
Intercellular Adhesion and Cancer Invasion: A Discrete Simulation Using the Extended Potts Model
16
J. theor. Biol. (2002) 216, 85–100 doi:10.1006/jtbi.2001.2522, available online at http://www.idealibrary.com on Intercellular Adhesion and Cancer Invasion: A Discrete Simulation Using the Extended Potts Model Stephen Turner and Jonathan A. Sherratt Centre for Theoretical Modelling in Medicine, Department of Mathematics, Heriot-Watt University, Edinburgh EH14 4AS, UK (Received on 4 January 2001, Accepted in revised form on 18 December 2001) We develop a discrete model of malignant invasion using a thermodynamic argument. An extension of the Potts model is used to simulate a population of malignant cells experiencing interactions due to both homotypic and heterotypic adhesion while also secreting proteolytic enzymes and experiencing a haptotactic gradient. In this way we investigate the influence of changes in cell–cell adhesion on the invasion process. We demonstrate that the morphology of the invading front is influenced by changes in the adhesiveness parameters, and detail how the invasiveness of the tumour is related to adhesion. We show that cell–cell adhesion has less of an influence on invasion compared with cell–medium adhesion, and that increases in both proteolytic enzyme secretion rate and the coefficient of haptotaxis act in synergy to promote invasion. We extend the simulation by including proliferation, and, following experimental evidence, develop an algorithm for cell division in which the mitotic rate is explicitly related to changes in the relative magnitudes of homotypic and heterotypic adhesiveness. We show that although an increased proliferation rate usually results in an increased depth of invasion into the extracellular matrix, it does not invariably do so, and may, indeed, cause invasiveness to be reduced. r 2002 Elsevier Science Ltd. All rights reserved. 1. Introduction The characteristics of a differentiated cell are normally tightly controlled by a variety of genetic, local and hormonal controls. When this control is lost and a cell begins to divide excessively, break contact with its neighbours, break down its surrounding extracellular matrix and migrate into the extracellular medium, the host is at risk of developing a malignant tumour. Such tumours are aggressive, have a high metabolic rate, can be hormonally active, and are able to invade surrounding healthy tissue and spread elsewhere in the bodyFa process known as metastasis. In Fig. 1, we show a histological slide of an invading tumour: the malignant cells at the top of the image are more densely packed than the regions of surrounding tissue. In addition, a finger of cells from the main tumour mass has broken through the basement membrane and has begun to invade the sur- rounding stroma. Should these cells continue to invade in this way and in addition evade destruction by the immune system, they may enter the host’s bloodstream or lymphatics, extravasate at a distant site, and establish secondary colonies with devastating conse- quences for the wellbeing of the host and the likelihood of therapeutic intervention being successful. Invasive cells are less adhesive, more highly mobile, more metabolically active, and more 0022-5193/02/$35.00/0 r 2002 Elsevier Science Ltd. All rights reserved.
-
Upload
stephen-turner -
Category
Documents
-
view
222 -
download
0
Transcript of Intercellular Adhesion and Cancer Invasion: A Discrete Simulation Using the Extended Potts Model

J. theor. Biol. (2002) 216, 85–100doi:10.1006/jtbi.2001.2522, available online at http://www.idealibrary.com on
Intercellular Adhesion and Cancer Invasion:A Discrete Simulation Using the Extended Potts Model
Stephen Turner and Jonathan A. Sherratt
Centre for Theoretical Modelling in Medicine, Department of Mathematics, Heriot-Watt University,Edinburgh EH14 4AS, UK
(Received on 4 January 2001, Accepted in revised form on 18 December 2001)
We develop a discrete model of malignant invasion using a thermodynamic argument. Anextension of the Potts model is used to simulate a population of malignant cells experiencinginteractions due to both homotypic and heterotypic adhesion while also secreting proteolyticenzymes and experiencing a haptotactic gradient. In this way we investigate the influence ofchanges in cell–cell adhesion on the invasion process. We demonstrate that the morphologyof the invading front is influenced by changes in the adhesiveness parameters, and detail howthe invasiveness of the tumour is related to adhesion. We show that cell–cell adhesion has lessof an influence on invasion compared with cell–medium adhesion, and that increases in bothproteolytic enzyme secretion rate and the coefficient of haptotaxis act in synergy to promoteinvasion. We extend the simulation by including proliferation, and, following experimentalevidence, develop an algorithm for cell division in which the mitotic rate is explicitly relatedto changes in the relative magnitudes of homotypic and heterotypic adhesiveness. We showthat although an increased proliferation rate usually results in an increased depth of invasioninto the extracellular matrix, it does not invariably do so, and may, indeed, cause invasivenessto be reduced.
r 2002 Elsevier Science Ltd. All rights reserved.
1. Introduction
The characteristics of a differentiated cell arenormally tightly controlled by a variety ofgenetic, local and hormonal controls. When thiscontrol is lost and a cell begins to divideexcessively, break contact with its neighbours,break down its surrounding extracellular matrixand migrate into the extracellular medium, thehost is at risk of developing a malignant tumour.Such tumours are aggressive, have a highmetabolic rate, can be hormonally active, andare able to invade surrounding healthy tissueand spread elsewhere in the bodyFa processknown as metastasis. In Fig. 1, we show ahistological slide of an invading tumour: the
0022-5193/02/$35.00/0
malignant cells at the top of the image are moredensely packed than the regions of surroundingtissue. In addition, a finger of cells from the maintumour mass has broken through the basementmembrane and has begun to invade the sur-rounding stroma. Should these cells continue toinvade in this way and in addition evadedestruction by the immune system, they mayenter the host’s bloodstream or lymphatics,extravasate at a distant site, and establishsecondary colonies with devastating conse-quences for the wellbeing of the host and thelikelihood of therapeutic intervention beingsuccessful.Invasive cells are less adhesive, more highly
mobile, more metabolically active, and more
r 2002 Elsevier Science Ltd. All rights reserved.

