
Improvement of thermal stability, rheological and ... school presentations... · Improvement of...
17
Improvement of thermal stability, rheological and mechanical properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy Racha Al-Itry a, b , Khalid Lamnawar a, c , Abderrahim Maazouz a, b, d, * a Université de Lyon, F-69361 Lyon, France b CNRS, UMR 5223, INSA Lyon, Ingénierie des Matériaux Polymères, F-69621 Villeurbanne, France c CNRS, UMR 5259, INSA Lyon, LaMCoS, Laboratoire de Mécanique des Contacts et des Structures, Groupe de Recherche Pluridisciplinaire en Plasturgie, F69621, France d Hassan II Academy of Science and Technology, 10 100 Rabat, Morocco article info Article history: Received 31 December 2011 Received in revised form 23 April 2012 Accepted 18 June 2012 Available online 16 July 2012 Keywords: Biodegradable polymers Thermal stability Chain extender Reactive extrusion Viscoelastic properties Molecular weight measurements abstract The aim of this study has been to gain a fundamental understanding of the mechanisms and conditions governing thermal degradation of poly (lactic acid) (PLA), poly (butylene-adipate-co-terephtalate) (PBAT) and their blends upon processing conditions. Thermal degradation of biodegradable PLA and PBAT was investigated firstly by thermal analysis and size-exclusion chromatography (SEC). It is shown that neat polymers degrade upon processing hence the decrease of the molecular weight, rheological and mechanical properties. Secondly, the reactive extrusion of polymers was performed with various amounts of chain extension/branching agent, containing nine Glycidyl methacrylate (GMA) functions, named Joncryl. The incorporation of this multi-functional oligomer showed an improvement of their thermal stability. SEC and intrinsic viscosity measurements of these modified PLA and PBAT confirmed the increase of viscosity and molecular weight probably related to the formation of extended and branched chains. Rheological investigation of extended/branched PLA and PBAT as well as their modified PLA/PBAT (80/20) (wt/wt) blends with various concentrations of GMA reactive functions exhibited higher viscosity and storage modulus compared to the unmodified samples. This increase becomes more pronounced as the concentration of Joncryl increases. Viscoelastic properties were assessed and related to the molecular structure of modified polymers. Hence, the mechanisms of degradation, chain extending with GMA functions and their competition have been proposed. The effect of reactive compatibilization on the PLA/PBAT blends has been confirmed using transmission electron microscopy (TEM), scanning electron microscopy (SEM) observations and tensile tests by the improvement of phase dispersion and the increase of both Young’s modulus and strain at break. Ó 2012 Elsevier Ltd. All rights reserved. 1. Introduction Natural polymers, biopolymers and synthetic polymers based on annually renewable resources are the basis for the twenty-first- century portfolio of sustainable, eco-efficient plastics. The interest on these polymers is considerable in the prospect to decrease the world resources in oil and in a concern to limit the contribution of plastics to the waste disposal. The subject is vast and hence it is a daunting task to summarize their remarkably rich and multi- faced area related to these biopolymers fields. Polylactide or poly(lactic acid) (PLA) is the front runner in the emerging bio-plastics market with the best availability and the most attractive cost structure. PLA is linear, aliphatic thermoplastic polyester with rigidity and clarity similar to polystyrene (PS) and poly (ethylene terephthalate) (PET). It is used for different appli- cations ranging from medical to packaging, resorbable and biodegradable under industrial composting conditions [1]. It can be produced by condensation directly from its basic building block lactic acid, which is derived by fermentation of sugars from carbohydrate sources (corn, sugarcane, tapioca.). But, higher molecular weights of PLA are achieved by ring-opening- polymerization (ROP) of cyclic lactide dimer [2]. Generally, PLA is made into useful items using thermal processes like injection molding and extrusion. Therefore, its rheological properties, especially its shear viscosity, have important effects on thermal processes. Despite all its advantages, some properties of PLA such as inherent brittleness, poor melt strength, narrow processing window and low thermal stability pose considerable scientific challenges and limit their large scale-applications (film blowing or * Corresponding author. CNRS, UMR 5223, Ingénierie des Matériaux Polymères, INSA Lyon F-69621 Villeurbanne, France. E-mail address: [email protected] (A. Maazouz). Contents lists available at SciVerse ScienceDirect Polymer Degradation and Stability journal homepage: www.elsevier.com/locate/polydegstab 0141-3910/$ e see front matter Ó 2012 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.polymdegradstab.2012.06.028 Polymer Degradation and Stability 97 (2012) 1898e1914
Transcript of Improvement of thermal stability, rheological and ... school presentations... · Improvement of...

at SciVerse ScienceDirect
Polymer Degradation and Stability 97 (2012) 1898e1914
Contents lists available
Polymer Degradation and Stability
journal homepage: www.elsevier .com/locate/polydegstab
Improvement of thermal stability, rheological and mechanical properties of PLA,PBAT and their blends by reactive extrusion with functionalized epoxy
Racha Al-Itry a,b, Khalid Lamnawar a,c, Abderrahim Maazouz a,b,d,*
aUniversité de Lyon, F-69361 Lyon, FrancebCNRS, UMR 5223, INSA Lyon, Ingénierie des Matériaux Polymères, F-69621 Villeurbanne, FrancecCNRS, UMR 5259, INSA Lyon, LaMCoS, Laboratoire de Mécanique des Contacts et des Structures, Groupe de Recherche Pluridisciplinaire en Plasturgie, F69621, FrancedHassan II Academy of Science and Technology, 10 100 Rabat, Morocco
a r t i c l e i n f o
Article history:Received 31 December 2011Received in revised form23 April 2012Accepted 18 June 2012Available online 16 July 2012
Keywords:Biodegradable polymersThermal stabilityChain extenderReactive extrusionViscoelastic propertiesMolecular weight measurements
* Corresponding author. CNRS, UMR 5223, IngénieINSA Lyon F-69621 Villeurbanne, France.
E-mail address: [email protected]
0141-3910/$ e see front matter � 2012 Elsevier Ltd.http://dx.doi.org/10.1016/j.polymdegradstab.2012.06.0
a b s t r a c t
The aim of this study has been to gain a fundamental understanding of the mechanisms and conditionsgoverning thermal degradation of poly (lactic acid) (PLA), poly (butylene-adipate-co-terephtalate) (PBAT)and their blends upon processing conditions. Thermal degradation of biodegradable PLA and PBAT wasinvestigated firstly by thermal analysis and size-exclusion chromatography (SEC). It is shown that neatpolymers degrade upon processing hence the decrease of the molecular weight, rheological andmechanical properties. Secondly, the reactive extrusion of polymers was performed with variousamounts of chain extension/branching agent, containing nine Glycidyl methacrylate (GMA) functions,named Joncryl. The incorporation of this multi-functional oligomer showed an improvement of theirthermal stability. SEC and intrinsic viscosity measurements of these modified PLA and PBAT confirmedthe increase of viscosity and molecular weight probably related to the formation of extended andbranched chains. Rheological investigation of extended/branched PLA and PBAT as well as their modifiedPLA/PBAT (80/20) (wt/wt) blends with various concentrations of GMA reactive functions exhibited higherviscosity and storage modulus compared to the unmodified samples. This increase becomes morepronounced as the concentration of Joncryl increases. Viscoelastic properties were assessed and relatedto the molecular structure of modified polymers. Hence, the mechanisms of degradation, chain extendingwith GMA functions and their competition have been proposed. The effect of reactive compatibilizationon the PLA/PBAT blends has been confirmed using transmission electron microscopy (TEM), scanningelectron microscopy (SEM) observations and tensile tests by the improvement of phase dispersion andthe increase of both Young’s modulus and strain at break.
� 2012 Elsevier Ltd. All rights reserved.
1. Introduction
Natural polymers, biopolymers and synthetic polymers basedon annually renewable resources are the basis for the twenty-first-century portfolio of sustainable, eco-efficient plastics. The intereston these polymers is considerable in the prospect to decrease theworld resources in oil and in a concern to limit the contribution ofplastics to the waste disposal. The subject is vast and hence it isa daunting task to summarize their remarkably rich and multi-faced area related to these biopolymers fields.
Polylactide or poly(lactic acid) (PLA) is the front runner in theemerging bio-plastics market with the best availability and the
rie des Matériaux Polymères,
(A. Maazouz).
All rights reserved.28
most attractive cost structure. PLA is linear, aliphatic thermoplasticpolyester with rigidity and clarity similar to polystyrene (PS) andpoly (ethylene terephthalate) (PET). It is used for different appli-cations ranging from medical to packaging, resorbable andbiodegradable under industrial composting conditions [1]. It canbe produced by condensation directly from its basic building blocklactic acid, which is derived by fermentation of sugars fromcarbohydrate sources (corn, sugarcane, tapioca.). But, highermolecular weights of PLA are achieved by ring-opening-polymerization (ROP) of cyclic lactide dimer [2]. Generally, PLA ismade into useful items using thermal processes like injectionmolding and extrusion. Therefore, its rheological properties,especially its shear viscosity, have important effects on thermalprocesses. Despite all its advantages, some properties of PLA suchas inherent brittleness, poor melt strength, narrow processingwindow and low thermal stability pose considerable scientificchallenges and limit their large scale-applications (film blowing or

Table 1Main characteristics of the used polymers.
Characteristics of PLA Characteristics of PBAT
Grade: PLA4032D Commercial name:Ecoflex FBX7011
Supplier: NatureWorks Supplier: BASFMelting point: 168 �C Melting point: 110e120 �CGlass transition temperature: 62 �C Glass transition
temperature: �30 �CD-Lactide: 2% Density: 1.25g/ccDensity: 1.25g/cc Average molecular
weight ¼ 40,000 g/molAverage molecular weight ¼ 100,000 g/mol
Characteristics of Joncryl
Functionality: f ¼ 9Glass transition temperature: 54 �CEEW (epoxy equivalent weight): 285 g/molMolecular weight (Mw) w 6800 g/mol
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e1914 1899
injection molding) [3,29,30]. Recently, some developments inmodification of PLA including copolymerization, blending, com-pounding and additives were highlighted to improve its relevantproperties [4e7]. It has been reported that the thermal degrada-tion of PLA, upon thermal processing, predominantly consists ofrandom main-chain scission and unzipping depolymerizationreaction [8]. The random degradation reaction involves hydrolysis,oxidative degradation, cis-elimination, intramolecular and inter-molecular trans-esterification [9e11]. Depending on processingconditions, one of these several undesired reactions is dominant.For example, trans-esterification was found as a dominant degra-dation mechanism of PLA and high temperatures (above 200 �C)leading to the formation of cyclic oligomers [12]. Moreover, themelt degradation of PLA at temperatures above 180 �C was studied[13]. A random main-chain scission in such temperatures seems tobe responsible of the melt degradation. Almost all the active chain-end groups, residual catalysts, residual monomers and otherimpurities enhance the thermal degradation of PLA [14]. Asa consequence, an undesired molecular weight reduction andweight loss occur from 180 �C to 220 �C. It is proved that themodification of the polymer to get long and branched structures byusing chain extenders is an efficient approach to control thedegradation of PLA [7e15]. Consequently, many authors haveshown the interest of using a chain extender which is able toreconnect cleaved chains by increasing molecular weight of poly-mer, strength of melt and also can be used as a reactive compati-bilizer in blends [16,17,31]. The chain extenders are generally poly-functional, thermally stable and easily available. Some of theextenders reported in literature are di and multi-functionalepoxides [18], diisocyanate compound [19], dianhydride [20], bis-oxazolines, tris (nonyl-phenyl) phosphate (TNPP) [7] and poly-carbodiimide (PCDI). Multi-functional epoxy compounds havebeen extensively used as chain extenders for polyesters. In thiscase, the epoxy functions can react with both nucleophilic endgroups eOH and eCOOH of PLA leading to high molecular weightpolymer. Branched but less cross-linked PLA could be obtained formore than 1.5%wt of a multifunctional epoxide [21]. The branchedpolymers not only increase the molecular weight, but have manydifferent properties compared with linear polymers. In the light ofprevious works, the addition of such a chain extender with the aimof enhancing the extrusion and injection foamability of PLA wasinvestigated [22,32]. With the same objective, a similar chainextender on amorphous and crystalline PLA(s) was also studied[23].
