
Interaction energy of DNAbase pairs and aminoacid...
22
Interaction energy of DNA-base pairs and amino-acid pairs: DFT, DFTB and WFT calculations Tomáš Kubař , Petr Jurečka, Jirka Černý, Honza Řezáč, Michal Otyepka, Haydée Valdés and Pavel Hobza* Institute of Organic Chemistry and Biochemistry Czech Academy of Sciences Prague * [email protected]
-
Upload
duongxuyen -
Category
Documents
-
view
216 -
download
3
Transcript of Interaction energy of DNAbase pairs and aminoacid...

Interaction energyof DNAbase pairs and aminoacid pairs:
DFT, DFTB and WFT calculations
Tomáš Kubař, Petr Jurečka, Jirka Černý,Honza Řezáč, Michal Otyepka, Haydée Valdés
and Pavel Hobza*
Institute of Organic Chemistry and BiochemistryCzech Academy of Sciences
Prague* [email protected]
Biomolecules
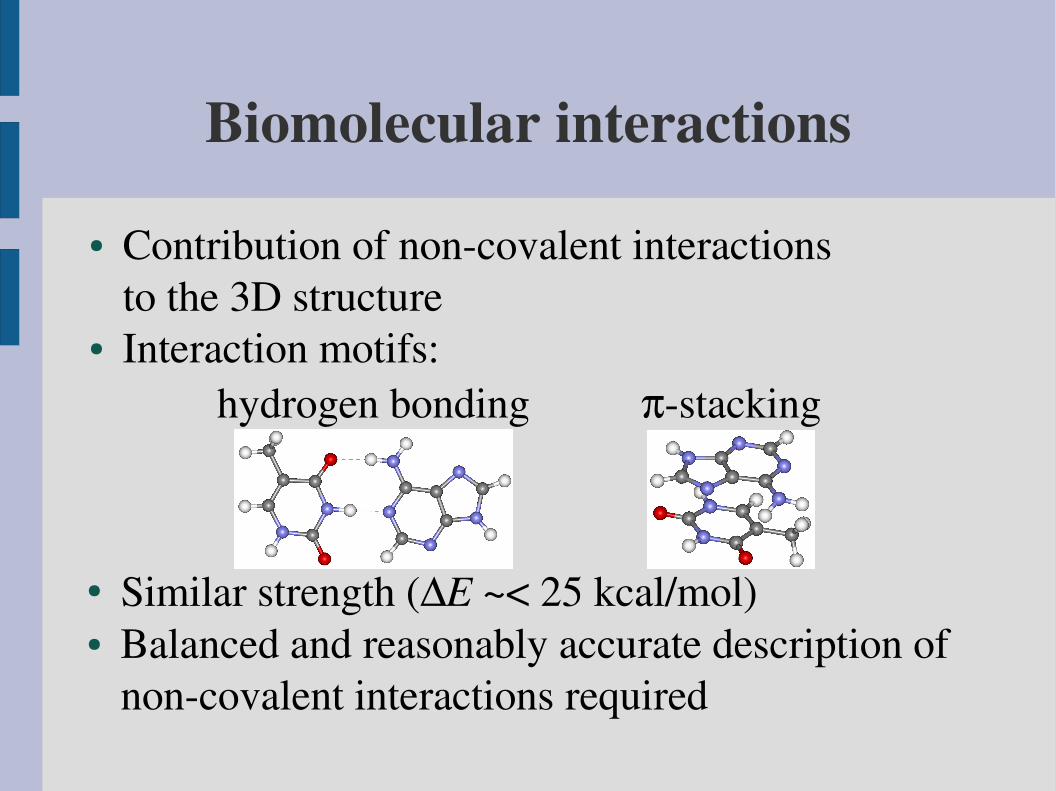
Biomolecular interactions
● Contribution of noncovalent interactionsto the 3D structure
● Interaction motifs:hydrogen bonding stacking
● Similar strength (E ~< 25 kcal/mol)● Balanced and reasonably accurate description of
noncovalent interactions required

