
Incidence, complexity and diversity of simple sequence repeats across potexvirus genomes
8
Incidence, complexity and diversity of simple sequence repeats across potexvirus genomes Chaudhary Mashhood Alam a , Avadhesh Kumar Singh b , Choudhary Sharfuddin a , Safdar Ali b, ⁎ a Department of Botany, Patna University, Bihar 800005, India b Department of Biomedical Sciences, SRCASW, University of Delhi, Vasundhara Enclave, New Delhi 110096, India abstract article info Article history: Accepted 4 January 2014 Available online 13 January 2014 Keywords: Potexvirus Simple sequence repeats Imperfect Microsatellite Extraction Correlation studies An in-silico analysis of simple sequence repeats (SSRs) in genomes of 32 species of potexviruses was performed wherein a total of 691 SSRs and 33 cSSRs were observed. Though SSRs were present in all the studied genomes their incident frequency ranged from 11 to 30 per genome. Further, 10 potexvirus genomes possessed no cSSRs when extracted at a dMAX of 10 and wherein present, the highest frequency was 3. SSR and cSSR incidence, relative density and relative abundance were non-significantly correlated with genome size and GC content sug- gesting an ongoing evolutionary and adaptive phase of the virus species. SSRs present primarily ranged from mono- to tri-nucleotide repeat motifs with a greatly skewed distribution across the coding and non-coding regions. Present work is an effort for the undergoing compilation and analysis of incidence, distribution and variation of the viral repeat sequences to understand their evolutionary and functional relevance. © 2014 Elsevier B.V. All rights reserved. 1. Introduction A sizeable number of satellite sequences (mega-, mini- and micro-) have been characterized from different species (Jeffreys et al., 1998) and their presence had been established in coding regions as well. They may be present as a part of the transcriptome or in the vicinity of the coding regions. The repeat sequences are known to be responsible for the reg- ulation of gene expression at transcriptional and translational level or even by gene silencing (Li et al., 2004; Rocha et al., 2002; Vergnaud and Denoeud, 2000). At the clinical level, expansion and contraction of these repeats are established risk factors for Huntington's disease, Myo- tonic dystrophy and several others genetic diseases (Borstnik and Pumpernik, 2002; Di Prospero and Fischbeck, 2005; Dushlaine et al., 2005; Richards, 2001; Sutherland and Richards, 1995). Simple sequence repeats (SSRs), also called as micro- or mini- satellites, are tandem repetitions of relatively short DNA motifs. Varia- tion in microsatellite distribution, complexity and copy number is high, owing to their unstable nature due to strand slippage and unequal recombination. This ensures a pivotal role in generating genomic diver- sity within and across species; which in turn provides a platform for evolution and adaptability (Deback et al., 2009; Kashi and King, 2006). Though their role in gene regulation has been the focus of studies for some time now and has been elucidated in few cases (Kashi and King, 2006; Usdin, 2008), but understanding their implications at functional and evolutionary level is still at a nascent stage. Different factors like ge- nome features such as size and GC content are known to influence the occurrence and complexity of microsatellites (Coenye and Vandamme, 2005; Dieringer and Schlotterer, 2003; Kelkar et al., 2008). However, this correlation is not universal and therefore, a single priority rule can- not be forged for predicting their occurrence and density. Moreover, variable length of microsatellites owing to differential copy numbers affects the local DNA structure or the encoded proteins and can be one of the means for regulating/altering the expression pro- file of the respective genes (Mrazek et al., 2007). Their presence in the realms of the virus world has been long debated but with several re- ports of their incidence and analysis in viruses like Hepatitis C virus (HCV) and Human cytomegalovirus (HCMV) (Chen et al., 2009; Picone et al., 2005) the onus now has shifted to understanding their enigma in the smallest entities of the living world. Based on the presence of interruptions, microsatellites may be interrupted, pure, compound, interrupted compound, complex and interrupted complex (Chambers and MacAvoy, 2000). Present study primarily focuses on pure and compound microsatellites (two or more microsatellites adjacent to each other). Their presence has been report- ed in diverse taxa across viruses, prokaryotes and eukaryotes (Chen et al., 2012; Gur-Arie et al., 2000; Kofler et al., 2008). Furthermore, the compound microsatellites constitute ~ 10% of SSRs in human genome (Weber, 1990), including highly polymorphic compound repeats such as (dC-dA)n(dG-dT)n (Bull et al., 1999). Other eukaryotic genomes like Macaca mulatta, Mus musculus and Rattus norvegicus have 4–25% of compound microsatellites (Kofler et al., 2008). An exhaustive study of the diversifications in satellite sequences would provide insight into the imperfections and evolution of microsatellites. Gene 537 (2014) 189–196 Abbreviations: SSR, simple sequence repeat; cSSR, compound simple sequence repeat; IMEx, Imperfect Microsatellite Extraction; RD, relative density; RA, relative abundance; RDRP, RNA dependent RNA polymerase; TGB, triple gene block; CP, coat protein. ⁎ Corresponding author. Tel.: +91 11 22623503; fax: +91 11 22623504. E-mail addresses: [email protected], [email protected] (S. Ali). 0378-1119/$ – see front matter © 2014 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.gene.2014.01.007 Contents lists available at ScienceDirect Gene journal homepage: www.elsevier.com/locate/gene
Transcript of Incidence, complexity and diversity of simple sequence repeats across potexvirus genomes

Gene 537 (2014) 189–196
Contents lists available at ScienceDirect
Gene
j ourna l homepage: www.e lsev ie r .com/ locate /gene
