

Molecular Biophysics III – dynamics Molecular Dynamics Simulations - 01/13/05
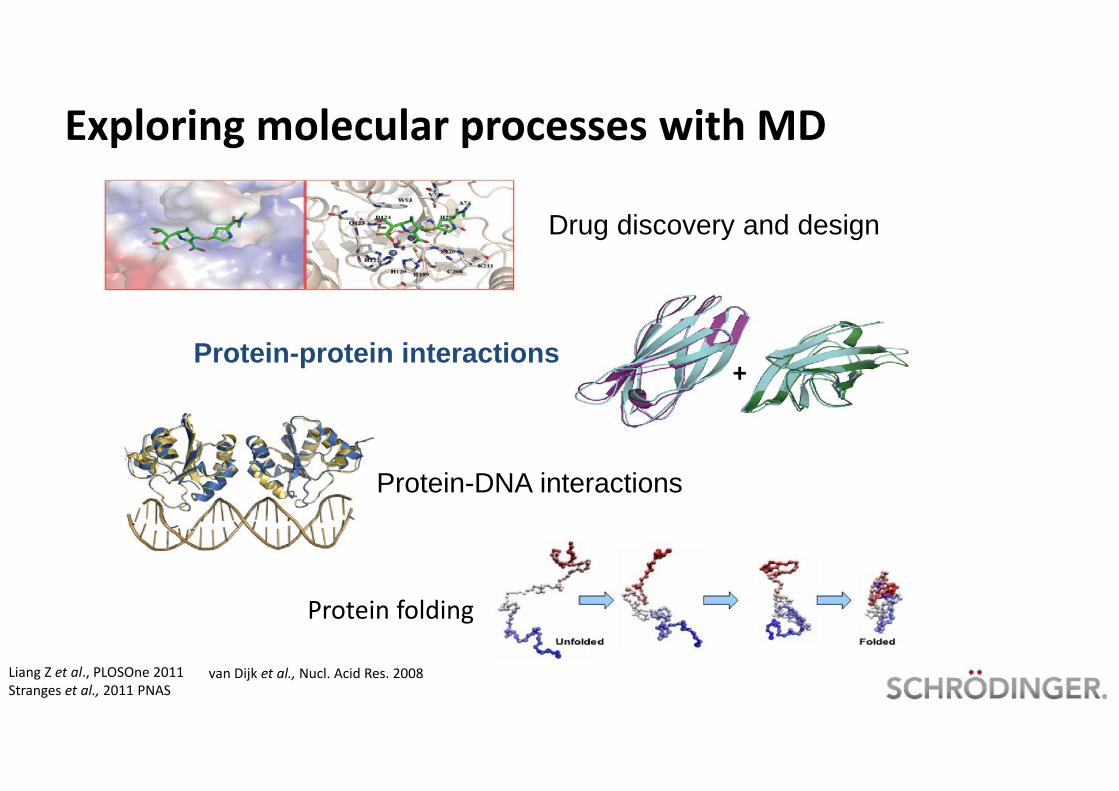
Exploring molecular processes with MD
Drug discovery and design
Liang Z et al., PLOSOne 2011Stranges et al., 2011 PNAS
+Protein-protein interactions
Protein-DNA interactions
van Dijk et al., Nucl. Acid Res. 2008
Protein folding
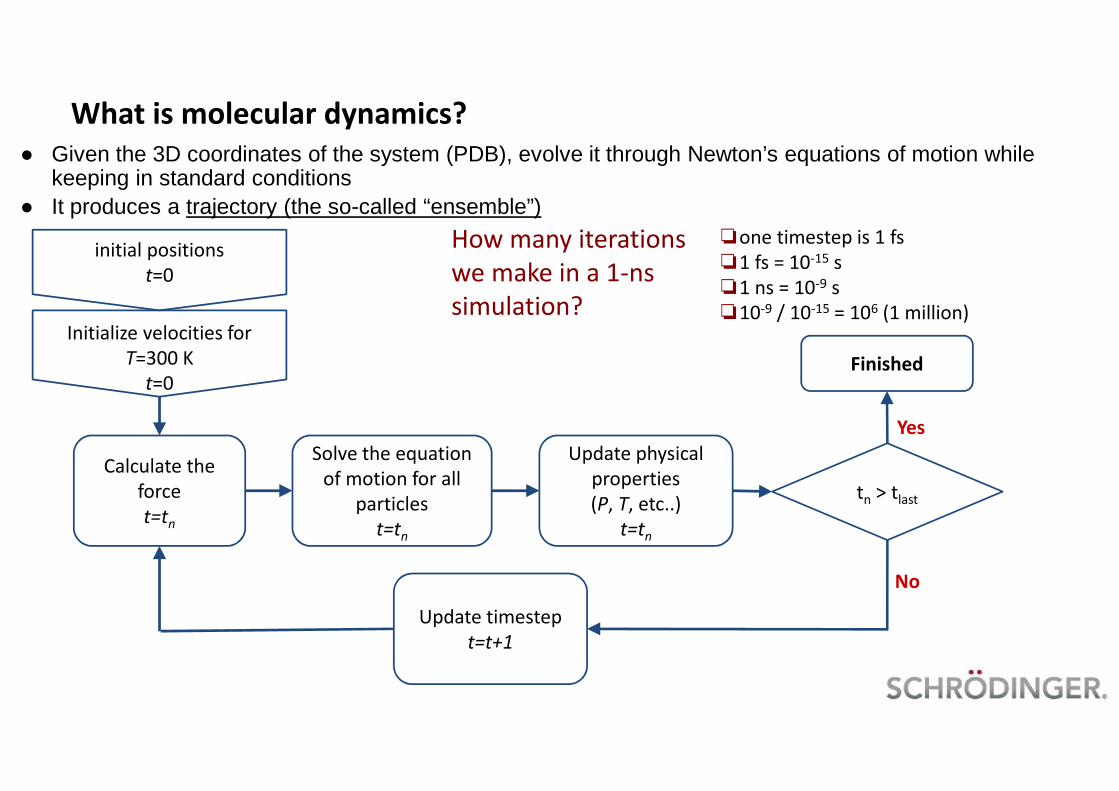
What is molecular dynamics?
initial positionst=0
Calculate theforcet=tn
Initialize velocities forT=300 K
t=0
Solve the equationof motion for all
particlest=tn
Update physicalproperties(P, T, etc..)
t=tn
tn > tlast
Finished
Yes
No
Update timestept=t+1
How many iterationswe make in a 1-nssimulation?
❏one timestep is 1 fs❏1 fs = 10-15 s❏1 ns = 10-9 s❏10-9 / 10-15 = 106 (1 million)
● Given the 3D coordinates of the system (PDB), evolve it through Newton’s equations of motion whilekeeping in standard conditions
● It produces a trajectory (the so-called “ensemble”)

