
Inhibition of motor neuron death in vitro and in vivo by a...
14
RESEARCH ARTICLE Inhibition of motor neuron death in vitro and in vivo by a p75 neurotrophin receptor intracellular domain fragment Dusan Matusica 1,2 , Fabienne Alfonsi 1 , Bradley J. Turner 3 , Tim J. Butler 1 , Stephanie R. Shepheard 4 , Mary-Louise Rogers 4 , Sune Skeldal 1 , Clare K. Underwood 1 , Marie Mangelsdorf 1 and Elizabeth J. Coulson 1, * ABSTRACT The p75 neurotrophin receptor (p75 NTR ; also known as NGFR) can mediate neuronal apoptosis in disease or following trauma, and facilitate survival through interactions with Trk receptors. Here we tested the ability of a p75 NTR -derived trophic cell-permeable peptide, c29, to inhibit p75 NTR -mediated motor neuron death. Acute c29 application to axotomized motor neuron axons decreased cell death, and systemic c29 treatment of SOD1 G93A mice, a common model of amyotrophic lateral sclerosis, resulted in increased spinal motor neuron survival mid-disease as well as delayed disease onset. Coincident with this, c29 treatment of these mice reduced the production of p75 NTR cleavage products. Although c29 treatment inhibited mature- and pro-nerve-growth-factor-induced death of cultured motor neurons, and these ligands induced the cleavage of p75 NTR in motor-neuron-like NSC-34 cells, there was no direct effect of c29 on p75 NTR cleavage. Rather, c29 promoted motor neuron survival in vitro by enhancing the activation of TrkB-dependent signaling pathways, provided that low levels of brain-derived neurotrophic factor (BDNF) were present, an effect that was replicated in vivo in SOD1 G93A mice. We conclude that the c29 peptide facilitates BDNF-dependent survival of motor neurons in vitro and in vivo. KEY WORDS: ALS, BDNF, TrkB, Motor neuron, p75NTR, NGFR, Survival INTRODUCTION The selective death of motor neurons is the central pathology of neurodegenerative diseases such as amyotrophic lateral sclerosis and spinal muscular atrophy (Murray et al., 2010). Motor neurons are also vulnerable following axonal injury, with motor neuron dysfunction resulting in paralysis (Branco et al., 2007). During development, motor neurons are dependent on growth factors such as the neurotrophins for their survival and function. In developing motor neurons, brain-derived neurotrophic factor (BDNF) and neurotrophins (NT) 3 and 4 mediate cell survival through their cognate Trk receptors, TrkB or C (Kaplan and Miller, 2000). Similarly, after nerve lesion and in animal models of motor neuron disease, BDNF and NT3 can protect and repair motor neurons (Yuen and Mobley, 1995; Wiese et al., 2004; Henriques et al., 2010). During development and following injury, the p75 neurotrophin receptor ( p75 NTR ; also known as NGFR) has been shown to mediate motor neuron death (Seeburger et al., 1993; Lowry et al., 2001; Dechant and Barde, 2002; Ibanez and Simi, 2012) in response to nerve growth factor (NGF) (Sedel et al., 1999; Ernfors, 2001) or its immature precursor (proNGF), released by activated astrocytes (Pehar et al., 2004; Domeniconi et al., 2006). However, p75 NTR is a multifunctional receptor for both mature and pro-neurotrophins, capable of regulating different biological effects in response to its ligands (Roux and Barker, 2002; Blochl and Blochl, 2007; Underwood and Coulson, 2008). In addition to its death-signaling capability, p75 NTR also acts as a co-receptor that can assist Trk receptors to bind their ligands with higher affinity and specificity, thereby enhancing their trophic functions (Hempstead et al., 1991; Barker, 2007). An important mechanism in this process appears to be the generation of biologically active receptor fragments following enzymatic cleavage of p75 NTR (Ceni et al., 2010; Kommaddi et al., 2011; Matusica et al., 2013). In a two-step process, p75 NTR is enzymatically initially cleaved by an α-secretase (ADAM17) that removes the extracellular domain of the receptor, leaving a membrane-bound C-terminal fragment. This fragment is subsequently cleaved by γ-secretase, releasing the intracellular domain into the cytoplasm (Jung et al., 2003; Kanning et al., 2003). The intracellular domain contains two regions that mediate intracellular signaling through protein–protein interactions: the juxtamembrane domain, and a tumor-necrosis-factor-receptor- like death domain (Roux and Barker, 2002; Skeldal et al., 2011). Increased metalloprotease cleavage of p75 NTR has been associated with the death of sciatic neurons following injury (DiStefano et al., 1993; Murray et al., 2003), and evidence of increased levels of p75 NTR proteolytic cleavage has been reported using urine analysis in both SOD1 transgenic mice and people living with amyotrophic lateral sclerosis (Shepheard et al., 2014). Elevated levels of the C-terminal and intracellular domain fragments have also been reported in the degenerating motor axons of SOD1 G93A transgenic mice (Perlson et al., 2009), and both p75 NTR cleavage fragments have been linked to death signaling (Skeldal et al., 2011). Although the nature of p75 NTR fragment signaling appears to be cell-type specific (Vicario et al., 2015), these studies indicate that the proteolytic cleavage of p75 NTR might be a key mechanism by which p75 NTR mediates death signaling after motor neuron injury and disease. We have previously shown that the juxtamembrane region of p75 NTR has the ability to induce cell death when bound to the cell membrane (Coulson et al., 2000; Underwood et al., 2008). We also demonstrated that a soluble peptide mimic (c29) of the juxtamembrane intracellular domain fragment of p75 NTR can act as a dominant-negative inhibitor of p75 NTR death signaling (Coulson et al., 2000). Recently, we have shown that the intracellular domain fragment of p75 NTR and the c29 peptide are able to increase NGF survival signaling through interaction with TrkA receptors (Matusica Received 4 May 2015; Accepted 22 October 2015 1 Queensland Brain Institute, The University of Queensland, Brisbane, Queensland 4072, Australia. 2 Department of Anatomy & Histology, Centre for Neuroscience, Flinders University, GPO Box 2100, Adelaide, South Australia 5001, Australia. 3 The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Parkville 3052, Victoria 3051, Australia. 4 Department of Human Physiology, Centre for Neuroscience, Flinders University, GPO Box 2100, Adelaide, South Australia 5001, Australia. *Author for correspondence ([email protected]) 517 © 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864 Journal of Cell Science
Transcript of Inhibition of motor neuron death in vitro and in vivo by a...

RESEARCH ARTICLE
Inhibition of motor neuron death in vitro and in vivo by a p75neurotrophin receptor intracellular domain fragmentDusan Matusica1,2, Fabienne Alfonsi1, Bradley J. Turner3, Tim J. Butler1, Stephanie R. Shepheard4,Mary-Louise Rogers4, Sune Skeldal1, Clare K. Underwood1, Marie Mangelsdorf1 and Elizabeth J. Coulson1,*
ABSTRACTThe p75 neurotrophin receptor (p75NTR; also known as NGFR) canmediate neuronal apoptosis in disease or following trauma, andfacilitate survival through interactions with Trk receptors. Here wetested the ability of a p75NTR-derived trophic cell-permeable peptide,c29, to inhibit p75NTR-mediated motor neuron death. Acute c29application to axotomized motor neuron axons decreased cell death,and systemic c29 treatment of SOD1G93A mice, a common model ofamyotrophic lateral sclerosis, resulted in increased spinal motorneuron survival mid-disease as well as delayed disease onset.Coincident with this, c29 treatment of these mice reduced theproduction of p75NTR cleavage products. Although c29 treatmentinhibited mature- and pro-nerve-growth-factor-induced death ofcultured motor neurons, and these ligands induced the cleavage ofp75NTR in motor-neuron-like NSC-34 cells, there was no direct effectof c29 on p75NTR cleavage. Rather, c29 promoted motor neuronsurvival in vitro by enhancing the activation of TrkB-dependentsignaling pathways, provided that low levels of brain-derivedneurotrophic factor (BDNF) were present, an effect that wasreplicated in vivo in SOD1G93A mice. We conclude that the c29peptide facilitates BDNF-dependent survival of motor neurons in vitroand in vivo.
KEY WORDS: ALS, BDNF, TrkB, Motor neuron, p75NTR, NGFR,Survival
INTRODUCTIONThe selective death of motor neurons is the central pathology ofneurodegenerative diseases such as amyotrophic lateral sclerosisand spinal muscular atrophy (Murray et al., 2010).Motor neurons arealso vulnerable following axonal injury, with motor neurondysfunction resulting in paralysis (Branco et al., 2007). Duringdevelopment, motor neurons are dependent on growth factors such asthe neurotrophins for their survival and function. In developingmotor neurons, brain-derived neurotrophic factor (BDNF) andneurotrophins (NT) 3 and 4 mediate cell survival through theircognate Trk receptors, TrkB or C (Kaplan and Miller, 2000).Similarly, after nerve lesion and in animal models of motor neurondisease, BDNF and NT3 can protect and repair motor neurons (Yuenand Mobley, 1995; Wiese et al., 2004; Henriques et al., 2010).
During development and following injury, the p75 neurotrophinreceptor (p75NTR; also known as NGFR) has been shown to mediatemotor neuron death (Seeburger et al., 1993; Lowry et al., 2001;Dechant and Barde, 2002; Ibanez and Simi, 2012) in response tonerve growth factor (NGF) (Sedel et al., 1999; Ernfors, 2001) or itsimmature precursor (proNGF), released by activated astrocytes(Pehar et al., 2004; Domeniconi et al., 2006). However, p75NTR is amultifunctional receptor for both mature and pro-neurotrophins,capable of regulating different biological effects in response to itsligands (Roux and Barker, 2002; Blochl and Blochl, 2007;Underwood and Coulson, 2008). In addition to its death-signalingcapability, p75NTR also acts as a co-receptor that can assist Trkreceptors to bind their ligands with higher affinity and specificity,thereby enhancing their trophic functions (Hempstead et al., 1991;Barker, 2007). An important mechanism in this process appears to bethe generation of biologically active receptor fragments followingenzymatic cleavage of p75NTR (Ceni et al., 2010; Kommaddi et al.,2011; Matusica et al., 2013).
In a two-step process, p75NTR is enzymatically initially cleaved byan α-secretase (ADAM17) that removes the extracellular domain ofthe receptor, leaving a membrane-bound C-terminal fragment. Thisfragment is subsequently cleaved by γ-secretase, releasing theintracellular domain into the cytoplasm (Jung et al., 2003; Kanninget al., 2003). The intracellular domain contains two regions thatmediate intracellular signaling through protein–protein interactions:the juxtamembrane domain, and a tumor-necrosis-factor-receptor-like death domain (Roux and Barker, 2002; Skeldal et al., 2011).Increased metalloprotease cleavage of p75NTR has been associatedwith the death of sciatic neurons following injury (DiStefano et al.,1993; Murray et al., 2003), and evidence of increased levels ofp75NTR proteolytic cleavage has been reported using urine analysis inboth SOD1 transgenic mice and people living with amyotrophiclateral sclerosis (Shepheard et al., 2014). Elevated levels of theC-terminal and intracellular domain fragments have also beenreported in the degenerating motor axons of SOD1G93A transgenicmice (Perlson et al., 2009), and both p75NTRcleavage fragments havebeen linked to death signaling (Skeldal et al., 2011). Although thenature of p75NTR fragment signaling appears to be cell-type specific(Vicario et al., 2015), these studies indicate that the proteolyticcleavage of p75NTR might be a key mechanism by which p75NTR
mediates death signaling after motor neuron injury and disease.We have previously shown that the juxtamembrane region of
p75NTR has the ability to induce cell death when bound to thecell membrane (Coulson et al., 2000; Underwood et al., 2008). Wealso demonstrated that a soluble peptide mimic (c29) of thejuxtamembrane intracellular domain fragment of p75NTR can act asa dominant-negative inhibitor of p75NTR death signaling (Coulsonet al., 2000). Recently, we have shown that the intracellular domainfragment of p75NTR and the c29 peptide are able to increase NGFsurvival signaling through interaction with TrkA receptors (MatusicaReceived 4 May 2015; Accepted 22 October 2015
1Queensland Brain Institute, The University of Queensland, Brisbane, Queensland4072, Australia. 2Department of Anatomy & Histology, Centre for Neuroscience,Flinders University, GPO Box 2100, Adelaide, South Australia 5001, Australia.3The Florey Institute of Neuroscience and Mental Health, University of Melbourne,Parkville 3052, Victoria 3051, Australia. 4Department of Human Physiology, Centrefor Neuroscience, Flinders University, GPO Box 2100, Adelaide, South Australia5001, Australia.
