in - Shodhganga : a reservoir of Indian theses @...
47
T here has been intense interest in the coordination compounds of unsaturated sulphur donor chelating ligands, dithiocarbamates, and their related molecules from chemists, physicists, biologists and theoreticians alike owing to their interesting chemical properties and possible wide applications." Interest in molecular structural investigations and chemical studies of these metal chelates covers a full gamut of areas ranging from general considerations of metal-sulphur bonding and the formation of four- membered chelate rings to the employment of these ligands in inorganic qualitative analysis,j their practical application in organic synthe~is,~ medi~ine,~ and biol~gy,~ and their uses as vulcanisatiorl accelerators,' floatation agents,"' fungicides, pesticides" radiation protectors,12 antioxidantslbd photostabilisers of polymers.'-' Their role in material science has also been quite significant. The interesting low spin + high spin cross-over phenomenon was first reported in an iron(1II)dithiocarbamate complex.l5 There are several metal dithiocarbamate complexes with bridging sulphur centres whch are known to participate actively in super exchange phenomenon imparting novel magnetic properties to these systems.16 In this chapter, the interesting ligation characteristics of dithiocarbamates and structural features of their various transition metal complexes in general and the coordination chemistry and stereochemistry of copper complexes in particular, along with the scope of the present work which covers the
Transcript of in - Shodhganga : a reservoir of Indian theses @...

T here has been intense interest in the coordination compounds of
unsaturated sulphur donor chelating ligands, dithiocarbamates, and their
related molecules from chemists, physicists, biologists and theoreticians
alike owing to their interesting chemical properties and possible wide
applications." Interest in molecular structural investigations and chemical
studies of these metal chelates covers a full gamut of areas ranging from
general considerations of metal-sulphur bonding and the formation of four-
membered chelate rings to the employment of these ligands in inorganic
qualitative analysis,j their practical application in organic synthe~is,~
medi~ine,~ and b io l~gy ,~ and their uses as vulcanisatiorl accelerators,'
floatation agents,"' fungicides, pesticides" radiation protectors,12
antioxidantslbd photostabilisers of polymers.'-' Their role in material
science has also been quite significant. The interesting low spin + high spin
cross-over phenomenon was first reported in an iron(1II)dithiocarbamate
complex.l5 There are several metal dithiocarbamate complexes with bridging
sulphur centres whch are known to participate actively in super exchange
phenomenon imparting novel magnetic properties to these systems.16 In this
chapter, the interesting ligation characteristics of dithiocarbamates and
structural features of their various transition metal complexes in general and
the coordination chemistry and stereochemistry of copper complexes in
particular, along with the scope of the present work which covers the

relatively unattended aspect of the primary amine derived dithiocarbamates
are discussed.
1.1 Dithiocarbamates as ligands
The ligand system such as dithiocarbamates, xanthates,
dithiophosphates, dithiophosphinates, and dithiocarbimates are all referred
to as 1,l-dithiolates. Various nucleophiles (Z- or 212-) are capable of attacking
carbondisulphide to form 1,l-dithiolates (la) and (lb).
Metal ions can react readily with (la) and (lb) to yield complexes with the
possibility of the two sulphur atoms getting bound to the same metal,
forming a four-membered chelate ring. A wide variety of ligands can be
made available by merely varying Z as shown in Table 1.1.
Nucleophilic attack of secondary or primary amines on CS2 in alkaline
medium is known to generate R2N-CS2-M+, which can be considered as salt of
carbamodithioic acid.'
RzNH + CS2 + MOH -+ RzNCSSM + H20
The free dithiocarbamic acids are relatively unstable and only a very few
have been isolated.'7 The simplest member of the series, I-IlNCiSH can be
obtained as an unstable crystalline solid by acidification of a concentrated
solution of the ammonium salt.1"

Table 1.1. Major types of 1,l-dithioluto ligands
=Composition / Structure I Name 1
RzNCSi
S /- RS-C<t Q Thioxanthate
S
S /-
R--C<. @ Dithiocarboxy late S
ROCSz
S /-
R2NZC<. 0 S
The disubstituted dihocarbamates are more stable, although they
decompose under acidic conditions according to the equation,
Dithiocarbamate
S /-
KO-C;. o S
CS2-
R2PS2
- -
(R0)2152
-
R~AsSZ~
(R0)2AsS<
Xanthate
/ so S=C\
so R S \ /F
P I 0 R' \-i
RO S \ /-
P I 0 RO' \k R S
\ /C
As;. 0 R' S
RO, /.? A?. O
RO' S
Trithiocarbonate
Dithiophosphinate
Dithiophosphate
Dithioarsinate
Dithioarsenate

R ~ N C S ~ ~ carbamodithioate anion popularly known as dithiocarbamate ions
(Dtc), have function which does not, however, behave quite like the
sulphur analogue of carboxylate (COO-) moiety. The difference is brought
about mainly by the N atom which is directly connected to the carbon of CS2.
In a detailed IR study, Chatt et al. showed that a significant contribution from
structure (2c) was necessary in order to describe the electronic structure of
dithiocarbamate~.'~
Their conclusion is based on the presence of a strong absorption peak,
the 'thioureide ion' band, in the 1542-1480 cm-' region of the IR spectrum
which is observed for all of the dithiorarbarnic acid drrivativc.~. The
contribution of resonance form (2c) to the structure of Dtc ligands and
complexes was offered as a possible explanation for the varying antifungal
activities of these compounds.20 What becomes evident from the contribution
from these various resonance structures is that there is an extended
conjugation encompassing at least four atoms. This imparts special property
to the molecule including stability. Evidence for delocalisation over the four-
atom skeleton S2CN of the dithiocarbamate system was observed by
Colapictro et al. in the short C-S and C-N distances of 1.720 and 1.344 A respectively, in the structure of N ~ ( S ~ C N ( C Z & ) ~ ] . ~ H ~ O . ~ ~ The effects of the
conjugated n system are also seen in the hyperfi~w interactions observed in

the NMR spectra of certain alkyl and aryl dithiocarbamate complexes of
iron(II1). The double bond character of the C-N bond in dithiocarbamate
complexes should result in hindered rotation of the NR2 group. This effect
was observed in the Mo(RzDtc).l(NO) complex. 'I'he NMR spectrum of t h s
compound is consistent with non-equivalent alkyl groups on the NR2 moiety
as a result of hindered rotation.22
Dith~ocarbamates are versatile ligands capable of coordinating in a
variety of forms.'' The diverse nature of its ligation characteristics is
presented in (3) where the ligand moiety can act as a monodentate, chelating
bidentate or as a multidentate to two or even three metal centres.
\ s 'i-M \ /S-M \ />\ \ /* \ \ /> N ; e C 4S N-C, M NI;-;C, M / \;/ / \-/ / \- / N-C' / \ S, S , S-M /N -C\ S-M
M M
Almost all transition and non-transition metal ions exhibit strong
coordinating affinity to the dithiocarbamate moiety and there does not seem
to be any ring-strain instability for four-membered cyclic structure of the
resulting complexes. Excellent reviews ar- available on ligation
characteristics of dithiocarbamates and covering structural aspects of the
wide variety of their metal ~omplexes .~ -~ Unlike many other donor atoms the
sulphur centres in Dtc stabilise polynuclear complexes with mixed valence
state. Another important characteristic of the Dtc ligand is its ability to
accommodate metal ions in unusual oxidation stc>tes, especially remarkable
being its potentiality to stabilise transition metal ions in higher oxidation
states.2,' The stability of complexes with unusually high or low oxidation

I
states of the central metal atom depends largely OIL the possibility for charge
levelling by o-bonding and x-back bonding. While in the case of normal
1,l-dithiolates these two electronic affects of the sulphur atoms are
considered to be of the same order of magnitude, in the dithiocarbamato
ligands an additional x-electron flow from the nitrogen atom to the sulphur
atom via a planar delocalised n-orbital system would make them strong
electron donors able enough to accommodate metal ions at higher oxidation
states. In contrast to the transition elements, the main group element
dithiocarbamates often have asymmetrical metal-sulphur bonds due to the
lack of p.-d, interaction. In these compounds the a-bonds are responsible for
metal-sulphur interaction. High oxidation states for these dithiocarbamates
are only found when high electron density is brought upon to the metal by
o-donating groups. For instance, MeSb(R2Dtc)z exists whereas Me2ISb(R2Dtc)z
does n0t.I'
It is not surprising that dithiocarbamato compounds with copper in the
oxidation state +3 are stable; instead it must be regarded as unexpected that
Cu(I) dithiocarbamato complexes exist The latter complexes are not simply
monomeric, but they are tetrameric or polymeric metal cluster compounds.
Obviously, the stability must be attributed to the metal-metal bond rather
than the stabilising effect of the ligand. The same holds good for the
hexameric Ag(1) and dimeric Au(I) dithiocarbamates. In all other
dithiocarbamate complexes in which the metal has a low oxidation state the
existence of this type of compounds is due to other, low-oxidation-number
stabilising ligands. Examples NO+ in Fe(EtzDtc)z(NO)z and CO in
Fe(E tzDtc)(CO)r .>+

Table 1.2. Stable uridcrflcrflon states of some metal ions in their dithiocarbamate cc~npleues
Metal dithiocarbamate species
The strong donating abilities of the Dtc ligands are lost when the
nitrogen is bound to aryl groups as in an aromatic system like diphenyl
dithiocarbamate. The C-N stretching frequency which is located around
1500 cm-1 in the al~phatic dithiocarbamates is seen lowered in the aromatic
dithiocarbamates." It is found that the lone pair of the nitrogen atom in
dithiocarbamate complex becomes progressively more important for the
donation of electrons, the higher the oxidation state of the metal."
The metal-sulphur distances are consistently longer in the
1,l-dithiolato systems than in the bis 1,Zdithiolene (substituted and
unsubstituted ethene-1,2-dithiolates (4a) and benzene-1,2-dithiolates (4b)
complexes.