Why molecular dynamics?
• time let us observe events happening at molecular time scales• have full solvation, temperature and pressure effects
Timefs (10-15s) ps (10-12s) ns (10-9s) µs (10-6s) ms (10-3s) s
Folding(tertiary struct.)Breathing motions
Bond vibrations
Folding(secondary struct)
Channel gating
Protein aggregationLigand binding
How long can we simulate?
Get more from your crystal:

Desmond: state-of-the-art MD engine• Developed by D.E. Shaw research to run on Anton (special purpose
machine)• Implements standard fixed-charges force field (Amber, Charmm,
OPLS) and solvation models TIP3P, SPC/E, TIP4P. No covalent bondbreaking.
Schrödinger enhancements:• Support for OPLS3 force-field (improved protein and nucleic acids
force field + enhancement on ligand parametrization)• Integration with Maestro modeling environment• Wealth of analysis tools for protein and protein/ligand interactions• Extends FEP and enhanced sampling modules• Watermap

Desmond molecular dynamics on CPU● Single/double precision● Scalable, can run on many CPUs● Best cross-node performance is with fast interconnect
(+Linux VM)
*caveat: Desmond requires a Linuxenvironment to run, analyses can beperformed also on the othersupported architectures

Desmond molecular dynamics on GPUDesmond on GPU
● only NVIDIA cards supported (Driver v346.59 or newer)● single precision only● cannot run across multiple cards
~3-4 GB of memory(less than 400K atomsystems)
~12 GB in memory
*Standard support does not cover consumer-level GPU cardssuch as GeForce GTX cards.
Gamer’s card(GTX series)
Professional card(Tesla series)
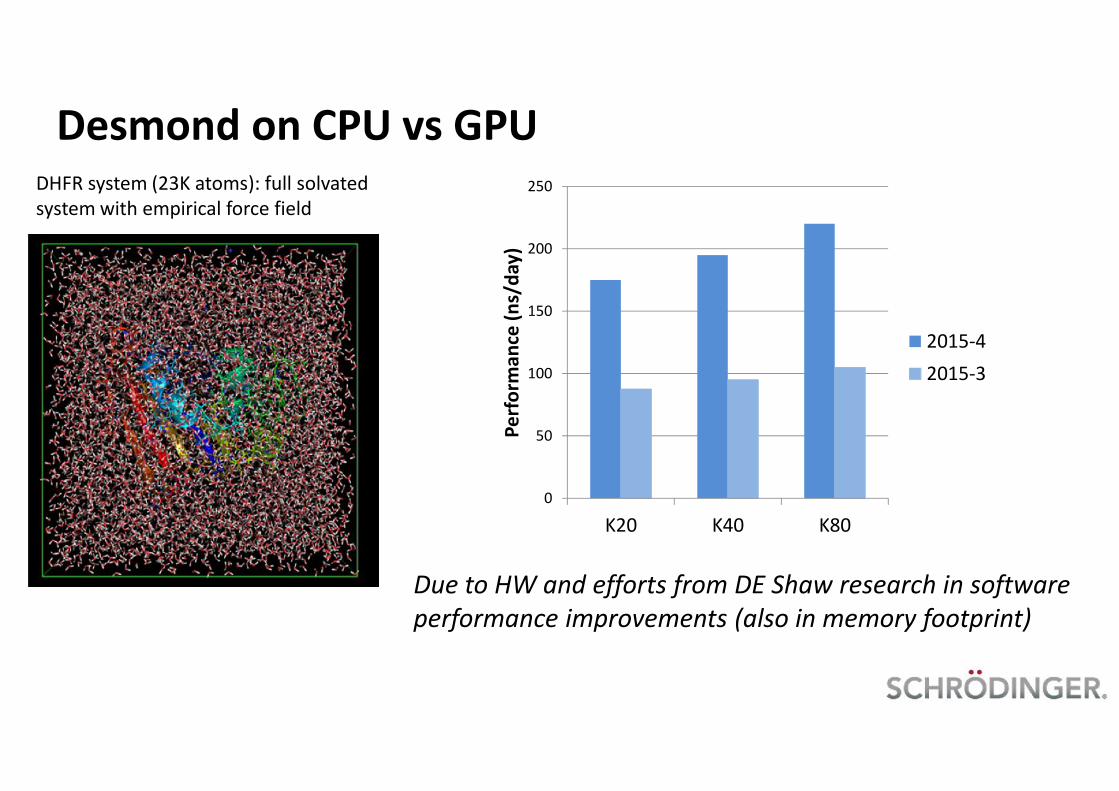
Desmond on CPU vs GPUDHFR system (23K atoms): full solvatedsystem with empirical force field
350
Due to HW and efforts from DE Shaw research in softwareperformance improvements (also in memory footprint)
0
50
100
150
200
250
K20 K40 K80Pe
rfor
man
ce (n
s/da
y)
2015-42015-3
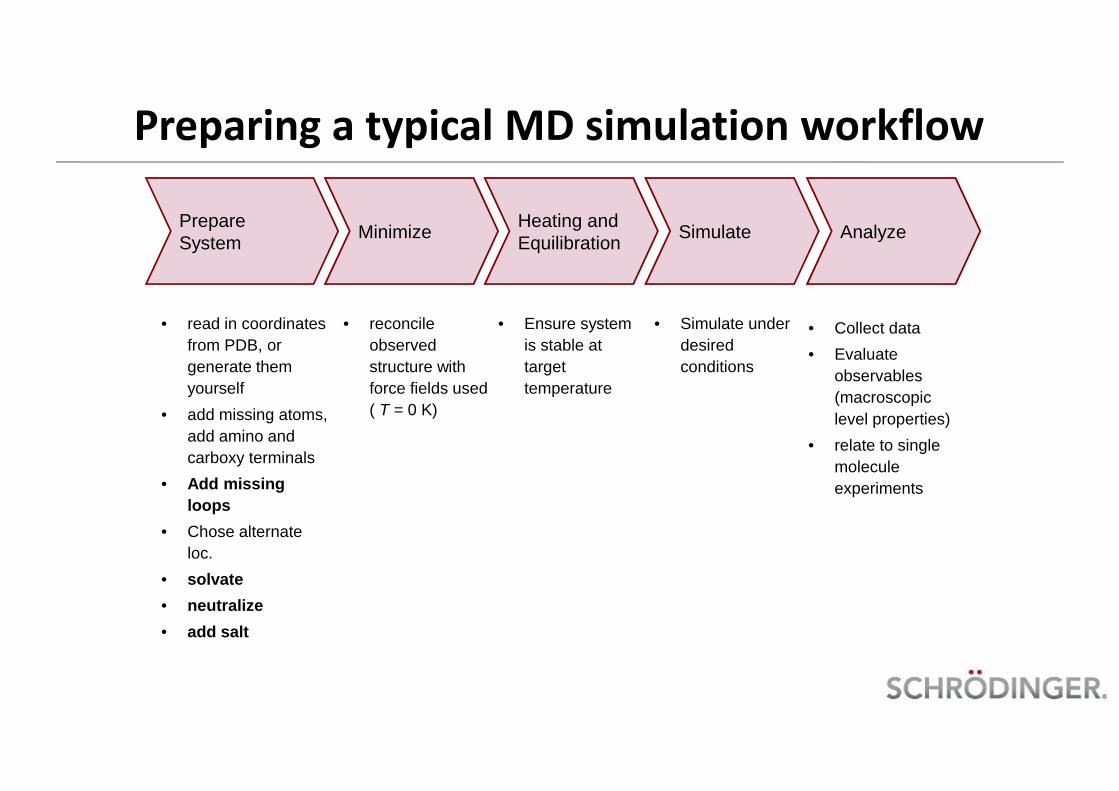
Preparing a typical MD simulation workflow
PrepareSystem Minimize Heating and
Equilibration Simulate Analyze
• read in coordinatesfrom PDB, orgenerate themyourself
• add missing atoms,add amino andcarboxy terminals
• Add missingloops
• Chose alternateloc.
• solvate• neutralize• add salt
• reconcileobservedstructure withforce fields used( T = 0 K)
• Ensure systemis stable attargettemperature
• Simulate underdesiredconditions
• Collect data• Evaluate
observables(macroscopiclevel properties)
• relate to singlemoleculeexperiments

