
stacks.stanford.eduhn589zc6302/Susan_Vleck... · I certify that I have read this dissertation and...
253
THE ROLE OF GLYCOPROTEIN H IN VARICELLA-ZOSTER VIRUS PATHOGENESIS A DISSERTATION SUBMITTED TO THE DEPARTMENT OF MICROBIOLOGY AND IMMUNOLOGY AND THE COMMITTEE ON GRADUATE STUDIES OF STANFORD UNIVERSITY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Susan Elizabeth Vleck June 2010
-
Upload
nguyencong -
Category
Documents
-
view
214 -
download
0
Transcript of stacks.stanford.eduhn589zc6302/Susan_Vleck... · I certify that I have read this dissertation and...

THE ROLE OF GLYCOPROTEIN H
IN VARICELLA-ZOSTER VIRUS PATHOGENESIS
A DISSERTATION
SUBMITTED TO THE
DEPARTMENT OF MICROBIOLOGY AND IMMUNOLOGY
AND THE COMMITTEE ON GRADUATE STUDIES
OF STANFORD UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS
FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
Susan Elizabeth Vleck
June 2010

http://creativecommons.org/licenses/by-nc-nd/3.0/us/
This dissertation is online at: http://purl.stanford.edu/hn589zc6302
© 2010 by Susan Elizabeth Vleck. All Rights Reserved.
Re-distributed by Stanford University under license with the author.
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 United States License.
ii

I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Ann Arvin, Primary Adviser
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Jeffrey Glenn
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Harry Greenberg
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Karla Kirkegaard
Approved for the Stanford University Committee on Graduate Studies.
Patricia J. Gumport, Vice Provost Graduate Education
This signature page was generated electronically upon submission of this dissertation in electronic format. An original signed hard copy of the signature page is on file inUniversity Archives.
iii

ABSTRACT
Glycoprotein H (gH) plays an essential role in virus binding, entry and fusion
of the Herpesviridae. Varicella-zoster virus (VZV) is an important human pathogen
that causes varicella (chicken pox) and herpes zoster (shingles). VZV gH function has
not be analyzed in depth. gH function was demonstrated to be important for VZV
pathogenesis in skin xenografts in vivo by administration of anti-gH mAb 206, a
conformation dependent neutralizing antibody. Antibody administration prevented
infection in 42% of skin xenografts, and reduced virus replication and lesion formation
in the remaining skin xenografts. Antibody binding to gH altered gH localization
following endocytosis, preventing gH trafficking to the trans-Golgi network for virus
secondary envelopment. Antibody binding to gH within the virus envelope resulted in
internalization of virus particles, possibly for targeted degradation.
Deletion of ORF 37, which encodes gH, demonstrated that gH was essential
for VZV pathogenesis. Mutational analysis demonstrated that the N-terminus of the
protein formed a structural epitope required for efficient VZV pathogenesis in vivo.
Several neutralizing anti-gH antibodies target this epitope. A region of the C-terminus
was required for VZV pathogenesis, and for efficient virus-induced cell-cell fusion.
Predicted α-helices that might act as heptad repeats or fusion peptides were also
required for gH function and VZV pathogenesis. Cysteine residues were important for
gH maturation and transport, and possibly for correct expression of gH on the cell
surface. Altogether, these studies demonstrate the importance of structural and
functional domains for gH-dependent fusion and VZV pathogenesis.
iv

ACKNOWLEDGEMENTS
I would like to thank a number of people who have contributed to my success
in lab and in life. First and foremost is Ann Arvin, who has been an excellent advisor
and a wonderful example of a successful woman in science. Her guidance has been
instrumental to my achievements over the years. I would also like to thank members of
the Arvin lab, including Stefan Oliver, Marvin Sommer, Jaya Rajamani, Mike
Reichelt, Leigh Zerboni, Xibing Che, Li Wong, and Nandini Sen, as well as past
members Anne Schaap-Nutt, Jeremy Jones, Barbara Berarducci, Teri Slifer, Vasavi
Ramachandran, Reija Matheson, Yibing Wang, Vaishali Chaudhuri, Chai-Chi Ku, and
Makeda Robinson, for their camaraderie and friendship over the years. Stefan has
been a wonderful mentor and has helped me greatly when it comes to experiments. I
definitely thank him for his patience! Marvin has assisted me numerous times. Jaya
has been extremely helpful with xenograft experiments.
Special thanks go to my committee members and other people in the Stanford
community and elsewhere for their help. My committee, Jeffrey Glenn, Harry
Greenberg, Karla Kirkegaard, and Peter Sarnow, have all offered excellent advice and
support. My collaborators, Carol Jones, James Zehnder and Charles Grose, have all
contributed to my work in essential ways. I’d also like to thank people throughout the
Microbiology and Immunology and Pediatrics departments for their guidance in
navigating through Stanford: Nancy Greguras, Kelly Nelson, Bonda Lewis, Wanapa
Veeraprasit, Mary Jeanne Olivia, Julie Wong, Nancy Magee, and Mayumi Beppu.
A large amount of thanks goes out to my fellow classmates, especially the
thirteen others who started this adventure with me: Paul Bryson, Leremy Colf, Emily
v

Deal, Robin Deis Trujillo, Drew Hotson, Jon Jones, Josephine Lee, Gwen Liu, Jeff
Margolis, Reija Matheson, Kosta Pajcini, Poornima Parameswaran and Elizabeth
Ponder. These years would have been much harder without their support and
friendship, and I wish them all the best of luck as we move on from Stanford.
I’d also like to thank a special group of people who have been the best of
friends and who have kept me sane over the years. Gar Wilson, Jason Goldman-Hall,
Cera Renault, Neil and Susi Berrington, Emily Johnston and Matt Smith are all
wonderful people, and their friendship means a great deal to me.
My family has been wonderfully supportive and encouraging during this
experience. My in-laws, Pat and Bernice, have always let me know how proud they
are of me, which I appreciate so much. My sister Annie has become one of my best
friends as we’ve made the transition from kids at home to grownups with “real” lives.
She’s always full of support and encouragement for me. My grandparents Joe and
Nancy have also expressed so much love and pride in my accomplishments. Finally,
my parents, Carol and David, have provided the best possible support for my life in
science by being excellent role models themselves and by always encouraging me. I
thank them all for the unconditional love and support.
Finally, I want to thank the person who has come to mean the most to me, my
husband Jonathan. His love, friendship and support have carried me through so many
hard times. He is always there for me when I need him, never more so than during
these last several months, and I thank him from the bottom of my heart.
vi

TABLE OF CONTENTS
ABSTRACT .................................................................................................................. iv
ACKNOWLEDGEMENTS ...........................................................................................v
TABLE OF CONTENTS .............................................................................................vii
LIST OF ILLUSTRATIONS AND TABLES.............................................................xiii
CHAPTER I. A BRIEF OVERVIEW OF THE FAMILY HERPESVIRIDAE..............1
1.1. Herpesvirus taxonomy.........................................................................................2
1.2. Herpesvirus classification and biological properties...........................................3
1.2.1. Herpesviridae................................................................................................3
1.2.2. Alphaherpesvirinae.......................................................................................4
1.2.3. Betaherpesvirinae .........................................................................................5
1.2.4. Gammaherpesvirinae ....................................................................................6
1.3. Herpesvirus evolution..........................................................................................6
1.4. Human herpesviruses ..........................................................................................7
CHAPTER II. VARICELLA-ZOSTER VIRUS ..........................................................13
2.1. Varicella-zoster virus ........................................................................................14
2.2. VZV pathogenesis in the host ...........................................................................14
2.3. VZV epidemiology............................................................................................16
2.4. Varicella and zoster prevention and treatment ..................................................17
2.4.1. Varicella vaccine ........................................................................................17
2.4.2. Drug treatment............................................................................................17
2.4.3.Varicella prophylaxis...................................................................................18
2.5. Immune response to VZV .................................................................................19
2.6. VZV immunomodulation of the host response .................................................20
2.7. VZV virion ........................................................................................................22
2.7.1. Core and genome ........................................................................................22
vii

2.7.2. Capsid .........................................................................................................23
2.7.3. Tegument ....................................................................................................23
2.7.4. Envelope .....................................................................................................24
2.8. VZV replication cycle .......................................................................................24
2.8.1. Attachment and entry..................................................................................24
2.8.2. Genome replication and protein synthesis..................................................25
2.8.3. Assembly and egress ..................................................................................26
2.9. VZV studies in the SCID mouse model in vivo ................................................28
CHAPTER III. VARICELLA-ZOSTER VIRUS ORF37 GLYCOPROTEIN H ........31
3.1. Glycoprotein H ..................................................................................................32
3.2. VZV gH and gL processing and interaction......................................................32
3.3. gH and gL processing and interaction in other herpesviruses...........................34
3.3.1. Alphaherpesviruses.....................................................................................34
3.3.2. Gammaherpesviruses..................................................................................38
3.3.3. Betaherpesviruses .......................................................................................38
3.4. VZV gH function...............................................................................................39
3.4.1. Binding and entry .......................................................................................39
3.4.2. Fusion .........................................................................................................39
3.5. gH and gL function in other herpesviruses .......................................................42
3.5.1. Binding .......................................................................................................42
3.5.2. Entry ...........................................................................................................43
3.5.3. Signaling cascades associated with gH ligand binding and virus entry .....45
3.5.4. Fusion .........................................................................................................47
3.5.5. Capsid egress from the nucleus ..................................................................52
3.5.6. Virion assembly..........................................................................................53
3.6. gH and immunity...............................................................................................54
viii

CHAPTER IV. ANTI-GLYCOPROTEIN H ANTIBODY IMPAIRS THE PATHOGENICITY OF VARICELLA-ZOSTER VIRUS IN SKIN XENOGRAFTS IN THE SCID MOUSE MODEL.................................................................................58
4.1. Abstract .............................................................................................................59
4.2. Introduction .......................................................................................................60
4.3. Materials and Methods ......................................................................................63
4.3.1. Cells and virus ............................................................................................63
4.3.2. Preparation, inoculation and harvest of skin xenografts in SCID mice......63
4.3.3. Treatment of SCIDhu mice with anti-gH antibody ....................................64
4.3.4. Infectious plaque assay...............................................................................64
4.3.5. Plaque neutralization assay.........................................................................65
4.3.6. ELISA.........................................................................................................65
4.3.7. Immunohistochemistry of skin xenograft sections.....................................66
4.3.8. Quantitative PCR........................................................................................66
4.3.9. VZV DNA in situ hybridization .................................................................66
4.3.10. Antibody treatment of pOka infected HELF cultures in vitro..................67
4.3.11. Electron microscopy .................................................................................67
4.3.12. Confocal microscopy................................................................................68
4.4. Results ...............................................................................................................70
4.4.1. Treatment with the mAb 206 at 6 hpi significantly reduced the number of skin xenografts infected with VZV ......................................................................70
4.4.2. VZV titer was significantly reduced following treatment with the anti-gH mAb 206 ...............................................................................................................70
4.4.3. VZV genome copies in skin xenografts were reduced following treatment with the anti-gH mAb 206 ....................................................................................73
4.4.4. Kinetics of anti-gH mAb 206 accumulation and clearance........................74
4.4.5. VZV lesion spread in skin xenografts was reduced following treatment with the anti-gH mAb 206 ....................................................................................75
ix

4.4.6. Spread and replication of VZV was decreased in vitro after treatment with the anti-gH mAb 206 ............................................................................................76
4.4.7. Anti-gH mAb 206 localized to intracellular vacuoles, virus particles on the cell surface and virus particles within infected cells ............................................78
4.4.8. mAb 206 was not trafficked to the TGN but colocalized with the endocytic pathway.................................................................................................................79
4.5. Discussion .........................................................................................................81
CHAPTER V. VZV GLYCOPROTEIN H AND L: ANALYSIS OF PROTEIN CONSERVATION AND PREDICTED FUNCTIONAL MOTIFS ............................96
5.1. Introduction .......................................................................................................97
5.2. Materials and Methods ......................................................................................98
5.2.1. Sequences ...................................................................................................98
5.2.2. Sequence conservation analysis..................................................................98
5.2.3. Prediction algorithms to identify functional motifs....................................98
5.3. Glycoprotein H protein sequence conservation...............................................102
5.3.1. Conservation of VZV gH protein sequence compared to gH sequences from the human herpesviruses and from the subfamily Alphaherpesvirinae .....102
5.3.2. Conservation of the gH protein sequence among sequenced VZV clinical and laboratory isolates ........................................................................................104
5.4. Prediction of functional motifs in VZV gH.....................................................105
5.4.1. Signal sequence and transmembrane domain...........................................105
5.4.2. Glycosylation............................................................................................106
5.4.3. Phosphorylation ........................................................................................107
5.4.4. Heptad repeats and fusion peptides ..........................................................108
5.4.5. YXXφ motif..............................................................................................110
5.5. Glycoprotein L protein sequence conservation ...............................................113
5.5.1. Conservation of VZV gL protein sequence compared to gL sequences from the human herpesviruses and from the subfamily Alphaherpesvirinae..............113
5.5.2. Conservation of the VZV gL protein sequence among sequenced VZV clinical and laboratory isolates ...........................................................................114
x

5.6. Prediction of functional motifs in VZV gL .....................................................116
5.6.1. Signal sequence and transmembrane domain...........................................116
5.6.2. Glycosylation............................................................................................117
5.6.3. Phosphorylation ........................................................................................117
5.6.4. YXXφ motif..............................................................................................117
5.6.5. Highly conserved residues........................................................................117
�5.6.6. gL residues possibly involved in the interaction with gH .....................118
CHAPTER VI. MUTATIONAL ANALYSIS OF STRUCTURAL AND FUNCTIONAL MOTIFS OF VARICELLA-ZOSTER VIRUS GLYCOPROTEIN H....................................................................................................................................152
6.1. Abstract ...........................................................................................................153
6.2. Introduction .....................................................................................................155
6.3. Materials and Methods ....................................................................................159
6.3.1. Analysis of potential functional motifs ....................................................159
6.3.2. Cells ..........................................................................................................160
6.3.3. Construction of pOka cosmids with mutations in ORF37[gH] ................161
6.3.4. Construction of pOka-DX bacterial artificial chromosomes (BACs) with mutations in ORF371[gH] ..................................................................................162
6.3.5. Repair of pOka-BACs with mutations in ORF371[gH] ...........................163
6.3.6. Transfection, virus isolation and DNA extraction....................................164
6.3.7. Excision of the MiniF- vector from the pOka BACs as determined by PCR............................................................................................................................164
6.3.8. Construction of expression plasmids containing ORF37 and ORF60......165
6.3.9. Transfection of expression plasmids ........................................................167
6.3.10. Virus titration and replication kinetics ...................................................168
6.3.11. Antibody treatment of pOka infected HELF cultures in vitro................168
6.3.12. Confocal microscopy of transfected and infected cells ..........................169
6.3.13. Preparation of cell lysates from transfected or infected cells for immunoprecipitation and Western blot analysis of gH ......................................170
xi

6.3.14. Protein identification by mass spectrometry ..........................................171
6.3.15. Replication of gH mutant viruses in skin xenografts in SCID mice ......172
6.4. Results .............................................................................................................173
6.4.1. Targeted disruption of predicted structural and functional domains in VZV gH .......................................................................................................................173
6.4.2. Deletion of ORF37[gH] or mutation of HR2, HR3, αH1, αH2 or FPNG are lethal ...................................................................................................................174
6.4.3. Point substitutions and the HR1 mutation alter the gH glycosylation state and secondary structure ......................................................................................175
6.4.4. Point substitutions and the HR1 mutation do not disrupt gH localization in vitro.....................................................................................................................178
6.4.5. Neutralization of VZV spread by the SG3 antibody ................................179
6.4.6. Point substitutions and the HR1 mutation do not affect virus replication kinetics in melanoma cells in vitro .....................................................................180
6.4.7. gH-S47A, gH-T751A and gH-HR1 reduce VZV virulence in skin xenografts in vivo ...............................................................................................181
6.4.8.gH-C540A, gH-C575A, gH-HR2, gH-HR3, gH-αH1, gH-αH2 and gH-FPNG mutations disrupt gH maturation.............................................................182
6.4.9. gH-C540A, gH-C575A, gH-HR2, gH-HR3, gH-αH1, gH-αH2 and gH-FPNG mutations are expressed but are not transported to the cell surface ........184
6.5. Discussion .......................................................................................................186
Chapter VII. SUMMARY ..........................................................................................212
Chapter VIII. REFERENCES ....................................................................................220
xii

LIST OF ILLUSTRATIONS AND TABLES
Figure 1.1. Six classes of genome organization in the family Herpesviridae ..............10
Table 1.1. The human herpesviruses ............................................................................12
Figure 2.1. VZV-induced syncytia formation in vitro and in vivo ...............................30
Table 3.1. Compilation of published mutations made in VZV gH and gL ..................57
Figure 4.1. VZV infection of human skin xenografts in SCIDhu mice treated with anti-gH mAb 206 for 0-12 days or 4-12 days post inoculation ...........................................87
Figure 4.2. Replication of VZV in human skin xenografts in SCIDhu mice treated with anti-gH mAb 206 for 0-12 days or 4-12 days post inoculation....................................88
Figure 4.3. The formation of lesions in VZV-infected human skin xenografts treated with the anti-gH mAb 206 for 0-12 days or 4-12 days post inoculation......................89
Figure 4.4. Cell-cell spread of VZV in vitro in the presence of the anti-gH mAb 206 90
Figure 4.5. VZV replication in vitro in the presence of the anti-gH mAb 206 ............91
Figure 4.6. Virus particle formation and the localization of mAb 206 in infected cells in vitro ..........................................................................................................................92
Figure 4.7. Localization of gH and mAb 206 relative to VZV gE and ORF23 proteins in fibroblasts in vitro ....................................................................................................93
Figure 4.8. Localization of gH and mAb 206 relative to EEA1 and Vps4 in fibroblasts in vitro ..........................................................................................................................94
Figure 4.9. A schematic of mechanisms for antibody disruption of gH function and trafficking and potential effects on the pathogenesis of VZV skin infection in vivo ...95
Table 5.1. gH sequence similarity among the human herpesviruses..........................120
Table 5.2. gH sequence identity among the human herpesviruses.............................121
Figure 5.1. Alignment of human herpesvirus gH sequences......................................123
Table 5.3. gH sequence similarity among the Alphaherpesvirinae............................124
Table 5.4. gH sequence identity among the Alphaherpesvirinae ...............................126
Figure 5.2. Alignment of alphaherpesvirus gH sequences .........................................130
Table 5.5. Conservation of gH between VZV laboratory and clinical isolates ..........131
Table 5.6. Predicted signal peptide and transmembrane regions in gH .....................133
xiii

xiv
Table 5.7. Predicted glycosylation sites in VZV gH ..................................................134
Table 5.8. Predicted phosphorylation sites in gH.......................................................135
Table 5.9. gL sequence similarity among the human herpesviruses ..........................138
Table 5.10. gL sequence identity among the human herpesviruses ...........................139
Figure 5.3. Alignment of human herpesvirus gL sequences ......................................140
Table 5.11. gL sequence similarity among the Alphaherpesvirinae ..........................141
Table 5.12. gL sequence identity among the Alphaherpesvirinae .............................143
Figure 5.4. Alignment of alphaherpesvirus gL sequences .........................................145
Table 5.13. Conservation of gL between VZV laboratory and clinical isolates ........147
Table 5.14. Predicted glycosylation sites in VZV gL ................................................149
Table 5.15. Predicted phosphorylation sites in gL .....................................................150
Table 6.1. Primers used for the mutagenesis of ORF37.............................................197
Figure 6.1. Predicted structural and functional motifs in VZV gH and substitutions used to disrupt them....................................................................................................201
Figure 6.2. Analysis of gH maturation and virus protein in lysates from infected melanoma cells ...........................................................................................................202
Figure 6.3. gH localization and syncytia formation in melanoma cells .....................204
Figure 6.4. Inhibition of pOka spread by anti-gH mAb 206 and SG3 .......................206
Figure 6.5. pOka and mutant gH virus replication kinetics in melanoma cells in vitro....................................................................................................................................207
Table 6.2. Wild type and mutant gH virus replication kinetics in skin xenografts in vivo .............................................................................................................................208
Figure 6.6. Analysis of gH maturation from lysates of transfected HEK-293 cells...209
Figure 6.7. Localization of transiently expressed gH in HEK-293 cells....................210
Table 7.1. Summary of gH mutations and the resulting phenotypes..........................218

CHAPTER I
A BRIEF OVERVIEW OF THE FAMILY HERPESVIRIDAE
1

1.1. Herpesvirus taxonomy
Herpesviruses infect nearly all animals, and the order Herpesvirales was
created by the International Committee on Taxonomy of Viruses (ICTV) to encompass
the diversity found among these viruses (59). This order is separated into three
families. The family Herpesviridae contains the mammal, bird and reptile viruses. The
Alloherpesviridae family contains the fish and frog viruses, while the
Malacoherpesviridae family includes the bivalve viruses.
Within the Herpesviridae family, three subfamilies exist (39). These are the
Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae. The
Alphaherpesvirus subfamily is further divided into the genera Simplexvirus,
Varicellovirus, Mardivirus, and Iltovirus. As of 2002, the Simplexvirus genus
contained ten viruses, including two that infect humans: Human Herpesvirus 1, or
Herpes simplex virus 1 (HHV1/HSV1) and HHV2, or HSV2. The Varicellovirus
genus contains seventeen viruses, including HHV3, or Varicella-zoster virus (VZV).
One additional virus is tentatively classified as a varicellovirus. The Mardivirus genus
includes three viruses and the Iltovirus genus includes one virus. Neither of these
genera currently includes a herpesvirus that infects humans. One virus in the
Alphaherpesvirus subfamily is not yet classified at the genus level.
The Betaherpesvirus subfamily contains three genera (39). The
Cytomegalovirus genus contains four viruses, including HHV5, or human
cytomegalovirus (HCMV). Two additional viruses are tentatively classified in this
genus. The Muromegalovirus genus contains two viruses. The Roseolovirus genus
2

contains two viruses, HHV6 and HHV7. The Betaherpesvirus subfamily has two
additional virus members that have not been classified at the genus level.
The Gammaherpesvirus subfamily contains the Lymphocryptovirus and
Rhadinovirus genera (39). The Lymphocryptovirus genus contains eight viruses,
including HHV4, or Epstein-Barr virus (EBV). The Rhadinovirus genus contains
fourteen viruses, plus an additional four that are tentatively classified in this genus. It
includes HHV8, or Kaposi’s sarcoma-associated herpesvirus (KSHV). One virus is
classified as a Gammaherpesvirus with no additional genus classification.
The Herpesviridae family contains one additional unassigned subfamily with
one virus member, the unassigned genus Ictalurivirus with one member, and fifty-one
viruses that do not have a subfamily or genus classification (39). Most of these
unclassified viruses infect reptiles.
1.2. Herpesvirus classification and biological properties
1.2.1. Herpesviridae
Herpesvirus virions are composed of a core, a capsid, a tegument and an
envelope (38). The herpesvirus genome is a linear, double-stranded DNA genome.
Genomes in this family are 120,000-220,000 nucleotides (nt) long and have a guanine
+ cytosine (G+C) content of 35-75%. The genome encodes structural and non-
structural proteins that are located in the capsid, tegument and envelope. The core is
constructed of a filament spool that the genome is wrapped around. The ends of these
fibers are anchored to the capsid. The capsid is round with icosahedral symmetry, and
consists of 162 capsomeres. Herpesvirus capsids have a diameter of 100-110
3

nanometers (nm). Capsids can be penetrated by stain and might appear dark at the
center. The tegument layer does not display any structure. Proteins within the
tegument are present in variable amounts, and might be arranged asymmetrically
between the capsid and the envelope. The envelope contains distinct surface
projections, or spikes, that evenly cover the surface of the virus particle. The virion
envelope contains lipids of host origin, which are derived from nuclear or host cell
membranes. The exact composition of these lipids is not presently known. Virions are
120-200 nm in diameter and have a buoyant density in cesium chloride of 1.15-1.29
grams per centimeter3 (g/cm3). Incomplete particles lacking a capsid shell and DNA
core are common.
There are four biological properties that are common to all herpesviruses
studied to date (190). First, they all encode an array of enzymes involved in nucleic
acid metabolism, DNA synthesis and processing. Second, during the virus replication
cycle, the synthesis of viral DNA and capsid assembly takes place in the nucleus,
while the final processing of the virion takes place in the cytoplasm. Third, production
of infectious virus progeny results in the destruction of the infected cell. Last, all
herpesviruses are able to establish latency in their natural host.
1.2.2. Alphaherpesvirinae
The Alphaherpesviruses have a buoyant density of 1.23 to 1.29 g/cm3 in
cesium chloride (33). The genome is 120,000-180,000 nt, with a G+C content of 67-
68%. The genome contains terminally redundant sequences that are reiterated
internally in inverted form (Figure 1.1). Two or four isomeric forms of the genome
4

exist, and multiple isomeric forms can be packaged into one virion. The
Simplexviruses have a buoyant density of 1.27 g/cm3 and a 152,000 nt genome (30).
The Varicelloviruses have a buoyant density of 1.27-1.29 g/cm3 and a 125,000 nt
genome (32). Two isomeric forms of the Varicellovirus genome predominate.
Alphaherpesviruses have a variable host range and a relatively short replication
cycle (18-20 hours for HSV) (190, 203). They rapidly spread in culture and efficiently
destroy the cells that become infected. They establish latency predominantly in the
sensory ganglia.
1.2.3. Betaherpesvirinae
The Betaherpesviruses have a 180,000-250,000 nt genome with a G+C content
of 56% (36). The genome has terminally redundant sequences that are repeated
internally (Figure 1.1). The Cytomegaloviruses have a 200,000 nt genome (34). The
Roseoloviruses have a buoyant density of 1.273 g/cm3 and a 200,000 nt genome (35).
Betaherpesvirus have a restricted host range (163, 190). They have a long
replication cycle (48 to 72 hours for HCMV) and a slow progression of infection in
vitro. A characteristic of infected cells is cytomegalia, or enlargement of infected cells.
The virus establishes latency in secretory glands, lymphoreticular cells, kidneys and
other tissues.
5

1.2.4. Gammaherpesvirinae
The Gammaherpesviruses have a 170,000 nt genome with a 56% G+C content
(37). The genome has terminally redundant sequences that are repeated (Figure 1.1).
No buoyant density for the Gammaherpesviruses is available from the ICTV database.
Gammaherpesvirus host range is restricted to the family or order to which the
natural host belongs (190). These viruses replicate in vitro in lymphoblastoid cells,
and some replicate in epithelial or fibroblast cells. Virus infection is usually specific
for T or B lymphocytes, and latency is established in the lymphoid tissue.
1.3. Herpesvirus evolution
Phylogenetic analysis of common genes from viruses within the Herpesviridae
family has suggested that the three subfamilies arose 220-180 million years before
present, with the tree root originating between the alphaherpesviruses and the
precursor to the beta- and gammaherpesviruses (159). Phylogenetic studies have
suggested that the major sublineages within these subfamilies arose 80-60 million
years before present, and speciation of the mammalian alphaherpesvirus sublineages
occurred within the last 80 million years. This would place the speciation event around
the same time of the great radiation of placental mammals, which is proposed to have
taken place 80-60 million years before present (158). Based on this time frame and
phylogenetic analysis showing that branching patterns of herpesviruses mimic the
branching patterns of their hosts on the mammalian tree, it has been hypothesized that
herpesviruses coevolved with their hosts.
6

1.4. Human herpesviruses
Eight herpesviruses are known to infect humans (Table 1.1) (190, 237). Herpes
simplex virus 1 and 2 (HSV1 and -2) are the causative agents of oral-labial and genital
herpes, respectively (203). HSV1 is the most intensely studied herpesvirus. HSV
infection general occurs when an infected individual has close, personal interaction
with an uninfected individual and the virus comes into contact with mucosal surfaces
or abraded skin. HSV1 infection generally occurs in the oropharynx, and latency is
established in the trigeminal ganglion. HSV2 infection can occur following genital
contact and the virus establishes latency in the sacral ganglia. HSV can infect the
central nervous system (CNS) and cause severe neurological damage, such as HSV
encephalitis, or it can establish latency. Reactivation leads to lesions on the skin and
mucocutaneous sites.
Varicella-zoster virus (VZV) is the causative agent of varicella (chicken pox)
and herpes zoster (shingles) (52). It is most closely related to HSV among the human
herpesviruses. VZV infects epithelial cells, T lymphocytes and the dorsal root ganglia.
Varicella is characterized by fever and a vesicular rash, and epidemics among
susceptible individuals commonly occur in winter and spring. Herpes zoster is also
characterized by a vesicular rash, which can be accompanied by severe pain. VZV is
transmitted by contact with fluids from vesicular lesions or by inhalation of
aerosolized particles.
Epstein-Barr virus (EBV) causes infectious mononucleosis, and is also an
oncolytic virus associated with Burkitt’s lymphoma and nasopharyngeal carcinoma, as
well as some sarcomas (133, 200). EBV is spread orally and is widespread in all
7

human populations. Primary infection in young children is usually asymptomatic, but
primary infection during adolescence or early adulthood might result in infectious
mononucleosis. EBV replicates in epithelial cells, but latency is established in B
lymphocytes. In vitro, EBV infection of B lymphocytes can transform cells into
lymphoblasts capable of long-term proliferation. EBV has a large, conserved domain
that is homologous to VZV open reading frames (ORFs) 28 to 59, which includes
several enzymes necessary for DNA replication, the thymidine kinase gene, and
glycoproteins B (gB) and gH.
Human cytomegalovirus (HCMV) is found worldwide, and is commonly
associated with opportunistic disease in immunocompromised patients (163).
Transmission in utero to the fetus can cause congenital birth defects. HCMV is
transmitted via infected bodily secretions and infects macrophages, dendritic cells,
endothelial cells and epithelial cells. Fibroblasts are commonly used to study the virus
in vitro, and lab passaged strains are typically noninfectious in primary cells due to
accumulated mutations. An example of this is the loss of the genes encoding UL129-
131, which encode protein products that associate with glycoprotein H (gH) and are
required for entry into epithelial and endothelial cells.
Human herpes virus 6 and -7 (HHV6 and -7) are ubiquitous throughout adult
populations (245). Transmission routes are not well understood, but are thought to be
horizontal from individuals in close contact. HHV6B and HHV7 cause exanthem
subitum, or childhood roseola infantum, characterized by a febrile rash, while HHV6A
has not been clearly associated with a particular disease to date. HHV6 reactivation
can be a risk following bone marrow or solid organ transplantation. It might be
8

associated with neoplasia or lymphoproliferative disorders such as acquired
immunodeficiency syndrome (AIDS). It has also been suggested that it is associated
with multiple sclerosis. HHV6 infects T lymphocytes, natural killer cells, dendritic
cells and peripheral blood mononuclear cells (PBMCs). HHV7 is more restricted,
infecting PBMCs or T lymphocytes.
Kaposi’s sarcoma-associated herpesvirus (KSHV) is the most recently
discovered human herpesvirus, and is the causative agent of Kaposi’s sarcoma (KS)
(90). KS is characterized as an angioproliferative and inflammatory lesion, and is the
most common neoplasm complication in AIDS patients. KSHV is also linked to
primary effusion lymphoma, commonly found in end-stage AIDS patients, and
Castleman’s disease, both of which are proliferative disorders associated with B
lymphocytes. In Western Europe and the United States, KSHV appears to spread
predominantly by sexual transmission, and prevalence is very low. In the
Mediterranean and Africa, where prevalence is higher, transmission might occur
vertically from infected parents, or might occur horizontally during childhood.
Salivary exchange is thought to account for most of this transmission.
9

