Hd: Unknown Hp: drop-in allele 2 Hd: sconosciuto con genotipo 1,2 e no drop-in c = probabilità di drop in di un allele c = 0.05 p1=0.21 p2=0.21
LR = Pr(E|S) cp2 c Pr(E|U) 2p1p2 2p1
≈ = = 0.12

2 1 3
Evidence Suspect Hp: Suspect Hd: Unknown
Esempio: LR considerando drop out e drop in
Affinché sia vera Hp: gli alleli 1 e 3 hanno subito drop out l’allele 2 ha subito drop in
P(dropin)=c, P(dropin allele 2)=cp2
Affinché sia vera Hd lo sconosciuto può avere qualsiasi genotipo:
P(E|Hp)=P(drop-out 1)*P(drop-out 3) *P(dropin 2)= d*d*cp2
P(E|Hd)=p2
2(1-c)(1-d’)+2p2pQ(1-d)d(1-c) + pQ
2*d’*cp2
Q = ogni allele diverso da quello presente nella traccia Assumendo che il locus abbia 5 alleli, ciascuno con frequenza pi: Q={2,3,4,5} e pQ=p2+p3+p4+p5
LR = d*d*cp2
p22(1-c)(1-d’)+2p2pQ(1-d)d(1-c) + pQ
2*d’*cp2
Genotipo Dropout Dropin Probabilità del genotipo
2,2 (1-d') (1-c) p22
2,Q (1-d)d (1-c) 2p2pQ
Q,Q d' cp2 pQ2

Effetto della P(dropout), della P(dropin) e delle frequenze alleliche su LR
2
1 3
Traccia
Sospettato
p2=0.1, pQ=0.9, d=0.3, d’=0.32, c=0.05
LR = 0.3*0.3*0.05*0.1
0.12(1- 0.32)(1-0.05)+2*0.1*0.9(1-0.3)0.3(1-0.05)+0.92*0.05*0.1
= 0.01
p2=0.5, pQ=0.5, d=0.3, d’=0.32, c=0.05
LR = 0.3*0.3*0.05*0.5
0.52(1- 0.32)(1-0.05)+2*0.5*0.5(1-0.3)0.3(1-0.05)+0.52*0.05*0.5
= 0.007
p2=0.5, pQ=0.5, d=0.8, d’=0.82, c=0.5
LR = 0.8*0.8*0.5*0.5
0.52(1- 0.82)(1-0.5)+2*0.5*0.5(1-0.8)0.8(1-0.5)+0.52*0.5*0.5
= 1.28

1) Probabilistic methods following the ‘basic model’ can be used to evaluate the evidential weight of DNA results considering drop-out and/or drop-in. 2) Estimates of drop-out and drop-in probabilities should be based on validation studies that are representative of the method used. 3) The weight of the evidence should be expressed following likelihood ratio
principles. 4) The use of appropriate software is highly recommended to avoid hand
calculation errors.

Necessario un software!!!!!
LRmix studio
Software analoghi: likeLTD (D. Balding) e FST (NYOCME, Mitchell et al)
Gli esempi considerati sono molto semplificati (1 solo contributore, 1 solo sistema)
Considerando più contributori e più sistemi i calcoli diventano
più complicati approccio probabilistico
Semi -continuous models

Continuous LR models
Software: • TrueAllele (Perlin et al. JFS, 2011) • STRmix (Taylor et al., FSIG, 2013) • EuroForMix (Bleka Ø et al., FSIG 2016) Includono dati come: altezza/area dei picchi, bilanciamento degli
eterozigoti (PHR), % stutters e il rapporto tra contributori (Mx)
Più informazioni numerosi calcoli complicati e modelli di simulazione
Non è possibile il confronto con il calcolo fatto a mano


