Incidence, complexity and diversity of simple sequence repeats acrosspotexvirus genomes
Chaudhary Mashhood Alam a, Avadhesh Kumar Singh b, Choudhary Sharfuddin a, Safdar Ali b,⁎a Department of Botany, Patna University, Bihar 800005, Indiab Department of Biomedical Sciences, SRCASW, University of Delhi, Vasundhara Enclave, New Delhi 110096, India
Abbreviations: SSR, simple sequence repeat; cSSR, comIMEx, Imperfect Microsatellite Extraction; RD, relative dRDRP, RNA dependent RNA polymerase; TGB, triple gene⁎ Corresponding author. Tel.: +91 11 22623503; fax: +
E-mail addresses: [email protected], [email protected]
0378-1119/$ – see front matter © 2014 Elsevier B.V. All rhttp://dx.doi.org/10.1016/j.gene.2014.01.007
a b s t r a c t
a r t i c l e i n f oArticle history:Accepted 4 January 2014Available online 13 January 2014
Keywords:PotexvirusSimple sequence repeatsImperfect Microsatellite ExtractionCorrelation studies
An in-silico analysis of simple sequence repeats (SSRs) in genomes of 32 species of potexviruses was performedwherein a total of 691 SSRs and 33 cSSRs were observed. Though SSRs were present in all the studied genomestheir incident frequency ranged from 11 to 30 per genome. Further, 10 potexvirus genomes possessed nocSSRswhen extracted at a dMAXof 10 andwherein present, the highest frequencywas 3. SSR and cSSR incidence,relative density and relative abundancewere non-significantly correlatedwith genome size and GC content sug-gesting an ongoing evolutionary and adaptive phase of the virus species. SSRs present primarily ranged frommono- to tri-nucleotide repeat motifs with a greatly skewed distribution across the coding and non-codingregions. Present work is an effort for the undergoing compilation and analysis of incidence, distribution andvariation of the viral repeat sequences to understand their evolutionary and functional relevance.
© 2014 Elsevier B.V. All rights reserved.
1. Introduction
A sizeable number of satellite sequences (mega-, mini- and micro-)have been characterized fromdifferent species (Jeffreys et al., 1998) andtheir presence had been established in coding regions as well. Theymaybe present as a part of the transcriptome or in the vicinity of the codingregions. The repeat sequences are known to be responsible for the reg-ulation of gene expression at transcriptional and translational level oreven by gene silencing (Li et al., 2004; Rocha et al., 2002; Vergnaudand Denoeud, 2000). At the clinical level, expansion and contraction ofthese repeats are established risk factors for Huntington's disease, Myo-tonic dystrophy and several others genetic diseases (Borstnik andPumpernik, 2002; Di Prospero and Fischbeck, 2005; Dushlaine et al.,2005; Richards, 2001; Sutherland and Richards, 1995).
Simple sequence repeats (SSRs), also called as micro- or mini-satellites, are tandem repetitions of relatively short DNA motifs. Varia-tion in microsatellite distribution, complexity and copy number ishigh, owing to their unstable nature due to strand slippage and unequalrecombination. This ensures a pivotal role in generating genomic diver-sity within and across species; which in turn provides a platform forevolution and adaptability (Deback et al., 2009; Kashi and King, 2006).
Though their role in gene regulation has been the focus of studies forsome time now and has been elucidated in few cases (Kashi and King,
pound simple sequence repeat;ensity; RA, relative abundance;block; CP, coat protein.91 11 22623504.m (S. Ali).
ights reserved.
2006; Usdin, 2008), but understanding their implications at functionaland evolutionary level is still at a nascent stage. Different factors like ge-nome features such as size and GC content are known to influence theoccurrence and complexity of microsatellites (Coenye and Vandamme,2005; Dieringer and Schlotterer, 2003; Kelkar et al., 2008). However,this correlation is not universal and therefore, a single priority rule can-not be forged for predicting their occurrence and density.
Moreover, variable length of microsatellites owing to differentialcopy numbers affects the local DNA structure or the encoded proteinsand can be one of the means for regulating/altering the expression pro-file of the respective genes (Mrazek et al., 2007). Their presence in therealms of the virus world has been long debated but with several re-ports of their incidence and analysis in viruses like Hepatitis C virus(HCV) and Human cytomegalovirus (HCMV) (Chen et al., 2009; Piconeet al., 2005) the onus now has shifted to understanding their enigmain the smallest entities of the living world.
Based on the presence of interruptions, microsatellites may beinterrupted, pure, compound, interrupted compound, complex andinterrupted complex (Chambers and MacAvoy, 2000). Present studyprimarily focuses on pure and compound microsatellites (two or moremicrosatellites adjacent to each other). Their presence has been report-ed in diverse taxa across viruses, prokaryotes and eukaryotes (Chenet al., 2012; Gur-Arie et al., 2000; Kofler et al., 2008). Furthermore, thecompound microsatellites constitute ~10% of SSRs in human genome(Weber, 1990), including highly polymorphic compound repeats suchas (dC-dA)n(dG-dT)n (Bull et al., 1999). Other eukaryotic genomeslike Macaca mulatta, Mus musculus and Rattus norvegicus have 4–25%of compound microsatellites (Kofler et al., 2008). An exhaustive studyof the diversifications in satellite sequences would provide insight intothe imperfections and evolution of microsatellites.

190 C.M. Alam et al. / Gene 537 (2014) 189–196
Also, viral microsatellites have the potential for generating genomicdiversity and phenotypic changes (Li et al., 2004). Their presence andpossible functional significance in plant viruses have been recognizedonly recently (George et al., 2012; Xiangyan et al., 2011). Here, we sys-tematically analyzed the occurrence, size, and density of differentmicrosatellites in the highly divergent potexviruses, a possible modelfor understanding functional aspects, evolutionary relationships, andadaptation to divergent hosts.