*Author for correspondence ([email protected])
517
© 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

et al., 2013). This in turn leads to increased neurite outgrowth at lowNGF concentrations, and enhanced survival of sympathetic neuronsundergoing growth factor withdrawal (Matusica et al., 2013). Herewe investigated whether c29 plays a similar role in TrkB-dependentcells, determining the role of p75NTR cleavage in neurotrophin-induced motor neuron death and exploring whether c29 can inhibitthis process in vitro and in vivo.
RESULTSc29 prevents the death of axotomized ulnar motor neuronsand delays disease progression of SOD1G93A-induced motorneuron diseaseIt is clear that a balance between Trk-receptor-mediated survival andp75NTR-mediated cell death signaling pathways regulates motorneuron survival both during development and following injury ordisease. In particular, the role of these competing neurotrophinsignaling pathways has been well studied in motor neuronsfollowing axotomy and in degenerative conditions includingamyotrophic lateral sclerosis. Therefore, to determine if c29 iscapable of inhibiting motor neuron cell death in vivo, we first usedthe injury paradigm of ulnar nerve axotomy model in which 50% ofthe ipsilateral motor neurons typically die, and where bothdownregulation of p75NTR by antisense oligonucleotides (Lowryet al., 2001) and BDNF application (Yuan et al., 2000) have beenshown to promote motor neuron survival. Following ulnar nerveaxotomy of newborn rat pups, c29 or protein transduction domain(PTD) vehicle-fused control peptide was applied to the cut nervestump in gel foam. After five days, the number of live motor neuronswas determined by stereological counts following histologicalprocessing of the spinal cords. In the axotomized animals treatedwith control peptide, axotomy resulted in a loss of ∼45% of cholineacetyl transferase (ChAT)-positive motor neurons in the ipsilateralcervical enlargement (C7–C8) compared with the contralateral side(Fig. 1). By contrast, the number of surviving motor neurons in thespinal cord of animals treated with c29 was significantly higher,with the average neuronal loss being less than 15% that of thecontralateral side (Fig. 1).As PTD-fused peptides and proteins given to mice systemically
have previously been shown to cross the blood–brain barrier(Schwarze et al., 2000), we confirmed that biotinylated-c29 peptidereached the spinal neurons after intraperitoneal injection of wild-typemice. In mice sacrificed one hour after 5 mg/kg peptide injection,immunostaining for biotin was observed in grey matter of the spinal
cord in c29-treated animals but not in controls (non-biotinylated-peptide-treated animals) (Fig. 2A,B). Next, we tested the effect ofsystemic c29 treatment in the SOD1G93A mouse model ofamyotrophic lateral sclerosis. c29 was administered chronically(5 mg/kg/day) to the mice from 9 weeks of age by means of asubcutaneous osmotic minipump, and the course of disease wasmonitored. No significant difference between c29-treated mice andvehicle-treated animals was observed in terms of lifespan (Fig. 2C).However, c29-treated mice had a delayed disease onset, takingsignificantly longer to lose 10% of their body weight (P<0.01;median SOD1+vehicle: 31 days; SOD1+c29: 42 days; Fig. 2D) andc29-treated animals maintained their performance on the rotarodbetween 100 and 125 days, the time frame during which theperformance of the vehicle-treated animals declined dramatically(Fig. 2E). Furthermore, at this time, the number of motor neurons inthe spinal cord of c29-treated SOD1G93A animals was significantlyhigher than in vehicle-treated animals (Fig. 2F,G), and fewer cellsappeared atrophic (Fig. 2G).
p75NTR cleavage associates with neurotrophin-induced celldeath in vivo and in vitroAs upregulation of p75NTR expression, proteolysis and deathsignaling are associated with motor neuron degeneration, weasked whether the reduced neuronal loss was associated withaltered p75NTR cleavage and/or expression by measuring the levelsof p75NTR extracellular domain (ECD) present in the urine ofSOD1G93A animals over the course of the disease. In SOD1G93A
animals, the level of p75NTR ECD present in urine was significantlyhigher than in C57BL/6J (wild-type) healthy controlspresymptomatically from 60 days of age and throughout diseaseprogression (Fig. 3A). However, in SOD1G93A animals treated withc29, the level of p75NTR ECD present in the urine between 80 and120 days of age was significantly lower than that found in the urineof vehicle-treated animals (Fig. 3B). Together, these results indicatethat c29 can potentiate the survival of injured or dying motorneurons in vivo, which corresponds to lowered p75NTR expressionand/or cleavage.
To determine whether cleavage of p75NTR is important forneurotrophin-mediated motor neuron death, we asked whetherneurotrophins stimulate the cleavage of p75NTR using the motor-neuron-like NSC-34 cell line. As previously reported for othercell types, treatment of cultures with PMA, which activatesmetalloproteases (Ni et al., 2001), stimulated the cleavage of
Fig. 1. c29 prevents the death of motor neurons after axotomy. (A) Percentage cervical motor neuron number, relative to contralateral motor neuron number,five days after unilateral axotomy of the ulnar nerve of newborn rats. c29 but not vehicle (PTD) peptides resulted in significant protection from axotomy-inducedmotor neuron loss. N=6 animals per group, mean±s.d.; ***P<0.001; two-tailed t-test. (B) Representative micrographs showing Nissl-stained cervical motorneurons within in the ventral horn of rat pups following unilateral ulnar nerve axotomy and treatment with control peptide (PTD) or c29 as indicated. Scale bars:100 µm.
518
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

p75NTR, resulting predominantly in the generation of its intracellulardomain fragment. This cleavage could be prevented with themetalloprotease inhibitor TAPI-2, whereas treatment of cells withthe γ-secretase inhibitor Compound E together with PMA resultedin accumulation of the C-terminal p75NTR fragment (Fig. 3C) (Junget al., 2003; Kanning et al., 2003; Zampieri et al., 2005). Treatmentof NSC-34 cells with BDNF or proBDNF produced predominantlyintracellular domain fragments of p75NTR whereas either NGF orproNGF treatment predominantly resulted in generation of theC-terminal fragment (Fig. 3C).The ability of NGF and the proneurotrophins to induce
p75NTR-mediated cell death in the presence of low concentrationsof the survival factor BDNF was then tested. Treatment of
E13 TrkB-dependent motor neuron cultures with NGF (Fig. 4A,D) or proNGF (Fig. 4C), resulted in significant neuronal deathover 48 h, whereas proBDNF treatment had no effect (Fig. 4C).NGF-induced death was prevented by treatment with the p75NTR
cleavage inhibitor TAPI-2 (Fig. 4A). Taken together, theseresults suggest that cleavage of p75NTR resulting in accumulationof the C-terminal fragment is associated with death signaling inmotor neurons.
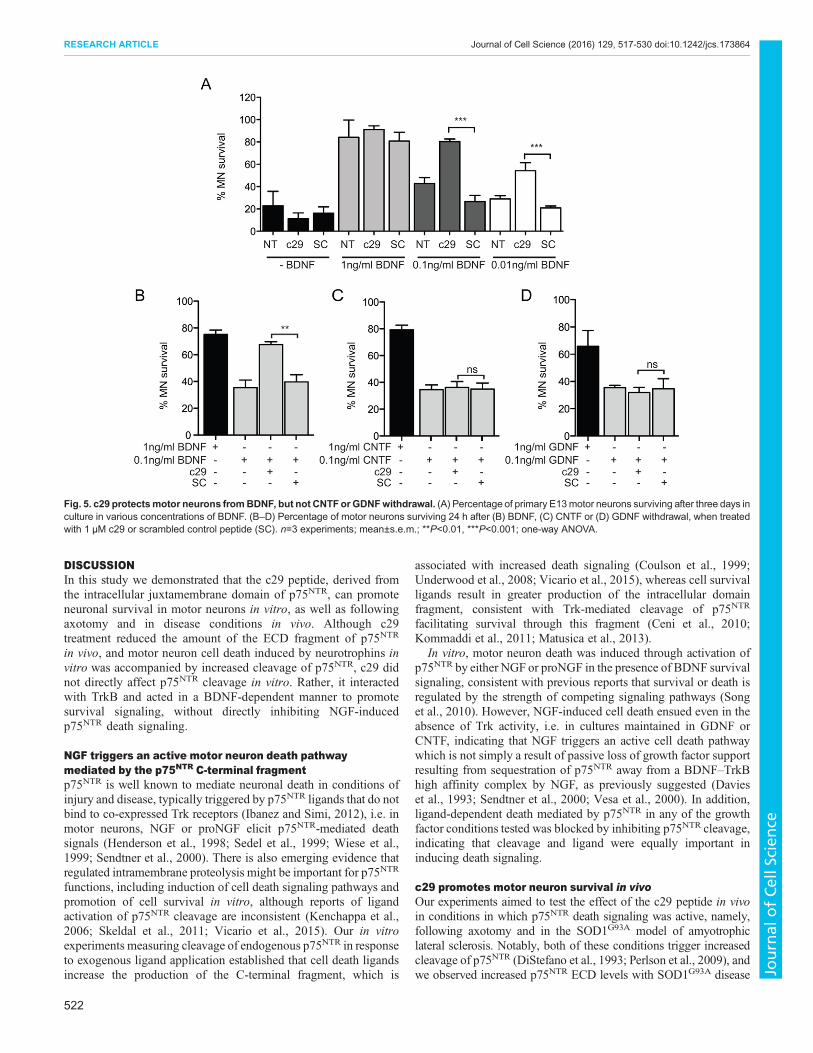
c29 promotes survival of BDNF-maintained neuronsWe next asked whether c29 inhibits the (pro)NGF-induced death ofmotor neurons. Co-treatment of motor neuron cultures with c29 atthe time of proNGF or NGF addition resulted in significantly
Fig. 2. c29 prevents the death of motor neurons in SOD1G93Amice. (A,B) Immunostaining for biotin in spinal cords of animals 1 h after intraperitoneal injectionof 5 mg/kg non-biotinylated scrambled peptide (A) or biotinylated c29 peptide (B), representative ofN=3 animals per treatment. Scale bars: 250 µm. (C–E) Rate ofsurvival (C), average body weight (D), and rotarod performance (E) of SOD1G93A mice treated by means of subcutaneous pumps with either vehicle (black) or∼5 mg/kg/day c29 (red) from ∼63 days of age until death. N=8 animals per treatment group, mean±s.e.m.; *P<0.05, one-way ANOVA with Sidak’s multiplecomparison post-hoc test. (F) Average number of motor neuron cell bodies per coronal section of the lumbar spinal cord of 116 day-old mice treated with c29 orvehicle. Mean±s.e.m., N=4 animals per group, **P<0.01; two-tailed t-test. (G) Representative images of spinal motor neurons in 112-week-old SOD1G93A micetreated with c29 or vehicle. Black arrows, atrophic neurons; red arrows, non-atrophic neurons. Scale bars: 50 µm.