Simulation setup: 2GMX example

Simulation setup: 2GMX example
For this specificexercise select chainA, create a new entryfrom it, and processonly this one

Protein reliability report

Protein preparation wizard
You might want to disable thisoption to preserve the crystalwater molecules
You might want to enable these if thereare missing loops. Not needed here.
Go!

Remove sulfates: usedfro crystallization butnot present inphysiological conditions(there is no problem inparametrizing themthough in OPLS3)
Protein preparation wizard
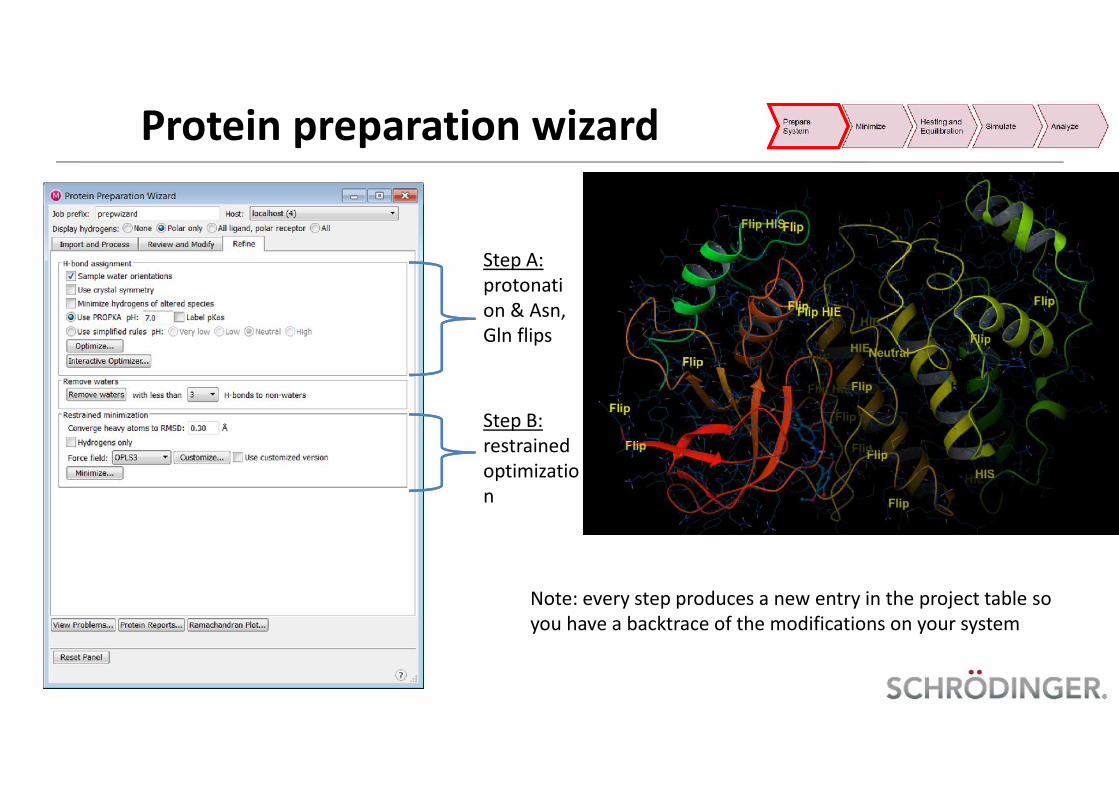
Step A:protonation & Asn,Gln flips
Step B:restrainedoptimization
Note: every step produces a new entry in the project table soyou have a backtrace of the modifications on your system
Protein preparation wizard
Protein preparation wizard
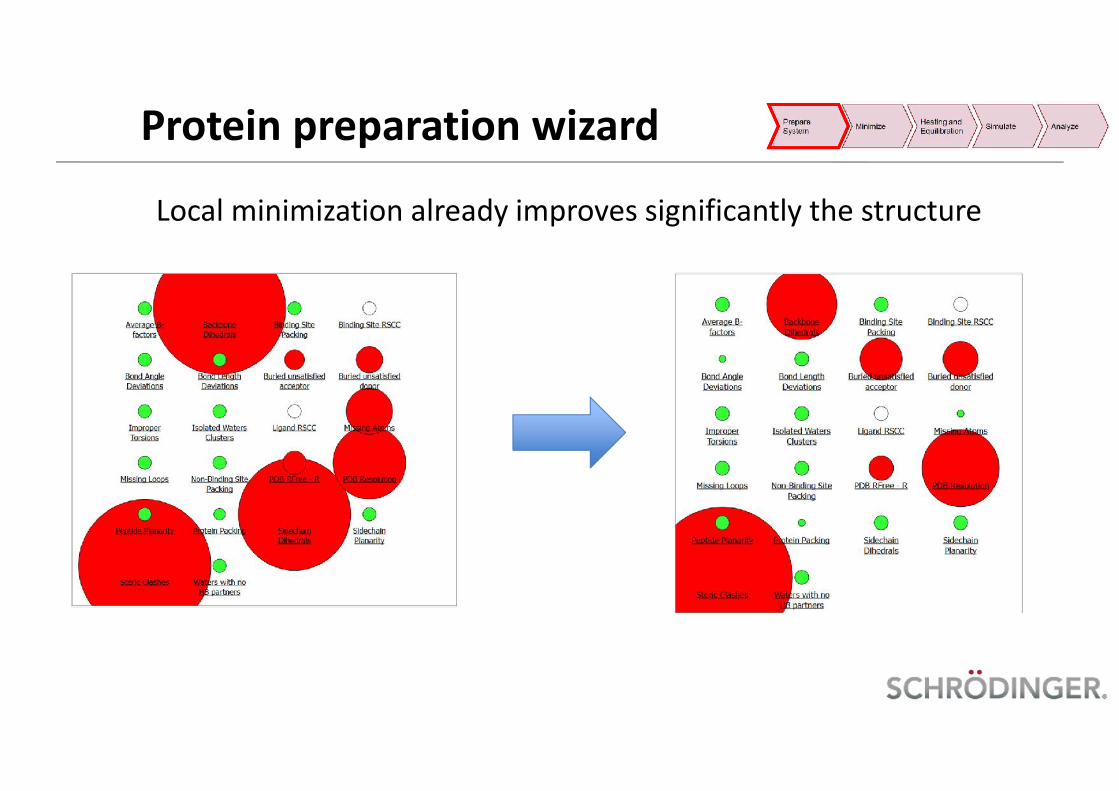
Local minimization already improves significantly the structure