Figure 1.1. Six classes of genome organization in the family Herpesviridae
(Adapted from Pellet and Roizman, 2007.)
Genomes in the family Herpesviridae are grouped into six classes (190). Class A
genomes have left and right terminal repeats (LTR and RTR). HHV6
(Gammaherpesvirus, Roseolovirus) is the typical example of this genome
organization. Class B genomes have terminal sequences that are repeated numerous
times at each terminus of the genome. Saimiriine herpesvirus (SaHV)
(Gammaherpesvirus, Rhadinovirus) is the typical example of this class. Class C
genomes have terminal repeat sequences and four internal repeats (R1-R4). EBV
(Gammaherpesvirus, Lymphocryotovirus) is the classic example of this genome type.
Class D genomes have terminal and internal repeats (TRL and IRL) flanking a unique
long (UL) region and internal and terminal repeats (IRS and TRS) flanking a unique
short (US) region. The US inverts with respect to the UL while the UL rarely inverts,
10

resulting in two isomers. VZV (Alphaherpesvirus, Varicellovirus) represents a typical
genome in this class. Class E genomes have inverted repeat regions (a, b and b’, a’)
flanking a UL region and inverted repeat regions (c and c’) flanking a US region. The
“a” repeat regions can be present in numerous copies. Both the short and long
components can invert relative to one another, resulting in four DNA isomers. HSV
(Alphaherpesvirus, Simplexvirus) is the typical example of this genome structure.
Class F genomes have not had terminal repeat regions identified. Tupaia herpesvirus
Betaherpesvirus) is an example of this genome class. (
11
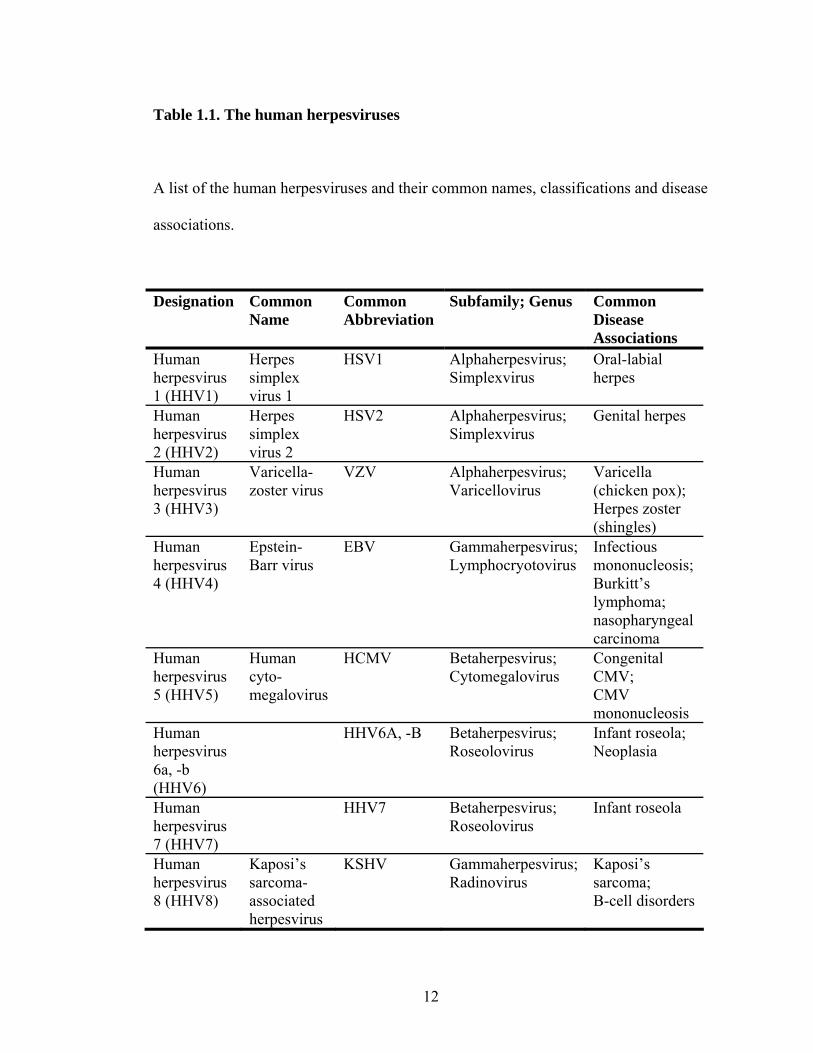
Table 1.1. The human herpesviruses
A list of the human herpesviruses and their common names, classifications and disease
associations.
Designation Common Name
Common Abbreviation
Subfamily; Genus Common Disease Associations
Human herpesvirus 1 (HHV1)
Herpes simplex virus 1
HSV1 Alphaherpesvirus; Simplexvirus
Oral-labial herpes
Human herpesvirus 2 (HHV2)
Herpes simplex virus 2
HSV2 Alphaherpesvirus; Simplexvirus
Genital herpes
Human herpesvirus 3 (HHV3)
Varicella-zoster virus
VZV Alphaherpesvirus; Varicellovirus
Varicella (chicken pox); Herpes zoster (shingles)
Human herpesvirus 4 (HHV4)
Epstein-Barr virus
EBV Gammaherpesvirus; Lymphocryotovirus
Infectious mononucleosis; Burkitt’s lymphoma; nasopharyngeal carcinoma
Human herpesvirus 5 (HHV5)
Human cyto-megalovirus
HCMV Betaherpesvirus; Cytomegalovirus
Congenital CMV; CMV mononucleosis
Human herpesvirus 6a, -b (HHV6)
HHV6A, -B Betaherpesvirus; Roseolovirus
Infant roseola; Neoplasia
Human herpesvirus 7 (HHV7)
HHV7 Betaherpesvirus; Roseolovirus
Infant roseola
Human herpesvirus 8 (HHV8)
Kaposi’s sarcoma-associated herpesvirus
KSHV Gammaherpesvirus; Radinovirus
Kaposi’s sarcoma; B-cell disorders
12

CHAPTER II
VARICELLA-ZOSTER VIRUS
13

2.1. Varicella-zoster virus
Varicella-zoster virus (VZV) is a Varicellovirus in the subfamily
Alphaherpesvirinae (32). VZV is most closely related to Cercopithecine herpesvirus 9
(CeHV9; also called Simian Varicella virus or SVV) (101). It is also closely related to
Equid herpesvirus 1 (EHV1) and Suid herpesvirus 1 (SHV1; also called Pseudorabies
virus or PRV), all within the Varicellovirus genus. No other Varicelloviruses infect
humans. Among the human herpesviruses, VZV is most closely related to Herpes
simplex virus 1 and 2 (HSV1 and -2), which are also Alphaherpesviruses.
2.2. VZV pathogenesis in the host
VZV transmission is thought to occur via inhalation of aerosolized respiratory
droplets or through contact with vesicular fluid from skin lesions on an infected
individual (102, 156). Inoculation occurs in the upper respiratory tract or in the
conjunctiva. The average incubation period from exposure to the development of
varicella rash is 14-16 days, with a range of 10-21 days. VZV is thought to spread
from the respiratory tract to the regional lymph nodes, and then from these sites to
cutaneous epithelial cells. Infected tonsil T cells expressing skin homing markers are
infected by the virus, and this might be the mode of transmission from the regional
lymph nodes to the skin (145). Alternatively, based on the identification of viral
proteins in capillary endothelial cells, virus from infected T cells might infect these
cells and then spread to adjacent epithelial cells (176). In the skin, infection causes
vasculitis followed by the formation of multinucleated epithelial cells and the
destruction of the epidermal basement membrane (227). This results in the typical
14

legions seen in varicella rash. VZV is released from the host and transmitted to the
next individual in the vesicular fluid from these lesions or from upper respiratory
secretions that become aerosolized.
VZV has a tropism for T cells and skin epithelial cells during primary
infection, but it might also spread to other tissues if replication is not controlled by the
host immune response (52). Virus can spread to the lungs, liver, central nervous
system (CNS), as well as other tissues or organs. Infection of the epithelial cells in the
pulmonary alveoli can result in varicella pneumonia. CNS infection can result in
meningoencephalitis, most commonly seen in immunocompromised patients with
prolonged T cell-associated viremia. Adrenal infection, which is common in fatal,
disseminated varicella, might result from infection of the adrenal glands or the
kidneys.
VZV can spread to the sensory ganglia, possibly hematogenously or by
neuronal axon infection at the site of mucocutaneous lesions followed by anterograde
neuronal transport (52, 56, 109). The virus establishes latency in these neurons.
Clinical reactivation presents as herpes zoster, a vesicular rash with lesions similar to
those during primary varicella. Unlike the disseminated rash typical in varicella, the
zoster rash is usually confined to a single sensory dermatome (116). This rash might
be more extensive in immunocompromised patients, and reactivation might also result
in cell-associated viremia, with the accompanying potential for dissemination to the
various organs and tissues as described for primary varicella (73).
15

2.3. VZV epidemiology
VZV is a ubiquitous human pathogen with worldwide distribution. The
majority of people are infected before or during adolescence in temperate climates or
by adulthood in tropical climates (156). Before the vaccine was introduced in 1995,
90-92% of cases in the United States occurred in patients less than 15 years of age.
Approximately 4 million cases were reported each year, which equaled the birthrate at
the time. The majority of hospitalizations relating to varicella occurred in children less
than one year of age or persons older than 20 years of age (105). Serious
complications were least common in patients 1-9 years of age, and the lowest fatality
rate was in patients 1-4 years of age. Between 1970 and 1994, the average annual
mortality rate attributed to varicella was 0.4 deaths per 1 million people. The most
frequent varicella complications resulted from secondary bacterial infections, varicella
pneumonia or neurological symptoms (76). In a study of children with cancer, patients
not treated with antiviral drugs had a mortality rate of 7% (74). Varicella pneumonia
developed in 28% of patients, and was associated with a 25% mortality rate.
The herpes zoster risk is estimated at 3.2-4.2 cases per 1000 people per year,
causing approximately 1 million cases annually, but risk increases to 10 cases per
1000 people aged 60+ years (111, 116). The lifetime risk of people who live to 85
years of age is estimated at 50%. The most common complication in herpes zoster is
post-herpetic neuralgia (PHN), characterized by severe pain. PHN increases in
frequency with age, from 3-4% in patients age 30-49 up to 34% in patients more than
80 years of age (7, 111, 116, 242).
16

There is a higher risk of zoster in immunosuppressed patients than in
immunocompetent patients (52). The incidence of zoster following a bone marrow
transplant is 25%, and 15% within five years of a renal transplant. Zoster is also more
common in patients with leukemia, Hodgkin’s or non-Hodgkin’s lymphoma or small
cell carcinoma of the lung. In patients infected with the human immunodeficiency
virus (HIV), the incidence of herpes zoster is 15-25 times higher than in the general
population (29, 97).
2.4. Varicella and zoster prevention and treatment
2.4.1. Varicella vaccine
The VARIVAX® varicella vaccine was created as a live attenuated virus
vaccine made from the Oka strain of the virus (108, 144, 156, 220, 222). It was
licensed in 1995 for use in the United States in healthy people over the age of 12
months. Following the introduction of the vaccine in the United States, surveillance in
three locations by the Centers for Disease Control indicated that cases of varicella had
declined 80+%, with similar declines in hospitalizations. By 1999-2001, the mortality
rate associated with varicella had dropped to <0.15 deaths per 1 million people.
2.4.2. Drug treatment
Acyclovir, a synthetic nucleoside analog that inhibits human herpesvirus
replication, is used to treat varicella (52, 156). Administered within 24 hours of the
onset of rash, acyclovir can diminish clinical severity of varicella in
immunocompromised patients, reducing dissemination and cutaneous disease, thereby
17

limiting the risk of secondary bacterial infection (196). It is also used to treat patients
experiencing varicella pneumonia. The Food and Drug Administration (FDA) has
approved oral acyclovir for treatment of otherwise healthy patients within 24 hours of
the onset of rash, which can reduce the duration of fever and the appearance of new
lesions (156). It has not been determined if acyclovir treatment can reduce the
incidence of complications associated with varicella. The American Academy of
Pediatricians, however, does not consider oral acyclovir to have sufficient benefits for
routine administration, and instead only recommends treatment of patients with a high
risk of severe varicella.
In cases of herpes zoster, especially in immunocompromised patients,
acyclovir administered within 72 h of virus reactivation can reduce the severity of
disease (111). The FDA has also approved famciclovir and valacyclovir, two other
nucleoside analogs, for the treatment of herpes zoster. These drugs have been
demonstrated to reduce the amount of viral shedding and lesion formation, the time to
rash healing, and the severity and duration of pain associated with herpes zoster. They
also might reduce the risk of PHN. PHN can be treated with corticosteroids, which
reduce pain and time to rash healing, in combination with the nucleoside analog drugs.
2.4.3.Varicella prophylaxis
Acyclovir has not been shown to be an effective prophylactic in
immunocompromised people who are exposed to VZV, and it is recommended that
these patients be treated with varicella-zoster immunoglobulin (VZIG) (52, 156).
VZIG administered within 96 h after exposure, preferably within 48 h, can reduce
18

VZV morbidity and mortality during initial infection (248). However, administration
of VZIG following the appearance of a rash is not effective at modulating the disease
course. VZIG is also recommended for use in treating infants whose mothers
developed varicella around the time of birth (156, 161). If the mother develops
varicella rash more than five days before giving birth, antibodies acquired
transplacentally can protect infants from varicella, but if the mother developed
varicella five days before to two days after, VZIG treatment is recommended. VZIG
does not always prevent infection in these infants, but it does limit the morbidity and
mortality associated with VZV infection.
2.5. Immune response to VZV
The innate immune response to VZV consists of upregulation of the interferon
(IFN) and nuclear factor kappa B (NF-κB) signaling pathways in epithelial cells (126,
147). Natural killer (NK) cells are capable of lysing VZV-infected cells and of
producing granulysin, which enhances infected cell death in vitro and inactivates
intracellular virus (112). These systemic and local innate immune responses are
thought to be capable of controlling virus spread and replication until the adaptive
immune response is activated.
IgG, IgM and IgA antibodies to VZV can usually be detected within three days
of the appearance of varicella rash (24, 64). These antibodies are directed against
multiple viral proteins, including gB, gC, gE, gH, gI, immediately early protein 62
(IE62) and IE63, and antibodies to gB, gH, gE and gI neutralize the virus in the
presence or absence of complement. Monoclonal antibody to gH has been shown in
19

vitro to block viral replication and spread, and might have similar effects in an
infected host (53, 66, 202). Early detection of antibodies does not correlate with the
severity of disease, but early T cell response within 24-72 h of the appearance of a
rash does correlate with milder symptoms (10). Cellular immunity has been shown to
be important for terminating cell-associated viremia and replication in skin, and a lack
of VZV-specific T cell proliferation correlates with persistent viremia and
dissemination throughout the host (10). Memory immunity is thought to be required to
maintain the virus in a latent state or to prevent symptomatic reactivation. While no
correlation between decreasing titers of IgG antibodies and the occurrence of zoster
has been shown, increasing susceptibility to zoster in the elderly or immunosuppressed
does correlate with diminishing in vitro T cell proliferation in response to VZV
antigens.
2.6. VZV immunomodulation of the host response
VZV has a number of mechanisms by which it modulates the innate and
adaptive immune response. VZV downregulates major histocompatibility complex
(MHC) class I expression on human fibroblasts and T lymphocytes by interfering with
transport of MHC class I molecules through the Golgi and to the cell surface (1). This
might limit recognition by CD8+ T lymphocytes, impairing immunosurveillance
during viral dissemination to the skin via infected CD4+ T lymphocytes. It might also
provide a mechanism by which VZV evades immune surveillance during reactivation
and the establishment of herpes zoster.
20

During initial infection in skin, the IFN-α response to virus is inhibited in
infected skin cells but is upregulated in neighboring uninfected cells (147). The VZV-
induced block in infected cells is thought to allow the virus to replicate, while the
upregulated IFN-α in neighboring cells is thought to restrict cell-cell spread of the
virus. It has been suggested that the long incubation time of VZV (10-21 days) is the
time required for VZV to overcome this innate response and spread to form syncytia
that eventually result in lesions on the skin surface. VZV also prevents nuclear factor
kappa B (NF-κB) from translocating to the nucleus in infected cells (126).
VZV infection inhibits MHC class II expression in human fibroblasts and in
dermal and epidermal cells in human skin biopsies (2). VZV interferes with IFN-γ
dependent transcription of the MHC class II DR-α gene by reducing Jak2 and Stat1α
protein levels, which are required for signal transduction from the IFN-γ receptor, and
by reducing transcript levels of CIITA, a MHC class II transactivator, and interferon
response factor 1 (IRF-1). VZV-specific T cells appear 24 to 72 h after the appearance
of varicella lesions, and it has been suggested that VZV replication is required to
sensitize these T cells to respond to infection (2, 52). Since the CD4+ T cell response
is mainly Th-1 type and IFN-γ is the major cytokine produced in response to infection,
it has been proposed that the viral immunomodulatory effect on MHC class II results
in slower clonal expansion of VZV-specific CD4+ T cells (2, 125).
21

2.7. VZV virion
The VZV virion is composed of a core, nucleocapsid, tegument and envelope
(31). The virion is 120-200 nm in diameter, depending on the amount of tegument that
is incorporated.
2.7.1. Core and genome
The core, a fibrillar cage, contains one copy of a linear, double-stranded DNA
genome. The genome is approximately 125 kbp in length and contains both terminal
and internal repeat regions. Of the six possible genome arrangements in the
herpesvirus family, VZV exemplifies Class D genomes (see Chapter 1.2.2 and Figure
1.1) (52). The genome is organized into a unique long region (UL, 105 kbp) flanked
by terminal repeat long and internal repeat long regions (TRL and IRL, each 0.9 kbp),
and a unique short region (US, 5.2 kbp) flanked by an internal repeat short and a
terminal repeat short region (IRS and TRS, each 7.3 kbp). Both orientations of the US
region are commonly found, but one form of the UL region occurs much more
frequently, and thus two isomeric forms of the genome predominate. The genome
overall has a G+C content of 46%, but the repeat regions have significantly higher
G+C contents. The TRS and IRS regions have 59% G+C and the TRL and IRL have
68% G+C. The genome also contains five small repeat regions of varying length with
a high G+C content.
Seventy unique genes are encoded by the VZV genome, and three of these are
duplicated in the IRS and TRS regions of the genome (52, 157). Approximately 40
genes are common to all herpesviruses, including those that encode enzymes and
22

structural proteins. Most VZV genes have homologs in HSV, the most closely related
human herpesvirus, with the exception of open reading frames (ORFs) 1, 2, 13, 32, 57
and S/L. ORFs 1, 2, 32 and 57 have homologs in the varicellovirus EHV1, while ORF
13 has a homolog in the gammaherpesvirus Kaposi’s sarcoma-associated herpesvirus
(KSHV).
2.7.2. Capsid
The VZV genome and core are contained within an icosahedral capsid that is
indistinguishable from that of other herpesviruses (31). The icosahedral capsid is
composed of 162 capsomeres, and is 100-110 nm in diameter. ORF23, a conserved
capsid protein, is dispensable in vitro but required in vivo (47). It functions to transport
the major capsid protein, ORF 40, to the nucleus. Immunofluoresence studies with
antibodies to ORF 23 can be used to identify nucleocapsids within and on the nuclear
envelope (199). Few studies have been conducted on the VZV capsid proteins other
than ORF23, but based on homology, ORFs 20, 33, 33.5, 40 and 41 are also likely to
encode capsid proteins.
2.7.3. Tegument
The tegument, an amorphous protein structure between the capsid and the
envelope, contains multiple VZV proteins (31). The tegument structure is not clearly
defined, and proteins within this structure are present in variable amounts. They might
also be asymmetrically distributed between the capsid and the envelope. The tegument
includes proteins encoded by ORFs 4, 10, 47, 62, 63 and 66 (52). Studies in HSV have
23

indicated that tegument proteins interact with both capsid and envelope proteins,
thereby facilitating assembly of the enveloped virus particle, and similar interactions
might take place during VZV assembly (51, 69, 104, 129, 179).
2.7.4. Envelope
The virus envelope is derived from host cell membranes that appear to
originate from the rough endoplasmic reticulum (ER), cytoplasmic vesicles, or the cell
surface (31). Lipids present in the envelope originate in host nuclear or cell
membranes. Numerous viral glycoproteins are incorporated into the envelope and
appear as densely dispersed small spikes that cover the virion surface. VZV encodes
nine glycoproteins, named gB, gC, gE, gH, gL, gK, gI, gM and gN.
2.8. VZV replication cycle
2.8.1. Attachment and entry
The VZV replication cycle takes 9-12 hours (199). The first step in VZV
replication is attachment to the cell. VZV glycoprotein B (gB) can bind to heparan
sulfate proteoglycans to facilitate VZV attachment (123). VZV gE has been shown to
bind insulin-degrading enzyme (IDE), and it is possible that IDE might facilitate VZV
entry (149). VZV gB has been reported to bind to myelin-associated glycoprotein
(MAG), which might act as a receptor on neuronal cells (218).
Following attachment, VZV entry requires membrane fusion, either directly
between the virus envelope and the plasma membrane, or endocytosis followed by
fusion of the virus envelope with the endosomal membrane. The minimum
24

complement of glycoproteins required for fusion in most herpesviruses is gB, gH and
gL. Monoclonal antibodies to gH have been shown to inhibit VZV entry, cell-cell
spread and syncytia formation, and antibodies to gB, gE and gI also can neutralize
VZV infectivity, indicating that these glycoproteins play a role in virus entry and cell-
cell spread (53, 66, 202). Several VZV fusion studies have suggested gH and gL or gB
and gE can facilitate cell-cell fusion when expressed from a vaccinia virus or
transfected into cells infected with a vaccinia virus (66, 154). These results suggests
that VZV is the only herpesvirus to not require gB, gH and gL as a minimum
complement of glycoproteins necessary for fusion, but it is possible that the vaccinia
virus infection facilitates the induction of fusion by the VZV glycoproteins. Another
study has recently shown that in the presence of myelin associated glycoprotein
(MAG), gB, gH and gL are required for fusion, similar to results from studies of other
herpesviruses (218). Expression of gE on the cells used in the fusion assay decreased
the relative amount of fusion, suggesting that only VZV gB, gH and gL are needed for
efficient virus-induced cell-cell fusion.
2.8.2. Genome replication and protein synthesis
Following entry, the unenveloped capsid is transported to the nuclear surface
(52). Mechanisms of capsid transport and the release of its contents into the nucleus
are not yet understood. VZV gene synthesis is likely initiated by tegument proteins,
such as IE4 and IE62, which translocate to the nucleus. Protein synthesis of immediate
early genes, such as IE62, begins within one hour of infection (199). VZV ORFs 4, 61,
62 and 63 are identified as IE proteins, and they all serve to regulate transcription of
25

IE, early and late gene promoters during the cascade of virus transcription and
translation (61, 62, 77, 121, 171, 192).
VZV early genes encode a DNA polymerase and several DNA binding
proteins that function in genome replication (52). During replication, the linear
genome circularizes, aided by complimentary unpaired nucleotides at each 3’ end of
the genome (58, 216). Replication likely takes place by a rolling-circle mechanism,
and concatemers are cleaved to generate linear DNA genomes, which can be packaged
into nucleocapsids. DNA replication compartments form by 4 hours post infection
(hpi) (199).
VZV also encodes additional enzymes, such as a thymidine kinase and other
protein kinases that are involved in posttranslational modification of virus proteins
(52). Late genes include tegument proteins, such as ORF 10, and nucleocapsid
proteins, such as ORF 33.5, as well as the glycoproteins (52). However, recent
evidence has demonstrated that gE synthesis takes place around 4 hours post infection
(hpi), which might be earlier than the kinetics of other late gene products (199).
2.8.3. Assembly and egress
Although the replication kinetics for VZV have been studied, there is little
evidence for how VZV particles are assembled. Capsids form by 9 hpi, and mature
VZV particles can be detected by 9-12 hpi (199). The assembly process involves
packaging DNA into the nucleocapsid, transport of the capsid to the cytoplasm, and
assembly of the capsid, tegument and envelope layers. This appears to be an efficient
process in vivo based on high levels of infectivity associated with vesicle fluids and
26

aerosolized virus particles (52). However, in vitro, many particles lack a dense core
and do not appear to be fully assembled. Additionally, virions remain closely
associated with the cell from which they are produced.
Assembly might involve cellular pathways. The autophagy marker L3BC, an
isoform of the microtubule-associated protein 1 light chain 3 (LC3), is upregulated late
in VZV infection, and autophagosomes are detected in cultured cells and in skin
biopsies from human zoster vesicles (221). Vps4, an ATPase that functions in both the
autophagy pathway and the multivesicular body (MVB) pathway, interacts with HSV
gB, and these pathways are utilized for envelopment and egress of HSV (45, 189).
While the autophagy pathway might be involved in degradation of VZV particles that
accumulate in cytoplasmic vacuoles, it is possible that VZV might utilize components
of the autophagy and MVB pathways for envelopment and egress of virus particles.
VZV envelopment is thought to occur via primary envelopment,
deenvelopment, and reenvelopment, or secondary envelopment (91). Studies in two
closely related alphaherpesviruses, HSV and PRV, also support this model of
envelopment (212, 240). Primary envelopment of the capsid takes place as it cross the
inner nuclear membrane. This particle is deenveloped as it crosses into the cytoplasm
through the outer nuclear membrane or the rough endoplasmic reticulum (ER)
membrane. The viral glycoproteins are processed in the trans-Golgi network (TGN)
and expressed on the cell surface, then endocytosed back into the cell and targeted to
the TGN, where they cover cisternae. Tegument proteins interact with both the
glycoprotein tails that point into the cytosol and with the capsid proteins. The naked
nucleocapsid then invaginates into these cisternae, resulting in secondary envelopment
27

of the virion and localization within a Golgi-derived vesicle. These vesicles are next
directed to pre-lysosomes and either degraded further or directed to late endosomes
that can fuse with the cell membrane and release the enveloped virions.
Following egress, VZV remains highly cell-associated in vitro, and thus most
studies are done using cell-associated virus instead of cell-free virus. VZV is typically
propagated by transferring infected cells onto uninfected cells and allowing the virus
to spread. The virus can be propagated in various cell lines, such as the human
melanoma cell line Mel39, or in primary human cells, such as human embryonic lung
fibroblasts (HELF). A hallmark of VZV infection in vitro is the formation of syncytia,
or giant multinucleated cells (Figure 2.1A). It is generally accepted that this occurs via
mechanisms linked to cell-cell fusion, although little is known about the actual
mechanisms (53). VZV forms syncytia in skin as well as in cultured cells. The
extensive formation of multi-nucleated giant cells representing fusion of epidermal
cells is readily apparent in SCIDhu skin xenografts (Figure 2.1B) and in biopsies of
varicella and herpes zoster skin lesions during natural infection (164).
2.9. VZV studies in the SCID mouse model in vivo
VZV is infectious almost primarily in humans, and no adequate animal model
exists to study VZV. Studies in vivo have utilized the engraftment of human tissue into
severe combined immunodeficient (SCID) mice. The SCID model has been used to
study VZV infection of skin, T cells and ganglia in vivo in human xenografts. Human
fetal skin is implanted subcutaneously as full-thickness grafts, while T cells are
implanted as coimplants of human fetal thymus and liver tissue (termed the thy-liv
28

model) under the kidney capsule (164). Fetal dorsal root ganglia (DRG) are also
implanted under the kidney capsule (249). These xenografts are allowed to implant
and become vascularized, at which point they can be infected to study VZV
replication in various cell types. Since SCID mice are immunodeficient (27), this
model allows analysis of VZV replication in the absence of adaptive immunity.
Xenografts are generally inoculated using infected human fibroblasts. Infected
T cells injected into the tail vein of mice can also be used to inoculate skin xenografts.
Infection in skin xenografts generally peaks at 14-21 days post infection (dpi) as the
virus spreads from the epidermal layer through the basement membrane and into
dermal tissue (165). T cell infection in the thy-live model peaks at 14 dpi and the virus
infects both CD4+ and CD8+ T cells (164). In the DRG model, virus replicates in
neurons and satellite cells following inoculation, but around 56 dpi, persistence has
been shown to occur (249). The virus is considered to be in a persistent state in the
DRG if IE62 and IE63 but not gB transcripts are detected.
29

Figure 2.1. VZV-induced syncytia formation in vitro and in vivo
(A) Melanoma cells infected with pOka were fixed 48 hours post infection and
stained for virus gH (red) (mAb SG3 mouse anti-gH, Biodesign, Saco, ME), the
cellular protein trans-Golgi network 46 (green) (AHP500 polyclonal sheep anti-
TGN46, AbD Serotec, Oxford, UK) and nuclei (blue) (HOECHST 33342, Molecular
Probes, Carlsbad, CA). (B) A SCID skin xenograft infected with pOka was harvested
7 days post infection and stained for gE (red) (mAb 8612 mouse anti-gE, Millipore,
Temecula, CA) and counterstained with hematoxylin (blue) to show nuclei.
Magnification, (A) 40X and (B) 100X.
30

CHAPTER III
VARICELLA-ZOSTER VIRUS ORF 37
GLYCOPROTEIN H
31

3.1. Glycoprotein H
Glycoprotein H (gH) is the second most conserved glycoprotein in
herpesviruses. In all herpesviruses in which it has been extensively studied, gH is
essential for virus replication. All gH proteins appear to require glycoprotein L (gL) as
a chaperone. gH has been shown to play a role in virus binding, entry and fusion in
multiple herpesviruses, and gL has also been suggested to play a role in fusion. The
majority of gH studies have been conducted in Herpes simplex virus (HSV). Very
little investigation has focused on Varicella-zoster virus (VZV) gH function, and
therefore most of what is assumed about VZV gH function is based on studies of other
herpesviruses.
3.2. VZV gH and gL processing and interaction
VZV open reading frame (ORF) 37 encodes an 841-amino acid (aa) protein
designated glycoprotein H (gH). gH is reported to be a type-1 transmembrane protein,
based on the presence of a single hydrophobic region near the C-terminus (160). VZV
gH is a 118 kDa highly glycosylated protein that is classified as a late protein in VZV
infection. It contains a predicted signal sequence and transmembrane region, as well as
multiple glycosylation sites. Treatment with various enzymes indicates that gH
contains N-linked glycomoieties but no O-linked glycomoieties (166). It has been
reported that the unglycosylated precursor of gH is 79 kDa, although the predicted
molecular mass of gH based on the amino acid sequence is 94 kDa. gH is first
processed in the endoplasmic reticulum (ER), where high-mannose oligosaccharides
are added, possibly as a co-translational event. This results in the expected 94 kDa
32

precursor gH, or pre-gH. Pre-gH is then transported to the Golgi, where some of the
glycomoieties are processed to sialated complex oligosaccharides, resulting in the
mature, glycosylated 118 kDa protein.
All gH proteins require a chaperone, glycoprotein L (gL), for correct
processing and transport. In VZV, ORF 60 encodes gL. VZV gL is a 159 aa, 18 or 19
kDa protein with one complex N-linked glycomoieties (65, 66, 153). When gH is
expressed without gL, only the 98 kDa pre-gH is present in cells, but when gH and gL
are co-expressed, the 118 kDa gH is identified, along with the 18 and 19 kDa gL
forms (65, 66, 78, 153). VZV gL is fully processed in the absence of gH, indicating
that it is able to undergo transport from the ER to the Golgi and mature on its own.
Expressed alone, gL localizes to the ER and the cis or medial Golgi, but when
expressed with gH, distribution occurs throughout the cell. VZV pre-gH forms a
complex with the 18 kDa form of gL, but the proteins are not covalently linked and do
not contain any intermolecular disulfide bonds. It has been reported that gL does not
contain a signal sequence or transmembrane region, but does have an ER-targeting
motif at aa 71-86 (66). However, gL has no ER-retention signal, so it has been
suggested that gL is processed through the ER and the cis/medial Golgi, then is
targeted back to the ER, where it forms a complex with pre-gH to chaperone gH
transport from the ER to Golgi for further processing. From there, gH is trafficked to
the cell surface, while gL might recycle back to the ER or might be degraded.
gL contains five cysteines, four of which are important for maintaining
structure (65, 66). When C21 was substituted to glycine, no effect was seen on gH
(65). When C48, C79, C146 or C158 were deleted or substituted to glycine, gL was
33

able to interact with gH but only the 94 kDa pre-gH was synthesized (Table 3.1) (65,
66). gH was either not expressed on the cell surface or was only observed in a patchy
distribution instead of a diffuse expression over the whole cell surface. This indicated
that these gL mutants were not able to facilitate maturation and cell surface expression
of gH. Similarly, if gH was expressed with either glycoprotein E (gE) or I (gI), gH was
expressed on the surface but exhibited a patchy distribution. This patchy gH was
identified as 94 kDa pre-gH by immunoprecipitation studies, indicating that gE, gI or
some mutant gL proteins can facilitate transport of gH from the ER to the cell surface,
but not from the ER to the Golgi. Additionally, mutations in gH to create a basic
cytoplasmic tail resulted in a gH protein able to transport to the cell surface by itself,
but again the distribution was patchy and only pre-gH was generated. The 94 kDa pre-
gH is not functional, even when expressed with gE or gI.
3.3. gH and gL processing and interaction in other herpesviruses
Among the herpesviruses, there are differences in the interaction between gH
and gL. One difference is whether gL is fully processed and transported in the absence
of gH. A second difference is whether or not the gH/gL interaction involves an
intermolecular disulfide bond. Additionally, the mature protein products differ in their
sizes and glycosylation states.
3.3.1. Alphaherpesviruses
In herpes simplex virus 1 (HSV1), UL22 encodes gH and UL1 encodes gL.
UL22 encodes a predicted 90 kDa protein with a signal sequence, transmembrane
34

region, a large extracellular N-terminal domain with several predicted N-glycosylation
sites and a short C-terminal cytoplasmic tail (40, 98). Translation of UL22 produces a
glycosylated 110 kDa gH. gH processing includes the addition of α2,3- and α2,6-
linked sialic acid residues, and sialic acid residues on HSV1 gH, gB and gD are
required for virus entry (223).
HSV1 gL has a predicted signal peptide, and is first synthesized as a 25 kDa
non-glycosylated precursor (120). N-linked glycomoieties are added to create a 30
kDa protein, which is then modified further by the addition of O-linked glycomoieties,
resulting in a 40 kDa protein. In the absence of gH, gL is only processed into the 30
kDa protein, and very little gL is found on the cell surface. gH is not expressed on the
cell surface in the absence of gL, indicating the gH/gL interaction is needed for
maturation and transport of both proteins. The two proteins are not linked by a
disulfide bond, but remain noncovalently associated following processing.
HSV2 gH is able to traffic to the surface when expressed alone if aa 19-47
following the signal sequence are deleted (42). Deletion of aa 19-28 results in a gH
protein that requires gL for transport and function, and deletion of aa 19-64 or aa 19-
72 results in a gH protein that requires gL for transport to the surface but remains
nonfunctional. Thus, it was proposed that residues 19-47 contain an ER retention
signal that would keep gH in the ER until gL was present for transport of gH to the
Golgi and then the cell surface. In agreement with this hypothesis, alanine substitution
of R39, Y41, W42 or D44, residues typically found in ER retention signals, resulted in
some gH trafficking to the cell surface in the absence of gL. Deletion of the N-
terminal 48 amino acids in HSV1 gH did not result in gL-independent transport of gH
35

to the cell surface, indicating that different residues might be responsible in HSV1 gH
for retaining the protein in the ER.
HSV1 gH aa 259-387 have been suggested to play a role in transport of gH/gL
to the surface, with aa 259-323 playing a role in gH binding to gL (44). It has also
been suggested that the minimal amino acids in gH that interact with gL are aa 300-
473 (238). The minimal HSV1 gL residues required for correct gH processing,
transport and function appear to be aa 1-161 (143). Deletion of aa 169-224 resulted in
decreased gL expression but gH was fully functional. Some gL truncation mutants
bound to gH but did not promote gH trafficking to the surface, indicating that binding
is a separate function from trafficking. Additionally, conformation-dependent
antibodies recognized gH, indicating that these truncation mutants were capable of
promoting gH maturation but not cell-surface expression.
HSV1 gL contains five cysteine residues. C44, C76, C149 and C160 are
required for correct gH/gL maturation (43). The HSV1 gL R156, R158 and C160 are
all important for gH/gL interaction (142). Substitution of C160 with alanine resulted in
gL that bound gH but could not facilitate cell surface expression. Substitution of gL
R158 to neutral alanine reduced gH cell surface expression and fusion and abolished
virus entry into cells, but mutation to lysine, a positively charged residue, resulted in
nearly wild-type levels of expression, fusion and entry. Substitution of R156A resulted
in decreased cell fusion and virus entry. This demonstrated the importance of
positively charged residues in these locations for intramolecular interactions in
maintaining gL structure or conformation.
36