Potexviruses have positive-strand RNA genomes ranging from 4 to7 kb, which encodes for five open reading frames (ORFs). Virus particlesmostly occur in the cytoplasm and occasionally in the nuclei. They pos-sess methylguanosine cap at 5′ end and poly (A) tail at 3′ end (Huanget al., 2004; Huisman et al., 1988). The first ORF encodes the viral RNAdependent RNA polymerase (RDRP) required for replication, followedby three overlapping genes known as the triple-gene block (TGB 1, 2and 3) needed for virus cell-to-cell movement (Verchot-Lubicz, 2005)andORF endswith viral coat protein (CP)which is essential for virion as-sembly and virus cell-to-cell movement (Huisman et al., 1988; SantaCruz et al., 1998). Infection by these viruses causes chlorosis, necrotic le-sion, decreased leaf size, mosaic and ringspot symptoms inmonocotyle-donous and dicotyledonous plants. They lead to severe infection topotato, papaya, tomato, strawberry and someornamental plants causingsevere loss of revenue worldwide. They are transmitted through sap in-oculation and don't need any vector for its transmission but spreadsthrough contact between infected plants. Owing to presence of almostall species in India few of them like Mint virus X, Papaya mosaic virusand Potato virus X are candidates for infective genetics research in plants.
2. Materials and methods
2.1. Genome sequences
Potexvirus genus belonging to the family Alphaflexiviridae has 37known species according to the ninth report of the International
Table 1Overview of simple microsatellites in complete potexvirus genome sequences.
S. no Name Accession number
P1 Allium virus X FJ670570P2 Alstroemeria virus X AB206396P3 Alternanthera mosaic virus GQ179646P4 Asparagus virus 3 AB304848P5 Bamboo mosaic virus AF018156P6 Cactus virus X AF308158P7 Cassava common mosaic virus U23414P8 Clover yellow mosaic virus D29630P9 Cymbidium mosaic virus JQ860108P10 Foxtail mosaic virus M62730P11 Hosta virus X JQ911698P12 Hydrangea ringspot virus AY707100P13 Lagenaria mild mosaic virus AB546335P14 Lettuce virus X AM745758P15 Lily virus X AJ633822P16 Malva mosaic virus FZ416760P17 Mint virus X AY789138P18 Narcissus mosaic virus AY225449P19 Nerine virus X HQ166713P20 Opuntia virus X AY366209P21 Papaya mosaic virus D13957P22 Pepino mosaic virus JX866666P23 Phaius virus X AB353071P24 Plantago asiatica mosaic virus AB360796P25 Potato aucuba mosaic virus S73580P26 Potato virus X M72416P27 Schlumbergera virus X AY366207P28 Strawberry mild yellow edge virus AJ577359P29 Tamus red mosaic virus JN389521P30 Tulip virus X AB066288P31 White clover mosaic virus X06728P32 Zygocactus virus X AY366208
Committee on the Taxonomy of Viruses (ICTV) (King et al., 2012). Ofthe 37 species, complete genome sequence of 32 potexviruses availableat NCBI (http://www.ncbi.nlm.nih.-gov/) was assessed and analyzed forsimple and compoundmicrosatellites. The accession number of the stud-ied sequences, the species and their GC content has been summarized inTable 1. The genome size ranged from ~3.8 kb (P13) to 7.1 kb (P1).
2.2. Microsatellite identification and investigation
The search for microsatellites was performed employing the‘Advance-Mode’ of IMEx with parameters as reported for HIV (Chenet al., 2012); as in Type of Repeat: perfect; Repeat Size: all; MinimumRepeat Number: 6, 3, 3, 3, 3, 3; Maximum distance allowed betweenany two SSRs (dMAX) is 10. Other parameters were set as default.
2.3. Statistical analysis
We used Microsoft Office Excel 2007 for statistical analysis. Linearregression was used for correlation studies.
3. Results
3.1. Incident frequency of SSRs and cSSRs
Genome wide scan for microsatellites across 32 potexviruses re-vealed a total of 691 SSRs and 33 cSSRs (Table 1, SupplementaryTable 1). The presence of SSRs was observed in all the genomes buttheir incident frequency varied ranging from 11 (P20) to 30 (P4). Com-pound cSSRs are defined as two adjacent SSRs separated by less than apredefined maximum distance dMAX (Kofler et al., 2008). Tenpotexvirus genomes lacked cSSR at dMAX of 10 and in genomes harbor-ing cSSRs, themaximum observed frequency was 3 (P3 and P24). Inter-estingly, these variations are not directly correlatedwith genome size as
Genome size GC content SSR RA RD
7118 50.6 22 3.09 21.077009 51.4 26 3.71 26.686607 51.7 29 4.39 31.636935 55 30 4.33 28.556366 50.62 23 3.61 23.566614 51.17 24 3.63 24.196376 47.16 26 4.08 27.297015 49.42 27 3.85 25.236224 48.8 20 3.21 20.736151 52.41 18 2.93 18.706431 52.48 16 2.49 16.336185 58.02 26 4.20 29.913860 49.06 12 3.11 22.287212 54.63 23 3.19 22.325823 53.46 17 2.92 19.236858 44.8 23 3.35 22.165914 59.18 19 3.21 21.476956 47.25 26 3.74 24.736581 48.97 19 2.89 18.696663 49.87 11 1.65 10.966656 47.92 24 3.61 27.496412 40.72 14 2.18 13.575816 50.91 12 2.06 12.906102 56.58 29 4.75 31.637059 43.63 20 2.83 18.707568 47.97 26 3.44 23.396633 47.13 31 4.67 31.965970 53.11 13 2.18 13.746495 48.72 19 2.93 18.636056 57.11 18 2.97 19.655845 44.37 28 4.79 32.516624 53.56 20 3.02 19.93

191C.M. Alam et al. / Gene 537 (2014) 189–196
in neither the smallest genome (P13) has the least incidence nor thelargest genome (P1) accounts for the highest incident frequency.