Meanwhile, blending PLA with other flexible polymers (PCL,PBS, PBAT.) was investigated [24e26]. In our study, Poly(butylene-adipate-co-terephtalate) (PBAT) is selected. It isaliphaticearomatic copolyester, which is fully biodegradable,a flexible plastic designed for film extrusion. In the view of its hightoughness and biodegradability, PBAT is considered as a goodcandidate for the toughness of PLA [27]. Based on previous work onPLA/PBAT blends, it was discussed the incorporation of Glycidylmethacrylate (GMA) functions in such a blend in order to investi-gate the mechanical properties of PLA/PBAT/GMA (80/20/GMA)blends [24]. The surprising elongation at break value (3%) is ob-tained for PLA/PBAT/GMA blends. It is lower than those of PLA(4.5%) and PBAT (500%) respectively. The better understanding ofthe polymer stability is an interesting approach to explain thisstrange obtained result. Despite the interesting nature of the kindof research, few efforts have been dedicated to the study of thethermal degradation of PBAT and no study was dedicated to theeffect of chain extender on the PBAT under processing. Compared toPLA, the onset temperature of thermal degradation started muchlater for PBAT [28]. It was also reported that no significant chainmodification of PBAT occurred upon processing up to 200 �C [14].
This paper represents the approach to obtain thermo-stable PLAand PBAT polymers upon melt processing and investigates theeffect of their thermal stability on the structural, rheological,mechanical and morphological properties of modified PLA, PBATand their compatibilized blends. Hence, the mechanisms ofdegradation, chain extension with epoxy functions and theircompetition will be discussed.
2. Experimental section
2.1. Materials and methods
2.1.1. Polymers usedPolylactic acid, PLA4032D, was purchased from Natureworks. It
is a semi-crystalline material containing 2% of D-LA. Poly (butylene-adipate-co-terephtalate), PBAT, was supplied by BASF. Commercialavailable multifunctional epoxide, Joncryl ADR�-4368, supplied byBASF, a highly polydisperse oligomer presenting chains bearinghigh number average functionality. Table 1 summarizes the maincharacteristics of the polymers used in our study. The chemicalstructures of PLA and PBAT and Joncryl were shown in Fig. 1.
2.1.2. ProcessingBefore compounding, both PBAT and PLA pellets were dried
under vacuum at 80 �C for 12 h to remove moisture. A PRISM TTW16/25D corotating twin-screw extruder (TSE) with a screw diam-eter of 16 mm (Thermo Electron Polylab System Rhecord RC400P)was used for i) the processing of the neat polymers at differenttemperatures, shear rate and residence time, ii) the reactiveextrusion to investigate the role of various amount of Joncryl oncontrolling the degradation of both PLA and PBAT iii) the elabora-tion of uncompatibilized and compatibilized PLA/PBAT blends. ThePRISM 16 mm twin screw extruder is presented in Fig. 2. It hada clam shell barrel design with a length to diameter ratio of 25:1.The screw configuration matched that in a larger 51 mm diametertwin screw extruder. The mixing was conducted under a nitrogenatmosphere to limit the thermo-oxidative and hydrolysis degra-dation at a rotation speed of 40 rpm for 3 min. The temperatureprofile was set to 140,190,190,180 and 180 �C from the feed zone tothe die. After melt blending, each extrudate was quenched in a coldwater bath and granulated. After a 12 h drying stage performed at80 �C under vacuum, the blend granules were molded into disks of25 mm diameter with a thickness of about 2 mm. These disks weresubsequently dried in a vaccum at 40 �C oven overnight beforeanalysis.

O
O
R5R4
O O
R6
R2R1
R3
zyx
O
O
CH
CH3
O H
n
OH
OH
n p
O O
O CH 2 O4CH 2 4 O H
4
OO
O
CH 2
a: PLA
b: PBAT
: General structure of the styrene–acrylic multi-functional oligomeric chain extenders. Where R1–R5 are H, CH3, a higher alkyl group, or combinations of them; R6 is an alkyl group, and x, y and z are each between 1 and 20.
c
Fig. 1. Chemical structure of (a): PLA, (b): PBAT and (c): Joncryl ADR�-4368.
Table 2Compositions of modified polymers, compatibilized and uncompatibilized PLA/PBAT blends (a) and (b).
Sample name Wt% of neatpolymer in the blends
Wt% of Joncrylin the blends
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e19141900
Various amounts of Joncryl i.e. from 0 to 1 wt% were used forPLA, PBAT and PLA/PBAT (80/20) blends. Notations and composi-tions of the studied materials are given in Table 2.
2.1.3. Molecular weight measurements2.1.3.1. Intrinsic viscosity measurements. The intrinsic viscosity wasmeasured using an Ubbelhode-type capillary viscometer at 25 �C.Chloroform was used as solvent. The flow time was measured atdifferent concentrations (0.5g/100 mle0.25g/100 mle0.16g/100 mle0.125g/100 ml) for each sample. The introduction of thesolution was done carefully and quickly in the viscometer whichis integrated in the adequate visco’clock� system in order to avoidevaporation of the chloroform and keep constant the concentrationof the solution. The experiments are checked more than threetimes.
2.1.3.2. Size-exclusion chromatography. The average molecularweight, the average molecular number and the polydispersityindex of PLA, as well as for PBAT, before and after processing atdifferent temperatures were measured using size exclusion chro-matography (SEC). SEC analyses were performed in a VARIANprostar chromatograph, made of RHEODYNE injector, two Mixed-PL gel columns (G4000 HXL to G1000 HXL) with porosity about50e100,000 A� and an RI-101 refractive index detector. Mono-disperse poly (styrene) was used as calibration standards. PLA and
Fig. 2. The 16 mm diameter corotating twin-screw extruder. The numbers 1e8 at thetop of the extruder indicate the sampling positions. All dimensions are given in mm:a ¼ 9 * 16 mm þ 1 * 8 mm forwarding conveying elements b ¼ 14 * 4 mm Kneadingblocks (60� offset) c ¼ 4 * 16 mm forwarding conveying elements d ¼ 18 * 4 mmKneading blocks (60� offset) e ¼ 4 * 14 mm forwarding conveying elements plusf ¼ Vent ports (closed).
PBAT solutions were prepared as 5mg/ml in THF, with a flow rate of0.5 ml/min.
2.1.4. Rheological investigations in the molten stateLow amplitude shear measurements (LAOS) were investigated
with an Advanced Rheometric Expansion system (ARES) usinga parallel plates geometrywith a plate diameter of 25mmand a gapinferior to 2 mm. The heated chamber at different temperatureswas continuously purged with nitrogen to avoid thermal degra-dation. Prior to experiments, dynamic strain sweep tests witha maximum angular frequency amplitude of 100 rad/s was per-formed. Hence, the strain value is set at 5% to validate the linearviscoelastic region.
The following rheological measurements were carried out:
a) Dynamic time sweep test to verify the thermal stability of neatPLA and PBAT at lower angular frequency in linear regime
b) Dynamic frequency sweep tests over an angular frequencyrange of 0.05e100 rad/s at fixed strain amplitude.
Neat & modified PLAPLA_0 100 0PLA_0.25 100 0.25PLA_0.5 100 0.5PLA_1 100 1Neat & modified PBATPBAT_0 100 0PBAT_0.25 100 0.25PBAT_0.5 100 0.5PBAT_1 100 1
Sample name PLA (wt%) PBAT (wt%) Joncryl (wt%)
PLA_PBAT_0 80 20 0PLA_PBAT_0.25 80 20 0.25PLA_PBAT_0.5 80 20 0.5PLA_PBAT_1 80 20 1

R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e1914 1901
2.1.5. Thermal properties2.1.5.1. Thermal gravimetric analysis (TGA). TGA measurementswere performed using a TGA-Q500 thermogravimetric analyzerfrom TA instruments. Samples of 10 mg were heated from roomtemperature to 550 �C at 10 �C/min under nitrogen atmosphere.The effect of the environment was firstly validated and degradationwas more pronounced under air. For the clarity purpose, onlyresults under nitrogen will be shown here.
2.1.5.2. Differential scanning calorimetry (DSC). The thermal prop-erties of neat and processed PLA, PBAT and their blends wereinvestigated with the help of a differential scanning calorimeterQ10� DSC (TA) equipped with a liquid nitrogen cooling system(LNCS). The DSC cell was constantly purged with nitrogen at a flowrate of 50 mL/min. A set of heating/cooling ramps was carried outfollowing a three step process; the samples were firstly heated to200 �C and kept in the molten state for 2 min to erase the thermalhistory of the material. They were then cooled down to 25 �C at10 �C/min to evaluate the ability of PLA component to crystallizeupon cooling. After cooling treatment, the samples were heatedback to 200 �C at 10 �C/min. The percent crystallinity was calcu-lated upon the second heating by using the following equation(Eq. (1)):
Xc ð%Þ ¼ 100*DHm
f *DHNm
(1)
Where “DHm” is the measured heat of fusion, “f” is the weightfraction of PLA or PBAT in the blend and “DHN
m ” is the enthalpy offusion for a crystal having infinite crystal thickness (93 J g�1 for PLAand 114 J g�1 for PBAT).
2.1.6. Fourier transformation infrared spectroscopy (FTIR)Infrared spectra of neat and processed PLA and PBAT
were obtained with an FTIR Nicolet iS10 thermo scientific spec-trometer in attenuated total reflectance (ATR) mode with adiamond crystal collecting 32 scans. Each spectrum was obtainedwithin the range of 4000e650 cm�1 with a wavelength resolu-tion of 4 cm�1
.