Fig. 1. A high-power photomicrograph from a micro-invasive tumour of the cervix, with a finger of invading cellsprotruding through the basement membrane at the bottomleft of the image. (courtesy of Michigan State UniversityCollege of Human Medicine (http://www.echt.chm.msu.edu/index.htm)).
S. TURNER AND J. A. SHERRATT86
highly mitotic than normal cells. The series ofchanges leading a normal cell to becomemalignant and invasive are related to each other,and possibly occur in a stepwise manner, witheach mutation following the next (Stetler-Stevenson et al., 1993). These mutations affectthe cell’s adhesiveness, its ability to secretematrix degrading enzymes, and its capacity foruncontrolled proliferation.Unmutated cells express receptors on their
surfaces involved in adhesion to both other cellsand the extracellular matrix: the integrins,cadherins, Ig superfamily and CD44 are allinvolved (Stetler-Stevenson et al., 1993). Follow-ing a pro-invasive mutation, cell–cell adhesionwill be reduced, but cell–medium adhesionwill be increased (Huang & Ingber, 1999). Therelative increase in the expression of cell surfacereceptors involved in cell–medium adhesion (inparticular the) avb3 integrins) causes increasedbinding of the appropriate ECM ligands.Through subsequent intracellular mediated cas-cades, this binding may stimulate the secretion ofECM degrading enzymes (Seftor et al., 1992),which is one of the next steps in the progressiontowards the invasive phenotype. In addition, ithas been argued (Huang & Ingber, 1999) thatthis binding is also implicated in promotingmitosis through a similar mechanism.The secretion of proteolytic enzymes by the
mutated cells is essential for the invasion of the
extracellular matrix for two reasons: the dissolu-tion of the ECM provides a space into which thecells can move, and it also provides gradients inboth diffusible and fixed ECM proteins whichthe cells can use to direct themselves (Murphy &Gavrilovic, 1999). The matrix metalloprotei-nases (MMPs) are the most important groupof ECM degrading enzymes (Stetler-Stevensonet al., 1993). Experimental work has suggestedthat it is not actually the malignant cells whichsecrete the enzymes, but stromal fibroblastsadjacent to the tumour surface, which do sofollowing stimulation by collagenase stimulatoryfactor secreted by the mutated cells (Nabeshimaet al., 1991). In addition, the activity of theenzymes is kept in check by the activity ofopposing anti-proteases secreted by nearbyhealthy tissue: in particular, the tissue inhibitorsof metalloproteinases (TIMPs) (Testa, 1992).Both of these findings would explain as to whythe activity of the proteolytic enzymes occursalmost exclusively in a narrow region of spaceadjacent to the tumour surface.In addition to altered adhesiveness and the
capacity to secrete proteolytic enzymes, invasivecells also exhibit changes in their motility. Thiscan be classified into both random movement(chemokinesis) and directed movement alonggradients of either diffusible materials (chemo-taxis) or fixed substrates (haptotaxis). Due to thedissolution of the extracellular matrix and themovement of the cells into it, the cells findthemselves in a less dense milieu compared withthe interior of the solid tumour. Correspond-ingly, their random movement is increased. Inaddition, the reduction or abscence of cell–cellcontacts by invasive cells may be an independentfactor in increasing random movement througha reduction in contact inhibition (Jones &Walker, 1997). It has been demonstrated thatthe microscopic processes involved in chemo-taxis and haptotaxis are independent of eachother (Aznavoorian et al., 1990). However, thetwo processes are microscopically similar: theextra binding of ECM material (whether fixed ordiffusible) on one side of a cell compared to theother causes the cell to extend a pseudopod inthe direction of increased density and increasethe number of bonds in that direction, inconjunction with a reduction in the number of

INTERCELLULAR ADHESION AND CANCER INVASION 87
bonds on the opposite side (Condeelis et al.,1992). Through coordinated contraction of thecytoskeleton, the cell then pulls itself up theECM gradient.Hence, the invasive phenotype corresponds to
alterations in adhesiveness, enzyme secretionrate, motility, and mitotic rate, and theseprocesses are all intimately linked. Our indepen-dent understanding of each of these microscopicphenomena is not matched, however, by adetailed understanding of their relative effects.Mathematical modelling provides a means bywhich we may quantify the various phenomenainvolved in the invasion process, and investigatetheir interactions with relative ease comparedwith conventional ‘‘benchtop’’ experimentation.By using experimental results for the variousmicroscopic quantities involved, we may buildup models which reveal the relationships be-tween cellular and biochemical parameters andmacroscopic phenomena.Previous modelling techniques for the invasion
process have included using sets of coupledreaction–diffusion equations for the cells andimportant groups of extracellular proteins andnutrients (Anderson et al., 2000; Chaplain, 1995;Orme & Chaplain, 1997; Perumpanani et al.,1996). The inclusion of adhesion has provenproblematic in this type of model, however,although there have been some attempts. (Byrne& Chaplain, 1996; Byrne, 1997). In addition, thereaction–diffusion approach makes the inclusionof the stochastic behaviour of individual cellsdifficult to treat. In the study of the invasionprocess, the explicit inclusion of the behaviour ofindividual cells is of some importance as second-ary tumours can evolve following the breakingfree of even a single cell from the primary tumour.In this study, we approach the modelling of
invasion in a manner which has a realistictreatment of adhesion at its core. The extendedPotts model (Graner & Glazier, 1992) which weuse here is similar to previous work in themodelling of benign avascular tumour growth(Stott et al., 1999; Drasdo et al., 1995), but is inthis case developed further for application tomalignant invasion. The simulation models anindividual cell as occupying a defined region of asquare lattice, and then employs a stochasticenergy minimization technique to display the
evolution of the cell mass over time. By biasingthe capacity of the cells to interact with theirneighbours, the extracellular medium, and gra-dients of extracellular protein concentrations, anaccurate representation of the invasion processcan be developed.In the following section, we describe in detail
the mathematical and computational under-pinnings of the model, and go on to presentresults which make clear the interrelationshipsbetween the various parameters which quantifydifferent properties of the invasive phenotype.We show that changes in cell–cell and cell–medium adhesion strength can have a majorinfluence on the morphology of the invadingfront of cells. In addition, we quantify theinvasiveness of the tumour in terms of themaximum depth of invasion after a given time,and go on to show the close relationship betweenthis and the adhesion parameters as well as otherparameters in the model. In Section 4, we extendthe model to include proliferation, and, basedon experimental evidence (Huang & Ingber,1999), we develop a term for the mitotic ratewhich explicitly relates this rate to changes inadhesiveness.The model of the invasion process which we
develop not only allows the important stochasticaspect of individual cell behaviour to be takeninto account; it also allows the close relationshipbetween changes in cell adhesion and theaggressiveness of a tumour’s invasive potentialto be made explicit.
2. The Model
We model a collection of biological cells byattaching to each lattice point (i; j) of a squarelattice a label sij : Adjacent lattice sites with thesame value sij are defined to lie within the samecell. We model the interactions of cell surfaceswith each other explicitly by defining couplingconstants Jsijsi0 j0 ; the size of which quantifies thestrength of the interaction between adjacentlattice points with differing values of sij :Physically, the J’s correspond to the total energyinvolved in the specified interaction, which isproportional both to the number of cell surfacereceptors involved in the interaction and thebinding energy of the involved ligands with their