3.2. Relative abundance and relative density
Owing to the variant incident frequencieswe looked into the relativeabundance (RA) and relative density (RD) for microsatellites. RA is the
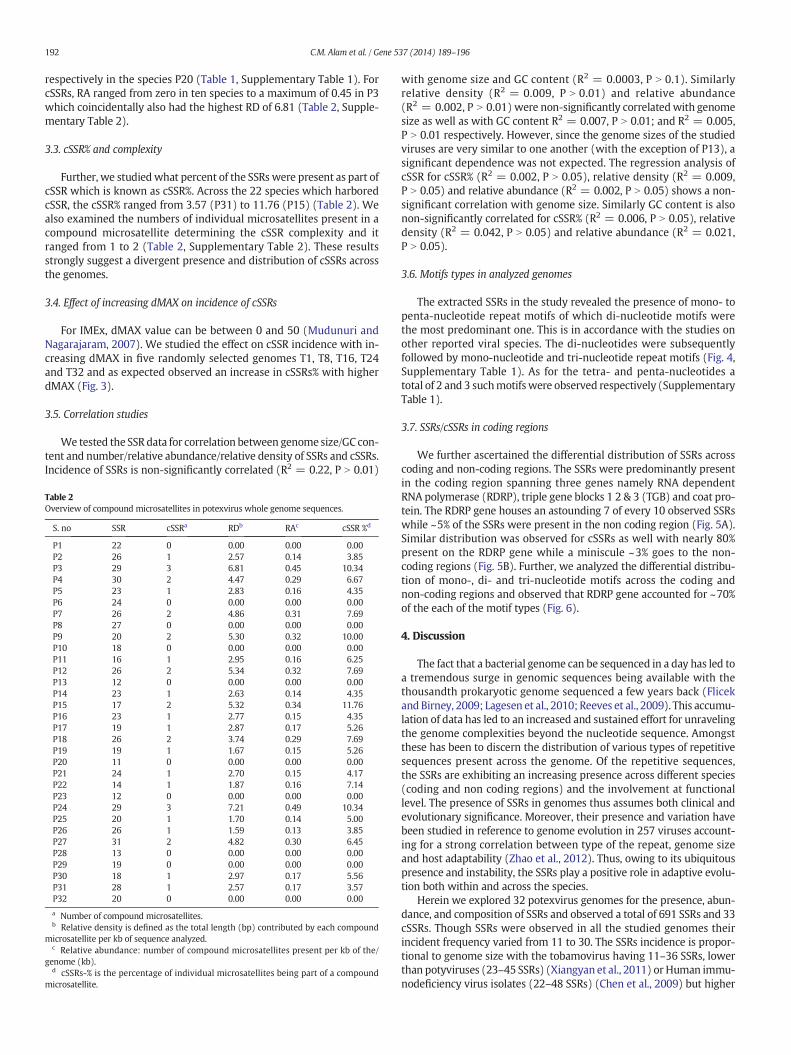
Fig. 1. Analysis of SSRs (A) distribution of SSRs; (B) relative abundance: SSRs present pe
number of microsatellites present per kb of the genome whereas RD isthe presence of microsatellites per kb of the genome. The distributionof RA and RD for SSRs and cSSRs across the genomes has been summa-rized in Tables 1, Fig. 1B-C and Table 2, Fig. 2B-C respectively.
For SSRs, P31 had the highest RA and RD of 4.79 and 32.51 respec-tively closely followed by P27 with respective values of 4.67 and31.96. The least observed values for RA and RD were 1.65 and 10.06
r kb of genome; (C) relative density: Total length covered by SSR per kb of genome.
192 C.M. Alam et al. / Gene 537 (2014) 189–196
respectively in the species P20 (Table 1, Supplementary Table 1). ForcSSRs, RA ranged from zero in ten species to a maximum of 0.45 in P3which coincidentally also had the highest RD of 6.81 (Table 2, Supple-mentary Table 2).
3.3. cSSR% and complexity
Further, we studiedwhat percent of the SSRs were present as part ofcSSR which is known as cSSR%. Across the 22 species which harboredcSSR, the cSSR% ranged from 3.57 (P31) to 11.76 (P15) (Table 2). Wealso examined the numbers of individual microsatellites present in acompound microsatellite determining the cSSR complexity and itranged from 1 to 2 (Table 2, Supplementary Table 2). These resultsstrongly suggest a divergent presence and distribution of cSSRs acrossthe genomes.
3.4. Effect of increasing dMAX on incidence of cSSRs
For IMEx, dMAX value can be between 0 and 50 (Mudunuri andNagarajaram, 2007). We studied the effect on cSSR incidence with in-creasing dMAX in five randomly selected genomes T1, T8, T16, T24and T32 and as expected observed an increase in cSSRs% with higherdMAX (Fig. 3).
3.5. Correlation studies
We tested the SSR data for correlation between genome size/GC con-tent and number/relative abundance/relative density of SSRs and cSSRs.Incidence of SSRs is non-significantly correlated (R2 = 0.22, P N 0.01)
Table 2Overview of compound microsatellites in potexvirus whole genome sequences.