2.1.7. Mechanical propertiesMechanical tests were carried out using an Instron machine
under room temperature (23 �C) with a cross head speed at 5 mm/min. The specimen dimension was 20 � 4 � 2 (in mm) denotingrespectively the length, breadth and the height samples. Prior totesting, the samples were dried in a vaccum oven at 60 �C for only2 h and kept in sealed dessicators until tests.
2.1.8. Morphological propertiesTo investigate the phase morphology of PLA/PBAT (80/20)
blends with various amounts of Joncryl, fractured surfaces at roomtemperature of the blends were observed by transmission electronmicroscopy (TEM) after being gold coated. Scanning electronmicroscopy was applied to characterize the morphology ofunmodified PLA/PBAT (80/20) blend. The cryo-fractured samplesurface was with thin layer gold, and then scanned at an acceler-ating voltage of 8 kV.
2.1.9. Acid value analysisTitration of the acid value of neat and processed PLA and PBAT
has been performed with the help of an automatic Mattler-ToledoT70 titrator. The solution was obtained by dissolving 3 g ofpolymer in 60 ml of Chloroform. KOH in MeOH solution(2.03 * 10�2 mol L�1) was used for the titration of the carboxylicgroups. A PH sensor was used to follow the evolution of the solution
acidity and to determinate the equivalent volume Ve as follow(Eq. (2)):
A:V ¼ Ve � CðKOHÞ �MðKOHÞm
(2)
where Ve (mL) is the equivalent volume corresponding to theequilibrium PH plateau, C (KOH) is the molar concentration(mol L�1) of KOH, M (KOH) is the molar mass (g mol�1) of KOH andm is the weight (g) of the dissolved polymer.
3. Results and discussions
3.1. Highlighting of PLA, PBAT thermal degradation: effect ofprocessing conditions
In this paragraph, the thermal stability of PLA and PBAT uponprocessing was investigated; for both polymers, a processing underan increased temperature (180,190 and 200 �C) and/or a higherresidence time (more than 3 min) exhibits a lower final torque, aswell as a reduced viscosity, making the melt more flowing. More-over, it was noted that nitrogen atmosphere is more effective inpreventing polymer degradation compared to air atmosphere.
The processing parameters were thus set in order to minimizethe thermal degradation of the studied polymers while maintain-ing a high quality of extrudate. Consequently, a processingtemperature below 200 �C, an inert atmosphere and a lower resi-dence time (3 min) were required.
Hence, the effect of these processing conditions on the thermalstability of neat polymers has been investigated by the molecularweight and intrinsic viscosity measurements at different steps ofthe process. The relative observations and the degradation mech-anism for both PLA and PBAT will be discussed in the next sections.
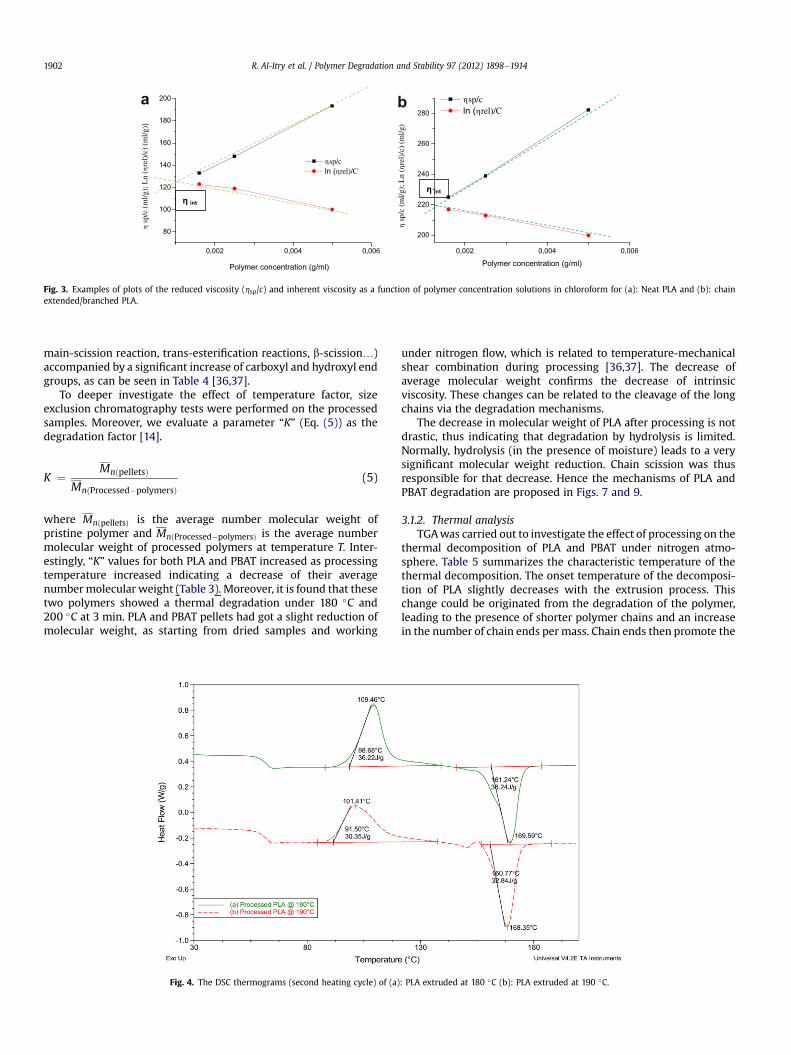
3.1.1. Molecular weight measurements3.1.1.1. Intrinsic viscosity. As it is common, the value of the intrinsicviscosity provides a measure of the ability of an isolated andGaussian polymer single chain to increase the viscosity of thesolvent in the absence of intermolecular interactions betweenpolymer molecules. The intrinsic viscosity, [h] was determined byextrapolation of reduced and inherent viscosities to zero concen-tration (cf. Fig. 3). These two viscosities were plotted respectivelyby using Huggins (Eq. (3)) [33] and Kraemer (Eq. (4)) [34,35]approaches:
hred ¼ hspC
¼ 1C*
�t � t0t0
�¼ ½h� þ K 0½h�2*C (3)
hinh ¼ Ln ðhrelÞC
¼ 1C*
�Ln
tt0
�¼ ½h� � K 00½h�2*C (4)
where “t” and “t0” denote respectively the efflux time of the solutionand solvent, K0 the Huggins constant and K00 the Kramer constant.The magnitude of K0 increasing can be related to the breadth of themolecular weight distribution or branching of the solute. K0 valuesfor PLA (K0 ¼ 0.3) and PBAT (K0 ¼ 0.93) showed that the chloroformrepresents a good solvent for the studied polyesters, indicating thatthe hydrodynamic volume of the polymer chain shrinks withfavorable interaction between polymer and solvent [35].
Intrinsic viscosities values of different solution in chloroform aresummarized in Table 3. The reported results show a decrease onintrinsic viscosity when both PLA and PBAT were extruded during3 min. These changes can be related to the cleavage of the chains toshorter ones via the degradation mechanisms (hydrolysis, random
0,002 0,004 0,006
80
100
120
140
160
180
200
Polymer concentration (g/ml)
sp/c ln ( rel)/C
sp/
c (m
l/g)
; Ln
(re
l)/c
) (m
l/g))
0,002 0,004 0,006
200
220
240
260
280
sp/c ln ( rel)/C
sp/
c (m
l/g);
Ln
(re
l)/c
) (m
l/g)
Polymer concentration (g/ml)
a b
Fig. 3. Examples of plots of the reduced viscosity (hsp/c) and inherent viscosity as a function of polymer concentration solutions in chloroform for (a): Neat PLA and (b): chainextended/branched PLA.
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e19141902
main-scission reaction, trans-esterification reactions, b-scission.)accompanied by a significant increase of carboxyl and hydroxyl endgroups, as can be seen in Table 4 [36,37].
To deeper investigate the effect of temperature factor, sizeexclusion chromatography tests were performed on the processedsamples. Moreover, we evaluate a parameter “K” (Eq. (5)) as thedegradation factor [14].
K ¼ MnðpelletsÞMnðProcessed�polymersÞ
(5)
where MnðpelletsÞ is the average number molecular weight ofpristine polymer and MnðProcessed�polymersÞ is the average numbermolecular weight of processed polymers at temperature T. Inter-estingly, “K” values for both PLA and PBAT increased as processingtemperature increased indicating a decrease of their averagenumbermolecular weight (Table 3). Moreover, it is found that thesetwo polymers showed a thermal degradation under 180 �C and200 �C at 3 min. PLA and PBAT pellets had got a slight reduction ofmolecular weight, as starting from dried samples and working
Fig. 4. The DSC thermograms (second heating cycle) of (a)
under nitrogen flow, which is related to temperature-mechanicalshear combination during processing [36,37]. The decrease ofaverage molecular weight confirms the decrease of intrinsicviscosity. These changes can be related to the cleavage of the longchains via the degradation mechanisms.
The decrease in molecular weight of PLA after processing is notdrastic, thus indicating that degradation by hydrolysis is limited.Normally, hydrolysis (in the presence of moisture) leads to a verysignificant molecular weight reduction. Chain scission was thusresponsible for that decrease. Hence the mechanisms of PLA andPBAT degradation are proposed in Figs. 7 and 9.
3.1.2. Thermal analysisTGAwas carried out to investigate the effect of processing on the
thermal decomposition of PLA and PBAT under nitrogen atmo-sphere. Table 5 summarizes the characteristic temperature of thethermal decomposition. The onset temperature of the decomposi-tion of PLA slightly decreases with the extrusion process. Thischange could be originated from the degradation of the polymer,leading to the presence of shorter polymer chains and an increasein the number of chain ends per mass. Chain ends then promote the
: PLA extruded at 180 �C (b): PLA extruded at 190 �C.