The reason for this difference is directly related to the electronic
structures of the ligand systems and also the way the ligand molecular
orbitals interact with the metal valence orbitals upon complexation. In
Figure 1.1, simplified pictures of the molecular orbitals of the two basic
ligand systems, viz., ethene-1,2-dithiolate a ~ d dithiocarbamate are
pre~ented.~ The 3n, function in the dithiolene ligands possesses correct
symmetry and appropriate energy to interact strongly with the metal d.
function to produce more stable, extensively delocalised molecular orbitals in
the metal complexes. Moreover, whereas the 3x, functions are filled when
the 1,Zdithiolene ligands are in their classical dianion formulation, the
orbitals are empty when the ligands are in their htghly oxidised dithione
formulation. It is possible, for these x functions to, therefore, serve as
acceptor orbitals, thus giving these ligand systems the x-acid character. This
type of interaction is not possible in complexes of the 1,l-dithiolato ligand
system such as dithiocarbamate because of the change in symmetry of 3n, of
SzCNHi. It is important to note that the x-acid character results primarily
from the 3n, function delocalised over the S-C-C-S backbone and not from
the vacant d orbitals of sulphur. If the use of the sulphur d orbitals were of
greater importance one would expect to see little change in metal-ligand n
bonding in going from the 1,2-dithiolene complexes to the 1,l-dithiolato
system. Thus the dithiocarbamate ligands exhibit little of the x acidity which
would serve to enhance the electrophilicity of, for example, the d9 Cu(Q ions
in their complexes. As a result, while the Cu(I1) dithiocarbamates are subject
to considerable axial ligation with coordinating Lewis bases, the adducts
formed by them are relatively unstable and cannot be isolated.2

a C-N
a a O ' C - N
Energy
Figure 1.1. A simplified representation of the zv nnzolecular orbitals of the hvo basic ligand systems ethene-1,2-dithiolate, s~c&?-, and dithiwarbamate, SzCNHi.
Normal coordinate analysis of some transition metal dithiocarbamates
have been reported.26-z Absorptions of diagnc~stic value occur in three
regions in the IR spectra. The 1450-1550 cm-1 region is associated primarily
with the thioureide vibration and is attributed largely to the v(CN) vibration

of SzC-NR2 bond. An increase in the double bond character of the C-N bond
(2) results in higher frequencies for this vibration.' A nearly linear correlation
is found in a plot of the v(CN) versus the methylene proton resonance from
lH-NMR data (6, ppm) on symmetric Et2Dtc complexes. An increase of the
partial positive charge on the nitrogen results in a desheilding of the
N-bonded methylene proton. But 6(CH2) and v(CN) are not interdependent
in complexes with asymmetric Et2Dtc bonding.2y An observed decrease in
v(CN) in the sequence R = Me > Et > Pr - Bu, is generally paralleled by the
calculated sequence obtained by increasing the po nt mass of the alkyl group
only. To reproduce the observed sequence, however, it was found necessary
to decrease the C-N force constant (f) in the seqLence Me > Et > Pr - Bu.
These results indicate that both electronic and kinematic effects are important
in determining V(CN).'~ According to Chatt et nl. the energy of the v(CN)
band falls roughly into groups according to the probable arrangements of
sulphur atoms around the central metal atom, the order of decreasing
frequency being planar > tetrahedral > octahedral > distorted octahedral >
pyramidal.19h The v(CN) band is known to undergo a blue shift in the
dithiocarbamato complexes with bidentate or multidentate bonding mode,
while for unidentate coordination this stretching is seen to be shifted towards
lower wavenumbers or remains unchanged at the value of the free
dithiocarbamate ammonium salt30
A second region between 950 and 1050 cm-I is associated with v(CS)
vibration and has been used effectively in differentiating between
monodentate and bidentate R2Dtc ligands. Two absorptions in the region of
1050-950 cm-1 is a diagnostic criterion for asymmetrically bound RzDtc
group.3' This criterion that distinguishes mo.~odentate from bidentate
bonding is shown to be valid, provided comparison is made between
complexes containing the same alkyl groups. It appears that the splitting of
the v(CS) vibration also should occur with unsymmetric bidentate bonding.
Monodentate bonding should be assumed only if the splitting exceeds
20 cm-l.Z8 Small splittings (-15 cm-1) of the v(CN) vibration also should occur
for unsymmetrical bidentate or monodentate binding of the RzDtc ligands.

Because of the ~nherently large width of the v(CIQ)-Emd W m a y not be,
however, observed." The absorptions in 300-400 cm-1 region is associated
with M-S vibrations.
By analysing the IR spectra of a numtc?r of bis(dithiocarbamat0)
palladium(II) complexes Sceney and Magee concluded33 that bands appear
and disappear in the region of v(CS) absorption in a completely random
fashion. Their intensity also vary without any apparent order. It appears
therefore, that v(CS) vibrational modes must be highly coupled with other
modes and are very sensitive to environmental changes.
1.2 Structural features of dithiocarbamate complexes
With 'pure' dithiocarbamate complexes in which no other coordinating
ligands are present, four definite structural types are obser~ed .~
(c) (4 (5)
(The four basic structure types observed for 'pure' dithiocarbamate complexes (a) the square planar coordination geometly, (b) the five coordinate dimer (c) the four coordinate dimer (d) the octahedral coordination geometty: (0) metal, (0) sulphur, ( 0 ) carbon, (0) nitrogen)

The first type (5a) is that of an essentially planar coordination
geometry which is found for all the structurally known bis complexes of
Ni(I1). In the complexes Ni(SzCNHz)z, Ni(S2CNEt;)z and Ni(SCN(n-GH7)z)z
the SC and C-N distances average 1.70 and 1.34 A respectively confirming
that the all the canonical forms (2a-2c) contribute to the electronic structures
of the ligands."~s The average SNi-S bond angle of 79" indicates the
magnitude of deviation from perfect square coordination. The average
intraligand S S distance is a very short 2.85 A and the average interligand S S
distance is 3.41 A. These values contrast sharply with the near equality of the
corresponding intra and interligands values in the monomeric bis
1,Zditluolene structures in which the planar arrangement is essentially
square.
Blauuw et RI. synthesised complexes of stoichiometry
AuX(SCN(~-(;HY)~) by the reaction of [Au(SzCN(n-GHY)~)] with various
halogens.17 The structure determination of this complex showed that it
consists of planar Au(LU) cations of [Au(SzCN(nGHr)2)2]+ and linear Au(1)
anion [AuBrz]( ).jX The structure of the interesting monodithiocarbamate
Cu(III) complex [CuBr2(SCN(nGHs)z)] reveals a strictly planar coordination
about the central metal.3y
Two basic types of dimeric structures have been observed for the bis
complexes containing dithiocarbamate ligands systems. The first structure
type is that of the five coordinate dimer observed in dithiocarbamate
complexes of copper(I1). The coordination in both the Cu(I1) dimers,
CUZ[SZCN(C~&)CI)Z]~ and C U ~ [ S Z C N ( ~ - G H ~ ) ~ ] ~ is best described as square
pyramidal.'"*l One of the Cu-S basal distances is slightly but significantly
larger than 2.312 A (of the other three) by 0.03 A and involves the sulphur
which serves as the apical atom to the centrosymmetrically related copper
atom. In the half dimer unit, the Cu atom is displaced out of the plane of the
four basal sulphur atoms by 0.26 A. The dimer linhagcs of 2.81 A (Cu-S) are
very weak and the [Cu(SzCNRz)z]z complex is found lo have a normal

monomeric molecular weight in such non polar solvents as benzene and
The dimeric arrangement found with such Cu(II)(RzDtc)2
complexes is pre-empted in Cu(MePhDtc)z, because of the orientation of the
phenyl rings. These rings are nearly normal to tl e plane of the rest of the
molecule, an orientation that appears to be dictated by the steric interactions
of the adjacent methyl s~bst i tuents .~~
The bridging dimer linkage has shrunk in the zinc complexes
Znz[S2CN(CH?)z)4 and Z~~[SZCN(CZ&)~]~ to a value of 2.383 A while the one
Zn-S basal distarre involving the bridging sulphur has increased frorn
2.342A to a value of 2.851 A.44.45 The primary function of one of the
dithiocarbarnate ligands has clearly changed from that of a chelating agent to
that of a bridging group. The coordination geometry about the Zn atom is
severely distorted and the four shortest Zn-S distances are directed to the
corners of a severely distorted tetrahedron (6).
(6)
[Idealised shucture of the Zn(Me2Dtc)z complex: (0) zinc, (0) sulphur, ( 0 )
carbon, (0) nitrogen]
The apparent strengthening of the dimer linkage is still not great
enough to maintain i t in solution. The molecular weight data indicate the
presence of monomers in non polar solvents such as benzene and
chlorofo~m.'~ The structure of Cdz(SzCNEtz)4 is also seen similar to that for
the zinc dimer.ib The two seemingly different structure types of the dimers
are still closely related to one another. Further, sbuctures in the intermediate
region between the two limiting geometries have also been obse r~ed .~

Structural studies on tris-dithiocarbamate complexes like
Ru(SCNMe2)3, Cr(SzCNMe& and Fe(SzCN(nE&,)& show the different
transition metal ions to possess significantly distorted octahedral
coordination geometries, the distortion being in the direction of triogonal
p r i ~ m . ~ ~ , ~ V o r example in the iron complex the sulphur donors are arranged
in two parallel equilateral triangles, one of which is rotated 32" (twist angle)
relative to the other (As shown in (7) the twist angle 0 behveen the upper and
lower triangles is 60" for octahedron and 0" for triogonal prism)
[Projection of the upper and lower hiangles of a geometry between an octahedron and Lriogonal prism. Definition of the twist angle O]
The distraction can be considered to be resulting from the strain of the
four-membered chelate rings and the relatively small 'bite angle' of the
1,l-dithiolato ligand system.
Metals like titanium, vanadium, thorium and niobium form complexes
of the compositions M(RzDtc)t. The crystal structure of Ti(Et2Dtc)r has been
determi11ed.4~ The complex contains eight coordinate Ti(1V) and chelating
Et~Dtc ligands. The coordination geometry of TiSs core is very close to
dodecahedral.