S. no SSR cSSRa RDb RAc cSSR %d
P1 22 0 0.00 0.00 0.00P2 26 1 2.57 0.14 3.85P3 29 3 6.81 0.45 10.34P4 30 2 4.47 0.29 6.67P5 23 1 2.83 0.16 4.35P6 24 0 0.00 0.00 0.00P7 26 2 4.86 0.31 7.69P8 27 0 0.00 0.00 0.00P9 20 2 5.30 0.32 10.00P10 18 0 0.00 0.00 0.00P11 16 1 2.95 0.16 6.25P12 26 2 5.34 0.32 7.69P13 12 0 0.00 0.00 0.00P14 23 1 2.63 0.14 4.35P15 17 2 5.32 0.34 11.76P16 23 1 2.77 0.15 4.35P17 19 1 2.87 0.17 5.26P18 26 2 3.74 0.29 7.69P19 19 1 1.67 0.15 5.26P20 11 0 0.00 0.00 0.00P21 24 1 2.70 0.15 4.17P22 14 1 1.87 0.16 7.14P23 12 0 0.00 0.00 0.00P24 29 3 7.21 0.49 10.34P25 20 1 1.70 0.14 5.00P26 26 1 1.59 0.13 3.85P27 31 2 4.82 0.30 6.45P28 13 0 0.00 0.00 0.00P29 19 0 0.00 0.00 0.00P30 18 1 2.97 0.17 5.56P31 28 1 2.57 0.17 3.57P32 20 0 0.00 0.00 0.00
a Number of compound microsatellites.b Relative density is defined as the total length (bp) contributed by each compound
microsatellite per kb of sequence analyzed.c Relative abundance: number of compound microsatellites present per kb of the/
genome (kb).d cSSRs-% is the percentage of individual microsatellites being part of a compound
microsatellite.
with genome size and GC content (R2 = 0.0003, P N 0.1). Similarlyrelative density (R2 = 0.009, P N 0.01) and relative abundance(R2 = 0.002, P N 0.01) were non-significantly correlatedwith genomesize as well as with GC content R2 = 0.007, P N 0.01; and R2 = 0.005,P N 0.01 respectively. However, since the genome sizes of the studiedviruses are very similar to one another (with the exception of P13), asignificant dependence was not expected. The regression analysis ofcSSR for cSSR% (R2 = 0.002, P N 0.05), relative density (R2 = 0.009,P N 0.05) and relative abundance (R2 = 0.002, P N 0.05) shows a non-significant correlation with genome size. Similarly GC content is alsonon-significantly correlated for cSSR% (R2 = 0.006, P N 0.05), relativedensity (R2 = 0.042, P N 0.05) and relative abundance (R2 = 0.021,P N 0.05).
3.6. Motifs types in analyzed genomes
The extracted SSRs in the study revealed the presence of mono- topenta-nucleotide repeat motifs of which di-nucleotide motifs werethe most predominant one. This is in accordance with the studies onother reported viral species. The di-nucleotides were subsequentlyfollowed by mono-nucleotide and tri-nucleotide repeat motifs (Fig. 4,Supplementary Table 1). As for the tetra- and penta-nucleotides atotal of 2 and 3 suchmotifswere observed respectively (SupplementaryTable 1).
3.7. SSRs/cSSRs in coding regions
We further ascertained the differential distribution of SSRs acrosscoding and non-coding regions. The SSRs were predominantly presentin the coding region spanning three genes namely RNA dependentRNA polymerase (RDRP), triple gene blocks 1 2 & 3 (TGB) and coat pro-tein. The RDRP gene houses an astounding 7 of every 10 observed SSRswhile ~5% of the SSRs were present in the non coding region (Fig. 5A).Similar distribution was observed for cSSRs as well with nearly 80%present on the RDRP gene while a miniscule ~3% goes to the non-coding regions (Fig. 5B). Further, we analyzed the differential distribu-tion of mono-, di- and tri-nucleotide motifs across the coding andnon-coding regions and observed that RDRP gene accounted for ~70%of the each of the motif types (Fig. 6).
4. Discussion
The fact that a bacterial genome can be sequenced in a day has led toa tremendous surge in genomic sequences being available with thethousandth prokaryotic genome sequenced a few years back (FlicekandBirney, 2009; Lagesen et al., 2010; Reeves et al., 2009). This accumu-lation of data has led to an increased and sustained effort for unravelingthe genome complexities beyond the nucleotide sequence. Amongstthese has been to discern the distribution of various types of repetitivesequences present across the genome. Of the repetitive sequences,the SSRs are exhibiting an increasing presence across different species(coding and non coding regions) and the involvement at functionallevel. The presence of SSRs in genomes thus assumes both clinical andevolutionary significance. Moreover, their presence and variation havebeen studied in reference to genome evolution in 257 viruses account-ing for a strong correlation between type of the repeat, genome sizeand host adaptability (Zhao et al., 2012). Thus, owing to its ubiquitouspresence and instability, the SSRs play a positive role in adaptive evolu-tion both within and across the species.