1 10 100
1500
2000
2500
3000
3500
Eta*
(Pa.
s)
(rad/s)
PLA_T180 PLA_T190 PLA_200
1 10 100
1000
1500
2000
2500
3000
35004000
Eta*
(Pa.
s)
(rad/s)
PLA_T180 PLA_T190 PLA_200
1 10 1001000
2000
3000
4000
5000
60007000
Eta*
(Pa.
s)
(rad/s)
PBAT_T180 PBAT_190 PBAT_200
a
b
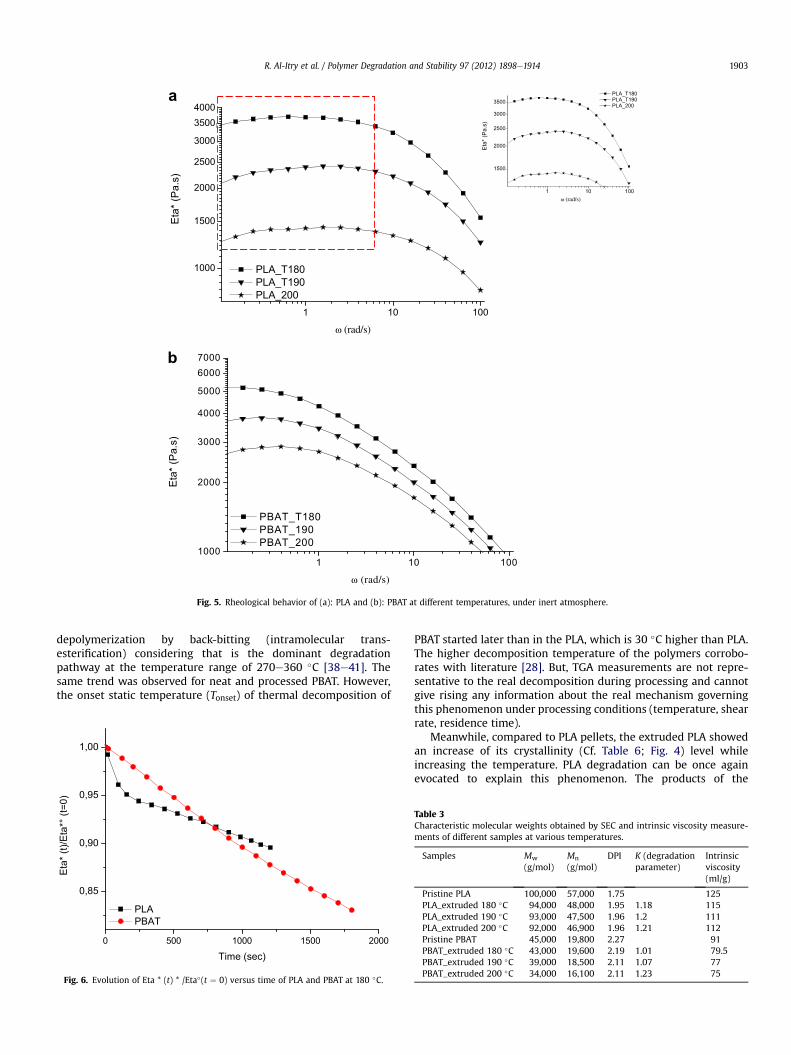
Fig. 5. Rheological behavior of (a): PLA and (b): PBAT at different temperatures, under inert atmosphere.
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e1914 1903
depolymerization by back-bitting (intramolecular trans-esterification) considering that is the dominant degradationpathway at the temperature range of 270e360 �C [38e41]. Thesame trend was observed for neat and processed PBAT. However,the onset static temperature (Tonset) of thermal decomposition of
0 500 1000 1500 2000
0,85
0,90
0,95
1,00
Eta*
(t)/E
ta*°
(t=0
)
Time (sec)
PLA PBAT
Fig. 6. Evolution of Eta * (t) * /Eta�(t ¼ 0) versus time of PLA and PBAT at 180 �C.
PBAT started later than in the PLA, which is 30 �C higher than PLA.The higher decomposition temperature of the polymers corrobo-rates with literature [28]. But, TGA measurements are not repre-sentative to the real decomposition during processing and cannotgive rising any information about the real mechanism governingthis phenomenon under processing conditions (temperature, shearrate, residence time).
Meanwhile, compared to PLA pellets, the extruded PLA showedan increase of its crystallinity (Cf. Table 6; Fig. 4) level whileincreasing the temperature. PLA degradation can be once againevocated to explain this phenomenon. The products of the
Table 3Characteristic molecular weights obtained by SEC and intrinsic viscosity measure-ments of different samples at various temperatures.
Samples Mw
(g/mol)Mn
(g/mol)DPI K (degradation
parameter)Intrinsicviscosity(ml/g)
Pristine PLA 100,000 57,000 1.75 125PLA_extruded 180 �C 94,000 48,000 1.95 1.18 115PLA_extruded 190 �C 93,000 47,500 1.96 1.2 111PLA_extruded 200 �C 92,000 46,900 1.96 1.21 112Pristine PBAT 45,000 19,800 2.27 91PBAT_extruded 180 �C 43,000 19,600 2.19 1.01 79.5PBAT_extruded 190 �C 39,000 18,500 2.11 1.07 77PBAT_extruded 200 �C 34,000 16,100 2.11 1.23 75

Table 4Acid value of neat and processed PLA and PBAT.
Samples Acid value (mgKOH/g polymer)
Pristine PLA 1.3PLA_extruded 180 �C 1.41Pristine PBAT 0.45PBAT_extruded 180 �C 0.588
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e19141904
degradation (Oligomers for example) would then act as nucleatingagent allowing a reduced cold crystallization temperature [36]. It isshown that the thermal analysis was corroborated with molecularweight measurements which highlighted a reduction on the
PLA CH
CH3
O
C O CH
CH3
O
C O PLA
OH H
PLA CH
CH3
O
C OHOH
P
+
PLA CH
CH3
O
C O CHCH2 O
C O PLA
H
Proposed hydrolytic mechanism of PLA
β-C-H Hydrogen transfer of PLA
a
b
Fig. 7. Proposed degradatio
Fig. 8. FT-IR spectra of nea
average molecular weight. However, DSC was not efficient to showthe thermal degradation of PBAT.
Therefore, in order to deeply understand the thermal degrada-tion of PLA and PBAT, the rheological properties of both polymerswere investigated.
3.1.3. Rheological investigationThe rheological investigation is used as a suitable tool to eval-
uate the thermal stability of PLA and PBAT upon processing atincreasing temperature from 180 �C to 200 �C. It is shown in Fig. 5that the viscosity values of PBAT and PLA decreased from 5000 Pa sto 2800 Pa s and from 3500 Pa s to 1250 Pa s respectively. Accordingto the literature [39], the decrease in viscosity corresponds to a loss
CH
CH3
O
C O PLA
LA CH
CH3
C O CH
CH3
O
C O PLA
OH OH
C
O
OH CH2C+
Acid Vinylic terminated ester
n mechanisms of PLA.
t and processed PLA.

OH
n
O O
O CH2 O4CH
O
CH2 O
p
CH2
O
CH2 CH
H
OCH2CH2
H
H
O
OCH 2 CH2 CH2CHHO
O
CH2CH2
CH2 CH2 +
OH
n
O O
O CH 2 O4CH
O
CH 2 O
p
CH 2
O
CH 2 CH
H
OCH 2CH 2
H
H
OH
n p
O O
O CH 2 O4CH 2 4 O H
4
OO
O
CH 2
OH H
OH
n
O O
O CH 2 O4 H CH 2 4OH
OCH 2
4
O
p
O H+
Proposed hydrolytic mechanism of PBAT
Main chain scissions degradation
-C-H Hydrogen transfer of PBAT
a
b
c
Fig. 9. Proposed degradation mechanisms of PBAT.
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e1914 1905
in molecular weight. This was caused mainly by the breaking downof the inter- and intra-molecular forces increasing the mobility ofchains. Moreover, the processing temperature had larger influenceon the viscosities values of PLA during time, which decreased fromat constant frequency. The viscosityefrequency curves of PLA meltsdisplay a maximum instead of a horizontal plateau. This unusualbehavior can be related to a thermal degradation process at longresidence times in the rheometer despite using nitrogen [62].Furthermore, the complex viscosity, normalized by its initial values
Table 5Thermogravimetric analysis for pristine and processed polymers.
Samples Tonset(�C)
T10(�C)
Tmaximum
degradation (�C)
Samples Tonset(�C)
T10 (�C) Tmaximum
degradation
(�C)
PLA_Pellets 328 354 383 PBAT_Pellets 356 393 424Processed PLA 300 343 381 Processed PBAT 343 382 425
at t ¼ 0, is presented as a function of time for PLA and PBAT, asshown in Fig. 6. For these two polymers, a significant reduction inviscosity over time was observed. This drastic decrease alsoconfirms the possible thermal degradation occurring duringprocessing.
3.1.4. Mechanism of thermal degradation of neat PLA and PBATThermal instability of PLA at high temperatures in the extrusion
process has been broadly investigated [40,42,43,46]. Numerous
Table 6Crystallinity* values or Pristine PLA, processed PLA at 180 �Cand at 190 �C.
Samples Xc (%)
PLA pellets 1.7PLA_extruded 180 �C 35PLA_extruded 190 �C 39

Fig. 10. FT-IR spectra of neat and processed PBAT.
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e19141906
researches reveal that the degradation reactions can occur duringprocessing. Their identification seems tobecomplicated.On theotherhand, few efforts have been dedicated to the study of the thermaldegradation of PBAT. It was reported that no significant chain modi-fication of PBAT occurred upon processing up to 200 �C [14].
In case of polyesters, many several degradationmechanisms canhappen depending on the processing conditions. Hydrolysis of theester linkage, water-based degradation mechanism, randomlytakes place in the polymer. The rate of such a mechanism dependson water concentration, acid or base catalyst, morphology of thepolymer and temperature [40e44]. Based on the study performedby Witzke (1997), hydrolysis is the dominant degradation mecha-nism at a temperature between 150 and 215 �C [45]. Other studyshowed that the thermal degradation was also found to proceed byrandommain-chain scission at temperatures above 180 �C [13]. Thechain end groups contribute also to the degradation. Their contri-bution is more pronounced in the temperature range of270�Ce360 �C [41]. Gupta et al. [8] reported that, in polyesters case,there are three possible linkages i.e. carboneOxygen ether linkage(beHeC hydrogen transfer), Carbonyl CarboneCarbon linkage andCarbonyl CarboneOxygen linkage which can undergo scissionlinkages. The Carbonyl CarboneOxygen linkagewould be cleaved inthe early stage of PLA thermal degradation. Tranesterification, cis-elimination and thermo-oxidative reaction can not occur in ourcase; Trans-esterification mechanism was found as a dominant
Table 7Comparison between the reaction rate of carboxylic acid/epoxide andhydroxyl/epoxide.
Reactive pair Reaction rate
Carboxylic acid/epoxide 18Primary hydroxyl/epoxide 1.2Secondary hydroxyl/epoxide 1
degradation mechanism of PLA at high temperatures (above200 �C), leading to the formation of cyclic oligomers [12,41,51]. Cis-elimination and thermo-oxidative reactions also occur at hightemperature range (300e500 �C) [12,41]. Based on these observa-tions, the possible mechanisms of the thermal degradation of PLAas well as PBAT in our study were determined, as shown in Figs. 7and 9 [49]. They include hydrolysis and random main-chain scis-sion (a b-C-H hydrogen transfer reaction, leading to a vinyl esterand acid end groups was presented).