The monovalent metal ions Cu(I), Ag(I) and Au(1) are known to form
an interesting series of polymeric compounds with the general formula
[M(S2CNR2)In where n is the degree of polymerity. When M is Au(I), n equals
2; when M is Cu(I), n equals 4; and when M is Ag(I), n is found to be 6. In all
these metal clusters the oxidation state of the metal atom is +1.50.51 Cotton has
pointed out the necessity of low formal oxidation states for the metal ion in
order to achieve the formation of the M-M bond.52 Many sulphur containing
ligands favour cluster formation because these ligands can very effectively
delocalise and distribute the charges within the molecules. Along with this
levelling of charges the possibility of inter ligand S-S bonding may also play
an important role. Because of the great interest in cluster compounds and
metal-metal bonding in general, the structural studies of a large number of
complex compounds containing Dtc ligand systems have been made in detail.
The interesting and pertinent features in discussing these structures concern
the number and arrangement of metal atoms in the cluster, the metal-metal
distances, and the coordination of the sulphur atom around each metal atom.
In [Au(S2CNR2)]2 complex, the two Au(1) ions, which are separated by
only 2.76 A, are each coordinated in a linear arr.~ngement by two sulphur
atoms from different Dtc ligands. The two Dtc ligands can be considered to
be serving as bridging ligands between the Au(1) ions. The structure is such
that a twofold axis of symmetry passes through the two Au(1) ions and the
linear AuSz coordination is perpendicular to the metal-metal axis of
symmetry. Another twofold axis passes through the C-N bonds of both of the
Dtc ligands and the overall symmetry for the [AU~(S~CN(~€~H,)~] complex
can be taken as D2 or 222.s3,X The Au-Au distance is shorter than the
corresponding value found in the structure of the metal. Raman studies
indicate a contribution of resonance structures involving the entities shown
in (8).s5
Introduction 16
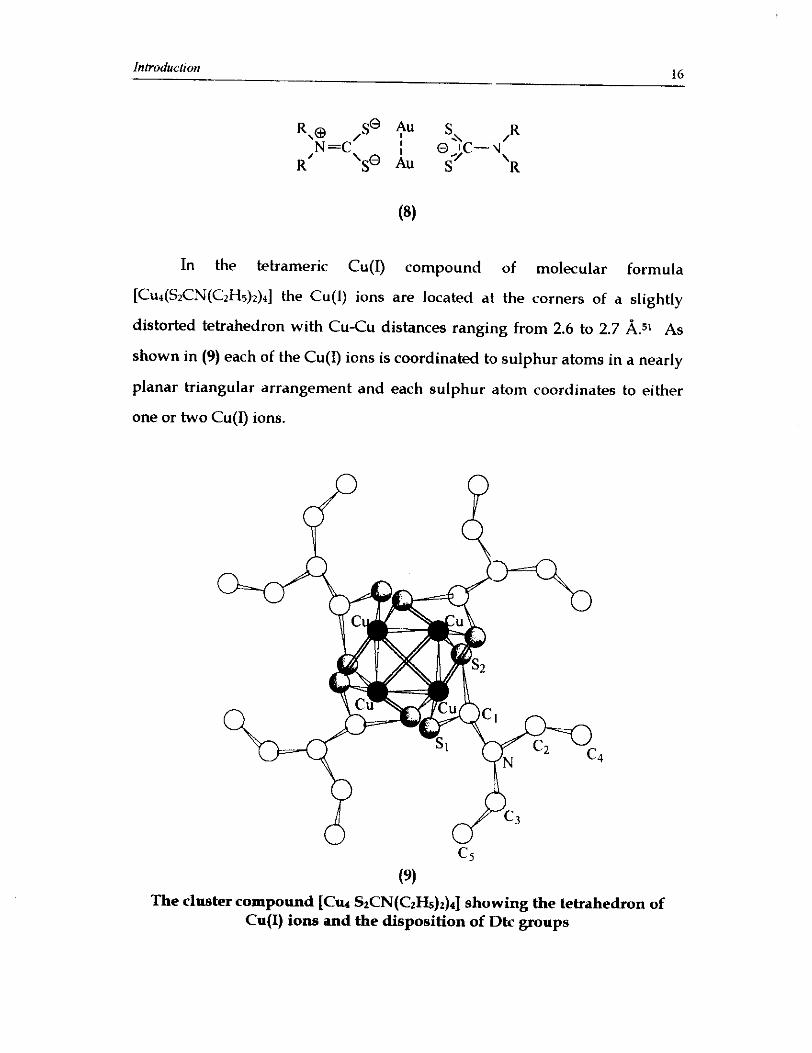
In the tetrameric Cu(1) compound of molecular formula
[CU~(SZCN(CZH~)Z)~] the Cu(1) ions are located at the corners of a slightly
distorted tetrahedron with Cu-Cu distances ranging from 2.6 to 2.7 A.51 AS
shown in (9) each of the Cu(1) ions is coordinated to sulphur atoms in a nearly
planar triangular arrangement and each sulphur atom coordinates to either
one or two Cu(1) ions.

Hesse also examined the structure of the hexameric
[Ag6(SzcN(C~tt)~)h] c o m p l e ~ . ~ The metal atoms I :Irm a somewhat distorted
octahedron with six comparatively short and six longer edges. The short
edges correspond to metal-metal distances, r hich are comparable or
somewhat longer than those in the metallic phase of silver. The long edges
form two centrosymmetrically related triangles in the silver octahedron.
Outside each of the other six faces of the octahedron one Dtc ligand is
situated, linked to the silver atoms of the face by silver-sulphur coordination,
one of the sulphur atoms is linked to one and the other to two silver atoms.
The silver coordination is threefold but not planar, the metal atoms being
situated 'inside' the plane of the coordinating sulphur atoms.
When the N-substituted alkyl groups are small, the hexameric Ag(I)
complex is very insoluble in organic solvents and appears to have some
polymeric properties. However, when more bulky alkyl substituents are
employed in the Dtc ligands the hexanuclear compounds show little tendency
to form higher polymers.
13 Copper--coordination chemistry
Copper occurs in a range of oxidation states 0 to 4. The Cu(0) and
Cu(1V) states are extremely limited. The Cu(II1) oxidation state is less
uncommon, and has been characterised for a few compounds including
copper dithiocarbamate ~omplexes.l,~,57,58 The Cu(I) and Cu(I1) oxidation
states are the most abundant for the metal. Cu(I1) is the more stable of the two
under normal conditions and form a wide variety of simple compounds and
complexes. The chemistry of Cu(1) is very much less extensive than that of
Cu(I1) and a number of accounts occur which describe the chemistry of
simple compounds of Cu(I) with less emphasis on the formation of its
cornple~es.~~~5~-61 The realisation that a copper(I) species may be involved as
the precursor of the silent partner in the type 111 copper p r ~ t e i n s ~ ~ ~ ~ has
resulted in a renaissance in the coordination chemistry of copper(1)

compounds which is reflected in the amount of space given to the chemistry
of copper(I) and (11) in the 'Advanced Inorganic Chemistry' of Cotton and
W i l k i n s ~ n . ~ ~ In the first edition of the book in 1952, more space was devoted
to copper(II) than to copper(I), while in the fourth edition the space allocation
is reversed.
1.3.1 Copper(11)compleres
Some of the salient aspects of Cu(I1) complexes are worth mentioning
here.
Copper(I1) forms complexes with coordination numbers four, five and
six, the latter being predominant A significant number of 7 and 8 coordinate
geometries also occur. Unlike other first row transition metal ions, the
copper(1I) complexes are characterised by a variety of distortions.65,66
Majority of six coordinate copper(II) complexes involve an elongated
tetragonal or rhombic octahedral structure, with a few involving a
compressed tetragonal structure. The tetrahedral geometry of Cu(I1) ion
always involves a significant compression along the S4 symmetry axis. Only
the square planar geometry is regular for Cu(II) ions, but even here it
involves a slight tetrahedral distortion. Copper(I1) ions with five
coordination rarely possess a regular square pyramidal geometry; it generally
undergoes both an elongation and a triogonal in-plane distortion," or less
frequently, a tetrahedral distortion. The triogonal bipyramidal geometry of
Cu(II) may be regular, but is frequently distorted towards a square pyramidal
stereochemistry.
Generally, Cu(L1) complexes are blue or green due to d-d electronic
transition causing absorption in the 600-900 nm regions. If there is a strong

charge transfer band spreading to the visible region the' complex appears red
or brown. Since copper(I1) ion is subjected to Jahn-Teller distortion and a
regular octahedral complex is not formed, the formal E, and Tzg terms get
splitted. The spectra do not usually correspond to the simple ZE, -+ 2T2g excitation67 but rather to one based on altered multiple states as shown
in (10).
(10)
[Splitting of 2Eg and 2T~g states in Cu(II)]
Tetragonal copper(Il) complexes are expected to show the transitions
~BI, + 2A~, 2B~, + 2B2, and ~BI, 2E, but bands due to these transitions
usually overlap to give often one broad absorption band.m.~~~
Four<oordinate Cu(I1) complexes are common, but the strict
tetrahedral or square planar stereochemistries are rare. Intermediate
stereochemistry of approximate D z ~ symmetry is more usual and four
transitions (between the d-orbitals) may be observed.b7b,7u The spectra of such
complexes often show two or three more or less resolved bands below about
20000 cm I. The polar~sahon properties of these bands have been studied in
some detail in certa~n cases assisting in the assignments of the transitions
1nvolved.67a "

1.3.2 Copper(1~0mplwolion characteristics
The Cu(1) and Cu(II) ions can readily form complexes in which the
cations act as Lewis acids and the ligands as Lewis bases. While Cu(I1) is
generally considered as borderline based acid, Cu(1) clearly behaves as a soft
acid and the nature of stability of the ligand to Cu(I) is that of a soft base with
class (b) behaviour (Table 1.3).72 In general the halides can form a wide range
of complexes, with the C1, Br and I ions predominant, but with very few
examples of the F ion acting as a ligand.73 Among the 0, S, N and P donor
ligands the 0 and N ligands dominate the chemistry of Cu(1I) while S and P
ligands are more frequent in Cu(I) chemistry.74~7~ 'This reversal of ligand role
is also influenced by the reducing properties of many S, I' and I ligands and
the ready reduction of the Cu(I1) ion (Equation 1.2) to the stable Cu(1) species
with these ligands.
Table 1.3. Hard and sop acid-base classification of copper ion and ligands
The hard-soft concept accounts for the following reactions and product
formations."
(a) CuCl~.H20 + KC1 So2(aq'q) ) K[CuC12]
Hard
(b) CuBrz + KBr -% K[CuBrz] lxrll
Border line
C ~ P P ~ ~ ( I I ) (a) Acid
(b) Base
(d) K[Cu(CN)z] Cuz+(aq) + KCN -t Cuz(CN)l excess
son C ~ P P ~ ~ ( I )
F- < C I ~ < B C ~ < l
O < < S = Se = Te
N < < P > A s > S b > B i
F~ > CI > Br- > I
0 ;.> S > Se > Te
N >> P > As> Sb> Bi

The electrode potentials of the reactions (1.1) and (1.2) readily lead to
the disproportionation as in equation (1.3) shown ldow.57
C U ~ + ~ , ) + ~ -+ CU+(,+ Eo = 0.15 V
2Cu+(,,, 4 Cu" + CU~+(.,), EQ = 0.37 V
Consequently the concentration of the Cu+ ion in aqueous solution is
extremely low (K for equation (1.3) being of the order of lo6) compared with
the very hgh stability of the Cu2+(aq) cation. For this reason water is rarely
found as a ligand to Cu(I), but is a common ligand in copper(1I) chemistry.74
The electrode potentials of reactions (1.1-1.3) get readily modified by
complex formation with appropriate ligands (Table 1.4).75
Table 1.4. (a) Reduction potentials of some Cu(II)/Cu(I) couples und (b) [Cu (Il)]/llcu (I)? ratios
(a) -
Cu(ll)lCu(l) Couple
C N ~ -
r c1- In laccasse
In ceruloplasmin
Loxalate < -0.2 I I 'en - HzN(CHz)zNH2; ' M e e n - HzN(CHz)3NHz; *Mesen - HzN(CH&NHz
bipy I:':: Glvcinate
0.15
0.12
-0.01
-0.16
-
-