HSV1 gH contains eight cysteines, and HSV2 gH contains 7 cysteines, with a
substitution of an arginine for the third cysteine at aa 404 (43). In the N-terminal
region of HSV1 gH, the four cysteines are located at C90, C258, C404 and C429.
Mutation of these residues does not disrupt gH/gL maturation, transport or function. In
HSV2 gH, however, mutation of the three cysteines at aa 90, 258 and 429 results in
normal synthesis and processing, but reduced levels of fusion, especially with
mutation of C258 or C429, suggesting that these two cysteines form a disulfide bond.
The four C-terminal cysteines are located at C554, C589, C652 and C706. Mutation of
C554 or C589 results in disruption of the HSV1 gH/gL complex formation and
incomplete maturation of gH, since antibodies that recognize conformation-dependent
epitopes can no longer detect these mutants. Additionally, these mutants are not
detected on the cell surface and are therefore likely not functional in fusion. Mutation
of C652 or C706 results in proteins that are detected on the cell surface but have only
low levels of fusion and infectivity complementation. It appears that C554 and C589
are important for HSV1 gH structure, while C652 and C706 are important for HSV1
gH function.
Pseudorabies virus (PRV), a closely related non-human alphaherpesvirus to
VZV, encodes a 686 aa gH protein with a predicted molecular weight of 72 kDa (139).
The addition of N-linked carbohydrates results in a mature 85 kDa protein (141). PRV
gL is a 156 aa protein containing a signal sequence and possibly a C-terminal
transmembrane region (137). PRV gL is not N-glycosylated, but the addition of O-
linked glycomoieties results in a shift from 17-18 kDa to 20 kDa. Localization of PRV
gL to virions is dependent on gH. PRV gL is required for virus penetration and
37

efficient cell-cell spread, but gH is incorporated into the virion in the absence of gL
(138). This gH is not fully processed and the progeny virions are not infectious,
indicating that gL is required for complete maturation and function of gH, or possibly
that gL itself plays a role in fusion. gH incorporation into the virion might occur
without gL or might result from gH interactions with other viral proteins, similar to
effects seen with gE or gI on VZV gH that result in immature, non-functional gH
being expressed on the cell surface (66, 138).
3.3.2. Gammaherpesviruses
Epstein-Barr virus (EBV) gH is encoded by the BXLF2 ORF, and is an 86 kDa
protein containing a short cytoplasmic tail and several hydrophobic regions predicted
to interact with membranes (113). EBV gH requires gL for processing and transport
(246). However, gL appears to be processed and trafficked normally when expressed
alone, indicating that this process does not require gH (150). EBV gL is encoded by
the BKRF2 ORF, and is a 137 aa protein with no predicted transmembrane region and
three predicted N-glycosylation sites. While the two proteins do form a complex, no
intermolecular disulfide bond is present.
3.3.3. Betaherpesviruses
Human cytomegalovirus (HCMV) gH (UL75) requires gL (UL115) for correct
processing and transport (130, 214). The betaherpesvirus gH/gL complex differs from
the alpha- and gammaherpesvirus in that an intermolecular disulfide bond covalently
links gH and gL. Human herpesvirus 6 (HHV6) gH and gL are also linked by a
38

disulfide bond (6). HHV6 gH contains fourteen cysteine residues, twelve of which are
conserved in various alpha- or gammaherpesviruses. It has been suggested that the
cysteines specific to the betaherpesviruses are involved in the disulfide gH-gL bond
that is not found in alpha- and gammaherpesviruses. In support of this hypothesis, the
N-terminal 230 aa of HHV6 gH are required for interaction with gL, and this region
includes the cysteine residues specific to the betaherpesviruses.
3.4. VZV gH function
3.4.1. Binding and entry
No studies have been done on VZV gH binding and entry. This is due in part to
the cell-associated nature of VZV, which makes production of cell-free virus at high
titers difficult. To date, most VZV studies have been conducted with cell-associated
virus. Two studies have indicated gH, gL and gB play roles in VZV fusion (detailed
below), which suggests that one or more of these proteins likely functions to bind a
cellular receptor. VZV gB has been shown to bind myelin-associated glycoprotein
(MAG), which is expressed on neuronal cells (218). No receptor for gH has been
identified.
3.4.2. Fusion
In VZV, fusion has been studied using two different assays. Early studies used
a vaccinia virus assay. A vaccinia virus containing the T7 polymerase was used to
infect cells, and this was followed by the transfection of plasmids containing the gH
and gL genes under the control of the T7 promoter (66). In other studies, the gH and
39

gL genes were incorporated into the vaccinia virus genome, which was then used to
infect cells (154). In either context, gH and gL expressed were capable of causing cell-
cell fusion and the formation of syncytia. Within this same assay, gB and gE were also
capable of promoting cell-cell fusion and syncytia formation, and gB expressed alone
resulted in the formation of very small syncytia. While it is likely that these
glycoproteins play a role in VZV-induced cell-cell fusion, all other herpesviruses
appear to require a minimum of gB, gH and gL to induce fusion. It is possible that
some vaccinia virus protein is contributing to the fusion observed in this assay.
A more recent study utilized a luciferase fusion assay (218). In this assay,
expression vectors containing glycoproteins and luciferase under the control of the T7
promoter or a T7 polymerase were transfected into effector and target cell populations,
respectively. These populations were mixed and luciferase was quantified as an
indicator of fusion. This assay has demonstrated that gH, gL and gB, as well as the gB
cellular receptor MAG, are all required for fusion. Inclusion of gE in this assay
reduced the levels of fusion. Although the data from this study are not identical to the
previous studies using the vaccinia virus assay, the data is consistent with data from
other herpesvirus studies that indicate gH, gL and gB are all needed for fusion.
Additionally, both the vaccinia virus and the luciferase assays indicate that gH is a
component of the VZV fusion machinery.
gH endocytosis has been shown to be important in fusion. gH contains a
YXXφ endocytosis motif (where Y is a tyrosine, X is any amino acid, and φ is any
bulky hydrophobic amino acid) at aa 835-838 in the cytoplasmic tail (185).
Substitution of tyrosine at aa 835 to alanine or deletion of the 835YNKI838 motif by
40

inserting a stop codon in place of the serine at position 830 (S830Stop) results in no
gH endocytosis and instead, gH accumulation on the cell surface of transfected cells
(Table 3.1). gH endocytosis appears to be efficient and continuous, and is clathrin-
dependent but antibody-independent. gH is endocytosed within transfected cells, cells
infected with a vaccinia virus expressing gH and gL, and within the context of VZV
infection. During infection, gH is endocytosed and localized to the center of syncytia,
where it colocalizes with markers for the trans-Golgi network (TGN). An YXXφ motif
is conserved among most herpesvirus gH cytoplasmic tails, but is not found in HSV1,
HSV2 or EBV.
When gH endocytosis is blocked using either the Y835A or S830Stop mutants,
more cell-cell fusion occurs compared to wild type gH (186, 187). This was based on
bigger syncytia size quantitated using the Kolmogorov-Smirnov point counting
method and the vaccinia virus assay. These endocytosis mutants still need gL for
transport and function, and when they are expressed with gE, a patchy cell-surface
localization and non-functional gH still results, indicating that they are not capable of
mediating their own transport or complete maturation. Additionally, mAb 206, a
monoclonal antibody that detects mature, conformation-dependent gH and which has
been shown to inhibit gH-induced fusion, still inhibits fusion in these mutants,
indicating that no new fusogenic domain has been created (53, 186, 202).
gE is capable of mediating some endocytosis of the endocytosis-deficient
mutant Y835A, and in a cell-cell fusion assay, this resulted in reduced syncytia size
similar to that seen with wild-type gH (186). gH has been shown to interact with gE
both in cells transfected with expression vectors and in virions (155, 186). Anti-gH
41

antibodies coimmunoprecipitate gE, and gE-gH complexes are identified in virions.
Additionally, gE contains a tyrosine-based endocytosis motif and is endocytosed in a
manner similar to gH (4, 182, 185). This endocytosis motif might explain how gE
interaction with the gH endocytosis mutant results in internalization of gH. From these
data, it has been proposed that endocytosis of gH is a mechanism by which VZV
might regulate cell surface expression of gH and thus virus-induced cell-cell fusion.
Furthermore, gE contains a TGN targeting motif (155). VZV secondary envelopment
takes place in the TGN, and the gE/gH interaction might provide a mechanism by
which gH is targeted to the TGN.
3.5. gH and gL function in other herpesviruses
3.5.1. Binding
Receptors have been identified for HSV and HCMV gH, and gH binding to
these receptors might be involved in cell signaling events during virus entry.
Expression of HSV1 gH on the cell surface prevented HSV infection, and this
expression was independent of gL (211). This suggested that gH might be interacting
with a receptor, thereby blocking virus entry. Using gH bound to the Fc portion of
human IgG1, it has been demonstrated that the gH-Fc/gL heterodimer binds to integrin
αvβ3, and expression of this integrin on CHO cells resulted in gH-Fc/gL binding to
the cells (184). HSV gH contains a RGD integrin-binding motif, and substitution of
RGD to RGE abolished binding of gH-Fc/gL to integrin-expressing CHO cells.
Antibodies against the integrin αv or β3 subunits also blocked binding, but did not
block HSV1 plaque formation. Mutation of RGD to RGE did not affect virus binding
42

or entry (83). This suggested that either gH binding to αvβ3 integrins was not
absolutely required for these events, or gH can interact with integrins through an
additional binding motif.
HCMV gH has also been shown to interact with αvβ3 integrins (232). The
αvβ3 integrins are required for HCMV infection. Antibodies to these integrin subunits
preincubated with cells or soluble αvβ3 ligands preincubated with virus resulted in an
inhibition of infection. A peptide corresponding the RGD motif of HCMV gH did not
block infection, indicating this interaction might involve other gH domains.
HCMV gH also has been reported to interact with other cellular receptors.
Expression of HCMV gH and human basic fibroblast growth factor receptor (FGFR)
resulted in mature gH expressed on the cell surface, suggesting that gH might interact
with FGFR on the cell surface during infection (214). gH also appears to interact with
a novel protein named CMV fusion receptor (CMVFR), a 341 aa, 92.5 kDa protein
with two potential membrane-spanning domains and several nuclear localization
signals (NLS) (17, 18). These NLS have been proposed to aid in capsid translocation
to the nucleus. Cells transfected with this protein are susceptible to HCMV entry,
while cells preincubated with recombinant gH or virus preincubated with anti-gH
antibodies are not able to fuse.
3.5.2. Entry
In some viruses, gH and gL form complexes with virus surface proteins or
glycoproteins that appear to determine cell-specific infectivity and possibly entry
routes into the cell. In EBV, where the luciferase-based cell fusion assay has been
43

utilized, gH and gL form a complex with gp42, and this tripartite complex is required
to infect B cells (150). gp42 interacts with MHC class II on B cells to promote EBV
attachment and entry (215). The presence of gp42 in the gH/gL complex inhibits
infection of epithelial cells, which only require the gH/gL complex for entry (234).
These three proteins have a stoichiometry of 1:1:1 within this complex, and gp42 aa
36-81 are involved in binding to gH/gL (134, 135).
In HCMV, gH and gL form complexes with several different proteins. One
complex is formed between gH, gL and glycoprotein O (gO) (118). Another complex
is formed by gH/gL and UL131/UL130/UL128, or UL131-128 (234). The
gH/gL/UL131-128 complex is required for infection of epithelial and endothelial cells,
while the gH/gL/gO complex is not (205, 234). Some data indicates that the presence
of the UL131-128 proteins in the gH complex causes a conformational change in gB,
which is also required for HCMV entry, that results in entry through direct fusion
between the virus envelope and the plasma membrane (188). However, other data
suggest that entry into endothelial and epithelial cells involves endocytosis of the
virus, followed by low pH-dependent fusion between the viral envelope and the
endosomal membrane (205, 207). In contrast, entry into fibroblasts does not require
UL131-128 and occurs by pH-independent direct fusion between the viral envelope
and the plasma membrane (55, 207). gO is not absolutely required for growth in
fibroblasts and is not incorporated into the envelope, but is required for efficient
replication and spread (114, 243). gO interacts with gH/gL in the endoplasmic
reticulum (ER) to promote transport to the Golgi, and a gO-null virus had very low
levels of gH/gL in the envelope (206, 243). This suggested that gO acted as a
44

chaperone to ensure gH/gL complex incorporation into the virus envelope, resulting in
both gH/gL and gH/gL/UL131-128 complexes.
Human herpesvirus 6 and 7 (HHV6 and -7) both have gO homologs that form
complexes with gH and gL (169, 208). A second complex composed of gH, gL and
glycoprotein Q (gQ) has been identified in HHV6 virions (168). gQ acts as a viral
ligand for CD46, suggesting CD46 can act as a receptor for HHV6 (170). The
gH/gL/gO complex in HHV6 does not interact with CD46, and therefore the separate
tripartite complexes might play different roles in entry, possibly by altering tropism of
the virus (169).
3.5.3. Signaling cascades associated with gH ligand binding and virus entry
gH has been reported to be involved in signaling cascades during virus entry.
HSV entry, but not binding, has been associated with rapid tyrosine phosphorylation
of cellular proteins, indicating activation of cell signaling transduction pathways might
be associated with entry events associated with ligand binding (197). gH peptides
corresponding to aa 602-615, 618-632 and 682-701 induce the c-Jun N-terminal
kinase (JNK) pathway, which regulates stress responses such as inflammation and
apoptosis (88). Phosphorylation of JNK starts by 3 minutes post infection and lasts at
least 10 minutes, but returns to baseline by 60 minutes post infection. These gH
peptides do not activate the extracellular signal-regulated kinase-1 or -2 (ERK-1/2)
pathway, which is involved in cell growth and differentiation, or the mitogen-activated
protein kinase MEK-1/2 or p38. The rapid induction of a JNK-specific
45

phosphorylation signal transduction pathway might be required for the virus infection
and replication.
Calcium (Ca2+) signaling also occurs following HSV entry. Membrane Ca2+
increases following virus envelope interactions with heparan sulfate
glycosaminoglycan chains on snydecan-1, -2 and nectin-1 coreceptors. Ligand binding
to αvβ3 integrin, presumably by gH, and inositol 1,4,5-triphosphate receptors (IP3R)
results in global release of Ca2+ from the ER and entry of the virus (48, 50). The
interactions that triggered membrane Ca2+ increases were not sufficient for entry of the
virus, but interactions that triggered global Ca2+ release are sufficient. Inhibitors of
IP3R that block ER Ca2+ release also inhibited HSV infection and phosphorylation of
focal adhesion kinase (FAK). FAK phosphorylation is important for transport of HSV
capsids to the nucleus (49). Thus, HSV gH interaction with αvβ3 integrins might be
important for global release of Ca2+ and signaling events.
HCMV gH interaction with αvβ3 integrins also causes signaling cascades
(232). gH binding induced tyrosine phosphorylation of the β3 subunit within 5
minutes of infection. gB also induced tyrosine phosphorylation of epidermal growth
factor receptor (EGFR), a gB receptor, within several minutes of infection (232, 233).
These tyrosine phosphorylation events resulted in activation of Src and
phosphatidylinositol-3 kinase (PI3K), the respective downstream targets of αvβ3
integrins and EGFR. αvβ3 integrins translocated to lipid rafts following gH binding,
where they interacted with EGFR to produce a synergistic increase in downstream
signaling. This signaling appears to reduce RhoA activation and RhoA downstream
signaling, which might affect cell cytoskeleton rearrangement.
46

HCMV gH and gB also induce Sp1 and NF-κB at a post-binding step (247).
This might account for expression of the transcription factors c-jun, c-myc, and c-fos
following infection. Activation of these cellular transcription factors likely contributes
to the immediate induction of various virus gene products. Antibodies to gH or gB
inhibited induction of these transcription factors, and binding of an anti-idiotypic
monoclonal antibody to the CMVFR, which antigenically mimics gH binding to the
receptor, upregulated Sp1 and NF-κB. This antibody was also capable of inducing
CMVFR stimulation of IP3, causing global release of Ca2+, and of inducing
phosphorylation of CMVFR, suggesting gH binding is also capable of these inducing
these events, which might be required for virus entry (131, 132).
3.5.4. Fusion
Fusion has been much more extensively studied in herpesviruses other than
VZV, especially HSV1. Using the luciferase fusion assay described above, HSV
glycoprotein D (gD), gB and gH/gL have been shown to be necessary and sufficient to
induce cell-cell fusion (226). Fusion requires that all these proteins be expressed in the
same cell, and that the target cell contains a gD receptor (194). During virus entry,
HSV1 gD binds various herpesvirus receptors. gB and gH are hypothesized to play
central roles in virus-induced fusion, and recent evidence suggests gL might be more
than just a gH chaperone and might play a role in fusion. When only gD, gH and gL
are expressed on cells, hemifusion results, where the outer lipid membrane mixes but
cell content is not mixed (217). Expression of gD and gB does not result in
hemifusion, but expression of all four glycoproteins results in full lipid and content
47

mixing. From these data, a sequential model for fusion has been proposed, where gD
is required for receptor binding, gH is required for hemifusion formation, and gB is
required for complete fusion.
HSV1 gD interacts with gB and with gH/gL, and gB and gH/gL interact (12,
13, 92). gD aa 240-260 are important for gD-gB interaction, and aa 260-310 are
important for gD-gH/gL interaction. gB and gH/gL appear to interact independently of
fusion (11). Coimmunoprecipitation studies have indicated that the herpesvirus entry
mediator (HVEM), gD and gH form a complex, which might be the complex that
functions in hemifusion (193).
Various residues and motifs in HSV1 gH have been shown to be important for
fusion. Complete deletion of the cytoplasmic tail and transmembrane region and
replacement with glycosylphosphatidylinositol-addition sequences abolished the
ability of gH to promote cell-cell fusion, indicating that these regions play a role in
cell-cell fusion (127). Mutation of a 830SVP832 motif in the cytoplasmic tail of gH in a
UL27-gB syn mutant virus reduced syncytia formation, but progeny virions containing
mutated gH were still infectious, indicating differences in the domains of gH required
for virus-cell and cell-cell fusion (244). The V831 residue is conserved in all gH
cytoplasmic tails except in VZV and PRV, where it is replaced by a serine. Several
conserved residues within the HSV1 gH transmembrane region, namely A808, S809
and G812, are important for fusion (110). All three of these residues fall on the same
side of a predicted transmembrane α-helix, and it is possible that they play a role in
destabilizing the membrane during fusion or interact with other molecules in the
membrane.
48

Multiple studies have suggested that HSV gH contains heptad repeats and α-
helices, which are typical elements of a Class I fusion protein. The fusion peptide is an
α-helix that consists mainly of hydrophobic residues, which associate or insert into the
membrane of the target cell, causing the protein to act as a bridge between the virion
envelope and the cell plasma membrane (54, 124). Insertion of the fusion peptide into
the cell plasma membrane initiates fusion. Heptad repeats are often found in the C-
terminal region of the protein. They consist of groups of seven amino acids with
specifically spaced hydrophobic residues. Heptad repeats form α-helices, and multiple
α-helices interact to form coiled-coils. In these structures, the hydrophobic amino
acids are buried inside the α-helix. Multiple heptad repeats can interact to form dimers
or trimers of coiled-coils, and this interaction causes a conformational change in the
protein that brings the virus membrane and the cell plasma membrane into closer
proximity to promote fusion.
Membrane α-helices predicted to form fusion peptides have been identified at
aa 377-397 and aa 512-532 in HSV1 gH (93, 94). Both of these regions can convert a
secreted protein into a membrane-bound protein, and mutation or deletion of these
regions abrogates virus infectivity and cell-cell fusion. Mimetic peptides of these
regions are capable of inducing fusion of lipid vesicles, and of enhancing herpesvirus
infection, presumably by destabilizing membranes to allow more virus entry into cells.
Prediction of helices in other herpesvirus indicates that the first α-helix fusion peptide
is positionally conserved, and replacement of the HSV1 α-helix sequence with the
predicted VZV α-helix results in a functional gH protein.
49

HSV1 gH heptad repeats are predicted at aa 443-471 and aa 556-585 (89, 95,
96, 209). Mutations predicted to abolish the helical nature of these heptad repeats, also
abolished virus infectivity and cell-cell fusion. Mimetic peptides inhibit virus
infectivity during entry, presumably by interacting with the opposite heptad repeats
within the protein and preventing virus heptad repeat interaction and conformational
change. In circular dichroism studies, both peptides adopted random coil structures
within an aqueous solution, but in the presence of trifluoroethanol (TFE), which
mimics the environmental transition from water to a membrane interface, both
peptides adopted α-helix formations. Additionally, the peptides were capable of
interacting with one another, similar to the formation of coiled-coils. This interaction
abrogated the block on virus entry that the individual peptides exerted, presumably
because peptides were not available to interact with the individual heptad repeats
within the protein.
Human cytomegalovirus (HCMV) gH has also been reported to contain a
heptad repeat predicted to form α-helical coiled-coils (151). A peptide corresponding
to the heptad repeat adopted random coil configurations in aqueous solution in circular
dichroism studies. No TFE studies have been done to assess coiled-coil conformation
of these peptides in a membrane interface-mimicking environment. The peptide was
able to block virus infection of cells if virus was pretreated or if virus and peptide
were added to cells concurrently. The peptide did not block virus attachment to cells
but did block virus entry.
Peptide studies of other regions of HSV1 gH have implicated aa 220-262, aa
381-420 (containing the first predicted fusion peptide), aa 493-537 and aa 626-644 in
50

membrane interactions and fusion (84, 85). These peptides can induce fusion of lipids,
assume α-helical structures in the presence of TFE, and block virus infection. The aa
626-644 peptide was particularly effective at inducing membrane fusion, and the
hydrophobic residues within this region are important for this function (87).
The pre-transmembrane region of HSV1 gH contains a positionally-conserved
glycosylation motif (784NGT786) within the highly conserved 779LLLFPNGTV787
sequence (83). The T786 residue was substituted with alanine without effect,
indicating glycosylation of the N784 residue does not play a role in virus infection.
Peptide studies of the region from aa 761-805 have indicated that this region is a
helical domain that might be capable of partitioning membranes and disrupting lipid
bilayers, thereby promoting membrane destabilization during fusion (86).
Interestingly, peptides corresponding to this region can inhibit virus infectivity
following pre-exposure of the virus to the peptide, while peptides corresponding to
heptad repeats can only inhibit if present during virus entry.
A role for gL in fusion other than chaperoning gH maturation and transport has
recently been studied in HSV and EBV. The first evidence for a role of gL in fusion
came from studies demonstrating that antibodies to HSV1 gL were not capable of
inhibiting virus entry and infection but were capable of inhibiting virus-induced cell-
cell fusion following infection, resulting in a reduction in plaque size (178). This
suggested that gL might play different roles in virus entry and cell-cell fusion.
Substitution of HSV1 gL R156 to alanine did not affect gH localization, but did reduce
cell fusion and virus entry, again suggesting that gL might be a functional component
of the gH/gL heterodimer (142). Linker insertion mutagenesis of HSV1 gL at P48,
51

R55 and T157 resulted in wild type levels of gL cell surface expression, but reduced
gH cell surface expression and aberrant gH/gL heterodimer formation, as well as
reduced or abrogated gL function in cell fusion and virus infectivity complementation
(67). Although these insertions might have affected the conformation of gH or the
gH/gL heterodimer, it was also suggested that they might affect a functional domain of
gL. However, neither of these studies was able to completely separate a role of gL as a
chaperone for gH from a role of gL in fusion.
EBV gL has also been proposed to play a role in cell fusion (195). EBV gL
shares a high sequence homology with Rhesus Lymphocryptovirus (Rh-LCV) gL, and
substitutions in EBV to mimic the Rh-LCV resulted in normal processing, transport
and cell surface expression of gH and gL but abrogated fusion of B lymphocytes.
Residues K54 and Q94 were identified as critical for EBV fusion in a luciferase assay.
This suggested that EBV gL plays a role in fusion other than as a chaperone. While
this investigation did not demonstrate any reduction of gH maturation, transport or cell
surface expression, it remains possible that gH was not functional, and further studies
are likely needed to clearly define gL roles as a chaperone and as a component of the
fusion machinery.
3.5.5. Capsid egress from the nucleus
HSV1 gH has been suggested to play a role in fusion and nuclear egress of
viral nucleocapsids. gB and gH accumulated in the nuclear envelope and on
perinuclear virions (70). Infection with a mutant virus lacking both gB and gH resulted
in nuclear herniations that contained enveloped capsids. It has been suggested that gB
52

and gH act in a redundant fashion to mediate fusion and nuclear egress. This data also
supports the assembly hypothesis that nucleocapsids are enveloped as they cross the
inner nuclear membrane, deenveloped as they cross the outer nuclear membrane, and
then are subsequently reenveloped within the cytoplasm. In pseudorabies virus (PRV),
however, no double deletion combination of gB, gH, gL and gD resulted in any defect
in nuclear egress, and no glycoprotein localization was identified on the nuclear
membrane, suggesting differences in mechanisms of nuclear egress among the
alphaherpesviruses (136).
3.5.6. Virion assembly
gH plays a role in virion assembly. In HSV1, the cytoplasmic tail of gH
interacts with VP16, a tegument protein (104). Five specific amino acids in the
cytoplasmic tail of gH were required for binding, and the interaction was temperature-
dependent. The temperature dependence of this interaction resulted from the
disorganized form the gH tail assumed at 37°C, the temperature at which VP16
binding occurred (129). At lower temperatures, such as 4°C, the tail was structured,
and it has been suggested that this prevents the gH tail from interacting with VP16.
From this, it was hypothesized that the unstructured nature of the gH cytoplasmic tail
at physiologically relevant temperatures allowed it to adopt multiple conformations,
and thus interact with multiple binding partners, during virion assembly. gH-VP16
interaction might facilitate incorporation of gH and VP16 into the virion, although
there are likely many redundant interactions between glycoproteins and tegument
53

proteins that result in complete virion formation, as a mutant gH incapable of binding
VP16 did not prevent VP16 incorporation into the virion.
3.6. gH and immunity
gH is one of the major antibody targets during herpesvirus infection. Patients
with primary or recurrent VZV, or patients who are immune to VZV, have humoral
and cellular immunity against gH (9). In a study of seven HSV-1 positive adults, all
seven had a gH/gL-specific IgG response and a gH/gL-specific T cell response, likely
a CD4+, Th-1 response (239). The predominant antibody from patients in the acute
phase of EBV infectious mononucleosis is directed against gH (198).
Antibodies to gH effectively neutralize VZV and other herpesviruses during
penetration but not adsorption, indicating an essential role for gH in the fusion of viral
and cellular membranes but not in initial attachment of the virus to the cell (82, 162,
202). VZV anti-gH mAb 206, which recognizes a conformation-dependent epitope on
mature, glycosylated gH, neutralizes virus entry and cell-cell spread (53, 166, 186,
202). Normal syncytia were visible at 48 hpi by scanning electron microscopy, but if
mAb 206 was added to media 90 minutes post infection, no syncytia were visible at 48
hpi. If media was then changed to remove antibody, syncytia were visible after a
further 48 h, indicating a need for new gH synthesis in VZV replication and spread.
Treatment with mAb 206 also reduced the number of infected nuclei, the percentage
of infected nuclei within syncytia, and the mean number of nuclei within syncytia. In a
neutralization assay, a humanized recombinant version of mAb 206 had 2400-fold
increase in biological activity compared to a standard VZIG prep on a mg per mg basis
54

(63). Studies of this humanized antibody have indicated that multimerization of the
single-chain antibody are important for neutralization, which suggests that steric
hindrance of gH might be required to block fusion. This steric hindrance could occur
via antibody mechanisms of aggregation or crosslinking, or through increased avidity
of a multimeric antibody compared to a monomeric antibody. No epitope for either the
original mAb 206 or the humanized version has been identified.
The V3 human mAb is another neutralizing antibody directed against VZV gH
(219). This antibody is capable of neutralizing at least five different VZV strains and
inhibiting spread. It mediates antibody-dependent cellular cytotoxicity but not
complement-dependent cytotoxicity of infected cells. Other anti-gH neutralizing
antibodies include mAb 9C6, mAb 258, and mAb 366 (166). mAb 258 neutralizes in a
complement-independent manner and recognizes a conformation-dependent epitope
on immature, glycosylated and mature, glycosylated gH.
Inoculation of mice with a vaccinia virus expressing VZV gH and gL induces
specific complement-independent antibodies that are capable of neutralizing VZV in
vitro (148, 173). Neutralizing antibodies are not produced in mice inoculated with
vaccinia virus expressing only gH or gL alone. In HSV, a recombinant DNA vaccine
containing HSV1 gB, gH and gL genes protected mice from HSV (46). Passive
administration of the antibody LP11, which recognizes a conformation-dependent
epitope on HSV gH, also protected mice from HSV challenge when administered 24
hours post inoculation (80). Mice immunized with a truncated form of HSV1 gH and
gL and then challenged with HSV had small primary lesion, no secondary zosteriform
and survived challenge (191). Inoculation with a vaccinia virus expressing HSV gH
55

and gL protected mice against HSV infection of the ganglia, but produced only weak
levels of neutralizing antibody and virus clearance was only slightly enhanced (28).
Inoculation of mice with a HSV gH-negative virus grown on a complementing
cell line resulted in a self-limiting infection (71). Anti-HSV humoral and T cell
responses were identified, and these mice were resistant to challenge with a wild-type
virus. The gH-negative HSV infection produced higher antibody titers and a better
delayed-type hypersensitivity response, and thus greater protection, than an inactivated
vaccine to HSV-1. This mutant also resulted in no virus dissemination since virus
particles lacking gH are noninfectious. It was suggested that this virus might be
capable of establishing latency but the virus would not be pathogenic if reactivated.
56

Table 3.1. Compilation of published mutations made in VZV gH and gL
A compilation of published reported mutations in VZV gH and gL, and their effects on
gH maturation and expression. “+”, equivalent to wild type. “+/-“, less than wild type.
“++”, more than wild type. “P”, patchy gH surface expression resulting from
immature gH that is transported to the surface. All work was done using transiently
expressed proteins from expression plasmids or vaccinia virus infection.
Mutation Ref Predicted domain
gH/gL interaction
gH maturation
gH surface expression
Fusion
-gL +gL -gL +gL gH Wild type + - + - + + Y835A (185) Endocytosis
motif + + ++
S830Stop (185, 218)
Endocytosis motif
+ + ++ +
S830K, L832S, E834K
(65) Basic tail + - + p +
gL Wild type + - + - + + C21G (65) Cysteine + + + G33A (65) Helix + +/- +/- C48G (65) Cysteine + - p C79G (66) Cysteine + - p ΔC146 (66) Deletion of
13aa (C146 and C158)
+ - p
57

CHAPTER IV
ANTI-GLYCOPROTEIN H ANTIBODY IMPAIRS THE
PATHOGENICITY OF VARICELLA-ZOSTER VIRUS IN SKIN
XENOGRAFTS IN THE SCID MOUSE MODEL
Susan E. Vleck, Stefan L. Oliver, Mike Reichelt, Jaya Rajamani, Leigh Zerboni, Carol
Jones, James Zehnder, Charles Grose and Ann M. Arvin
Forward: This chapter is a version of an article published in the Journal of Virology,
Vol. 84, 2010. S.E.V. performed experiments, data analysis and manuscript
preparation. S.L.O. advised on experiment design and data analysis. M.R. performed
the immuno-electron microscopy. J.R. and L.Z. assisted with the SCID mouse
experiments. C.J. performed the quantitative PCR analysis in the lab of J.Z. C.G.
provided the mAb 206 and advised on the manuscript. These experiments were done
under the direction of A.M.A.
This work was supported by training grants from the National Institutes of
Health, R01 AI 020459 and P01CA49605. S.E.V. received support from T32
GM007279 and T32 AI07328.
We thank Nafisa Ghori, Department for Microbiology & Immunology,
Stanford University, for assistance with transmission electron microscopy, and Dr.
Barbara Berarducci for technical help and scientific discussion.
58

4.1. Abstract
Varicella-zoster virus (VZV) infection is usually mild in healthy individuals
but can cause severe disease in immunocompromised patients. Prophylaxis with
varicella-zoster immunoglobulin can reduce the severity of VZV if given shortly after
exposure. Glycoprotein H (gH) is a highly conserved herpesvirus protein with
functions in virus entry and cell-cell spread and is a target of neutralizing antibodies.
The anti-gH mAb 206 neutralizes VZV in vitro. To determine the requirement for gH
in VZV pathogenesis in vivo, mAb 206 was administered to SCID mice with human
skin xenografts inoculated with VZV. Anti-gH antibody given at 6 hours post
infection significantly reduced the frequency of skin xenograft infection by 42%.
Virus titers, genome copies and lesion size were decreased in xenografts that became
infected. In contrast, administering anti-gH antibody four days post infection
suppressed VZV replication but did not reduce the frequency of infection. The
neutralizing anti-gH mAb 206 blocked virus entry, cell fusion or both in skin in vivo.
In vitro, mAb 206 bound to plasma membranes and to surface virus particles.
Antibody was internalized into vacuoles within infected cells, associated with
intracellular virus particles, and colocalized with markers for early endosomes and
multivesicular bodies but not the trans-Golgi network. The mAb 206 blocked spread,
altered intracellular trafficking of gH and bound to surface VZV particles, which
might facilitate their uptake and targeting for degradation. As a consequence, antibody
interference with gH function would likely prevent or significantly reduce VZV
replication in skin during primary or recurrent infection.
59