S. TURNER AND J. A. SHERRATT88
receptors. Hence, the ‘‘energy’’ contained in theinteractions between cells can be defined by theHamiltonian
H ¼Xij
Xi0j0
Jsij ;si0 j0 : ð1Þ
Here, the (i; j) terms describe points on thelattice: the first summation runs over all of thelattice points, the second over the eight nearestneighbours of (i; j). If sij ¼ si0j0 ; then the twolattice points (i; j) and (i;0 j0) are both inside thesame cell, and their contribution to the surfaceinteraction energy is zero: hence, in this case,Jsij ;si0 j0 ¼ 0: If we assume that there are a limitednumber or cell types (for example, two: corre-sponding to normal and malignant cells), andthat the interactions between them are indepen-dent of the actual cell but merely on its type,then we can simplify the model by introducingan additional cell label tðsijÞ: Then for eachlattice point (i; j) we have two labels: sij ; which isthe label identifying the corresponding cell, andtsij ; which is that particular cell’s type. Then theHamiltonian becomes
H ¼Xij
Xi0j0
JtðsijÞ;tðsi0 j0 Þf1� dsij ;si0 j0 g: ð2Þ
The Kronecker delta term ensures that Jsij ;si0 j0 ¼ 0for sij ¼ si0j0 ; and is required because differentcells of the same type do have a surfaceinteraction. In fact, the simulations in this paperinvolve only a single cell type; however, interac-tions between the cells and the medium aremodelled in the same way as cell–cell interac-tions, using a coupling coefficient Jcell2ECM : Theinteraction between the cells and the ECM whichgive rise to haptotactic movement are treatedseparately, and the inclusion of this interactionin the model is discussed below. This Hamilto-nian is a modification of the Potts model, whichis used to model the interactions betweenadjacent ‘‘spins’’ on a lattice, and Hamiltoniansof this type traditionally have been used to studythe dynamics of magnetic materials, spin glassesand neural networks (Wu, 1982).Physically, both the growth and mechanical
deformation of cells requires energy. We takethis into account by including an elastic termlðvs � VT Þ
2: in this term, VT is the ‘‘target’’
volume of the cell (the volume to which it wouldrelax in the absence of external forces and in thepresence of adequate nutrition), and deforma-tions which expand or compress the cell above orbelow this value require energy (Stott et al.,1999; Mombach & Glazier, 1996). Hence, thetotal energy term is now:
H ¼Xij
Xi0j0
JtðsijÞ;tðsi0 j0 Þf1� dsij ;si0 j0 g
þXs
lðvs � VT Þ2;
ð3Þ
where the summation over s runs over the totalnumber of cells in the lattice.In order to simulate the sorting of which are
interacting energetically according to the aboveHamiltonian, we use the Metropolis algorithm(Metropolis et al., 1953; Metropolis & Ulam,1949). Here, we select a site (i; j) at random, thenselect a nearest neighbour (i0; j0) also at random.If (i0; j0) and (i; j) lie in the same cell (i.e.sij ¼ si0j0), then we repeat the random selection.However, if the two lattice points lie in differentcells, then we investigate the effect on the surfaceenergy of copying the parameters for sði;jÞ andtðsði;jÞÞ into (i0; j0). If the resulting change in totalsurface energy DH is negative, then we accept thecopy and change the values for (i0; j0) accord-ingly; conversely, if the energy change is positive,then we accept the change with Boltzmann-weighted probability expð�DH=bÞ: Hence,
pðsij-si0j0 Þ ¼1 if DHp0;
e�DH=b if DH40:
(ð4Þ
The parameter b influences the likelihood ofenergetically unfavourable events taking place:the higher b; the more out-of-equilibrium thesystem will be. Also, the greater will bethe random motility of the cells, and the greaterthe area across which they will move in a givenspace of time. Hence, the parameter b isphysically analogous to the diffusion coefficientD in the diffusion equation. In applications ofthe Potts model in physics, b generally corre-sponds to the temperature.The above model describes cell-sorting
phenomena and has already been extensively

INTERCELLULAR ADHESION AND CANCER INVASION 89
studied (Mombach & Glazier, 1996; Mombach,1999; Mombach et al., 1993, 1995; Stott et al.,1999). However, we have taken this modelforward to describe the phenomenon of malig-nant invasion by extending it to include hapto-taxis and proteolytic enzyme secretion, asdescribed in the introduction.Haptotaxis is included in our model by
attaching to each lattice point a parameter fij ;which corresponds to the local extracellularmatrix (ECM) protein concentration alonggradients of which the cells are moving (suchproteins include fibrin, vitronectin, and someof the collagen family; Stetler-Stevenson et al.,1993). When evaluating the energy changecorresponding to a site copy, we amend themodel by introducing a bias into the energychange proportional to the fibrin concentrationgradient ( fi0j0 � fij). The energy change DH isthen given by
DHij ¼ DH1ij þ DH2ij þ kHð fi0j0 � fijÞ; ð5Þ
where DH1ij is the surface energy change, andDH2ij the mechanical energy change [DH1 andDH2 are as in eqn (3)]. The parameter kHinfluences the strength of haptotaxis relative tothe other parameters in the model, and the ratioof b to kH quantifies the relative effects ofrandom and directed motility, respectively. Thisterm is similar to the term used by Savill &Hogeweg (1997), who used it in their studies ofchemotaxis in Dictyostelium discoideum aggrega-tion. In reality, it is conceivable that for verysteep ECM gradients, the cell velocity willsaturate and will not be increased by increasingDf still further. However, assuming a linearrelationship between Df and cell velocity is anadequate first approximation for typical invasivebehaviour (Murphy & Gavrilovic, 1999; Perum-panani et al., 1998). In our simulations, there isassumed to be an even concentration of fibrinacross the domain initially, and the synthesis ofECM proteins is neglected.The secretion of proteolytic enzymes by
malignant cells gives rise to ECM gradientswhich change with time. To model this phenom-enon, we change the ECM concentration at eachlattice point with each Monte-Carlo times step: ifa lattice point is occupied by a cell then the rate
of dissolution of the ECM protein at that pointis higher compared with another region of thelattice which is not occupied by a cell. Weassume that this dissolution gives rise to anexponential decay of the ECM protein concen-tration with time. Hence,
fijðtþ 1Þ
¼ fijðtÞ
e�ki if lattice point occupied
by a cell;
e�kn if adjacent pointðsÞ occupied;
0 empty space
8>>>><>>>>:
ð6Þ
with ki4kn to indicate the higher rate ofECM degradation. An implicit assumption inthis model is that proteolysis takes placeexclusively in the region of the tumour surface.This is in agreement with experimental evidence(Murphy & Gavrilovic, 1999), which has shownclearly that the activity of ECM degradingenzymes is strongly localized to this area, asdescribed in Section 1. In our simulations wedefined ki to be twice the value of kn to take intoaccount the increased concentration of secretedenzymes in the areas of the extracellular mediumwhich lie beneath the cells (in 2D). The absolutevalues of ki and kn are of less qualitativeimportance in the simulation than their relativevalue ki=kn; as it is this ratio which willdetermine the steepness of the haptotacticgradient produced.The choice of the absolute values for the
coefficients in eqn (3) is also of less importancethan their relative values. When each site changeis attempted, a value of DH will be evaluatedand, if positive, the dynamics of the simulationwill be controlled by the value of DH relative tob eqn (4). Hence, the choice of coefficients in themodel is determined by the relative influencewhich cell–cell adhesion, cell deformability, andhaptotaxis have over the simulation.
3. Results
3.1. SIMULATION DETAILS
The simulations are conducted on a 200 200square grid, with each cell initially defined tooccupy a set number of lattice points, equal to