Computational methods
● Hydrogenbonding interaction– electrostatic and chargetransfer origin– good description by all methods (HF, DFT, …)
● electronstacking interaction– London dispersive forces– HF and DFT approaches fail (correlation missing or
incorrect)– MP2 overestimates Eint, CCSD(T) desirable – basisset dependence (polarization and diffuse
functions, completebasisset limit)

Computational methods
● Correct prediction of the entire PES necessary,not only the interaction energy in minima
● Reason: minimization and dynamics● Example: minimization of the G–C stacked
complex using DFT (X3LYP/ccpVTZ)
● Error: no minimum on the PES for the stack – this complex is unstable and Hbonding prevails

Computational methods
● Modern QCh offers DFT– reliable except the treatment of dispersion energy – excellent timing (use of RI / density fitting)– needs adaptation to describe nonbonded interactions
1. Modification of the XC density functional2. Full DFT calculation with empirical correction for
dispersion energy (DFTD)3. Densityfunctional tightbinding with empirical
contribution of dispersion energy (SCCDFTBD)

DFTbased approaches – 1
● Modification of the XC density functional– blending of various functionals– eg. Zhao, Schultz, Truhlar
● J Chem Theor Comput 2, 364 (2006)– reliable interaction energy in the minima– the inclusion of exactexchange functional – decreased
speed– the exchange functional drives the complex formation
– incorrect distance dependence of energy for the dispersionbound complexes (r–1 or r–3 instead of r–6)

DFTbased approaches – 2
● Full DFT calculation with empirical correction to dispersion energy (DFTD)– eg. Grimme
● J Comp Chem 25, 1463 (2004)– our implementation – Jurečka, Černý, Hobza, Salahub
● J Comp Chem, in press – r–6 function damped at middletoshort distances
r (nm)
E(kcal/mol)

DFTbased approaches – 2
● Full DFT calculation with empirical correction to dispersion energy (DFTD)– our implementation – Jurečka, Černý, Hobza, Salahub
● J Comp Chem, in press – r–6 function damped at middletoshort distances– no extra computational cost– RI approximation in DFT leads to favorable timing– (2) parameters – fitting on extensive CCSD(T) data
extrapolated to the CBS limit– testing on a large & balanced class of complexes– best performance – with TPSS / TZVP– facility to calculate normalmode frequencies

DFTbased approaches – 3
● Densityfunctional tightbinding– selfconsistent charges and empirical contribution of
dispersion energy– first introduction of empirical Edisp into a DFTlike
framework – Elstner, Hobza, Frauenheim, Suhai, Kaxiras
● J Chem Phys 114, 5149 (2001)– (surprisingly) good results– extremely fast compared to other approaches– for nonbonded complexes, no failure found yet– slightly inaccurate in the description of Hbonding
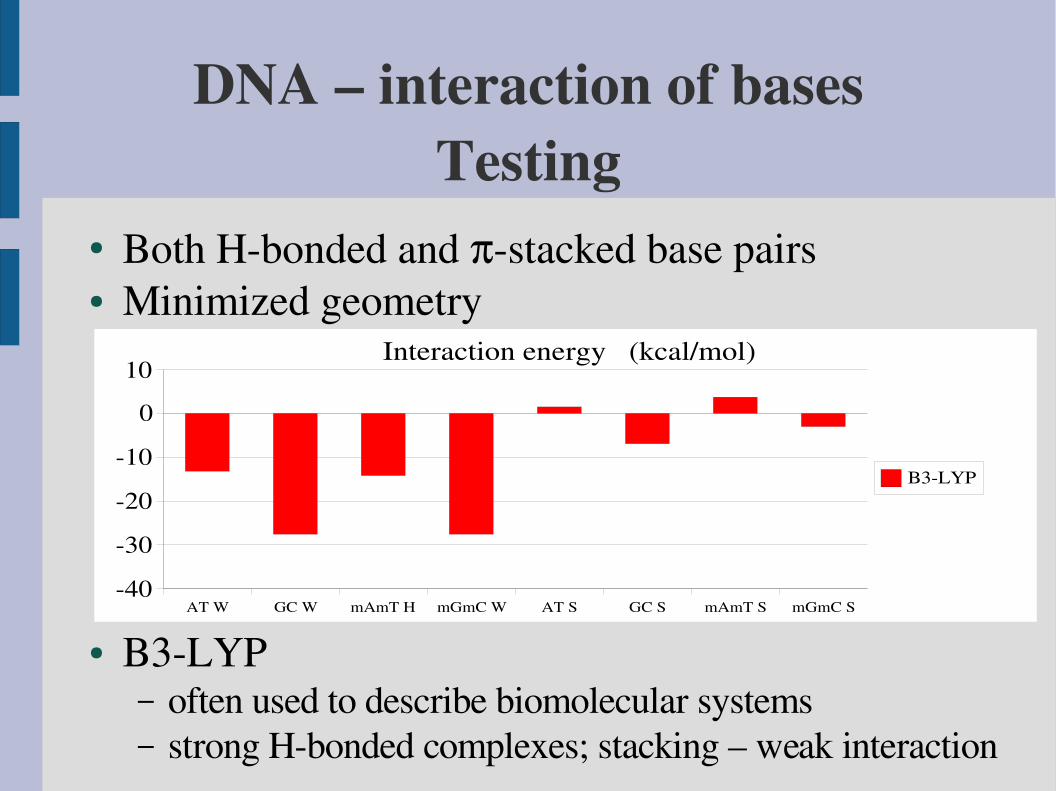
DNA – interaction of basesTesting
● Both Hbonded and stacked base pairs● Minimized geometry
AT W GC W mAmT H mGmC W AT S GC S mAmT S mGmC S40
30
20
10
0
10Interaction energy (kcal/mol)
B3LYP
● B3LYP– often used to describe biomolecular systems– strong Hbonded complexes; stacking – weak interaction