4.2. Introduction
Varicella-zoster virus (VZV) causes chicken pox (varicella) upon primary
infection. Lifelong latency is established in neurons of the sensory ganglia, and
reactivation leads to shingles (herpes zoster) (8). Disease is usually inconsequential in
immunocompetent people, but can be severe in immunocompromised patients. The
current prophylaxis for these high-risk individuals exposed to VZV is high titer
immunoglobulin to varicella-zoster virus administered within 96 hours of exposure.
This prophylaxis does not always prevent disease, but the severity of symptoms and
mortality rates are usually reduced (156).
Glycoprotein H (gH) is a type 1 transmembrane protein that is required for
virus-cell and cell-cell spread in all herpesviruses studied (66, 79, 107, 140).
Glycoprotein H is an important target of the host immune system. Individuals who
have had primary VZV or herpes simplex virus (HSV), the most closely related human
alphaherpesvirus, have humoral and cellular immunity against gH (8, 239).
Immunization of mice with a recombinant vaccinia virus expressing VZV gH and its
chaperone, glycoprotein L (gL), induced specific antibodies capable of neutralizing
VZV in vitro (148, 173). Immunization of mice with purified HSV gH/gL protein
resulted in the production of neutralizing antibodies and protected mice from HSV
challenge (28, 191), and administration of an HSV anti-gH monoclonal antibody
protected mice from HSV challenge (80). Antibodies to HSV and Epstein-Barr virus
(EBV) gH effectively neutralize during virus penetration but not adsorption in vitro,
indicating an essential role for gH in the fusion of viral and cellular membranes but
not in initial attachment of the virus to the cell (82, 162).
60

Anti-gH mAb 206, an immunoglobulin G1 (IgG1) antibody which recognizes a
conformation-dependent epitope on the mature glycosylated form of gH, neutralizes
VZV infection in vitro in the absence of complement (166). The mAb 206 inhibits
cell-cell fusion in vitro based on a reduction in the number of infected cells and the
number of infected nuclei within syncytia, and appears to inhibit the ability of virus
particles to pass from the surface of an infected epithelial cell onto a neighboring cell
via cell extensions (53, 166, 186). When infected cells were treated with mAb 206 for
48 h post infection (hpi), virus egress and syncytia formation were not apparent, but
were evident within 48 h after removal of the antibody, suggesting that the effect of
the antibody was reversible and that there was a requirement for new gH synthesis and
trafficking to produce cell-cell fusion. Conversely, non-neutralizing antibodies to
glycoproteins E (gE) and I (gI), as well as an antibody to immediate-early protein 62
(IE62), had no effect on VZV spread (202).
Like other herpesviruses, VZV entry into cells is presumed to require fusion of
the virion envelope with the cell membrane or endocytosis followed by fusion. One of
the hallmarks of VZV infection is cell fusion and formation of syncytia (53). Cell
fusion can be detected as early as 9 hpi in vitro, although VZV spread from infected to
uninfected cells is evident within 60 minutes (199). In vivo, VZV forms syncytia
through its capacity to cause fusion of epidermal cells. Syncytia are evident in biopsies
of varicella and herpes zoster skin lesions during natural infection and in SCIDhu skin
xenografts (164). VZV gH is produced, processed in the Golgi and trafficked to the
cell membrane, where it might be involved in cell-cell fusion (65, 153, 166).
Glycoprotein H then undergoes endocytosis and is trafficked back to the trans-Golgi
61

network (TGN) for incorporation into the virion envelope (91, 155, 185). As VZV is
highly cell-associated in vitro, little is known about the glycoproteins required for
entry, but VZV gH is present in abundance in the skin vesicles during human infection
with chickenpox and zoster (236).
Investigating the functions of gH in the pathogenesis of VZV infection in vivo
is challenging because it is an essential protein and VZV is species-specific for the
human host. The objective of this study was to investigate the role of gH in VZV
pathogenesis by establishing whether antibody-mediated interference with gH function
could prevent or modulate VZV infection of differentiated human tissue in vivo using
the SCIDhu mouse model. The effects of antibody administration at early and later
times after infection were determined by comparing infectious virus titers, VZV
genome copies and lesion formation in anti-gH antibody treated xenografts. In vitro
experiments were performed to determine the potential mechanism(s) of mAb 206
interference with gH during VZV replication, virion assembly and cell-cell spread.
The present study has implications for understanding the contributions of gH to VZV
replication in vitro and in vivo, the mechanisms by which production of antibodies to
gH by the host might restrict VZV infection, and the use of passive antibody
prophylaxis in patients at high risk of serious illness caused by VZV.
62

4.3. Materials and Methods
4.3.1. Cells and virus
Human melanoma cells and human embryonic lung fibroblasts (HELF) were
grown at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal
bovine serum (Gemini Bio-Products, Woodland, CA), nonessential amino acids
(melanoma cells only) (100μM; Omega Scientific, Inc., Tarzana, CA), and antibiotics
(Penicillin, 100 U/ml; Streptomycin, 100 μg/ml; Invitrogen, Carlsbad, CA). Wild-type
recombinant VZV pOka was generated in melanoma cells using the cosmid system
(175) and propagated in HELF cultures.
4.3.2. Preparation, inoculation and harvest of skin xenografts in SCID mice
Skin xenografts were prepared in homozygous CB-17scid/scid mice, using human
fetal skin tissue obtained according to federal and state regulations (164). Animal
protocols complied with the Animal Welfare Act and were approved by the Stanford
University Administrative Panel on Laboratory Animal Care. Human tissues were
obtained, in accordance with state and federal regulations, from Advanced Bioscience
Resources (Alameda, CA). VZV pOka-infected HELF cultures were used to inoculate
the xenografts. Infectious virus titer was determined at the time of inoculation by 10-
fold serial dilution on melanoma cells. Skin xenografts were harvested at 7, 14, 21, 28,
35 and 42 days post infection (dpi). Half of each xenograft was stored in 4%
paraformaldehyde for histology; half was homogenized and resuspended in 1 ml PBS
for virus titration and DNA extraction. Serum was prepared from blood collected from
63

mice at the time of tissue harvest and allowed to coagulate at room temperature (RT),
then centrifuged at 6000 × g for 20 minutes.
4.3.3. Treatment of SCIDhu mice with anti-gH antibody
Anti-gH mAb 206 is an IgG1 complement-independent neutralizing antibody
that recognizes a conformational epitope on mature glycosylated gH (166). Either 100
μl PBS containing 25 μg mAb 206 or 100 μl PBS was administered to mice
intraperitoneally every four days starting 6 hours post infection (hpi). Mice were
treated with antibody beginning 6 h or 4 dpi and repeated doses were given every four
days through 12 dpi. The two antibody-treated groups consisted of mice treated with
antibody at 6 hpi, 4 days, 8 days and 12 dpi (Ab-0-12 group), or 4 days, 8 days and 12
dpi (Ab-4-12 group). PBS was administered at time points when antibody was not
given out to 42 dpi. A control group (PBS group) was given PBS at all comparable
time points. The number of xenografts evaluated at each time point was: 7-21 dpi,
n=11-12; 28 dpi, Ab-0-12, n=5, Ab-4-12 and PBS, n=11-12; 35-42 dpi, n=5-6.
4.3.4. Infectious plaque assay
Melanoma cells were seeded in a 24-well plate and inoculated in triplicate with
0.1 ml of a 10-fold serial dilution of xenograft homogenate or the inoculum virus to be
titered. For the titration of virus from homogenates, media was changed 24 h after
inoculation. Cells were cultured for 5 days, and plaques were stained with anti-VZV
polyclonal serum. Titer was analyzed using a Student’s t test to determine if a
statistically significant difference (p≤0.05) in titer existed. Number of xenografts
64

positive for virus was analyzed using a Fisher’s exact test to determine if a statistically
significant difference (p≤0.05) existed.
4.3.5. Plaque neutralization assay
Melanoma cells were seeded in a 24-well plate and inoculated in triplicate with
0.1 ml of 10 PFU/ml pOka in the absence (10 PFU/ml) or presence of 0.1 mL of a 10-
fold dilution of xenograft homogenate (10 PFU/ml + “group”). Media was changed
after 24 h, and plates were incubated for 5 days. Plates were stained as above and titer
was analyzed using a Student’s t test to determine if anti-gH antibody within the
homogenate neutralized the 10 PFU/ml inoculum. 10 PFU/ml was considered the
baseline infection and any statistically significant reduction below 10 PFU/ml was
considered to be neutralization of the baseline infection by antibody within the
homogenate. Increase above the baseline 10 PFU/ml resulted from virus within the
homogenate infecting the monolayer.
4.3.6. ELISA
The IgG1 mAb 206 in mouse serum was measured using a Mouse IgG1 ELISA
Quantitation Kit from Bethyl Laboratories, Inc. (Montgomery, TX) following the
manufacturer’s recommended protocol. Briefly, plates were coated with Capture
Antibody and blocked with Postcoat Solution. Sera samples were diluted 1:100 and
1:1000 in duplicate and incubated for 60 minutes at RT. Horseradish peroxidase
conjugate and tetramethylbenzidine (TMB) with an acid stop were used to detect the
presence of IgG1 antibody. Plates were read in a SpectraMax 190 (Molecular Devices,
65

Sunnyvale, CA). IgG1 concentration was determined from a standard curve, range
250-3.9 ng/ml, analyzed using a four-parameter logistic curve fit, as recommended by
the manufacturer.
4.3.7. Immunohistochemistry of skin xenograft sections
Mouse anti-gE antibody (mAb 8612, Millipore, Temecula, CA) was used at
1:2000 to detect VZV lesions in sectioned xenografts. Slides were developed using an
alkaline phosphatase-based enzyme detection method (Millipore, Temecula, CA) with
PermaRed substrate (VWR, West Chester, PA). Slides were counterstained with
hematoxylin. Xenografts were examined using an Axiovert 200 microscope (Zeiss).
4.3.8. Quantitative PCR
DNA was isolated from xenograft homogenates using DNAzol (Gibco-BRL,
Grand Island, NY), following the manufacturer’s protocol. VZV genome copy number
was assessed using primers/probes to detect ORF31 (encoding glycoprotein B),
ORF62 (encoding IE62) and ORF63 (encoding IE63), as previously reported (249).
Each gene target was measured in duplicate and the mean of each was used to
determine genome copies.
4.3.9. VZV DNA in situ hybridization
The VZV-specific DNA probe was prepared as previously described (199).
Paraffin-embedded sections were deparaffinated and incubated with proteinase K
(Roche Proteinase K, Indianapolis, IN, in proteinase K buffer: 0.1 M Tris HCl pH 7.5,
66

150 mM NaCl, 12.5 mM EDTA) for 10 minutes at 37°C and then dried completely.
Hybridization mix (15 μL) was added to each section, covered with a glass coverslip,
denatured for 10 minutes at 95°C and then hybridized overnight at 60°C. Sections
were washed twice in 2x SSC and once in 0.2x SSC for 10 minutes at 50°C, then
blocked for 30 minutes in DIG blocking solution (Roche, Indianapolis, IN). Sections
were incubated in anti-DIG monoclonal antibody (Roche, Indianapolis, IN) (1:50 in
blocking solution) for 1 h at 37°C, then secondary anti-mouse-DIG antibody (1:100 in
blocking solution) for 1 h at 37°C to amplify the DIG signal. Finally, sections were
incubated with anti-DIG-AP (1:200 in blocking solution) for 1 h at 37°C followed by
NBT-BCIP staining to detect the VZV DNA specific signal. Sections were
counterstained in methyl green and imaged using an Axiovert 200 microscope (Zeiss).
4.3.10. Antibody treatment of pOka infected HELF cultures in vitro
Following a protocol adapted from Rodriguez et al. (202), 1x106 HELF/well
were seeded in 6-well plates 24 h prior to inoculation. Monolayers were inoculated
with log10 3 PFU pOka for 90 minutes at 37°C. Media was changed, then 25 μg of
anti-gH mAb 206 was added and repeated at 24 h intervals. Mock cells received only
media. HELF cultures were harvested for titration and DNA extraction at 24 h
intervals.
4.3.11. Electron microscopy
HELF monolayers were seeded in 10 cm dishes containing glass coverslips,
infected with pOka and treated with antibody, as detailed above. After 48 h, the cells
67

on the coverslips were washed in PBS, fixed and processed for transmission electron
microscopy (TEM) as previously described (47), although without the use of
propylene oxide in the dehydration steps. The remaining cells were washed in PBS
and collected for cryo-EM. For cryo-EM, treated and untreated samples were fixed in
4% PFA with 0.1% glutaraldehyde in phosphate buffer (0.1 M, pH 7.2), washed in
PBS and infiltrated in 2.3 M sucrose overnight at 4°C. The samples were then
mounted on pins for cryo-ultramicrotomy and frozen in liquid nitrogen. Ultrathin
cryosections (80 nm) were prepared with a diamond knife (Diatome, Hatfield, PA) at -
130°C using an ultramicrotome (Ultracut, Leica) equipped with a cryosectioning
chamber. Thawed cryosections were transferred to Formvar- and carbon-coated EM-
grids (Nickel) within a drop of 2.3 M sucrose, washed in PBS, immunogold-labeled
and counterstained with 0.5% uranylacetate in 2% methylcellulose for 10 min on ice.
For immunogold labeling, thawed cryosections were blocked with 1x DIG blocking
solution. The mouse monoclonal antibody mAb 206, used for treating the samples,
was detected by incubation with 1:100 diluted polyclonal rabbit anti-mouse antibody
(Cappel Laboratories) for 1 h at RT followed by protein-A gold (15nm) incubation for
30 min at RT.
4.3.12. Confocal microscopy
HELF cultures were infected with pOka and treated with antibody or mock-
treated as detailed above. At 48 hpi, cells were fixed in 4% paraformaldehyde and
blocked with PBS containing 10% donkey serum and 0.1% Triton X-100. Cellular
localization of VZV proteins was performed using primary antibodies to VZV proteins
68

gH (SG3 monoclonal mouse anti-gH, Biodesign, Saco, ME), ORF23 (rabbit
polyclonal) (47) and gE (rabbit polyclonal) (122), and to cellular proteins trans-Golgi
network 46 (TGN46) (AHP500 polyclonal sheep anti-TGN46, AbD Serotec, Oxford,
UK), early endosome antigen 1 (EEA1) (NB300-502 rabbit anti-EEA1, Novus
Biologicals, Littleton, CO) and vacuolar protein sorting 4 (Vps4) (sc-32922 rabbit
anti-Vps4, Santa Cruz Biotechnology, Inc, Santa Cruz, CA). Secondary antibodies
used were FITC-coupled donkey anti-sheep (Jackson ImmunoResearch, West Grove,
PA), Alexafluor 555-coupled donkey anti-mouse (Molecular Probes, Carlsbad, CA),
Alexafluor 647-donkey anti-rabbit (Molecular Probes, Carlsbad, CA) and HOECHST
33342 (Molecular Probes, Carlsbad, CA). Confocal microscopy was performed using
a Zeiss LSM510 confocal microscope equipped with two-photon excitation.
69

4.4. Results
4.4.1. Treatment with the mAb 206 at 6 hpi significantly reduced the number of skin
xenografts infected with VZV
To investigate the effect of anti-gH neutralizing antibody on VZV
pathogenesis in vivo, skin xenografts were inoculated with pOka at a titer of log10 5.3
PFU/ml and mice were administered intraperitoneally either mAb 206 or PBS. Of 56
xenografts from mock-treated mice (PBS group), 70% were positive for VZV (Figure
4.1). Only 28% of the xenografts were positive for virus from the mice treated with the
anti-gH mAb 206 at 6 hpi (Ab-0-12 group), significantly less than the number positive
in the PBS group (p≤0.01; Fisher’s exact test). In contrast, anti-gH mAb 206 treatment
at 4 dpi (Ab-4-12 group) did not significantly reduce the number of infected skin
xenografts compared to the PBS group (p=0.33; Fisher’s exact test), as 60% of the
xenografts were positive for infectious virus. The number of virus-positive xenografts
in the Ab-0-12 group was also significantly reduced compared to the Ab-4-12 group
(p≤0.01; Fisher’s exact test), indicating that early (6 hpi) treatment with mAb 206 was
capable of preventing VZV infection in 42% of skin xenografts but delaying treatment
until 4 dpi failed to prevent infection in any skin xenografts.
4.4.2. VZV titer was significantly reduced following treatment with the anti-gH mAb
206
In the PBS group, mean virus titer peaked by 28 dpi with a 77-fold increase in
titer compared to day 7 (Figure 4.2A). Mean virus titers were unchanged by 35 dpi,
but fell by 42 dpi. The 30-fold decrease from 28 to 42 dpi was consistent with the
70

depletion of cells permissive for infection in the skin xenograft as a consequence of
VZV replication.
Administration of mAb 206 starting at 6 hpi (Ab-0-12 group) significantly
reduced the titer of recoverable virus at 7 to 35 dpi compared to the PBS group
(p≤0.01; Student’s t test) with a 7- to 70-fold difference between the two groups. By
35dpi, 67% of the Ab-0-12 xenografts were positive for virus, the maximum
percentage in this group. This was the only time point at which >50% of the
xenografts were positive, compared with >50% of xenografts positive for virus in the
Ab-4-12 and PBS groups at 4 and 5 of the time points, respectively (Figure 4.1). By
42 dpi, the maximum titer reached in the Ab-0-12 group was 88-fold higher than the
titer at day 7. The Ab-0-12 titer at 42 dpi was equivalent to the mean titer in the PBS
group at 21 dpi, showing a considerable delay in reaching this titer in the Ab-0-12
group. The peak titer in the PBS group occurred at 28 dpi whereas the maximal titer in
the Ab-0-12 group was delayed by at least 14 days, and peak titer in this group might
have occurred at an even later time point. Thus, antibody treatment at 6 hpi not only
reduced the number of skin xenografts that became infected with pOka, but was
associated with lower virus titers in those that became infected both during and after
antibody treatment until 35 dpi, 23 days after the final antibody dose was
administered.
Virus titers peaked in the Ab-4-12 group by 35 dpi, with a 33-fold increase
from 7 dpi. Mean virus titers were significantly reduced at 7 to 28 dpi compared to the
PBS group (p≤0.02; Student’s t test) and virus was recovered from the majority of skin
xenografts in the Ab-4-12 group from day 21 onward. The time to the highest virus
71

titer was delayed by one week compared to the PBS group and occurred at least one
week earlier than the Ab-0-12 group. Treatment with mAb 206 was associated with
lower virus titers during the administration period, as was seen in the Ab-0-12 group,
and until 28 dpi, 16 days after the last antibody dose was administered. Virus titers
increased with kinetics similar to the PBS titers when antibody treatment was
suspended. This data suggested that passive treatment with anti-gH mAb 206 has
limitations for control of an established VZV infection.
It was conceivable that the homogenates from skin xenografts might have
contained neutralizing antibody that would interfere with plaque formation in the virus
titer assay when the xenograft homogenates were tested for infectious virus. A plaque
reduction assay was performed in which melanoma cells were inoculated with 10
PFU/ml pOka in the presence or absence of xenograft homogenate. Under these
conditions, detection of less than 10 PFU/ml would indicate neutralization of plaque
formation by antibody within the homogenate, while a minimum titer of 10 PFU/ml
would indicate that antibody within the homogenate did not prevent plaque formation.
As expected, virus titers for xenograft homogenates from the PBS group were ≥10
PFU/ml at each time point, with the increased titer being attributable to virus present
in the homogenate (Figure 4.2B). Evidence of neutralizing activity was not seen for
xenograft homogenates from either of the mAb 206-treated groups, as virus titers at
early time points remained at approximately 10 PFU/ml but increased at later time
points. The lack of plaque neutralization of the 10PFU/ml demonstrated that any
residual mAb 206 present within the homogenate did not disrupt plaque formation
within the virus titer assay.
72

4.4.3. VZV genome copies in skin xenografts were reduced following treatment with
the anti-gH mAb 206
As expected from the increase in viral titers for the PBS group, viral genome
copies increased 794-fold from 7 dpi to 42 dpi in the xenografts from which infectious
virus was recovered (Figure 4.2C). Xenografts that did not yield infectious virus had a
mean genome copy number of log10 5-6 copies per μg human DNA throughout the
experiment, with no increase over time. Xenografts that did not become infected from
either of the anti-gH mAb 206-treated groups had similar levels of DNA. This was
likely to be residual VZV DNA from the inoculum, and these data were not included
in the average copy number for each group.
The mean genome copy number in the infected xenografts from mice treated
with mAb 206 at 6 hpi (Ab-0-12 group) followed the same trend as virus titer.
Genome copies increased 40-fold from 7 dpi to 42 dpi (Figure 4.2C). Mean genome
copy numbers were significantly lower at 14 to 42 dpi compared to infected xenografts
from the PBS group (p≤0.05; Student’s t test). Similar to the Ab-0-12 group, treatment
of mice with mAb 206 starting at 4 dpi (Ab-4-12 group) significantly reduced the
mean genome copy number at each time point, except at 14 dpi, when compared to the
PBS group (p≤0.01; Student’s t test). There was a 501-fold increase from 7 dpi to 42
dpi (Figure 4.2C). The significantly lower genome copies in both mAb 206 treatment
groups compared to the PBS group was highly likely to be caused by the reduced virus
spread as determined by virus titer.
73

4.4.4. Kinetics of anti-gH mAb 206 accumulation and clearance
As expected, the PBS group had extremely low levels of serum IgG1
throughout the duration of the 42-day experiment. SCID mice are known to
spontaneously develop partial immune reactivity as they age, resulting in low levels of
serum IgG in approximately 87% of 2-3 month old mice (177). The highest
concentration of IgG1 in the PBS group at 14 dpi was not significantly higher than at
other times points (p≥0.3; Student’s t test) (Figure 4.2D). The low levels of IgG1 were
considered to be background.
The serum levels of IgG1 in mice administered mAb 206 starting at 6 hpi (Ab-
0-12 group) were significantly higher than levels in mock-treated mice (p≤0.02;
Student’s t test), and therefore above background, at all time points (Figure 4.2D).
IgG1 levels in both mAb 206-treated groups peaked by 14 dpi, and then decreased over
time to 42 dpi. The Ab-0-12 group had significantly higher IgG1 levels than the Ab-4-
12 group at 7, 14 and 42 dpi (p≤0.02; Student’s t test). IgG1 levels in the Ab-4-12
group were significantly higher compared to the PBS group at 7-35 dpi (p≤0.01;
Student’s t test). The decrease in serum antibody levels correlated with the increase in
viral titer and genome copies in both antibody-treated groups. The additional dose of
mAb 206 for the Ab-0-12 group at 6 hpi resulted in higher levels of serum IgG1, likely
leading to a greater reduction of viral titers and genome copy numbers.
74

4.4.5. VZV lesion spread in skin xenografts was reduced following treatment with the
anti-gH mAb 206
Lesions were identified in almost all xenografts from which infectious virus
was recovered. Syncytia were found in all lesions. Xenografts from mock-treated mice
had small epidermal lesions at 7 dpi, with only slight disruption of the basement
membrane and penetration of virus into the dermis (Figure 4.3A). At 14 dpi, lesions
extended into the dermis, and by 21 dpi, lesions extended throughout most of the
xenograft (Figure 4.3B & C). In the majority of xenografts examined at 35 and 42 dpi,
the entire xenograft had become infected and had extensive necrosis, as nuclei were
not seen with the hematoxylin counterstain (Figure 4.3E & F). This correlates with the
decrease in titer in the PBS group at these time points.
A single lesion was identified among twelve xenografts from the Ab-0-12
group at 7 dpi (Figure 4.3G). This lesion was contained within the epidermis and was
smaller than the lesions in PBS xenografts at 7 dpi (Figure 4.3A). Lesions were not
seen at 14 dpi, but at 21 dpi, several lesions, smaller than those seen in the PBS group
at 7 dpi, were identified (Figure 4.3H and I). These lesions were contained within the
epidermis or hair follicles, and only small syncytia were seen. Lesions were not seen
at 28 dpi, but by 35 and 42 dpi, lesions extended into the dermis and were
approximately the size of those seen in PBS xenografts at 14 dpi (Figure 4.3J-L).
Small lesions were identified in the epidermis or hair follicles of the xenografts
from the Ab-4-12 group at 7 and 14 dpi (Figure 4.3M and N). At 21 dpi, lesions
extended into the dermis (Figure 4.3O). Lesion size continued to increase until, by 35
and 42 dpi, the majority of the tissue was infected (Figure 4.3P-R). Genome-positive
75

cells, identified by in situ hybridization to detect VZV genomic DNA, correlated with
protein-positive cells in both Ab-4-12 and PBS xenografts at 14 dpi (data not shown).
Overall, lesion size increased in infected xenografts in all groups over time, but the
increase was delayed in both groups administered anti-gH mAb 206, with a more
pronounced delay in the Ab-0-12 group.
4.4.6. Spread and replication of VZV was decreased in vitro after treatment with the
anti-gH mAb 206
The mAb 206 was previously shown to neutralize cell-cell spread (53, 166,
186, 202), but effects on VZV titer and genome copy number have not been
determined. To confirm the neutralizing activity, HELFs were inoculated with pOka
and cultured in the presence or absence of antibody. Small plaques were seen in
HELFs infected with pOka in mock-treated cultures at 24 hpi, increasing in size until
96 hpi, when nearly all cells were infected (Figure 4.4A-D). Treatment of HELFs with
mAb 206 (Ab-96) reduced infection to single cells in the monolayer at 24 hpi, with
only a few small plaques visible at 48 hpi. A small increase was seen in plaque size at
72 hpi, but no further increase was observed at 96 hpi (Figure 4.4E-H). The plaques
seen at 72 and 96 hpi were not much larger than the plaques at 24 hpi in the mock-
treated cells, and the overall increase between 24 hpi and 72-96 hpi was much less
than that seen from 24 to 48 hpi in the mock-treated cells, indicating extremely
inefficient spread of the virus in the presence of mAb 206.
In the absence of mAb 206 (mock-treated cells), the mean virus titer peaked at
72 hpi, and the titer increased 73-fold from 24 hpi (Figure 4.5A). Genome copies
76

increased 251-fold during the 96-hour period (Figure 4.5B). Virus titer and genome
copy number did not significantly increase over the first 48 h in HELFs continuously
treated with mAb 206 (Ab-96 cells), but increased significantly (p≤0.01; Student’s t
test) between 48 and 72 hpi (Figure 4.4A). No further increase was seen between 72
and 96 hpi (Figure 4.5A and B). The peak titer in the Ab-96 wells at 72-96 hpi was 12-
fold higher than at 24 hpi. At all time points, titers in Ab-96 wells were significantly
lower than the titers in mock-treated wells (p≤0.01; Student’s t test) and genome copy
numbers remained statistically equivalent to the mock-treated cells at 24 hpi. The
slight increase in titer and genome copies occurred at the same time as the small
increases in plaque size.
Virus titers were significantly lower than those in mock-treated HELFs at all
time points (p≤0.02; Student’s t test) in cells treated with mAb 206 for only 24 h (Ab-
24) or 48 h (Ab-48) following inoculation (Figure 4.5A). Genome copy number in Ab-
24 wells was significantly lower compared to mock-treated cells at 48 hpi (p=0.01;
Student’s t test) (Figure 4.5B). The virus titer increased 440-fold and genome copies
increased 122-fold over the 96 hours in HELFs treated for 24 h. At 48 and 72 hpi, the
titer was statistically equivalent to mock-treated cells at 24 hpi and 48 hpi (p≥0.13;
Student’s t test), but by 96 hpi was significantly lower than mock at 72 hpi (p≤0.04;
Student’s t test). A similar trend was seen for HELFs treated with mAb 206 for 48 hpi.
Virus titers increased 199-fold and genome copies increased 89-fold between 24 and
96 hpi. The virus titer at 96 hpi was statistically equivalent to mock at 48 hpi, and
genome copies were statistically equivalent at 48-96 hpi to the mock values 24 h prior.
The in vitro data reflected what was seen in vivo with neutralizing activity against
77

pOka causing decreased virus spread, titers and genome copies, but this effect only
persisted while antibody was present.
4.4.7. Anti-gH mAb 206 localized to intracellular vacuoles, virus particles on the cell
surface and virus particles within infected cells
Normal nucleocapsids and enveloped virus particles were found on mock-
treated HELFs infected with pOka (Figure 4.6A). Some virus particles were observed
in aggregates on the surface of cells. Nucleocapsids and enveloped virus particles were
also identified in HELFs infected with pOka and treated with mAb 206, and were
identical to those from mock-treated cells (Figure 4.6B). Thus, mAb 206 did not
appear to disrupt virion assembly.
Based on immunogold EM analysis, the anti-gH mAb was enriched in
numerous endosome-like structures within treated cells (Figure 4.6C-D). Gold
particles within endosomes were identified more frequently within infected cells than
in neighboring uninfected cells, where only one or two gold particles per cell were
found, compared to much more dense labeling in infected cells (Figure 4.6D). The
limited detection of mAb 206 in uninfected but treated HELFs might have been due to
passive uptake of the anti-gH antibody from media or minimal background labeling
with unbound gold particles. The increased uptake within infected cells suggested that
mAb 206 bound to cell surface gH and was internalized. The mAb 206 localized to the
surface of infected cells (Figure 4.6E), and to virus particles (Figure 4.6E-G). These
antibody-virus complexes were observed on the cell surface and within the cell near
the plasma membrane, indicative of endocytosis.
78

4.4.8. mAb 206 was not trafficked to the TGN but colocalized with the endocytic
pathway
Glycoprotein H localized to the cell surface of infected HELFs and to the trans-
Golgi network (TGN) at 48hpi, as determined by confocal microscopy (Figure 4.7A
and C). The TGN is thought to be the site of virus particle secondary envelopment
(91). Glycoprotein H often colocalized with gE both in the TGN and on the surface of
infected HELFs (Figure 4.7A). Glycoprotein H also colocalized with a major capsid
protein, ORF23 (Figure 4.7C). It has previously been demonstrated that ORF23 is
found predominantly in the nuclei of infected cells (47), but ORF23 staining on the
surface of cells or near nuclear membranes has been associated with incoming virions
clustered on the plasma membrane or capsids along the nuclear envelope (199).
Therefore, in this study, ORF23 staining that did not colocalize with the nuclear
marker but did colocalize with gH was identified as representing enveloped virions
clustered on the surface of infected cells, in agreement with the small aggregates of
virion particles or individual virions lining the surface of infected cells as seen by
electron microscopy.
Similar to the gH localization by confocal microscopy, mAb 206 localized to the
cell surface and colocalized with gE and ORF23 on infected cell surfaces, but did not
localize to the TGN (Figure 4.7B and D). Colocalization between ORF23 and gH or
mAb 206 on the cell surface was confirmed in non-permeabilized cells (data not
shown). The similarities between gH and mAb 206 localization demonstrated that
mAb 206 bound to gH on the surface of infected cells and on virions, but the lack of
colocalization between mAb 206 and the TGN was markedly different compared to
79

the localization of gH within the TGN. In treated uninfected cells, minimal mAb 206
was seen, mostly localizing to the surfaces of cells (Figure 4.7B and D).
Glycoprotein H and mAb 206 often had a punctate distribution pattern within the
cytoplasm of infected cells. Some but not all gH or mAb 206 colocalized with the
early endosome marker EEA1 (Figure 4.8A and B, arrowheads). This indicated that
vesicles labeled with mAb 206 by immunogold EM were likely to be endosomes. The
gH and mAb 206 also colocalized with Vps4, a marker for the multivesicular body
(MVB) pathway, in somewhat diffuse patches or punctae within cells (Figure 4.8C
and D, arrowheads). The colocalization with EEA1 and Vps4 indicated that the
endocytic and MVB pathways were involved in trafficking of both gH and the mAb
206-bound gH, albeit to different destinations as the mAb 206-gH complex did not
reach the TGN while gH did.
80

4.5. Discussion
VZV glycoprotein H is predicted to play an essential role in VZV virulence
and is a known target of the humoral immune system during infection (8). The present
study of the SCIDhu mouse model showed for the first time that gH contributes to
VZV infection and cell-cell spread of virus in skin xenografts in vivo, and confirmed
this contribution in vitro. The low levels of persistent VZV replication and spread
observed in some VZV-infected xenografts in vivo and in HELF cells in vitro in the
presence of anti-gH antibody indicate that passive antibody interference with gH
functions has some limitations. This residual spread might have resulted from
incomplete binding of antibody to all functional gH. Alternatively, other VZV
glycoproteins might be capable of mediating some virion-cell or cell-cell fusion in the
absence of functional gH. It has been suggested that VZV gH and gL or gB and gE
might induce fusion (66, 154), although if so, VZV would be the only herpesvirus
investigated to date that does not require both gB and gH for this event.
This report also demonstrated for the first time that the administration of anti-
gH antibody was effective at preventing or reducing VZV pathogenicity in human
skin. Since these animals are immunodeficient (27), this inhibitory effect could be
assessed in the absence of a polyclonal B cell response to multiple viral proteins and
without VZV-specific cell-mediated immunity. The prevention of infection might be
attributed to a block on virus attachment and entry into cells. The reduced
pathogenicity in xenografts that became infected might be attributed to the block in
spread of virus from cell-to-cell, inhibition of canonical gH trafficking and potential
virus particle degradation. The mAb 206 binds to a conformation-dependent epitope
81

on mature glycosylated gH, and neutralization of VZV is complement-independent
(166). Two potential mechanisms for mAb 206 neutralization of VZV are inhibition of
receptor binding and attachment or inhibition of fusion. The antibody might also
disrupt post-entry events such as interactions between virus proteins and trafficking of
proteins or virion particles (Figure 4.9).
Neutralizing antibodies to herpesviruses block attachment by physically
blocking interaction and preventing binding of virus proteins to cellular receptors
(174, 180, 241). A VZV gH receptor has not been identified, and the cell-associated
nature of VZV prevents study of VZV attachment steps. If gH interaction with a
cellular protein is required for attachment, neutralizing antibody could disrupt binding
and prevent infection (Figure 4.9, Step 1).
EBV, human cytomegalovirus (HCMV) and human herpesvirus-6 and -7
(HHV-6 and -7) gH/gL form a complex with additional glycoproteins. Some of these
complexes appear to determine cell-specific infectivity, receptor specificity, or the
route of virus entry into the cell (169, 208, 231, 235). No interactions with cellular
proteins have been identified for VZV gH, but antibody binding to gH could disrupt or
prevent formation of a protein complex required for receptor binding, thereby
preventing infection (Figure 4.9, Step 1).
Neutralizing anti-gH mAb might mask functional domains of VZV gH,
preventing fusion and entry. VZV gH and gL can mediate fusion between cell
membranes when expressed in vitro in a vaccinia vector (66). Many anti-HSV gH
neutralizing antibodies block virus penetration and prevent entry (82). HSV gH is
required for hemifusion, and one mAb specifically blocks this fusion step (217).
82

