
Imaging end points for monitoring neuroprotection in Parkinson's disease
10
Imaging End Points for Monitoring Neuroprotection in Parkinson’s Disease David J. Brooks, MD, DSc, FRCP, FMedSci In this review, the potential role of positron emission tomography and single-photon emission computed tomography as biological markers for following the progression of Parkinson’s disease (PD) is discussed, and their value for assessing the efficacy of putative neuroprotective agents in PD is considered. It is concluded that functional imaging provides a valuable adjunct to clinical assessment when judging the efficacy of neuroprotective approaches to PD. Ann Neurol 2003;53 (suppl 3):S110 –S119 The pathological hallmark of Parkinson’s disease (PD) is degeneration of pigmented and other brainstem nu- clei in association with the formation of neuronal Lewy inclusion bodies 1 Loss of cells from the substantia nigra in PD results in profound dopamine depletion in the striatum, lateral nigral projections to putamen being most affected. 2 Loss of cells from the median raphe, locus ceruleus, and nucleus basalis leads to lesser reduc- tions in striatal and cortical serotonin, noradrenaline, and acetylcholine. 3 The pathology of PD also involves association cortex, especially anterior cingulate and pa- rietotemporal areas. 4 Microglial activation has been documented as a response to the degenerative process. 5 Currently, treatments for PD are symptomatic and aimed at either restoring dopaminergic tone or block- ing excessive glutamatergic or cholinergic activity. The eventual hope for effective treatment of PD, however, is the development of neuroprotective and restorative therapies. Currently, drug licensing authorities require a clini- cal primary end point when assessing the efficacy of a putative neuroprotective agent. The ideal end point has yet to be defined though time to a particular event, such as requirement of dopaminergic medication when patients are treatment naive, has been widely used. This approach is not ideal because the end point is subjective and not all patients are prepared to delay onset of symptomatic medication. An alternative strat- egy is longitudinal assessment of patients with either subjective clinical rating scales and/or objective timed motor tasks. Because PD is a condition that manifests with bradykinesia, rigidity, tremor, postural instability, and autonomic dysfunction, these features can all be present with different degrees of severity and progress at different rates in individuals. 6 It is, therefore, essen- tial that when patients are randomized into treatment arms selection bias is avoided. In addition, clinical rat- ing scales tend to emphasize particular aspects of the condition. The mostly widely accepted is the Unified Parkinson’s Disease Rating Scale (UPDRS). 7 This is a sensitive scale that has undergone extensive validation and has been found to have acceptably low interob- server variability. 8 Its disadvantages are that it is sub- jective, somewhat duplicative, and unduly emphasizes bradykinesia. Objective timed motor tasks that have been pro- posed for the measurement of the severity of PD in- clude repetitive upper limb pronation-supination, arm movement between two targets, the Purdue peg-board test of finger dexterity, and timed standing-walking- sitting. 9 Currently, it is not clear which of these tasks is the most sensitive for assessing disease progression, and patients can develop later difficulties completing the pronation-supination and finger-dexterity tasks. 10 In a recent study of disease progression, we found that of these four tests only the standing-walking-sitting test was sensitive to disease progression over 18 months. 11 The biggest problem one faces when using clinical rating scales to assess the efficacy of a neuroprotective agent in PD, however, is confounding symptomatic ef- fects due to either the neuroprotective drug itself or dopaminergic agents added over time to maintain pa- tient function. This problem was highlighted by the DATATOP trial where the putative neuroprotective agent, L-deprenyl, had a mild but significant symptom- atic effect confounding interpretation of the find- ings. 12,13 A way around this problem may be to use a wash-out or wash-in design 14 ; however, optimal wash- From the MRC Clinical Sciences Centre and Division of Neuro- science, Faculty of Medicine, Imperial College, Hammersmith Hos- pital, London, United Kingdom. Published online Mar 24, 2003, in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/ana.10480. Address correspondence to Dr Brooks, Hartnett Professor of Neu- rology. MRC Cyclotron Building, Hammersmith Hospital, Du Cane Road, London W12 0NN, United Kingdom. E-mail: [email protected] S110 © 2003 Wiley-Liss, Inc.
-
Upload
david-j-brooks -
Category
Documents
-
view
213 -
download
1
Transcript of Imaging end points for monitoring neuroprotection in Parkinson's disease

Imaging End Points for MonitoringNeuroprotection in Parkinson’s Disease
David J. Brooks, MD, DSc, FRCP, FMedSci
In this review, the potential role of positron emission tomography and single-photon emission computed tomography asbiological markers for following the progression of Parkinson’s disease (PD) is discussed, and their value for assessing theefficacy of putative neuroprotective agents in PD is considered. It is concluded that functional imaging provides avaluable adjunct to clinical assessment when judging the efficacy of neuroprotective approaches to PD.
Ann Neurol 2003;53 (suppl 3):S110–S119
The pathological hallmark of Parkinson’s disease (PD)is degeneration of pigmented and other brainstem nu-clei in association with the formation of neuronal Lewyinclusion bodies1 Loss of cells from the substantia nigrain PD results in profound dopamine depletion in thestriatum, lateral nigral projections to putamen beingmost affected.2 Loss of cells from the median raphe,locus ceruleus, and nucleus basalis leads to lesser reduc-tions in striatal and cortical serotonin, noradrenaline,and acetylcholine.3 The pathology of PD also involvesassociation cortex, especially anterior cingulate and pa-rietotemporal areas.4 Microglial activation has beendocumented as a response to the degenerative process.5
Currently, treatments for PD are symptomatic andaimed at either restoring dopaminergic tone or block-ing excessive glutamatergic or cholinergic activity. Theeventual hope for effective treatment of PD, however,is the development of neuroprotective and restorativetherapies.
Currently, drug licensing authorities require a clini-cal primary end point when assessing the efficacy of aputative neuroprotective agent. The ideal end point hasyet to be defined though time to a particular event,such as requirement of dopaminergic medication whenpatients are treatment naive, has been widely used.This approach is not ideal because the end point issubjective and not all patients are prepared to delayonset of symptomatic medication. An alternative strat-egy is longitudinal assessment of patients with eithersubjective clinical rating scales and/or objective timedmotor tasks. Because PD is a condition that manifestswith bradykinesia, rigidity, tremor, postural instability,and autonomic dysfunction, these features can all bepresent with different degrees of severity and progress
at different rates in individuals.6 It is, therefore, essen-tial that when patients are randomized into treatmentarms selection bias is avoided. In addition, clinical rat-ing scales tend to emphasize particular aspects of thecondition. The mostly widely accepted is the UnifiedParkinson’s Disease Rating Scale (UPDRS).7 This is asensitive scale that has undergone extensive validationand has been found to have acceptably low interob-server variability.8 Its disadvantages are that it is sub-jective, somewhat duplicative, and unduly emphasizesbradykinesia.