In this way the concentration of Cu(I) in aqueous solution can be
significantly increased by complex formation such as in [CuC12]- and
[Cu(CN)2] anion by addition of an excess of tht. appropriate ligand. This
strategy can be made use of the preparation of various Cu(1) complexes.
Alternatively, Cu(I) can be brought into nonaqueous solution in which the
solvent is a good ligand for Cu(1) and forms complex. Such complex solution,
(for example, [Cul(NC-Me)r]X), can be used for the preparation of other Cu(1)
complexes by reacting with suitable ligands.
1.3.3 Copper(1) complexes-stereochemistry
Cu(1) can exist as mononuclear, bi-nuclear, tri-nuclear, tetra, penta and
exanuclear complexes. Even octa and decanucl~ar complexes have been
Many of the Cu(1) complexes are polymeric in nature and
possess chain and ribbon structures and infinite three dimensional lattices. In
all these complexes copper has the relatively low coordination numbers of
two, three and four and very rarely five.
In the solid state the stereochemish-y of Cu(1) in its mononuclear
complexes as determined by X-ray crystallography is dominated by four
coordination. The four- coordinate Cu(1) complexes are generally tetrahedral.
A significant number of three and two coordinate complexes are also known;
very few five coordinate complexes exist and six coordination or above is
unknown. This contrasts with the predominance of six coordination in the
chemistry of Cu(I1) and the absence of two or three coordination in the solid
state and with the formation of a significant number of seven and eight
coordinate geometries.65,m

Binuclear complexes form a significant class of Cu(I) complexes
involving bridgmg by one or two, but not three Iigand atoms or groups. In
practice the bridging role is most common for halide ions, especially iodides.
The resulting structures mainly involve a symmetrical arrangement of two
single atom bridges with either trigonal (llb), tetrahedral ( l la) and mixed
trigonal-tetrahedral (llc) copper stere~chemistries."~
(a) (b) (c) Tetrahedral Trigonal Mixed trigonal tetrahedral
[X = Brrdg~rig lrganJ whrc h may be (I) slngle anron, e.g., C1; (11) polyatomrc anlon, e.g., NCS; (111) organlc link]
(11)
[Stereochemishy of binuclear Cu(1) complexes]
Trinuclear complexes of Cu(I) are much less common than either
mononuclear or dinuclear complexes and occur ns a linear arrangement of
bridged Cu(1) atoms, as a triangular arrangement ar as a planar or puckered
six numbered ring with bridging X groups (12).W-%
x, lX\ /x\ f X Cu Cu , \ , \
X X X X
(a) (b) (c)
Linear Triangular Planar hexagorlal ring Puckered hexagonal ri
(12)
[Stereochemistry of hinuclear Cu(1) complexes)
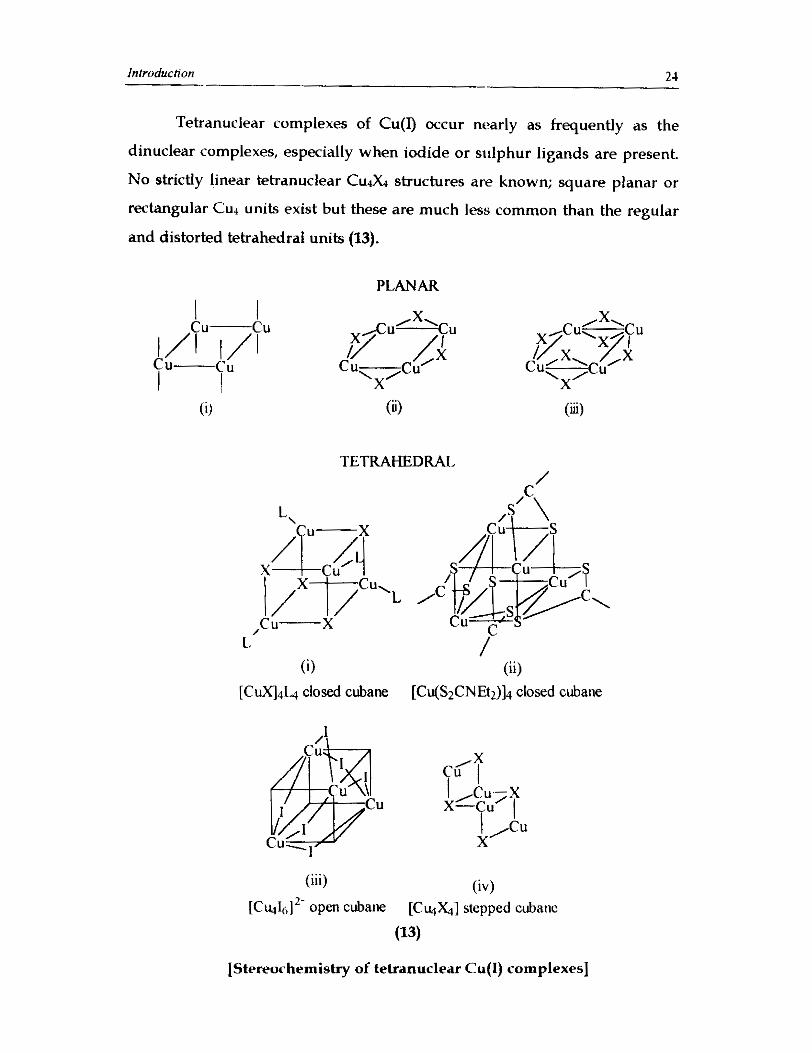
Tetranuclear complexes of Cu(1) occur nearly as frequently as the
dinuclear complexes, especially when iodide or si~lphur ligands are present.
No strictly linear tetranuclear C u A structures are known; square planar or
rectangular Cu4 un~ts exist but these are much less common than the regular
and distorted tetrahedral units (13).
PLANAR
( i ) (4 (i)
TETRAHEDRAL 0 /
s*\
X flLt7\
1 ,x+.cu\L /+q->':\ Cu- X cu-
L ' / (ii)
[CUX]4L4 closed cubane [Cu(S2CNEt2)I4 closed cubane
(iii) . .
[ cuJ I~ ]~ - open cubane [C4X4] stepped cubane
(13)
[Stereochemistry of tetranuclear Cu(1) complexes]

Introduction 25
The tetrahedral Cu,X4 is the most common tetranuclear Cu(1) species
and primarily occurs as the 'cubane' structure C U J X L where X is C1-, Br or
r.",m A relatively complex cubane type C U ~ S ~ units occurs in [Cu(S2CNEtz)]4
in which one of the S atoms constitutes the Cu& and the second S of each
S2CNEt2 group symmetrically bridges two copper atoms on the four side-face,
Cu-Cu pairs.5' Pentanuclear and hexanuclear conlplexes are only of limited
occurrence.
The Cu(1) complexes so far described are characteristic with their
relatively low coordination numbers of two, three and four. Such low
coordination numbers are conducive57 to bridging ligand functions and hence
to infinite lattices involving chain (or ribbon), sheets and three dimensional
lattices.&Y In (14) are shown the varieties of structures possible for polymeric
copper(I) complexes.
One of the most structurally intriguing complexes of Cu(1) is one with
the CuOzCMe composition. Its structure retains the unique acetate ligand
bridging role of the,Cu(II) complex, but this is now restricted to planar
dimer~c un~ts, llnked by further oxygen br~dges into a staggered linear
polymer whlch 1s planar overall (16a).W If Cu-Cu distance of 2.556(2) A is
considered non-bonding, the Cu03 chromophore is unusual as it has a T
shaped structure that involves two angles much less than 120°, namely 80.4
and 110.5", with a t h ~ r d 0-Cu-0 angle nearly linear at 170.1". The electron
diffraction structure of anhydrous cuprous acetate has been reported to
~nvolve a drmeric planar sbucture related to the above solid state structure
with relatively shorter Cu-Cu distance of 2.491 A (Figure 1.12b).y1

CllAlN AND RIBBON STRUC'I'URI:~
S~ngk churn
(1) (h) Two-coordinate Three-cwrdnate--pla~lar or stepped
\ ,Cu, ,Cu, ,Cu
C u \ ,X\ ,X, / Cu, ,Cu, ,Cu
x ' x "(' x \ % (") (\I) (w)
Four-coordhate Stepped straight 'Twisted step
(U) (W I h u h k nbbon Pkuuir
INFINITE. TWO-DIMLXSIONAL SkEETS
( 1) (u) INFlNl fE TIIREE-DIMENSIONAL IATTICHS
X X X I I I
,cu-cu*cu, X X X X X X
(14) [Stereochemistry of polymeric copper (I) complexes]

TH3 YH3 '9-c\9 7"' 'cu ---- u. A,
9'c'~ I T P P 0 \ 0 'cu...-- 7"---"7" 7 I F \ CHi 0,
O'F'O
C C H3
C H3
(a) (b)
(16)
[(a) Polymeric Cu(1) acetate and (b) Dimeric Cu(1) acetate]
1.3.4 Redox properties of copper(l)/(l) systems
Redox propert~es of Copper(I)/(II) systems are associated with three
p r o c e s s e ~ : ~ , ~ ~ , ~ ~ (a) electrolytic oxidation or reductions (Eqn. 1.4); (b)
disproportionation (Eqn. 1.5); (c) the oxidation of copper(1) with molecular
ultimately yielding water (Eqn. 1.6).
Electrolytic reduction of Cu(1) to copper metal is readily carried out as the
second step in the electrolytic refining of copper metal and in the copper
plating process. Although the disproportion reaction is favourable in aqueous
solution, the low solubility of the Cu(1) species in water renders this reaction
less important than the oxidation to Cu(I1). Complex formation in solution
notably increases the solubility but also increases the stability of the Cu(1)
species to the disproportionation process. Due to this the coordination
chemistry of the copprr(I)/(II) redox process is primarily concerned with the

reduction of copper(I1) to copper(I), a process that has been already discussed
(Table 1.4).
There are a number of reports of many organic molecules being
reduced by cupric ions to form copper(I) complexes.% Trimethyl phosphine
sulphide, for example, reacts with Cu(I1) salts in acetone medium forming
Cu(I) complexes with compositions [Cu(Me3PS)CI], and [Cu(Me3PS)3] X(X- =
CIOI- or BF4). X-ray crystal studies show the complex to contain three
t,oordinate ~ o p p e r ( I ) . ~ , ~ ~
CuClz.2HzO is known to get reduced by dithiobiuret (17) in a 95%
ether-ethanol solution to give, as one of the products, an olive green material
with structure (IS).*
This is an unusual example of a complex bearing a positively charged
ligand moiety.
Interesting copper(1) complexes have also been reported to form on
addition of copper(I1) salt to a refluxing solution of excess ligand 1-
methylimidazoline-2(3H)-thione (meimtH) in acetone-acetonitrile. The
complexes species formed are [Cu(meimtH)j]N03 and [Cu:(meimtH)6](BF~)z
as well as [Cuz(meimeH),-1%. 3H20 from aqueous acetone.lw"J2