Herein we explored 32 potexvirus genomes for the presence, abun-dance, and composition of SSRs and observed a total of 691 SSRs and 33cSSRs. Though SSRs were observed in all the studied genomes theirincident frequency varied from 11 to 30. The SSRs incidence is propor-tional to genome size with the tobamovirus having 11–36 SSRs, lowerthan potyviruses (23–45 SSRs) (Xiangyan et al., 2011) or Human immu-nodeficiency virus isolates (22–48 SSRs) (Chen et al., 2009) but higher
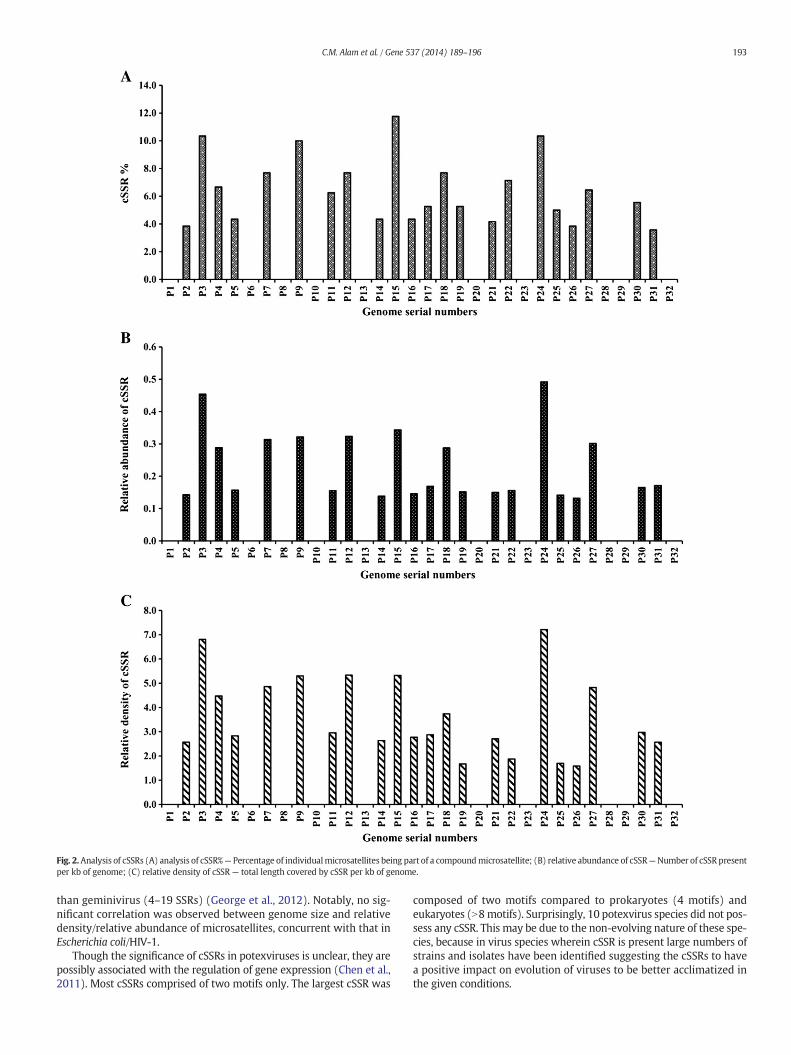
Fig. 2.Analysis of cSSRs (A) analysis of cSSR%— Percentage of individualmicrosatellites being part of a compoundmicrosatellite; (B) relative abundance of cSSR—Number of cSSR presentper kb of genome; (C) relative density of cSSR — total length covered by cSSR per kb of genome.
193C.M. Alam et al. / Gene 537 (2014) 189–196
than geminivirus (4–19 SSRs) (George et al., 2012). Notably, no sig-nificant correlation was observed between genome size and relativedensity/relative abundance of microsatellites, concurrent with that inEscherichia coli/HIV-1.
Though the significance of cSSRs in potexviruses is unclear, they arepossibly associated with the regulation of gene expression (Chen et al.,2011). Most cSSRs comprised of two motifs only. The largest cSSR was
composed of two motifs compared to prokaryotes (4 motifs) andeukaryotes (N8motifs). Surprisingly, 10 potexvirus species did not pos-sess any cSSR. This may be due to the non-evolving nature of these spe-cies, because in virus species wherein cSSR is present large numbers ofstrains and isolates have been identified suggesting the cSSRs to havea positive impact on evolution of viruses to be better acclimatized inthe given conditions.
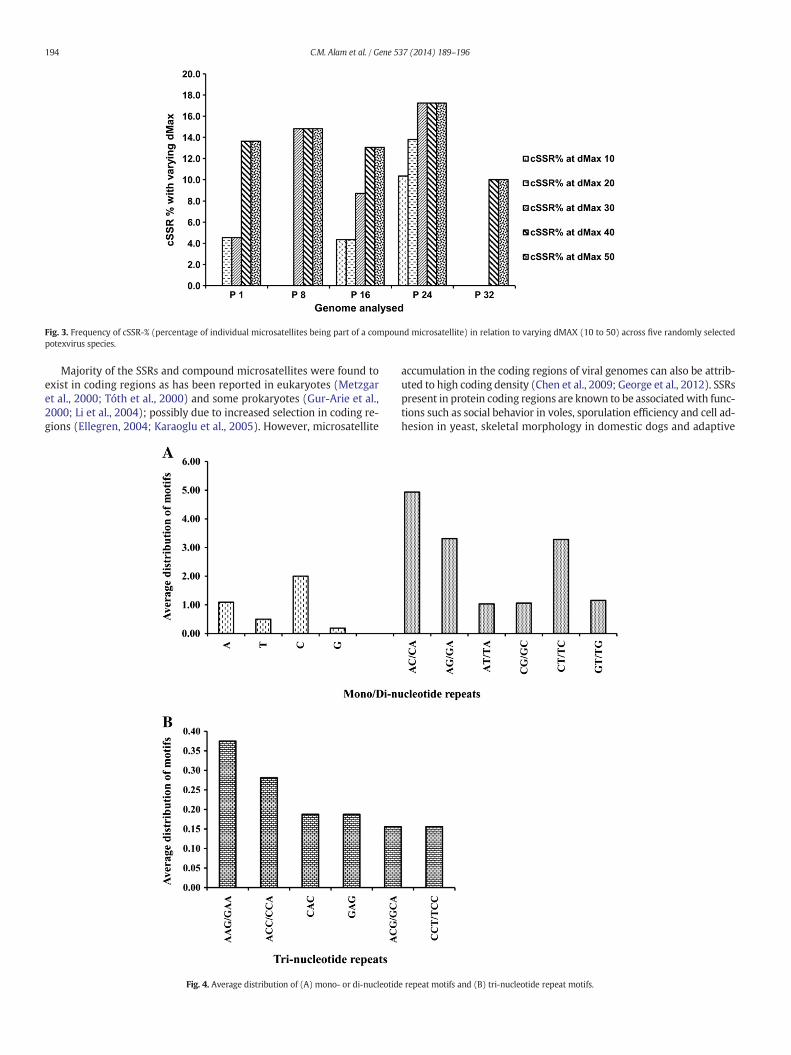
Fig. 3. Frequency of cSSR-% (percentage of individual microsatellites being part of a compound microsatellite) in relation to varying dMAX (10 to 50) across five randomly selectedpotexvirus species.