Objective timed motor tasks that have been pro-posed for the measurement of the severity of PD in-clude repetitive upper limb pronation-supination, armmovement between two targets, the Purdue peg-boardtest of finger dexterity, and timed standing-walking-sitting.9 Currently, it is not clear which of these tasks isthe most sensitive for assessing disease progression, andpatients can develop later difficulties completing thepronation-supination and finger-dexterity tasks.10 In arecent study of disease progression, we found that ofthese four tests only the standing-walking-sitting testwas sensitive to disease progression over 18 months.11
The biggest problem one faces when using clinicalrating scales to assess the efficacy of a neuroprotectiveagent in PD, however, is confounding symptomatic ef-fects due to either the neuroprotective drug itself ordopaminergic agents added over time to maintain pa-tient function. This problem was highlighted by theDATATOP trial where the putative neuroprotectiveagent, L-deprenyl, had a mild but significant symptom-atic effect confounding interpretation of the find-ings.12,13 A way around this problem may be to use awash-out or wash-in design14; however, optimal wash-
From the MRC Clinical Sciences Centre and Division of Neuro-science, Faculty of Medicine, Imperial College, Hammersmith Hos-pital, London, United Kingdom.
Published online Mar 24, 2003, in Wiley InterScience(www.interscience.wiley.com). DOI: 10.1002/ana.10480.
Address correspondence to Dr Brooks, Hartnett Professor of Neu-rology. MRC Cyclotron Building, Hammersmith Hospital, DuCane Road, London W12 0NN, United Kingdom.E-mail: [email protected]
S110 © 2003 Wiley-Liss, Inc.

out or wash-in times of putative neuroprotective anddopaminergic drugs are still uncertain. There is also aninherent assumption that the neuroprotective effect ofthe trial drug remains constant throughout the diseasecourse, whereas it may be most effective in the armtreated earliest. In reality, complete wash-out of dopa-minergic agents is poorly tolerated by PD patients oncetheir condition has become established.
Finally, it is likely that symptoms do not begin inPD until nigral cell loss reaches a certain criticalthreshold, possibly 50%.2 If this is so, nigral pathologywill be significant when symptoms and signs are stillmild. In this situation, clinical measures of disease pro-gression are likely to underestimate the rate of furthercell loss and are more likely to provide a measure ofthe failure of compensatory mechanisms, such as in-creased dopamine turnover, to maintain locomotor sta-tus in the face of nigral cell loss. In support of thisviewpoint, a recent longitudinal clinical study on therate of progression of 237 L-dopa–treated PD patientswith a mean disease duration of 6.8 years at entry re-ported only a 1.5% annual increase in motor UPDRSscore.6
Functional Imaging ApproachesGiven the above difficulties, it has been proposed thatin vivo functional imaging might provide a biologicalmarker for measuring the severity and progression ofPD, therefore providing solutions to the problems con-founding clinical trials of neuroprotection. In theory,both positron emission tomography (PET) and single-photon emission computed tomography (SPECT) canprovide in vivo an objective measure of disease severityin PD as reflected by loss of neurotransmitter function.Given the known pathology of PD, these modalitiespotentially could follow loss of dopamine, serotonin,noradrenaline, and acetylcholine terminal function andmonitor the inflammatory microglial response to cellloss. In addition, reductions in resting cortical glucosemetabolism and blood flow activation signals could bedetected.
If such surrogate markers are to be used, the tracersselected need to provide adequate signal and have areproducible uptake. Reproducibility is likely to beaided if tracer time activity curve characteristics allowtransformation of all scans into standard stereotaxicspace so minimizing artefact caused by changes in headposition and variations in region placement betweenscans. The biomarker clearly should reflect a biologicalprocess that changes with progression of PD and wherethe clinical status of the patient withdrawn from med-ication is correlated to tracer signal. Uptake of the im-aging tracer used should not be affected by the putativeneuroprotective agent or by symptomatic treatmentsfor PD. Scans should be read in a blinded fashion andideally with an independent centralized assessment.
18F-Dopa Positron Emission Tomography18F-6-fluorodopa (18F-dopa) PET is a marker of pre-synaptic dopaminergic terminal function and reflectsdopa transport into the terminal, dopa decarboxylaseactivity, and dopamine storage capacity.16 A single hu-man study has reported that striatal 18F-dopa influxconstants (Ki) correlated with subsequent postmortemdopaminergic cell density in the substantia nigra. Thisreport involved a group of six patients, three with pro-gressive supranuclear palsy and single cases of PD,amyotrophic lateral sclerosis, and Alzheimer’s disease.17
The interval between the scan and death ranged from 5to 62 months. An 18F-dopa PET study in monkeysvariably unilaterally lesioned with intracarotid injec-tions of the nigral toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) demonstrated significantcorrelations between striatal Ki values and striatal do-pamine levels, tyrosine hydroxylase and dopa decarbox-ylase activity, and nigral cell counts.18 Yee and col-leagues also found a correlation between striatal Kivalues and striatal and nigral dopamine levels and dopadecarboxylase activity, although in vivo estimations ofthe latter were 10-fold lower than in vitro measure-ments probably reflecting an influence of transport re-strictions.19,20 There was poor correlation with nigralcell counts, but this may have been because their ani-mals were only mildly lesioned compared with thestudy by Pate.18 There was only a 10 to 35% nigralcell loss and no overt parkinsonism was evident. Thereis also evidence to support vesicular storage of 18F-dopamine in terminals.21,22 Given this, it appears rea-sonable to use 18F-dopa PET as an indirect in vivomeasure of striatal dopamine levels and dopa decarbox-ylase activity, although it may be less accurate in re-flecting nigral dopamine cell counts, particularly inearly PD.
Several cross-sectional PET studies in PD haveshown that putamen 18F-dopa uptake correlates cross-sectionally with UPDRS motor scores, in particular,with bradykinesia ratings.23,24 Our in-house currentmean test–retest variability of putamen 18F-dopa influxconstant (Ki) measurements in normal subjects is now2% with a state-of-the-art three-dimensional PET cam-era and the ability to accurately coalign three-dimensional image data sets.25 This increases to 5% forearly PD in which putamen 18F signals are halved.
The influence of dopaminergic medication on stria-tal 18F-dopa uptake is still being investigated. Acuteoral administration of clinical doses of L-dopa appearsto have no significant effect on measured striatal 18F-dopa Ki values.26 Restless leg syndrome patients whohave been chronically exposed to L-dopa or dopamineagonists show normal levels of striatal 18F-dopa up-take27 (N. Pavesi, D. J. Brooks, unpublished observa-tions). These findings all suggest that 18F-dopa PET isan acceptable biomarker of disease progression in PD.