Deletion or mutation of predicted HSV gH heptad repeats and α-helical coiled-coils
disrupts virus infectivity and cell-cell fusion (93, 96). Mimetic peptides of the α-
helices interact with lipid membranes (84, 93). When the HSV α-helix-1 was replaced
with the positionally conserved α-helix from VZV, the resulting chimeric gH was
capable of promoting cell-cell fusion and rescuing infectivity of an HSV gH-negative
virus, indicating that VZV gH contains a functional α-helix capable of mediating
fusion (94). Thus, the reduced number of infected skin xenografts and cell-cell spread
during mAb 206 treatment could potentially have resulted from inhibition of virus
entry into cells (Figure 4.9, Step 1 and 2).
Glycoprotein trafficking from the plasma membrane to the Golgi can occur via
clathrin-coated vesicles (26).VZV gH endocytosis is antibody-independent but
clathrin-dependent (185), and this report has demonstrated that anti-gH mAb 206
binds gH and is internalized with gH (Figure 4.9, Step 2). The mAb 206 and gH
colocalized with EEA1, a marker for early endosomes, and Vps4, a marker for
multivesicular bodies (MVB), indicating that mAb 206 and gH underwent endocytosis
and sorting via the MVB pathway. Vps4 is required for transport of endocytosed
proteins between pre-vacuolor endosomes and vacuoles (15). Vps4 is also required for
autophagy, presumably for autophagosome-endolysosome fusion (204). Endocytosed
proteins can be sorted to the Golgi via the endocytic recycling pathway or late
endosomes. The colocalization between gH and Vps4 might occur during one of the
sorting steps as gH is targeted to the TGN. Alternatively, the association of gH and
Vps4 might indicate that VZV uses the MVB pathway for assembly and egress of
virus particles, as has been suggested following studies of HSV gB colocalization with
83

Vps4 during envelopment and egress (45). The mAb 206-gH complexes were not
targeted to the TGN. The lack of these complexes in the TGN might have resulted
from the experimental system, but they might also demonstrate that the antibody
disrupts gH trafficking, although the complexes did travel through endosomes and the
MVB pathway. VZV gH interacts with gE on the plasma membrane and in virions,
and it has been suggested that this interaction results in the targeting of gH to the TGN
for secondary envelopment via the TGN-targeting motif of gE (155, 186). Binding of
mAb 206 to gH particles expressed on the surface of infected cells does not disrupt
endocytosis of gH, but the resulting mAb-gH complex might not interact with other
virion proteins, resulting in the altered trafficking of gH to sites other than the TGN
(Figure 4.9, Step 3). These complexes colocalize with Vps4, similar to gH, but rather
than sorting to the TGN, they could be sorted through late endosomes to lysosomes or
autophagosomes, which are induced during VZV infection (221).
The mAb 206 might also direct gH complexes to be degraded by interacting
with Fc receptors on the surface of infected cells. Antigen-antibody complexes bound
to Fc receptors on human fibroblasts undergo endocytosis and are targeted for
degradation, and these fibroblast Fc receptors mainly interact with monomeric
immunoglobulins, especially IgG1 (57, 81). Skin keratinocytes have also been shown
to express functional Fc receptors that interact with IgG (224). Antibodies against
pseudorabies virus (PrV) gD and gB expressed on monocytes induce antibody-
dependent, clathrin-dependent endocytosis of the glycoproteins mediated by tyrosine-
based endocytosis motifs (72, 75, 228). VZV gH contains a functional tyrosine-based
endocytosis motif (185). Thus, mAb 206 would not have to disrupt gH protein
84

interactions, but instead could interact with cellular Fc receptors in order to alter
trafficking of gH and target it for degradation via the Vps4/MVB pathway and sorting
to either lysosomes or autophagosomes (Figure 4.9, Step 3).
Immunogold EM analysis of mAb 206 localization within treated infected cells
demonstrated that mAb 206 bound to gH on virus particles on the cell surface, which
resulted in neutralization of these particles and prevention of virus spread from cell to
cell (Figure 4.9, Step 4). The mAb 206-labeled virions were found within cells,
suggesting that labeled particles on the surface of infected cells were internalized. A
mAb 206 interaction with Fc receptors could direct not only the internalization and
degradation of gH, but also of virions containing gH in their envelope (Figure 4.9,
Step 5).
Interferon production is activated in human epidermal cells in VZV-infected
skin xenografts and modulates the progression of lesion formation (146). The
experiments reported here with anti-gH antibody showed that infection was
suppressed during treatment but progressed to complete destruction of the skin tissue
when anti-gH antibody was cleared from the circulation. Together, these findings
indicate that the combination of the innate interferon response of epidermal cells and
passively administered antibody is not sufficient to eliminate VZV when replication
has been initiated in skin. This suggests that an effective cell-mediated immune
response to VZV is likely to be necessary to resolve primary VZV infection. Herpes
zoster as a result of VZV reactivation has been suggested to result in part from a
decreased cellular immune response as patients age, even though the humoral immune
response remains high (8). The observations presented here suggest that pre-existing
85

anti-gH antibodies might reinforce the innate IFN response and slow the progression
of cell-cell spread for an interval that allows for clonal expansion of memory VZV-
specific T cells that make IFNγ and stimulate B cells and cytotoxic T lymphocytes to
respond to VZV reactivation.
Anti-gH antibody administration to SCIDhu mice with pOka-infected skin
xenografts prevented infection or reduced pathogenicity if infection had been
established. This mirrors the prophylaxis of immunocompromised patients, who are
administered varicella-zoster immunoglobulin (VZIG) as soon as possible after
exposure, up to 96h post-exposure to the virus (156). Neutralization assays
demonstrated that humanized mAb 206 had a biological activity that was 2400-fold
that of the standard VZIG preparation (63). Administration of VZIG does not
consistently prevent varicella following exposure but can ameliorate the severity of the
disease, although severe disease and death can still occur. The data presented here
demonstrated that administration of anti-gH antibody immediately after inoculation
prevented infection, but delaying treatment by 4 days resulted only in suppression of
infection, emphasizing the need to give prophylaxis to exposed patients as soon after
exposure as possible. This has potential clinical relevance because maintaining the
supply of VZIG has been challenging, and anti-gH monoclonal antibody might be
developed as an alternative for prophylaxis of patients at risk of severe primary VZV
infection.
86

Figure 4.1. VZV infection of human skin xenografts in SCIDhu mice treated
with anti-gH mAb 206 for 0-12 days or 4-12 days post inoculation
The timeline of treatment is shown above the percentage of xenografts from which
infectious virus was recovered. Skin xenografts were inoculated with pOka-infected
HELF at a titer of log10 5.3 PFU/ml, and mice were administered anti-gH mAb 206
(solid line, administration time points indicated with “A”) or PBS (dashed line,
administration time points indicated with “P”). “PBS” designates the control group
treated with PBS. “Ab-0-12” designates the group treated with mAb 206 at 6 hpi, 4
days, 8 days and 12 dpi. “Ab-4-12” designates the group treated with mAb 206 at 4
days, 8 days and 12 dpi. Xenografts were collected at 7-day intervals up to 42 dpi. The
total percentage of infected xenografts over the 42-day interval is shown on the right.
87

Figure 4.2. Replication of VZV in human skin xenografts in SCIDhu mice treated
with anti-gH mAb 206 for 0-12 days or 4-12 days post inoculation
(A) Mean virus titers recovered from VZV-positive xenografts. (B) Neutralization
capacity of mAb 206 within xenograft homogenates against a standard inoculum of 10
PFU/ml of VZV. (C) Mean VZV genome copy number per μg human DNA from
VZV-positive xenografts measured by quantitative PCR for ORFs 31 (gB), 62 (IE62)
and 63 (IE63). Each gene target was measured in duplicate and the mean of each was
used to determine genome copies. (D) Concentration of IgG1 in the sera of mAb 206-
or PBS-treated mice. Standard error of the mean is shown on all graphs.
88

Figure 4.3. The formation of lesions in VZV-infected human skin xenografts
treated with the anti-gH mAb 206 for 0-12 days or 4-12 days post inoculation
Lesions in skin xenografts inoculated with pOka-infected HELF were identified by
VZV gE expression (red), and were counterstained with hematoxylin (blue).
Representative lesions are shown at each time point: 7 dpi, A, G, M. 14 dpi, B, H, N.
21 dpi, C, I, O. 28 dpi, D, J, P. 35 dpi, E, K, Q. 42 dpi, F, L, R. The total number of
xenografts with lesions is shown in the lower left of each panel. Lesions were
identified in xenografts from the PBS (A-F) and Ab-4-12 (M-R) groups at all time
points. Lesions were only observed in xenografts from the Ab-0-12 group (G-L) at 7,
21, 35 and 42 dpi. Representative uninfected xenografts are shown for the Ab-0-12
group at 14 and 28 dpi. Syncytia were seen in all lesions. Magnification: 50X.
89
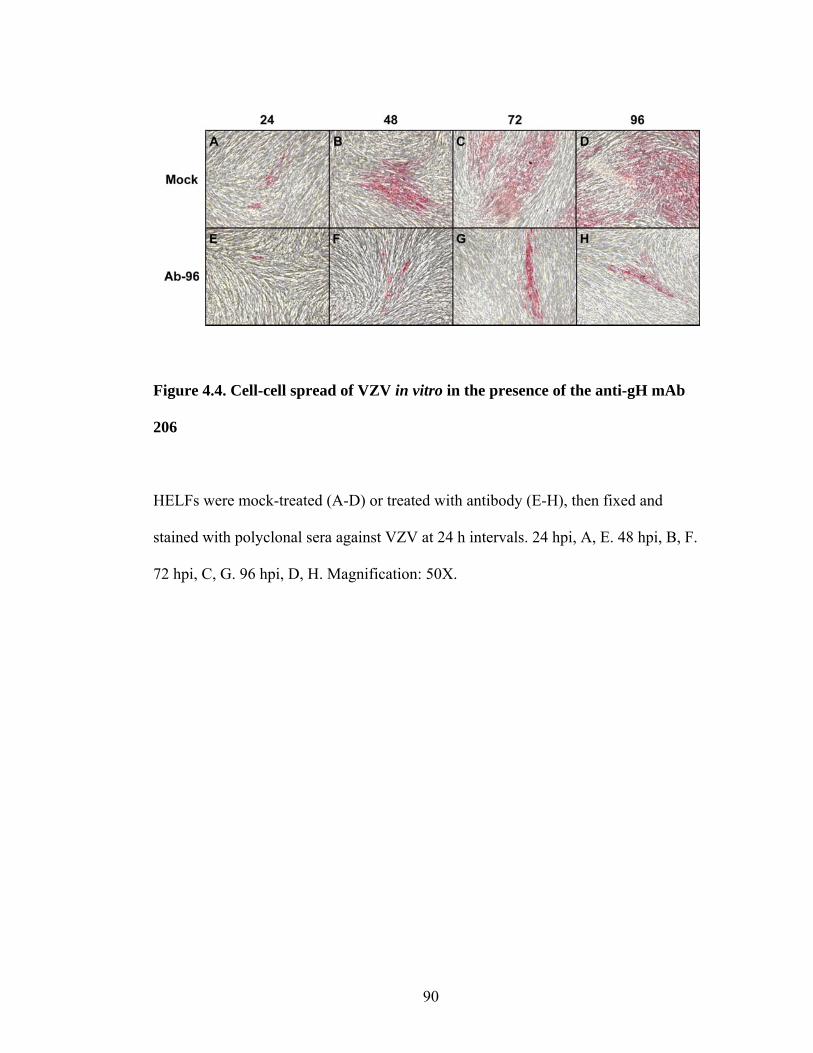
Figure 4.4. Cell-cell spread of VZV in vitro in the presence of the anti-gH mAb
206
HELFs were mock-treated (A-D) or treated with antibody (E-H), then fixed and
stained with polyclonal sera against VZV at 24 h intervals. 24 hpi, A, E. 48 hpi, B, F.
72 hpi, C, G. 96 hpi, D, H. Magnification: 50X.
90

Figure 4.5. VZV replication in vitro in the presence of the anti-gH mAb 206
HELF cells were inoculated with pOka and grown in media without antibody (Mock)
or media supplemented with mAb 206 for 24, 48 or 96 h (Ab-24, Ab-48 or Ab-96,
respectively). Cells were harvested at 24 h intervals for 96 h. (A) Mean titer in HELF
cultures. (B) Mean genome copy number based on quantitative PCR for ORFs 31
(gB), 62 (IE62) and 63 (IE63) per ng human DNA. Each gene target was measured in
duplicate and the mean of each was used to determine genome copies. Standard error
of the mean is shown on both graphs.
91

Figure 4.6. Virus particle formation and the localization of mAb 206 in infected
cells in vitro
HELFs inoculated with pOka were mock-treated (A) or treated with antibody (B-G),
and were fixed at 48 hpi. (A and B) TEM of infected and untreated (A) or treated (B)
cells. Arrowheads and right inset boxes show virion particles on the cell surface.
Arrows and left inset boxes show nucleocapsids. (C-F) Immunogold labeling of mAb
206 in treated cells. Arrows indicate mAb 206 on the cell surface or within vacuoles.
Arrowheads indicate mAb 206 on virus particles. (D) Infected cell (top) with mAb 206
labeling next to uninfected cell (bottom) lacking dense mAb 206 labeling.
Magnification: A-B, 10,000X; C-F, scale bars: 0.2μm.
92
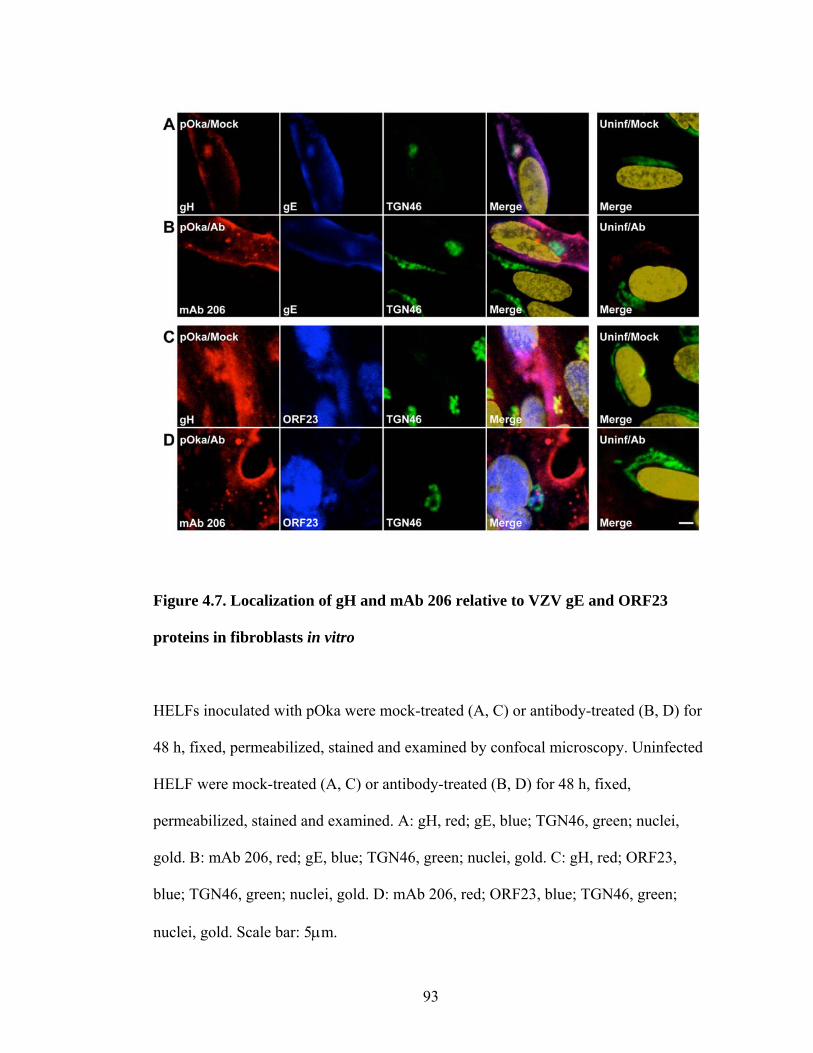
Figure 4.7. Localization of gH and mAb 206 relative to VZV gE and ORF23
proteins in fibroblasts in vitro
HELFs inoculated with pOka were mock-treated (A, C) or antibody-treated (B, D) for
48 h, fixed, permeabilized, stained and examined by confocal microscopy. Uninfected
HELF were mock-treated (A, C) or antibody-treated (B, D) for 48 h, fixed,
permeabilized, stained and examined. A: gH, red; gE, blue; TGN46, green; nuclei,
gold. B: mAb 206, red; gE, blue; TGN46, green; nuclei, gold. C: gH, red; ORF23,
blue; TGN46, green; nuclei, gold. D: mAb 206, red; ORF23, blue; TGN46, green;
nuclei, gold. Scale bar: 5μm.
93

Figure 4.8. Localization of gH and mAb 206 relative to EEA1 and Vps4 in
fibroblasts in vitro
HELFs inoculated with pOka were mock-treated (A-D, K-N) or antibody-treated (F-I,
P-S) for 48 h, fixed, permeabilized, stained and examined by confocal microscopy.
Uninfected HELF were mock-treated (E, O) or antibody-treated (J, T) for 48 h, fixed,
permeabilized, stained and examined. Arrowheads highlight colocalization. A: gH,
red; EEA1, blue; TGN46, green; nuclei, gold. B: mAb 206, red; EEA1, blue; TGN46,
green; nuclei, gold. C: gH, red; Vps4, blue; TGN46, green; nuclei, gold. D: mAb 206,
red; Vps4, blue; TGN46, green; nuclei, gold. Scale bar: 5μm.
94

Figure 4.9. A schematic of mechanisms for antibody disruption of gH function
and trafficking and potential effects on the pathogenesis of VZV skin infection in
vivo
(1) Anti-gH antibody present soon after inoculation can bind and neutralize virus
particles on the inoculum cells, preventing VZV attachment or entry into skin
epidermal cells. (2) Antibody present after infection has been established can bind gH
expressed on the infected cell surface, preventing cell-cell fusion. (3) These antibody-
gH complexes are internalized, and antibody-bound gH is not targeted for secondary
envelopment and incorporation into virions, but instead targeted for degradation. (4)
Antibody binds to gH on the envelope of surface virus particles, blocking envelope
fusion with neighboring cell membranes and inhibiting cell-cell spread of the virus. (5)
Antibody-virion complexes are internalized and targeted for degradation.
95

CHAPTER V
VZV GLYCOPROTEIN H AND L:
ANALYSIS OF PROTEIN CONSERVATION AND
PREDICTED FUNCTIONAL MOTIFS
96

5.1. Introduction
This chapter has several purposes. The first is to provide an analysis of
glycoprotein H (gH) and glycoprotein L (gL) protein sequence conservation among
the human herpesviruses, alphaherpesviruses and sequenced Varicella-zoster virus
(VZV) isolates. Conservation studies among the herpesviruses have been done, mostly
at the genome level between two or three viruses. Additionally, single nucleotide
polymorphisms (SNPs) have been used to analyze VZV isolates originating from
around the globe (230). However, no analysis has focused exclusively on gH or gL
protein sequence conservation among the Herpesviridae. The second purpose of this
chapter is to analyze possible functional motifs within gH and gL using bioinformatics
prediction algorithms. It has been demonstrated that gH and gL are glycosylated, and
that gH has a functional endocytosis motif (65, 66, 153, 166, 185). VZV gH has been
suggested to have heptad repeats and α-helices that are positionally conserved relative
to these motifs in Herpes simplex virus (HSV) (94, 95). Currently, though, a thorough
analysis of possible functional motifs within gH and gL has not been done. The aim of
this chapter, therefore, is to provide both an analysis of gH and gL protein sequence
conservation, as well as a description of possible functional motifs within gH and gL.
97

5.2. Materials and Methods
5.2.1. Sequences
Sequences were obtained from the National Center for Biotechnology
Information (NCBI) database. The VZV parent Oka (pOka) gH (accession number
AAK19939) and gL sequences (accession number AAK19941) were used for
sequence comparison and for prediction of structural and functional motifs. For
determination of protein sequence similarity and identity, only published sequences
were analyzed, and where possible, the gH and gL sequences from the same virus
isolate were used in sequence analysis. All gH and gL VZV isolate sequences were
available from the NCBI database but not all were published.
5.2.2. Sequence conservation analysis
Sequences were aligned using ClustalX 1.0. Similarity and identity were
calculated from the aligned sequences using GeneDoc, available from the National
Resource for Biomedical Supercomputing. GeneDoc was used to highlight residues of
interest. The NCBI algorithm Basic Local Alignment Search Tool (BLAST) was also
used to align VZV gH and gL with all available sequences in the NCBI database and
to identify regions of these proteins that closely align with other herpesvirus gH and
gL sequences (5).
5.2.3. Prediction algorithms to identify functional motifs
Multiple algorithms were used to analyze the location of a signal peptide and
any transmembrane regions. Many signal and transmembrane domain prediction
98

programs use neural networks and hidden Markov models to predict these sequences.
Neural network modeling is a method that uses an artificial neural network employing
simple elements to predict global patterns, and it can be used in classification of
pattern or sequence recognition. A hidden Markov model is a statistical model in
which it is assumed that the entity being modeled is a Markov process in an
unobserved state. A Markov process, a probability theory from Andrei Markov, is a
random phenomenon that has a specific property. In a hidden Markov models, the
state of the entity is not known but the output from the entity is, and there is a
probability distribution of possible outcomes.
The SignalP 3.0 Server, a prediction server available on the Center for
Biological Sequence Analysis (CBS) website hosted by the Technical University of
Denmark, predicts signal sequences based on both neural networks and hidden
Markov models trained on eukaryotes (19). TMpred is a transmembrane prediction
program available from the European Molecular Biology Network (EMBnet), hosted
by the Swiss Institute of Bioinformatics. TMpred predicts transmembrane helices 17-
33 amino acids (aa) in length based on statistical analysis and comparison to a
database of naturally occurring transmembrane proteins, called TMbase (115). The
Biocomputing Unit in the Department of Biology at the University of Bologna
provides TRAMPLE, a web server with multiple algorithms for the prediction of
transmembrane protein sequences (68). The algorithms used described briefly. The
Signal Peptide Predictor, trained on eukaryotes, uses two neural networks, one to
predict the signal peptide and one to predict the cleavage site. The ENSEMBLE
algorithm predicts α-helices based on an ensemble of neural networks and hidden
99

Markov models. The HTMR, Psi Kyte-Doolittle and Kyte-Doolittle algorithms use a
neural network-based predictor based on evolutionary information derived from non-
redundant data sets of protein sequences and from the Kyte-Doolittle hydrophobicity
scale. The PRODIV_TMHMM_0.91 and TMHMM_2.0 algorithms predict
transmembrane helices using hidden Markov models. The MEMSAT algorithm
predicts transmembrane regions and secondary structure based on the topology of the
sequence compared to the topology of well-characterized proteins.
Potential glycosylation residues were identified using five algorithms. The
Center for Biological Sequence Analysis (CBS) server, hosted by the Technical
University of Denmark, provides NetNGlyc 1.0, NetOGlyc 3.1, and YinOYang 1.2.
NetNGlyc 1.0 predicts NX(S/T) glycosylation sites using artificial neural networks
(106). NetOGlyc 3.1 predicts O-glycosylation with mucin-type N-acetylglucosamine
(GalNAc) based on mammalian neural networks (106). YinOYang 1.2 predicts O-β-
N-acetylglucosamine (GlcNAc) attachment sites in eukaryotic proteins based on
neural network predictions (128). The Eukaryotic Linear Motif (ELM) predicts
functional sites in eukaryotic proteins based on patterns (100). Motif Scan uses hidden
Markov models to predict functional motifs (183).
Potential phosphorylation sites were identified using five algorithms, including
those available from ELM and Motif Scan. The CBS server provides NetPhos 2.0 and
NetPhosK. NetPhos 2.0 uses neural networks to predict phosphorylation of serine,
threonine and tyrosine residues (22). NetPhosK 1.0 produces neural network
predictions of kinase-specific eukaryotic protein phosphorylation sites (23). The
KinasePhos algorithm, available from the Integrative Systems Biology Laboratory at
100

the Institute of Bioinformatics and Systems Biology in Taiwan, uses hidden Markov
modeling to predict the substrate and kinase class (117).
Potential coiled coils were predicted using the COILS algorithm, available
from EMBnet, which compares a given sequence to a database of known coiled-coils
and derives a similarity score. Analysis was done with a 14, 21 or 28 amino acid
window and a MTIDK matrix (152). A weighted scan, where the hydrophobic “a” and
“d” positions within the 7 aa comprising the heptad were given equal weight to the
remaining five hydrophilic positions, and an unweighted scan, where all residues are
given equal weight, was done. This algorithm can predict false positives within highly
charged sequences even if they lack heptad periodicity and coiled-coil forming
potential. A drop of more than 20-30% from the unweighted to the weighted score
might indicate a false positive.
Predicted α-helices were identified using several algorithms available from
TRAMPLE (68). These were the ENSEMBLE, HTMR, Psi Kyte-Doolittle and Kyte-
Doolittle algorithms, described above, and the Secondary Structure Predictor
algorithm, which predicts α-helix, β-strand and random coils from neural network
computation and profile generation based on soluble globular proteins.
The ELM algorithm also predicted other motifs in gH and gL, including the
YXXφ motif.
101

5.3. Glycoprotein H protein sequence conservation
5.3.1. Conservation of VZV gH protein sequence compared to gH sequences from the
human herpesviruses and from the subfamily Alphaherpesvirinae
There are eight human herpesviruses: VZV, HSV1 and HSV2 in the subfamily
Alphaherpesvirinae, Human cytomegalovirus (HCMV), Human herpesvirus 6 (HHV6)
and Human herpesvirus 7 (HHV7) in the subfamily Gammaherpesvirinae, and
Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) in the
subfamily Betaherpesvirinae. For a review, please see Chapter I, Section 1.4.
Glycoprotein H is the second most conserved herpesvirus glycoprotein, and
much of the gH conservation is in the C-terminus of the protein (99). Based on gH
protein sequences, the most closely related viruses among the eight human herpesvirus
are HSV1 and HSV2, with 85% similarity and 78% identity, and HHV6 and HHV7
with 63% similarity and 38% identity (Table 5.1 and 5.2 and Figure 5.1).
The VZV gH protein sequence shares 38% similarity and 20-21% identity with
HSV1 and HSV2, the only other human alphaherpesviruses, over the whole protein.
VZV gH has 21-25% similarity and less than 20% identity with the human beta- and
gammaherpesviruses. Only identity or similarity above 20% was considered valid.
The VZV gH protein sequence has higher similarity and identity to the human
herpesviruses across smaller sequences, identified using BLAST. VZV gH aa 148-841
have 41% similarity and 25% identity to aa159-815 in HSV1, and aa 207-814 have
41% similarity and 25% identity to aa 220-815 of HSV2 (5). Although the entire VZV
gH protein has only 23% similarity to Epstein-Barr virus (EBV) gH, aa 481-797 have
39% similarity to aa 396-678 in EBV. The BLAST algorithm does not identify any
102

regions of significant similarity or identity between VZV gH and the remaining human
herpesviruses. This emphasizes that although certain regions of gH are highly
conserved, as is gH function in virus binding, entry and fusion, the overall gH protein
sequence is not highly conserved among the human herpesviruses.
The gH protein sequence is only slightly more conserved among the
Alphaherpesvirus subfamily than among the human herpesviruses. The
Alphaherpesvirinae subfamily contains four genera: Varicellovirus (including VZV),
Simplexvirus, Mardivirus and Iltovirus. For a review, please see Chapter I, Section
1.1. VZV gH has 71% similarity and 53% identity to Cercopithecine herpesvirus 9, or
Simian Varicella virus (CeHV9/SVV) (Table 5.3 and 5.4 and Figure 5.2). It has 34-
48% similarity and 22-30% identity with other alphaherpesviruses in the
Varicellovirus genus, including those that infect bovine, feline, canine, or equine
species. Among this genus, VZV gH has the lowest homology, 34% similarity and
22% identity, with Suid herpesvirus 1, or Pseudorabies virus (SHV1/PRV). However,
when VZV and PRV gH protein sequences are compared using the BLAST algorithm,
aa 229-832 in VZV gH have 50% identity and 32% homology to aa 140-680 in PRV.
VZV gH has 26-37% similarity and 20-22% identity with most of the
alphaherpesviruses in the Simplexvirus genus (Table 5.3 and 5.4 and Figure 5.2). It
does not have an identity higher than 20% when compared to Saimiriine herpesvirus 1
(SaHV1). VZV gH shares 35-38% similarity and 20% identity with the Mardiviruses,
and 27-30% similarity but no identity above 20% with the Iltoviruses. There appears
to be little variation in sequence similarity among these families, again illustrating that
gH function is likely conserved although the protein sequence varies.
103

5.3.2. Conservation of the gH protein sequence among sequenced VZV clinical and
laboratory isolates
The VZV gH sequence from multiple laboratory and clinical isolates have been
reported. When these were aligned and compared to the VZV pOka gH sequence, five
amino acid changes were identified (Table 5.5). Among the laboratory isolates, the
pOka and vaccine Oka (vOka), as well as the vaccine isolates VariVax and VarilRix,
which were derived from pOka, are identical. One Ellen laboratory isolate contains a
R71K mutation. Three Dumas isolates and one Ellen isolate contain mutations at
L269P and K700R. The L269 and K700 amino acids are common to phylogenetic
clades originating in Asia, and the K71 amino acid is common to North
American/European clades (230).
Among the clinical isolates, eight of twenty-two sequences are identical to the
pOka sequence. Five isolates contain the R71K mutation, similar to the Ellen isolate.
Of these, one contains an additional S319A mutation and a second contains additional
S319A and T639A mutations. One clinical isolate is identical to Dumas with L269P
and K700R mutations, while seven clinical isolates contain only the mutation at
K700R. One clinical isolate, originating in Morocco, has a unique N551H mutation.
104