194 C.M. Alam et al. / Gene 537 (2014) 189–196
Majority of the SSRs and compound microsatellites were found toexist in coding regions as has been reported in eukaryotes (Metzgaret al., 2000; Tóth et al., 2000) and some prokaryotes (Gur-Arie et al.,2000; Li et al., 2004); possibly due to increased selection in coding re-gions (Ellegren, 2004; Karaoglu et al., 2005). However, microsatellite
Fig. 4. Average distribution of (A) mono- or di-nucleotid
accumulation in the coding regions of viral genomes can also be attrib-uted to high coding density (Chen et al., 2009; George et al., 2012). SSRspresent in protein coding regions are known to be associatedwith func-tions such as social behavior in voles, sporulation efficiency and cell ad-hesion in yeast, skeletal morphology in domestic dogs and adaptive
e repeat motifs and (B) tri-nucleotide repeat motifs.

Fig. 5. Differential distribution of (A) SSRs (%) and (B) cSSRs (%) in coding/non-coding regions of potexvirus.
195C.M. Alam et al. / Gene 537 (2014) 189–196
divergence in barley and wheat populations (Kashi and King, 2006).Though the information aboutmicrosatellite sequences is increasing ex-ponentially our understanding of the complexities involved in its
Fig. 6. Distribution of mono-, di- and tri-nucleotide SSR mot
diversity and resultant impact on the functional attributes is far fromsatisfactory. An in depth analysis of viral repetitive sequences in corre-lation to their evolution, host adaptability and regulatory mechanisms
ifs (%) across coding/non-coding regions of potexvirus.

196 C.M. Alam et al. / Gene 537 (2014) 189–196
may provide the platform for ascertaining its impact on genome diver-sity, complexities and function.
5. Conclusion
The complete relevance of our findings would help in the under-standing of the functional and evolutionary significance of viral repeatsequences. However, repetitive sequences are accepted hot spots forrecombination with recombination enzymes having high affinity fordi-nucleotide repeat sequences (Biet et al., 1999). The distribution biasobserved for SSR and cSSR at three loci namely, RDRP, TGB and coat pro-tein suggests their involvement in recombination, leading to sequencediversity and ultimately host adaptation. More detailed study of com-pound microsatellites in divergent viral genomes will pave the wayfor understanding complex biological features such as changes in viru-lence and their emergence as new epidemics.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.gene.2014.01.007.
Conflict of interests
The authors declare that they have no conflicts of personal, commu-nication or financial interests.
Acknowledgments
We thank the Department of Botany, Patna University and Depart-ment of Biomedical Sciences, ShaheedRajguruCollege of Applied Sciencesfor Women, University of Delhi for the financial and infrastructuralsupport provided.
References
Biet, E., Sun, J., Dutreix, M., 1999. Conserved sequence preference in DNA binding amongrecombination proteins: an effect of ssDNA secondary structure. Nucleic Acids Res.27, 596–600.
Borstnik, B., Pumpernik, D., 2002. Tandem repeats in protein coding regions of primategenes. Genome Res. 12, 909–915.
Bull, L.N., Pabon-Pena, C.R., Freimer, N.B., 1999. Compound microsatellite repeats: practi-cal and theoretical features. Genome Res. 9, 830–838.
Chambers, G.K., MacAvoy, E.S., 2000. Microsatellites: consensus and controversy. Comp.Biochem. Physiol. Biochem. Mol. Biol. 126, 455–476.
Chen, M., et al., 2009. Similar distribution of simple sequence repeats in diverse com-pleted Human Immunodeficiency Virus Type 1 genomes. FEBS Lett. 583,2959–2963.
Chen, M., et al., 2011. Compound microsatellites in complete Escherichia coli genomes.FEBS Lett. 585, 1072–1076.
Chen, M., Tan, Z., Zeng, G., Zhuotong, Z., 2012. Differential distribution of compoundmicrosatellites in various Human Immunodeficiency Virus Type 1 complete genomes.Infect. Genet. Evol. 12, 1452–1457.
Coenye, T., Vandamme, P., 2005. Characterization of mononucleotide repeats in se-quenced prokaryotic genomes. DNA Res. 12, 221–233.
Deback, C., et al., 2009. Utilization of microsatellite polymorphism for differentiating her-pes simplex virus type 1 strains. J. Clin. Microbiol. 47, 533–540.
Di Prospero, N.A., Fischbeck, K.A., 2005. Therapeutic development for triplet repeat ex-pansion diseases. Nat. Rev. Genet. 6, 756–765.
Dieringer, D., Schlotterer, C., 2003. Two distinct modes of microsatellite mutation pro-cesses: evidence from the complete genomic sequences of nine species. GenomeRes. 13, 2242–2251.
Dushlaine, C.T.O., Edwards, R.J., Park, S.D., Shields, D.C., 2005. Tandem repeat copy numbervariation in protein-coding regions of the human genes. Genome Biol. 6, R69.