The hydrolytic degradation of PLA and PBAT is primarily due tohydrolysis of the ester linkages, which occurs more or lessrandomly along the backbone of the polymer. It requires thepresence of water according to the following reaction (Reactions(7a) and (9a)). In PBAT case water can also react with the Carbonylfunctions close to the benzene ring.
FTIR spectra of neat and processed PLA are represented in Fig. 8.According to Ibrahim et al. very strong and broad peaks in the range3200e3600 cm�1 are due to stretching vibrations of OeH groupsand the two peaks at around 2800e3000 cm�1 are clearly associ-ated with the symmetric stretching vibration of the axial CH groupsin saturated hydrocarbons. The lower intensity peaks in the range2100e2300 cm�1 are also related to OeH stretching vibrations incarboxyl acids groups. The intensive peak originating from C]Ostretching vibrations is located at around 1720 cm�1. Many weakerpeaks in the range 1250e1050 cm�1 are assigned to CeO from
Table 8K0 values for neat and modified polymers.
Samples K0 Samples K0
PLA_0 0.3 PBAT_0 0.93PLA_0.25_stable 0.42 PBAT_0,25_stable 1.8PLA_0.5_stable 0.5 PBAT_0,5_stable 1.44PLA_1_stable 0.54 PBAT_1_stable 3.42
O
O CH2 CH2 CH2CH HOO
CH2CH2
CH2 CH2
O
O
OH
CHCH2
OH
n
O O
O CH2 O4CH
O
CH2 O
p
CH2
O
CH2 CH
H
OCH2CH2
H
H
JONCRYL
O
OR5
(a)
OH
CH
OO
O
CH2
Degradation
Vinylic ester Acid end groups
(b)
O
PBAT
O
JONCRYL
O
OR5
O
+
O
CH
CH
OOn
OO O
OH
O
O
H H
O
O CH
(a)
JONCRYL
O
OR5
OC
O
OH CH C
Acid End Groups
(b)
JONCRYL
OH
CH O
PLA
Degradation
+
Vinylic terminated ester
Predicted mechanism of the reaction between PLA and Joncryl
Predicted mechanism of the reaction between PBAT and Joncryl
a
b
Fig. 11. Predicted reaction between polyesters and epoxy functions.
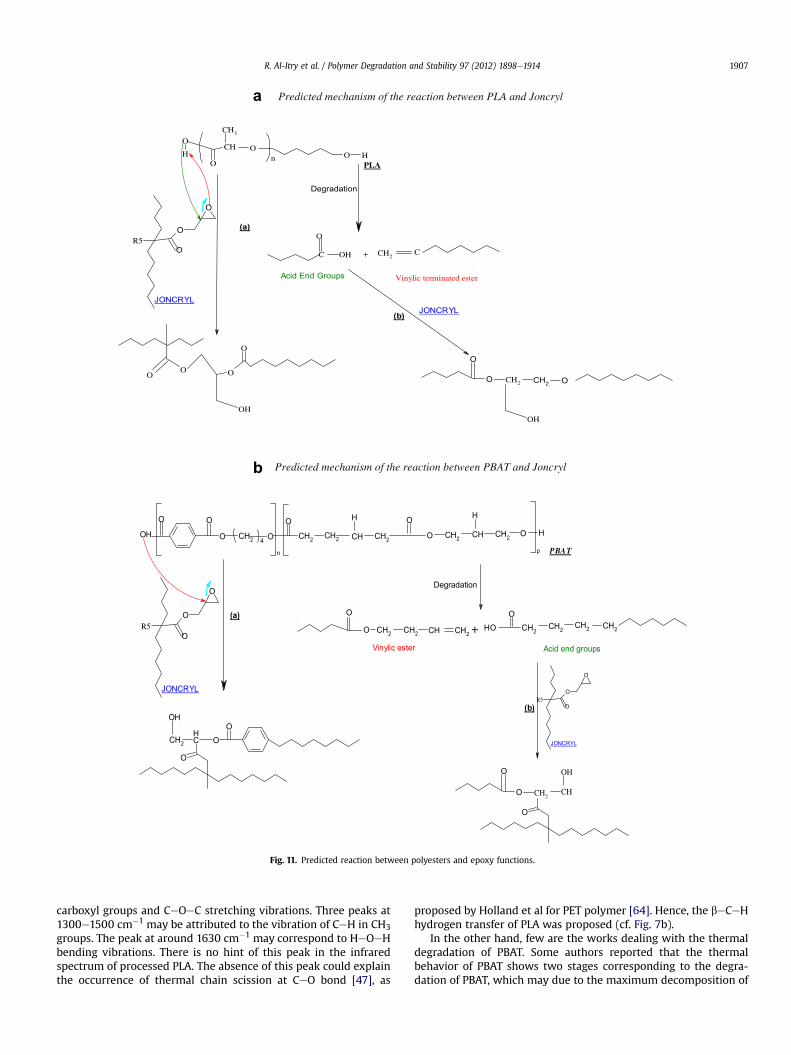
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e1914 1907
carboxyl groups and CeOeC stretching vibrations. Three peaks at1300e1500 cm�1 may be attributed to the vibration of CeH in CH3groups. The peak at around 1630 cm�1 may correspond to HeOeHbending vibrations. There is no hint of this peak in the infraredspectrum of processed PLA. The absence of this peak could explainthe occurrence of thermal chain scission at CeO bond [47], as
proposed by Holland et al for PET polymer [64]. Hence, the beCeHhydrogen transfer of PLA was proposed (cf. Fig. 7b).
In the other hand, few are the works dealing with the thermaldegradation of PBAT. Some authors reported that the thermalbehavior of PBAT shows two stages corresponding to the degra-dation of PBAT, which may due to the maximum decomposition of
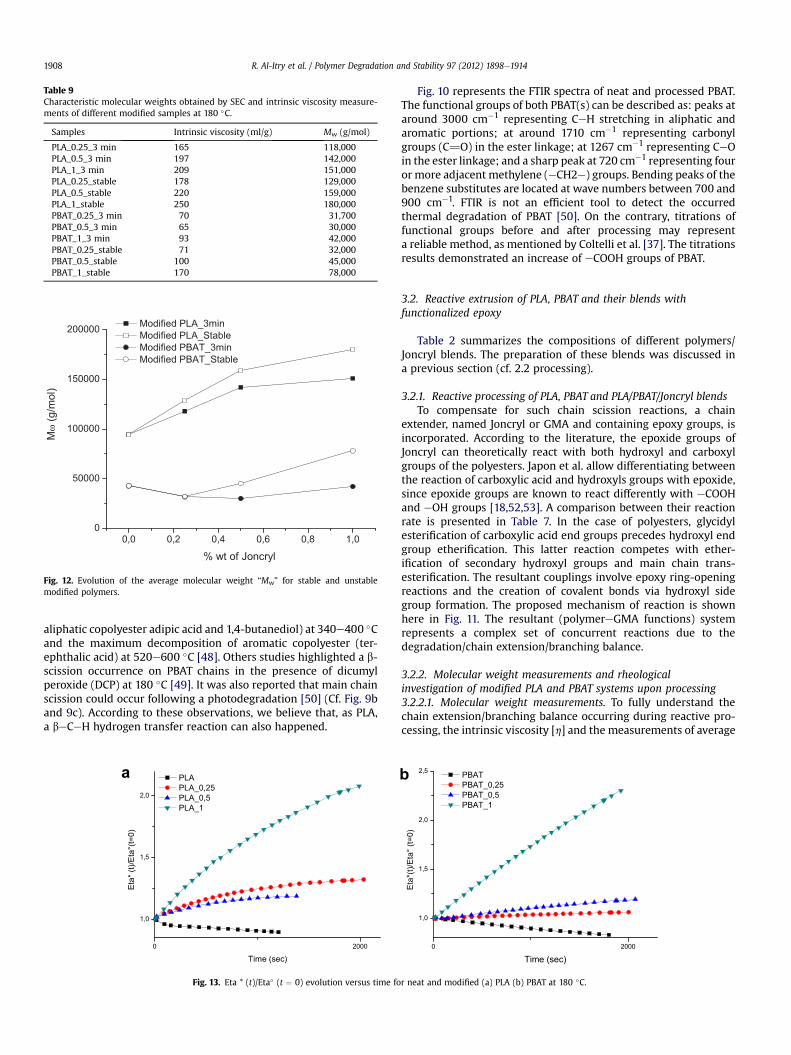
Table 9Characteristic molecular weights obtained by SEC and intrinsic viscosity measure-ments of different modified samples at 180 �C.
Samples Intrinsic viscosity (ml/g) Mw (g/mol)
PLA_0.25_3 min 165 118,000PLA_0.5_3 min 197 142,000PLA_1_3 min 209 151,000PLA_0.25_stable 178 129,000PLA_0.5_stable 220 159,000PLA_1_stable 250 180,000PBAT_0.25_3 min 70 31,700PBAT_0.5_3 min 65 30,000PBAT_1_3 min 93 42,000PBAT_0.25_stable 71 32,000PBAT_0.5_stable 100 45,000PBAT_1_stable 170 78,000
0,0 0,2 0,4 0,6 0,8 1,00
50000
100000
150000
200000
M (g
/mol
)
% wt of Joncryl
Modified PLA_3min Modified PLA_Stable Modified PBAT_3min Modified PBAT_Stable
Fig. 12. Evolution of the average molecular weight “Mw” for stable and unstablemodified polymers.
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e19141908
aliphatic copolyester adipic acid and 1,4-butanediol) at 340e400 �Cand the maximum decomposition of aromatic copolyester (ter-ephthalic acid) at 520e600 �C [48]. Others studies highlighted a b-scission occurrence on PBAT chains in the presence of dicumylperoxide (DCP) at 180 �C [49]. It was also reported that main chainscission could occur following a photodegradation [50] (Cf. Fig. 9band 9c). According to these observations, we believe that, as PLA,a beCeH hydrogen transfer reaction can also happened.
00020
1,0
1,5
2,0
Eta*
(t)/E
ta°(
t=0)
Time (sec)
PLA PLA_0,25 PLA_0,5 PLA_1
a
Fig. 13. Eta * (t)/Eta� (t ¼ 0) evolution versus time fo
Fig. 10 represents the FTIR spectra of neat and processed PBAT.The functional groups of both PBAT(s) can be described as: peaks ataround 3000 cm�1 representing CeH stretching in aliphatic andaromatic portions; at around 1710 cm�1 representing carbonylgroups (C]O) in the ester linkage; at 1267 cm�1 representing CeOin the ester linkage; and a sharp peak at 720 cm�1 representing fouror more adjacent methylene (eCH2e) groups. Bending peaks of thebenzene substitutes are located at wave numbers between 700 and900 cm�1. FTIR is not an efficient tool to detect the occurredthermal degradation of PBAT [50]. On the contrary, titrations offunctional groups before and after processing may representa reliable method, as mentioned by Coltelli et al. [37]. The titrationsresults demonstrated an increase of eCOOH groups of PBAT.