Brooks: Monitoring Neuroprotection in PD S111

Vingerhoets and colleagues28 were the first to dem-onstrate that the decline of whole striatal 18F-dopa up-take in PD is more rapid than in age-matched normalcontrols. Subsequently, Morrish and colleagues re-ported an average 12% annual decline in baseline pu-tamen 18F-dopa influx constant (Ki) for a group of 17PD patients with a mean clinical disease duration of 40months.11 An age-matched group of controls showedno significant change in putamen 18F-dopa uptake.This study subsequently was extended to include a co-hort of 32 PD patients with early disease and reportedan overall 9% annual decline of the baseline specificputamen 18F-dopa uptake over 18 months.29 Themean total UPDRS score increased from 29 to 37,and, on average, 18F-dopa uptake in the putamen de-creased by 7.4% of the normal mean value for eachincrease of 10 points on the total UPDRS scale. As-suming a linear relationship between decline in puta-men 18F-dopa uptake and disease duration and no ef-fect of treatment on progression, a preclinical windowfor PD of 6 � 3 years was estimated, clinical symp-toms arising after a 30% loss of terminal dopaminergicfunction.29
The above findings29 are in good agreement with amore recently reported longitudinal 18F-dopa PETstudy involving seven de novo PD patients later treatedwith L-dopa.30 Over a 5-year interval, these patientsshowed a mean annual 10% loss of anterior and 12%loss of posterior putamen 18F-dopa uptake. Morrish’sfindings are also in line with a cross-sectional study byFearnley and Lees,29 which correlated postmortem cellcount data with estimated clinical disease duration be-fore death. These workers found an exponential rela-tionship between nigral cell counts and premortemclinical disease duration and estimated that in PD thereis a mean preclinical disease period of 4.6 years. Theycalculated that nigral cell loss in PD occurs at approx-imately 10 times the rate of loss associated with agingand by onset of symptoms there is a 48% nigral cellloss, the disease itself being responsible for a 30% neu-ronal loss.
The dissociation between the 30% terminal functionloss at onset of symptoms in PD estimated by longitu-dinal 18F-dopa PET studies and the 50% nigral cellloss extrapolated by Fearnley and Lees2 from cross-sectional postmortem cell counts probably reflects that18F-dopa PET is primarily reflecting terminal dopa de-carboxylase activity that may well become relatively up-regulated in remaining terminals to increase dopamineturnover and so compensate for cell loss in early dis-ease. In support of this suggestion, it is generally agreedthat there is a loss of nigral cells with aging, yet mostcross-sectional 18F-dopa PET studies of normal volun-teers have not shown any consistent aging effect.31,32 Itfollows then, as concluded from animal data, that 18F-dopa PET does not provide a strict measure of cell
count but rather reflects an aspect of dopaminergic ter-minal function. 18F-dopa PET progression studies, likeclinical studies of progression, are as much examiningthe failure of compensatory mechanisms as nigral cellloss. However, prevention of deterioration of symp-toms via maintenance of compensating pharmacologymay be as important a mechanism of neuroprotectionas prevention of further cell loss.
123I-�-CIT Single-Photon Emission ComputedTomographyDopamine terminal function also can be assessed invivo with 123I-�-CIT (2 beta-carboxy-methoxy-3 beta-4-iodophenyl-tropane) SPECT, a marker of dopaminetransporter (DAT) binding.33 In cross-sectional studies,striatal 123I-�-CIT uptake correlates with locomotordisability off medication in PD.34,35 Mean test–retestreproducibility for striatal specific 123I-�-CIT binding(V3��) has been reported to be 13% in normal volun-teers and 17% in PD, albeit with earlier generationSPECT cameras.36 Marek and colleagues37 used 123I-�-CIT SPECT to investigate reductions in striatalDAT binding in patients with early PD. Striatal uptakeof 123I-�-CIT was reduced by 53% contralateral and38% ipsilateral to the clinically symptomatic side ineight hemi-PD patients compared with mean striataluptake in age-matched healthy subjects. As with 18F-dopa uptake, the reduction in 123I-�-CIT uptake inPD was greater in the putamen than in the caudate.Innis and colleagues have demonstrated that 6 weeks ofexposure to clinical doses of either L-dopa or selegilinedoes not appear to affect striatal 123I-�-CIT uptake.38
Marek and colleagues39 examined the change in stri-atal 123I-�-CIT uptake in sequential SPECT scans toassess the rate of progression of DAT loss in 32 pa-tients with early PD over a 4-year period. SequentialSPECT scans showed a mean annual decline in striatal123I-�-CIT uptake of 11% compared with 0.8% inage-matched healthy controls (p � 0.001). Cross-sectionally, striatal 123I-�-CIT uptake in PD correlatedwith clinical severity in patients withdrawn from med-ication and rated with the UPDRS. However, the an-nual percentage loss of 123I-�-CIT striatal uptake didnot correlate with the annual loss in measures of clin-ical function. Assuming a linear relationship betweenloss of striatal 123I-�-CIT uptake and disease durationand no effect of treatment on disease progression, theseresearchers have extrapolated that PD has a mean5-year preclinical window.40 Such 123I-�-CIT SPECTfindings are in good agreement with 18F-dopa PETdata despite the different pharmacological processes be-ing monitored.29 In contrast with dopa decarboxylaseactivity, which is probably relatively upregulated in theface of degeneration of dopamine neurons, DAT den-sity has been reported to be downregulated in the re-serpinized rat, presumably to maintain synaptic dopa-
S112 Annals of Neurology Vol 53 (suppl 3) 2003

mine levels. 123I-�-CIT SPECT therefore in fact mayoverestimate nigral cell loss.
More recently, 123I-FP-CIT SPECT also has beendeveloped as a DAT marker. It differentiates clinicallyprobable essential tremor from PD41 and its striataluptake correlates inversely with severity of PD.42 123I-FP-CIT has the advantage that its specific uptake canbe measured 3 hours after administration, whereas onehas to wait 24 hours to assess specific 123I-�-CIT bind-ing. It is also licensed for use in Europe.
11C-Dihydrotetrabenazine Positron EmissionTomography11C-dihydrotetrabenazine (11C-DTBZ) PET is amarker of vesicular monoamine transporter (VMAT2)function in dopamine terminals. In rat studies, striatal3H-methoxy-TBZ and 3H-TBZ uptake do not appearto be altered by in vivo exposure to L-dopa or bro-mocriptine.43 Using 11C-DTBZ PET, Frey and col-leagues reported a 0.8% annual loss of specificVMAT2 binding in normal subjects. PD patientsshowed a 61% reduction in specific DTBZ binding inthe putamen and 43% in the caudate nucleus.44 Theauthors concluded that 11C-DTBZ PET was a suitablemarker for objective quantification of nigrostriatal in-tegrity, including evaluation of PD progression and itspossible therapeutic modification.
Using PET, Lee and colleagues45 have compared stri-atal 18F-dopa, 11C-DTBZ, and 11C-methylphenidateuptake reflecting dopa decarboxylase activity, VMAT2transporter binding, and plasma membrane DA trans-porter density, respectively. Three consecutive PET scanswere performed in three-dimensional mode with 35 PDpatients and 16 age-matched, normal controls. 18F-dopainflux constants (Ki) were relatively preserved comparedwith binding potentials (Bmax/Kd) for 11C-DTBZ in theparkinsonian striatum, whereas 11C-methyl-phenidatebinding was reduced more than that of 11C-DTBZ. Theauthors concluded that, compared with striatal VAMT2binding, dopa decarboxylase activity is relatively upregu-lated, whereas the plasma membrane DA transporter isrelatively downregulated in patients with PD.