Build the simulation box
* If you do not see the “Applications” panel you need to go to Task->Applications view
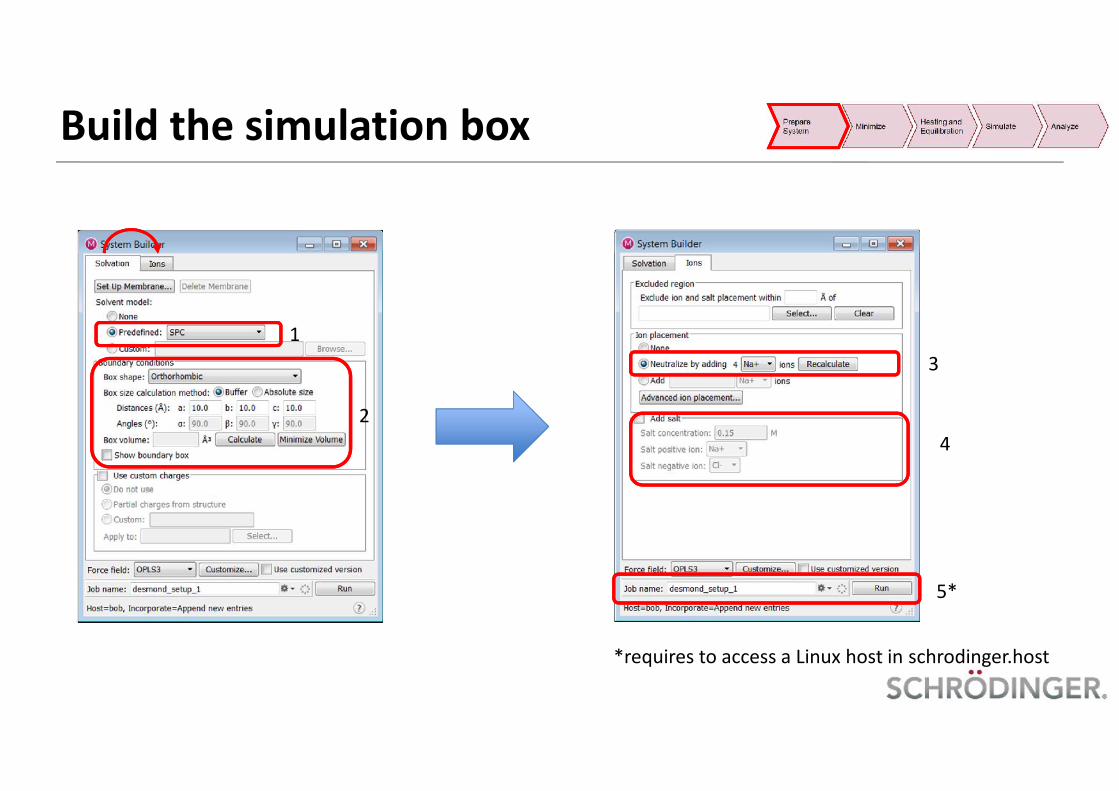
Build the simulation box
1
2
3
4
5*
*requires to access a Linux host in schrodinger.host
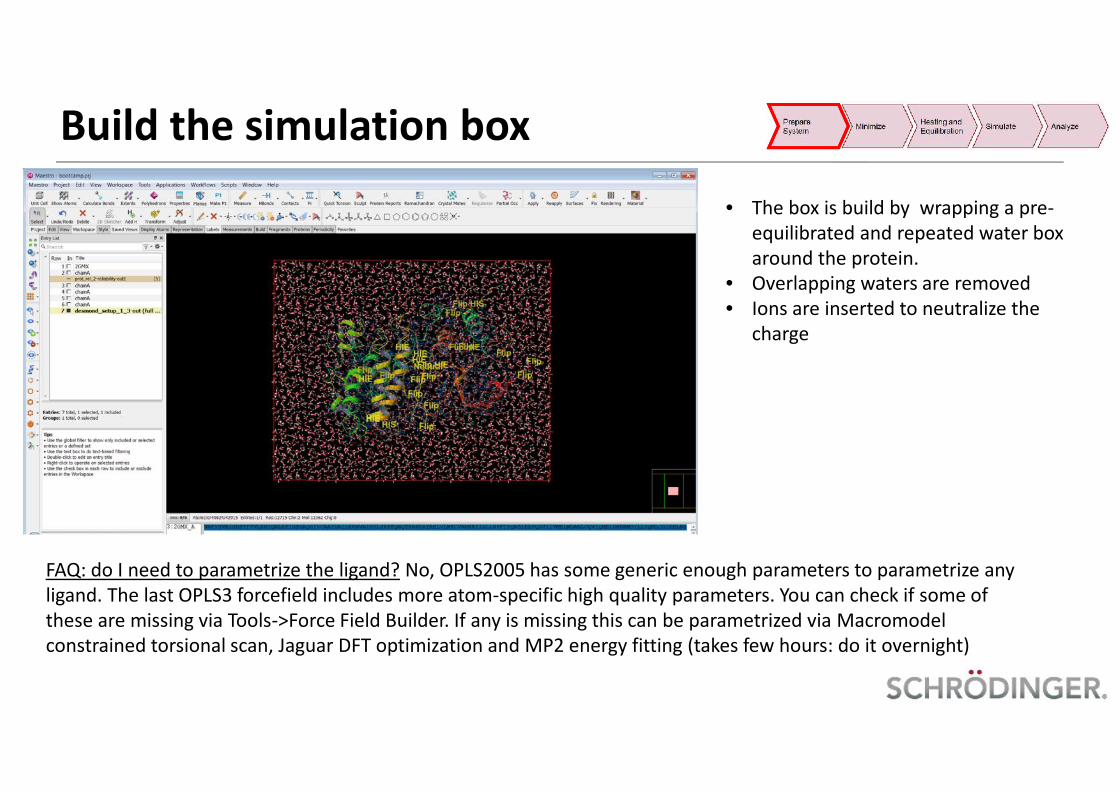
• The box is build by wrapping a pre-equilibrated and repeated water boxaround the protein.
• Overlapping waters are removed• Ions are inserted to neutralize the
charge
FAQ: do I need to parametrize the ligand? No, OPLS2005 has some generic enough parameters to parametrize anyligand. The last OPLS3 forcefield includes more atom-specific high quality parameters. You can check if some ofthese are missing via Tools->Force Field Builder. If any is missing this can be parametrized via Macromodelconstrained torsional scan, Jaguar DFT optimization and MP2 energy fitting (takes few hours: do it overnight)
Build the simulation box

Run the simulation
1
2
3
1. Take the system which is on screen (must be the one youprepared with the “System Builder”)
2. Optional: relax prior to production. Need always to dowhen you start with a PDB structure
3. MD expert section: ensembles but also positional restraints

Caveat emptor:MD runs can take days therefore it is not advisable to run straightfrom Maestro since the job control can be lost when Maestro isclosed for example.Best practice:1. Set up your run through the Molecular Dynamics interface2. Set up your machine trough “cog wheel”-> Job Settings (where
you can choose a machine that has GPU for example, as set inthe schrodinger.host file) and press “OK” (not “Run”!)
3. Write the output files in your current working directory (you setthis at the beginning with Project->Change Directory): “cogwheel” -> Write
4. Transfer the files produced (desmond_md_job_1.msj,desmond_md_job_1.cms and desmond_md_job_1.cfg) on themachine you selected for Job Settings
5. Use the command at the bottom of desmond_md_job_1.msj tolaunch it
6. Once finished, bring the output on your machine and load it inMaestro
A
B
Scale up to 10 ns fora therm
Beware: smallnumber = lot ofdata!
Run the simulation