3.2. Reactive extrusion of PLA, PBAT and their blends withfunctionalized epoxy
Table 2 summarizes the compositions of different polymers/Joncryl blends. The preparation of these blends was discussed ina previous section (cf. 2.2 processing).
3.2.1. Reactive processing of PLA, PBAT and PLA/PBAT/Joncryl blendsTo compensate for such chain scission reactions, a chain
extender, named Joncryl or GMA and containing epoxy groups, isincorporated. According to the literature, the epoxide groups ofJoncryl can theoretically react with both hydroxyl and carboxylgroups of the polyesters. Japon et al. allow differentiating betweenthe reaction of carboxylic acid and hydroxyls groups with epoxide,since epoxide groups are known to react differently with eCOOHand eOH groups [18,52,53]. A comparison between their reactionrate is presented in Table 7. In the case of polyesters, glycidylesterification of carboxylic acid end groups precedes hydroxyl endgroup etherification. This latter reaction competes with ether-ification of secondary hydroxyl groups and main chain trans-esterification. The resultant couplings involve epoxy ring-openingreactions and the creation of covalent bonds via hydroxyl sidegroup formation. The proposed mechanism of reaction is shownhere in Fig. 11. The resultant (polymereGMA functions) systemrepresents a complex set of concurrent reactions due to thedegradation/chain extension/branching balance.
3.2.2. Molecular weight measurements and rheologicalinvestigation of modified PLA and PBAT systems upon processing3.2.2.1. Molecular weight measurements. To fully understand thechain extension/branching balance occurring during reactive pro-cessing, the intrinsic viscosity [h] and the measurements of average
00020
1,0
1,5
2,0
2,5
Eta*
(t)/E
ta° (
t=0)
Time (sec)
PBAT PBAT_0,25 PBAT_0,5 PBAT_1
b
r neat and modified (a) PLA (b) PBAT at 180 �C.

1 10 100
1000
10000
Com
plex
vis
cosi
ty E
ta* (
Pa.s
)
(rad/s)
DFS180_PLA4032D DFS180_PBAT
Fig. 14. Complex viscosity versus angular frequency (at 180 �C) for the neat PLA andPBAT.
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e1914 1909
molecular weight have been carried out. The incorporation of GMAfunctions into PLA and PBAT during processing significantlyincreases the Mw and the intrinsic viscosity (Table 9; Fig. 12). Thechange of molecular weight of studied systems was a directevidence for chain extension which can be considered as the
1 10 100
100
1000
10000
PLA_T180 PLA_0,25_T180 PLA_0,5_T180 PLA_1_T180
Eta*
(Pa.
s)
ω (rad/s)
a b
Fig. 15. The complex viscosity (a) and the storage modulus (b) angular frequency dependelibrium state.
0111,0
2000
4000
6000
H (
Pa.
s
(sec)
PBAT PBAT_0,25 PBAT_0,5 PBAT_1
a b
Fig. 16. Relaxation spectrum of neat and m
recuperation of the decrease of the molecular weight during meltextrusion, which is more pronounced for PLA. The measurementsof intrinsic viscosity corroborate thus the obtained one of SECexperiments. We believe that the increase of K0 values with theincorporation of reactive GMA functions is caused by the lowersolubility of polymer chains, probably due to branching chain (cf.Table 8) [35]. In other words, the polymeresolvent interactions aredecreasing, but the polymerepolymer interactions, or association,are increasing with the ionic strength. It is clearly evident to noticethat the branching reactions at such Joncryl concentrations did notreach the point of heavy cross-linking or gel formation. Recently,some authors [21] reported that when the ratio of Joncryl reached1.5 wt%, the sample was slightly cross-linking. It was also showedthat the reaction of crosslinking of epoxy functions starts at hightemperatures [54].
A careful study of rheological behavior and thermal stability ofthese materials will provide more informations for processing PLA/PBAT blends in proper conditions and thus avoid the irreversiblestructure changes under non optimizedmelt processing conditions.
3.2.2.2. Rheological investigations of modified PLA and PBAT sys-tems. As discussed before, the results of rheological tests of neatPLA and PBAT at 180 �C demonstrated a monotonous decreasingviscosity over time, highlighting therefore the occurrence ofthermal degradation. This latter phenomenon is related to thedecrease of intrinsic viscosity and the average molecular weight.Strong differences in the rheological response of polymer matrix
1 10 100
1000
10000
100000
PLA_T180 PLA_0,25_T180 PLA_0,5_T180 PLA_1_T180
G' (
Pa)
ω (rad/s)
nce at 180 �C for neat and modified PLA with chain extender after reaching the equi-
0111,00
2000
4000
6000
H (
Pa.s
sec
PLA PLA_0,25 PLA_0,5 PLA_1
odified polymers (a): PBAT (b): PLA.
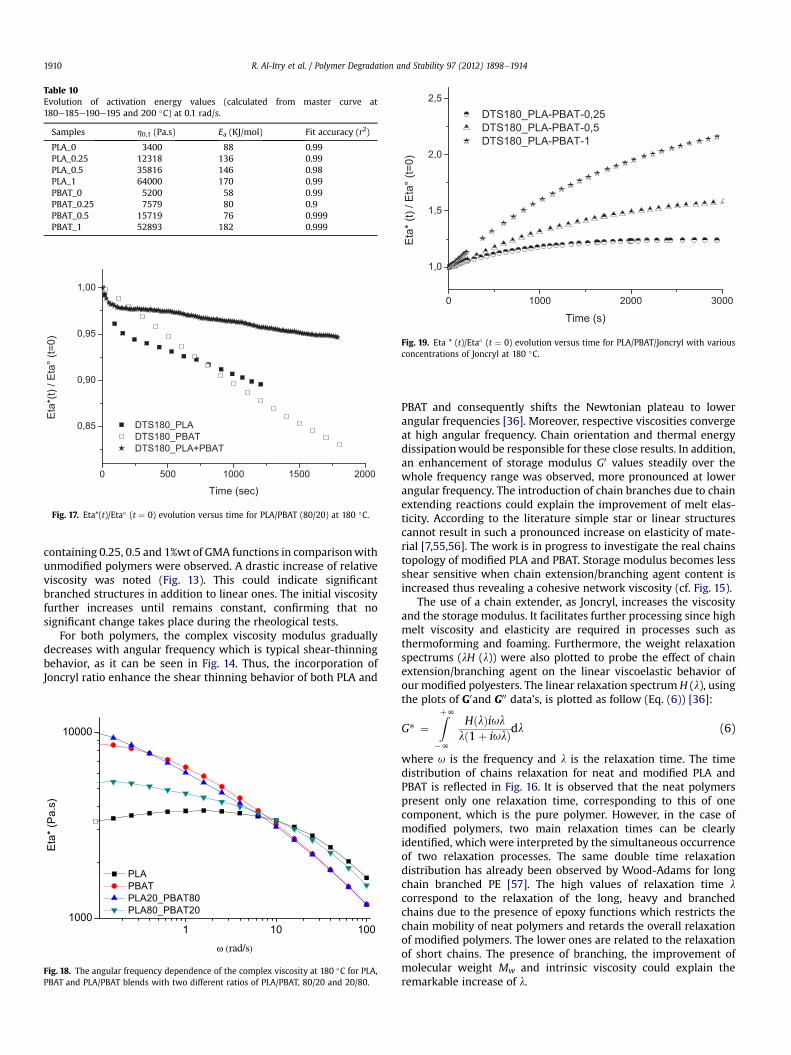
Table 10Evolution of activation energy values (calculated from master curve at180e185e190e195 and 200 �C) at 0.1 rad/s.
Samples h0,1 (Pa.s) Ea (KJ/mol) Fit accuracy (r2)
PLA_0 3400 88 0.99PLA_0.25 12318 136 0.99PLA_0.5 35816 146 0.98PLA_1 64000 170 0.99PBAT_0 5200 58 0.99PBAT_0.25 7579 80 0.9PBAT_0.5 15719 76 0.999PBAT_1 52893 182 0.999
0 500 1000 1500 2000
0,85
0,90
0,95
1,00
DTS180_PLA DTS180_PBAT DTS180_PLA+PBAT
Eta*
(t) /
Eta°
(t=0
)
Time (sec)
Fig. 17. Eta*(t)/Eta� (t ¼ 0) evolution versus time for PLA/PBAT (80/20) at 180 �C.
0 1000 2000 3000
1,0
1,5
2,0
2,5 DTS180_PLA-PBAT-0,25 DTS180_PLA-PBAT-0,5 DTS180_PLA-PBAT-1
Eta*
(t) /
Eta
° (t=
0)
Time (s)
Fig. 19. Eta * (t)/Eta� (t ¼ 0) evolution versus time for PLA/PBAT/Joncryl with variousconcentrations of Joncryl at 180 �C.
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e19141910
containing 0.25, 0.5 and 1%wt of GMA functions in comparisonwithunmodified polymers were observed. A drastic increase of relativeviscosity was noted (Fig. 13). This could indicate significantbranched structures in addition to linear ones. The initial viscosityfurther increases until remains constant, confirming that nosignificant change takes place during the rheological tests.
For both polymers, the complex viscosity modulus graduallydecreases with angular frequency which is typical shear-thinningbehavior, as it can be seen in Fig. 14. Thus, the incorporation ofJoncryl ratio enhance the shear thinning behavior of both PLA and
1 10 1001000
10000
Eta*
(Pa.
s)
rad/s
PLA PBAT PLA20_PBAT80 PLA80_PBAT20
Fig. 18. The angular frequency dependence of the complex viscosity at 180 �C for PLA,PBAT and PLA/PBAT blends with two different ratios of PLA/PBAT, 80/20 and 20/80.
PBAT and consequently shifts the Newtonian plateau to lowerangular frequencies [36]. Moreover, respective viscosities convergeat high angular frequency. Chain orientation and thermal energydissipationwould be responsible for these close results. In addition,an enhancement of storage modulus G0 values steadily over thewhole frequency range was observed, more pronounced at lowerangular frequency. The introduction of chain branches due to chainextending reactions could explain the improvement of melt elas-ticity. According to the literature simple star or linear structurescannot result in such a pronounced increase on elasticity of mate-rial [7,55,56]. The work is in progress to investigate the real chainstopology of modified PLA and PBAT. Storage modulus becomes lessshear sensitive when chain extension/branching agent content isincreased thus revealing a cohesive network viscosity (cf. Fig. 15).