S. TURNER AND J. A. SHERRATT90
the cell’s target area [VT in eqn (3), set in allsimulations to be 20 pixels]. At each iteration ofthe energy minimization procedure, the energeticeffect of copying the parameters for a siteinto one of its neighbours, or of vacating thatsite, are examined as described in the precedingsection.The choice of parameter values in the simula-
tions is determined by the relationship whichwe wish to study. For reasons discussed inSection 2, the relative values of the parametersare of greater importance than their absolutevalues. Recall that the Hamiltonian described ineqn (5) has the form
DH ¼ DH1 þ DH2 þ DH3; ð7Þ
where DH1 is the energy changes associatedwith alterations in cell–cell adhesion, DH2 isthe energy change due to mechanical deforma-tion, and DH3 is the energy changes due tomovement along ECM gradients. Should wewish to examine the effect of doubling theimportance of haptotaxis relative to cell–celladhesion, e.g. we choose parameter values whichdouble DH3 relative to DH1 and run thesimulation. Similarly, to examine the importanceof the other terms in the model to promot-ing invasion, we alter the parameter values sothat the three terms in the Hamiltonian scaleaccordingly.To quantify the effect of the different para-
meters in the model on the invasion process, weconcentrate on the parameter dmax; whichcorresponds to the maximum depth of invasion(in pixels) on the grid. Biologically, dmax is anappropriate parameter to study as the maximumdepth of invasion corresponds to the clinicalseverity of the disease, the likelihood of meta-stasis having occurred, and the options forclinical management. For example, in the clinicalmanagement of melanoma, the lesion is surgi-cally removed and examined microscopically. Insuch an examination, the clinician attempts toestablish whether the lesion has breachedthe basement membrane and, if so, the depthof invasion into the dermis in millimetres. Thelatter quantity is termed the Breslow thickness: itis an important clinical parameter in estimatingthe likelihood of metastasis having occurred and,
consequently, the severity of the disease andthe likelihood of therapeutic intervention beingsuccessful in curing the patient (Graham-Brown& Burns, 1996).Our initial conditions throughout consist of a
layer of cells at the top of the grid 10 cells thickwith an initial (target) volume of 20 pixels. Theboundary conditions are zero flux at the top andbottom of the grid and periodic at the left andright sides. This corresponds to a spatiallyextended lesion which is invading from anepithelial cell lining down through its basementmembrane and into the surrounding stroma.Zero flux boundary conditions at the top areappropriate as cell masses of this type are usuallylocalized in the epithelial boundary layers coat-ing a lumen, so there is no tissue for the cells tomove into in that direction. Periodic boundaryconditions laterally are also appropriate in oursimulation as the model is intended to examine asection through a spatially extended lesion muchlarger in size than could reasonably be modelledon our grid. Assuming that the cells are of a sizeof the order of 10mm, the domain correspondsto a physical size of around 0.4mm. Even thesmallest detectable malignant lesion has a spatialextent considerably greater than this, whichunderlines the fact that the domain should beregarded as only a part of a much larger lesion.Periodic boundary conditions laterally helpreduce boundary effects. Zero flux conditionsat the base are arbitrarily set as such, as this isthe limit of the validity of dmax in our model;hence, we stop the simulation before the bottomof the grid is reached.Simulations were run using a wide range of
parameter values, and the model was foundrobustly to reproduce the phenomenon of‘‘fingering’’ across a wide range of these values.This phenomenon is illustrated in Fig. 2. Thesurface appearance of many malignancies hasthis morphology, and it corresponds to diseaseseverity: benign tumours are smooth, whereasaggressive malignancies are ‘‘ragged’’. There is acorrelation between this ‘‘raggedness’’ and thetumour’s invasive potential: studies of photo-micrographs of tumour surfaces have succeededin demonstrating self-similarity at differentlength scales, and have noticed a relationshipbetween the fractal (Hausdorff) dimesion of
In Fig. 4, we illustrate the dependence of dmaxon Jc2c for two values of Jc2ECM : As we can see,there is a significant gap between the two linesacross the full range of Jc2c; in contrast to Fig. 3.
Fig. 2. Showing the characteristic appearance of thesimulated tumour after 1500 Monte-Carlo time steps, withstrands or ‘‘fingers’’ of cells extending into the ECM. This‘‘fingering’’ phenomenon is typical of an invasive cell mass.In biopsy specimens, the maximum depth of invasion ofmalignant cells into the ECM is an important clinicalindicator of disease severity. (Parameters used in the figure:Jc2c ¼ 3; Jc2ECM ¼ 6; kH ¼ 40; t=1500, n=2000.)
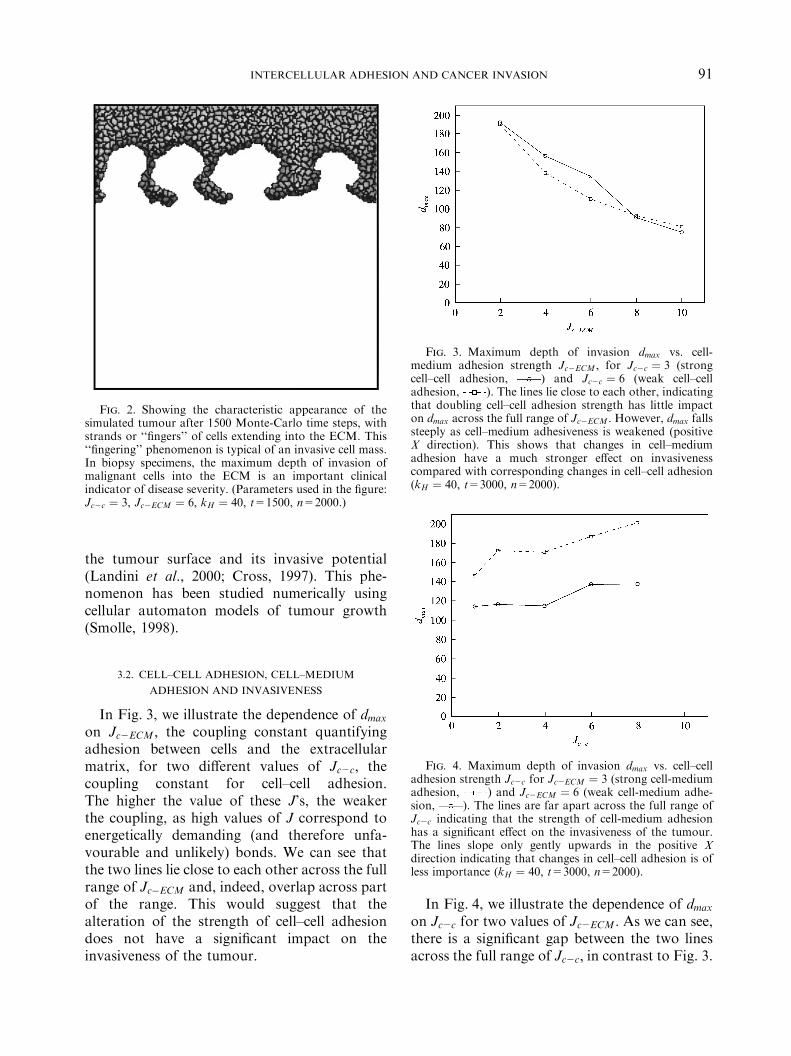
Fig. 3. Maximum depth of invasion dmax vs. cell-medium adhesion strength Jc2ECM ; for Jc2c ¼ 3 (strongcell–cell adhesion, ) and Jc2c ¼ 6 (weak cell–celladhesion, ). The lines lie close to each other, indicatingthat doubling cell–cell adhesion strength has little impacton dmax across the full range of Jc2ECM : However, dmax fallssteeply as cell–medium adhesiveness is weakened (positiveX direction). This shows that changes in cell–mediumadhesion have a much stronger effect on invasivenesscompared with corresponding changes in cell–cell adhesion(kH ¼ 40; t=3000, n=2000).
Fig. 4. Maximum depth of invasion dmax vs. cell–celladhesion strength Jc2c for Jc2ECM ¼ 3 (strong cell-mediumadhesion, ) and Jc2ECM ¼ 6 (weak cell-medium adhe-sion, ). The lines are far apart across the full range ofJc2c indicating that the strength of cell-medium adhesionhas a significant effect on the invasiveness of the tumour.The lines slope only gently upwards in the positive Xdirection indicating that changes in cell–cell adhesion is ofless importance (kH ¼ 40; t=3000, n=2000).
INTERCELLULAR ADHESION AND CANCER INVASION 91
the tumour surface and its invasive potential(Landini et al., 2000; Cross, 1997). This phe-nomenon has been studied numerically usingcellular automaton models of tumour growth(Smolle, 1998).
3.2. CELL–CELL ADHESION, CELL–MEDIUM
ADHESION AND INVASIVENESS
In Fig. 3, we illustrate the dependence of dmaxon Jc2ECM ; the coupling constant quantifyingadhesion between cells and the extracellularmatrix, for two different values of Jc2c; thecoupling constant for cell–cell adhesion.The higher the value of these J’s, the weakerthe coupling, as high values of J correspond toenergetically demanding (and therefore unfa-vourable and unlikely) bonds. We can see thatthe two lines lie close to each other across the fullrange of Jc�ECM and, indeed, overlap across partof the range. This would suggest that thealteration of the strength of cell–cell adhesiondoes not have a significant impact on theinvasiveness of the tumour.