519
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience
reduced cell death of cultures grown in BDNF (Fig. 4B–D). Thescrambled control peptide had no significant effect on either celldeath or survival, and c29 had no effect on proBDNF-treated motorneurons (Fig. 4C).To determine whether c29 was acting by affecting p75NTR
cleavage, NSC-34 cells were treated with NGF or proNGF togetherwith c29 or scrambled control peptide. No difference in the level ofp75NTR cleavage was observed in cells treated with the peptides(Fig. 4E), indicating that c29 was more likely to be affecting thebalance of survival signaling downstream of p75NTR activation.To determine whether c29 was acting to prevent p75NTR-
mediated death signaling or promote TrkB-mediated signaling,neurons cultured in ciliary neurotrophic factor (CNTF) or glial cellline-derived neurotrophic factor (GDNF) instead of BDNF weretreated with NGF, which also resulted in a 40–50% reduction insurvival, similar to that induced in BDNF conditions. The cleavageinhibitor TAPI-2 significantly inhibited NGF-induced cell death inCNTF- and GDNF-supported cultures (Fig. 4F,G). However, c29did not inhibit the NGF-induced death of CNTF- or GDNF-dependent neurons (Fig. 4F,G). These results demonstrate thatNGF-induced death signaling relies on a cleaved form of p75NTR,that its death signaling is independent of TrkB signaling, and,finally, that c29 does not appear to act in a dominant-negativemanner in vitro to inhibit p75NTR signaling, but instead acts bypromoting neuronal survival only in the presence of BDNF.
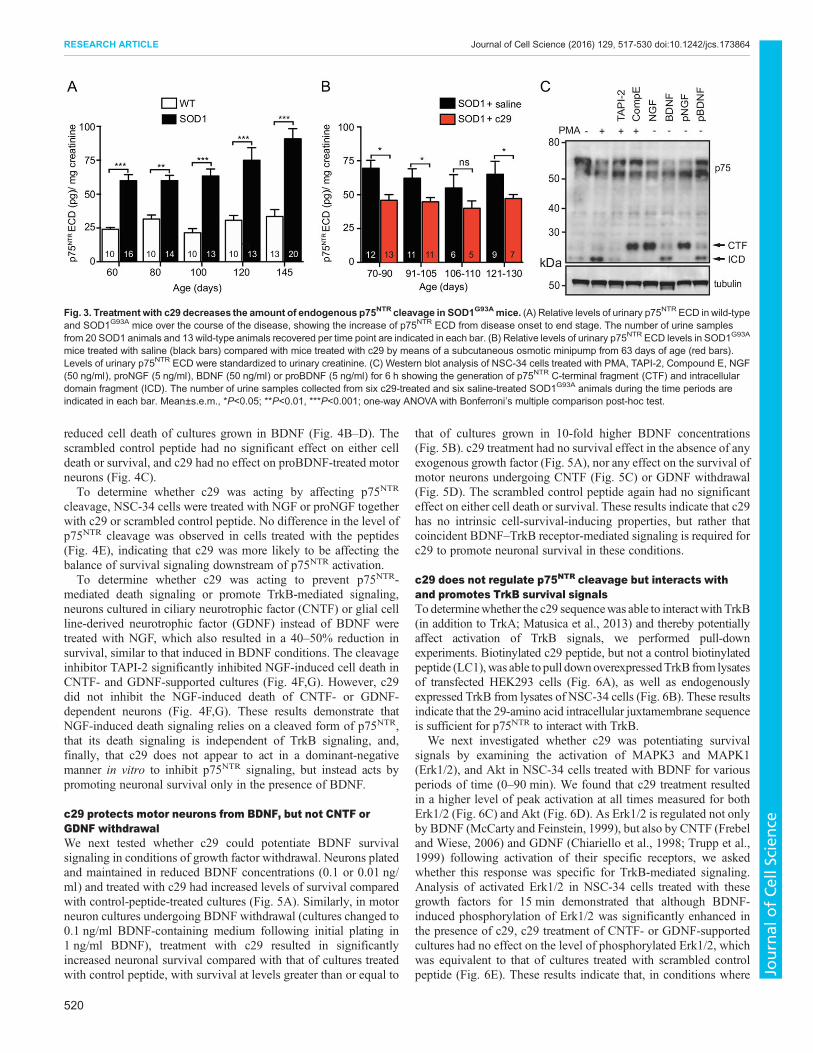
c29 protects motor neurons from BDNF, but not CNTF orGDNF withdrawalWe next tested whether c29 could potentiate BDNF survivalsignaling in conditions of growth factor withdrawal. Neurons platedand maintained in reduced BDNF concentrations (0.1 or 0.01 ng/ml) and treated with c29 had increased levels of survival comparedwith control-peptide-treated cultures (Fig. 5A). Similarly, in motorneuron cultures undergoing BDNF withdrawal (cultures changed to0.1 ng/ml BDNF-containing medium following initial plating in1 ng/ml BDNF), treatment with c29 resulted in significantlyincreased neuronal survival compared with that of cultures treatedwith control peptide, with survival at levels greater than or equal to
that of cultures grown in 10-fold higher BDNF concentrations(Fig. 5B). c29 treatment had no survival effect in the absence of anyexogenous growth factor (Fig. 5A), nor any effect on the survival ofmotor neurons undergoing CNTF (Fig. 5C) or GDNF withdrawal(Fig. 5D). The scrambled control peptide again had no significanteffect on either cell death or survival. These results indicate that c29has no intrinsic cell-survival-inducing properties, but rather thatcoincident BDNF–TrkB receptor-mediated signaling is required forc29 to promote neuronal survival in these conditions.
c29 does not regulate p75NTR cleavage but interacts withand promotes TrkB survival signalsTo determinewhether the c29 sequencewas able to interactwith TrkB(in addition to TrkA; Matusica et al., 2013) and thereby potentiallyaffect activation of TrkB signals, we performed pull-downexperiments. Biotinylated c29 peptide, but not a control biotinylatedpeptide (LC1),was able to pull downoverexpressedTrkB from lysatesof transfected HEK293 cells (Fig. 6A), as well as endogenouslyexpressed TrkB from lysates of NSC-34 cells (Fig. 6B). These resultsindicate that the 29-amino acid intracellular juxtamembrane sequenceis sufficient for p75NTR to interact with TrkB.
We next investigated whether c29 was potentiating survivalsignals by examining the activation of MAPK3 and MAPK1(Erk1/2), and Akt in NSC-34 cells treated with BDNF for variousperiods of time (0–90 min). We found that c29 treatment resultedin a higher level of peak activation at all times measured for bothErk1/2 (Fig. 6C) and Akt (Fig. 6D). As Erk1/2 is regulated not onlyby BDNF (McCarty and Feinstein, 1999), but also by CNTF (Frebeland Wiese, 2006) and GDNF (Chiariello et al., 1998; Trupp et al.,1999) following activation of their specific receptors, we askedwhether this response was specific for TrkB-mediated signaling.Analysis of activated Erk1/2 in NSC-34 cells treated with thesegrowth factors for 15 min demonstrated that although BDNF-induced phosphorylation of Erk1/2 was significantly enhanced inthe presence of c29, c29 treatment of CNTF- or GDNF-supportedcultures had no effect on the level of phosphorylated Erk1/2, whichwas equivalent to that of cultures treated with scrambled controlpeptide (Fig. 6E). These results indicate that, in conditions where
Fig. 3. Treatment with c29 decreases the amount of endogenous p75NTR cleavage in SOD1G93Amice. (A) Relative levels of urinary p75NTR ECD in wild-typeand SOD1G93A mice over the course of the disease, showing the increase of p75NTR ECD from disease onset to end stage. The number of urine samplesfrom 20 SOD1 animals and 13 wild-type animals recovered per time point are indicated in each bar. (B) Relative levels of urinary p75NTR ECD levels in SOD1G93A
mice treated with saline (black bars) compared with mice treated with c29 by means of a subcutaneous osmotic minipump from 63 days of age (red bars).Levels of urinary p75NTR ECD were standardized to urinary creatinine. (C) Western blot analysis of NSC-34 cells treated with PMA, TAPI-2, Compound E, NGF(50 ng/ml), proNGF (5 ng/ml), BDNF (50 ng/ml) or proBDNF (5 ng/ml) for 6 h showing the generation of p75NTR C-terminal fragment (CTF) and intracellulardomain fragment (ICD). The number of urine samples collected from six c29-treated and six saline-treated SOD1G93A animals during the time periods areindicated in each bar. Mean±s.e.m., *P<0.05; **P<0.01, ***P<0.001; one-way ANOVA with Bonferroni’s multiple comparison post-hoc test.
520
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

growth factors are limited, c29 enhances survival signaling and thusneuronal survival in a BDNF–TrkB-dependent manner.Finally, to establish whether c29 could activate BDNF-mediated
survival signaling within the spinal cords of SOD1G93A mice,16-week-old SOD1G93A animals were treated acutely with c29 orcontrol peptide. SOD1G93A mice treated with the control peptide hadvery low levels of phosphorylated TrkB (Fig. 7A), Erk1/2 and Akt(Fig. 7B), and CREB (Fig. 7C), in comparison to the levels in lysates
from wild-type animals. However, the activity of these survival-signaling proteins was significantly increased by acute c29 treatmentin comparison to that in control peptide-treated animals (Fig. 7A–C).Conversely, levels of cleaved (activated) caspase 3 in SOD1G93A
spinal cord lysates were reduced by c29 treatment compared with thecontrol treatment (Fig. 7D). Together, these findings indicate that c29can enhance neuronal survival in a BDNF–TrkB-dependent mannerin vitro and in diseased motor neurons in vivo.
Fig. 4. c29 promotes survival of BDNF-maintained neurons. (A,B) Percentage of primary E13 motor neurons surviving after three days in culture in 1 ng/mlBDNF and 50 ng/ml NGF, with or without the addition of TAPI-2 (A) or c29 or scrambled (SC) control peptides (B). (C) Percentage of motor neurons surviving afterthree days in culture in BDNF and proNGFor proBDNF, with or without the addition of c29 or scrambled peptides. (D) DIC micrographs of motor neurons culturedfor three days in the presence of 1 ng/ml BDNF or 1 ng/ml BDNF and 50 ng/ml NGF with or without c29 treatment. Scale bar: 50 µm. (E) Representativewestern blot of NSC-34 cell lysates 6 h after treatment with PMA and TAPI-2, Compound E, NGF or proNGF, with or without c29 or scrambled peptide (SC).(F,G) Percentage of primary E13 motor neurons surviving after three days in culture in CNTF (F) or GDNF (G) and NGF with or without co-treatment with TAPI-2,c29 or scrambled peptide. n=3 experiments per condition; mean±s.e.m.; **P<0.01, ***P<0.001; one-way ANOVA.
521
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience
DISCUSSIONIn this study we demonstrated that the c29 peptide, derived fromthe intracellular juxtamembrane domain of p75NTR, can promoteneuronal survival in motor neurons in vitro, as well as followingaxotomy and in disease conditions in vivo. Although c29treatment reduced the amount of the ECD fragment of p75NTR
in vivo, and motor neuron cell death induced by neurotrophins invitro was accompanied by increased cleavage of p75NTR, c29 didnot directly affect p75NTR cleavage in vitro. Rather, it interactedwith TrkB and acted in a BDNF-dependent manner to promotesurvival signaling, without directly inhibiting NGF-inducedp75NTR death signaling.