DNA – interaction of basesTesting
● Both Hbonded and stacked base pairs● Minimized geometry
AT W GC W mAmT H mGmC W AT S GC S mAmT S mGmC S40
30
20
10
0
10Interaction energy (kcal/mol)
B3LYP
CCSD(T)/CBS
● B3LYP incorrect for stacked complexes,there is strong attraction

DNA – interaction of basesTesting
● Both Hbonded and stacked base pairs● Minimized geometry
AT W GC W mAmT H mGmC W AT S GC S mAmT S mGmC S40
30
20
10
0
10Interaction energy (kcal/mol)
B3LYP
DFTDSCCDFTBD
CCSD(T)/CBS
● Generally perfect performance of D methods● Slightly weaker Hbonding by SCCDFTBD

DNA – interaction of basesApplication
● Analysis of 128 doublehelical DNA octamers● Interaction energy for 3 types of base pairs
– Hbonded, intra and interstrand stacked● Massive amount of calculation
– efficiency is crucial● Avg. total interaction energy per octamer:
● Idea of „relative importance“ of these interactions● Methods compare well
kcal/mol DFTD DFTBDHydrogen bonded −204.2 −160.8Intrastrand stacked −78.0 −86.1Interstrand stacked −21.3 −20.5
Řezáč et al., Chem Eur J, submitted