That the quantity being measured does not directlyreflect dopamine cell body densities per se does not, ofcourse, mean that 18F-dopa PET and 123I-�-CITSPECT cannot be used to study the effects of neuro-protection. As discussed before, both these approachesprovide an objective marker of dopamine terminalfunction which correlates with disease severity. Theytherefore can be used to objectively measure diseaseprogression, although changes in tracer uptake in partmay be reflecting a change in the compensatory reac-tion to the disease process rather than overt cell loss.
Parkinson’s Disease Progression and DopamineAgonistsAlthough L-dopa remains the most effective symptom-atic treatment of PD, in vitro studies have suggestedthat it might be neurotoxic to remaining dopaminergicneurons.46 Dopamine agonists exert their antiparkinso-nian effect by directly stimulating dopamine receptors.Their early use as a substitute for L-dopa avoids theproduction of hydrogen peroxide and hydroxyl freeradicals from oxygenation of exogenous dopamine toDOPAC by monoamine oxidase B. In addition, theyact on presynaptic dopamine D2 autoreceptors, therebysuppressing endogenous dopamine synthesis, release,and turnover. They also increase levels of natural anti-oxidants such as glutathione and superoxide dis-mutase.47 It has, therefore, been speculated that dopa-mine agonists may exert a disease-modifying effect inPD by reducing oxidative stress.48
Using 18F-dopa PET, Whone and colleagues49 havestudied in vivo the relative rates of progression of denovo PD in a group of 186 patients randomized 1 to 1in a double-blind fashion to either the nonergot dopa-mine D2 agonist, ropinirole, or L-dopa therapy. PDpatients had serial 18F-dopa PET scans performed atone of six different centers (three North American andthree European) at baseline and again at 2 years. Theprimary end point was percentage reduction in puta-men 18F-dopa influx constant (Ki) in patients with aclinical diagnosis of PD supported by baseline 18F-dopa PET. Seventy-five percent of patients completedthe study in each of the two treatment arms.
In this trial, the PET scan data were centrally trans-formed into standard stereotactic space to normalizebrain position and shape. Scans then were analyzedwith a standardized template of regions of interest(ROIs) defined on a normal F-dopa template and withstatistical parametric mapping to localize and comparepeak voxel changes between groups without any as-sumption as to their location. Seventy-three percent ofthe ropinirole and 74% of the L-dopa patients com-pleted the 2-year study. The mean daily doses after 2years were 12.2 (6.1, SD) mg ropinirole and 558.7(180.8, SD) mg L-dopa. Fourteen percent of ropiniroleand 5% of L-dopa patients required open rescue withopen L-dopa. Improvement in Unified Parkinson’s Dis-ease Rating Scale motor score was six points greatertaking L-dopa than ropinirole after 2 years, but ropini-role was associated with a significantly lower incidenceof dyskinesias (3.3%) than L-dopa (25.3%; p � 0.0002).
Figure 1 shows changes in 18F-dopa uptake in onepatient over a 2-year period. Blinded review of baselinePET identified 11% of subjects as having normal cau-date and putamen 18F-dopa uptake; therefore, thispopulation was considered separately. Central ROIanalysis of putamen 18F-dopa uptake in PD cases withappropriate baseline PET findings showed significantly
Brooks: Monitoring Neuroprotection in PD S113
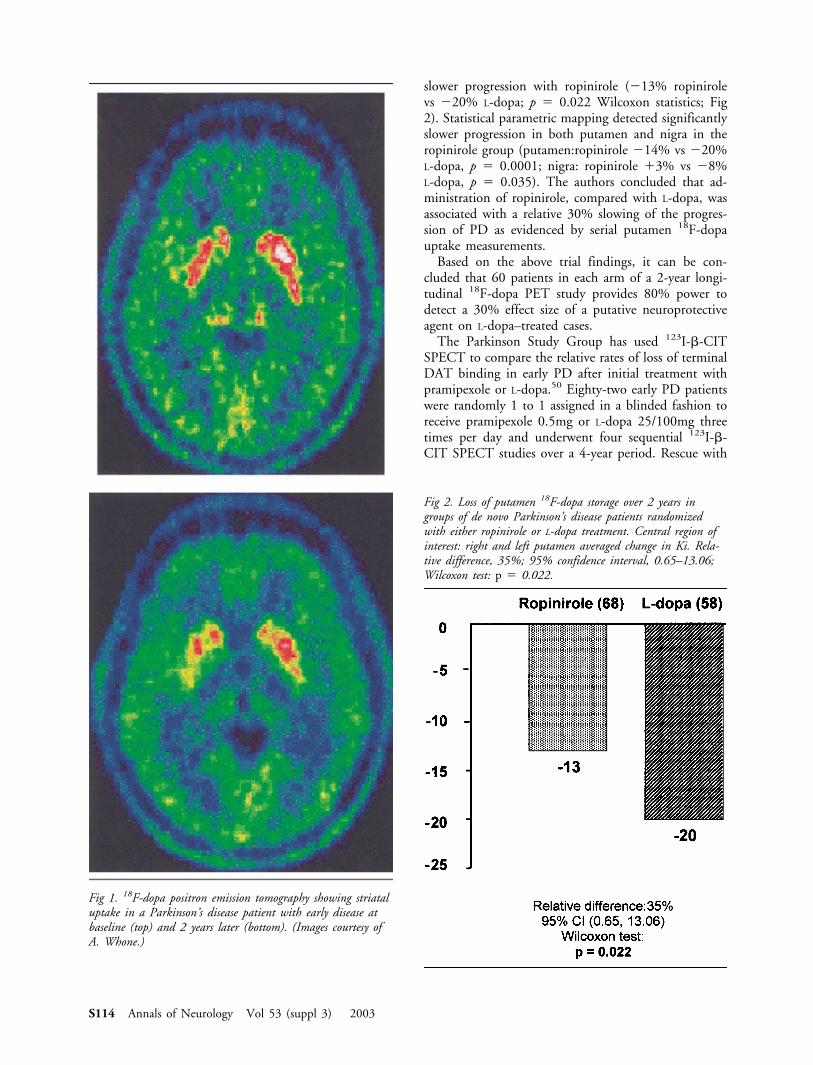
slower progression with ropinirole (�13% ropinirolevs �20% L-dopa; p � 0.022 Wilcoxon statistics; Fig2). Statistical parametric mapping detected significantlyslower progression in both putamen and nigra in theropinirole group (putamen:ropinirole �14% vs �20%L-dopa, p � 0.0001; nigra: ropinirole �3% vs �8%L-dopa, p � 0.035). The authors concluded that ad-ministration of ropinirole, compared with L-dopa, wasassociated with a relative 30% slowing of the progres-sion of PD as evidenced by serial putamen 18F-dopauptake measurements.
Based on the above trial findings, it can be con-cluded that 60 patients in each arm of a 2-year longi-tudinal 18F-dopa PET study provides 80% power todetect a 30% effect size of a putative neuroprotectiveagent on L-dopa–treated cases.