NGF triggers an active motor neuron death pathwaymediated by the p75NTR C-terminal fragmentp75NTR is well known to mediate neuronal death in conditions ofinjury and disease, typically triggered by p75NTR ligands that do notbind to co-expressed Trk receptors (Ibanez and Simi, 2012), i.e. inmotor neurons, NGF or proNGF elicit p75NTR-mediated deathsignals (Henderson et al., 1998; Sedel et al., 1999; Wiese et al.,1999; Sendtner et al., 2000). There is also emerging evidence thatregulated intramembrane proteolysis might be important for p75NTR
functions, including induction of cell death signaling pathways andpromotion of cell survival in vitro, although reports of ligandactivation of p75NTR cleavage are inconsistent (Kenchappa et al.,2006; Skeldal et al., 2011; Vicario et al., 2015). Our in vitroexperiments measuring cleavage of endogenous p75NTR in responseto exogenous ligand application established that cell death ligandsincrease the production of the C-terminal fragment, which is
associated with increased death signaling (Coulson et al., 1999;Underwood et al., 2008; Vicario et al., 2015), whereas cell survivalligands result in greater production of the intracellular domainfragment, consistent with Trk-mediated cleavage of p75NTR
facilitating survival through this fragment (Ceni et al., 2010;Kommaddi et al., 2011; Matusica et al., 2013).
In vitro, motor neuron death was induced through activation ofp75NTR by either NGF or proNGF in the presence of BDNF survivalsignaling, consistent with previous reports that survival or death isregulated by the strength of competing signaling pathways (Songet al., 2010). However, NGF-induced cell death ensued even in theabsence of Trk activity, i.e. in cultures maintained in GDNF orCNTF, indicating that NGF triggers an active cell death pathwaywhich is not simply a result of passive loss of growth factor supportresulting from sequestration of p75NTR away from a BDNF–TrkBhigh affinity complex by NGF, as previously suggested (Davieset al., 1993; Sendtner et al., 2000; Vesa et al., 2000). In addition,ligand-dependent death mediated by p75NTR in any of the growthfactor conditions tested was blocked by inhibiting p75NTR cleavage,indicating that cleavage and ligand were equally important ininducing death signaling.
c29 promotes motor neuron survival in vivoOur experiments aimed to test the effect of the c29 peptide in vivoin conditions in which p75NTR death signaling was active, namely,following axotomy and in the SOD1G93A model of amyotrophiclateral sclerosis. Notably, both of these conditions trigger increasedcleavage of p75NTR (DiStefano et al., 1993; Perlson et al., 2009), andwe observed increased p75NTR ECD levels with SOD1G93A disease
Fig. 5. c29 protectsmotor neurons fromBDNF, but not CNTFor GDNFwithdrawal. (A) Percentage of primary E13motor neurons surviving after three days inculture in various concentrations of BDNF. (B–D) Percentage of motor neurons surviving 24 h after (B) BDNF, (C) CNTF or (D) GDNF withdrawal, when treatedwith 1 µM c29 or scrambled control peptide (SC). n=3 experiments; mean±s.e.m.; **P<0.01, ***P<0.001; one-way ANOVA.
522
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

progression, as previously observed in human amyotrophic lateralsclerosis (Shepheard et al., 2014). As a result of c29 treatment, weobserved a significant improvement in motor neuron survival afteraxotomy and in SOD1G93A animals, and in the latter model, reducedlevels of p75NTR ECD, and a coincident improvement in rotarodperformance and a delay in disease-induced weight loss, suggestingthat the functional outcomes could be the result of reduced p75NTR
activation and cell death signaling. However, the observed reductionin the level of p75NTR ECD might simply reflect a slower diseaseprogression. Although p75NTR expression and the generation of thecleavage fragment are upregulated in motor neurons coincident withaxonal degeneration, p75NTR is also upregulated in oligodendrocytes,Schwann cells and microglia following nerve injury (reviewed inMeeker and Williams, 2014). Therefore, it is not clear from ourexperiments whether the p75NTR ECD is derived frommotor neuronsand/or other cell types and whether it reflects basal levels of p75NTR
cleavage accompanying broader expression, or increased cleavagespecifically reflecting activation of p75NTR degenerative processes.The reduction in the level of p75NTR ECD afforded by c29 treatmentis therefore difficult to interpret in isolation.Although we cannot rule out the possibility that c29 acted to
prevent p75NTR death signaling in vivo, our in vitro experimentsstrongly indicate that it did not directly affect the level of p75NTR
cleavage or block p75NTR death signaling. Firstly, no effect on thelevel of NGF- or proNGF-induced cleavage of p75NTR wasobserved following treatment of cultures with c29, nor did c29treatment affect the level of NGF-induced death of neurons culturedin the absence of BDNF, even though p75NTR cleavage inhibitorspromoted neuronal survival in the same conditions. Rather, our datasuggest that c29 treatment, by keeping motor neurons alive orhealthier for longer (as discussed below), indirectly affected otherdisease mechanisms, including reducing the upregulation and/orcleavage of p75NTR. Therefore, the specific process being measuredby quantifying p75NTR ECD levels remains unclear. Nonetheless, asp75NTR ECD levels increased with disease progression and c29treatment resulted in reduced steady-state levels of p75NTR ECD inmid disease, we suggest that further validation of this fragment as abiomarker of motor neuron disease severity is warranted.
c29 promotes motor neuron survival in vitro but requirescoincident TrkB activationAlthough we hypothesized that c29 might act to block p75NTR deathsignaling, our results consistently demonstrated that c29 onlypromotes motor neuron survival when coincident with TrkBactivation in vitro. Specifically: (i) c29 overcame NGF-induceddeath signaling in neurons cultured in BDNF but not in neuronscultured in CNTF; (ii) in low concentrations of BDNF that wouldnot otherwise support motor neuron survival, co-treatment ofcultures with c29 promoted levels of survival at or above that ofcultures in 10-fold higher BDNF concentrations, but had no effecton cultures grown in low concentrations of CNTF or GDNF; and(iii) c29 treatment facilitated both increased activation of andlonger-lived survival signaling in motor-neuron-like NSC-34 cells,but only in cultures grown in the presence of BDNF. Thisconclusion is consistent with our in vivo findings.In the axotomy paradigm, an exogenous supply of BDNF is
reported to promote neuronal survival (Sendtner et al., 1992; Boydand Gordon, 2001; Murray et al., 2010). Thus, given that at least lowendogenous levels of neurotrophins are present at the axotomizedcell bodies, c29 could have acted by enhancing the survivalsignaling of locally produced BDNF. Similarly, previous work hasindicated that BDNF is upregulated in neurons, glia and immune
cells within the spinal cord following injury (Li et al., 2006, 2009;Xin et al., 2012), which might allow c29 to act in motor neurons.Indeed, acute treatment of SOD1G93A animals with c29 resulted inenhanced phosphorylation of TrkB, Akt, Erk1/2 and CREB withinthe spinal cord coincident with reduced levels of cleaved caspase 3,which was not induced by control peptide treatment. Our currentresults therefore provide strong evidence to support the idea that c29enhances BDNF-induced TrkB survival signaling both in vitro andin vivo, which in turn blocks death signals.
Role of the p75NTR juxtamembrane (c29) region in TrksignalingOne characterized role of p75NTR is its ability to increase ligand-binding affinity for the TrkA receptor and increase neuronal survival(Hempstead et al., 1991; Lee et al., 1994;Horton et al., 1997; Espositoet al., 2001), and it is widely accepted that p75NTR also interacts withother Trk receptors with equivalent effect (Hempstead et al., 1991;Davies et al., 1993; Huber and Chao, 1995; Bibel et al., 1999; Vesaet al., 2000; Ceni et al., 2010; Kommaddi et al., 2011). Although theprecise mechanism responsible for this outcome remains elusive(Barker, 2007), the transmembrane and juxtamembrane regions ofp75NTR have been implicated in this interaction (Hempstead et al.,1991; Bibel et al., 1999; Esposito et al., 2001; Iacaruso et al., 2011),and more recent studies also suggest that cleavage of p75NTR plays animportant role in this process (Ceni et al., 2010; Kommaddi et al.,2011; Matusica et al., 2013). Our observation that a soluble peptidemimicking the juxtamembrane region of p75NTR is able to enhancethe function of TrkA (Matusica et al., 2013) andTrkB (herein) accordswith the previous studies. In particular, the high conservation of thejuxtamembrane region of p75NTR supports the idea that this domain isessential for p75NTR function (Hutson and Bothwell, 2001). We havepreviously shown its importance in p75NTR-mediated death signalingwhen attached to the membrane as well as demonstrating that thefunction of this domain differs following intramembranous cleavage(Coulson et al., 2000;Matusica et al., 2013), i.e. cleavage of p75NTR toits intracellular domain fragment is a requirement for enhancing NGFaffinity for TrkA in vitro, and this can be reproduced using the c29peptide (Matusica et al., 2013).
Proposed mechanism of c29 actionAs the current findings using c29 in vitro mirror our recent work inTrkA-dependent neurons (Matusica et al., 2013), we propose thatc29 interacts with all Trk receptors to facilitate ligand binding.Increased numbers of ligand-bound TrkB receptors per cell allowactivation of significantly more receptors than are normallyactivated in equivalent concentrations of neurotrophins (unless theconcentrations were already saturating). Thus, the threshold forBDNF survival and trophic signaling per neuron would be reachedsooner and/or be higher in the presence of c29. In the context of ourexperiments, the resulting increased TrkB survival signaling wassufficient to promote survival, including overriding coincidentp75NTR-mediated death signals. Although recent work hassuggested that TrkB expression causes a more severe SOD1disease phenotype (Zhai et al., 2011), it appears that this deleteriouseffect is mediated by the truncated TrkB isoform T1 (Yanpallewaret al., 2012). TrkB.T1 lacks the intracellular kinase domain and,unlike full-length TrkB, is upregulated in motor neurons in SOD1animals (Zhang and Huang, 2006). Thus, the effect of promotingmotor neuron survival in SOD1 animals by c29 can be explained byselective enhancement of full-length TrkB trophic signaling, whichcan counteract p75NTR-induced death (Casaccia-Bonnefil et al.,1996; Yoon et al., 1998; Friedman, 2000).
