
Immunodeficiencies
28
Immunodeficiencies Martin Liška
-
Upload
colette-hammond -
Category
Documents
-
view
23 -
download
1
description
Immunodeficiencies. Martin Liška. Basic immunological terms. Endocrine system. Immune system provides 3 basic functions: Defense against infection Homeostasis – elimination of old or impaired cells Immunological surveillance - elimination of mutated cells. Immune system. Nervous system. - PowerPoint PPT Presentation
Transcript of Immunodeficiencies

Immunodeficiencies
Martin Liška
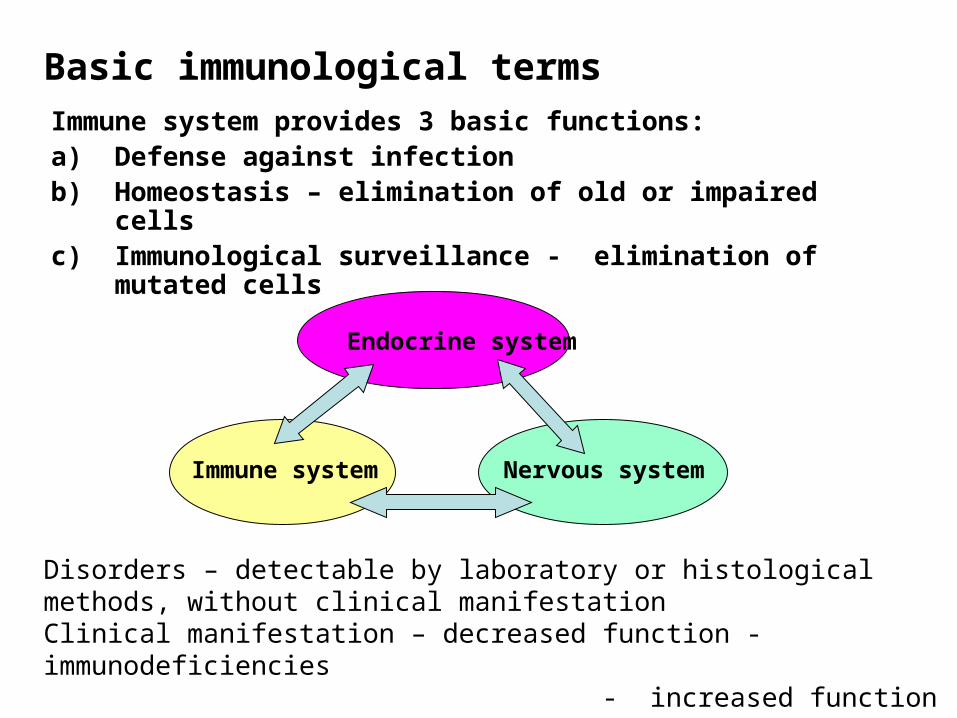
Basic immunological termsImmune system provides 3 basic functions:a) Defense against infectionb) Homeostasis – elimination of old or impaired cellsc) Immunological surveillance - elimination of mutated cells
Endocrine system
Immune system Nervous system
Disorders – detectable by laboratory or histological methods, without clinical manifestationClinical manifestation – decreased function - immunodeficiencies - increased function – allergy, autoimmunity

Immunodeficiencies
• Humoral – innate immunity - complement, MBL acquired immunity – immunoglobulins (B lymphocytes)
• Cell mediated immunity – innate immunity – phagocytes - acquired immunity – T lymphocytes
• Primary – congenital, genetically defined, symptoms predominantly at an early age
• Secondary – the onset of symptoms at any age chronic diseases the effects of irradiation immunosuppression surgery, injuries stress

Immunodeficiencies – critical life periods in respect to symptoms onset
• Newborn age - severe primary disorders of cell mediated immunity
• 6 mth. – 2 yrs. – severe humoral immunodeficiencies cong./transient
• 3 - 5 yrs. – transient and selective humoral immunodeficiencies, secondary immunodeficiencies
• 15 – 20 yrs. – hormonal instability, thymus involution, life-style changes, some typical infections first symptoms of CVID
• Middleage – often excessive workload, stress first symptoms of autoimmune disorders (also immunodeficiency)
• Advanced and old age – rather symptoms of severe secondary immunodeficiencies,
repercussion of functional disorders