The Parkinson Study Group has used 123I-�-CITSPECT to compare the relative rates of loss of terminalDAT binding in early PD after initial treatment withpramipexole or L-dopa.50 Eighty-two early PD patientswere randomly 1 to 1 assigned in a blinded fashion toreceive pramipexole 0.5mg or L-dopa 25/100mg threetimes per day and underwent four sequential 123I-�-CIT SPECT studies over a 4-year period. Rescue with
Fig 1. 18F-dopa positron emission tomography showing striataluptake in a Parkinson’s disease patient with early disease atbaseline (top) and 2 years later (bottom). (Images courtesy ofA. Whone.)
Fig 2. Loss of putamen 18F-dopa storage over 2 years ingroups of de novo Parkinson’s disease patients randomizedwith either ropinirole or L-dopa treatment. Central region ofinterest: right and left putamen averaged change in Ki. Rela-tive difference, 35%; 95% confidence interval, 0.65–13.06;Wilcoxon test: p � 0.022.
S114 Annals of Neurology Vol 53 (suppl 3) 2003

open L-dopa was permitted, and the primary outcomevariable was the percentage of change from baseline instriatal 123I-�-CIT uptake. 123I-�-CIT SPECT showedthat the percentage of loss in striatal tracer uptake frombaseline was significantly reduced in the group initiallytreated with pramipexole compared with the group ini-tially treated with L-dopa: 7.1 � 9.0% versus 13.5 �9.6% at 22 months p � 0.004; 10.9 � 11.8% versus19.6 � 12.4% at 34 months, p � 0.009; and 16.0 �13.3% vs 25.5 � 14.1% at 46 months, p � 0.01. Thepercentage of loss from baseline in striatal 123I-�-CITuptake was correlated with the change from baseline inUPDRS at the 46-month evaluation (r � 0.40; p �0.001). The authors concluded that patients treatedinitially with pramipexole demonstrated a relative 40%reduction in the rate of loss of striatal 123I-�-CIT up-take compared with those treated initially with L-dopaover a 4-year period.
The above PET and SPECT findings both supportthe notion that treatment with dopamine agonists rel-atively slows the rate of loss of striatal dopamine ter-minal function by 30 to 40% in early PD patientscompared with L-dopa therapy. The two studies bothhighlight the potential for functional imaging to deter-mine the efficacy of putative neuroprotective agents inPD and relate the status of dopamine terminal func-tion to clinical end points of PD progression.
Nondopaminergic MarkersAs mentioned in the introduction, other neurotrans-mitter systems as well as dopamine degenerate in PD,and these too potentially can be monitored with PETand SPECT. Serotonin HT1A sites are found presyn-aptically on the serotonin cell bodies in the median ra-phe, and 11C-WAY100635 PET, a marker of serotoninHT1A binding, can provide in vivo information on thestatus of serotonergic function in PD. Doder and col-leagues have demonstrated an average 25% loss of me-dian raphe HT1A binding in established PD, individ-ual levels of which correlate significantly with severityof rest tremor.51
Noradrenergic transporter (NAT) binding can beimaged along with DAT binding using the nonselectiveligand 11C-RTI-32. Thalamic binding of this ligandprimarily reflects noradrenergic terminal function, andthis is preserved in nondepressed PD but significantlyreduced to 60% of normal in depressed cases.67
Cholinergic terminal function can be imaged withvesamicol tracers.52,53 To date, there have been no re-ported 18F-fluoroethoxybenzovesamicol or 11C-vesamicol PET or 123I-vesamicol SPECT studies inPD.
Cortical Metabolism in Parkinson’s DiseaseThe prevalence of clinical dementia is twofold higherin PD than in the general population, estimates rang-
ing between 10 to 40% and the incidence increasingwith age.54 Pathological processes responsible for cog-nitive impairment in PD patients include the presenceof coexistent Alzheimer’s pathology and cortical Lewybodies.55 However, specific cognitive deficits can berecognized in clinically nondemented PD patients evenearly in the course of the disease, including impairmentof visuospatial capacity, attentional control, planning,short-term memory, and immediate recall.56–58 Not allpatients with PD and cognitive impairment go on todevelop frank dementia. Risk factors for dementia atdiagnosis include being elderly, rapidly progressive mo-tor disability, poor verbal fluency, and reduced abilityon visuospatial subtests of the Wechsler adult intelli-gence scale.58
Fig 3. An 11C-PK11195 positron emission tomography imageshowing increased microglial activation in the nigra (arrow,bottom) and pallidum (arrow, top) of a Parkinson’s diseasepatient. (Images courtesy of R. Banati.)
Brooks: Monitoring Neuroprotection in PD S115

18F-fluorodeoxyglucose (18FDG) PET has demon-strated reduced glucose metabolism in frontal and tem-poroparietal association areas in Alzheimer’s demen-tia,59 PD with dementia,60–62 and dementia withLewy bodies.63 Recently, Hu and colleagues have used18FDG PET to show absolute bilateral reductions inglucose metabolism in posterior parietal and temporalcortical gray matter in nondemented PD patients withestablished disease.64 18FDG PET may, therefore, pro-vide useful predictors of future cognitive impairment ina subset of PD patients who go on to develop demen-tia and provide a marker of the efficacy of putativeneuroprotective agents.
Neuroinflammation11C-PK11195 PET is an in vivo marker of neuroin-flammation.65 This isoquinoline ligand binds to pe-ripheral benzodiazepine sites expressed by the mito-chondria of activated microglia, the natural immunedefense of the brain. It is now possible to demonstratein vivo the presence of increased binding in the sub-stantia nigra and pallidum of PD patients66 (Fig 3).This opens the intriguing future possibility of beingable to monitor the immune component of this degen-erative disorder as a marker of disease activity and pos-sibly to detect beneficial antiinflammatory effects ofneuroprotective agents.
ConclusionIn summary, we have shown that functional imagingstudies can offer an objective method of assessing dis-ease progression in PD, so avoiding several of the prob-lems associated with clinical studies. PET and SPECTstudies, however, provide markers of nerve terminalfunction rather than cell density and so, like clinicalassessments, are potentially influenced by compensa-tory mechanisms. Because of the heterogeneity in PD,PET studies of putative neuroprotective agents requirelarge enough cohorts to provide the power to allow forthe wide between-subject variability in the rate of pro-gression. Despite this, they should provide a valuableadjunct to clinical data in the assessment of neuropro-tective and restorative therapies in PD.
References1. Jellinger K. The pathology of parkinsonism. In: Marsden CD,
Fahn S, eds. Movement disorders 2. London: Butterworths,1987:124–165.
2. Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substan-tia nigra regional selectivity. Brain 1991;114:2283–2301.
3. Agid Y, Javoy-Agid F, Ruberg M. Biochemistry of neurotrans-mitters in Parkinson’s disease. In: Marsden CD, Fahn S, eds.Movement disorders 2. London: Butterworths, 1987:166–230.
4. Byrne E, Lennox G, Lowe J, Godwin-Austin RB. Diffuse Lewybody disease: clinical features in 15 cases. J Neurol NeurosurgPsychiatr 1989;52:709–717.
5. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive micro-glia are positive for HLA-DR in the substantia nigra of Parkin-son’s and Alzheimer’s disease brains. Neurology 1988;38:1285–1291.
6. Louis DE, Tang MX, Cote L, et al. Progression of parkinsoniansigns in Parkinson’s disease. Arch Neurol 1999;56:334–337.