5.4. Prediction of functional motifs in VZV gH
VZV gH is thought to play a role in virus entry and fusion, and might play a
role in other aspects of VZV infection and replication, such as receptor binding, cell
signaling, nucleocapsid egress or virus assembly (see Chapter III). Identification of
possible functional motifs within VZV gH will likely contribute to identification of
areas that can be targeted for mutational analysis. To date, the only functional motif
identified within VZV gH is a YXXφ endocytosis motif at 835YNKI838 (185).
5.4.1. Signal sequence and transmembrane domain
gH has been reported to be a type-1 transmembrane protein, which is a protein
with the N-terminus in the extracellular space, a single transmembrane region, and the
C-terminus in the cytoplasm (60). However, this has only been inferred from the
sequence of gH. A number of transmembrane regions were identified in gH using
prediction algorithms outlined in Section 5.2.3 (Table 5.6). It is possible that gH
contains several transmembrane sequences and is not a type-1 transmembrane protein.
However, prediction agreement among these algorithms is mostly found in the area of
the signal peptide and of a single transmembrane region at the C-terminus of gH.
It is possible that the additional predicted regions are not actually
transmembrane segments. Instead, they might be α-helices with a hydrophobic nature
that reside in the extracellular space, possibly in the center of the protein structure and
thus are not exposed to the environment. Indeed, the TRAMPLE server cautions that
the provided algorithms might “detect isolated or locally distributed membrane α-
helices, due to eventual common features or chemical-physical properties carried by
105

this kind of secondary structures”. One problem with the neuronal network and hidden
Markov models is that they detect sequences with common characteristics, but the
stringency of the modeling might result in the identification of additional sequences. If
this is the case and gH is a type-1 transmembrane protein, the consensus signal
sequence is predicted to be in the first twenty amino acids and the transmembrane
region is predicted to span approximately 25 to 30 aa at the C-terminus, originating
around aa 800. Several of the remaining predicted transmembrane sequences are
discussed below (see sections 5.2.4 and 5.2.6).
5.4.2. Glycosylation
gH is thought to originate as a 79 kDa protein that is processed to a 94 kDa
precursor and then a 118 kDa mature protein (166). The processing to the 94 kDa form
takes places in the endoplasmic reticulum (ER), where high mannose oligosaccharides
are added, and the processing to the 118 kDa form requires transport to the Golgi
apparatus, where some high mannose oligosaccharides are further processed into
complex-type oligosaccharides. All of these glycomoieties are thought to be N-linked
oligosaccharides.
Many proteins that are secreted or expressed on the cell surface are highly
glycosylated. N-linked glycosylation results from the attachment of oligosaccharides
to a nitrogen residue on the side chain of an asparagine residue within the recognition
motif NX(S/T), where X is any amino acid except proline and S/T is a serine or
threonine residue (20). O-linked glycosylation occurs when oligosaccharides are
linked to an oxygen atom in the side chain of serine or threonine. N-linked
106

glycosylation takes place initially in the ER and can be modified in the Golgi. O-
linked glycosylation takes place in the Golgi. The addition of oligosaccharides can
determine protein folding and targeting. It has been suggested that N- and O-linked
glycosylation might facilitate targeting of transmembrane or secreted glycoproteins to
the apical membrane of epithelial cells (119).
A number of possible glycosylation sites were identified (Table 5.7). Some of
these sites (T16 and N18) are within the predicted signal peptide and might not be
glycosylated. The S319A amino acid change in some strains of VZV would abolish
the predicted glycosylation motif at N317, possibly resulting in a protein with less
glycosylation. Only two O-linked glycosylation sites were predicted. gH does contain
a number of predicted O-linked β-N-acetylglucosamine sites (GlcNAc), which are
normally found on nuclear or cytoplasmic proteins (41). Proteins with GlcNAc
modifications are involved in transcription, ubiquitination, cell cycle regulation and
stress responses. GlcNAc modification, similar to phosphorylation, can cycle on and
off to alter protein activation of function, and many GlcNAcylated sites might also be
targeted by Serine/Threonine kinases. While gH is not a nuclear or cytoplasmic
protein, it has been suggested to play a role in cell signaling, and it is possible that
GlcNAc sites contribute to this function. However, enzymatic digests of gH have
suggested that it does not contain any O-linked glycomoieties (166).
5.4.3. Phosphorylation
Phosphorylation can play an important role in protein interaction and signaling
pathways. Although it has been reported that gH is not phosphorylated (167), a
107

number of phosphorylation sites were predicted in gH (Table 5.8). Of note, residues
T28, T127, S131, T286 and T841, which were all predicted to be GlcNAcylated (see
Section 5.2.2), are also predicted to be phosphorylated. The majority of these sites fall
in the predicted ectodomain of gH, assuming gH is a Type-1 transmembrane protein
(see Section 5.2.1), and phosphorylation of the protein ectodomain is rare. However,
the Marburg glycoprotein GP has been reported to be phosphorylated at a serine
residue in the ectodomain during protein processing in the Golgi apparatus (210). This
phosphorylation likely occurs via luminal protein kinases, such as casein kinase II
(CKII) or Golgi casein kinase. gH has several of predicted CKII phosphorylation sites.
5.4.4. Heptad repeats and fusion peptides
Class I fusion proteins are often cleaved and from trimers that interact with the
cell membrane and induce fusion of the virus and cell membranes. Influenza
hemagglutinin is a typical example of this class (54). A class I fusion protein has two
distinct components: fusion peptides and heptad repeats. Fusion peptides are α-helices
composed of hydrophobic residues. These peptides are exposed during the fusion
event, and insert themselves into the cell membrane. Heptad repeats are tandem
repeats of 14, 21 or 28 amino acids composed of groups of seven amino acids. Within
these seven amino acids, hydrophobic residues are located in position “a” and “d” and
hydrophilic or polar residues are located at positions “b”, “c”, “e”, “f” and “g”. These
heptad sequences take on the secondary structure of α-helices with the hydrophobic
residues arranged in a stripe around the helix. These α-helices then interact to form the
coiled-coil, and this interaction buries the hydrophobic residues inside the coiled-coil
108

structure. Multiple heptad repeats can interact to form a dimer or trimer of coiled-
coils, and this interaction can cause conformational changes in a protein (124). The
conformational change induced by the heptad repeats results in a newly folded protein
where the transmembrane anchor, and therefore the virus membrane, and the fusion
peptide, and therefore the cell membrane, are in close proximity to aid fusion.
VZV gH has not been shown to trimerize or be cleaved, but it is predicted to
have some of the canonical components of Class I fusion proteins: heptad repeats and
hydrophobic α-helices. Possible heptad repeats were identified using COILS. One
heptad repeat was identified in the N-terminus of gH. A 21 aa window identified aa
31-51 with an unweighted score of 0.021 (out of 1) and a weighted score of 0.199. A
14 aa window identified aa 38-51 with an unweighted score of 0.302 and a weighted
score of 0.461. Two heptad repeats were identified in the middle of the gH sequence.
A 21 aa window identified aa 389-409 with an unweighted score of 0.005 and a
weighted score of 0.299, while a 14 aa window identified aa 391-404 with an
unweighted score of 0.076 and a weighted score of 0.238. A 21 aa window identified
the second region as aa 447-467 with an unweighted score of 0.002 and a weighted
score of 0.20, but a 14 aa window identified aa 454-468 with an unweighted score of
0.103 and a weighted core of 0.224. The prediction scores for these heptad repeats do
not decrease from the unweighted to the weighted score, indicating that these regions
likely form heptad repeats.
The second component of a class I fusion protein are α-helices that can act as
fusion peptides. The Secondary Structure Predictor from TRAMPLE predicts that aa
362-385 and aa 500-519/524-539 form α-helices. Further evidence for the α-helical
109

nature of these possible fusion peptides, as well as the predicted heptad repeat regions,
is provided by analysis of transmembrane regions in gH (see section 5.4.1 and Table
5.6). ENSEMBLE, HTMF, Psi Kyte-Doolittle and Kyte-Doolittle algorithms predict
the first α-helix. ENSEMBLE and Kyte-Doolittle algorithms predict the second α-
helix. The second heptad repeat is predicted to be an α-helix with a hydrophobic
nature by the ENSEMBLE and HTMF algorithms. This provides evidence that these
regions are α-helical and hydrophobic in nature, and possibly form coiled-coils or
fusion peptides. It also provides evidence that the predicted transmembrane regions
corresponding to these peptides might not be true transmembrane regions, supporting
the hypothesis that gH is a Type-1 transmembrane protein with multiple, isolated α-
helical sequences but only a single transmembrane pass.
5.4.5. YXXφ motif
gH contains an YXXφ endocytosis motif that is required for gH endocytosis
(185, 186). This endocytosis is clathrin-dependent, and mutation or deletion of this
835YNKI838 motif causes gH to accumulate on the cell surface in transfected cells,
increasing the amount of syncytia in a vaccinia virus fusion assay. This motif is also
predicted by ELM to act as a sorting signal that interacts with the μ subunit of adaptor
protein (AP) complexes. Adaptor proteins recruit clathrin to form clathrin-coated
vesicles, and specific adaptor proteins determine cargo selection for these vesicles (25,
172, 201). AP-1, AP-2, AP-3 and AP4 have all been shown to interact with YXXφ
motifs. This interaction can result in internalization, lysosomal targeting and
110

basolateral targeting of cargo proteins. AP-1A transports cargo between endosomes
and the trans-Golgi network (TGN). AP-1B and AP-4 transport cargo from the TGN
to the basolateral membrane. AP-2 directs cargo from the membrane to endosomes.
AP-3A and AP-4 transport cargo from the TGN to lysosomes or lysosome-related
organelles. AP-3B directs cargo from endosomes to lysosomes or lysosome-related
organelles. Interaction between gH and adaptor proteins might direct the transport of
gH from the TGN to the membrane, and might direct endocytosed gH from the surface
back to the TGN for incorporation into the virion envelope and secondary
envelopment. This might also explain why gH endocytosis is clathrin-dependent.
5.4.6. Highly conserved residues
Although conservation of gH among the herpesviruses is not extremely high, it
is possible that short sequences that are important for structure or function of gH are
highly conserved. The eight human herpesvirus and twenty-one alphaherpesvirus gH
sequences used to analyze gH sequence similarity and identity were further analyzed
for any regions of high conservation. Among the human herpesviruses, the cysteine
residues are highly conserved (Figure 5.1). VZV C327, C724 and C727 are conserved
in the beta- and gammaherpesviruses. VZV C540, C575, C647 and C703 are
conserved across all eight viruses. VZV C827 in the cytoplasmic tail is conserved in
HSV2, HCMV and HHV6. A motif surrounding C575 is also highly conserved:
573(S/T)PCxxxxPxD582 is present in all eight viruses. Y834 is present in all human
herpesvirus gH sequences. A highly conserved glycosylation motif, 783NG(T/S)V786, is
located near the transmembrane region in all eight viruses. The larger
111

778(V/L)LLFPNGTV786 motif is conserved in all three human alphaherpesviruses. The
leucines or similar residues are also conserved in the other human herpesviruses, but
not the phenylalanine or proline. Of particular note, the amino acids in this region are
predicted to form a hydrophobic α-helix by Kyte-Doolittle modeling from the
TRAMPLE server (see section 5.2.1 and Table 5.4).
Similar conservation is found among the Alphaherpesvirinae. C327 was found
only in VZV and not in any other alphaherpesvirus sequence (Figure 5.2).
Interestingly, this cysteine was conserved in the human beta- and gammaherpesviruses
(Figure 5.1). VZV C540, C575 and C703 are conserved in every alphaherpesvirus
sequence. VZV C647 is identified in every sequence except in SHV1/PRV. VZV
C724 and C727 are conserved in every Varicellovirus, Mardivirus and Iltovirus
sequence. These are also conserved in Bovine herpesvirus 2 (BoHV2) and SaHV1 of
the Simplexviruses. Again, the 573(S/T)PCxxxxPxD582 is highly conserved, as is a
motif surrounding C540, 539(M/L)C(T/S)541, L506, Y782, and Y834. The
778(V/L/M)(L/M/I)(L/M)(F/Y)PNGT(V/I)786 sequence is conserved across the
Varicelloviruses, Simplexviruses and the Mardiviruses. It was not conserved in the
Simplexvirus SaHV1 or in the Iltoviruses.
Conservation of numerous cysteines, the motifs surrounding them, and of the
hydrophobic region predicted to form an α-helix, among both the human herpesvirus
and alphaherpesvirus sequences likely indicates that these regions are important for
proper gH structure and function.
112

5.5. Glycoprotein L protein sequence conservation
5.5.1. Conservation of VZV gL protein sequence compared to gL sequences from the
human herpesviruses and from the subfamily Alphaherpesvirinae
Glycoprotein L is not highly conserved, in part due to the differences in gL
protein sequence length (Table 5.9 and 5.10, Figure 5.3). EBV has the shortest gL
sequence with 137 aa, while HCMV has the longest, with 278 aa. Most of the
additional sequence in the longer gL protein sequences occurs at the C-terminus of the
protein. As with gH, the most closely related gL sequences are HSV1 and HSV2, with
74% similarity and 64% identity, and HHV6 and HHV7, with 56% similarity and 38%
identity. HSV1 and HSV2 are identical in length, while HHV6 and HHV7 only differ
by 4 amino acids in length. VZV gL is 64 amino acids shorter than the other human
alphaherpesviruses. The two human betaherpesviruses differ by 30 amino acids, while
the gammaherpesvirus HCMV is 20 and 24 amino acids longer than HHV6 and
HHV7, respectively.
VZV gL has only 24% similarity to HSV1 and HSV2, the other human
alphaherpesviruses, and 20-22% similarity with some of the human beta- and
gammaherpesviruses across the entire protein sequence. It shares no identity higher
than 20% with any of the other human herpesviruses. The BLAST algorithm identifies
VZV gL aa 40-122 as 46% similar and 27% identical to aa 35-118 in HSV1, and aa
40-111 as 51% similar and 28% identical to aa 35-107 in HSV2.
Among the Alphaherpesviruses, gL is somewhat more conserved (Table 5.11
and 5.12 and Figure 5.4). The Simplexviruses, with the exception of SaHV1, are 224-
233 aa in length, and the Mardiviruses are 190-214 aa in length. The Varicelloviruses
113

are shorter, ranging from 156 to 174 aa in length. The Iltoviruses, characterized only
by Psittacid herpesvirus 1 (PsHV1), are shorter still, at 102 aa in length. VZV gL was
61% similar and 40% identical to CeHV9/SVV, but only 21-33% similar to the
remaining Varicelloviruses. It is 20-27% similar to the Simplexviruses, and 27-31%
similar to the Mardiviruses. VZV gL has no identity above 20% compared to any other
Varicelloviruses, except Equine herpesvirus 4 (EHV4), or to any Simplexviruses or
Mardiviruses. It has no similarity or identity above 20% to PsHV1.
5.5.2. Conservation of the VZV gL protein sequence among sequenced VZV clinical
and laboratory isolates
VZV gL has only three amino acid changes among published sequences from
laboratory and clinical isolates (Table 5.13). Using the VZV pOka gL sequence
(Accession number AAK19941) for comparison, the first amino acid difference is
deletion of M10 in the majority of gL sequences. This methionine is only present in
six isolates other than pOka: two vaccine isolates derived from pOka, one clinical
isolate from Los Angeles, California, USA in the 1990s, one clinical isolate from
Morocco in 2000 and two isolates of unknown origin. The second difference, and most
common amino acid substitution, is from threonine to alanine at aa 108 (aa 108 in
isolates with M10, aa 107 in other isolates). T108 is present in every isolate that also
contains the M10 residue, with the exception of the clinical isolate from Morocco and
from one of the two clinical isolates of unknown origin. All other clinical and
laboratory isolates have an alanine residue at this position. Finally, the third amino
acid difference is at S65 (S64 in sequences lacking M10). All VZV isolates, including
114

pOka gL, have a serine at this position, with the exception of a single isolate of
unknown origin, which has a leucine residue.
Conservation of gL among strains does not imply conservation of gH among
strains. For example, while the LAX-1, VariVax, and VarilRix gH and gL sequences
are identical to the pOka gH and gL sequences, the HHV3-M2DR isolate from
Morocco has an identical sequence compared to pOka gL, but a single point
substitution when compared to pOka gH. Additionally, the Ellen and Dumas isolates
have identical gL sequences, but differ from each other in gH sequence at three amino
acids.
115

5.6. Prediction of functional motifs in VZV gL
Analysis of VZV gL has indicated that the mature protein contains one high-
mannose or hybrid-type N-linked oligosaccharide and several important cysteine
residues (65, 66, 153). The single glycomoiety alters the Mr of the protein from 14
kDa to 18 or 19 kDa. The 18 kDa form of gL interacts with gH, while the role of the
19 kDa form is unknown. C48, C79, C146 and C158, but not C21, are all thought to
be involved in gL structure relating to interaction with gH and gH maturation. gL has
not been extensively studied in VZV or in other herpesviruses, as it has been difficult
to separate the role of gL as gH chaperone from any role gL might play in virus
pathogenesis.
5.6.1. Signal sequence and transmembrane domain
VZV gL is predicted to have a 21-25 amino acid signal peptide based on a
number of algorithms. C21 is part of the 21-25 amino acid signal peptide that might be
cleaved off of gL and this would explain why mutation of this residue does not affect
gH/gL interaction or gH processing. The inserted methionine present in the gL
sequence of some VZV isolates is also within this predicted peptide, which indicates
that this deletion might have little or no effect on the fully processed gL protein. It is
possible that cleavage of the signal peptide converts the 19 kDa gL to the 18 kDa gL,
and that only the cleaved form of gL is capable of interacting with gH.
The only algorithm to predict additional transmembrane regions was the
TRAMPLE Kyte-Doolittle algorithm, which predicted transmembrane regions at aa
116

39-53 and 71-86. All other prediction algorithms failed to identify any potential
transmembrane regions.
5.6.2. Glycosylation
gL contains a single predicted N-linked glycosylation motif at N66 (Table
5.14). It also contains a number of predicted GlcNAc sites. The S65A mutation
present in one gL isolate would abolish one of the predicted GlcNAc sites.
5.6.3. Phosphorylation
gL has a number of predicted phosphorylation sites (Table 5.14). No analysis
has been done on the phosphorylation state of this protein, and it is possible that it is
phosphorylated. Substitution of T108 with alanine in the majority of VZV isolates
other than pOka would disrupt the predicted phosphorylation of T108.
5.6.4. YXXφ motif
gL 101YVTL104 is an YXXφ motif commonly thought to act as an endocytosis
signal or as a tyrosine-based sorting signal that interacts with the μ subunit of adaptor
proteins. Although gL is not likely to be anchored in the cell membrane by a
transmembrane, and thus has no cytoplasmic tail, the YVTL motif might act as a
sorting signal to help direct gL transport within clathrin-coated vesicles inside the cell.
5.6.5. Highly conserved residues
A single valine, V75, is conserved in all human herpesvirus gL sequences. No
other region is highly conserved, although most homology between gL sequences
117

occurs in the central region of the protein sequence, with the size differences occurring
due to additional sequences, mostly in the C-termini. VZV C48, C79 and C146 are
conserved in HSV1 and HSV2.
Among the Alphaherpesviruses, VZV C48 is conserved among all
Varicelloviruses except SHV1/PRV, as well as among the Simplexviruses,
Mardiviruses, and Iltoviruses. VZV C79 residue is conserved in all sequences, and
C146 and C158 are also conserved in most sequences except SHV1/PRV, SaHV1 and
PsHV1. The single VZV N-linked glycosylation site is only conserved in EHV4 and
EHV9, which both have an NXT motif in the same region as the VZV NXS
glycosylation motif. The sequence 40VxxI(I/Y)xxxC48 is conserved across most
sequences, as are the sequences 73GIxVKxxC79, 85ILM87,
94AxWxNPYVxxxGLxQxV110, and 147GCV149.
�5.6.6. gL residues possibly involved in the interaction with gH
VZV gL is thought to interact with gH in the endoplasmic reticulum (ER), and
transport gH to the Golgi (66). It has been proposed that the proteins dissociate and gH
is then transported to the cell surface, while gL is either recycled back to the ER or is
degraded. Part of the rationale for this hypothesis originated from the observation that
prediction algorithms could not identify a gL signal peptide at the time, and from
studies using gL antibodies, now no longer available. One gL antibody, R-60,
precipitated pre-gH but not mature gH, and did not detect gL on the cell surface (66).
This antibody was manufactured by expressing gL aa 99-159 as a β-
galactosidase/ORF60 fusion protein. A second antibody, directed against a peptide
118

composed of gL aa 111-121 coupled to keyhole limpet haemocyanin, designated 60A,
immunoprecipitated the 19 kDa form of gL but not the 18 kDa form or gH, which only
forms a complex with the 18 kDa form (153). However, interaction between these two
glycoproteins and coexpression on the surface is commonly observed in other
herpesviruses. It is possible that the 60A antibody binds an epitope that is masked
when gH and gL interact. It is also possible that the R-60 antibody binds an epitope
that is accessible when gL interacts with pre-gH, but becomes masked when gH is
fully processed and mature.
There is potential, then, that gH and gL interact and are expressed together on
the cell surface despite evidence from gL antibody studies. VZV gL is not predicted to
contain a transmembrane region (see Section 5.4.1) and thus might not be inserted into
the cell membrane, but it might be anchored to the cell surface based on interaction
with gH. This hypothesis then suggests that gL aa 111-121, and possible aa 99-159,
are involved in the interaction between mature gH and gL or are masked by
glycomoieties on gH when gH and gL interact. In support of this hypothesis, HSV gL
aa 1-161 are required for gH/gL interaction, and the central region of gL contains the
most conservation among the human herpesviruses and the alphaherpesvirus gL
sequences (see Section 5.5 and 5.6.6) (143).
119

Table 5.1. gH sequence similarity among the human herpesviruses
Conservation of gH protein sequences based on amino acid similarity among the eight
human herpesvirus sequences. Similarity was identified using GeneDoc following
sequence alignment by ClustalX 1.0. αHV, alphaherpesvirus. βHV, betaherpesvirus.
γHV, gammaherpesvirus. HHV3/VZV, isolate pOka, AAK1939. HHV1/HSV1, isolate
F, AAG17895. HHV2/HSV2, isolate HG2, NP_044491. HHV4/EBV, isolate B96-8,
CAA24797. HHV8/KSHV, AAL91720. HHV5/HCMV, AAA45946. HHV6, isolate
pZVB43, AAB19779. HHV7, isolate JI, AAB64293. All sequences have been
published.
αHV βHV γHV
HH
V3/
VZV
HH
V1/
HSV
1
HH
V2/
HSV
2
HH
V4/
EBV
HH
V8/
KSH
V
HH
V5/
HC
MV
HH
V6
HH
V7
HHV3/VZV 38% 38% 23% 22% 21% 23% 25%
HHV1/HSV1 85% 21% 22% 23% 26% 27%
αHV
HHV2/HSV2 21% 20% 24% 26% 26%
HHV4/EBV 38% 27% 31% 32%
βHV
HHV8/KSHV 27% 29% 31%
HHV5/HCMV 43% 43%
HHV6 63%
γHV
HHV7
120

Table 5.2. gH sequence identity among the human herpesviruses
Conservation of gH protein sequences based on amino acid identity among the eight
human herpesvirus sequences. Identity was identified using GeneDoc following
sequence alignment by ClustalX 1.0. αHV, alphaherpesvirus. βHV, betaherpesvirus.
γHV, gammaherpesvirus. HHV3/VZV, isolate pOka, AAK1939. HHV1/HSV1, isolate
F, AAG17895. HHV2/HSV2, isolate HG2, NP_044491. HHV4/EBV, isolate B96-8,
CAA24797. HHV8/KSHV, AAL91720. HHV5/HCMV, AAA45946. HHV6, isolate
pZVB43, AAB19779. HHV7, isolate JI, AAB64293. Only identity of 20% or higher is
reported. All sequences have been published.
αHV βHV γHV
HH
V3/
VZV
HH
V1/
HSV
1
HH
V2/
HSV
2
HH
V4/
EBV
HH
V8/
KSH
V
HH
V5/
HC
MV
HH
V6
HH
V7
HHV3/VZV 20% 21%
HHV1/HSV1 78%
αHV
HHV2/HSV2
HHV4/EBV 20%
βHV
HHV8/KSHV
HHV5/HCMV 21%
HHV6 38%
γHV
HHV7
121

122

Figure 5.1. Alignment of human herpesvirus gH sequences
Human herpesvirus gH sequences (Table 5.1 and 5.2) were aligned using ClustalX 1.0
and imported into GeneDoc for analysis. All sequences are compared to VZV pOka
gH (AAK19939). “.”, identical amino acid to VZV pOka gH. “-“, gap in alignment.
Light gray, dark gray and black shading indicates areas of varying conservation,
designated by GeneDoc. VZV pOka gH NX(S/T) glycosylation sites are highlighted in
blue. Cysteine residues are highlighted in red. The conserved hydrophobic region
roximal to the transmembrane region is highlighted in green and blue. p
123

Table 5.3. gH sequence similarity among the Alphaherpesvirinae
Conservation of gH protein sequences based on amino acid similarity among the
alphaherpesvirus sequences. Similarity was identified using GeneDoc following
sequence alignment by ClustalX 1.0. HHV3/VZV, isolate pOka, AAK1939.
CeHV9/SVV, AAG27211. EHV1, isolate V592, AAS45924. EHV4, isolate 1942,
BAA03379. EHV9, isolate P19, BAH02465. CHV1, Canid herpesvirus 1, isolate
Australian, AAK51057. FHV1, Felid herpesvirus 1, AAB27840. BoHV5, isolate
SV507/99, NP_954926. BoHV1, isolate Cooper, CAA41677. SHV1/PRV,
CAA41678. HHV1/HSV1, isolate F, AAG17895. HHV2/HSV2, isolate HG2,
NP_044491. CeHV1/McHV1, Macacine herpesvirus 1, isolate E2490, NP_85188.
CeHV16/HVP2, Papiine herpesvirus 2, isolate X313, YP_443868. BoHV2,
AAK55404. SaHV1, VGBE11. GaHV2, Gallid herpesvirus 2, AAP13936. GaHV3,
isolate HPRS24, BAB16531. MeHV1, Meleagrid herpesvirus 1, AAB27146. GaHV1,
CAA74689. PsHV1, isolate 97-0001, AAQ73700. All sequences have been published.
124

125

Table 5.4. gH sequence identity among the Alphaherpesvirinae
Conservation of gH protein sequences based on amino acid identity among the
alphaherpesvirus sequences. Identity was identified using GeneDoc following
sequence alignment by ClustalX 1.0. HHV3/VZV, isolate pOka, AAK1939.
CeHV9/SVV, AAG27211. EHV1, isolate V592, AAS45924. EHV4, isolate 1942,
BAA03379. EHV9, isolate P19, BAH02465. CHV1, isolate Australian, AAK51057.
FHV1, Felid herpesvirus 1, AAB27840. BoHV5, isolate SV507/99, NP_954926.
BoHV1, isolate Cooper, CAA41677. SHV1/PRV, CAA41678. HHV1/HSV1, isolate
F, AAG17895. HHV2/HSV2, isolate HG2, NP_044491. CeHV1/McHV1, isolate
E2490, NP_85188. CeHV16/HVP2, isolate X313, YP_443868. BoHV2, AAK55404.
SaHV1, VGBE11. GaHV2, AAP13936. GaHV3, isolate HPRS24, BAB16531.
MeHV1, AAB27146. GaHV1, CAA74689. PsHV1, isolate 97-0001, AAQ73700.
Only identity of 20% or higher is reported. All sequences have been published.
126

127
128
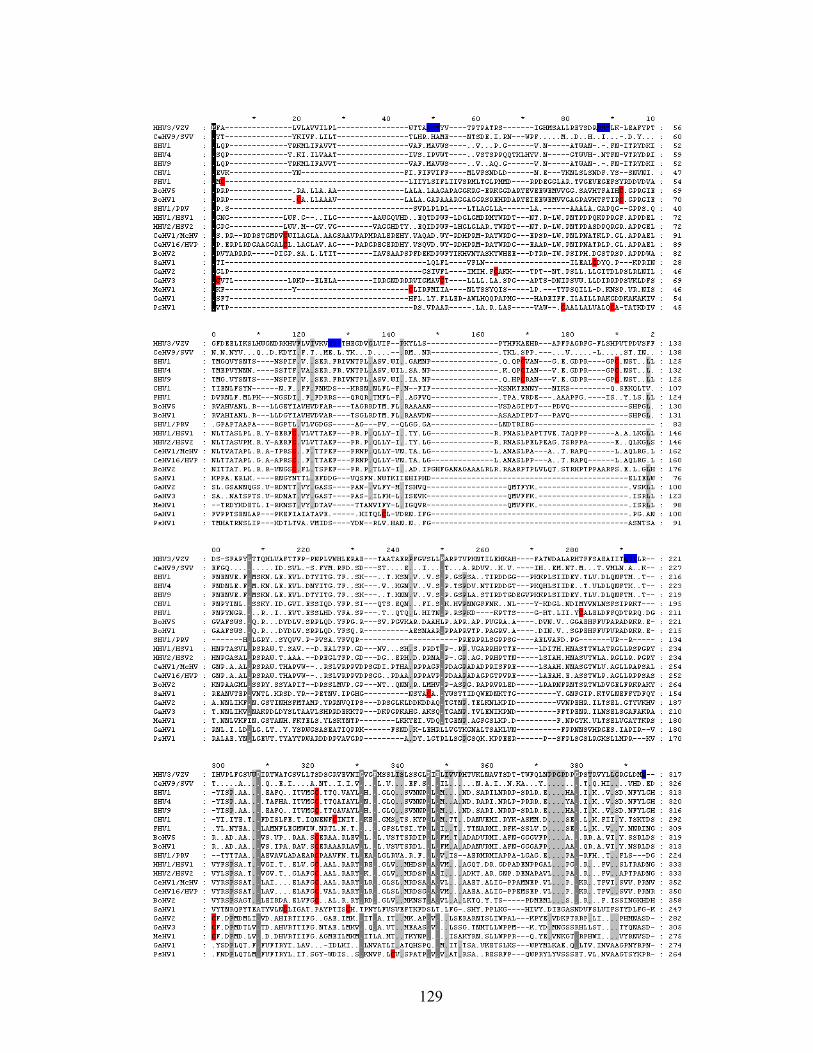
129

Figure 5.2. Alignment of alphaherpesvirus gH sequences
Alphaherpesvirus gH sequences (Table 5.3 and 5.4) were aligned using ClustalX 1.0
and imported into GeneDoc for analysis. All sequences are compared to VZV pOka
gH (AAK19939). “.”, identical amino acid to VZV pOka gH. “-“, gap in alignment.
Light gray, dark gray and black shading indicates areas of varying conservation,
designated by GeneDoc. VZV pOka gH NX(S/T) glycosylation sites are highlighted in
blue. Cysteine residues are highlighted in red. The conserved hydrophobic region
proximal to the transmembrane region is highlighted in green and blue.
130

Table 5.5. Conservation of gH between VZV laboratory and clinical isolates
Compilation of amino acid changes in gH sequences from laboratory and clinical
isolates submitted to the National Center for Biotechnology Information as compared
to VZV pOka gH (Accession number AAK19939).
Accession # Isolate Location; Date 71 269 319 551 639 700 AAK19939 Oka Japan; 1970s R L S N T K Q775J3 Oka Vaccine strain AAY57720 VariVax Japan; 1970s AAY57649 VarilRix Japan; 1970s
AAG32551 Ellen Georgia, USA; 1960s K
AAK50445 Ellen Georgia, USA; 1960s P R
NP_040160 Dumas Netherlands; 1970 P R CAA27920 Dumas Netherlands; 1970 P R P09260 Dumas Netherlands; 1970 P R
AAK19965 LAX1
Los Angeles, California, USA; 1990s
ABE67146 CA123 California, USA AAP32866 1140 Singapore; 1990s AAP32852 1419 Singapore; 1990s AAP32847 1242 Singapore; 1990s AAP32842 681 Singapore; 1990s AAP32837 529 Singapore; 1990s AAP32832 376 Singapore; 1990s
AAK19957 80-2 Pennsylvania, USA; 1982 K
CAI44880 HJO Germany K AAK01042 Iceland Iceland; 1990s K
AAP32857 V8 Bangkok, Thailand; 1980s K A
AAP32862 Z24 Bangkok, Thailand; 1980s K A A
131
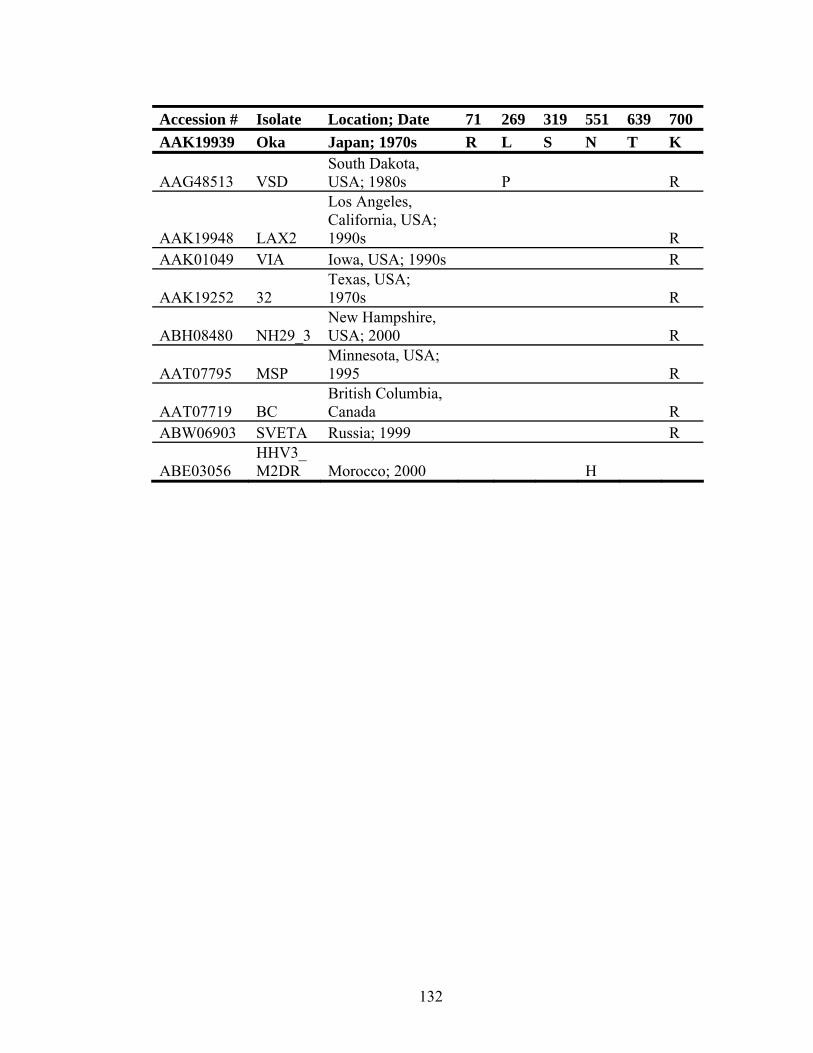
Accession # Isolate Location; Date 71 269 319 551 639 700 AAK19939 Oka Japan; 1970s R L S N T K
AAG48513 VSD South Dakota, USA; 1980s P R
AAK19948 LAX2
Los Angeles, California, USA; 1990s R
AAK01049 VIA Iowa, USA; 1990s R
AAK19252 32 Texas, USA; 1970s R
ABH08480 NH29_3 New Hampshire, USA; 2000 R
AAT07795 MSP Minnesota, USA; 1995 R
AAT07719 BC British Columbia, Canada R
ABW06903 SVETA Russia; 1999 R
ABE03056 HHV3_M2DR Morocco; 2000 H
132
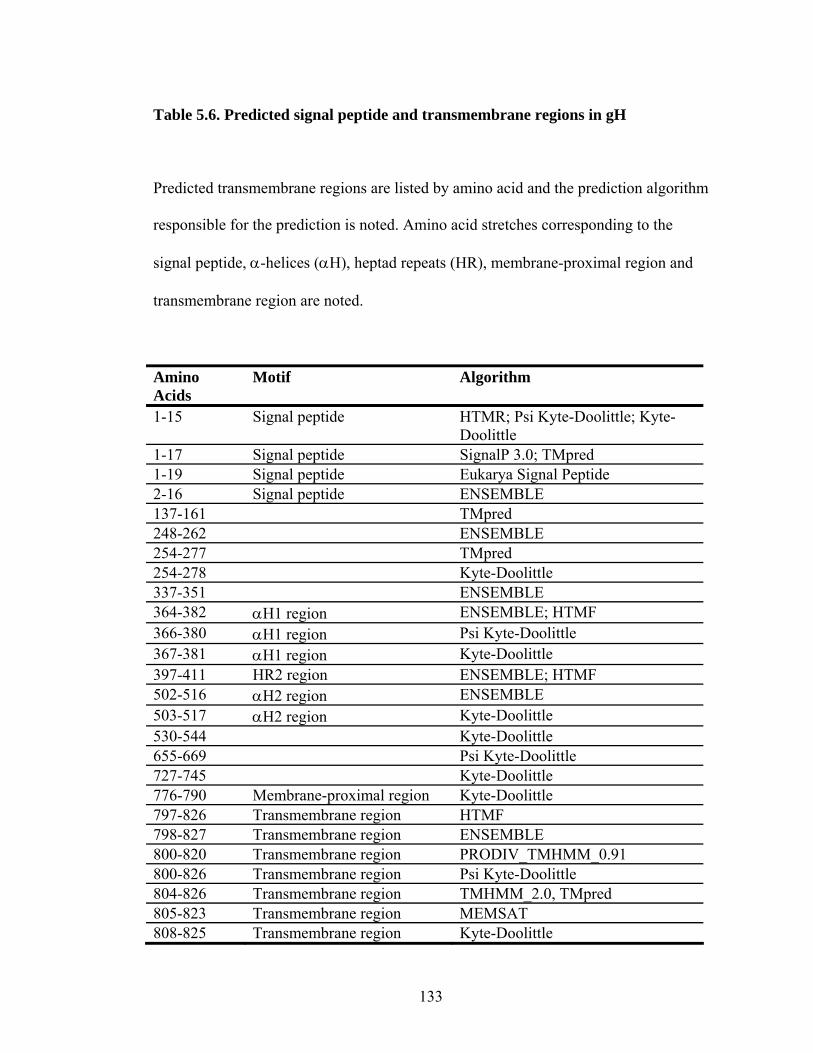
Table 5.6. Predicted signal peptide and transmembrane regions in gH
Predicted transmembrane regions are listed by amino acid and the prediction algorithm
responsible for the prediction is noted. Amino acid stretches corresponding to the
signal peptide, α-helices (αH), heptad repeats (HR), membrane-proximal region and
transmembrane region are noted.
Amino Acids
Motif Algorithm
1-15 Signal peptide HTMR; Psi Kyte-Doolittle; Kyte-Doolittle
1-17 Signal peptide SignalP 3.0; TMpred 1-19 Signal peptide Eukarya Signal Peptide 2-16 Signal peptide ENSEMBLE 137-161 TMpred 248-262 ENSEMBLE 254-277 TMpred 254-278 Kyte-Doolittle 337-351 ENSEMBLE 364-382 αH1 region ENSEMBLE; HTMF 366-380 αH1 region Psi Kyte-Doolittle 367-381 αH1 region Kyte-Doolittle 397-411 HR2 region ENSEMBLE; HTMF 502-516 αH2 region ENSEMBLE 503-517 αH2 region Kyte-Doolittle 530-544 Kyte-Doolittle 655-669 Psi Kyte-Doolittle 727-745 Kyte-Doolittle 776-790 Membrane-proximal region Kyte-Doolittle 797-826 Transmembrane region HTMF 798-827 Transmembrane region ENSEMBLE 800-820 Transmembrane region PRODIV_TMHMM_0.91 800-826 Transmembrane region Psi Kyte-Doolittle 804-826 Transmembrane region TMHMM_2.0, TMpred 805-823 Transmembrane region MEMSAT 808-825 Transmembrane region Kyte-Doolittle
133