523
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience
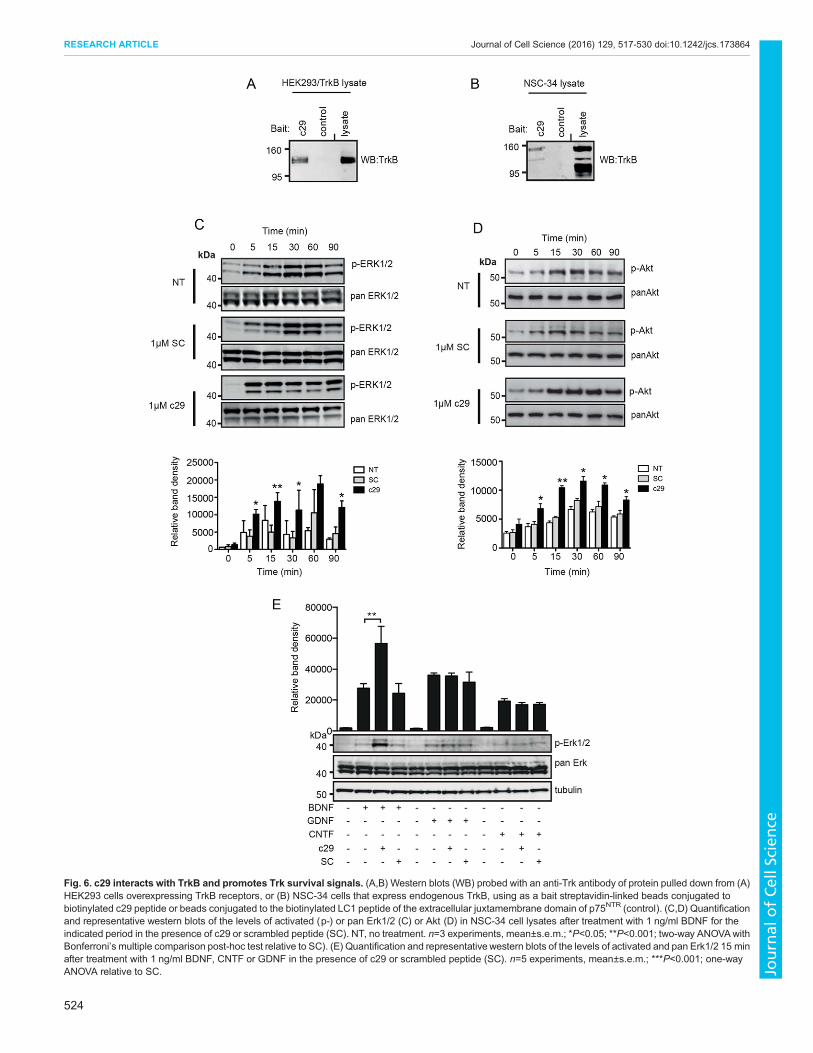
Fig. 6. c29 interacts with TrkB and promotes Trk survival signals. (A,B) Western blots (WB) probed with an anti-Trk antibody of protein pulled down from (A)HEK293 cells overexpressing TrkB receptors, or (B) NSC-34 cells that express endogenous TrkB, using as a bait streptavidin-linked beads conjugated tobiotinylated c29 peptide or beads conjugated to the biotinylated LC1 peptide of the extracellular juxtamembrane domain of p75NTR (control). (C,D) Quantificationand representative western blots of the levels of activated (p-) or pan Erk1/2 (C) or Akt (D) in NSC-34 cell lysates after treatment with 1 ng/ml BDNF for theindicated period in the presence of c29 or scrambled peptide (SC). NT, no treatment. n=3 experiments, mean±s.e.m.; *P<0.05; **P<0.001; two-way ANOVAwithBonferroni’s multiple comparison post-hoc test relative to SC). (E) Quantification and representative western blots of the levels of activated and pan Erk1/2 15 minafter treatment with 1 ng/ml BDNF, CNTF or GDNF in the presence of c29 or scrambled peptide (SC). n=5 experiments, mean±s.e.m.; ***P<0.001; one-wayANOVA relative to SC.
524
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

Alternatively or additionally, c29 could also have acted in vivoto promote motor neuron survival through TrkC (or TrkA), whichis upregulated in the longer-lived motor neurons in motor neurondisease patients (Duberley et al., 1997; Nishio et al., 1998). UnlikeBDNF, exogenous delivery of the TrkC ligand NT3 has shownpromise in slowing disease progression and maintaining motorneuron–muscle connectivity in a motor neuron disease mousemodel (specifically the pmn mouse; Haase et al., 1998). Given thatc29 acts in a similar fashion in vitro to promote both TrkA andTrkB signals, it would be surprising if c29 did not affect TrkC inthe same manner. Thus, the ability of c29 to promote motorneuron survival in vivo can be explained by our in vitro findingsthat Trk receptor function (TrkB and/or TrkC) is enhanced inneurons in conditions of low-level neurotrophin exposure oractivation of p75NTR death signaling. Even though c29 treatmentdid not alter the lifespan of the aggressive SOD1G93A model mice,it remains to be determined whether earlier treatment (e.g. prior towhen p75NTR cleavage is first evident), a different dose, a morebiostable mimetic, or a targeted treatment route of c29, eitheralone or in combination with other candidate therapeutics, wouldprovide any additional benefit to animals undergoing motorneuron degeneration.
In summary, our results show that regulated intramembraneproteolysis of p75NTR plays an important role in (pro)NGF-mediated motor neuron death, which can be induced even in thepresence of coincident survival signals. We also demonstrate that apeptide mimetic of the p75NTR intracellular domain (c29), thatinteracts with TrkB receptors, potentiates the survival of motorneurons in a BDNF- and TrkB-dependent manner, under conditionsof growth factor withdrawal as well as active ligand killing, byredressing the neurotrophin signaling imbalance in favor of trophicsupport.
MATERIALS AND METHODSc29 and control peptide synthesisA 29 amino acid peptide of the juxtamembrane ‘Chopper’ domain (Coulsonet al., 2000) (c29: KRWNSCKQNKQGANSRPVNQTPPPEGEKL) and arandomly scrambled version (SC: SKGQVCRNQPGQNKPEPANKSWK-ETPLRN) were synthesized as N-terminal fusions to a non-naturallyoccurring TAT-like protein transduction domain (PTD) peptide(YARAAARNARA) (Ho et al., 2001) using t-boc chemistry then purifiedusing reverse phase HPLC by Dr James I. Elliott (Yale University). Foraxotomy experiments, PTD was conjugated to c29 by means of a disulfidelink (Coulson et al., 2000) by Auspep. No difference in functionality wasseen between the two conjugation methods in cell culture assays, and noeffects were seen in cells treated with PTD peptide alone, peptides withoutcarrier or disulfide-linked peptides that had been reduced with DTT. Forimmunoprecipitation experiments, the unconjugated c29 peptide waslabeled on the amino terminus with biotin through a six-carbon spacer.
Fig. 7. Treatment with c29 enhances TrkB-mediated signaling.Representative western blots (and quantification) of spinal cord lysates frompositive control (wild-type, WT)mice and 16-week-old SOD1G93A (SOD1)miceacutely treated with c29 or scrambled peptide (SC). Blots are probed for (A)phosphorylated TrkB (Ser 478; arrow), pan Trk; (B) phosphorylated (p-)Akt(Ser473) and pan Akt, phosphorylated Erk1/2 (Thr202/Tyr204) and pan Erk1/2; (C) p-CREB (Ser133; arrow) or pan CREB or (D) cleaved (cl-) caspase 3(arrow). The loading control for total protein levels is tubulin or actin. Relativedensitometry quantification of western blot bands indicate that the relativelevels of phosphorylated Akt, Erk1/2 and TrkB are higher in mice treated withc29 whereas levels of activated caspase 3 in SOD1G93A mice are reduced byc29 treatment. N=4 wild-type, N=4 c29-treated SOD1G93A animals, and N=3SC-treated SOD1G93A animals; *P<0.05, **P<0.01, ***P<0.001; one-wayANOVA with Bonferroni’s multiple comparison post-hoc test.
525
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

Except for these immunoprecipitation experiments, ‘c29’ refers to the PTD-conjugated form of the c29 peptide. The biotinylated control peptidemimicking the p75NTR extracellular juxtamembrane domain LC1(H-RGTTDNLIGGSC-NH2) was manufactured by Auspep.
Motor neuron axotomyThe Institutional Animal Ethics Committee approved all experimentsinvolving the use of animals, in accordance with the Australian Code ofPractice for the Care and Use of Animals for Scientific Purposes. Ulnarnerve axotomy was performed on newborn Wistar rat pups as previouslydescribed (Lowry et al., 2001). Peptides (PTD–c29 and PTD only;100 nM) were applied by means of a 1 mm3 cube of pluronic gel foam thatwas sutured to the proximal axonal stump (Cheema et al., 1996). After5 days the animals were perfused with a 4% solution of paraformaldehydein phosphate buffer. Cervical spinal cords were removed, after which serialsections were cut at 40 µm, mounted onto gelatin-coated slides and stainedin 0.1% Cresyl Violet. Details of the counting procedure have beendescribed previously (Lowry et al., 2001). Briefly, motor neurons in thespinal cord displaying a prominent nucleolus were counted in every fifthsection. The percentage neuronal survival after axotomy was calculatedbased on the nucleolar number on the axotomized side as a percentage ofthe nucleolar number in the contralateral side. Data were analyzed by two-tailed t-test.
SOD1 animal experimentsTreatmentsMale and female transgenic mice with the G93A human SOD1 mutation(SOD1G93A; line B6SJL-TgN [SOD1-G93A] 1Gur; Jackson Laboratories),and in some cases age-matched C57Bl6 mice, were used. Some cohortsof mice were administered peptides by a single intraperitoneal injection(5 mg/kg) and sacrificed 60–90 min later by perfusion (for histology) orcervical dislocation (biochemical analyses). Alternatively, mice were treatedsystemically with peptides starting from 9 weeks of age (the time at whichp75NTR expression is upregulated in spinal motor neurons; Lowry et al.,2001). The animals were randomly assigned a treatment group andimplanted with a 28-day subcutaneous osmotic minipump (Alzet model1004) delivering ∼5 mg/kg/day PTD–c29 (flow rate: 0.11 µl/h) or vehicleand the pumps were replaced monthly until death or euthanasia.
Behavioral analysesMice were weighed weekly until close to end stage when they weremonitored daily. Urine discharged during weighing was collected andfrozen. Testing of motor function using a rotarod device began at 10 weeksof age. Each weekly session consisted of three trials on the elevatedaccelerating rotarod (Ugo Basile) beginning at 5 rpm and accelerating to20 rpm over 20 s for a total duration of 3 min. To determine mortalityin a reliable and humane fashion, we used an artificial end point, definedas 80% minimum original weight threshold. The moribund micewere scored as ‘dead’ and were euthanized. Survival analysis of theSOD1G93A transgenic mice was performed using a log-rank (Mantel–Cox)test and analysis of motor neuron cell numbers in spinal cord sections wasperformed using a Student’s t-test. Between-group analyses of weight wereperformed by repeated measure ANOVA followed by a Sidak’s test ofanalysis of the differences between groups. A two-tailed Mann–Whitneytest was used to compare the time it took mice to lose 10% of their maximalweight. Rotarod data were analyzed by ANOVA with a Sidak’s multiplecomparison test.
Biochemical analysis of p75NTR extracellular domain fragmentUrine was collected from SOD1G93A and wild-type mice during weighing,using metabolic cages or from the bladder after euthanasia. However,insufficient urine for analysis was obtained from some mice at each timepoint. For peptide-treated mice, urine was collected from six c29-treatedand six saline-treated SOD1 animals on an at least weekly basis. Foranalysis, we pooled data obtained from consecutive samples such thatmany data points contain data derived from two samples from the sameanimals but collected a week or less apart. A sandwich ELISA was used to
measure the extracellular domain fragment of p75NTR (p75NTR ECD) inurine as previously described (Shepheard et al., 2014). Briefly, ELISA plates(96-well plates, Costar Corning) were coated for 18 h with anti-p75NTR ECDMLR1 (4 µg/ml in 25 mM sodium carbonate 25 mM, sodium hydrogencarbonate, 0.01% thimerosol, pH 9.6) at 4°C.Wells were blockedwith samplebuffer comprising phosphate buffered saline (PBS), 2%bovine serum albumin(BSA; Sigma) and 0.01% thimerosol, pH 7.4, for 1 h at 37°C. Recombinanthuman mouse p75NTR ECD (control antigen, aa:20–243; R&D Systems) andurine samples were diluted in sample buffer and incubated for 20 h at roomtemperature in the antibody coatedwells. For detection,wellswere incubated ingoat anti-p75NTR ECD (R&D Systems) for 1 h followed by a bovine anti goatIgG-HRP antibody (Jackson ImmunoResearch) for 1 h at room temperature.Visualizationwas bymeans of a peroxidase reaction developed using the colorreagent 3,3′,5,5′-tetramethylbenzidine (TMB; Bio-Rad) and stoppedwith 2 Msulfuric acid. Between steps, plates were washed 4 times with wash buffer(PBS, 0.05%Tween 20, 0.01% thimerosol, pH7.4). Plateswere read at 450 nmwith a PerkinElmer Victor-x4 Plate Reader. Mouse creatinine was used as astandard for urinary protein measurements and was assessed using a creatininedetection kit (Enzo Life Sciences) as previously described (Shepheard et al.,2014). p75NTR-ECD-specific ELISA results were analyzed by one-wayANOVA, with a Bonferroni’s multiple comparison test.