The use of a chain extender, as Joncryl, increases the viscosityand the storage modulus. It facilitates further processing since highmelt viscosity and elasticity are required in processes such asthermoforming and foaming. Furthermore, the weight relaxationspectrums (lH (l)) were also plotted to probe the effect of chainextension/branching agent on the linear viscoelastic behavior ofour modified polyesters. The linear relaxation spectrum H (l), usingthe plots of G0and G00 data’s, is plotted as follow (Eq. (6)) [36]:
G* ¼ZþN
�N
HðlÞiullð1þ iulÞdl (6)
where u is the frequency and l is the relaxation time. The timedistribution of chains relaxation for neat and modified PLA andPBAT is reflected in Fig. 16. It is observed that the neat polymerspresent only one relaxation time, corresponding to this of onecomponent, which is the pure polymer. However, in the case ofmodified polymers, two main relaxation times can be clearlyidentified, which were interpreted by the simultaneous occurrenceof two relaxation processes. The same double time relaxationdistribution has already been observed by Wood-Adams for longchain branched PE [57]. The high values of relaxation time l
correspond to the relaxation of the long, heavy and branchedchains due to the presence of epoxy functions which restricts thechain mobility of neat polymers and retards the overall relaxationof modified polymers. The lower ones are related to the relaxationof short chains. The presence of branching, the improvement ofmolecular weight Mw and intrinsic viscosity could explain theremarkable increase of l.

Fig 20: The angular frequency dependence
of (a): the complex viscosity (Eta*) and (b):
storage modulus (G’) versus angular
frequency at 180°C for unmodified and
modified PLA/PBAT (80/20) blends
1 10 1001
10
100
1000
10000
100000
PLA PBAT PLA_PBAT_0 PLA_PBAT_0,25 PLA_PBAT_0,5 PLA_PBAT_1
G' (
Pa)
ω (rad/s)
1 10 100
1000
10000
100000
DFS180_PLA-PBAT-0 DFS180_PLA-PBAT-0,25 DFS180_PLA-PBAT-0,5 DFS180_PLA-PBAT-1
Com
plex
vis
cosi
ty (P
a.s)
ω (Hz)
a
b
Fig. 20. The angular frequency dependence of (a): the complex viscosity (Eta*) and (b): storage modulus (G0) versus angular frequency at 180 �C for unmodified and modified PLA/PBAT (80/20) blends.
Fig. 21. Predicted reaction between Joncryl and polyesters.
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e1914 1911
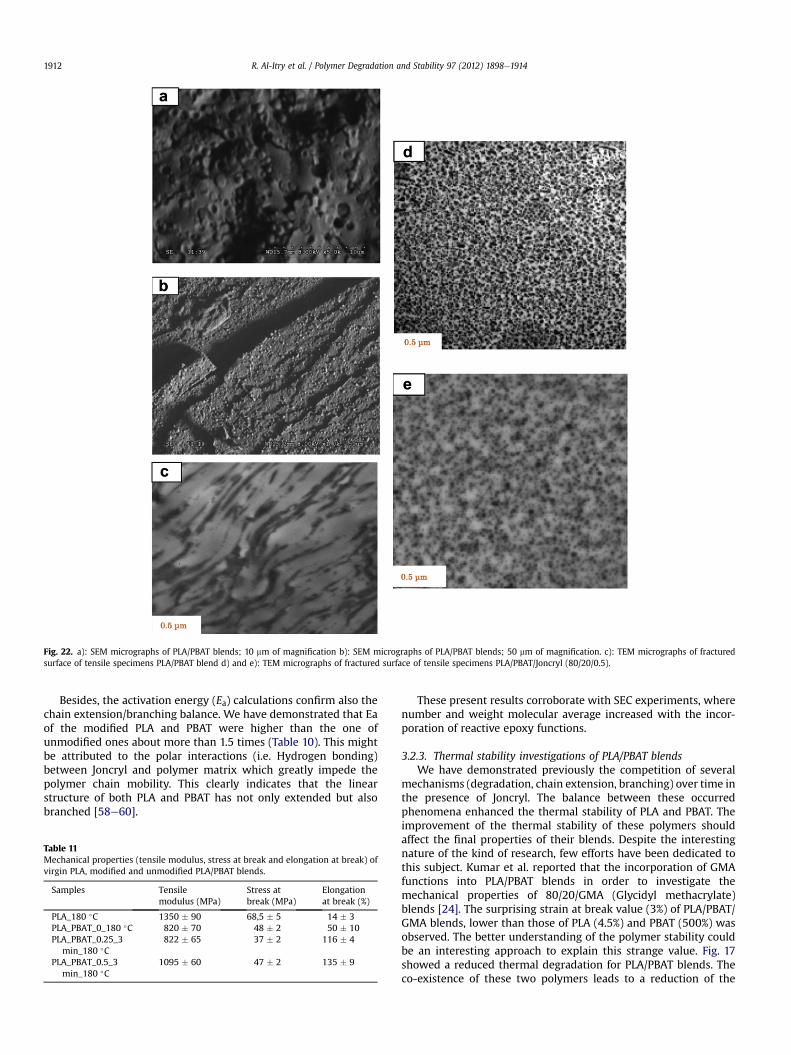
Fig. 22. a): SEM micrographs of PLA/PBAT blends; 10 mm of magnification b): SEM micrographs of PLA/PBAT blends; 50 mm of magnification. c): TEM micrographs of fracturedsurface of tensile specimens PLA/PBAT blend d) and e): TEM micrographs of fractured surface of tensile specimens PLA/PBAT/Joncryl (80/20/0.5).
R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e19141912
Besides, the activation energy (Ea) calculations confirm also thechain extension/branching balance. We have demonstrated that Eaof the modified PLA and PBAT were higher than the one ofunmodified ones about more than 1.5 times (Table 10). This mightbe attributed to the polar interactions (i.e. Hydrogen bonding)between Joncryl and polymer matrix which greatly impede thepolymer chain mobility. This clearly indicates that the linearstructure of both PLA and PBAT has not only extended but alsobranched [58e60].
Table 11Mechanical properties (tensile modulus, stress at break and elongation at break) ofvirgin PLA, modified and unmodified PLA/PBAT blends.
Samples Tensilemodulus (MPa)
Stress atbreak (MPa)
Elongationat break (%)
PLA_180 �C 1350 � 90 68,5 � 5 14 � 3PLA_PBAT_0_180 �C 820 � 70 48 � 2 50 � 10PLA_PBAT_0.25_3
min_180 �C822 � 65 37 � 2 116 � 4
PLA_PBAT_0.5_3min_180 �C
1095 � 60 47 � 2 135 � 9
These present results corroborate with SEC experiments, wherenumber and weight molecular average increased with the incor-poration of reactive epoxy functions.
3.2.3. Thermal stability investigations of PLA/PBAT blendsWe have demonstrated previously the competition of several
mechanisms (degradation, chain extension, branching) over time inthe presence of Joncryl. The balance between these occurredphenomena enhanced the thermal stability of PLA and PBAT. Theimprovement of the thermal stability of these polymers shouldaffect the final properties of their blends. Despite the interestingnature of the kind of research, few efforts have been dedicated tothis subject. Kumar et al. reported that the incorporation of GMAfunctions into PLA/PBAT blends in order to investigate themechanical properties of 80/20/GMA (Glycidyl methacrylate)blends [24]. The surprising strain at break value (3%) of PLA/PBAT/GMA blends, lower than those of PLA (4.5%) and PBAT (500%) wasobserved. The better understanding of the polymer stability couldbe an interesting approach to explain this strange value. Fig. 17showed a reduced thermal degradation for PLA/PBAT blends. Theco-existence of these two polymers leads to a reduction of the

R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e1914 1913
terminal eCOOH and eOH in the PLA molecular chain due toa condensation reaction [59]. Comparing to PLA, PLA/PBAT blendsshowed stronger shear-thinning behavior in the whole frequencyrange. This trend becomes more pronounced with the increasing ofPBAT contents as well PBAT present a more pronounced pseudo-plastic behavior (cf. Fig. 18) [63]. The complex viscosity of PLA/PBATblends increased significantly compared with pure PLA. This fullyproves the effectiveness of PBAT in improving melt viscosity andelasticity of PLA. But, binary unmodified PLA/PBAT blend exhibitthe typical morphological feature of incompatible systems witha poor interfacial adhesion betweenmatrix and dispersed phase (cf.SEM micrographs, Fig. 22).
However, it has demonstrated that the presence of the chainextension/branching agent in blends avoids any thermal degrada-tion and provides the stabilization of the blends, probably due toester linkage at the interface PLA/PBAT, as it can be seen in Fig. 19.The dependence of the complex viscosity (h*) versus angularfrequencyof PLA/PBAT blendswith various concentrations of Joncrylis shown in Fig.18. Different qualitative behaviors depending on theamount of Joncryl were shown (Cf. Fig. 20), indicating some chem-ical reactions taking place during the reactive blending process. Theshear-thinning tendency of PLA/PBAT melts becomes stronger withthe increasing of GMA functions content compared to neat PLA. Thisphenomenon can be attributed to the introduction of long chainbranches during the reactive extrusion and the improvementofmeltstability during processing [61]. At given oscillatory shear frequency,the complex viscosity of PLA/PBAT/Joncryl is higher than that of PLA/PBAT blends and generally increases with increasing the amount ofthe compatibilizer [40]. The enhancement of the dispersion, theincrease of the interaction among droplets and the stiffening of theinterface could explain this increase. Moreover, the improvedadhesion due to formation of chemical bonds between PLA-PBATand GMA functions was confirmed by TEM micrographs (Fig. 22).More information about the rheological behavior of the modifiedPLAePBAT blends can be given by the evolution of storage modulus(G0) (Fig. 20). The incorporation of 0.25%wtof GMA functions slightlychanges the behavior of PLAePBAT blends due to poor or insufficientcompatibilization between the matrix and the dispersed phase. Inthis case, PLA/PBAT blend still almost linear since there is no obvioustransition in dynamic modulus at low frequency. On the other hand,the transition of storage modulus at very low frequency for PLA/PBAT/GMA (80/20/0.5) and PLA/PBAT/GMA (80/20/1) indicates theappearance of some branched chains than linear ones. The mecha-nisms of interfacial reaction of PLA/PBAT blends with epoxy func-tions is demonstrated here (Fig. 21).
3.2.4. Mechanical properties of PLA/PBAT blendsOnce the reactive blends were prepared during 3 min (reaction
time), the tensile properties were being performed. The tensilemechanical properties (tensile modulus, stress at break and elon-gation at break) of PLA/PBAT blends are summarized in Table 11. Theresults showed that the brittle properties of PLA can be modified bythe addition of PBAT. The elongation at break increases from 14%(pure PLA) to 50% (PLA/PBAT blends) under processing conditionsunlike Kumar et al.’s study [24]. It is probably related to the presenceof soft elastomeric phase. The reducing of brittleness is also high-lighted by the decrease of the tensile modulus from 1350 MPa to820MPa. Hence, the elongation at break of PLA/PBAT (80/20) blendswith 0,25 and 0.5 %wt Joncryl achieved 116% and 135% respectively,higher than that for pure PLA (14%). The tensile modulus increaseswith the incorporation of GMA functions from 1350 MPa for PLApure to 1095 MPa for PLA/PBAT/0.5 which indicates a reactivitycontrol at the interface due to the formation of ester linkagesbetween PLA, PBAT and Joncryl. Therefore, the improvement ofmechanical properties like elongation at break and tensile modulus
was highlighted by the effect of compatibilization of Joncryl. Theobtained properties have been corroborated with TEMmicroscopy.