DNA – interaction of basesApplication
● Comparison of interaction energy due tointer and intrastrand stacking (DFTD, kcal/mol)
● Hypothesis: complementarity of stabilization stemming from these two interaction modes
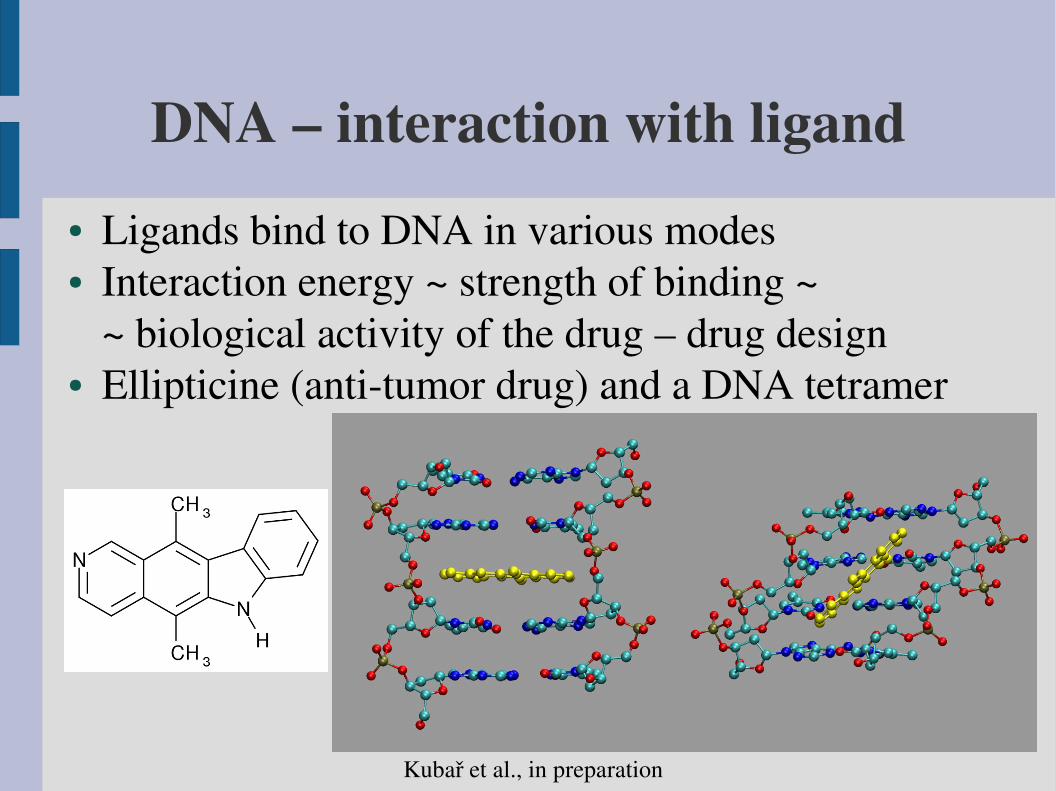
DNA – interaction with ligand● Ligands bind to DNA in various modes● Interaction energy ~ strength of binding ~
~ biological activity of the drug – drug design● Ellipticine (antitumor drug) and a DNA tetramer
Kubař et al., in preparation