The oxidation of copper(I) with molecular oxygen ultimately yielding
water (Eqn. 1.6), a process associated with the redox properties of copper(Q
system, merits attention. This is the process that is responsible for the air
sensitivity of copper(1) compounds, and equally for the catalytic role of
copper(I) in the oxidation of organic mole~ules . '~~~~* Copper(I), for example,
is the catalyst in the oxidation of dimethyl sulphoxide to dimethyl sulphone
using Cu(NCMe)dBF, and in the oxidative coupling of phenols to
q u i n ~ n e s . ' ~ ~ , ' ~
1.3.5 Biological copper
In biological copper systems the copper is involved in three basic
p r o ~ e s s e s : ~ Z - ~ , ~ . ~ ~ - ~ ~ ~ (1) electrolytic redox processes, involving copper(III)/(I),
(2) oxygen atom processes, involving the absorption of oxygen molecules and
their ultimate reduction to water,'"lW (Cu @)/(I) systems) and (3) transport
processes,* through which copper is absorbed and rejected by the body. In
general, copper(1) acts as the electron donorl'u for processes (1) and (2)
involving copper proteins, while in process (3) copper(I1) is known to be
primarily involved. As for the nature of interaction of copper(I) with protein
there is, however, surprisingly little information available. The copper(I) site
is generally considered to be a hydrophobic area in the protein and the
characteristically positive reduction potentials of the copper biological
systems suggest that the reduced state of the copper is compatible with stable
copper(1) stereochemistry. This is generally assumed to involve a distorted

tetrahedral geometry. The more positive reduct i~n potentials may also be
associated with the 'similarity' of stereochemistry in both the copper(I) and
(11) oxidation states and in this respect a common tetrahedral stereochemistry
might well be appropriate as this geometry is known to be stable for both the
oxidation states.
1.4 Copper complexes of dithiocarbamates-a review
The d~thiocarbamates derived from both primary and secondary
amines show comparable ligation characteristic^',^ with almost all metal ions.
It is worthwhile to have an overview on copper complexes of
dithlocarbarnales, whlch could throw light on the ligation characteristics and
nature of interachon of both N-monosubstituted and N-disubstituted
dithiocarbamates with copper ions.
The chemistry of univalent copper dithiocarbamates has been studied
in considerable detail. The Cu(I) complexes of the disubstituted
dithiocarbamates (derived from secondary amine) Cu(I) complexes are not
much stable and their solutions oxidise to the corresponding cupric
complex."' l'he complexes of monovalent copper can be obtained by the
following methods: (a) treatment of Cu(1) oxide with the sodium salts of the
ligands in an inert atmosphere, (b) oxidation of copper, bronze or metallic
copper with tetra alky l thiuram disulphides in chloroform or benzene, and (c)
reaction of alkali metal Dtc with a cuprous halide in aqueous c o n d i t i o n . ~ ~ ~ ~ ~ 7
Kkerstrom conducted detailed molecular weight studies on these complexes
and showed that Cu(1) dithiocarbamates usually exist as tetramers in CSz or
benzene solution. It appears that the size and structural characteristics of the
alkyl chains on the ligands play an important role in determining the degree
of p ~ l y m e r i t y . ~ ~ ~ ~ " ' ~ The crystal structure of (CuEkDtc), complex has been
determined."l The disubstituted dithiocarbamates of Cu(l1) are stable, watrr-

-- --
I I . : . , ';. insoluble compounds.~~5 'The these species along with
other similar complexes are already discussed in Section 1.2. Cu(II) reacts
quantitatively with Pb(RzDtc)z, Bi(R~Dtc)3 and Tl(RzDtc)? complexes to form
the deep brown-red Cu(R2Dtc)z complex. These re rctions have been used for
the indirect determination of Pb(II), Bi(II1) and TI(II1) ions.l16 Crystal structure
determination of the Cu(Et2Dtc)z has shown that it is a dimer with five-
coordinate Cu(I1) i o n ~ . ~ ' ~ , ~ l W e w r n a n et al. have shown that Cu(pyrrole Dtc)~
is isomorphous to the square planar Ni(pyrrole Dtc )~ and that the former is
monomeric with a square planar CuS4 chromophore."Y The tendency of the
pyrrole ring to preserve aromaticity makes the resonance form RZN+=CS,~-
quite unimportant in the structure of the ligand. Complexes of this ligand
including Cu(I1) complex, show the C-N vibration between 1250 and
1350 ~ m ~ ~ . l L "
The EPR spectra of Cu(R2Dtc)z complexes have been investigated in
considerable detnil.ljll2' Viinngard and Akerstrom examined the room
temperature EPK spectra of the Cu(Et2 Dtc)z, Cu(i-pr Dtc)2 and Cu(MePhDtc)z
complexes in benzene solution and found them 1.2 be almost ide1itical.'2~ A
typical spectrum consists of a quartet with g = 2.046 and A = (0.74 f 0.02)
cm-I. Patterson and Viinngard obtained the EPR spectra of polycrystalline
samples and calculated the parameters and evaluated the degree of covalency
of the Cu-S bonding in them.122 They concluded that Cu-S o-bond is
appreciably covalent. The EPK studies show that, in general, covalency in the
M-S bonds increases strongly in the order: Cu < Ag < Au and that the metal
3d character of the MO of the unpaired electron decreases from 50% in
Cu(EtzDtc)z to 26% in Ag(HzDtc)z and 15% in the homologous Au(II)
c o m p l e ~ . ~ ~ , ~ ~ ~
Interaction of Cu(Bu2Dtc)z with pip, py and n-hexylamine has been
studied by variable temperature EPR measurements. Evidence for the

formation of 1: I adducts was presented by Gorden and Rieger.'26 In a similar
study of EPR and absorption spectra of the Cu(pipDtc)? and Cu(EtBzDtc)~
complexes, Yordonov and Shopov observed the foi.mation of 1:2 adducts with
py having no evidence for the 1:l a d d ~ c t l ~ ~ This shows the influence of R2
group on the ability of the Cu(R2Dtc)z complexes to form base adducts of
varying compositions. Yordanov et al. studied thc interaction of Cu(1I) ions
with Ni(Et2Dtc)~ and Cu(Et2Dtc)z and detected the formation of CuX(Et2Dtc)'
and Cu(Et2Dtc)' spe~ies.lzJ2~
A detailed electrochemical study on sixteen different Cu(R2Dtc)z
complexes has been reported."' The oxidation of the Cu(R2Dtc) complex is
unambiguously defined as the one-electron reversible process.
The reduction of the Cu(R2Dtc)z complexes often appears quasi
reversible, presumably owing to a structural reorganisation of CuSd core of
the [Cu(RzDtc)?] product.
Although the monoanion could not be isolated the solutions containing
this species show reversible two one-electron oxidation step by cyclic
voltammetry. In general, for the reduction step given above the potentials are
more positive by about 1 volt than the corresponding potentials in the
reduction of Ni(II2Dtc)z complexes and illustrate convincingly the
~nvolvements of the metal ion in the reduction proc-ess.
Oxidation of the Cu(Bu2Dtc)z or [Cu(Buz Dtc)]4 by halogens results in
the formation of the diamagnetic square planar monomeric [Cu(BuzDtc)Xz]
complex (X = C1, Br).I3l Brinkhoff obtained [Cu(R2Dtc)2]+ cation by the
oxidation of the Cu(lI) complexes with iodine.132 The increase in the Cu-S
stretching frequency observed in the Cu(R2Dtc); cations (385, 410 cm-')

compared to that in the Cu(R2Dtc)z complexes (- 345, 370 cm-l) has been
attributed to the strengthening of the Cu-S bond as a result of removing an
electron from an antibonding orbital composed primarily of metal and
sulphur functions. The structure of the [Cu(Bu~Dtc)z]G complex shows a
Cu(II1) ion with a four coordinate planar geometry.llJ
M~xrd Cu(llI), Cu(l1) valence complexes of the general formula
[Cu3(Bu~Dtc)b] [M Br+ (M = Zn, Cd, Hg) were obtained by interacting
Cu(BuzDtc)2 with MBrz and Br2 in stoichiometric amounts.lw The X-ray crystal
structure of [Cu3(Bu2Dtc)6] [CdzBr6] shows a loosely held centrosymmetric
trimer.lS
Van der Leemput et al. prepared the paramagnetic Cuz(RzDtc)&z
complexes with copper in the oxidation states(I1) and (111). These compounds
were obtained by the interaction of CuCl or CuBr and b t d s in chloroform.
'The crystal structure of Cu2(H>Dtc)3Brz consists of alternaling Cu(H2Dtc): and
Cu(H2Dtc) Br; units in chains.lX
The reaction between CuBr and b t d s in CH2Cl2 in various molar ratios
resulted in a number of polynuclear copper complexes which can be
represented by the general formula [Cu(R2Dtc); ] [CU,,.~ Br,,](-) with n varying
as a function of R group. Values of n are 7, 5, 3 for n-Bu, n-Pr and Et
Refluxing [Cu(Et~Dtc)]4 in CHCb yields a compound Cu3(EtzDtc)zCI in
which copper is in the +1 oxidation state.Iw Sholozhenkin et 01. studied the
reaction of Cu(I1) with the ambidentate ligand, carboxydecyldiethyldithio-
carbamate.I3' CuL2,HzO [HL = E~zNC(S)S(CH~)IOCOOH] was prepared from
CuSO4 and NaL in aqueous solution. The complc~x in saturated chloroform
solution exists as CuSI chromophore and with dilution isomerises to CuS30,
CuS202 and Cu01 chromophores.