Peptide distribution and motor neuron survivalTo assess the biodistribution of c29 and motor neuron survival in treatedpostnatal day 115 SOD1G93A mice, animals were first anesthetized and thenintracardially perfused with PBS, followed by 4% paraformaldehyde inPBS. Spinal cords were dissected and post-fixed in 4% paraformaldehyde atroom temperature for 2 h.
Fixed spinal cord samples were cryoprotected overnight by immersion inPBSwith 20% sucrose, embedded in optimal cutting temperature compound(OCT; Tissue Tek, Raymond Lamb Ltd Medical Supplies) and frozen ondry ice. All spinal cord samples were stored at −80°C until needed, and thencryosectioned.
For biodistribution, 20 µm-thick lumbar spinal cord sections were stainedusing streptavidin-conjugated horseradish peroxidase. Motor neuronnumbers were determined on 20 µm-thick lumbar spinal cord sectionsstained with ChAT. Sections werewashed in 0.1 M phosphate buffer, pH 7.4and then incubated in 3% H2O2, 50% ethanol to inactivate endogenousperoxidase. Sections were subsequently washed in 0.1 M phosphate buffer,and incubated for 1 h at room temperature in blocking solution (phosphatebuffer, 10% horse serum, 0.1% Triton X-100). Primary antibody (goat anti-ChAT, 1:1000; Millipore, AB144P) was diluted in the above blockingbuffer and applied onto sections for overnight incubation at roomtemperature. Sections were then washed at room temperature in phosphatebuffer containing 0.1% Triton X-100 before incubation with biotinylatedsecondary antibody (1:1000; Jackson Immunoresearch Laboratories,#705-067-003) and avidin-biotin complex (ABC) reagents (Vector EliteKit, Vector Laboratories; 6 µl/ml avidin and 6 µl/ml biotin). Blackimmunoreactivity was revealed by a nickel-intensified diaminobenzidinereaction. Sections were finally washed in phosphate buffer and graduallydehydrated in ethanol and xylene before being mounted in DePex mountingmedium (VWR International). A total of 10 sections per mousewere analyzed. Motor neuron numbers were compared using a two-tailedt-test.
Spinal cord lysate preparationFollowing a 60 min treatment of wild-type C57Bl6 and SOD1G93A micewith c29 or control peptide delivered intraperitoneally, mice were killed bycervical dislocation and spinal cords were dissected immediately aftersacrifice and snap-frozen. The lumbar spinal cords were thawed anddissected in RIPA buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1%Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 1 mMsodium orthovanadate, 1 mM PMSF, 1 µM BB94 (Betimastat), 15 mMsodium fluoride, 40 mM β-glycerophosphate, 0.5 mM 4-deoxypyridoxine,1% (v/v) Roche Complete-Mini inhibitor cocktail stock and 1 mM DTT atpH 7.4. They were then lysed in 300 µl of the same buffer using a RetchTissue Lyser (Qiagen) for 3 min at 4°C. Lysates were centrifuged at16,000 g and pellets discarded. Protein estimation of all lysates was done
526
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

using the Pierce BCA protein assay kit (Thermo Fisher). Western blots wereperformed as described below.
Motor neuron isolation and cultureCultures of spinal motor neurons were prepared from embryonic day (E)13wild-type C57BL/6J mice and isolated by immunopanning as previouslydescribed (Wiese et al., 2010). Briefly, pregnant dams were sacrificed bycervical dislocation and the uterus removed by Cesarean section andplaced in neurobasal medium (Gibco). Embryos were removed from theuterine sac and the lumbar spinal cords were dissected on ice. Batches oftwo or three spinal cords were digested in 1 ml of 0.05% trypsin–ETDA(Gibco) for 10 min at 37°C in 15 ml tubes. To prevent further digestion,0.14 mg/ml trypsin inhibitor (Sigma) solution was added, and the tissuewas centrifuged at 100 g for 7 min. Spinal cords were trituated manuallyfor 2 min with a polished glass pipette, after which the suspension wasfiltered through a 100 μm sieve (Falcon). The remaining tissue was furthertriturated ∼8 times and filtered through another 100 μm sieve. Thecombined cell suspensions were then immunopanned by being plated ontoa 10 cm culture dish (Nunc) pre-coated with MLR-2 mouse anti-p75NTR
antibody (Rogers et al., 2006). The dish was pre-coated with 8 ml ofimmunopanning solution containing 1.5 μg/ml MLR-2 in 10 mM Tris-HCl, pH 9.5, for 2 h and washed with neurobasal medium before thespinal cord suspension was applied. After 45 min incubation, the dish waswashed three times with neurobasal medium and the remaining attachedcells were isolated from the plate by incubation for 1 min in 0.85 M KCl.The collected cell suspension was centrifuged at 400 g for 5 min at roomtemperature. The supernatant was removed and the cell pellet wasresuspended in 1 ml of neurobasal medium. Neurons were plated at adensity of 3000 live cells per well in 4-well culture dishes (Greiner) pre-coated with 0.0015% poly-ornithine (overnight at 4°C) and 3.75 μg/mllaminin (for 30 min at 37°C). Cells were allowed to adhere for 1 h at37°C, after which 2 ml of motor neuron culture medium was added[neurobasal medium supplemented with B27 (Life Sciences), 500 μMglutamax (Life Sciences), 10% horse serum (Linaris), 25 μMβ-mercaptoethanol (Merck) and 1 ng/ml recombinant human BDNF(Peprotech)]. Cells were maintained at 37°C in a humidified atmospherewith 5% CO2 for 24 h, after which the culture medium was replaced, livecells counted (day 1), and the experimental treatments administered. Phasecontrast images of motor neurons were taken using an Olympus (6IX81)microscope.
Neuronal survival assaysIn addition to BDNF, the following growth factors were used in primaryneuronal culture assays at concentrations indicated in individualexperiments: purified mouse 2.5S NGF (Biosensis), proNGF (SCILproteins GmbH), proBDNF (Biosensis), recombinant rat cilliaryneurotrophic factor (CNTF; R&D Systems) and recombinant human glial-derived neurotrophic factor (GDNF; Peprotech). For the neuronal survivalassays, BDNF, CNTF or GDNF was added at the time of plating asindicated. A concentration of 1 ng/ml for each trophic factor has previouslybeen determined to result in optimal rates of survival (Wiese et al., 1999). Inorder to count the same field at each time point, grids were etched onto theplates using an 18-gauge needle. Motor neurons were counted on day 1 (fourseparate fields for each well) and the same fields were recounted on day3. Cell survival was expressed as a percentage compared with survival inBDNF alone on day 1.
For assays investigating the effects of the induction of p75NTR cleavageby the metalloproteinase enhancer phorbol 12-myristate 13-acetate (PMA;200 nM), and blocking p75NTR cleavage with the selective non-competitivemetalloproteinase inhibitors TAPI-2 (200 nM, extracellular cleavage) andCompound E (20 µM, intracellular cleavage; Tian et al., 2002), thecompounds were added to established cultures on day 1 after the initial cellcounts and cells were then counted again on day 3.
To assess the effects of c29 peptide on motor neuronal survival in limitinggrowth factor concentrations, isolated spinal motor neurons were plated andtreated with BDNF, as indicated, in the presence of 1 µM PTD-linked c29 orscrambled peptide, then cultured for three days with the addition of fresh
BDNF and peptide treatments on day 2. For growth-factor-withdrawalassays, following initial plating in 1 ng/ml of BDNF, CNTF or GDNF,cultures were washed four times with serum-free neurobasal medium, afterwhich the medium was replaced with medium containing 0.1 ng/ml BDNF,CNTF or GDNF and 1 µM PTD-linked c29 or scrambled peptide, asindicated.
For assays investigating the effect of c29 on NGF-mediated motor neurondeath, cultures were grown in 1 ng/ml BDNF, CNTF or GDNF for one day,before being treated with 100 ng/ml 2.5S NGF and 1 µM PTD-linked c29 orscrambled peptide, as indicated. Motor neurons were counted on day 1 and,to assess the effects of various treatments, again on day 3.
Rates of survival were compared between conditions by ANOVA withBonferonni post-hoc testing.
Immortalized cell culture, pull-down experiments and westernblottingMycoplasma-free neuroblastoma×spinal cord (NSC-34) cells weremaintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco)supplemented with 10% FBS and 1% penicillin/streptomycin/glutaminesolution (PSG; Cashman et al., 1992). For experiments, cells were grownto 60% confluency in medium comprising 1:1 DMEM:Ham’s F12, plus1% N-2 supplement, 1% PSG, 1% modified Eagle’s medium non-essentialamino acids, and 1 µM retinoic acid (Sigma). The cells were allowed todifferentiate for up to three days, with the medium being replaced after48 h.
For p75NTR-shedding experiments, NSC-34 cells were cultured in poly-L-ornithine-coated 6-well plates, starved for 1 h in serum-free DMEM,and treated with 200 nM PMA, 200 nM TAPI-2, or 20 µM Compound E;50 ng/ml NGF, 5 ng/ml proNGF, 50 ng/ml BDNF or 5 ng/ml proBDNF for6 h. To inhibit degradation of the p75NTR intracellular domain fragment,cells were treated with 1 µM epoxomicin (Sigma) 1 h prior to the addition ofother compounds.
For experiments measuring phosphorylated TrkB, Akt, and Erk1/2, NSC-34 cells were differentiated for five days before being serum starved for 4 hin serum-free DMEM. Cells were then pretreated with 1 µM c29 orscrambled control peptide for 1 h prior to addition of 10 ng/ml of BDNFfollowed by treatments with growth factors as indicated for time intervalsranging from 1–90 min. Following treatments, cells were immediatelywashed with ice cold PBS, lyzed and prepared for SDS-PAGE analysis asdescribed below.
Cells were lyzed using chilled lysis buffer containing 10 mM Tris-HCl,pH 8.0; 150 mM NaCl; 2 mM EDTA; 1% NP-40; 1% Triton X-100;10% glycerol; 1 mM phenylmethanesulfonyl fluoride; 1 mM sodiumorthovanadate; 1 µM batimastat (BB-94) and 1% Roche protease inhibitorcocktail (Ceni et al., 2010).
HEK293 cells used for pull-down assays were routinely cultured inDMEM supplemented with 10% FBS, in 5% CO2 at 37°C. For experiments,cells were transfected with TrkB plasmid DNA using FuGENE 6transfection reagent (Roche Applied Science), lyzed 48 h aftertransfection and the lysates used for c29 pull-down experiments asdescribed below.
To perform pull-down assays, biotinylated c29 or LC1 control peptidewas bound to Dynabeads MyOne Streptavidin T1 (Invitrogen) beforeincubation with transfected HEK293 or NSC-34 cell lysates in the presenceof 2% BSA. Nonspecific binding was removed by washing the beads fivetimes in Tris-buffered saline (TBS) supplemented with 0.05% Tween 20,and the proteins were eluted by boiling samples in reducing sample buffer(20 mM dithioerythritol, 2.5% SDS).