S. TURNER AND J. A. SHERRATT92
We may conclude from these two figures,therefore, that a change in the strength of theadhesiveness between cells and the ECM has afar greater impact on the invasiveness of thetumour compared with a corresponding changein the strength of cell–cell adhesiveness.
3.3. CELL–CELL ADHESION, PROTEOLYTIC ENZYME
SECRETION RATE AND INVASIVENESS
In Fig. 5, we illustrate the relationship be-tween dmax and the protolytic enzyme secretionrate. The parameter n corresponds to thenumber of Monte-Carlo time steps after whichthe minimum value of ECM concentration willhave decreased to negligible levels (arbitrarily setto 0.01 with an initial ECM concentration of 1).Hence, a high value of n corresponds to a lowproteolytic enzyme secretion rate, as it has takenlonger for the fibrin to be degraded to negligiblelevels. We see that at high enzyme secretion rates(e.g. n¼1000), a reduction in cell–cell adhesive-ness from 2 to 6 results in a marked change indmax; whereas at low enzyme secretion rates (e.g.n¼6000), changes in cell–cell adhesiveness havelittle effect on dmax: We may conclude from thisthat genetic mutations which give rise to altera-tions in cell–cell adhesiveness properties during
Fig. 5. Maximum depth of invasion dmax vs. proteolyticenzyme secretion rate n for both strong (Jc2c ¼ 2; ) andweak (Jc2c ¼ 6; ) cell–cell adhesion. As the enzymesecretion rate decreases, the maximum depth of invasionalso decreases for both cell–cell adhesion strengths. How-ever, the decrease is greater for weak cell–cell adhesionexcept at low enzyme secretion rates (kH ¼ 40; t=3000,Jc2ECM ¼ 4).
the stepwise progression to malignancy onlyhave an effect in the presence of additionalmutations which give rise to the pronouncedsecretion of proteolytic enzymes. Taking intoaccount our discussion of Fig. 3, we mayconclude that a reduction in cell–cell adhesionand an increase in cell–ECM adhesion in thepresence of proteolytic enzyme secretion are themutations which produce the most invasivephenotype.
3.4. HAPTOTAXIS, PROTEOLYTIC ENZYME SECRETION
RATE AND INVASIVENESS
In Fig. 6, we illustrate the relationship be-tween invasion depth dmax and protease secretionrate (given by n) for two values of kH [thecoefficient of haptotaxis in eqn (5)]. We see thatchanges in the coefficient of haptotaxis andproteolytic enzyme secretion rate act in synergy:doubling kH from 20 to 40 doubles the influenceof haptotaxis over DH and gives rise to a markedchange in dmax for all values of n. However, ifthis change is coupled with a change in n (from2000 to 1000, for example), the increase isgreater than the sum of each of these changes.We may conclude, therefore, that geneticchanges which produce a greater sensitivity to
Fig. 6. Maximum depth of invasion dmax vs. proteolyticenzyme secretion rate n for both weak (kH ¼ 20; ) andstrong (kH ¼ 40; ) haptotaxis. Doubling kH results in asignificant increase in dmax for all n; however, if both n andkH are increased, the two changes act in synergy to promoteinvasion (t=3000, Jc2ECM ¼ 6; Jc2c ¼ 3).

INTERCELLULAR ADHESION AND CANCER INVASION 93
haptotactic gradients are in themselves effectiveat promoting invasion, but if these changes arecoupled to increases in the proteolytic enzymesecretion rate then the invasiveness of the cell ismarkedly increased.
3.5. MORPHOLOGY OF THE ADVANCING FRONT
Within some parameter ranges for kH ; Jc2cand n, the ‘‘fingers’’ which extend into the ECMsplit, with an advancing cell mass in frontmoving off into the ECM, and a retracting massbehind which remains with the main cell mass
Fig. 7. Illustrating the development of the advancing frontan advancing front of cells moving down through the extracelluinto the main tumour mass behind. (Here Jc2ECM ¼ 6; Jc2c ¼
(illustrated in Fig. 7). The explanation for this isrelated to the manner in which the advancingfront forms. Initially, slender fingers of cellsextend out into the ECM. Their tips thenbecome spread out laterally and join with theirneighbours to form an advancing mass of cells.This mass is anchored initially to the main cellmass by the fingers which gave rise to it. Thecells lying closest to the front of the invading cellmass experience a greater haptotactic pull thanthose behind them due to the steeper fibringradients there; however, the cells also experi-ence the effect of cell–cell adhesion (which may
with time. The invading cells split into two distinct colonies:lar matrix, and a retracting portion of cells being pulled back6; kH ¼ 40; n=2000.)

S. TURNER AND J. A. SHERRATT94
oppose the effect of haptotaxis). It is possible,therefore, for the fingers to break causing theadvancing cell mass to break contact with themain cell mass behind it thus allowing it toinvade the ECM without hinderance. The pointin the fingers at which the split will occur will bethe point at which the forces due to cell–celladhesion and haptotaxis balance. Cells in frontof this point will break free and move off into theECM, whereas those behind will be pulled backinto the primary cell mass. Figure 7 illustratesthe phenomenon of an advancing ‘‘front’’ ofcells, with a retracting mass behind.
4. The Inclusion of Proliferation
4.1. MODELLING PROLIFERATION
The invasive phenotype includes the develop-ment of changes in both random and directedmotility, changes in the adhesive properties ofmutated cells, and the secretion of proteolyticenzymes. In addition, malignant cells have ahigher proliferation rate relative to their un-mutated counterparts. Huang explains how theexcessive proliferation of malignant cells is aconsequence of changes in cell membrane sur-face receptor composition. Cells are triggered todivide by intracellular cascades which start whenmembrane-bound integrin receptors bind toextracellular matrix proteins. Malignant cellshave reduced cell–cell adhesiveness, but in-creased cell–ECM adhesiveness due to a changein the relative numbers of the correspondingreceptors (Huang & Ingber, 1999). In ourmodel, these adhesivenesses (and the corre-sponding number of cell surface receptors)is quantified through the coupling constantsJtðsi;jÞ: Hence, we can relate the proliferation rateof a given cell type to the relative values of itscoupling constants for cell–cell and cell–ECMinteractions.The life of most cells is divided into a series of
discrete steps which, in general, include shortperiods of division interspersed by long periodsof metabolic activity (Alberts et al., 1989).Conventionally, these are labelled G1 (duringwhich cells are metabolically active and producetheir protein products); S, during which cellsbegin to replicate their DNA; and M, duringwhich chromosomes are divided and the cells
undergo division. The length of G1 variesenormously between cell types: from very short(such as cell of the developing embryo) to verylong (such as hepatocytes) (Alberts et al., 1989).Some cells, such as neurons, are unable to divideand are said to be terminally differentiated:they are arrested in a state conventionallylabelled G0. Once a cell has entered the Sphase of division, the length of time taken toproceed through the S and M phases andterminate with the production of two daughtercells is fairly constant. However, although thereis an average length of G1 for a given cell type,there is a wide distribution of periods spent inG1, and for an individual cell the time of onset ofthe S phase will be stochastic. An experimentalinvestigation of cell division which supportsthese conclusions has been conducted by Smith& Martin (1973).Given this knowledge concerning the cell
cycle, we must assume that the time betweencell divisions has a stochastic distribution.Hence, in our simulation we implement thisunderstanding of the cell cycle as follows: at eachMonte-Carlo time step, the simulation runsthrough all of the cells and evaluates theprobability Ps of that cell dividing. Due to theassociation between the adhesion characteristicsof the cell and the mitotic rate, this probability isa function of both the J’s and the time since thecell last divided. Except for extremely rapidlydividing cells, those which recently divided willstill be growing and the likelihood of their re-entering the S phase is extremely small. Hence,we set a time interval Td during which the cell isprohibited from dividing. Once the time since thelast division exceeds Td ; we assume that theprobability of division slowly increases, andapproaches 1 as the time since last divisionbecomes very long. Hence, we use the followingfunctional form for Ps:
PsðtÞ ¼ fs½ktðsÞ; ðt� TsÞ�; ð8Þ
where
k ¼ Jc2ECM=Jc2c; ð9Þ
which is the ratio between the cell–ECMand cell–cell coupling constants for a cell oftype tðsÞ:

Fig. 8. The difference between the depth of invasionincluding proliferation and in the abscence of proliferation,dmax �do vs. a [defined in eqn (10)]. For a given range of a;the maximum depth of invasion is reduced despite thepresence of more cells. This is caused by the greater surfacearea of contact between cells at the tip of the advancingfront and the proliferating cell mass behind, as illustrated inFig. 9. (Here t=3000, Jc2ECM ¼ 6; Jc2c ¼ 6; kH ¼ 40).
INTERCELLULAR ADHESION AND CANCER INVASION 95
We propose the following functional form forf in eqn (8):
PsðtÞ ¼ fs½ktðsÞ; ðt� TsÞ�
¼0 if ðt� TsÞpTd ;
1
aðktðsÞ=ðt� TsÞÞ2 þ 1
if ðt� TsÞ > Td :
8><>:
ð10Þ
This form takes into account a dormant periodTd for a cells which divided at time Ts; as well asa gradually increasing probability of divisionafter this period has passed. A high value of kcorresponds to cells which have a high value ofJc2ECM=Jc2c: this corresponds to cells whichhave close adhesiveness to each other and less tothe medium, as creating a bond with a high Jrequires more energy than creating one with alower value. Hence, high k corresponds tonormal cells, and low k to malignant cells.In eqn (8), a low k causes an increased valueof f (and, hence an increased proliferationrate) compared with a low k. This correspondsin reality to malignant cells having a higherproliferation rate compared with normal cells.So this functional form explicitly takes into
account three observed phenomena concerningcell division: a period following cell divisionduring which cells will not divide again; astochastic distribution of times between division;and the connection between proliferation andchanges in adhesiveness.Biological cells divide through their centre of
mass along the axis of minimum length (Mom-bach & Glazier, 1996), so we implemented themechanics of cell division by determiningthe lattice point corresponding to the cell’scentre of mass, working out the lengths ofthe horizontal, vertical and diagonal diametersthrough it, and giving all of the latticepoints within the cell on one side of theminimum diameter a new value of s: The choiceof the parameter Td is fixed at 100 Monte-Carlotime steps: most of the simulations were stoppedafter 3000 Monte Carlo time steps and themaximum depth of invasion evaluated after thisperiod. As we mentioned above, the length oftime for which a cell remains in its G1 state
between divisions varies greatly from tissue totissue, and between different types of malig-nancy. Hence, our choice of Td represents avalue which is a compromise between very slowgrowing and very aggressive tumour types.
4.2. RESULTS INCLUDING PROLIFERATION
The alteration in dmax due to the inclusion ofproliferation in the simulations is illustrated inFig. 8, and the parameters chosen were the sameas those used in the simulation for Fig. 7.Intuitively, one expects proliferation to be pro-invasive, on the basis that the additional cellpopulation will facilitate invasion. However, aswe can see, within some regions of the parameterspace dmax is reduced due to proliferation. Theexplanation for this counter-intuitive result isrelated to our discussion of the morphology ofthe advancing front in Section 3. The front iscreated when fingers of cells invading the ECMjoin together to form an invading cell mass,when then breaks its contacts with the main cellmass behind it and moves on through the ECM.By including proliferation in the simulation, thefingers of cells which initially ‘‘anchor’’ thisadvancing front to the main cell mass are thicker(as the cells comprising them are dividing) and

S. TURNER AND J. A. SHERRATT96
also remain connected to the advancing front forlonger (due to cells being pushed forward bytheir dividing and growing neighbours). Theevidence for this is illustrated in Fig. 9, whichshows the effect of including proliferation on themorphology and depth of invasion of theinvading cells. This figure should be comparedwith Fig. 7: apart from the inclusion of prolif-eration, all other parameters are the same. Cellsat the front are spreading out laterally to forman invading cell mass, and this mass is connectedby long, thick fingers of cells to the main cellmass behind it. In the simulation with no
Fig. 9. The effect of including proliferation on the formatwith Fig. 7 in which proliferation was not included, but in whiarea of contact between cells at the tip of the advancing froninvasion. (Here Jc2ECM ¼ 6; Jc2c ¼ 6; kH ¼ 40; n=2000, a ¼
proliferation included, these fingers are notpresent after the same length of time: they havealready broken and the cells composing themhave been pulled (under the influence of cell–celladhesion) into either the cell mass in front of orbehind them (depending on their position infront of or behind the point in the string at whichthe effects of haptotaxis and cell–cell adhesionbalance). Hence, one of the potential effects ofincluding proliferation is to reduce the depth ofinvasion of a cell mass, although this possibilityoccurs only in a narrow region of the parameterspace.
ion of the advancing front. This image should be comparedch all other parameters were the same. The increased surfacet and the proliferating cell mass behind inhibits the rate of5 107).