Jean-Pierre Barbier et nl. have shown that irteraction of Cu(BF4)2.6H20
or Cu(ClO4)z. 6Hz0 with a solution of Cu(Et2Dtc); leads to the less common
disproportionation, copper(11) into copper(1II) and copper
4Cu1'(Et2Dtc)z +2Cu"(BFp)2 + 3CuU (EtzDtc)~ BF4 + Cu0I(EtzDtc)2 BF4
The formation of a very stable copper(1) complex, probably a cluster,
may be the driving force for this reaction.
Dani rt nl. have prepared and characterised bimetallic tetramorpholine
-4-carbodithioates of the composition MM'(med.t)+ where M = Cu(I1) or
Ni(II), M' = Zn(II), Cd(I1) or Hg(II) and medt = morpholine-4-carbo-
dithoate.1" These complexes are reported to exhibit enhanced antifungal
activity. Square planar stereochemishy around Cu(1I) and Ni(I1) has been
proposed for the polymeric complexes.
Yusuff studied the action of thionyl chloride on bis(dithiocarbamat0)
copper(1I) complex in benzene or CCl4 solution.la This provides a simple
alternative route to the synthesis of dichloro(dithiocarbamato) Cu(1II)
complexes. l 'he following reaction scheme has been proposed
Cu(Dtc)z + Cl2 -+ [Cu(Dtc)C12] + Dtc
The various types of copper dithiocarbamate complexes prepared so
far are presented rn Table 1.5.
Tuhle 1.5. Types of copper dithic~urbamute complexes found so fur r
Oxidation Number - I
I1
III
II, I
111, I
111, 11 -
Compound
[Cu(RzDtc)]4 Cu3(RzDtc)2X
[Cu(RzDtc)z]z [X Cu (R2 Dtc)]2
[Cu(RzDtc)z](+)X(-I XzCu(R2Dtc)
Cu(R2 Dtc)~ nCuBr C13Cu3 (RzDtc),
[Cu(R2Dtc)2+] [CU,.~ Br,] [Cu(R2Dtc)+2]~ [ C U ~ ~ r i - ]
[Cul(Rz ~ t c ) ? ] [MX;]z Cuz(RzDtc)+ 13 CUZ (R2Dtc)~ Br2

The types of copper dithiocarbamate complexes sun~marised above
shows the remarkable variety in the permutation of R2Dtc ligands, halide ions
and various oxidation states of copper.
1.4. I Copper complexes of dithiocarbamates derived from primary amines
During their investigations on the interi~ction of Cu(II) ion with
RHDk, Compin and Malatesta143J44 noted the generally instability of
Cu(RHDtc)z complexes. However, it was Cambi and Coriselli who first
reported that Cu(RHDtc)z complexes were unstable and rapidly decomposed
to the corresponding Cu(1) compounds."' Because of the very high
decomposition rate they could not isolate any Cu(RHDtc)z even if they are
formed. They reported that the decomposition of the complexes was
complicated. However, they claimed the formation of isothiocyanate during
the decomposilion.
Kawshik ! 111. synthesised a series of bis-N(p-ethoxyphenyl)
dithiocarbamate complexes of Cu(II), Zn(II), Cd(II), Sn(II), Ni(1I) and Pd(I1)
with composition, M[Eto-G&-NHCS2]2.'45 These complexes have been
characterised; the metal ligand ratio in all the complexes including the copper
complex was shown to be 1:2. Thermal studies of these complexes have been
carried out in static air atmosphere to dcterminc their mode of
decomposition. l n e t i c parameters have been determined.
Kaushik et nl. prepared another series of complexes,
bis-N(o-chloropheny1)dithiocarbamate and bis-N(p-chlorophenyl)
dithiocarbamate complexes of the stoichiometry M(CI-GK-NHCS2)z where
M is Cu(II), Zn(II), Cd(II) or Sn(Q.146 The complexes have been characterised
by physicochemical methods and the thermal behaviour of these complexes
In static air atmosphere has been studied.
Singh el ol. have synthesised metal complexes of 4-aminophenazone
dithiocarbamate. The formula of the complexes have been given as

(19)
where n is the oxidation state of the metal (M = C++, Fe3+, CuZt, Ni2+, Ce2+,
V02+, Zn2+, Hg2+ and TI'). Surprisingly magnetic moment of the copper
complex is given as 1.73 BM, corresponding to one unpaired electron.147
CuX (X = I, Br) or CuBrz reacted with HL (HL = RSC(S) NHPh, R = Me,
Bu) lo givv C'ui ., <'u\l3uSC(S)NFlI1h]. Will(.m: t- 1.1 ~ r l . synll~c~sisc~cl the
complexes and determined the crystal s t ru~ture ."~ For HL (R = Ph) no CuL
was obtained because of rapid elimination of PhNCS. Crystals of CuL
(R= Bu) are rhombohedra1 with one hexameric unit in the unit cell.
Matter rt r r l . prepared dimeric and polymeric Cu(1) complexes of
dithiocarbamate esters. Reactions of N-methyl Smethyl dithiocarbamate(L)
and N-phenyl S-methyl dithiocarbamate (L') with Cu(1) or Cu(1I) halides
yield [CuIL,z]z, [CuIL'&, [CuCIL'], and [CuIL'],,. Their structures have been
determined by single crystal X-ray crystallography. [CuILr]> and [CuIL1:]2 are
dimeric, Cu(1) being tetrahedrally coordinated. (CuCIL'], and [CuIL'],, are
polymeric with edge-linked tetrahedral coordination polyhedra of
Cus2cuXzCu.'~"
Iordanov rt d l . studied the interaction of some Cu(I1) chelate complexes
with NO and NO1 by EPIZ and electronic spwtroscopy.'~' 'The chelates were
Cu(acac)z, C:u(OX)>, C'u(Utc)~ and Cu(Dtp)z. (Hacac = acetyl acetone, 130X =
8-hydroxy quinoline, HDtc = H2NCSzH and HDtp = dithiophosphoric acid).
Weak reversible adducts were formed with NO, whereas NO: oxidise the
ligands in all complexes. The reaction of Cu(Dtc)z with NO2 proceeds through

Introduction 37 . .
m~xed l~gand complexes w ~ t h the participat~on of NO,-; Cu(N03)z and
d~sulphidc. front the l~gand be~ng the final products.
Salam r t r r l synthes~sed and character~sed some metal chelates of
ethylenedlam~ne-monodrth~ocarbamates. Cu2+, Ni2+, Znzt, Colt, Fez+ and Cd2+
formed complexes of the composition ML2 and Fe3+ and Colt formed ML?
(HL = HzN-CHJ-CH2-NH CSJ H) species.151
Mekil complexes of 1,3-propanediamine rnonodithiocarbamate were
prepared and characterised by Salam et nl. Divalent cations CuZ+, Ni2+, Mn2+,
Znz', Coz', Fez' and Cdz' formed ML2 and trivalent cations Feq+ and Co"'
formed ML.$(HL = H2N ( C H ~ ) Z - N H C S ~ H ) . ~ ~ ~
Enzymatic activity of copper, zinc superoxide dismutase (SOD), a
metalloprotein that catalyses superoxide radical disproportionation, involves
a cyclic Cu(lI)/Cu(l) redox process of the Cu(I1) i?n held at the active centre
in the protein. St.vcral copper (11) dithiocarbamates are known to exhibit
SOD-like activity.'jT.'3 The activity of such complexes depends on the
Cu(II)/Cu(I) rcdox process. Roberto Cao et nl. studied the SOD-like activity
of the copper(I1) complexes of the amino acids glycine, alanine, serine,
aspargine and glutatnic acid. In all these complexes copper is reported to be
existing as Cu(II) and the EPR inactivity is explained in terms of
antiferromagnetic in te ra~t i0ns . l~~
1.5 Scope and objectives
Conipin in 1920 noted the instability of Cu(I1) complexes of
dithiocarbamates derived from primary amines.143 In 1936 Cambi and
Coriselli noticed that Cu(SzCNHR)2 could not be isolated because of its rapid
decomposition to the Cu(I) species by some complicated mechanism. They
verified the presence of isothiocyanate in the decomposition products.ll'
Since 1936 there have been only very few reports, probably less than ten, on
copper complexrs with RHDtc ligands. In most ~f these reports the metal-

ligand ratio is stated to be 1 :2 the oxidation state of the metal being Cu(I1).
These reports contradict with the observation of Compin, and Cambi and
Coriselli.
The present investigation is to establish the true nature of the
interaction between Cu(II) ions and RHNCS;. The products of the reaction
between Cu(I1) ion and RHNCS; ion were carefully isolated and
characterised by elemental analysis, molecular mass determination, various
spectroscopic techniques and thermal analysis. In anticipation that the
dielectric of the reaction medium and the nature of the substituent on the N
atom of RHNCS, would dictate the nature and course of the reaction and
stereochemistry of the reaction products, interaction of Cu(1I) ions with a
range of RHNCS; ions with various alkyl, acyl and heterocyclic substituents
was studied in different solvents. With a view to evolve a strategy to achieve
redox-stabilised copper(I1) complexes of RHNCSz, the Dtc function was
anchored to a polymeric support and its interaction with Cu(l1) was
investigated mainly by electron spin resonance spectroscopy. The present
study broadly covers the following aspects:
(i) Various dithiocarbamates derived from primary amines are prepared
in solution condition by the nucleophilic addition of primary amines
on CSr in alkaline medium. Dithiocarbamates with a variety of
N-substituents-alkyl, simple aryl, heterocyclic, aralkyl, aryl with
either electron donating or electron withdrawing groups-are
prepared with a view to study the effect of N-substituents on the
course and nature of the reaction between Cu(1I) and RHDtc.
(ii) The primary amine-dithiocarbamates generated in solution are
allowed to interact with cupric ion in aqueous medium. The reaction
products, both in solid state and in solution are isolated and purified.
The compounds are systematically analysed by various analyl~cal and

spectral methods which suggest the occuirence of a redox process
during the interaction, forming polymeric copper(1) dithiocrbamates
and thiuram disulphides, RHNCS2-SzCHNK.
(iii) To establish the identity of the oxidised form of the ligands, thiuram
disulphides, formed on interaction of copper(I1) with Dtc, all the
dithiocarbamates are oxidised separately and individually with iodine
solution and their composition and spectral properties compared with
those obtained through copper(I1) interaction.
(iv) Since preparation of dithiocarbamate ligands and their complexation
with metal ions in ethanol medium have been reported, the generation
of various dithiocarbamates, RHDtc and their interaction with
copper(I1) are tried in ethanol medium also. The course of the reaction
is found to be different for N-alkyl and N-aryl substituents though the
occurrence of the expected redox process is confirmed.
(v) Since the reaction medium is found to affect the redox potential of a
process," the interactions are studied in TIIF and/or DMF expecting
that solvents with a lower polarity might stabilise the copper(1I) state.
The products are isolated in the pure state and analysed by various
physicochemical methods including molecular mass determination.
Though redox process does occur, structure and stereochemistry of the
resulting Cu(1) complexes are found to be different from those formed
in aqueous or alcoholic medium. Various oligomeric complexes,
dimers and tetramers, could be isolated.
(vi) Complexation with metal ions is expected to facilitate interesting
reactivity pattern of RHDtc. With this in view electrophilic
substitution, like benzoylation, is attempted on the various oligomeric
and polymeric Cu(1)RHDtc formed during the interaction studies. 'The
difference in the reactivity of the Cu(1) complexes towards

benzoylation is found helpful, to a great extent, to infer the structure
and stereochemistry of the complexes.
(vii) With a view to prepare cupric dithiocarbam:~te by inhibiting the redox
process, the ditluocarbamate function is immobilised by anchoring to a
polymer matrix. Three polymer bound dith ocarbamates derived from
ethylenediamine, p-aminophenol and o-antinophenol are separately
treated with Cuz+(aq). The polymer beads are analysed mainly by EPR
which indicates the redox stabilisation of copper(I1) during the
interaction.
(viii) Attempts are made to generate mixed ligand Cu(1I) complexes
involving N-monosubstituted Dtc on polymer matrix. Each of the
dithiocarbamate functionalised resins is treated separately and
individually with six different Cu(I1) complexes of Schiff bases,
P-diketones and 8-hydroxy quinoline. All the eighteen polymer metal
complexes generated are studied by EPR to characterise the complexes
formed on the polymer matrices.
(ix) To studv the effect of the N-substituent and slereochemistry of the
complex on the mode of decomposition and thermal stability, and also
to confirm the structure and compositiori proposed for the Cu(1)
complexes prepared by interaction of Cu(1.) with RNHCSk), thermal
analysis of the various dimeric, tetrameric and polymeric Cu(I)
dithiocarbamate complexes are carried out by TG and DTG. With the
help of the nine mechanism-based equations proposed by Satava
various kinetic and thermodynamic parameters are calculated from the
thermograms. Suitable mechanisms are proposed for all the major
decomposition stages.