DNA – interaction with a ligand
● Neutral and protonated ellipticine molecule bound to DNA
● Interaction energy: (kcal/mol)Ligand uncharged protonatedMethod DFT DFTD DFTBD DFT DFTD DFTBD
Intercalated −0.1 −198.3 −245.6 −255.4Minor groove −9.6 −204.9 −236.2 −242.7
−44.9 −48.0−39.9 −37.5
● Dispersionbound complex – DFT fails● DFTD and DFTBD: excellent agreement of results ● DFTBD much faster (1–2 min vs 11–37 hours);
slow convergence of both SCF and SCC
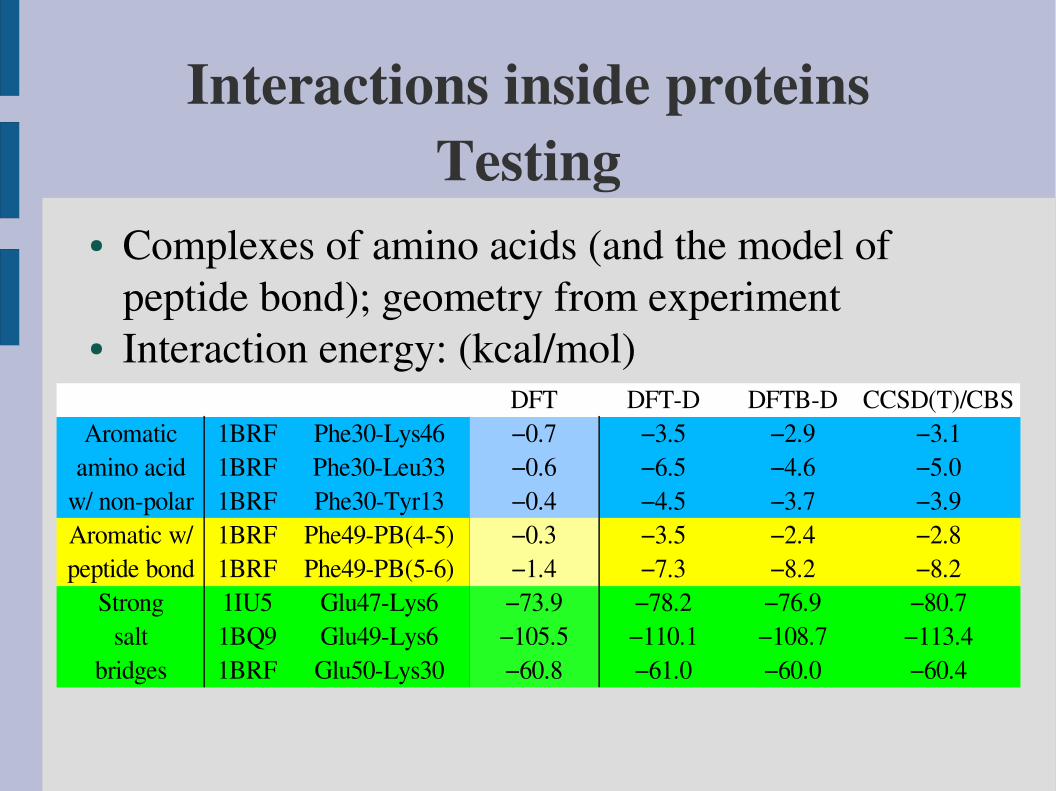
Interactions inside proteinsTesting
● Complexes of amino acids (and the model of peptide bond); geometry from experiment
● Interaction energy: (kcal/mol)DFT DFTD DFTBD CCSD(T)/CBS
Aromatic 1BRF Phe30Lys46 −0.7 −3.5 −2.9 −3.1amino acid 1BRF Phe30Leu33 −0.6 −6.5 −4.6 −5.0
w/ nonpolar 1BRF Phe30Tyr13 −0.4 −4.5 −3.7 −3.9Aromatic w/ 1BRF Phe49PB(45) −0.3 −3.5 −2.4 −2.8peptide bond 1BRF Phe49PB(56) −1.4 −7.3 −8.2 −8.2
Strong 1IU5 Glu47Lys6 −73.9 −78.2 −76.9 −80.7salt 1BQ9 Glu49Lys6 −105.5 −110.1 −108.7 −113.4
bridges 1BRF Glu50Lys30 −60.8 −61.0 −60.0 −60.4

INK4 tumor suppressor – hydrophobic core
● protein with a hydrophobic core consisting of 10 helices
● EDFTBD = −513 kcal/mol● interaction energy of every
pair of neighboring helices:
12 13 14 23 24 34 35 36 45 46 56 57 58 67 68 78 79 710 89 810 910125
100
75
50
25
0Interaction energy (kcal/mol)
DFTD
SCCDFTBD
Otyepka et al., J Phys Chem B 110, 4423 (2006)

● MD simulation of TrpGlyGly using SCCDFTBD● Goal – to find conformers coexisting in the gas phase
– to calculate IR spectra & compare with expt.● SCCDFTBD used for MD/Q● then, DFTD used for minimization (superior to MP2)
Small peptide – molecular dynamics
Valdés et al., J Phys Chem B 110, 6385 (2006)
DFT MP2DFTD

DFTD and SCCDFTBD
● Balanced and reliable description of the interactions
inside biomolecules
● No failure found yet
● Very efficient
– SCCDFTBD useful for MD simulations
● Universal
– DFTD works even for less common elements

Thanks further to …
Eva Mrázková, Dominik Horinek, Filip Ryjáček,
for control statement, Iwona Dąbkowska,
Jarda Rejnek, Jindra Fanfrlík, Kristýna Pluháčková,
Lada Bendová, Martin Kabeláč, Michal Hanus,
Míša Kolář, Petr Dobeš, Petr Sklenovský,
trinity at matrix, Vojta Klusák,
VOND (last but not least).


