Cell lysates, lumbar spinal cord lysates or pulled-down proteins wereelectrophoresed through 4–12% Bis-Tris buffered SDS gels (LifeSciences), transferred onto a PVDF or nitrocellulose membrane andprobed using the following antibodies: rabbit anti-p75NTR intracellulardomain #9992 (1:5000; a gift from Moses Chao), rabbit anti-TrkB(1:500; Biosensis, #R121-100), phospho-TrkB (Ser478) (1:250; a giftfrom Biosensis), mouse pan anti-Akt (1:2000; Cell SignalingTechnologies, #2920S), XP™ rabbit mAb rabbit anti-phosphorylatedserine 473 (Ser 473) Akt (1:2000; Cell Signaling Technologies, #4060),
527
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

XP™ rabbit mAb phospho-p44/42 MAP kinase (Erk1/2; Thr202 andTyr204) (1:2000; Cell Signaling Technologies, #4370), rabbit phospho-p44/42 MAP kinase (Erk1/2; Thr202 and Tyr204) (1:1000; Millipore,#05-797R), rabbit anti-pan p44/42 Erk1/2 (1:1000; Cell SignalingTechnologies, #9101), rabbit cleaved caspase-3 (Asp175) (1:1000; CellSignaling Technologies, #9661) and rabbit caspase-3 (8G10) (1:1000;Cell Signaling Technologies, #9665), CREB (48H2, #9197) and phospho-(Ser133) CREB (87G3, #9198) (1:1000; Cell Signaling Technologies),and mouse anti-beta-III tubulin (1:2000; Promega, #G7121). Themembranes were blocked for 1 h at room temperature in 0.1% Tween-20, and 0.02% NaN3 in TBS, pH 8.0, and either 4% skim milk powder forp75NTR detection, 3% BSA for phosphorylated Erk1/2 and Akt, or 1%BSA for phosphorylated TrkB, before being incubated overnight withprimary antibodies at 4°C. Membranes were then washed in TBS-Tween20 (TBST), and incubated for 1 h with LICOR donkey anti-rabbit 680 ordonkey anti-mouse 800 secondary antibody (1:50,000; Invitrogen, #925-68073, #926-32212) in TBS at room temperature after which they werewashed and imaged using the Gel Doc XR imaging system (Bio-Rad).Alternatively, blots were probed with appropriate species-specific HRP-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch,#115-035-003/ #111-035-003) and visualized with either SupersignalWest Femto Sensitivity Substrate (Pierce) or Clarity™ Western ECLsubstrate (Bio-Rad) using a Fujifilm ImageQuant™ LAS-4000 (FujiCorporation). For stripping and re-probing, western blot membranes weretreated with 10 ml of Restore™ PLUS Western Blot Stripping Buffer(Thermo Fisher) for 30 min at room temperature on a orbital shaker,washed three times with TBST and blocked with either 5% skim milkpowder or 3% BSA in TBST for 1 h. The membranes were thenincubated with appropriate primary and secondary antibodies asdescribed above.
Statistical analysesN represents the number of individual animals; n represents the number ofindividual cell culture experiments, each with four replicates. Stereology,rotarod and weight measurements, p75NTR ECD analysis and, for keyexperiments, the motor neuron culture counts were conducted byresearchers blinded to the treatment conditions. Densitometry analysisof relative protein levels on western blots was performed using ImageStudio Digits (Licor), Image Lab (Bio-Rad) and Fiji (ImageJ, NIH). Allrepresentative graphs measuring relative densitometry of proteins onwestern blots were calculated from three independent experiments, withbands normalized to loading controls (tubulin or actin) probed andexposed at the same time. Statistical analysis was performed usingGraphPad Prism (GraphPad Software) as described in each method.P<0.05 was considered statistically significant.
AcknowledgementsRobert Rush (Flinders University, Adelaide, Australia) kindly provided the TrkBantibody used for western blotting and immunoprecipitation experiments. MosesChao (Skirball Institute, New York, USA) kindly provided the 9992 p75NTR antibodyfor western blotting. We thank Stephan Weise and Michael Sendtner (WurzburgUniversity, Germany) for assistance in establishing the NGF-killing motor neuronassay. NSC-34 cells were kindly provided by Dr Neil Cashman (University of BritishColumbia). We thank other members of the Coulson laboratory past and present forhelpful discussions and Rowan Tweedale for critical reading of the manuscript andeditorial assistance.
Competing interestsPatents covering the use of c29 as a therapeutic in a range of neurological conditionsare held by the University of Queensland.
Author contributions
All authors executed and interpreted experiments and contributed to the draftingthe article. D.M., F.A., C.U. and E.J.C. conceived and designed the experiments andD.M. and E.J.C. revised the article.
FundingThis work was supported by the National Health and Medical Research Councilof Australia [Career Development Fellowship 569601 and project grant 10012610
to E.J.C.]; the Australian Research Council; and NuNerve Pty Ltd [Linkage grantLP10012610 to E.J.C.]; Goodenough and Wantoks bequest (to E.J.C and M.M.);The Australian Government (Postgraduate Award to C.K.U); a Ross MacleanFellowship (to M.M.); the Centre for Neuroscience, Flinders University and theFlinders Medical Centre Research Foundation (grants to D.M.); Australian RotaryHealth (Neville & Jeanne York Motor Neuron Disease PhD Scholarship toS.R.S); and the Motor Neuron Disease Reseach Instititue of Australia (RosalindNicholson Grant to M-L.R.).
ReferencesBarker, P. A. (2007). High affinity not in the vicinity? Neuron 53, 1-4.Bibel, M., Hoppe, E. and Barde, Y.-A. (1999). Biochemical and functional
interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 18,616-622.
Blochl, A. and Blochl, R. (2007). A cell-biological model of p75NTR signaling.J. Neurochem. 102, 289-305.
Boyd, J. G. and Gordon, T. (2001). The neurotrophin receptors, trkB andp75, differentially regulate motor axonal regeneration. J. Neurobiol. 49, 314-325.
Branco, F., Cardenas, D. D. and Svircev, J. N. (2007). Spinal cord injury: acomprehensive review. Phys. Med. Rehabil. Clin. N. Am. 18, 651-679, v.
Casaccia-Bonnefil, P., Carter, B. D., Dobrowsky, R. T. and Chao, M. V. (1996).Death of oligodendrocytes mediated by the interaction of nerve growth factor withits receptor p75. Nature 383, 716-719.
Cashman, N. R., Durham, H. D., Blusztajn, J. K., Oda, K., Tabira, T., Shaw, I. T.,Dahrouge, S. and Antel, J. P. (1992). Neuroblastoma×spinal cord (NSC) hybridcell lines resemble developing motor neurons. Dev. Dyn. 194, 209-221.
Ceni, C., Kommaddi, R. P., Thomas, R., Vereker, E., Liu, X., McPherson, P. S.,Ritter, B. and Barker, P. A. (2010). The p75NTR intracellular domain generated byneurotrophin-induced receptor cleavage potentiates Trk signaling. J. Cell Sci. 123,2299-2307.
Cheema, S. S., Barrett, G. L. and Bartlett, P. F. (1996). Reducing p75 nerve growthfactor receptor levels using antisense oligonucleotides prevents the loss ofaxotomized sensory neurons in the dorsal root ganglia of newborn rats.J. Neurosci. Res. 46, 239-245.
Chiariello, M., Visconti, R., Carlomagno, F., Melillo, R. M., Bucci, C., deFranciscis, V., Fox, G. M., Jing, S., Coso, O. A., Gutkind, J. S. et al. (1998).Signalling of the Ret receptor tyrosine kinase through the c-Jun NH2-terminalprotein kinases (JNKS): evidence for a divergence of the ERKs and JNKspathways induced by Ret. Oncogene 16, 2435-2445.
Coulson, E. J., Reid, K. and Bartlett, P. F. (1999). Signaling of neuronal cell deathby the p75NTR neurotrophin receptor. Mol. Neurobiol. 20, 29-44.
Coulson, E. J., Reid, K., Baca,M., Shipham,K.A., Hulett, S.M.,Kilpatrick, T. J. andBartlett, P. F. (2000).Chopper, a newdeath domain of thep75 neurotrophin receptorthat mediates rapid neuronal cell death. J. Biol. Chem. 275, 30537-30545.
Davies, A. M., Lee, K.-F. and Jaenisch, R. (1993). p75-deficient trigeminal sensoryneurons have an altered response to NGF but not to other neurotrophins. Neuron11, 565-574.
Dechant, G. and Barde, Y.-A. (2002). The neurotrophin receptor p75NTR: novelfunctions and implications for diseases of the nervous system. Nat. Neurosci. 5,1131-1136.
DiStefano, P. S., Chelsea, D. M., Schick, C. M. and McKelvy, J. F. (1993).Involvement of a metalloprotease in low-affinity nerve growth factor receptortruncation: inhibition of truncation in vitro and in vivo. J. Neurosci. 13,2405-2414.
Domeniconi, M., Hempstead, B. L. and Chao, M. V. (2006). Pro-NGF secretedby astrocytes promotes motor neuron cell death. Mol. Cell. Neurosci. 34,271-279.
Duberley, R. M., Johnson, I. P., Anand, P., Leigh, P. N. and Cairns, N. J.(1997). Neurotrophin-3-like immunoreactivity and Trk C expression in humanspinal motoneurones in amyotrophic lateral sclerosis. J. Neurol. Sci. 148,33-40.
Ernfors, P. (2001). Local and target-derived actions of neurotrophins during peripheralnervous system development. Cell. Mol. Life Sci. 58, 1036-1044.
Esposito, D., Patel, P., Stephens, R. M., Perez, P., Chao, M. V., Kaplan, D. R. andHempstead, B. L. (2001). The cytoplasmic and transmembrane domains of thep75 and Trk A receptors regulate high affinity binding to nerve growth factor.J. Biol. Chem. 276, 32687-32695.
Frebel, K. and Wiese, S. (2006). Signalling molecules essential for neuronalsurvival and differentiation. Biochem. Soc. Trans. 34, 1287-1290.
Friedman, W. J. (2000). Neurotrophins induce death of hippocampal neurons viathe p75 receptor. J. Neurosci. 20, 6340-6346.
Haase, G., Pettmann, B., Vigne, E., Castelnau-Ptakhine, L., Schmalbruch, H.and Kahn, A. (1998). Adenovirus-mediated transfer of the neurotrophin-3 geneinto skeletal muscle of pmn mice: therapeutic effects and mechanisms of action.J. Neurol. Sci. 160 Suppl. 1, S97-S105.
Hempstead, B. L., Martin-Zanca, D., Kaplan, D. R., Parada, L. F. and Chao, M. V.(1991). High-affinity NGF binding requires coexpression of the trk proto-oncogeneand the low-affinity NGF receptor. Nature 350, 678-683.
528
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

Henderson, C. E., Yamamoto, Y., Livet, J., Arce, V., Garces, A. anddeLapeyriere, O. (1998). Role of neurotrophic factors in motoneurondevelopment. J. Physiol. Paris 92, 279-281.
Henriques, A., Pitzer, C. andSchneider, A. (2010). Neurotrophic growth factors forthe treatment of amyotrophic lateral sclerosis: where do we stand? Front.Neurosci. 4, 32.
Ho, A., Schwarze, S. R., Mermelstein, S. J., Waksman, G. and Dowdy, S. F.(2001). Synthetic protein transduction domains: enhanced transduction potentialin vitro and in vivo. Cancer Res. 61, 474-477.
Horton, A., Laramee, G., Wyatt, S., Shih, A., Winslow, J. and Davies, A. M.(1997). NGF binding to p75 enhances the sensitivity of sensory and sympatheticneurons to NGF at different stages of development. Mol. Cell. Neurosci. 10,162-172.
Huber, L. J. and Chao, M. V. (1995). A potential interaction of p75 and trkA NGFreceptors revealed by affinity crosslinking and immunoprecipitation. J. Neurosci.Res. 40, 557-563.
Hutson, L. D. and Bothwell, M. (2001). Expression and function of Xenopus laevisp75NTR suggest evolution of developmental regulatory mechanisms. J. Neurobiol.49, 79-98.
Iacaruso, M. F., Galli, S., Martı, M., Villalta, J. I., Estrin, D., Jares-Erijman, E. A.and Pietrasanta, L. I. (2011). Structural model for p75NTR-TrkA intracellulardomain interaction: a combined FRET and Bioinformatics study. J. Mol. Biol. 414,681-698.
Ibanez, C. F. and Simi, A. (2012). p75 neurotrophin receptor signaling in nervoussystem injury and degeneration: paradox and opportunity. Trends Neurosci. 35,431-440.