7. Fahn S, Elton RL, and members of the UPDRS developmentcommittee. Unified Parkinson’s disease rating scale. In: Fahn S,Marsden CD, Goldstein M, eds. Recent developments in Par-kinson’s disease. New York: Macmillan, 1987:153–163.
8. Martinez-Martin P, Gil-Nagel A, Gracia LM, et al. UnifiedParkinson’s Disease Rating Scale: characteristics and structure.Mov Disord 1994;9:76–83.
9. Langston JW, Widner H, Goetz CG, et al. Core assessmentprogram for intracerebral transplantations (CAPIT). Mov Dis-ord 1992;7:2–13.
10. Lang AE, Benabid AL, Koller WC, et al. The Core AssessmentProgram for Intracerebral Transplantation. Mov Disord 1995;10:527–528.
11. Morrish PK, Sawle GV, Brooks DJ. An [18F]dopa PET andclinical study of the rate of progression in Parkinson’s disease.Brain 1996;119:585–591.
12. The Parkinson Study Group. Effect of deprenyl on the progres-sion of disability in early Parkinson’s disease. N Engl J Med1989;321:1364–1371.
13. Ward CD. Does selegiline delay the progression of Parkinson’sdisease? A critical reevaluation of the DATATOP study. J Neu-rol Neurosurg Psychiat 1994;57:217–220.
14. Leber P. Slowing the progression of Alzheimer disease: meth-odologic issues. Alzheimer Disease and Associated Disorders.1997;11(suppl 5):S10–S20.
15. Firnau G, Sood S, Chirakal R, et al. Cerebral metabolism of6-[18F]fluoro-L-3,4-dihydroxyphenylalanine in the primate.J Neurochem 1987;48:1077–1082.
16. Kuwabara H, Cumming P, Reith J, et al. Human striatalL-dopa decarboxylase activity estimated in vivo using6-[18F]fluorodopa and positron emission tomography: erroranalysis and application to normal subjects. J Cereb Blood FlowMetab 1993;13:43–56.
17. Snow BJ, Tooyama I, McGeer EG, et al. Human positronemission tomographic [18F]fluorodopa studies correlate withdopamine cell counts and levels. Ann Neurol 1993;34:324–330.
18. Pate BD, Kawamata T, Yamada T, et al. Correlation of striatalfluorodopa uptake in the MPTP monkey with dopaminergicindices. Ann Neurol 1993;34:331–338.
19. Yee RE, Huang SC, Stout DB, et al. Nigrostriatal reduction ofaromatic L-amino acid decarboxylase activity in MPTP-treatedsquirrel monkeys: in vivo and in vitro investigations. J Neuro-chem 2000;74:1147–1157.
20. Yee RE, Irwin I, Milonas C, et al. Novel observations withFDOPA-PET imaging after early nigrostriatal damage. MovDisord 2001;16:838–848.
21. Diffley DM, Costa JL, Sokoloski EA, et al. Direct observationof 6-fluorodopamine in guinea pig nerve microsacs by 19FNMR Biochem Biophys Res Commun 1983;110:740–745.
22. Deep P, Gjedde A, Cumming P. On the accuracy of an[18F]FDOPA compartmental model: evidence for vesicular stor-age of [18F]fluorodopamine in vivo. J Neurosci Methods 1997;76:157–165.
23. Morrish PK, Sawle GV, Brooks DJ. Clinical and [18F]dopaPET findings in early Parkinson’s disease. J Neurol NeurosurgPsychiat 1995;59:597–600.
24. Otsuka M, Ichiya Y, Kuwabara Y, et al. Differences in the re-duced 18F-dopa uptakes of the caudate and the putamen inParkinson’s disease: correlations with the three main symptoms.J Neurol Sci 1996;136:169–173.
S116 Annals of Neurology Vol 53 (suppl 3) 2003

25. Rakshi JS, Bailey DL, Morrish PK, Brooks DJ. Implementationof 3D acquisition, reconstruction, and analysis of dynamic [18F]fluorodopa studies. In: Myers R, Cunningham V, Bailey D,Jones T, eds. Quantification of brain function using PET. SanDiego: Academic Press, 1996:82–87.
26. Ceravolo R, Piccini P, Bailey DL, et al. 18F-dopa PET evi-dence that tolcapone acts as a central COMT inhibitor in Par-kinson’s disease. Synapse 2002;43:201–207.
27. Turjanski N, Lees AJ, Brooks DJ. Striatal dopaminergic recep-tor dysfunction in patients with restless legs syndrome: 18F-dopa and 11C-raclopride PET studies. Neurology 1999;52:932–937.
28. Vingerhoets FJG, Snow BJ, Langston JW, et al. Positron emis-sion tomographic evidence for progression of human MPTP-induced dopaminergic lesions. Ann Neurol 1994;36:765–770.
29. Morrish PK, Rakshi JS, Sawle GV, Brooks DJ. Measuring therate of progression and estimating the preclinical period of Par-kinson’s disease with [18F]dopa PET. J Neurol Neurosurg Psy-chiat 1998;64:314–319.
30. Nurmi EM, Ruottinen HM, Bergman J, et al. The rate of pro-gression in Parkinson’s disease: a [18F]dopa PET study. Neu-rology 1999;52(suppl 2):A91.
31. Sawle GV, Colebatch JG, Shah A, et al. Striatal function innormal aging: implications for Parkinson’s disease. Ann Neurol1990;28:799–804.
32. Eidelberg D, Takikawa S, Dhawan V, et al. Striatal F-18 dopauptake—absence of an aging effect. J Cereb Blood FlowMetabol 1993;13:881–888.
33. Laruelle M, Giddings SS, Zea-Ponce Y, et al. Methyl 3 beta-(4-[125I]iodophenyl)tropane-2 beta-carboxylate in vitro bind-ing to dopamine and serotonin transporters under “physiologi-cal” conditions. J Neurochem 1994;62:978–986.
34. Seibyl JP, Marek KL, Quinlan D, et al. Decreased single-photon emission computed tomographic [123I]�-CIT striataluptake correlates with symptom severity in Parkinson’s disease.Ann Neurol 1995;38:589–598.
35. Booij J, Tissingh G, Boer GJ, et al. [ 123I]FP-CIT SPECTshows a pronounced decline of striatal dopamine transporterlabelling in early and advanced Parkinson’s disease. J NeurolNeurosurg Psychiat 1997;62:133–140.
36. Seibyl JP, Marek K, Sheff K, et al. Test/retest reproducibility ofiodine-123-betaCIT SPECT brain measurement of dopaminetransporters in Parkinson’s patients. J Nucl Med 1997;38:1453–1459.
37. Marek K, Seibyl JP, Zoghbi SS, et al. [I-123] beta-CIT SPECTimaging demonstrates bilateral loss of dopamine transporters inhemiparkinsons disease. Neurology 1996;46:231–237.
38. Innis RB, Marek KL, Sheff K, et al. Effect of treatment withL-dopa/carbidopa or L-selegiline on striatal dopamine trans-porter SPECT imaging with [I-123]beta-CIT. Mov Disord1999;14:436–442.