Immunodeficiencies – major clinical features
• Antibodies - microbial infections (encapsulated bacterias)
respiratory - pneumonia, sinusitis, otitis
GIT – diarrhea• Complement – microbial infections (pyogenic), sepsis
various systems affection
edema (HAE) – C1-INH deficiency• T lymphocytes - bacterial, fungal, viral
GIT – diarrhoea
respiratory – pneumonia, sinusitis• Phagocytes - abscesses, recurrent purulent skin infections
granulomatous inflammations

The differentiation of pluripotent stem-cell

I. Primary immunodeficiencies – phagocytic cell defects
1/ Quantitative – decreased numbers of granulocytes –neutrophil elastase mutation
Congenital chronic agranulocytosis
Cyclic agranulocytosis (neutropenia)• Systemic manifestation, fevers in 3-weeks cycles in
cyclic form• Treatment with granulocyte growth factors, ATB

I. Primary immunodeficiencies – phagocytic cell defects
2/ Qualitative – phagocytes functional disorders, various enzyme deficits, inability of phagocytes to degrade the ingested material
Chronic Granulomatous Disease (CGD)• Approximately in 60% X-linked • Enzymatic inability to generate toxic oxygen metabolites (H2O2)
during oxygen consumption) - result of defect in neutrophilic cytochrome b (part of complex containing NADPH oxidase)
• Inability to kill bacteria such as Staph.aureus, Pseud.aeruginosa that produce the enzyme catalase
• Clinical features: granulomas in many organs• Treatment: long-term ATB administration
• Myeloperoxidase deficiency• Recurrent microbial infections, susceptibility to Candida albicans and
Staph.aureus infections

I. Primary immunodeficiencies – phagocytic cell defects
Chédiak-Higashi syndrome• Clinical features: recurrent, severe, pyogenic infections
(streptococcal, staphylococcal)• Defective intracellular killing of bacteria (neutrophils
contain abnormal giant lysosomesHyperIgE (Job’s) syndrome• Mutation of STAT3 gene• Recurrent “cold” staphylococcal abscesses, chronic
eczema, otitis media• Extremely high serum IgE levels• Treatment: ATBLAD syndrome – Leukocyte Adhesion Deficiency
(neutrophil adhesion molecules deficiency)• LAD I – integrins expression deficiency• LAD II – selectins expression deficiency

II. Primary immunodeficiencies – B cell disorders
Bruton’s X-linked hypogamaglobulinemia• Blockage in the maturation of pre-B lymphocytes into B lymphocytes
(tyrosine kinase defect)• Frequency 1: 50-100 thousands• Undetectable or very low serum levels of Ig• Clinical symptoms in 5-9 mth.of age• Pneumonia, pyogenic otitis, complicated sinusitis, increased occurrence
of pulmonary fibrosis• Treatment: life-long IVIG substitution
CVID – Common Variable ImmunoDeficiency• B cell functional disorder, mostly low levels of IgG and IgA• Frequency 1:100-200 thousands. Symptoms’ onset between 2nd and 3rd
decade• Recurrent respiratory tract infections (pneumonia)• Treatment: IVIG substitution

II. Primary immunodeficiencies – B cell disorders
Selective IgA deficiency• Disorder of B cell function • Frequency 1: 500-700, manifestation mostly at pre-school age• Recurrent mild/moderate infections (respiratory, GIT, urinary tract)
or asymptomatic• Risk of reaction to live attenuated vaccines or generation of anti-IgA
antibodies after a blood transfusion• Laboratory criterion: IgA < 0,07g/l (age > 4 years)
Selective IgG subclasses or specific IgG deficiency • B cell function disorder• Frequency ?• Onset of symptoms in childhood, mostly respiratory tract infections
caused by encapsulated bacteria (H.influenzae, Pneumococci) Transient hypogammaglobulinemia of infancy

III. Primary immunodeficiencies – T cell disorders
diGeorge syndrome• Disorder of prethymocytes maturation due to absence of thymus• Disorder of development of 3rd and 4th branchial pouch – absence of
parathyroid glands • Congenital heart diseases• Frequency 1 : 100 thousands• The onset of symptoms after the birth – hypocalcemic spasms and
manifestations of cong.heart disease• Immunodeficiency could be only mild, the numbers of T lymphocytes
later usually become normal• Treatment symptomatic

III. Primary immunodeficiencies – T cell disorders
Lymphoproliferative syndrome• Malignant proliferation of activated T lymphocytes• Hypogamaglobulinaemia, lymphoma• Its development is induced by EBV infectionChronic mucocutaneous candidiasis• Impaired ability of T cells to produce macrophage migration inhibiting
factor (MIF) in response to Candida antigenBare lymphocyte sy. • Disorder of antigen presentation – defect of MHC I and/or MHC II
expression leads to the dysfunction of CD4 and CD8 lymphocytesIFN-Receptor deficiency • Th1 lymphocytes differentiation disorder inability of intracellular killing of Mycobacteria (fatal BCG itis)

DiGeorge sy
MHC I, MHC II def. (bare lymphocyte sy), CD3 def.