Table 5.7. Predicted glycosylation sites in VZV gH
Potential residues at which glycosylation occurs are listed, along with the type of
glycosylation and the prediction algorithm.
*The S319A amino acid change in some isolates of VZV would abolish the predicted
glycosylation motif at N317.
**Proline follows arginine at these predicted N-linked glycosylation sites, making
glycosylation unlikely.
Residue Type Algorithm T16 O-GlcNAc YinOYang 1.2 N18 N-linked Motif Scan; NetNGlyc 1.0 T25 O-linked NetOGlyc 3.1 T28 O-GlcNAc YinOYang 1.2 N45 N-linked Motif Scan; NetNGlyc 1.0; ELM N83** N-linked NetNGlyc 1.0 T127 O-GlcNAc YinOYang 1.2 S131 O-GlcNAc YinOYang 1.2 T143 O-GlcNAc YinOYang 1.2 T184 O-linked NetOGlyc 3.1 N217 N-linked Motif Scan; NetNGlyc 1.0; ELM T286 O-GlcNAc YinOYang 1.2 N317* N-linked Motif Scan; NetNGlyc 1.0; ELM N499 N-linked Motif Scan; NetNGlyc 1.0; ELM N522 N-linked Motif Scan; NetNGlyc 1.0; ELM N560** N-linked NetNGlyc 1.0 N760 N-linked Motif Scan; NetNGlyc 1.0; ELM N783 N-linked Motif Scan; NetNGlyc 1.0; ELM T841 O-GlcNAc YinOYang 1.2
134

Table 5.8. Predicted phosphorylation sites in gH
Predicted phosphorylation sites and their possible kinase are listed, along with the
algorithm responsible for the prediction. PKA, Protein kinase A. PKC, Protein kinase
C. PKG, Protein kinase G. CKI, Casein kinase I. CKII, Casein kinase II. MAPK,
Mitogen-activated protein kinase. INSR, Insulin receptor kinase. GSK3, Glycogen
synthase kinase 3. EGFR, Epidermal growth factor receptor kinase. ATM, Ataxia
telangiectasia mutated kinase. Syk, Syk tyrosine kinase. cdc2, Serine/threonine cyclin-
dependent kinase 1, encoded by CDC2. cdk5, Cyclin-dependent kinase 5. RSK,
Ribosomal s6 kinase. DNAPK, DNA-dependent serine/threonine protein kinase. Src,
Src tyrosine kinase. Some algorithms predicted a given phosphorylation site twice
where noted (2X), with multiple possible kinases provided. NetPhos 2.0 does not
predict a kinase.
Residue Predicted Kinase Algorithm S20 NetPhos 2.0 Y21 EGFR NetPhos 2.0; NetPhosK T23 CKI, MAPK NetPhos 2.0; ELM (2X); KinasePhos T25 MAPK, cdk5 ELM, KinasePhos; NetPhosK T28 cdc2 NetPhosK S35 cdc2 NetPhosK S42 PKC NetPhos 2.0; Motif Scan S47 PKC, PKA, RSK NetPhos 2.0; Motif Scan; NetPhosK (2X) T56 CKII Motif Scan; NetPhosK T85 CKII, PKG Motif Scan; ELM; NetPhosK S101 ATM KinasePhos S102 MAPK ELM; NetPhosK S123 GSK3 ELM T127 GSK3, MAPK, cdk5 NetPhos 2.0; ELM (2X); KinasePhos;
NetPhosK (3X)
135

Residue Predicted Kinase Algorithm S131 CKII NetPhos 2.0; Motif Scan; ELM T142 PKC KinasePhos T151 PKC NetPhosK T169 cdc2 NetPhosK T184 PKA, PKC NetPhos 2.0, ELM; NetPhosK T189 CKII Motif Scan; ELM T207 PKA, PKC ELM; NetPhosK S210 CKI NetPhos 2.0, NetPhosK S218 PKC NetPhosK T219 PKC Motif Scan; NetPhosK S240 GSK3, PKC, cdc2 ELM; NetPhosK (2X) S247 PKC, cdc2 NetPhos 2.0; Motif Scan; NetPhosK S260 GSK3 ELM S261 PKA NetPhosK S264 CKI, cdc2 NetPhos 2.0; ELM; KinasePhos; NetPhosK S267 CKI ELM T279 PKC Motif Scan; NetPhosK T286 GSK3 ELM T290 CKI ELM S304 PKC NetPhos 2.0; Motif Scan S319 GSK3 ELM Y336 INSR NetPhosK S341 GSK3, cdc2 ELM; NetPhosK T351 PKC; PKA NetPhos 2.0; Motif Scan; ELM (2X);
KinasePhos; NetPhosK T352 CKII, PKG NetPhos 2.0; Motif Scan; NetPhosK S363 DNAPK NetPhosK Y364 Src NetPhos 2.0; NetPhosK Y365 Src, EGFR NetPhos 2.0; NetPhosK (2X) S379 CKI NetPhosK T385 PKA NetPhos 2.0; ELM Y405 INSR KinasePhos T418 GSK3 ELM S423 CKI ELM S431 ATM, DNAPK, cdc2 KinasePhos; NetPhosK (2X) Y451 NetPhos 2.0 S454 CKII Motif Scan; ELM; KinasePhos; NetPhosK S479 PKC NetPhosK S487 DNAPK NetPhosK T501 PKC Motif Scan S523 PKC Motif Scan
136
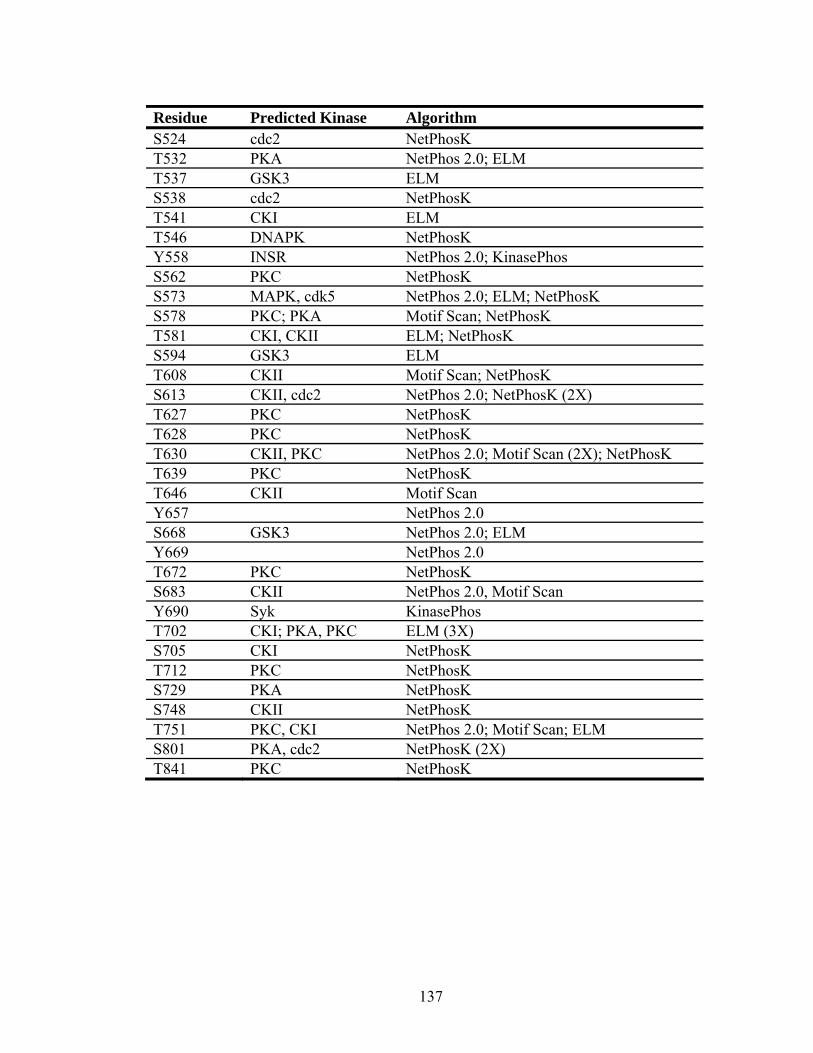
Residue Predicted Kinase Algorithm S524 cdc2 NetPhosK T532 PKA NetPhos 2.0; ELM T537 GSK3 ELM S538 cdc2 NetPhosK T541 CKI ELM T546 DNAPK NetPhosK Y558 INSR NetPhos 2.0; KinasePhos S562 PKC NetPhosK S573 MAPK, cdk5 NetPhos 2.0; ELM; NetPhosK S578 PKC; PKA Motif Scan; NetPhosK T581 CKI, CKII ELM; NetPhosK S594 GSK3 ELM T608 CKII Motif Scan; NetPhosK S613 CKII, cdc2 NetPhos 2.0; NetPhosK (2X) T627 PKC NetPhosK T628 PKC NetPhosK T630 CKII, PKC NetPhos 2.0; Motif Scan (2X); NetPhosK T639 PKC NetPhosK T646 CKII Motif Scan Y657 NetPhos 2.0 S668 GSK3 NetPhos 2.0; ELM Y669 NetPhos 2.0 T672 PKC NetPhosK S683 CKII NetPhos 2.0, Motif Scan Y690 Syk KinasePhos T702 CKI; PKA, PKC ELM (3X) S705 CKI NetPhosK T712 PKC NetPhosK S729 PKA NetPhosK S748 CKII NetPhosK T751 PKC, CKI NetPhos 2.0; Motif Scan; ELM S801 PKA, cdc2 NetPhosK (2X) T841 PKC NetPhosK
137

Table 5.9. gL sequence similarity among the human herpesviruses
Conservation of gL protein sequences based on amino acid similarity among the eight
human herpesvirus sequences. Similarity was identified using GeneDoc following
sequence alignment by ClustalX 1.0. αHV, alphaherpesvirus. βHV, betaherpesvirus.
γHV, gammaherpesvirus. HHV3/VZV, isolate pOka, AAK19941. HHV1/HSV1,
isolate F, ADD60021. HHV2/HSV2, isolate HG52, BAA01264. HHV4/EBV, isolate
B95-8, CAD53428. HHV8/KSHV, AAB62638. HHV5/HCMV, isolate Towne,
ACM48085. HHV6, isolate CRIPJ, AAZ81570. HHV7, isolate JI, AAC54744. All
sequences have been published.
αHV βHV γHV
HH
V3/
VZV
HH
V1/
HSV
1
HH
V2/
HSV
2
HH
V4/
EBV
HH
V8/
KSH
V
HH
V5/
HC
MV
HH
V6
HH
V7
HHV3/VZV 24% 24% 22% 20% 21% HHV1/HSV1 74% 20% 22% 29% αH
V
HHV2/HSV2 22% 23% 30% HHV4/EBV 34% 20%
βHV
HHV8/KSHV HHV5/HCMV 38% 43% HHV6 56% γH
V
HHV7
138

Table 5.10. gL sequence identity among the human herpesviruses
Conservation of gL protein sequences based on amino acid identity among the eight
human herpesvirus sequences. Identity was identified using GeneDoc following
sequence alignment by ClustalX 1.0. αHV, alphaherpesvirus. βHV, betaherpesvirus.
γHV, gammaherpesvirus. HHV3/VZV, isolate pOka, AAK19941. HHV1/HSV1,
isolate F, ADD60021. HHV2/HSV2, isolate HG52, BAA01264. HHV4/EBV, isolate
B95-8, CAD53428. HHV8/KSHV, AAB62638. HHV5/HCMV, isolate Towne,
ACM48085. HHV6, isolate CRIPJ, AAZ81570. HHV7, isolate JI, AAC54744. Only
identity of 20% or higher is reported. All sequences have been published.
αHV βHV γHV
HH
V3/
VZV
HH
V1/
HSV
1
HH
V2/
HSV
2
HH
V4/
EBV
HH
V8/
KSH
V
HH
V5/
HC
MV
HH
V6
HH
V7
HHV3/VZV HHV1/HSV1 64% αH
V
HHV2/HSV2 HHV4/EBV 20%
βHV
HHV8/KSHV HHV5/HCMV 22% HHV6 38% γH
V
HHV7
139

Figure 5.3. Alignment of human herpesvirus gL sequences
Human herpesvirus gL sequences (Table 5.9 and 5.10) were aligned using ClustalX
1.0 and imported into GeneDoc for analysis. All sequences are compared to VZV
pOka gL (AAK19941). “.”, identical amino acid to VZV gL. “-“, gap in alignment.
Light gray, dark gray and black shading indicates areas of varying conservation,
designated by GeneDoc. VZV pOka gL NX(S/T) glycosylation sites are highlighted in
lue. Cysteine residues are highlighted in red. b
140

Table 5.11. gL sequence similarity among the Alphaherpesvirinae
Conservation of gL protein sequences based on amino acid similarity among the
alphaherpesvirus sequences. Similarity was identified using GeneDoc following
sequence alignment by ClustalX 1.0. VV, Varicelloviruses. SV, Simplexviruses. MV,
Mardiviruses. IV, Iltoviruses. HHV3/VZV, isolate pOka, AAK19941. CeHV9,
AAG27236. EHV4, isolate NS80567, NP_045279. EHV9, isolate P19, BAH02487.
BoHV5, isolate SV507-99, NP_954948. BoHV1, isolate Cooper, AAC54555.
SHV1/PRV, isolate Kaplan, AAA18858. HHV1/HSV1, isolate F, ADD60021.
HHV2/HSV2, isolate HG52, BAA01264. CeHV1/McHV1, isolate E2490,
NP_851860. CeHV16/HVP2, isolate X313, YP_443847. SaHV1, I36810. GaHV2,
isolate GA, AAF66737. GaHV3, isolate HPRS24, NP_066830. MeHV1, isolate
FC126, AAG45738. PsHV1, isolate 97-0001, AAQ73745. Only similarity of 20% or
higher is reported. All sequences have been published.
141

142

Table 5.12. gL sequence identity among the Alphaherpesvirinae
Conservation of gL protein sequences based on amino acid identity among the
alphaherpesvirus sequences. Identity was identified using GeneDoc following
sequence alignment by ClustalX 1.0. VV, Varicelloviruses. SV, Simplexviruses. MV,
Mardiviruses. IV, Iltoviruses. HHV3/VZV, isolate pOka, AAK19941. CeHV9,
AAG27236. EHV4, isolate NS80567, NP_045279. EHV9, isolate P19, BAH02487.
BoHV5, isolate SV507-99, NP_954948. BoHV1, isolate Cooper, AAC54555.
SHV1/PRV, isolate Kaplan, AAA18858. HHV1/HSV1, isolate F, ADD60021.
HHV2/HSV2, isolate HG52, BAA01264. CeHV1/McHV1, isolate E2490,
NP_851860. CeHV16/HVP2, isolate X313, YP_443847. SaHV1, I36810. GaHV2,
isolate GA, AAF66737. GaHV3, isolate HPRS24, NP_066830. MeHV1, isolate
FC126, AAG45738. PsHV1, isolate 97-0001, AAQ73745. Only identity of 20% or
higher is reported. All sequences have been published.
143
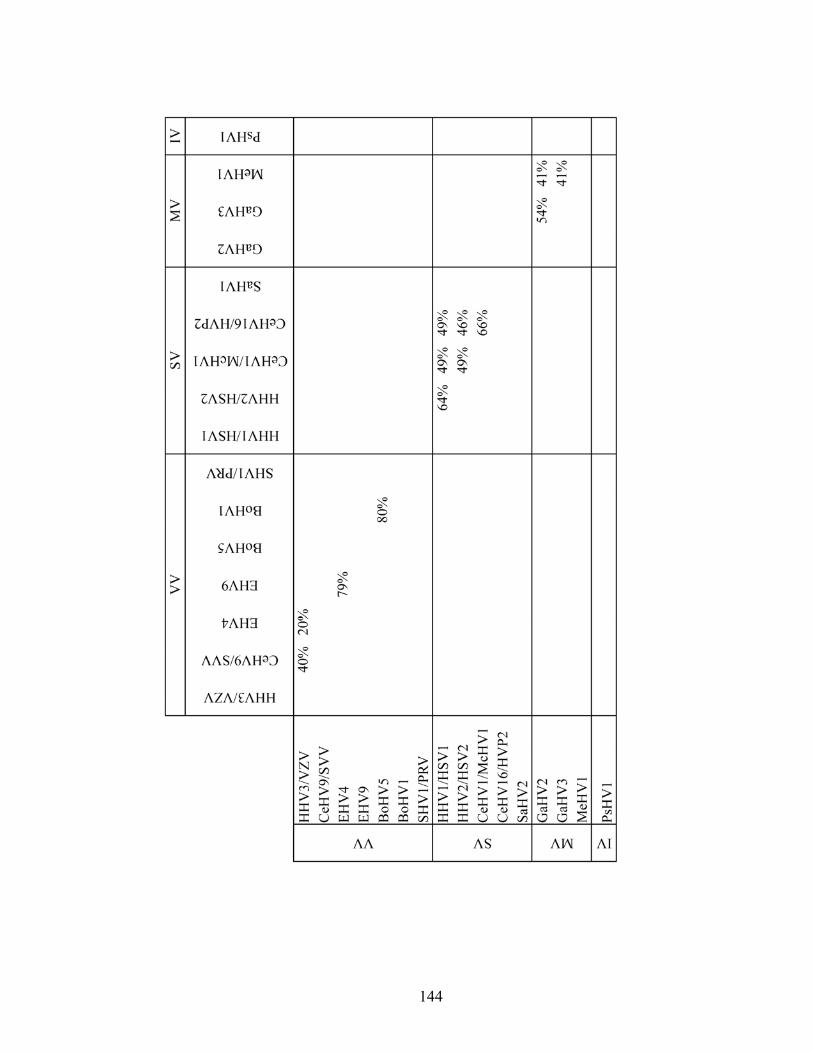
144

Figure 5.4. Alignment of alphaherpesvirus gL sequences
Alphaherpesvirus gL sequences (Table 5.11 and 5.12) were aligned using ClustalX 1.0
and imported into GeneDoc for analysis. All sequences are compared to VZV pOka
gL (AAK19941). “.”, identical amino acid to VZV gL. “-“, gap in alignment. Light
gray, dark gray and black shading indicates areas of varying conservation, designated
145

by GeneDoc. VZV pOka gL NX(S/T) glycosylation sites are highlighted in blue.
Cysteine residues are highlighted in red.
146
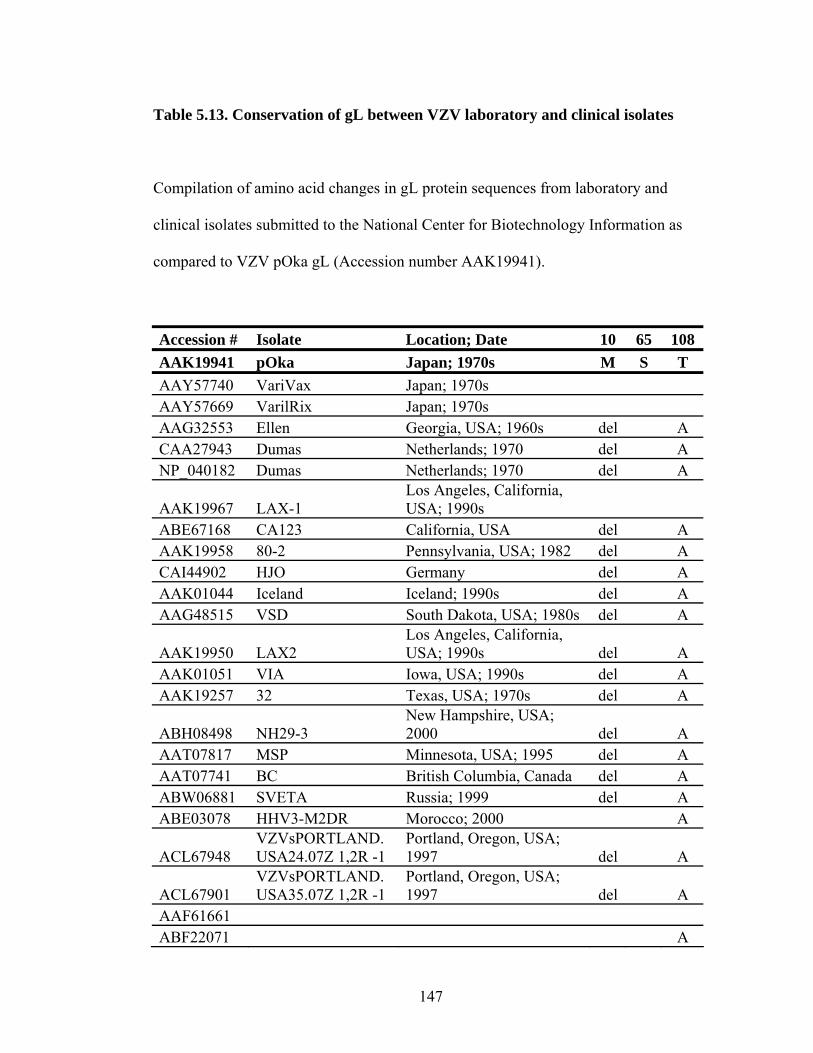
Table 5.13. Conservation of gL between VZV laboratory and clinical isolates
Compilation of amino acid changes in gL protein sequences from laboratory and
clinical isolates submitted to the National Center for Biotechnology Information as
compared to VZV pOka gL (Accession number AAK19941).
Accession # Isolate Location; Date 10 65 108 AAK19941 pOka Japan; 1970s M S T AAY57740 VariVax Japan; 1970s AAY57669 VarilRix Japan; 1970s AAG32553 Ellen Georgia, USA; 1960s del A CAA27943 Dumas Netherlands; 1970 del A NP_040182 Dumas Netherlands; 1970 del A
AAK19967 LAX-1 Los Angeles, California, USA; 1990s
ABE67168 CA123 California, USA del A AAK19958 80-2 Pennsylvania, USA; 1982 del A CAI44902 HJO Germany del A AAK01044 Iceland Iceland; 1990s del A AAG48515 VSD South Dakota, USA; 1980s del A
AAK19950 LAX2 Los Angeles, California, USA; 1990s del A
AAK01051 VIA Iowa, USA; 1990s del A AAK19257 32 Texas, USA; 1970s del A
ABH08498 NH29-3 New Hampshire, USA; 2000 del A
AAT07817 MSP Minnesota, USA; 1995 del A AAT07741 BC British Columbia, Canada del A ABW06881 SVETA Russia; 1999 del A ABE03078 HHV3-M2DR Morocco; 2000 A
ACL67948 VZVsPORTLAND.USA24.07Z 1,2R -1
Portland, Oregon, USA; 1997 del A
ACL67901 VZVsPORTLAND.USA35.07Z 1,2R -1
Portland, Oregon, USA; 1997 del A
AAF61661 ABF22071 A
147

Accession # Isolate Location; Date 10 65 108 AAK19941 pOka Japan; 1970s M S T ABF22290 del A ABF22217 del A ABF22144 del A ABF21998 del A ABF21925 del A ABF21852 del A ABF21779 del A ABF21633 del A ABF21560 del A ABF21706 del L A
148

Table 5.14. Predicted glycosylation sites in VZV gL
Potential residues at which glycosylation occurs are listed, along with the type of
glycosylation, and the prediction algorithm.
*The S65L amino acid change in some isolates of VZV would abolish the predicted
glycosylation motif at S65.
Residue Type Algorithm S39 O-GlcNAc YinOYang 1.2 S51 O-GlcNAc YinOYang 1.2 S65* O-GlcNAc YinOYang 1.2 N66 N-linked NetNGlyc 1.0; ELM S68 O-GlcNAc YinOYang 1.2 S129 O-GlcNAc YinOYang 1.2 S135 O-GlcNAc YinOYang 1.2 T136 O-GlcNAc YinOYang 1.2
149

Table 5.15. Predicted phosphorylation sites in gL
Predicted phosphorylation sites and their possible kinase are listed, along with the
algorithm responsible for the prediction. PKA, Protein kinase A. PKC, Protein kinase
C. CKI, Casein kinase I. CKII, Casein kinase II. MAPK, Mitogen-activated protein
kinase. GSK3, Glycogen synthase kinase 3. EGFR, Epidermal growth factor receptor
kinase. ATM, Ataxia telangiectasia mutated kinase. cdc2, Serine/threonine cyclin-
dependent kinase 1, encoded by CDC2. PIKK, Phosphatidyl inositol 3' kinase-related
kinase. Some algorithms predicted a given phosphorylation site twice (2X) where
noted, with multiple possible kinases provided. NetPhos 2.0 does not predict a kinase.
*The T108A substitution would abolish the predicted phosphorylation at T108.
Residue Predicted Kinase Algorithm S3 PKC Motif Scan S23 PKC NetPhosK T29 MAPK ELM; KinasePhos S39 NetPhos 1.0 S42 CKI, GSK3 ELM (2X); NetPhos 1.0 S52 CKII NetPhosK; ELM; NetPhos 1.0; Motif Scan S68 CKI, GSK3 ELM (2X) Y59 EGFR NetPhosK; NetPhos 1.0 S72 PKC NetPhosK T78 NetPhos 1.0 Y101 NetPhos 1.0 T103 PKC NetPhosK; KinasePhos; NetPhos 1.0; Motif Scan T108* PIKK ELM S110 CKII, PKC, ATM NetPhosK (2X); ELM; KinasePhos; NetPhos 1.0;
Motif Scan S117 CKII, cdc2 NetPhosK (2X) S135 GSK3 ELM
150

Residue Predicted Kinase Algorithm S138 CKI ELM T136 MAPK, cdk5,
GSK3, cdc2 NetPhosK (2X); ELM (2X); KinasePhos (3X); NetPhos 1.0
S138 CKI, MAPK KinasePhos (2X); NetPhos 1.0 S139 MAPK KinasePhos, NetPhos 1.0
151

CHAPTER VI
MUTATIONAL ANALYSIS OF STRUCTURAL AND FUNCTIONAL MOTIFS
OF VARICELLA-ZOSTER VIRUS GLYCOPROTEIN H
Susan E. Vleck, Stefan L. Oliver, Marvin Sommer, Jaya Rajamani and Ann M. Arvin
Forward: This chapter is a version of manuscript in preparation. S.E.V. performed
experiments, data analysis and manuscript preparation. S.L.O. designed and performed
the gH BAC mutagenesis, and advised on experiment design and data analysis. M.S.
assisted with cosmid mutagenesis. J.R. assisted with the SCID mouse experiments.
These experiments were done under the direction of A.M.A.
This work was supported by training grants from the National Institutes of
Health, R01 AI 020459 and P01CA49605. S.E.V. received support from T32
GM007279 and T32 AI07328.
152

6.1. Abstract
Glycoprotein H (gH) is highly conserved in the Herpesviridae and has been
demonstrated to play a role in binding, entry and fusion, but little is known about
Varicella-zoster virus (VZV) gH function. In these experiments, VZV gH was shown
to be essential by deletion of ORF37, which encodes gH, from the pOka isolate. To
determine the importance of structural and functional motifs predicted in VZV gH and
conserved in gH in the Alphaherpesvirinae, substitutions were made at glycosylation
motifs, cysteines, heptad repeats (HR), α-helices and a hydrophobic region. Alanine
mutations at N45 and S47 disrupted glycosylation of gH, resulting in a reduced Mr by
Western blot, and the S47A, but not the N45A, substitution caused a delayed growth
phenotype in vivo in the SCID mouse skin xenograft model of VZV pathogenesis.
Alanine substitutions at S42, T127, T351 and T751 and a threonine substitution at S47
did not affect gH maturation, localization or virus replication in vitro or in skin in vivo,
but the T751A substitution reduced syncytia formation. Amino acid substitutions were
generated that disrupted predicted HRs, α-helices, which are potential fusion peptides,
and a hydrophobic region. Substitutions that disrupted the helical nature of a predicted
HR at aa 38-51 had no effect in vitro but impaired virulence in skin in vivo, and
substitutions that disrupted the predicted HRs at aa 391-404 and 455-468 resulted in
reduced cell surface expression of gH and were lethal. Substitutions to disrupt the α-
helices at aa 362-385 and 500-539 and the hydrophobic region abolished gH
maturation and gH cell surface expression and were lethal. Alanine substitution of
C540 and C575 disrupted gH maturation and transport to the cell surface, and alanine
substitution of C647, C703 and C724 reduced cell surface expression. This study is
153

the first to analyze gH function using mutagenesis of the VZV genome, and presents
evidence for multiple functional domains within VZV gH.
154

6.2. Introduction
Varicella-zoster virus (VZV) is a human herpesvirus that causes chicken pox
(varicella) during primary infection and shingles (herpes zoster) following the
establishment of latency and virus reactivation. H (gH) is an envelope glycoprotein
that plays a role in virus binding, entry and fusion in many herpesviruses, but little is
known about VZV gH function. VZV gH is processed as a 94 kDa precursor protein
that remains in the endoplasmic reticulum unless glycoprotein L (gL), its chaperone, is
present (166). Four of the five cysteine residues in gL are required for the gH/gL
interaction although gH and gL are not covalently linked by an intermolecular
disulfide bond (65, 66). As a result of interaction with gL, gH is transported to the
Golgi and processed into a 118 kDa protein, which is then trafficked to the cell surface
(166). Monoclonal antibodies directed against gH expressed on the virus envelope or
the cell surface are able to neutralize VZV (166). Antibody administered shortly after
inoculation in vitro can prevent virus spread to neighboring cells, as well as the
formation of syncytia, which are multinucleated cells that result from virus-induced
cell-cell fusion (53, 66, 202, 229). Antibody administered shortly after inoculation of
skin xenografts can eliminate or reduce virus replication and spread in vivo (229).
These studies indicate that gH on the virion envelope or cell surface plays an
important role in virus spread and cell-cell fusion both in vitro and in vivo.
Following cell surface expression, gH is endocytosed and targeted back to the
trans-Golgi network, where it is incorporated into the virus envelope (155, 185).
Transient transfection studies have demonstrated that a 835YNKI838 endocytosis motif
in the cytoplasmic tail of gH is required for clathrin-dependent endocytosis. Deletion
155

or mutation of the endocytosis motif results in larger syncytia in a vaccinia virus
fusion assay, suggesting that endocytosis might serve as a mechanism to downregulate
gH cell surface expression, thereby reducing virus-induced cell-cell fusion. gH and gL
or gE and gB are capable of inducing cell-cell fusion in a vaccinia virus infection
system (66, 154). In a luciferase assay, myelin-associated glycoprotein (MAG), a
potential gB receptor, and gB, gH and gL are required for cell-cell fusion (218). The
presence of gE in the luciferase assay decreases the amount of fusion. Despite the
different results from these assays, VZV gH is required by both for cell-cell fusion.
In HSV1, gH is an essential protein that is required for the formation of
hemifusion, or mixing of the outer lipid layer of two membranes, and gH and gB are
required for complete fusion (79, 217). HSV1 gH contains eight cysteine residues
(43). Two of these cysteines are important for gH maturation and cell surface
expression, while two other cysteines are important for gH function in fusion and virus
infectivity. HSV1 gH has two heptad repeats and two α-helices, typical elements of
class I fusion proteins (84, 85, 89, 93-96). α-helices function as fusion peptides that
interact with the cell membrane, thereby causing the fusion protein to act as a bridge
between the virus envelope and the plasma membrane. Heptad repeats (HRs) are
formed from 14, 21 or 28 amino acids composed of clusters of 7 amino acids that can
form α-helices, which then form a coiled-coil. Together, two HRs interact to form a
trimer of hairpins, or a six-helix bundle, and this interaction results in a
conformational change within the fusion protein to bring the viral envelope and
cellular membrane into close proximity. Mutations in the regions of the HSV1 gH
heptad repeats or α-helices abrogate fusion and virus infectivity complementation (93-
156