INTERCELLULAR ADHESION AND CANCER INVASION 97
5. Discussion
The invasive phenotype includes genetic mu-tations which give rise to changes in cellularadhesiveness, the ability to secrete proteolyticenzymes, the ability to follow gradients of bothsoluble and fixed substrates, and an increasedproliferation rate (Stetler-Stevenson et al., 1993).These genetic changes may be linked, withalterations in the cell surface receptor composi-tion that are of fundamental importance in onephenotypic change also triggering the next(Seftor et al., 1992). These various processeshave been modelled separately, but the interac-tion between them all (and in particular theconsideration of cell–cell adhesiveness) has notbeen studied in detail.The mathematical modelling of cell–cell
interation has been addressed previously,although not in the same context as we haveinvestigated here. The process of juxtracrinesignallingFin which receptors on cell mem-branes bind to ligands on the membranes ofneighbouring cellsFhas been considered(Wearing et al., 2000; Monk, 1998; Collieret al., 2000), but in these models cells are fixedand immobile. This work is useful in the studyof development and carcinogenesis as at itscentre is the study of contact inhibition, inwhich a developing or mutated cell inhibits itsneighbours from following the same develop-mental pathway. However, in the study ofcancer invasion it would be necessary to extendthis work to allow the cells to move. Weliky &Oster (1990) have developed a model of cellmovement in which a 2D sheet of epithelial cellswere modelled as polygons which experienceddeforming forces from their neighbours throughtheir vertices. Although movement is possible inthis model, and adhesion is explicit within it,the inclusion of chemo- and haptotaxis wouldrequire an extension of it. In addition, thepossibility of cells breaking free from theirneighbours is not mentioned. The explicitinclusion of cell–cell adhesion in a continuousmodel of tumour growth has been investigatedpreviously (Byrne & Chaplain, 1996; Byrne,1997) in a model in which the curvature of thetumour surface is related to cell–cell adhesive-ness, and a study conducted of the stability of
the model solutions to radially asymmetricperturbations. This work is useful in illuminat-ing the possible morphologies of the tumoursurface and its dependence of the strength ofcell–cell adhesion; however, the possibility ofcells moving along gradients of extracellularmaterial is not included.The ability of cells to secrete proteolytic
enzymes and establish gradients of extracellularmatrix proteins along which cells may move hasalso been extensively modelled (Anderson et al.,2000; Chaplain, 1995; Perumpanani et al., 1996).The continuum approach has yielded usefulinsights into the interaction between some ofthe phenomena involved in the invasion process,and interesting results have been obtained fromtheir studyFin particular concerning the non-linear dependence of the velocity of a travellingwave of invading cells on the model’s parameters(Perumpanani et al., 1999). However, the use ofthe continuum approach overlooks the stochas-tic behaviour or individual cells, which is ofsome importance in the study of metastasis, andthe possibility of individual cells establishingsecondary colonies. Hybrid models in which cellsmodelled as discrete points and capable ofproliferation which are immersed in a contin-uous milieu of ECM proteins (Dallon & Othmer,1997) or nutrients and oxygen (Ferreira Jr. et al.,1998; Anderson et al., 2000) have been investi-gated. These models have yielded useful results,but the possible influence of cell–cell adhesion onthe conclusions drawn has not been extensivelystudied.The thermodynamic approach to modelling
invasion which we have used here is an extensionof techniques used to model the aggregation ofthe slime mould Dictyostelium discoideum (Savill& Hogeweg, 1997) and benign avascular tumourgrowth (Stott et al., 1999). It is an appropriateway of modelling the random component of cellmovement, as experimental evidence indicatesthat individual cells in a cell mass exhibitBrownian motion in the absence of chemical oradhesion gradients (Mombach & Glazier, 1996).Hence, the energy spectrum of the cells will havea Boltzmann distribution, and the cells willpossess a Maxwellian distribution of velocities.The minimization of the free energy associatedwith cell–cell and cell–medium interfaces drives

S. TURNER AND J. A. SHERRATT98
the reorganization of the aggregate (Mombach,1999) and, indeed, this is in agreement with aseries of experimental observations by Steinberg(Steinberg, 1962a–c) of sorting phenomena incollections of various types of cell. Theseexperiments support the idea of differentialadhesiveness as being the driving force behindmorphogenesis and, through drawing compar-isons with studies of fluid mixtures of differingviscosity, it is concluded that a thermodynamicapproach to cell sorting is highly physicallyrealistic. In our extension of these ideas, we haveincluded the directed component of cell move-ment by biasing the probability of a cell movingto a lattice point which causes it to move up agradient of ECM concentration, with the time-dependent concentration of ECM at each latticepoint being explicitly included in the simulation.The advantage of this approach is that bothdirected and random motility can be included ina manner closely in tune with experimentalobservations of the behaviour of individual cellsin an aggregate.The motivation for this work was to determine
the relative importance and interrelationshipsbetween some of the main parameters involvedin the invasion process, concentrating in parti-cular on the influence of changes in cell–celladhesiveness. We have demonstrated that, in ourmodel, changes in the adhesiveness between cellsand the extracellular medium has a greaterimpact on the invasiveness of the cell mass(using the maximum depth of invasion after agiven time as our index of invasiveness) com-pared with changes in cell–cell adhesiveness. Wehave also demonstrated that changes in cell–celladhesiveness have a very small influence oninvasiveness unless the protease expression rateis high. In addition, increases in both proteaseexpression rate and the coefficient of haptotaxisact in synergy to promote invasion. The inclu-sion of proliferation in the simulation showedthat the morphology of the invading cell masswas changed by this inclusion, usually resultingin the cells invading as a solid mass rather thanas a succession of ‘‘fingers’’ spreading out intothe ECM. However, for some regions of theparameter space, including proliferation resultedin a reduction in the invasiveness of the tumour,for reasons discussed in Section 4.2. In our
simulation, we have explicitly related the pro-liferation rate to changes in cell–cell and cell–medium adhesiveness as recent experimentalwork indicates that these changes are linked(Huang & Ingber, 1999). The possibility of anincreased proliferation rate resulting in a reduc-tion in a tumour’s invasive potential under somecircumstances has, to our knowledge, not pre-viously been considered. An experimental in-vestigation into this using in vitro assays wouldbe an interesting research study.It is appropriate to ask what the therapeutic
significance of our conclusions may be. Anti-invasive therapies which are currently attractingthe greatest interest include interfering with theability of malignant cells to follow chemotacticgradients (Perumpanani et al., 1998), andinhibiting the ability of tumours to establish avascular network to provide the proteins andnutrients essential for growth and metastasis(Anderson et al., 2000). In the former case, it hasbeen shown (Perumpanani & Byrne, 1999) thatin the presence of gradients of both soluble andfixed ECM protein gradients which the cells canfollow, inhibiting only chemotaxis could becounterproductive. In the latter case, the reduc-tion in the likelihood of invasion is a by-productof the growth limitation of the tumour. Ther-apeutic interventions aiming at modulating theadhesive properties of the tumour have notattracted much attention, but in the light of ourresults we can make some predictions about thepossible success of any such intervention whichmay be developed. Such therapy should concen-trate on strengthening cell–cell adhesion whileinhibiting cell–ECM adhesion, and could becoupled with an additional intervention whichinhibits the ability of the tumour to secreteproteolytic enzymes. In doing so, the cellswill be more inclined to remain within the bodyof the main cell mass, as they will be held therethrough being tightly bound to their neighboursand through the abscence of ECM gradients.The development of an intervention whichblocks the cell–ECM receptors and thus reducescell–ECM adhesion while failing to trigger theintracellular cascades which are believed topromote both proliferation and enzyme secre-tion may be an optimal strategy for inhibitingmalignant invasion.