39. Marek K, Innis R, van Dyck C, et al. [123I]beta-CIT SPECTimaging assessment of the rate of Parkinson’s disease progres-sion. Neurology 2001;57:2089–2094.
40. Marek KL, Innis RB, Seibyl J. Assessment of Parkinson’s dis-ease progression with �-CIT and SPECT imaging. Mov Disord1998;13(suppl 2):238–230.
41. Benamer TS, Patterson J, Grosset DG, et al. Accurate differen-tiation of parkinsonism and essential tremor using visual assess-ment of [123I]-FP-CIT imaging: the [123I]-FP-CIT StudyGroup. Mov Disord 2000;15:503–510.
42. Benamer HTS, Patterson J, Wyper DJ, et al. Correlation ofParkinson’s disease severity and duration with I-123-FP-CITSPECT striatal uptake. Mov Disord 2000;15:692–698.
43. Naudon L, Leroux-Nicollet I, Costentin J. Short-term treat-ments with haloperidol or bromocriptine do not alter the den-sity of the monoamine vesicular transporter in the substantianigra. Neurosci Lett 1994;173:1–4.
44. Frey KA, Koeppe RA, Kilbourn MR, et al. Pre-synaptic mono-aminergic vesicles in Parkinson’s disease and normal aging. AnnNeurol 1996;40:873–884.
45. Lee CS, Samii A, Sossi V, et al. In vivo positron emission to-mographic evidence for compensatory changes in presynapticdopaminergic nerve terminals in Parkinson’s disease. Ann Neu-rol 2000;47:493–503.
46. Alexander T, Sortwell CE, Sladek CD, et al. Comparison ofneurotoxicity following repeated administration of L-dopa,D-dopa, and dopamine to embryonic mesencephalic dopamineneurons in cultures derived from Fischer 344 and Sprague-Dawley donors. Cell Transplant 1997;6:309–315.
47. Iida M, Miyazaki I, Tanaka K, et al. Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole,a dopamine agonist. Brain Res 1999;838:51–59.
48. Olanow CW, Jenner P, Brooks D. Dopamine agonists and neu-roprotection in Parkinson’s disease. Ann Neurol 1998;44(suppl1):S167–S174
49. Whone AL, Remy P, Davis MR, et al. The REAL-PET study:slower progression in early Parkinson’s disease treated withropinirole compared with L-dopa. Neurology 2002;57(suppl 3).
50. Parkinson Study Group. Dopamine transporter brain imagingto assess the effects of pramipexole vs levodopa on Parkinsondisease progression. JAMA 2002;287:1653–1661.
51. Doder M, Rabiner EI, Turjanski N, et al. Functional imagingof tremor in Parkinson’s disease with [C- 11]-WAY 100635PET. Neurology 2001;56(suppl 3):A271–A272.
52. Mulholland GK, Wieland DM, Kilbourn MR, et al.[F-18]Fluoroethoxy-benzovesamicol, a PET radiotracer for thevesicular acetylcholine transporter and cholinergic synapses.Synapse 1998;30:263–274.
53. Shiba K, Mori H, Ikeda E, et al. The potential of radioiodi-nated (-)-m-iodovesamicol for diagnosing cholinergic deficit de-mentia. Nucl Med Biol 2001;28:261–264.
54. Brown RG, Marsden CD. Cognitive function in Parkinson’sdisease: from description to theory. TINS 1990;13:21–29.
55. Byrne EJ, Lowe J, Godwin-Austen RB, et al. Dementia of Par-kinson’s disease associated with diffuse cortical Lewy bodies.Lancet 1987;i:501.
56. Lees AJ, Smith E. Cognitive deficits in the early stages of Par-kinson’s disease. Brain 1983;106:257–270.
57. Brown RG, Marsden CD. Visuospatial function in Parkinson’sdisease. Brain 1986;109:987–1002.
58. Sagar HJ, Sullivan EV, Gabrieli JDE, et al. Temporal orderingand short-term memory deficits in Parkinson’s disease. Brain1988;111:525–539.
59. Foster NL, Chase TN, Mansi L, et al. Cortical abnormalities inAlzheimer’s disease. Ann Neurol 1984;16:649–654.
60. Kuhl DE, Metter EJ, Riege WH. Patterns of local cerebral glu-cose utilisation determined in Parkinson’s disease by the 18F-fluorodeoxyglucose method. Ann Neurol 1984;15:419–424.
61. Peppard RF, Martin WRW, Carr GD, et al. Cerebral glucosemetabolism in Parkinson’s disease with and without dementia.Arch Neurol 1992;49:1262–1268.
62. Vander-Borght T, Minoshima S, Giordani B, et al. Cerebralmetabolic differences in Parkinson’s and Alzheimer’s diseasematched for dementia severity. J Nucl Med 1997;38:797–802.
63. Minoshima S, Foster NL, Sima AA, et al. Alzheimer’s diseaseversus dementia with Lewy bodies: cerebral metabolic distinc-tion with autopsy confirmation. Ann Neurol 2001;50:358–365.
Brooks: Monitoring Neuroprotection in PD S117

64. Hu MTM, Taylor-Robinson SD, Chaudhuri KR, et al. Corticaldysfunction in non-demented Parkinson’s disease patients: acombined 31phosphorus MRS and 18 FDG PET study. Brain2000;123:340–352.
65. Banati RB, Myers R, Kreutzberg GW. PK (“peripheralbenzodiazepine”)-binding sites in the CNS indicate early anddiscrete brain lesions: microautoradiographic detection of[3H]PK11195 binding to activated microglia.. J Neurocytol1997;26:77–82.
66. Gerhard A, Banati RB, Cagnin A, Brooks DJ. In vivo imagingof activated microglia with [C-11]PK11195 positron emissiontomography (PET) in idiopathic and atypical Parkinson’s dis-ease. Neurology 2001;56(suppl 3):A270.
67. Remy P, Doder M, Lees AJ, Turjanski N, Hammers A, BrooksDJ. Depression in Parkinson’s disease is associated with im-paired catecholamine terminal function in the midbrain, thala-mus, and anterior cingulate cortex. Neurology 2002;58(suppl3):A488.
DiscussionSchapira: Thank you very much, David. Can I ask interms of using imaging measures in a neuroprotectionstudy, do you think it is possible, necessary, or practi-cal to use more than one marker to look at differentaspects of your patient population? For example, is itpossible to use fluorodopa PET alone to assess neuro-protection or do you think it might be useful to alsomeasure postsynaptic function with a D2 receptors ag-onist such as raclopride?
Brooks: I guess it depends what answer you want. Ifyou want a holistic view of what is going on you canutilize many of the different agents that I have de-scribed in the article. The reality is that you are con-strained to a certain degree by levels of radiation andwhat patients will find tolerable. I would say that ifyou want to demonstrate that an agent is neuroprotec-tive it might be very useful to look at markers of Lewybody formation (if we had them) or the inflammatoryresponse in addition to a more standard marker of do-pamine terminal function. In terms of radiation expo-sure, this is possible in principle. Expense and feasibil-ity are other problems. It may be better in practice totake small subgroups of patients and test whether aneuroprotective agent can affect microglial activation ina small subgroup while doing a more major study ofdopamine terminal function in the entire group.