References
D. Coucouvanis, Progress in lnorganic Chemistry, Vol. 11 (Ed., S. J. Lippard), John Wiley, New York, 1970.
R. Eisenberg, Progress in Inorganic Chemistry, Vol. 12 (Ed., S. J. Lippard), John Wiley, New York, 1970.
D. Coucouvanis, Progress in inorganic Chemistry, Vol. 26 (Ed., S. J. Lippard), John Wiley, New York, 1979.
V. F. Plyusnin, V. P. Crivin and S. V. Larionov, Coordination Chemistry Rer~ituis, 159, 121 (1997) and references therein.
I. star^, Cxtra~.tion ofchelates, Mir, Moscow, 1966, p. 392.
I . M. Blrko, A~ralytical Reagents. Dithiocarbant,-tes, Nauka, Moscow, 1984, p. 343.
M. V . Korablev, Derivative of Dithiocarbanline Acids, Chenlistry, Toxicology, Pharnrucology and Clinical Application, Belarus, Minsk, 1971, p. 152.
C. D. Thorn and R. A. Ludwig, The Dithiocarbamates and Related Compounds, Elsev~er, Amsterdam, 1962, p. 298.
C. A. Bloch, Organic Accelerators of Rubber Vulcanisation, Khimiya, Moscow, 1972, p. 559.
V. A . (;lrmhotsh~~, V . I . Klassen and I. N. Plaskin, Flotufion, Nedra, Moscow, 1973, p. 784.
D. Coucouvanis, Prog. Inorg. Chem., 11,223 (1970).
F . Yu. Rach~nsk~i, A. S. Mozhuchin, N. M. Slovachevskaya and L. I . Tank, Usp. Khlnl., 28, 1488 (1959).; J. Tomson, Protei,tion of Mammuliu from lowisation Irradiatiorrs, Atomizdat, Moscow, 1%4, p. 176.
G. Scott (Ed.), Dcuelopn~cnts in Polymer Stabilisation, Part I, Applied Science, London, 1979.
Y. L. Goldfarb, E. C. Ostapenko, V. B. Ivanov, A. F. Efremkin and V. L. Litv~nov, K ~ I N I . Geterotsikl Soedin., 1481 (1982).
A. bl. Ewald, I<. L. Martin, 1. C. Ross and A. t~ i . White, Proc. Roy. Soc. (London), A280, 235 (1964).
W. E. Hatfield, Inorg. Chem., 22, 833 (1983); L. Qian, P. Sing H. Ro, W. E. Hatfield, Inorg. Chenr., 29, 761 (1990).
E. Emmet Reld, Organic Chmristry of Bivalent Sulphur, Vol. IV, Chemical Publishing To., New York, 1962, p. 140.
E. Mulder, I. Prakt. Clvnl., 101,401 (1867).
(a) J. Chatt, I.. A. Duncason and L. M. Venanzi, Naturc, 177, 1042 (1956); (b) Suomen Kemistilehti, 298, 75 (1956).
C. J. M. Van der Kerk and H. L. Klopping Rcc. Trau. Chim., 71,1179 (1952).
M. Colaplelro, A . Domen~cano and A. Vaciago, Cllr>nl. Conrnlun., 572 (1968).

B. F. C. Johnson and K. H. Al-Obaide, Chetn. Con~nrun., 876 (1968).
J. Willemse, J. A. Cras, J. J. Steggerda, C. P. Keijzers, Structure and Bonding, Vol. 29, 1977.
J. A. Cras and J. Willemse, Recl. Trav. Chint. Pays-Bas, 97, 28 (1978).
D. Coucouvanls and J. P. F. Fackler Jr., Inorg. Ch~nr., 6, 2047 (1967).
K. Nakamoto, J . Fujita, R. A. Kondrate a n d Y. Morimoto, I . Chem. Phys., 39, 423 (1 963).
G. Durgaprasad, D. N. Sathyanarayana and C. C. Patel, Can. I . Chem., 47,631 (1969).
D. A. Brown, W. K. Glass and M. A. Burke, Spectrochinr. Acta, 32A, 137 (1976).
G. 5. Nikolov, Irwrg. Nucl. Cknr. Lett., 7, 1213 (1971).
C. O'Conner, J . D. Gilbert and G. Wilkinson, 1. Civnr. Soc., A, 84 (1969).
F. Bonatl and R. Ugo, I . Organonlet. Cht~n., 10, 257 (1967)
E. C. Alyea and 8. C. Ramaswamy, Inorg. Nucl. Qwnr. Lett., 6, 441 (1970).
C. G. Seeney and R. J. Magee, lnorg. Nucl. Chem. Lett., 10,323 (1974).
G. Gasparrl, M. Nardell1 and A. Villa, Acta Cryst., 23, 384 (1967).
M. Bonamso, G. Dessy, C. Mariani, A. Vaciago and L. Zambonelli, Acta Cryst., 19,619 (1965).
G. Peyronel and A. Pignedoli, Acta Cryst., 23, 398 (1967).
H. J. A. Blauuw, R. J. P. Nlvard and G. J. M. Van der Kerk, 1. Orgomet. Chcnl., 2, 236 (1964).
P. T. Beurskens, H. J. A. Blauuw, J. A. Cras and J. J. Steggerda, Inorg. Chem., 7,805 (1968)
P. T. Beurskens, 1. A. Cras and J. J. Sleggerda, lnorg. Chem., 7,810 (1968).
M. Bonarn~co, G. Dessy, A. Mugnoli, A. Vaciago and L. Zambonelli, Acta Cryst., 19, 886 (1965).
A. Plgnedol~ and G. Peyronel, Gazz. Chint. Ital., 92, 745 (1962).
G. D. 'Thorn and I<. A. Ludwig, T k Dithiocarbunratrs and Related Cortrpuurrds, Elsev~er, New York, 1%2.
J. M. Martin, P. W. G. Newman, B. W. Robinson and A. H. White, I . Clrem. Soc., Dalton Trans., 2233 (1972).
H. P. Klug. Arta Cryst., 21, 536 (1966).
M. Bonarnico, G. Muzzone, A. Vaziago and L. Zambonelli, Acta Cryst., 19, 898 (1965).
A. Donirn~cano, I.. Torelli, A. Vaciago and L. Zan~bonelli, I. Chenr. Sot.., Scr. A, 1351 (1968).
A. Domenicano, A. Vaciago, L. Zambonelli, F. 1,. Loader and L. M. Venanzi, Chcnr. Conlnrun., 476 (1966).

B. F. Hosklns and B. P. Kelley, Chem. Coninrun., 517 (1968).
(a) M. Cohpietro, A. Vaciago, D. C. Bradley, M. B. Hursthouse and I. F. Rendall, Chem. Commun., 743 (1970); (b) 1. Chtm. Soc., Dalton Trans., 1052 (1972).
S. Akerstrom, Arkiv Kemi, 14,387 (1959).
R. Hesse, Arkio Kenii, 20, 481 (1%3).
F. A. Cotton, Accounts Ckm. Res., 2, 240 (1x9).
R. Hesse, Adz'unces in the Chemistry of Coordination Conipounds (Eds., S. K~rschner), MacMlllan, New York, 1961, p. 324.
L. E. Sutton (Ed.), Cheni. Soc. Spic. Publ., No. 11, 53 (1958).
F. J. Farrell and T. G. Sp~ro, lnorg. Chem., 10,1606 (1971).
R. Hesse and L. Nilson, Acta Chem. Scand, 23, 825 (1%9).
A. F. Wells, Structural lnorganic Chemistry, 5" edn., Clarendon, Oxford, 1984.
J. Willemse, J. A. Cras, J. J. Steggerda and C. P. Keijzers, Struct. Bonding, 28, 83 (1976).
C. K. Jorgenson, Adsorption Spectra and Chemical Bonding, Academic, London, 1%2; Inorganic Complexrs, Academic, London, 1963.
F. A. Cotton and G. Wilkinson, Aduanccd Inorganic Chemistry, Wiley Intersoencr, New York, 1.1 edn., 1952; 4h edn., '1980.
R. W. G. Wyckoff, Crystal Strutures, 2nd edn., interscience, New York, 1965, Vol. 1.
J. Pe~sach, P. Atsen and W. E. Blumberg (eds.), The Biochemistry of Copper, Academic, New York, 1%6.
T. G. Spiro (ed.), Coppcr Proteins, Wiley International, New York, 1981; R. Lont~e, Copppcr I'roteins and Coppcr Enzyrncs, CRC Press, Uoca liaton, FL, 1984, Vol. 1-111.
H. S~gel (ed.), Metal Ions rn B~ological Systcnis, Dekker, New York, 1973-81, Vol. 1-13.
B. J. Hathway and D. E. Blllmg, Cwrd. Chern. Rev., 5, 143 (1970).
B. J. Hathway, Struct. Bonding (Berlin), 57, 55 (1984),
(a) M. A. H~tchman, Trans. Met. Chenl., 9, 1 (1984); (b) J. Ferguson, Prog. Inorg. Chcnr., 12, 159 (1970).
Y. N ~ s h ~ d a and S. K~da, Coord. Chem. Rev., 15,279 (1975).
R. Sreekala and K. K. M. Yusuff, Synth. React. lnorg. Met.-Org. Chen~., 24, 1773 (1994).
A. B. P. Lever, Inorganic Electronic Spec!roscopy, 2.d edn., Elsevier, Amsterdam, 1984.
N. 5. Hugh and R. J. M. Hobbs, Prog. lnorg. Clrenr., 10, 259 (1968)