INTERCELLULAR ADHESION AND CANCER INVASION 99
Many thanks to Dr Nick Savill for many usefuldiscussions and for both programming assistance andthe supply of the graphics routines used in this work.Thanks also to Prof. Des Johnson and Ms ChristinaCobbold for useful discussions. ST was supportedby an EPSRC research studentship, and JAS wassupported in part by an EPSRC advanced researchfellowship. We thank SHEFC for support (researchdevelopment grant 107).
REFERENCES
Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K.& Watson, J. D. (1989). Molecular Biology of the Cell,2nd Edn. New York: Garland Publishing Inc.
Anderson, A. R. A., Chaplain, M. A. J., Newman,E. L., Steele R. J. C. & Thompson, A. M. (2000).Mathematical modelling of tumour invasion and metas-tasis. J. theor. Med. 2, 129–154.
Aznavoorian, S., Stracke, M. L., Krutzsch, H.,Schiffman, E. & Liotta, L. A. (1990). Signal transduc-tion for chemotaxis and haptotaxis by matrix moleculesin tumor cells. J. Cell Biol. 110, 1427–1438.
Byrne, H. M. (1997). The importance of intercellularadhesion in the development of carcinomas. IMAJ. Math. Appl. Med. Biol. 14, 305–323.
Byrne, H. M. & Chaplain, M. A. J. (1996). Modelling therole of cell–cell adhesion in the growth and developmentof carcinomas. Math. Comp. Modell. 24, 1–17.
Chaplain, M. A. J. (1995). The mathematical modelling oftumour angiogenesis and invasion. Acta Biotheoret. 43,387–402.
Collier, J. R., McInerny, D., Schnell, S., Maini,P. K., Gavaghan, D. J., Houston, P. & Stern, C. D.(2000). A cell cycle model for somitogenesis: mathema-tical formulation & numerical simulation. J. theor. Biol.207, 305–316.
CondeElis, J., Jones, J. & Segall, J. E. (1992).Chemotaxis of metastatic tumour cells: clues to mechan-isms from the Dictyostelium paradigm. Cancer MetastasisRev. 11, 55–68.
Cross, S. S. (1997). Fractals in pathology. J. Pathol. 182,1–8.
Dallon, J. C. & Othmer, H. (1997). A discrete cell modelwith adaptive signalling for aggregation of Dictyosteliumdiscoideum. Philos. Trans. R. Soc. London B 352,391–417.
Drasdo, D.,Kree, R. &McCaskill, J. S. (1995). Monte-Carlo approach to tissue cell populations. Phys. Rev. E52, 6635–6657.
Ferreira Jr, S. C., Martins, M. L. & Vilela, M. J.(1998). A growth model for primary cancer. Physica A261, 569–580.
Graham-Brown, R. & Burns, T. (1996). Lecture Notes inDermatology, 7th Edn. Oxford: Black-well Sciences.
Graner, F. & Glazier, J. A. (1992). Simulation ofbiological cell sorting using a two-dimensional extendedPotts model. Phys. Rev. Lett. 69, 2013–2016.
Huang, S. & Ingber, D. E. (1999). The structural andmechanical complexity of cell-growth control. Nat. CellBiol. 1, E131–E138.
Jones, J. J. & Walker, R. A. (1997). Cell–cell andcell–stromal interactions in breast cancer (Review). Int.J. Oncol. 11, 609–616.
Landini, G., Hirayama, Y., Li, T.-J. & Kitano, M.(2000). Increased fractal complexity of the epithelial–connective tissue interface in the tongue of 4NQ0-treatedrats. Pathol. Res. Pract. 196, 251–258.
Metropolis, N. M. & Ulam, S. (1949). The Monte-Carlomethod. J. Am. Stat. Assoc. 44, 335–341.
Metropolis, N. A., Rosenbluth A. W., Rosenbluth,M. N. & Teller, A. H. (1953). Equation of statecalculation by fast computting machines. J. Chem. Phys.21, 1087–1092.
Mombach, J. C. M. (1999). Simulation of embryonic cellself-organisation: a study of aggregates with differentconcentrations of cell types. Phys. Rev. E 59, R3827–R3830.
Mombach, J. C. M., DeAlmeida, R. M. C., Iglesias,J. R. (1993). Mitosis and growth in biological tissues.Phys. Rev. E 48, 598–602.
Mombach, J. C. M. & Glazier, J. A. (1996). Single cellmotion in aggregates of embryonic cells. Phys. Rev. Lett.76, 3032–3035.
Mombach, J. C. M., Glazier, J. A., Glazier, R. C. &Zajac, M. (1995). Quantitative comparison betweendifferential adhesion models and cell sorting in thepresence and abscence of fluctuations. Phys. Rev. Lett.75, 2244–2247.
Monk, N. A. M. (1998). Restricted-range gradients andtravelling fronts in a model of juxtacrine cell relay. Bull.Math. Biol. 60, 901–918.
Murphy, G. & Gavrilovic, J. (1999). Proteolysis and cellmigration: creating a path? Curr. Opin. Cell Biol. 11,
614–621.Nabeshima, K., Lane, W. S. & Biswas, C. (1991). Partialsequencing and characterisation of the tumour cell-derived collagenase stimulatory factor. Arch. Biochem.Biophys. 285, 90–96.
Orme, M. E. & Chaplain, M. A. J. (1997). Two-dimensional models of tumour angiogenesis and anti-angiogenesis strategies. IMA J. Math. Appl. Med. Biol.14, 189–205.
Perumpanani, A. J. & Byrne, H. M. (1999). Extracellularmatrix concentration exerts selection pressure on invasivecells. Eur. J. Cancer 35, 1274–1280.
Perumpanani, A. J., Sherratt, J. A., Norbury, J. &Byrne, H. M. (1996). Biological inferences from amathematical model for malignant invasion. InvasionMetastasis 16, 209–221.
Perumpanani, A. J., Sherratt, J. A., Norbury, J. &Byrne, H. M. (1999). A two-parameter family oftravelling waves with a singular barrier arising from themodelling of extracellular mediated cellular invasion.Physica D 126, 145–159.
Perumpanani, A. J., Simmons, D. L., Gearing, A. J. H.,Miller, K. M., Ward, G., Norbury, J., Schneeman,M. & Sherratt, J. A. (1998). Extracellular matrix-mediated chemotaxis can impede cell migration. Proc.Roy. Soc. London B 265, 2347–2352.
Savill, N. J. & Hogeweg, P. (1997). Modelling morpho-genesis: from single cells to crawling slugs. J. theor. Biol.184, 229–235.
Seftor, R. E. B., Seftor, E. A., Gehlsen, K. R., Stet-ler-Stevenson, W. G., Brown, P. D., Ruoslahti, E. &

S. TURNER AND J. A. SHERRATT100
Hendris, M. J. C. (1992). Role of alpha-v-beta-3 integrinin human melanoma cell invasion. Proc. Natl Acad. Sci.U.S.A. 89, 1557–1561.
Smith, J.A. & Martin, L. (1973). Do cells cycle? Proc.Natl Acad. Sci. U.S.A. 70, 1263–1267.
Smolle, J. (1998). Fractal tumor stromal border in anonequilibrium growth model. Anal. Quant. Cytol.Histol. 20, 7–13.
Steinberg, M. S. (1962a). On the mechanism of tissuereconstruction by dissociated cells, I. population kinetics,differential adhesiveness, and the abscence of directedmigration. Proc. Natl Acad. Sci. U.S.A. 48, 1577–1582.
Steinberg, M. S. (1962b). Mechanism of tissue reconstruc-tion by dissociated cells II: time course of events. Science137, 762–763.
Steinberg, M. S. (1962c). On the mechanism of tissuereconstruction by dissociated cells III: free energyrelations and the reorganisation of fused, heteronomictissue fragments. Proc. Natl. Acad. Sci. U.S.A. 48,
1769–1776.
Stetler-Stevenson, W. G., Aznavoorian, S. & Liotta,L. A. (1993). Tumor cell interactions with the extracellularmatrix during invasion and metastasis. Ann. Rev. CellBiol. 9, 541–573.
Stott, E. L., Britton, N. F., Glazier, J. A. & Zajac, M.(1999). Stochastic simulation of benign avascular tumourgrowth using the Potts model.Math. Comput. Modell. 30,183–198.
Testa, J. (1992). Loss of metastatic phenotype by ahuman epidermoid carcinoma cell line hep-3 isaccompanied by increased expression of tissue inhibitorof matrix metalloproteinase-2. Cancer Res. 52,
5597–5603.Wearing, H. J., Owen, M. R. & Sherratt, J. A. (2000).Mathematical modelling of juxtacrine patterning. Bull.Math. Biol. 62, 293–320.
Weliky, M. & Oster, G. (1990). The mechanical basis ofcell rearrangement. Development 109, 373–386.
Wu, F. Y. (1982). The Potts model. Rev. Mod. Phys. 54,
235–268.









![Original Article Polymorphisms in the intercellular adhesion … · 2018. 8. 31. · cancer [25], breast cancer [26], gastric cancer [27] and so on. However, all available results](https://static.fdocuments.in/doc/165x107/60e1a5c6fa93d31335254a43/original-article-polymorphisms-in-the-intercellular-adhesion-2018-8-31-cancer.jpg)