Tatton: David, is it possible to look at NADH? Thiswould be particularly interesting because it is a markerof mitochondrial function, and there is evidence thatmitochondria are impaired in PD and that they are animportant source of proapoptotic signals. Is there aPET ligand for NADH?
Brooks: What you suggest is very interesting, butthere is no such ligand at the present time. One of theproblems in developing a labeled form of NADH or asimilar prodrug is assuring that it enters into the mi-tochondria and really reflects what that pool is doing. Idon’t think we have that technology at the present
time. I don’t know whether MR spectroscopy can de-tect NADH. Do you know, Flint?
Beal: I don’t think we can.Stocchi: I would like to ask an important practical
question. What can we do to establish that neuroim-aging surrogate markers are good end points thatwould be accepted by neurologists, the scientific com-munity, and regulatory authorities? Until we have de-fined such a surrogate, it may not be possible to deter-mine that a drug is neuroprotective and companiesmight not be willing to invest the time and money tostudy many promising new drugs?
Brooks: I think part of the problem is that currentfunctional imaging looks at one transmitter systemalone, unless you are looking at an overall metabolicprofile, and I guess the regulatory authorities feel thateven if you show that there is a loss in the rate ofdecline of dopamine terminal function they want toknow that this somehow translates into patients beingclinically better. In addition, it has not yet been estab-lished that any of the neuroimaging markers directlyreflects the number of surviving dopamine neurons orterminals. The FDA and CPMP have indicated thatthey currently will not accept these technologies as anend point for purposes of labeling. I don’t know whatneeds to be done to get them to accept that an imagingsurrogate marker is an index of nigrostriatal functionthat they would accept as an end point, and this re-mains an important problem as you have stated. Iwould be interested to hear Ken’s views on this.
Marek: I would agree. I think that there are nowwell-validated imaging measures that can provide anindex of what is happening with respect to dopamineneuronal degeneration and disease progression. The is-sue that remains unanswered is to what extent dochanges in this surrogate marker result in clinicallymeaningful changes in PD patients. Until we have es-tablished long-term correlations between changes inneuroimaging markers and clinical progression, regula-tors will have difficulty accepting surrogate end points.I think one option is to continue to follow patientswho have shown improvement on neuroimaging surro-gate markers to see if there is long-term benefit forclinical function. It may be that improvement in clin-ical status and neuroimaging measures of nigrostriatalfunction do not occur simultaneously.
Olanow: I think this a crucial point. I do not thinkthe problem is going to be so much to determine ifthere is a clinical benefit associated with one of thedrugs; either we will see a clinical change or we willnot. Frankly, you can have all the imaging changes inthe world and if you do not eventually get a clinicalcounterpart it will be difficult to convince physiciansthat the drug is a neuroprotective agent. I think thereal issue is that if you do see a clinical change, howcan you attribute it to a neuroprotective as opposed to
S118 Annals of Neurology Vol 53 (suppl 3) 2003

a symptomatic effect, at least in PD? Part of the prob-lem from the regulatory perspective is that regulatorshave taken the position that they don’t know if thechanges seen on neuroimaging are necessarily, in andof themselves, reflective of the underlying disease pro-cess and aren’t, for example, caused by some as yet un-identified pharmacological or drug-induced compensa-tory effect. I think that this is a crucial issue and onethat needs to be resolved. They have defined surrogateimaging markers in multiple sclerosis and to a certaindegree they are beginning to do so in Alzheimer’s dis-ease. We need to do the same in PD. One of thethings we may want to do is to extend the studies thatwere performed in Vancouver to determine to what ex-tent the changes seen with fluorodopa PET and �-CITSPECT reflect the number of surviving nigral neuronsand the integrity of the nigrostriatal system and deter-mine to what extent compensatory and pharmacologi-cal effects occur. I think, in the final analysis, it is go-ing to be necessary to demonstrate changes in bothclinical and neuroimaging markers to be confident thatwe have defined a neuroprotective effect.
Brooks: I think we will clearly have to do the studiesas you suggest. There is no easy way to answer theseproblems in PD patients because you can’t keep a pla-cebo group going for long enough, and you cannot relyon being able to count neurons in a predictable way.One could set this study up with the MPTP monkeymodel, but one of the big problems is being sure that2 years of exposure to different treatments has not af-fected, in some way, the imaging markers. It is alsoimportant to appreciate that the lesion models in mon-keys are not good models of PD, and there is notmuch if any progression in this model.
Olanow: Disease progression is not the issue here.The issue is, that when you see a change on fluorodopaor �-CIT uptake, what is it you are measuring? Whatdoes that tell you about the underlying state of thenigrostriatal system? You could, for example, look at arange of lesions of different severity in monkeys andcorrelate them with what you see on the scan. If youcould show a tight correlation, you can argue thatchanges in an imaging surrogate marker reflect the in-tegrity of the nigrostriatal system. You could even de-sign in components to assess for drug effect and tem-poral compensation. Small studies have been done, butI think a larger more definitive study is required.
Isacson: I think this is an important study, but it
may be very complex. For example, if you make a le-sion, you can get a progressive neuronal decline afteryou have stopped the toxin. On the other hand, themonkeys may show behavioral and even anatomic re-covery because they have a static lesion and not PDand may compensate. In some cases, they get improve-ment in their movement disorder, but the dopaminesystem continues to degenerate. In these circumstances,imaging markers may be a better marker of what isgoing on in the brain than clinical evaluation. I thinkit therefore is important to perform a study as you sug-gest so that we can have a more objective measure ofthe integrity of the nigrostriatal system than can beachieved with clinical assessment alone.
Olanow: You are talking about the fact that there arecompensatory mechanisms that can confound our clin-ical and perhaps even our imaging evaluations. This iswhat regulators are concerned about. When changesare reported, they don’t know how much of this iscompensatory, how much is real, how much is a directpharmacological consequence of the drug. What I amarguing for are experiments that can help to resolvethis.
Rascol: David, when you look at imaging measures ofnondopaminergic markers such as serotonin in PD pa-tients, do you see the same rate of progression as youdo with dopaminergic markers?
Brooks: That is an important question. We have notdone that type of study yet, but we will.
Beal: David Eidelberg has argued that imaging thefunctional basal ganglia network is an accurate way tofollow progression. What are your thoughts on that?
Brooks: I have not done that type of study. Davidhas shown, for instance, that glucose metabolism inPD is normal, but there is a covariance pattern that isabnormal between regions in the brain, and there isreduced activation of premotor regions that can benormalized with treatment. He maintains that youcould use changes in the network to monitor the neu-roprotective efficacy of a drug. What I don’t know iswhat the sensitivity and power of this type of study isand whether it is equivalent to what could be attainedwith markers that more directly measure dopaminergicfunction. There is no reason, in principal, why onecan’t run the two together. I think his approach is per-fectly valid. I am just not clear what the relative meritsof the different approaches are.
Brooks: Monitoring Neuroprotection in PD S119


