IV. Primary immunodeficiencies – combined defects of T and B cells
• SCID – Severe Combined ImmunoDeficiency• X-linked recessive or AR disease, combined disorder of humoral and cell mediated
immunity• Severe disorder (patients often die during first 2 years of life), symptoms’ onset soon
after the birth (severe diarrhoea, pneumonia, meningitis, BCGitis)• Immunological features: typically lymphopenia and thymus hypoplasia• Forms: AR form – often enzymatic deficiency (ADA, PNP) that leads to accumulation of metabolites toxic to DNA synthesis (lymphocytes) X-linked form – disorder of stem-cell Treatment: ATB, IVIG BMT is of critical significance in ADA deficiency the gene therapy was tested successfully
• Reticular dysgenesis• Stem-cell differentiation is blocked• Very rare• Symptoms immediately after the birth – severe diarrhoea, infections of deep layers• Fatal, BMT is the only treatment

SCID
Reticular dysgenessis

IV. Primary immunodeficiencies – combined defects of T and B cells
Ataxia teleangiectasia • Increased radiation induced chromosomal breakage, ataxia,
dilatation of small blood vessels (teleangiectasia)• Neurological disorders, immunodeficiency is not necessary (if
present, IgA deficiency or T lymphocytes function disorders are most common)
• Treatment: ATB, IVIG
Hyper IgM syndrome • CD40L expression disorder, poor cooperation of B and T cells,
impaired isotype switching, increased IgM levels
Wiskott-Aldrich syndrome • thrombocytopenia, eczema, recurrent infections (encapsulated
microbes), decreased IgM levels
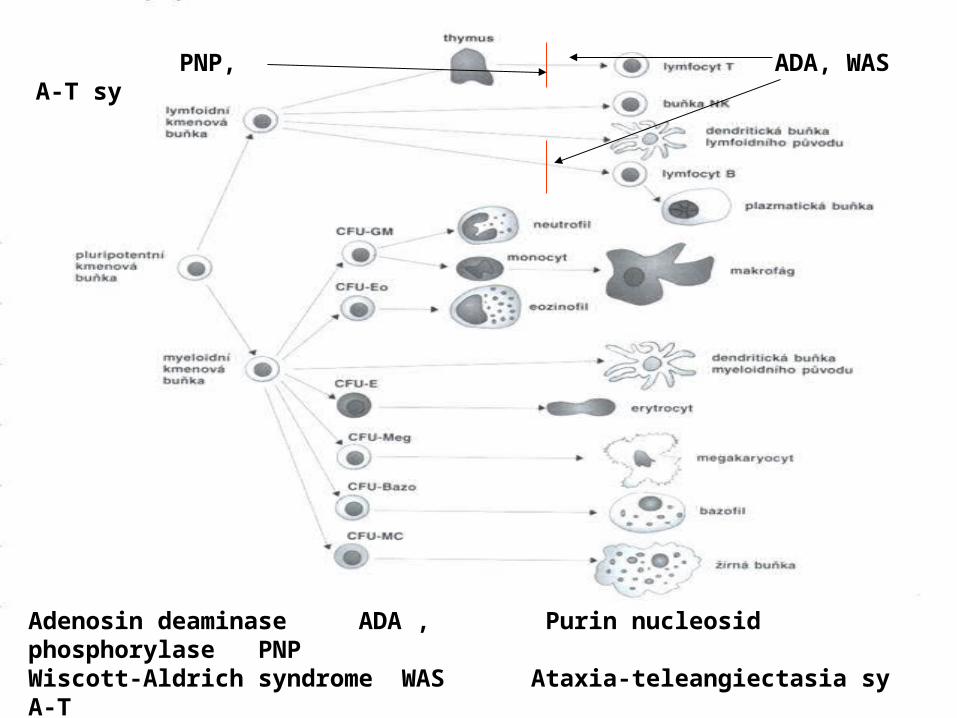
ADA, WAS PNP, A-T sy
Adenosin deaminase ADA , Purin nucleosid phosphorylase PNPWiscott-Aldrich syndrome WAS Ataxia-teleangiectasia sy A-T

V. Primary immunodeficiencies – complement system disorders
Deficiency of:• C1, C2, C3, C4 – impaired opsonization, susceptibility to
infections, autoimunity, SLE–like syndrome• C6, C7, C8, C9 – SLE–like syndrome, increased
susceptibility to neisserial infections• MBL deficiency – mannan binding lectin (lectin way of
complement activation), various infections, susceptibility to autoimmunity, association with allergy.