Ellegren, H., 2004. Microsatellites: simple sequences with complex evolution. Nat. Rev.Genet. 5, 435–445.
Flicek, P., Birney, E., 2009. Sense from sequence reads: methods for alignment and assem-bly. Nat. Methods 6 (Suppl. 11), S6–S12.
George, B., Mashhood, A.C., Jain, S.K., Sharfuddin, C., Chakraborty, S., 2012. Differential dis-tribution and occurrence of simple sequence repeats in diverse geminivirus genomes.Virus Genes 45, 556–566.
Gur-Arie, R., Cohen, C.J., Eitan, Y., 2000. Simple sequence repeats in Escherichia coli: abun-dance, distribution, composition, and polymorphism. Genome Res. 10, 62–71.
Huang, Y.L., Han, Y.T., Chang, Y.T., Hsu, Y.H., Meng, M., 2004. Critical residues for GTPmethylation and formation of the covalent m7GMP-enzyme intermediate in the cap-ping enzyme domain of bamboo mosaic virus. J. Virol. 78, 1271–1280.
Huisman, M.J., Linthorst, H.J., Bol, J.F., Cornelissen, J.C., 1988. The complete nucleotide se-quence of potato virus X and its homologies at the amino acid level with various plus-stranded RNA viruses. J. Gen. Virol. 69, 1789–1798.
Jeffreys, J., Murray, J., Neumann, R., 1998. High-resolution mapping of crossovers in humansperm defines a minisatellite-associated recombination hotspot. Mol. Cell 2, 267–273.
Karaoglu, H., Lee, C.M., Meyer, W., 2005. Survey of simple sequence repeats in completedfungal genomes. Mol. Biol. Evol. 22, 639–649.
Kashi, Y., King, D.G., 2006. Simple sequence repeats as advantageous mutators in evolu-tion. Trends Genet. 22, 253–259.
Kelkar, Y.D., Tyekucheva, S., Chiaromonte, F., Makova, K.D., 2008. The genome-wide deter-minants of human and chimpanzee microsatellite evolution. Genome Res. 18, 30–38.
King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., 2012. Virus Taxonomy: Classifica-tion and Nomenclature of Viruses. Ninth Report of the International Committee onTaxonomy of Viruses. Elsevier, San Diego.
Kofler, R., Schlotterer, C., Luschutzky, E., Lelley, T., 2008. Survey ofmicrosatellite clusteringin eight fully sequenced species sheds light on the origin of compoundmicrosatellites. BMC Genomics 9, 612.
Lagesen, K., Ussery, D.W., Wassenaar, T.M., 2010. Genome update: the 1000th genome —
a cautionary tale. Microbiology 156, 603–608.Li, Y.C., Korol, A.B., Fahima, T., Nevo, E., 2004.Microsatellites within genes: structure, func-
tion, and evolution. Mol. Biol. Evol. 21, 991–1007.Metzgar, D., Bytof, J., Wills, C., 2000. Selection against frameshift mutations limits micro-
satellite expansion in coding DNA. Genome Res. 10, 72–80.Mrazek, J., Guo, X., Shah, A., 2007. Simple sequence repeats in prokaryotic genomes. Proc.
Natl. Acad. Sci. U. S. A. 104, 8472–8477.Mudunuri, S.B., Nagarajaram, H.A., 2007. IMEx: imperfect microsatellite extractor. Bioin-
formatics 23, 1181–1187.Picone, O., Ville, Y., Costa, J.M., Rouzioux, C., Leruez-Ville, M., 2005. Human cytomegalovirus
(HCMV) short tandem repeats analysis in congenital infection. J. Clin. Virol. 32, 254–256.Reeves, G.A., Talavera, D., Thornton, J.M., 2009. Genome and proteome annotation: orga-
nization, interpretation and integration. J. R. Soc. Interface. 6, 129–147.Richards, R.I., 2001. Dynamicmutations: a decade of unstable expanded repeats in human
genetic disease. Hum. Mol. Genet. 10, 2187–2194.Rocha, E.P., Matic, I., Taddei, F., 2002. Over-expression of repeats in stress response genes:
a strategy to increase versatility under stressful conditions? Nucl. Acid Res. 30,1886–1894.
Santa Cruz, S., Roberts, A.G., Prior, D.A., Chapman, S., Oparka, K.J., 1998. Cell-to-cell andphloem-mediated transport of potato virus X. The role of virions. Plant Cell 10,495–510.
Sutherland, G.R., Richards, R.I., 1995. Simple tandem repeats and human genetic disease.Proc. Natl. Acad. Sci. U.S.A. 92, 3636–3641.
Tóth, G., Gáspári, Z., Jurka, J., 2000. Microsatellites in different eukaryotic genomes: surveyand analysis. Genome Res. 10, 967–981.
Usdin, K., 2008. The biological effects of simple tandem repeats: lessons from the repeatexpansion diseases. Genome Res. 18, 1011–1019.
Verchot-Lubicz, J., 2005. A new model for cell-to-cell movement of potexviruses. Mol.Plant Microbe Interact. 18, 283–290.
Vergnaud, G., Denoeud, F., 2000. Minisatellites: mutability and genome architecture. Ge-nome Res. 10, 899–907.
Weber, J.L., 1990. Informativeness of human (dC-dA)n. (dG-dT)n polymorphisms. Geno-mics 7, 524–530.
Xiangyan, Z., et al., 2011. Microsatellites in different Potyvirus genomes: survey and anal-ysis. Gene 488, 52–56.
Zhao, X., et al., 2012. Coevolution between simple sequence repeats (SSRs) and virus ge-nome size. BMC Genomics 13, 435.


