4. Conclusions
Throughout this work, the thermal degradation was firstlydemonstrated and quantified of the neat PLA and PBAT by usingthermal and rheological tools coupled to the measurements of theaverage molecular weight and intrinsic viscosity. Secondly, it wasshown that the reactive extrusion gives rise a more stable andmodified PLA and PBAT. Finally, the 80/20 blend was also preparedand its structural, rheological and improved mechanical propertieswas compared the uncompatibilized one and discussed in the basesof the real mechanisms of degradation/chain extension of thepolymers. Many observations were derived:
1. The assessment of thermal degradation for both PLA and PBATshowed that these two polymers degrade under temperature.The occurred thermal degradation of PLA and PBAT wasassessed by the decrease of molecular weight and intrinsicviscosity. Moreover, the present thermal degradation wasconfirmed by the rheological investigation, where the viscositydecreased at very high residence time.
2. The incorporation of a chain extension/branching agent(Joncryl ADR-4368), by reaction extrusion, into both PLA andPBAT showed an improvement of their thermal stability. Thisproperty is thus strongly dependent on the amount of GMAfunctions and on the reaction time because of the competingbranching and degradation reactions. It is demonstratedthroughout this paper that the epoxy reactive functions havebeen successfully used to increase the molar mass, intrinsicviscosity, shear thinning and elasticity during melt processingdue to the formation of branching chains.
3. Hence, the mechanisms of degradation, chain extending,branching with GMA functions and their competition havebeen proposed.
4. Rheological investigation of compatibilized PLA/PBAT (80/20)(wt/wt) blends with various amounts of chain extension/branching agent exhibited higher complex viscosity and higherstorage modulus. This increase becomes more pronounced asthe concentration of reactive epoxy functions increases.Compared to unmodified samples, the presence of reactiveepoxy functions showed an increase in the tensile modulus(from 820 MPa (PLA/PBAT blends) to 1095 MPa (PLA/PBAT/GMA)) and the elongation at break (from 50% (PLA/PBATblends) to 135% (PLA/PBAT/GMA)) which contradicts theresults presented by Kumar et al. where a low elongation atbreak value (3%) was obtained. However, in our study, theimproved mechanical properties indicate a reactivity control atthe interface due to the formation of ester linkages betweenPLA, PBAT and Joncryl. Meanwhile, the effect of reactive com-patibilization on the PLA/PBAT blends has been confirmed bytransmission electron microscopy (TEM) observations by theimprovement of phase dispersion and the increase of modulusand strain at break.
The work is in progress to (i) study the topology of modified PLAand PBAT and (ii) to quantify the role of Joncryl on the interfacialtension of modified and unmodified PLA/PBAT blends.
Acknowledgments
Many thanks to DGCIS, Ministry of Industry, for its financialsupport. The authors also express their appreciation to thereviewers for their meticulous review and assessment of this work.

R. Al-Itry et al. / Polymer Degradation and Stability 97 (2012) 1898e19141914
References
[1] Gupta A, Kumar V. Eur Polym J 2007;43(10):4053e74.[2] Garlotta D. J Polym Environ 2001;9(2):63e84.[3] Alexandre M, Dubois P. Mater Sci Eng 2000;28(1e2):1e63.[4] Anderson KS, Lim SH, Hillmyer MA. J Appl Polym Sci 2003;89(14):3757e68.[5] Martino VP, Jimenez A, Ruseckaite RA. J Appl Polym Sci 2009;112(4):2010e8.[6] Ogata N, Jimenez G, Kawai H, Ogihara T. J Polym Sci Part B 1997;35(2):389e96.[7] Lehermeier HJ, Dorgan JR. Polym Eng Sci 2001;41(12):2172e84.[8] Gupta MC, Deshmukh VG. Colloid Polym Sci 1982;260(5):514e7.[9] Kopinke FD, Remmler M, Mackenzie K, Moder M, Wachsen O. Polym Degrad
Stab 1996;53(3):329e42.[10] Taubner V, Shishoo R. J Appl Polym Sci 2001;79(12):2128e35.[11] Hyon SH, Jamshidi K, Ikada Y. Polym Int 1998;46(3):196e202.[12] Wachsen O, Platkowski K, Reichert KH. Polym Degrad Stab 1997;57(1):87e94.[13] Sodergard A, Nasman JH. Polym Degrad Stab 1994;46(1):25e30.[14] Signori F, Coltelli MB, Bronco S. Polym Degrad Stab 2009;94(1):74e82.[15] Mantia FL. Handbook of plastic recycling. Rapra Technology Limited; 2002.[16] Di Y, Iannace S, Di Maio E, Nicolais L. Macromol Mat Eng 2005;290(11):
1083e90.[17] Kylma J, Seppala JV. Macromol 1997;30(10):2876e82.[18] Villalobos M, Awojulu A, Greeley T, Turco G, Deeter G. Energy 2006;31(15):
3227e34.[19] Li BH, Yang MC. Polym Adv Technol 2006;17(6):439e43.[20] Awaja F, Daver F, Kosior E, Cser F. J Therm Anal Calorim 2004;78(3):865e84.[21] Yuanliang W, Chunhua F, Yongxiang L, Changshun R, Yaoyao Z, Ya F. J Wuhan
Univ Technol Mater Sci Ed 2010;25(5):774e9.[22] Pilla S, Kim Seong G, Auer George K, Gong S, Park Chul B. Polym Eng Sci 2009;
49(8):1653e60.[23] Mihai M, Huneault MA, Favis BD. Polym Eng Sci 2010;50(3):629e42.[24] Kumar M, Mohanty S, Nayak SK, Rahail Parvaiz M. Bioresour Technol 2010;
101(21):8406e15.[25] Biresaw G, Carriere CJ. J Polym Sci Part B 2002;40(19):2248e58.[26] Huneault MA, Li H. Polymer 2007;48(1):270e80.[27] Gu SY, Zhang K, Ren J, Zhan H. Carbohyd Polym 2008;74(1):79e85.[28] Ko SW, Hong MK, Park BJ, Gupta RK, Choi HJ, Bhattacharya SN. Polym Bull
2009;63(1):125e34.[29] Maazouz A, Lamnawar K, Mallet B. Front Sci Eng (Int J) 2011:1e44.
[30] Lamnawar K, Maazouz A, Mallet B. Patent; 2010. International patent C08J5/10, C08L67/00, FR2941702.
[31] Frenz V, Scherzer D, Villalobos M, Awojulu AA, Edison M, van der Meer R.BASF publication.
[32] Corre YM, Duchet J, Maazouz A, Reignier J. J Supercrit Fluids 2011;58(1):177e88.
[33] Huggins ML. J Am Chem Soc 1942;64(11):2716e8.[34] Kraemer EO. Ind Eng Chem Res 1938;30(10):1200e3.[35] Cho J, Heuzey MC, Begin A, Carreau PJ. J Food Eng 2006;74(4):500e15.[36] Corre YM, Duchet J, Maazouz A, Reignier J. Rheol Acta 2011;50(7e8):613e29.[37] Coltelli MB, Toncelli C, Ciardelli F, Bronco S. Polym Degrad Stab 2011;96(5):
982e90.[38] Hwang S-S, Hsu PP, Yeh J-M, Chang K-C, Lai Y-Z. Polym Comp 2009;30(11):
1625e30.[39] Ray SS, yamada K, Okamoto M, Ogami A, Ueda K. Macromol 2002;35(8):
3104e10.[40] Amar K, Mohanty M, Lawrence TD, editors. (vol. 1-Chapter 16). Michigan:
Taylor & Francis; 2005.[41] Kopinke FD, Mackenzie K. J Anal Appl Pyrolysis 1997;40e41:43e53.[42] Fan YJ, Nishida H, Tokiwa Y, Endo T. Polym Degrad Stab 2004;86(2):197e208.[43] Lucas N, Bienaime C, Belloy C, Queneudec M, Sylvestre F, Nava-Saucedo JE.
Chemosphere 2008;73(4):429e42.[44] Jong SJD, Arias ER, Rijkers DT, Nostrum CF, Hennink WE. Polymer 2001;42(7):
2795e802.[45] Witzke DR. Doctor of philosophy. Michigan: Michigan State University; 1997.[46] Ramkumar DHS, Bhattacharya M. Polym Eng Sci 1998;38(9):1426e35.[47] Harnnecker F, Derval dos Santos R, Lenz DM. J Polym Environ 2012;20(1):
237e44.[48] Nor Azowa I, Nazri MR, Wan Zin Wan Y, Jamaliah S. J Polym Res 2011;18(5):
891e6.[49] Kanzawa T, Tokumitsu K. J Appl Polym Sci 2011;121(5):2908e18.[50] Kijchavengkul T, Auras R, Rubino M. Polym Test 2008;27(1):55e60.[51] Carrasco F, Pagès P, Gámez-Pérez J, Santana OO, Maspoch ML. Polym Degrad
Stab 2010;95(2):116e25.[52] Japon S, Luciani A, Nguyen QT, Leterrier Y, Manson J-A. Polym Eng Sci 2001;
41(8):1299e309.[53] Lamnawar K, Maazouz A. Rheol Acta 2008;47(4):383e97.[54] Lamnawar K, Baudouin A, Maazouz A. Eur Polym J 2010;46(7):1604e22.[55] Jianye L, Lijuan L, Wei Y, Ruogu L, Runming L, Chixing Z. Polymer 2010;
51(22):5186e97.[56] Munari A, Pezzin G, Pilati F, Manaresi P. Rheol Acta 1989;28(1):25e9.[57] Wood-Adams P, Costeux S. Macromolecules 2001;34(18):6281e90.[58] Gu Shu- Y, Cun-Yang Z, Zhou K, Ren J. J Appl Polym Sci 2009;114(3):1648e55.[59] Yuan H, Liu Z, Ren J. Polym Eng Sci 2009;49(5):1004e12.[60] Munari A, Pezzin G, Pilati F. Rheol Acta 1985;24(5):534e6.[61] Zhang N, Wang. Q, Ren J, Wang L. J Mater Sci 2009;44(1):250e6.[62] Lopez AA, Sarasua JR, Verdu J, Colin X. Intern Polym Process 2007;XXI(5):
389e94.[63] Li. K, Peng J, Turng LS, Huang HX. Adv Polym Tech 2011;30(2):150e7.[64] Holland BJ, Hay JN. Polymer 2002;43(6):1835e47.


