A
Details of production run
Initial position andforcefield
List of all the steps ofminimization andtherm/press to do
Last line of msj contains the command youshould use on the remote machine (note:you should remove “#” and replaceC:\Users\DBranduardi\MyPrograms\Schrodinger2015-4_build14\utilities\multisimwith $SCHRODINGER/utilities/multisim )
Run the simulation
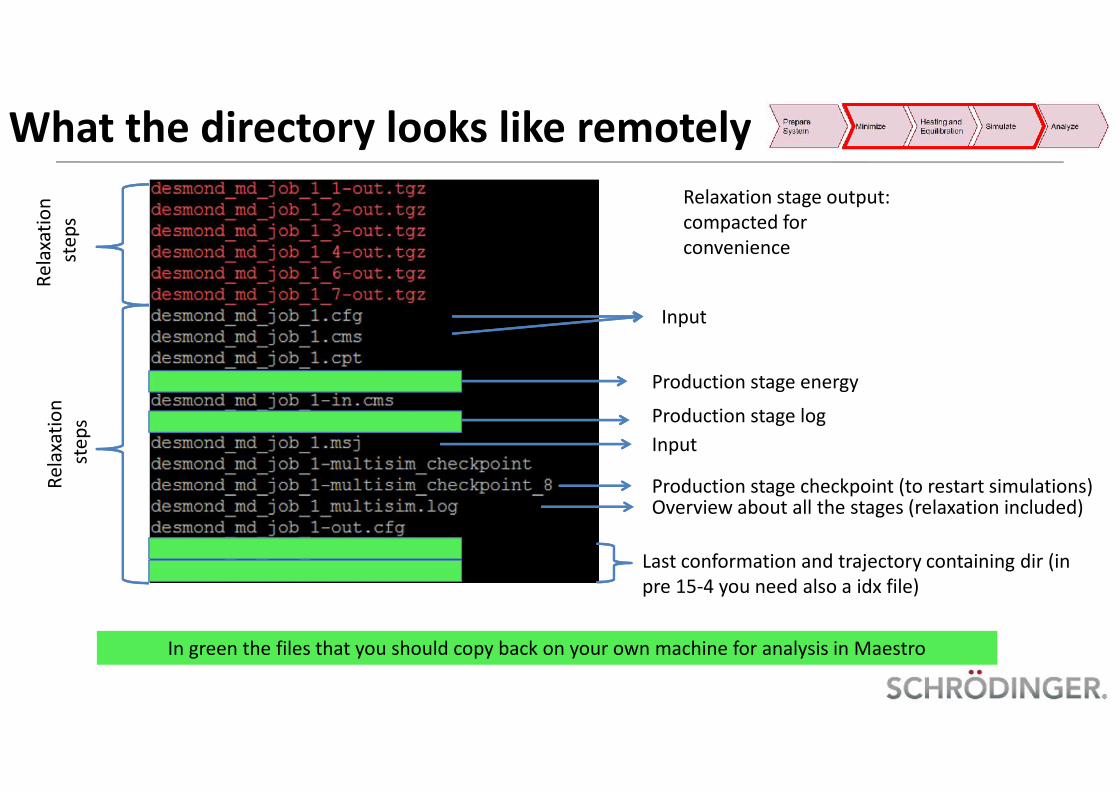
What the directory looks like remotelyRelaxation stage output:compacted forconvenience
Rela
xatio
nst
eps
Rela
xatio
nst
eps
Last conformation and trajectory containing dir (inpre 15-4 you need also a idx file)
Overview about all the stages (relaxation included)
Production stage log
Production stage energy
Production stage checkpoint (to restart simulations)
Input
Input