V. Primary immunodeficiencies – complement system disorders
Hereditary angioedema (HAE)
• Absence or functional deficiency of C1-inhibitor• Anaphylactoid reactions with skin and/or mucosal (oral, laryngeal,
gut) edemas caused by overproduced bradykinin• Injuries or surgical/stomatological operations are mostly the
triggering factor • Laryngeal edemas could be life-threatening, immediate treatment is
necessary !• Treatment: preventive – androgens, EACA immediate – C1-INH concentrate or fresh frozen plasma administration, icatibant• Secondary forms also exist !

Secondary immunodeficiencies
• Acute and chronic viral infections – infectious mononucleosis, influenza
• Metabolic disorders – diabetes mellitus, uremia• Autoimmune diseases – autoantibodies against immunocompetent
cells (neutrophils, lymphocytes); autoimmune phenomena also after administration of certain drugs (e.g. oxacilin, quinidine)
• Allergic diseases• Chronic GIT diseases• Malignant diseases (leukemia)• Hypersplenism/asplenia• Burn, postoperative status, injuries• Severe nutritional disorders• Chronic infections• Ionizing radiation• Drug induced immunodeficiencies (chemotherapy)• Immunosupression• Chronic stress• Chronic exposure to harmful chemical substances

Secondary immunodeficiencies
• Splenectomy – deficiency in generation of antibodies against encapsulated microorganisms (Pneumococci, Neisseria)
• A loss of immunoglobulins – nephrotic syndrome - lymphangiectasies• Lymphomas, myelomas, CLL
• Chronic fatigue syndrome• First, it is necessary to exclude all chronic diseases which can lead
to fatigue: • autoimunity • malignancy• focal infection• neurological disorders• metabolic disorders• depression

Secondary immunodeficiencies - A.I.D.S.
• Caused by retrovirus HIV 1 or HIV 2• Current incidence 40 mil.people, predominantly in central Africa,
5mil. of new infections per year, 3 mil. deaths per year • CZ:10/06 - HIV+ 904/248, AIDS 204/14, deaths 23/2 • Virus has a tropism for cells bearing CD4 surface marker (Th CD4+
lymphocytes); also affects macrophages and CNS cells• Viral genome transcribes into human DNA and infected cell provides
viral replication• Transmission: sexual intercourse contact with blood endouterine (mother – fetus, breast milk)• Phases: acute (flu-like sy) asymptomatic – several years, viral replication symptomatic – infections, autoimmunity, malignancy,
allergy final – systemic breakdown, opportune infections

A.I.D.S. - Treatment
• Reverse transcriptase inhibitors (zidovudine, dideoxyinosine, dideoxycytosine, 3-thiacytosine)
• Protease inhibitors - block the viral protease enzyme• Combined drug therapy• Antimicrobial agents

Substitution therapy with immunoglobulins
• Prepared by fractionation of pooled human plasma from huge amount of donors (1000)
• Examination of donors, inactivating procedures to minimize the risk of infection transmission
• Mainly IgG, minimal content of IgA• i.v. (Octagam, Gammagard) or s.c. use (Subcuvia, Gammanorm)

Indications
• Primary humoral immunodeficiencies
• IgG subclasses deficiencies, deficiencies of generation of specific antibodies, secondary humoral immunodeficiencies, combined immunodeficiencies

Dosage
• agamaglobulinemia – IVIG 400-600 mg/kg/month• regular substitution in outpatient’s room every 3-4 weeks• „home therapy“ (s.c.administration by infusion pump)

Adverse effects
• Allergic reactions, or even anaphylaxis [in minor reactions (chill, headache) only slowing down of infusion is usually needed]













![Immunodeficiencies [Autosaved]](https://static.fdocuments.in/doc/165x107/577cde971a28ab9e78af6d75/immunodeficiencies-autosaved.jpg)