M. N. Hughes, lnorganic Chemistry of Biologicrrl Systems, 2nd edn., Wiley, Chichester, 1981
D. J. (;ulliver, W. Levason and M. Webster, Inorg. Chlm. Acta, 52, 153 (1981).
W. E. Hatheld and R. Whyman, Trans. Met. Chenr., 47,5 (1969).
F. H. Jardrn, Adu. Inorg. C1lt.m. Radiochenr., 17, ll!. (1975).
J. P. Fackler, jr. and D. Coucouvanis, 1. Am. Clrm.. Soc., 88, 3913 (1966).
L. E. McCandlish, E. C. Bissell, D. Coucouvanis, J. P. Faciler and K. Knox, I . Am. Chem. Soc., 90,7357 (1968).
F. 1. Hollander and D. Coucouvanis, 1. Am. Chent. Soc., 99,6288 (1977).
P. bet^, B. Krebs and G. Henkel, Angew. Chem. Int. Ed. Engl., 23, 311 (1984)
8. J. Hathaway, Essays C h ~ m . , 2,61 (1971).
P. C. Healy, C. Pakawatcha~ and A. H. White, 1. Cliern. Soc., Dalton Trans., 1917 (1983).
M. R. Churchill and F. J. Rotella, lnorg. Chern., 18. 166 (1979).
H. Neg~ta, M. H~ura, Y. Kushi, M. Kuramoto a ~ d T. Okuda, Bull. Chem. Sor. Ipn., 54, 1247 (1981).
D. M. tio and I<. Bau, Inorg. Chenr., 22,4079 (1980)
N. Brescianl, N. Marsich, C. Nardin and L. Randaccio, Inorg. Chim. Acta, 10, 15 (1974).
G. Nardin, L. Kandaccio and E. Zangrando, 1. Cllenr. Soc., Dalton Trans., 2566 (1975).
M. R. Churchill and K. L. Kalra, Inorg. Chem., 13, 1899 (1974).
J. C. Dyason, P. C. Healy, L. M. Engelhardt, V. A. Patrick, C. L. Raston and A. H. White, J. Cllenr. Soc., Dalton Trans., 831 (1985).
D. M. Adams, lnorganic Solids, Wiley, London, 1574, Ch. 4.
R. D. blounts, T. Ogura and Q. Fermando, Inorg. Chenr., 13,802 (1974).
K. lijin~a, T. ltoh dnd S. Shibata, j. Chem. Soc., Dclton Tratts., 2555 (1985).
A. Massey, Co'unlprehensiue Inorganic Chemistry (Eds., J. C. Bailar, H. 1. Emeleus, R. S. Nyholm and A. F. Trotman-Dickinson), Pergamon, Oxford, 1973, Vol. 3, p. I .
K. D. Karlin and J. Zubieta (ed.), (a) Copper Coordirration Chrt~listry: Bioclrcnliclll and Inorganic Perspectir~es, Adenine Press, New York, 1983; (b) Biological and Inorganic Copper Chemistry, Adenine Press, New York, 1986.
M. C. Simmons, C. L .Metrill, L. 1. Wilson, L. A. Bottomley and K. M. Kadish, 1. Ckrn. Soc., Dalton Trans., 1827 (1980); C. L. Merrill, 1.. J. Wilson, T. J. Tharnann, 'T. M. Loehr, N. S. Ferris and W. Woodruff, I . Chcm. Soc. Dalton Trurrs., 2207 (1984).

Introduction 45
J. E. Bulkowski, P. L. Burk, M. F. Ludmann and J. A. Osborn, 1. Chem. Soc., Clrenr. Conrnrun., 498 (1977).
E. W. Ainscough and A. M. Brodie, Cwrd. Chem. Rev., 27,59 (1978)
J. A Thlethof, J. K. Statlck and D. W. Meek, Inor;. Chem., 1% 1170 (1973).
P. G. Eller and P W. R. Corheld, Chem. Cornmun., 105 (1971).
D. L. Dunnmg, B. L. Kmdberg, C. F. Plese, E. I{. Crtfhth and E. L. Amma, 1. Chenr. Soc. C k m . Comntun., 550 (1975).
A. R. Atkinson, D. J. Garolimer, A. R. W. Jackson and E. S. Raper, lnorg. Chinz. Acta, 98, 35 (1985).
E. S. Raper, J. R. Creighton, D. Robson, J. D. Wilson, W. Clegg and A. Milne, lnorg. Chinr. Arta, 143, 95 (1988).
E. R. Atkmson, D. J. Gardiner, A. R. W. Jackson, E. S. Raper, H. Dawes and N. P. C. Walker, lnorg. Chim. Acta, 100, 285 (1985).
H. Carnpp and A. D. Zuberbuhler, Chintia, 3 2 5 4 (1978)
A. E. Martell, Pure Appl. Chem., 55, 125 (1983); T. C. Splro, Metal Actiation o/ Dloxygen, W~ley Intersclence, New York, 1980.
G. Davis, M. A. El-Sayed and R. E. Fasano, lnorg. Clrint. Acta, 7l, 95 (1983).
A. El-Toukhy, G. Z. Cai, C. Davies, T. R. Gilbert, K. D. Onan and M. Veidis, 1. Am. Clwnr. Sot.., 106, 45% (1984).
E. Ochiat, Bioinorganic Chenristry: An Inroduction, Allyn and Bacon, Boston, 1977.
A. S. Brill, l'ransition Metals in Biochenlistry, Springer-Verlag, Berlin, 1977.
R. W. Hay, Hrurnorgunic Owrttistry, Wiley, Chichester, 1984.
N. Marsich, C. Nardin and L. Randaccio, lnorg. Chinr. Acta, 23, 131 (1977)
L. Cambi and C. Coriselli, Gazz. Chim. Ital., 66, 7 9 (1936).
V. Tamminen and E. Hjelt, Soumen Kemi, 23B, 39 (1950).
S. Akerstrom, Arta Clrenr. Scand., 19, 699 (1956).
H. Jorg, Qrenr. Ber., 60,1466 (1927).
M. Deleplne, Conrpt. Rmd., 144, 1125 (1907).
V. Sed~vek and V. Vasak, Chcnl. Listy, 46,607 (1952).
R. Bally, Contpt. Rend., 257,425 (1963).
M. Bonatnico, C. Dessy, A. Mugnoli, A. Vacelgo and L. Zambonelli, Acta Cryst., 19, 886 (1965).
P. W. C. Newman, C. I. Raston and A. H. White, /. Chem. Soc. Dallon Trans., 1332 (1973).
R. D. Bereman and D. Nalewajek, lnorg. Chenr., 11,638 (1972).
T. Vanngard and S. Akerstrom, Nature, 183 (1959).

Introduction 46
R. Petterson and T . Vanngard, Ankiv Kent~, 17,249 (1960).
H. R. Gersmann and J. D. Swalen, 1. Chenr. Phys., 36,12 (1962)
T . R. Reddy and R. Srmivasan, 1. Chem. Phys., 43,1404 (1965).
J . G. M. Van Rens, M. P. A. Viegers and E. de Boer, Chem. Phys. Lett., 14,113 (1 972).
B. J. Corden and P. H. Rieger, Inorg. Chenl., 10, 263 (1971).
N. D. Yordanov and D. Shopov, Inorg. Chinz. Acta, 5,679 (1971).
N . D. Yordanov, V. Terziev and D. Shopov, PI-oc. Bulg. Acad. Sci., 27, 1529 (1 974).
N. D. Yordanov and D. Shopov, 1. Inorg. Nucl. Chenz., 38,137 (1976).
A. R. Hendrickson, R. J. Martin and N. M. Rohde, Inorg. Chem., 11,638 (1972).
P. T. Beurskens, J . A. Cras and J. J. Steggerda, 1m:rg. Chtm., 7,810 (1968).
H. C. Brinkhoff, Recl. Trav. Chinl. Pays-Bas, 90, 377 (1971).
J. G. Wijnhoven, T. E. M. Van der Hark and P. T. Beurskens, 1. Cryst. Mol. Struct., 2, 189 (1972).
J. A. Cras, J. Willemse, A. W. Gal and B. G. M. C. Hummelink-Peters, Recl. Trav. Chim. Pays-Bas, %,78 (1971).
M . Bonamico, C. Dessy, V. Fares and L. Scavamuzza, 1. Chenl. Soc., Dalton Trans., 2250 (1975).
P. J. H. A. M. Van der Leemput, J. A. Cras, J. Willemse, P. T. Beurskens and F. Megnet, Recl. Trao. Chim. Pays-Bas., 95,191 (1976).
P. j. H. A. M . Van der Leemput, J. Willemse and J. A. Crds, Recl. Trav. Chinr. Pays-Bas., 95, 53 (1976).
V. 'l'arnmlnen and E. Hjelt, Sourrren Kenri., 823, 39 (1950).
P. M. Solohenkin, A. A. Gornostal, A. V. Ivanav, I . N. Cigima, Dokl. Akad. Nuuk. USSR, 314(1), 209 (1 990).
j. P. Barb~er, R. P. Hugel and C. Kappenstein, Inorg. Chinr. Acta, 77, 117 (1983).
C. M. Dani and A. K. Das, Ind. 1. Chem., 22A, 991 (1983).
K. K. M. Yusuff and E. J. Mathew, Synth. React. Inorg. Met.-Org. Chem., 22, 575 (1 992).
L. M. Compm, Bull. Soc. Chim., France, 27,464 (1920).
L. Malatesta, Gazz. Chim. Ital., 70, 541, 553 (1940).
N. K. Kaush~k, C. Chhahval and A. K. Sharma, Thernrochim. Acta, 58, 231 (1982).
N. K . Kaushlk, B. Bhushan and A. K. Sharma, Therrrrochinr. Acta., 76, 345 (1984).

H. B. Smgh, S. Maheshwari, S. Srivastava and V. Rani, Synth. React. Met.-Org. Chern., 12, 659 (1982).
J . Wtllense dnd W. P. Bosman, Reel. 1. R. Neth. Chenr. Soc., 102, 477 (1983).
Matter, Rawer, Schulbert, Klaus and Z. Naturforsch, Chem. Sci, 46,468 (1991).
N . lordmov, V. Terziov and B. Zhelyaskova, Inorg. Chinc. Acta, 58,213 (1982).
M . A. Salem and A. J. M. Begum, Chittagong Univ. Stud. Part I1,14,9 (1990).
M. A. Salem and M. Alauddin, Nucl. Sci. Appl. (Dhaka), 3, 77 (1991).
M. C. Appella, R. Totaro and F. T. Baran, Biol. Trace Elent. Res., 37,293 (1993).
P. Chautemps, G. Gellon, B. Morin, J. L. Pierre, C. Provent, S. M. Refait, C. G. Beguin, A. E. Marzouki, C. Serratrice and E. Saint-Aman, Bull. Soc. Chint. Fr., 131, 434 (1994).
R. Cao, N Irav~eso, A. Fragoso, R. Vlllanonga, A. Dtaz, M. E. Martmez, J. Alpmr and D. X West, 1. lnorg. Biochem., 66, 213 (1997).