In green the files that you should copy back on your own machine for analysis in Maestro
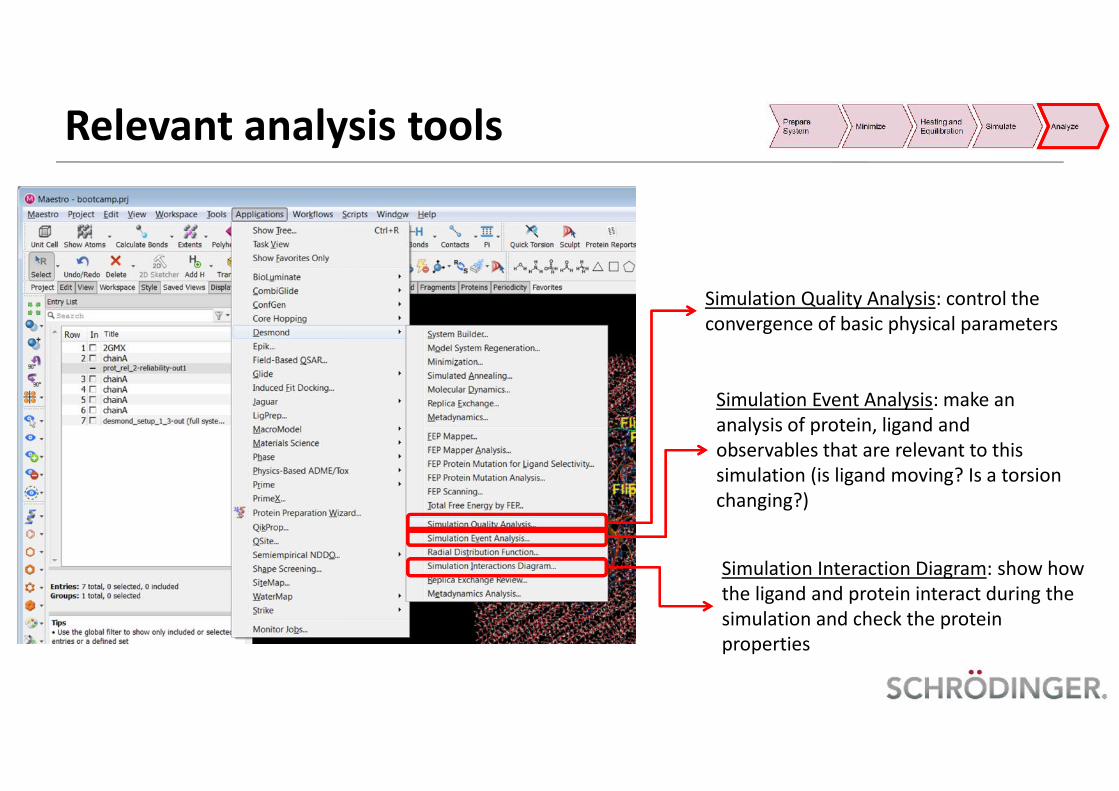
Relevant analysis tools
Simulation Quality Analysis: control theconvergence of basic physical parameters
Simulation Event Analysis: make ananalysis of protein, ligand andobservables that are relevant to thissimulation (is ligand moving? Is a torsionchanging?)
Simulation Interaction Diagram: show howthe ligand and protein interact during thesimulation and check the proteinproperties
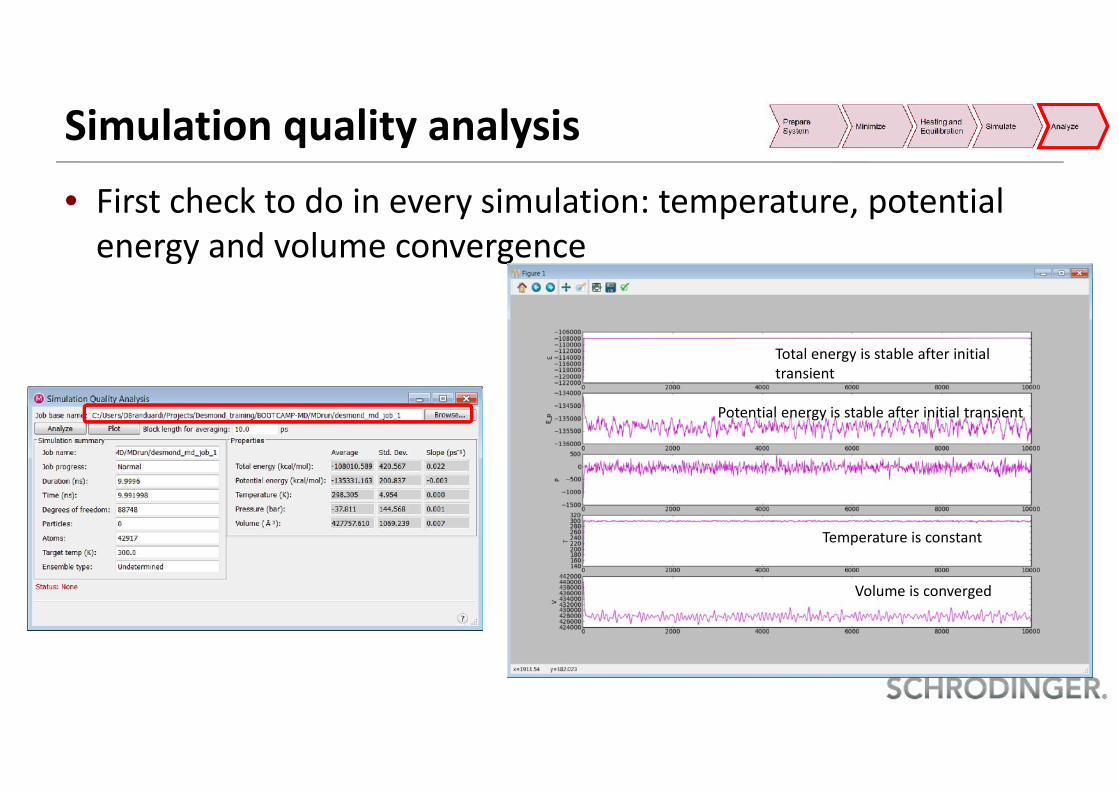
Simulation quality analysis• First check to do in every simulation: temperature, potential
energy and volume convergence
Total energy is stable after initialtransient
Potential energy is stable after initial transient
Temperature is constant
Volume is converged