Jung, K.-M., Tan, S., Landman, N., Petrova, K., Murray, S., Lewis, R., Kim, P. K.,Kim, D. S., Ryu, S. H., Chao, M. V. et al. (2003). Regulated intramembraneproteolysis of the p75 neurotrophin receptor modulates its association with theTrkA receptor. J. Biol. Chem. 278, 42161-42169.
Kanning, K. C., Hudson, M., Amieux, P. S., Wiley, J. C., Bothwell, M. andSchecterson, L. C. (2003). Proteolytic processing of the p75 neurotrophinreceptor and two homologs generates C-terminal fragments with signalingcapability. J. Neurosci. 23, 5425-5436.
Kaplan, D. R. and Miller, F. D. (2000). Neurotrophin signal transduction in thenervous system. Curr. Opin. Neurobiol. 10, 381-391.
Kenchappa, R. S., Zampieri, N., Chao, M. V., Barker, P. A., Teng, H. K.,Hempstead, B. L. and Carter, B. D. (2006). Ligand-dependent cleavage of theP75 neurotrophin receptor is necessary for NRIF nuclear translocation andapoptosis in sympathetic neurons. Neuron 50, 219-232.
Kommaddi, R. P., Thomas, R., Ceni, C., Daigneault, K. and Barker, P. A. (2011).Trk-dependent ADAM17 activation facilitates neurotrophin survival signaling.FASEB J. 25, 2061-2070.
Lee, K. F., Davies, A. M. and Jaenisch, R. (1994). p75-deficient embryonic dorsalroot sensory and neonatal sympathetic neurons display a decreased sensitivity toNGF. Development 120, 1027-1033.
Li, L., Xian, C. J., Zhong, J.-H. and Zhou, X.-F. (2006). Upregulation of brain-derived neurotrophic factor in the sensory pathway by selective motor nerve injuryin adult rats. Neurotox. Res. 9, 269-283.
Li, F., Li, L., Song, X.-Y., Zhong, J.-H., Luo, X.-G., Xian, C. J. and Zhou, X.-F.(2009). Preconditioning selective ventral root injury promotes plasticity ofascending sensory neurons in the injured spinal cord of adult rats–possibleroles of brain-derived neurotrophic factor, TrkB and p75 neurotrophin receptor.Eur. J. Neurosci. 30, 1280-1296.
Lowry, K. S., Murray, S. S., McLean, C. A., Talman, P., Mathers, S., Lopes, E. C.and Cheema, S. S. (2001). A potential role for the p75 low-affinity neurotrophinreceptor in spinal motor neuron degeneration in murine and human amyotrophiclateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2,127-134.
Matusica, D., Skeldal, S., Sykes, A. M., Palstra, N., Sharma, A. and Coulson,E. J. (2013). An intracellular domain fragment of the p75 neurotrophin receptor(p75NTR) enhances tropomyosin receptor kinase A (TrkA) receptor function.J. Biol. Chem. 288, 11144-11154.
McCarty, J. H. and Feinstein, S. C. (1999). The TrkB receptor tyrosine kinaseregulates cellular proliferation via signal transduction pathways involving SHC,PLCγ, and CBL. J. Recept. Signal Transduct. Res. 19, 953-974.
Meeker, R. and Williams, K. (2014). Dynamic nature of the p75 neurotrophinreceptor in response to injury and disease. J. Neuroimmune Pharmacol. 9,615-628.
Murray, S. S., Bartlett, P. F., Lopes, E. C., Coulson, E. J., Greferath, U. andCheema, S. S. (2003). Low-affinity neurotrophin receptor with targeted mutationof exon 3 is capable of mediating the death of axotomized neurons. Clin. Exp.Pharm. Physiol. 30, 217-222.
Murray, L. M., Talbot, K. and Gillingwater, T. H. (2010). Review: neuromuscularsynaptic vulnerability in motor neurone disease: amyotrophic lateral sclerosisand spinal muscular atrophy. Neuropathol. Appl. Neurobiol. 36, 133-156.
Ni, C.-Y., Murphy, M. P., Golde, T. E. and Carpenter, G. (2001). γ-Secretasecleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science294, 2179-2181.
Nishio, T., Sunohara, N. and Furukawa, S. (1998). Neutrophin switching inspinal motoneurons of amyotrophic lateral sclerosis. Neuroreport 9,1661-1665.
Pehar, M., Cassina, P., Vargas, M. R., Castellanos, R., Viera, L., Beckman, J. S.,Estevez, A. G. and Barbeito, L. (2004). Astrocytic production of nerve growthfactor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis.J. Neurochem. 89, 464-473.
Perlson, E., Jeong, G.-B., Ross, J. L., Dixit, R., Wallace, K. E., Kalb, R. G. andHolzbaur, E. L. F. (2009). A switch in retrograde signaling from survival to stress inrapid-onset neurodegeneration. J. Neurosci. 29, 9903-9917.
Rogers, M.-L., Atmosukarto, I., Berhanu, D. A., Matusica, D., Macardle, P. andRush, R. A. (2006). Functional monoclonal antibodies to p75 neurotrophinreceptor raised in knockout mice. J. Neurosci. Methods 158, 109-120.
Roux, P. P. and Barker, P. A. (2002). Neurotrophin signaling through the p75neurotrophin receptor. Prog. Neurobiol. 67, 203-233.
Schwarze, S. R., Hruska, K. A. and Dowdy, S. F. (2000). Protein transduction:unrestricted delivery into all cells? Trends Cell Biol. 10, 290-295.
Sedel, F., Bechade, C. and Triller, A. (1999). Nerve growth factor (NGF) inducesmotoneuron apoptosis in rat embryonic spinal cord in vitro. Eur. J. Neurosci. 11,3904-3912.
Seeburger, J. L., Tarras, S., Natter, H. and Springer, J. E. (1993). Spinal cordmotoneurons express p75NGFR and p145trkB mRNA in amyotrophic lateralsclerosis. Brain Res. 621, 111-115.
Sendtner, M., Holtmann, B., Kolbeck, R., Thoenen, H. and Barde, Y.-A. (1992).Brain-derived neurotrophic factor prevents the death of motoneurons in newbornrats after nerve section. Nature 360, 757-759.
Sendtner, M., Pei, G., Beck, M., Schweizer, U. and Wiese, S. (2000).Developmental motoneuron cell death and neurotrophic factors. Cell TissueRes. 301, 71-84.
Shepheard, S. R., Chataway, T., Schultz, D. W., Rush, R. A. and Rogers, M.-L.(2014). The extracellular domain of neurotrophin receptor p75 as a candidatebiomarker for amyotrophic lateral sclerosis. PLoS ONE 9, e87398.
Skeldal, S., Matusica, D., Nykjaer, A. and Coulson, E. J. (2011). Proteolyticprocessing of the p75 neurotrophin receptor: a prerequisite for signalling?:Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75NTR. Bioessays 33, 614-625.
Song, W., Volosin, M., Cragnolini, A. B., Hempstead, B. L. and Friedman, W. J.(2010). ProNGF induces PTEN via p75NTR to suppress Trk-mediated survivalsignaling in brain neurons. J. Neurosci. 30, 15608-15615.
Tian, G., Sobotka-Briner, C. D., Zysk, J., Liu, X., Birr, C., Sylvester, M. A.,Edwards, P. D., Scott, C. D. and Greenberg, B. D. (2002). Linear non-competitive inhibition of solubilized human gamma-secretase by pepstatin Amethylester, L685458, sulfonamides, and benzodiazepines. J. Biol. Chem. 277,31499-31505.
Trupp, M., Scott, R., Whittemore, S. R. and Ibanez, C. F. (1999). Ret-dependentand -independent mechanisms of glial cell line-derived neurotrophic factorsignaling in neuronal cells. J. Biol. Chem. 274, 20885-20894.
Underwood, C. K. and Coulson, E. J. (2008). The p75 neurotrophin receptor.Int. J. Biochem. Cell Biol. 40, 1664-1668.
Underwood, C. K., Reid, K., May, L. M., Bartlett, P. F. and Coulson, E. J. (2008).Palmitoylation of the C-terminal fragment of p75NTR regulates death signaling andis required for subsequent cleavage by gamma-secretase.Mol. Cell. Neurosci. 37,346-358.
Vesa, J., Kruttgen, A. and Shooter, E. M. (2000). p75 reduces TrkB tyrosineautophosphorylation in response to brain-derived neurotrophic factor andneurotrophin 4/5. J. Biol. Chem. 275, 24414-24420.
Vicario, A., Kisiswa, L., Tann, J. Y., Kelly, C. E. and Ibanez, C. F. (2015). Neuron-type-specific signaling by the p75NTR death receptor is regulated by differentialproteolytic cleavage. J. Cell Sci. 128, 1507-1517.
Wiese, S., Metzger, F., Holtmann, B. and Sendtner, M. (1999). The role ofp75NTR in modulating neurotrophin survival effects in developing motoneurons.Eur. J. Neurosci. 11, 1668-1676.
Wiese, S., Beck, M., Karch, C. and Sendtner, M. (2004). Signalling mechanismsfor survival of lesioned motoneurons. Acta Neurochirurgica 89, 21-35.
Wiese, S., Herrmann, T., Drepper, C., Jablonka, S., Funk, N., Klausmeyer, A.,Rogers, M.-L., Rush, R. and Sendtner, M. (2010). Isolation and enrichment ofembryonic mouse motoneurons from the lumbar spinal cord of individual mouseembryos. Nat. Protoc. 5, 31-38.
Xin, J., Mesnard, N. A., Beahrs, T., Wainwright, D. A., Serpe, C. J., Alexander,T. D., Sanders, V. M. and Jones, K. J. (2012). CD4+ T cell-mediatedneuroprotection is independent of T cell-derived BDNF in a mouse facial nerveaxotomy model. Brain Behav. Immun. 26, 886-890.
Yanpallewar, S. U., Barrick, C. A., Buckley, H., Becker, J. and Tessarollo, L.(2012). Deletion of the BDNF truncated receptor TrkB.T1 delays disease onsetin a mouse model of amyotrophic lateral sclerosis. PLoS ONE 7, e39946.
Yoon, S. O., Casaccia-Bonnefil, P., Carter, B. and Chao, M. V. (1998).Competitive signaling between TrkA and p75 nerve growth factor receptorsdetermines cell survival. J. Neurosci. 18, 3273-3281.
Yuan, Q., Wu, W., So, K.-F., Cheung, A. L. M., Prevette, D. M. andOppenheim, R. W. (2000). Effects of neurotrophic factors on
529
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience

motoneuron survival following axonal injury in newborn rats. Neuroreport 11,2237-2241.
Yuen, E.C. andMobley,W.C. (1995). Therapeutic applicationsof neurotrophic factorsin disorders of motor neurons and peripheral nerves.Mol. Med. Today 1, 278-286.
Zampieri, N., Xu, C.-F., Neubert, T. A. and Chao, M. V. (2005). Cleavage of p75neurotrophin receptor by alpha -secretase and gamma -secretase requiresspecific receptor domains. J. Biol. Chem. 280, 14563-14571.
Zhai, J., Zhou, W., Li, J., Hayworth, C. R., Zhang, L., Misawa, H., Klein, R.,Scherer, S. S., Balice-Gordon, R. J. and Kalb, R. G. (2011). The in vivocontribution of motor neuron TrkB receptors to mutant SOD1 motor neurondisease. Hum. Mol. Genet. 20, 4116-4131.
Zhang, J. and Huang, E. J. (2006). Dynamic expression of neurotrophic factorreceptors in postnatal spinal motoneurons and in mouse model of ALS.J. Neurobiol. 66, 882-895.
530
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 517-530 doi:10.1242/jcs.173864
Journal
ofCe
llScience


