A look to the trajectory• Import the *-out.cms file: this is linked to the trajectory
directory (do not move the relative position of the two)
-out.cms filecontains thepointer totrajectorydirectory.Trajectoryinformation isdenoted byIf you clickyou opentrakectorypanel
Play trajectory interactively andcenter your reference system. Youcan also use VMD for that.
Save frames for restarting, make movies

Simulation Events Analysis
• Drill down into the data by adding properties

Simulation Interactions Diagram• Analyze property of ligand, protein and protein ligand• Generate an automatic report in PDF format
RMSD
RMSFRMSF
Contacts ContactsSSE
Rot.Bonds
L-Props.

Simulation Interactions Diagram

Simulation Interactions Diagram
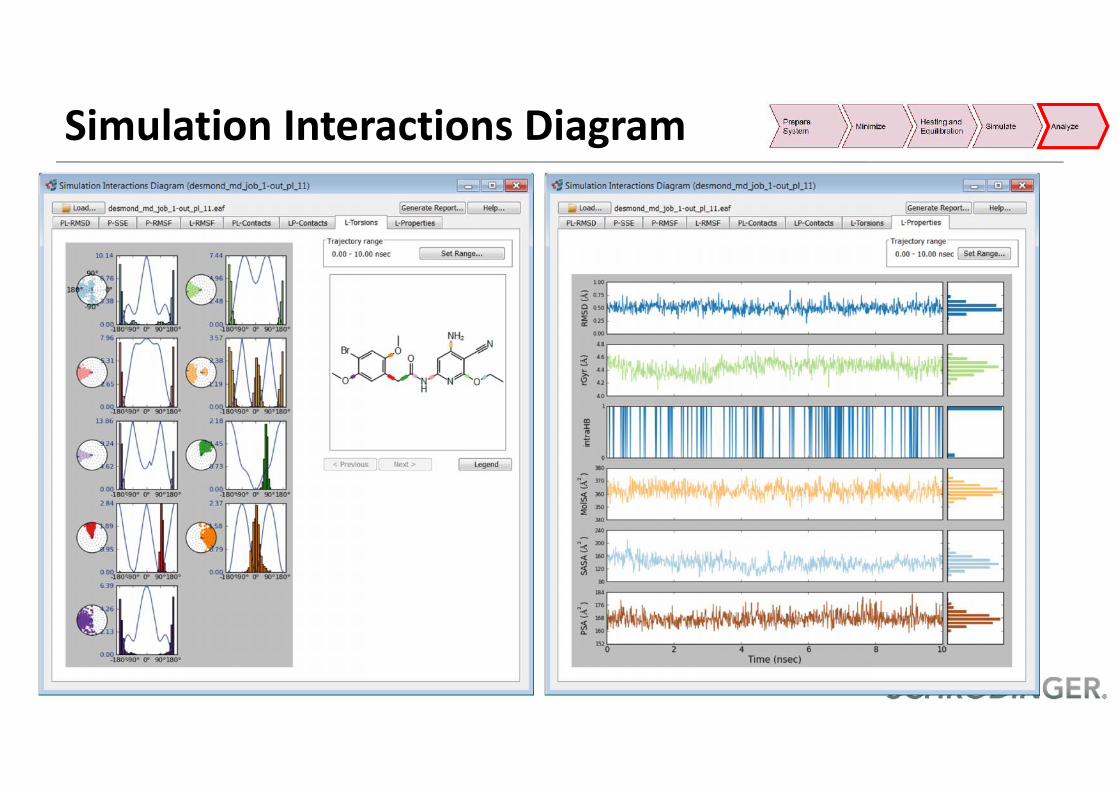
Simulation Interactions Diagram

MD ‘Modes’ with Desmond● Standard Mode● Free Energy Perturbation (FEP)
○Total free energy■ Absolute binding■ Absolute solvation
○Relative binding○Protein mutation
■ Ligand selectivity■ Protein-protein interactions■ Stability
● Enhanced sampling methods○Metadynamics○REMD○REST
● Mixed solvent simulations● Watermap
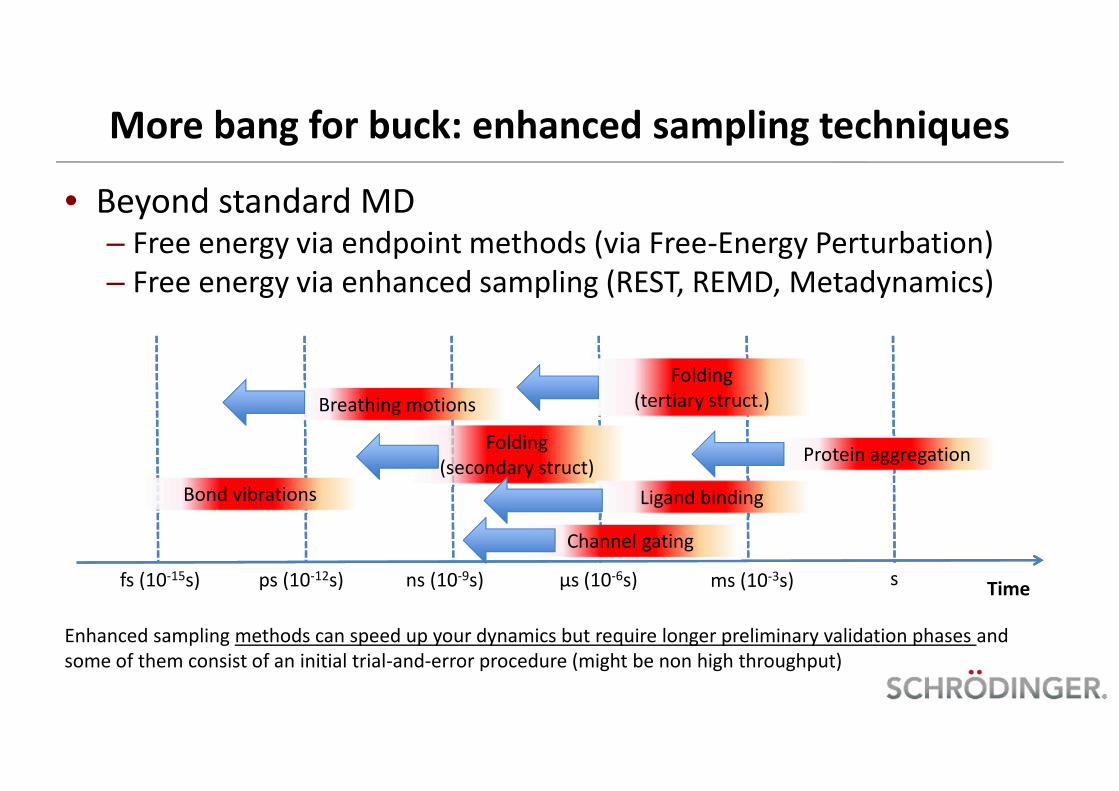
More bang for buck: enhanced sampling techniques
• Beyond standard MD– Free energy via endpoint methods (via Free-Energy Perturbation)– Free energy via enhanced sampling (REST, REMD, Metadynamics)
Timefs (10-15s) ps (10-12s) ns (10-9s) µs (10-6s) ms (10-3s) s
Folding(tertiary struct.)Breathing motions
Bond vibrations
Folding(secondary struct)
Channel gating
Protein aggregation
Ligand binding
Enhanced sampling methods can speed up your dynamics but require longer preliminary validation phases andsome of them consist of an initial trial-and-error procedure (might be non high throughput)

Conclusions• MD is fully integrated in the Schrödinger suite: protein
preparation is similar to any other modelling task• Building a suitable simulation box is just few clicks• Desmond GPU implementation allows you to achieve great
performance with just your workstation• MD protocol includes an effective thermalization phase,
completely transparent to the user• Effective analysis tools allow to judge the quality of simulation
and to trust your results providing valuable insights notavailable from the static structure

Acknowledgments• Dima Lupyan• Dan Robinson• Jas Bachoo• Woody Sherman
Thank you for yourattention!


