96). Peptides mimicking these HRs inhibit virus entry, adopt an α-helical
conformation in circular dichroism studies, and interact with one another, suggesting
that they are capable of forming a six-helix bundle within the gH protein (89, 95, 96).
Peptides corresponding to the α-helices can interact with membranes and induce lipid
mixing (84, 85, 93). They also adopt an α-helical conformation in circular dichroism
studies. Some studies have indicated that these peptides can enhance infection,
possibly by destabilizing membranes, but other studies have indicated that infectivity
is blocked at the step of virus attachment or entry.
Several other stretches of amino acids in HSV1 gH have also been shown to
interact with membranes. One particular region, a transmembrane-proximal stretch of
amino acids defined as HSV1 gH aa 776-802, is able to interact with membranes,
induce lipid mixing, and inhibit virus infectivity, although a glycosylation motif
within this region is not required (83, 86). Peptides mimicking other hydrophobic
regions of HSV1 gH, such aa 220-262 or aa 626-644 can also induce fusion, partition
membranes, and inhibit HSV infection (84, 85, 87). Specific residues within the HSV
transmembrane region, namely A808, S809 and G812, and the cytoplasmic tail,
especially 830SVP832, are also important for fusion, possibly because of their ability to
either act as helix breakers within the membrane or to interact with the membrane
(110, 244). These studies indicate that multiple regions within HSV-1 gH closely
associate with membranes and contribute to the initiation of hemifusion.
VZV gH might contain similar functional regions, but very little functional
analysis of VZV gH has been done, other than mutational analysis of the endocytosis
motif within the context of an expression vector or a recombinant vaccinia virus. No
157

mutations have been made in ORF37 in the context of the VZV genome. The objective
of this study was to determine whether gH was essential, as anticipated, and to analyze
predicted functional motifs of gH for contributions to VZV replication. gH mutations
that were compatible with virus replication were evaluated for pathogenesis in vitro
and in skin xenografts in the SCID mouse model in vivo. Mutations that were lethal
were analyzed using transient expression constructs to determine the effect of the
mutations on gH expression, maturation or localization. This study is the first to
analyze gH function using mutagenesis of the virus, and presents evidence for multiple
functional domains within VZV gH.
158

6.3. Materials and Methods
6.3.1. Analysis of potential functional motifs
The VZV parent Oka (pOka) gH sequence (accession number AAK19939) was
used to analyze bioinformatically predicted structural and functional motifs in gH.
Eight cysteines were identified within the gH sequence. The transmembrane region
was identified using algorithms available from TRAMPLE (68). These were the
ENSEMBLE algorithm, which predicts α-helices based on an ensemble of neural
networks and hidden Markov models, the HTMR, Psi Kyte-Doolittle and Kyte-
Doolittle algorithms, which use a neural network-based predictor based on
evolutionary information derived from non-redundant data sets of protein sequences
and from the Kyte-Doolittle hydrophobicity scale, and PRODIV_TMHMM_0.91,
TMHMM_2.0 predict transmembrane helices. The MEMSAT algorithm predicts
transmembrane regions and secondary structure based on the topology of the sequence
compared to the topology of well-characterized proteins. The TMpred algorithm,
which predicts membrane-spanning regions based on statistical analysis of TMbase, a
database of naturally occurring transmembrane proteins, was also utilized (115).
Potential coiled coils were predicted using the COILS algorithm with a 14, 21 or 28
amino acid window and a MTIDK matrix and comparing the prediction scores
between an unweighted and a weighted scan (152). Predicted α-helices were identified
using several algorithms available from TRAMPLE (68). These were the
ENSEMBLE, HTMR, Psi Kyte-Doolittle and Kyte-Doolittle algorithms and the
Secondary Structure Predictor algorithm, which predicts α-helix, β-strand and random
coils from neural network computation and profile generation based on soluble
159

globular proteins. Potential glycosylation residues were identified using NetNGlyc
1.0, which predicts NX(S/T) glycosylation sites using artificial neural network,
NetOGlyc 3.1, which predicts O-glycosylation with mucin-type N-
acetylgalactosamine (GalNAc) based on mammalian neural networks, YinOYang 1.2,
which predicts O-β-N-acetylglucosamine (GlcNAc) attachment sites in eukaryotic
proteins based on neural network predictions, the Eukaryotic Linear Motif (ELM),
which predicts functional sites in eukaryotic proteins based on patterns, and Motif
Scan, which uses hidden Markov models to predict functional motifs (100, 106, 128,
183). Potential phosphorylation sites were identified using ELM, Motif Scan, NetPhos
2.0, which uses neural networks to predict phosphorylation of serine, threonine and
tyrosine residues, NetPhosK, which produces neural network predictions of kinase-
specific eukaryotic protein phosphorylation sites, and KinasePhos, which uses hidden
Markov modeling to predict the substrate and kinase class (22, 23, 117). Published
sequences from twenty-one alphaherpesvirus sequences were aligned using ClustalX
1.0 in order to identify any region of gH with a high degree of conservation. The
accession numbers for the sequences used were as follows: AAK1939, AAG27211,
AAS45924, BAA03379, BAH02465, AAK51057, AAB27840, NP_954926,
CAA41677, CAA41678, AAG17895, NP_044491, NP_85188, YP_443868,
AAK55404, VGBE11, AAP13936, BAB16531, AAB27146, CAA74689, AAQ73700.
6.3.2. Cells
Human melanoma cells (Mel39) and human embryonic lung fibroblasts
(HELF) were grown at 37°C in culture media (MEM) supplemented with 10% fetal
160

bovine serum (Gemini Bio-Products, Woodland, CA), nonessential amino acids
(melanoma cells only) (100μM; Omega Scientific, Inc., Tarzana, CA), and antibiotics
(Penicillin, 100 U/ml; Streptomycin, 100 μg/ml; Invitrogen, Carlsbad, CA). HEK-293
cells were grown at 37°C in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO,
Carlsbad, CA) supplemented with 10% fetal bovine serum and antibiotics.
6.3.3. Construction of pOka cosmids with mutations in ORF37[gH]
The complete genome of the VZV parental Oka strain (pOka) is contained in
five overlapping SuperCos 1 cosmid vectors (Stratagene, La Jolla, CA), designated
pFsp73 (nucleotide [nt] 1-33128), pSpe14 (nt 21795-61868), pAfl17 (nt 53828-
79886), pAvr102 (nt 72298-96113) and pSpe23ΔAvrII (nt 94055-125123) (175).
ORF37 (nt 65935-66861) is located in the unique long region of the VZV genome,
contained within the pAfl17 cosmid. A KpnI fragment of approximately 15.5 kb (nt
57343-72902) containing the ORF37 sequence was cloned into the pNEB193 plasmid
(New England Biolabs, Ipswich, MA). In this plasmid, named pNEB193/pAfl-KpnI,
the SpeI (nt 5010) and SgrAI (nt 15853) sites, located 4067 nt upstream and 6776 nt
downstream of the ORF37 ATG, respectively, were unique sites and were used to
insert the ORF37 mutations into the cosmid. The pNEB193/pAfl-KpnI plasmid
contained unique BbvCI (nt 7387) and Bsu36I (nt 13361) sites, located 1690 nt
upstream and 4281 nt downstream of the ORF37 ATG, respectively. The
pNEB193/pAfl-KpnI plasmid was used as a template for PCR-based mutagenesis.
PCR mutagenesis using Taq (Invitrogen, Carlsbad, CA) was performed with two steps
of PCR to introduce the appropriate mutation in the BbvCI-Bsu36I fragment. The
161

strategy used for these substitution mutants has been previously described (16).
Briefly, in the first step, two PCR reactions were carried out using a 5’-outside
forward primer (2149-gH[37]-F) in combination with an internal mutagenic reverse
primer or a 3’-outside reverse primer (gH[37]4519-R) with an internal mutagenic
forward primer (Table 6.1). Internal mutagenic primers were designed in which the
mutation was inserted after the first 6-15 nt; the two internal mutagenic primers
overlap for approximately 20 nt. In the second step, the two PCR products were
combined and used as template for the second PCR. For this PCR, the two outside
primers were used, and a 6.67 kb fragment containing the mutation was amplified.
This fragment was cloned into pCR4-TOPO-TA (Invitrogen, Carlsbad, CA) and
sequenced to verify the mutation. The pCR4-TOPO-TA-gH plasmid containing the
6.67 kbp fragment with the substitution was digested with BbvCI and Bsu36I and the
fragment was inserted in pNEB193/pAfl-KpnI, replacing the wild-type BbvCI-Bsu36I
fragment. The pNEB193/pAfl-KpnI mutant plasmids were then digested with SpeI and
SgrAI and inserted into the pAfl17 cosmid, replacing the wild-type SpeI-SgrAI
fragment (nt 61941 to 72784 in pOka). These cosmids were sequenced to verify the
mutations.
6.3.4. Construction of pOka-DX bacterial artificial chromosomes (BACs) with
mutations in ORF371[gH]
Mutagenesis was performed using the self-excisable pOka-DX BAC as
described previously with modifications specific for ORF37[gH] (225). Briefly,
mutagenesis primers for recombination (Table 6.1) were used to amplify the Kanr gene
162

from the pKANS vector using AccuPrime Pfx (Invitrogen, Carlsbad, CA). The PCR
products were cloned into pCR4-TOPO-TA after the addition of adenosine overhangs
using recombinant Taq (Invitrogen, Carlsbad, CA). Clones were sequenced to
determine that the VZV-specific sequences did not have any unexpected deletions or
substitutions. The Kanr cassette flanked with the VZV sequences was amplified from
the pCR4.0 vectors using short primers (Table 4.1) to generate high yields of PCR
product. PCR products were gel purified (Qiagen, Inc., Valencia, CA) then 100ng was
used for recombination. After the red recombination steps to insert the mutations and
remove the Kanr cassette, BACs were purified using a Large-construct kit (Qiagen,
Inc., Valencia, CA). All purified BACs were digested with HindIII to ensure the
expected DNA fragments were present. In addition, BACs were sequenced directly
across the sites of mutagenesis to ensure that unexpected deletions or substitutions
were not present.
6.3.5. Repair of pOka-BACs with mutations in ORF371[gH]
Each of the ORF37 mutant BACs had the complete ORF deleted by red
recombination as described in the previous section using the primers [D37]F65895-
65915 and [D37]R68477-68497. To insert the wild type ORF37, a construct was
generated, pORF37-Kanr, which contained ORF37 coding for amino acids 1-434 and
362-841 of gH plus 111 nucleotides upstream and 82 nucleotides down stream of the
ORF. The pORF37-Kanr construct consisted of two regions of the pOka genome
(65824-67237 and 67016-68542; nucleotide locations were determined from the
pOka-DX BAC) that flanked the Kanr cassette. The ORF37-Kanr cassette was cloned
163

into pCR4-TOPO-TA and flanked by the PmeI restriction endonuclease site. The
restriction endonucleases NaeI and PmeI were used to digest 5ug of pORF37-Kanr
generating 20, 1499, 2,445 and 3,951 base pair (bp) DNA fragments. The 3,951 bp
fragment was gel purified and used for red recombination as previously described.
Purified BACs were used to recover infectious virus.
6.3.6. Transfection, virus isolation and DNA extraction
Recombinant viruses were isolated by transfection of human melanoma cells
(Mel39) with mutated pAfl17 cosmid and the four wild type cosmids or with the BAC
as previously described (213). Cosmid DNA was digested with AscI (the pAfl17
cosmid was also digested with AvrII), and 3 μg of pFsp73, pSpe14, pAfl17, and
pAvr102 and 1.5 μg of pSpe23ΔAvrII was cotransfected into Mel39 cells using 2 M
CaCl2 and HEPES-buffered saline. BAC DNA was transfected with the same method
using 4μg DNA total. Two clones of each mutant were transfected. Virus was
typically recovered 5-10 days post transfection. DNA was isolated from infected cells
using DNAzol (Gibco-BRL, Grand Island, NY), following the protocol as
recommended by the manufacturer, or using phenol-chloroform. PCR and sequencing
were performed to confirm that the expected mutations were present.
6.3.7. Excision of the MiniF- vector from the pOka BACs as determined by PCR
BAC-derived recombinant viruses were passed in HELFs until the MiniF-
sequences Cat (P1/P2, see Table 6.1) and SopA (P3/P4, see Table 6.1) were not
164

detectable by PCR as previously described (225). In addition, the primers gH[37]980-
F and gH[37]1395-R were used to detect ORF37 to determine the presence of virus.
6.3.8. Construction of expression plasmids containing ORF37 and ORF60
The wild-type gH and gL sequence and the mutant gH sequences were cloned
into the expression plasmid pCDNA3.1/V5-His TOPO TA (Invitrogen, Carlsbad, CA)
directly or from a pCR4-TOPO-TA vector. The vector pCR4-TOPO-TA-gH created
by amplifying ORF37 from the pNEB193/pAfl-KpnI plasmid using AccuPrime Pfx
(Invitrogen, Carlsbad, CA) and primers that inserted a HindIII site and a Kozak
sequence immediately prior to the ORF37 ATG start codon, and a second stop codon,
a BglII and a XbaI site following the ORF37 stop codon (Table 6.1). The PCR product
was cloned into pCR4-TOPO-TA after the addition of adenosine overhangs using
recombinant Taq (Invitrogen, Carlsbad, CA). This vector contained a unique HindIII
site 11 nt upstream of the ORF37 ATG, a unique BamHI site 897 nt downstream of
the ATG, a unique AccI site 1712 nt downstream of the ATG, a unique BstBI site
2375 nt downstream of the ATG and a unique XbaI site 2537 nt downstream of the
ATG. The pCDNA3.1-gH vector was created by amplifying gH from the
pNEB193/pAfl-KpnI plasmid using AccuPrime Pfx and primers that inserted a
HindIII site and a Kozak sequence immediately prior to the ORF37 ATG and a second
stop codon and a XhoI site immediately following the ORF37 stop codon. The
pCDNA3.1-gL vector was created by amplifying gL from the pSpe23ΔAvrII cosmid
using AccuPrime Pfx and primers that inserted a HindIII site and a Kozak sequence
immediately prior to the ORF60 ATG and a second stop codon and a XhoI site
165

immediately following the ORF60 stop codon. These PCR products were cloned into
pCDNA3.1 using the unique HindIII and XhoI sites. The pCDNA3.1-gH vector
contained a unique HindIII site 11 nt upstream of the ORF37 ATG, a unique BamHI
site 897 nt downstream of the ATG, a unique SwaI site 1493 nt downstream of the
ATG, a unique XhoI site 2530 nt downstream of the ATG, and a unique XbaI site
2536 nt downstream of the ATG. These vectors were sequenced to confirm that the gH
and gL sequence did not contain any unexpected deletions or substitutions.
Mutagenesis primers were used to insert amino acid changes into the ORF37
sequence (Table 6.1). Phosphorylated, adjoining primers and primers that included
unique restriction endonuclease sites were used to amplify the gH mutants from the
pCR4-TOPO-TA-gH plasmid. Each PCR product was digested with a single
restriction endonuclease, the vector was digested with two restriction endonucleases,
and a triple ligation was performed. The C575A and FPNG primers included either the
AccI site or the BstBI site, respectively, and were amplified as a single PCR product
that was then cloned into the vector directly using two unique. C327A PCR products
were inserted into pCR4-TOPO-TA-gH using HindIII and BstBI, then inserted into
pCDNA3.1-gH using BamHI and SwaI. C540A PCR products were inserted into
pCR4-TOPO-TA-gH using BamHI and BstBI, then inserted into pCDNA3.1-gH using
SwaI and XbaI. The C575A PCR product was inserted into pCR4-TOPO-TA-gH
using BamHI and AccI (contained within the C575A primer), then inserted into
pCDNA3.1-gH using SwaI and XbaI. C647A, C703A, and C724A PCR products were
inserted into pCDNA3.1-gH using BamHI and XhoI. C727A PCR products were
inserted into pCR4-TOPO-TA-gH using AccI and XbaI, then inserted into
166

pCDNA3.1-gH using SwaI and XbaI. HR2 and HR3 PCR products were inserted into
pCDNA3.1-gH using BamHI and XhoI. αH1 PCR products were inserted into pCR4-
TOPO-TA-gH using BamHI and AccI, then inserted into pCDNA3.1-gH using BamHI
and SwaI. αH2 PCR products were inserted into pCR4-TOPO-TA-gH using BamHI
and BstBI, then inserted into pCDNA3.1-gH using BamHI and SwaI. The FPNG PCR
product was inserted into pCR4-TOPO-TA-gH using AccI and BstBI (contained
within the FPNG primer), then inserted into pCDNA3.1-gH using SwaI and XbaI. All
vectors were sequenced to verify the presence of the expected mutation.
6.3.9. Transfection of expression plasmids
HEK-293 cells were seeded in regular media at a density of 1 x 106 cells per
cm2 24 h prior to transfection. Cells were transfected with a total of 0.5 μg DNA per
cm2. Cells transfected with multiple plasmids were transfected with equimolar
amounts of each plasmid. Lipofectamine2000 (Invitrogen, Carlsbad, CA) was used at
a 2:1 enzyme:DNA ratio following the manufacturer’s suggested protocol. Briefly,
cells were washed in Opti-MEM (Invitrogen, Carlsbad, CA) and then covered in Opti-
MEM. DNA and Opti-MEM or Lipofectamine2000 and Opti-MEM were mixed and
allowed to incubate at room temperature for five minutes. The Lipofectamine2000
mixture was then added to the DNA mixture and allowed to incubate at room
temperature for 30 minutes. This mixture was then added to cells, which were
incubated for 6 h at 37°C. After this time, media was replaced with regular HEK-293
media. 48 h after transfection, cells were fixed in 4% paraformaldehyde or lysed in
extraction buffer containing 0.1 M Tris-base (pH 8.8), 0.1 M NaCl, 5 mM KCl, 1 mM
167

CaCl2, 0.5 mM MgCl2, 1% sodium deoxycholate, 1% NP40 plus an EDTA-free
protease inhibitor cocktail (Roche, CA) (103).
6.3.10. Virus titration and replication kinetics
Virus titration was performed by 10-fold serial dilution on melanoma cells,
done in triplicate. For replication kinetics, 1 x 106 melanoma cells were seeded 24 h
prior to inoculation with 1 ml containing log10 3.0 PFU of virus. Virus was harvested
every 24 h for 6 days and titered in triplicate on melanoma cells for 6 days. All titer
plates were fixed at 3 dpi in 4% paraformaldehyde and stained with anti-VZV
polyclonal serum or a mixture of mouse monoclonal antibodies to VZV (Meridian
Life Sciences, Saco, ME). A 2-way analysis of variance (ANOVA) test was used to
determine if statistical differences (p≤0.05) existed between mutant and wild type
virus replication kinetics.
6.3.11. Antibody treatment of pOka infected HELF cultures in vitro
Following a protocol adapted from Rodriguez et al. (202), 1x106 HELF/well
were seeded in 6-well plates 24 h prior to inoculation. Monolayers were inoculated
with log10 3 PFU pOka for 90 minutes at 37°C. Media was changed, then 25 μg of
anti-gH mAb 206 or anti-gH SG3 was added and repeated at 24 h intervals. Mock-
treated cells received only media. Cells were fixed at 96 hours post infection (hpi) in
4% paraformaldehyde and stained with anti-VZV polyclonal serum.
168

6.3.12. Confocal microscopy of transfected and infected cells
HEK-293 cells were seeded in two-well chamber slides and transfected with
expression plasmids as detailed above. Melanoma cells were seeded at 3 x 105 cells
per well in 2-well chamber slides 24 h prior to inoculation with 300 PFU of virus. At
48 hpi, cells were fixed in 4% paraformaldehyde and blocked with PBS containing
10% donkey serum. If gH cell surface expression was to be analyzed in non-
permeabilized cells, blocking solution did not contain any Triton-X. If total gH
expression was to be analyzed, cells were blocked in solution containing 0.1% Triton-
X to permeabilized the cells. Staining for TGN46, a protein identified only in
permeabilized cells, was used as a control in non-permeabilized cells to ensure that no
permeabilization was present. Cellular localization of VZV proteins was performed
using four primary antibodies to VZV gH: mAb SG3 mouse anti-gH, Meridian Life
Sciences (Saco, ME), mAb 206 and mAb 258 (kind gifts of C. Grose) (166), and a
rabbit polyclonal antibody designated gH292-305. The SG3 antibody is an IgG1 antibody
that was created using Ellen isolate of VZV as an immunogen, according to the
manufacturer. mAb 206 and mAb 258 were created using pOka isolate of VZV as an
immunogen and are conformation-dependent, complement-independent neutralizing
antibodies (166). The gH292-305 antibody was produced as a rabbit antisera against a
synthetic peptide composed of gH amino acids (aa) 292-305. The antibody was
manufactured by GenScript (Piscataway, NJ). Primary antibodies were also used to
detect virus protein gE (rabbit polyclonal) (122) and cellular protein trans-Golgi
network 46 (TGN46) (AHP500 polyclonal sheep anti-TGN46, AbD Serotec, Oxford,
UK). Secondary antibodies used were FITC-coupled donkey anti-sheep (Jackson
169

ImmunoResearch, West Grove, PA), Alexafluor 488-coupled donkey anti-mouse
(Molecular Probes, Carlsbad, CA), Alexafluor 547-coupled donkey anti-rabbit
(Molecular Probes, Carlsbad, CA), Alexafluor 555-coupled donkey anti-mouse
(Molecular Probes, Carlsbad, CA), Alexafluor 647-coupled donkey anti-rabbit
(Molecular Probes, Carlsbad, CA), Alexafluor 647-coupled donkey anti-sheep
(Molecular Probes, Carlsbad, CA) and HOECHST 33342 (Molecular Probes,
Carlsbad, CA). Confocal microscopy was performed using a Zeiss LSM510 confocal
microscope equipped with two-photon excitation.
6.3.13. Preparation of cell lysates from transfected or infected cells for
immunoprecipitation and Western blot analysis of gH
Lysates were harvested from transfected HEK-293 cells as detailed above.
Infected cell lysates were prepared by seeding 1 x 105 melanoma cells per cm2 24
hours prior to inoculation with 1 x 102 PFU per cm2 of virus. Lysates were harvested
in extraction buffer at 48 hpi. Monoclonal antibody anti-gH SG3 (Biodesign, Saco,
ME) or no antibody was cross-linked to immobilized Protein G (Protein G Plus
UltraLink Resin, Thermo Scientific, Rockford, IL) as previously described (181).
Lysates were incubated with bound antibodies overnight, washed extensively, and
eluted with sodium dodecyl sulfate (SDS) sample buffer by incubating the beads at
100°C for 5 minutes. Denatured samples were resolved on SDS-polyacrylamide gels.
Proteins were transferred to Immobilon-P membranes (Millipore Biosciences,
Temecula, CA) and blocked with 5% BSA (mouse monoclonal antibodies) or dried
milk (rabbit polyclonal antibodies). gH was detected using two primary antibodies:
170

mouse mAb SG3 anti-gH (Meridian Life Sciences, Saco, ME) or the rabbit polyclonal
antibody designated gH292-305. Following Western blot detection of
immunoprecipitated gH, membranes were stripped, washed extensively in water, and
stained with GelCode Blue (Thermo Scientific, Rockford, IL), following the protocol
recommended by the manufacturer.
Virus lysates that were not immunoprecipitated were also resolved on pre-cast
SDS-polyacrylamide gels and transferred to Immobilon-P membranes for Western
blotting. These membranes were blocked as stated above, and probed with primary
antibodies to virus proteins gH (SG3), gE (mouse mAb 8612 anti-gE, Millipore,
Billerica, MA), IE63 (rabbit anti-IE63, a kind gift William Ruyechan), ORF23 (rabbit)
(47) and cellular α-tubulin (clone B-5-1-2 mouse anti-α-tubulin, Sigma, St. Louis,
MO). All primary antibodies were detected using horseradish peroxidase conjugated
antibodies to anti-mouse or anti-rabbit IgG and an ECL plus detection kit (GE
Healthcare Bio-Sciences Corp., Piscataway, NJ).
6.3.14. Protein identification by mass spectrometry
Melanoma cells were inoculated with pOka, infected cell lysates were
harvested 48 hours post infection and immunoprecipitated with anti-gH SG3 as
described above. Samples were separated by SDS-polyacrylamide gel electrophoresis,
and proteins were visualized by silver stain. Bands were excised and digested with
trypsin, and the proteins were identified using mass spectrometry performed by the
Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University Mass
Spectrometry (http://mass20spec.stanford.edu).
171

6.3.15. Replication of gH mutant viruses in skin xenografts in SCID mice
Skin xenografts were prepared in homozygous CB-17scid/scid mice, using human
fetal skin tissue obtained according to federal and state regulations (164). Animal
protocols complied with the Animal Welfare Act and were approved by the Stanford
University Administrative Panel on Laboratory Animal Care. Human tissues were
obtained, in accordance with state and federal regulations, from Advanced Bioscience
Resources (Alameda, CA). VZV-infected HELF cultures were used to inoculate the
xenografts. Infectious virus titer was determined at the time of inoculation by 10-fold
serial dilution on melanoma cells. Skin xenografts were harvested at 10 and 21 days
post infection (dpi). Half of each xenograft was homogenized and resuspended in 1 ml
PBS for virus titration and DNA extraction. Melanoma cells were seeded in a 24-well
plate and inoculated in triplicate with 0.1 ml of a 10-fold serial dilution of xenograft
homogenate. Media was changed 24 h after inoculation. Cells were cultured for 5
days, fixed in 4% paraformaldehyde and plaques were stained with anti-VZV
polyclonal serum or a mixture of mouse monoclonal antibodies against VZV
(Meridian Life Science, Saco, ME). Titer was analyzed using 2-way ANOVA to
determine if a statistically significant difference (p≤0.05) in titer existed. DNA was
extracted using DNAzol (Gibco-BRL, Grand Island, NY) following the
manufacturer’s recommended protocol or using phenol-chloroform. DNA was
amplified by PCR and sequenced to confirm that the expected mutation was present.
172

6.4. Results
6.4.1. Targeted disruption of predicted structural and functional domains in VZV gH
Analysis of VZV pOka gH (accession number AAK19939) for
bioinformatically predicted motifs was used to identify potential structural or
functional domains to investigate using targeted mutagenesis studies (Figure 6.1).
Eight cysteine residues were identified in gH. Agreement between a number of
algorithms (see Materials and Methods) predicts that the gH transmembrane region is
made up of amino acids in the region of aa 797-827. Based on this prediction, seven
cysteines are located in the gH ectodomain. These cysteines are C327, C540, C575,
C647, C703, C724 and C727, and each was substituted with alanine.
Coiled-coil domains that might act as potential heptad repeats (HR) were
identified at aa 38-51 (weighted score, 0.461 out of 1; designated HR1), aa 389-409
(weighted score, 0.299 out of 1; HR2), and aa 454-468 (weighted score, 0.224 out of
1; HR3). HR1 substitution of 38LrEY41 with GrGG decreased the coiled-coil prediction
score from 0.461 to 0.001. HR2 substitution of 399QL400 with AA decreased the
prediction score from 0.299 to 0.034. HR3 substitution of 456DEardqL462 with
AAardqA decreased the prediction score from 0.244 to 0.017. α-helices were
identified at aa 362-385 (designated αH1) and aa 500-539 (αH2). Since prolines and
aspartic acids act as helix breakers while alanines and leucines have high helix-
forming propensities, a region of αH1 was mutated from 368AAriA372 to PPriP and
αH2 was mutated from 505LFFA508 to DPPD. The double proline residues are
predicted to kink these helices.
173

Two predicted phosphorylation sites (S42 and S47) and one predicted N-linked
glycosylation site (N45) were identified within the predicted HR1 and were substituted
with alanine. The S47 residue was also substituted to threonine to maintain the
recognition motif for glycosylation of the N45 residue. Substitution of these residues
slightly altered the predicted HR1 score: S42A reduced the score to 0.368, N45A
increased the score to 0.468, S47A increased the score to 0.496, and S47T reduced the
score to 0.377. During cloning of the S47A substitution, an alanine substitution arose
at residue T127, a predicted O-N-acetylglucosamine site and phosphorylation site.
Each of these substitutions was analyzed separately, as was the double substitution
clone. T351 was a predicted phosphorylation site that was substituted with alanine.
The T751 residue was a predicted phosphorylation site adjacent to a highly-conserved
membrane-proximal region, and this residue was substituted with alanine. Alignment
of 21 alphaherpesvirus gH sequences demonstrated that the membrane-proximal
sequence (L/V/M)(L/M)(L/M)(F/Y)PNGT(V/I), located at aa 778-786 in VZV, was
highly conserved in the Varicelloviruses, Simplexviruses and Mardiviruses, but not
the Iltoviruses. N783 is a predicted glycosylation residue. This region is predicted to
form an α-helix. The highly conserved hydrophobic 781FPNG784 was substituted to
AAAA.
6.4.2. Deletion of ORF37[gH] or mutation of HR2, HR3, αH1, αH2 or FPNG are
lethal
Mutations in gH were incorporated into the VZV genome using the cosmid or
the BAC method. Transfection of either of two cosmid clones with point substitutions
174

gH-S42A, gH-N45A, gH-S47A, gH-S47T, gH-S47A/T127A, gH-T127A, gH-T351A
and gH-T751A resulted in viable mutant virus, as did transfection of BAC constructs
with mutations in gH-HR1.
Transfection of BAC clones with ORF37 deleted did not produce any virus,
indicating that gH is an essential protein for VZV pathogenesis. Transfection of either
of two BAC constructs with mutations in gH-HR2, gH-HR3, gH-αH1, or gH-αH2 or
gH-FPNG did not produce virus, indicating that these mutations were lethal. In order
to confirm that lethality was a direct result of the mutations in gH, the wild-type gH
sequence was restored and the transfection was repeated with the rescued clone. All of
these rescued viruses were viable, indicating that lethality was a direct consequence of
the deletion of or mutation in gH.
6.4.3. Point substitutions and the HR1 mutation alter the gH glycosylation state and
secondary structure
gH point substitution mutants and the HR1 mutant were assayed for gH
maturation in vitro. Lysates were harvested from infected cells at 48 hpi and gH
maturation was analyzed by immunoprecipitation with the anti-gH mAb SG3,
followed by SDS-polyacrylamide gel electrophoresis and Western blotting with the
SG3 antibody (Figure 6.2A). In wild type pOka infected cell lysate
immunoprecipitated with SG3, three distinct protein species were identified as gH.
The first was a small Mr band, with a calculated Mr of 100 kDa, which is likely the
same form of gH as the previously identified 94 kDa immature, glycosylated precursor
gH (166). The second was the expected 118 kDa mature, glycosylated gH product.
175

The third was a higher Mr band, calculated to be 130 kDa, which was observed only in
infected cell lysates immunoprecipitated with the SG3 antibody. Mass spectrometry
analysis of this 130 kDa protein species indicated that it was gH based on the detection
of four gH peptides (371IATSIFALSEMG382, 622TMMIFTTWTA631,
654NGEYVLILPAVQGHSYVIT672, and 679GLVYSLADVDVYNPISVVYLS699).
Western blot analysis of this same Immobilon-P membrane with the gH292-305 antibody
detected the 100 kDa gH and the mature, glycosylated 118 kDa gH, but not the high
Mr 130 kDa gH, indicating that the 292-305 epitope is masked, possibly by
glycomoieties, in the denatured form of this protein species.
gH-N45A and gH-S47A, as well as the double mutant gH-S47A/T127A,
exhibited a slight reduction in Mr of the mature, glycosylated gH protein species to
approximately 113 kDa, a loss of 5 kDa (Figure 6.2A). The Mr of the 100 kDa and the
130 kDa gH protein species were also reduced by approximately 5 kDa. This indicated
that the N45 residue was glycosylated and that the two alanine substitutions within the
recognition motif 45NMS47 disrupted this glycosylation. The gH-S47T was the same
Mr as the wild-type gH, indicating that no disruption in glycosylation resulted from
this substitution. No reduced Mr was apparent in the gH-T127A mutant, and the gH-
S47A/T127A double mutant was the same size as the single substitution mutants gH-
N45A and gH-S47A. The T127 residue might not be O-glycosylated, or deletion of the
O-linked oligosaccharide might not create a detectable difference in size.
Substitution of residues in this region of gH also disrupted the affinity of the
SG3 antibody. The gH-HR1 protein was not immunoprecipitated by the SG3 antibody
(Figure 6.2A). The SG3 antibody detected lower amounts of the gH-S42A, gH-N45A,
176

gH-S47A, gH-S47A/T127A and gH-T127A protein compared to wild type. Equivalent
protein amounts of the 100 kDa and 118 kDa forms of all gH proteins were detected
by the gH292-305 antibody, indicating that equal amounts of the gH proteins were
immunoprecipitated by the SG3 antibody. Protein amounts were also directly
determined by GelCode Blue staining of the nitrocellulose membranes, which
confirmed that equivalent protein amounts were present following
immunoprecipitation with the SG3 antibody (data not shown). The varying detection
might have resulted from alterations in how the gH mutant proteins denatured, thus
altering the presence of a structural epitope recognized by SG3. These data suggest
that the SG3 epitope is located in the region of the N-terminus that contains the
residues 38LREY41 and possibly S42, N45 and S47.
Both the SG3 and the gH292-305 antibodies detected the gH-T751A protein, but
the level of detection of this mutant gH protein in infected cell lysates appeared to be
lower than pOka gH levels (Figure 6.2A). In order to analyze whether gH protein was
decreased or if total virus protein was affected, infected cell lysates were analyzed
(Figure 6.2B). gH could not be detected directly from lysates, likely due to low levels
of total gH protein, but gE, immediate early protein 63 (IE63) and ORF23, a capsid
protein, were all detected. The amount of gE and ORF63 detected was equal to or
greater than that detected in the pOka infected cell lysate for all viruses. The amount
of ORF23 detected varied. This again confirms that the lower protein levels observed
in the lysates from cells infected with the gH N-terminus mutant viruses were a result
of differences in antibody affinity and not virus protein levels, while the differences
observed with respect to the gH-T751A protein might be due to slightly lower gH
177

protein levels, lowered affinity of antibodies for the gH-T751A protein, or degradation
of gH-T751A due to slight alterations in the protein.
6.4.4. Point substitutions and the HR1 mutation do not disrupt gH localization in vitro
gH localization during virus infection was examined in melanoma cells at 48
hpi using the SG3 antibody (Figure 6.3A). Syncytia, or large multinucleated cells,
were observed in monolayers infected with all viruses. Syncytia typically appear as
rosettes of nuclei, and a marker for the trans-Golgi network (TGN) accumulates in the
center of these rosettes. In infected cells and syncytia, gH localized to the cell surface
and throughout the cytoplasm, where it colocalized with the TGN marker. The TGN is
the site of both gH processing and maturation, and of secondary envelopment (91,
155). gH is targeted to the TGN following endocytosis from the cell surface. The
gH292-305 antibody did not detect any gH protein in infected cells, indicating that this
epitope is masked during infection, likely by protein folding in the 94 kDa and 118
kDa forms or by protein folding and additional glycomoieties in the 130 kDa form.
gH localization was normal in gH-S42A, gH-N45A, gH-S47A, gH-S47T, gH-
T127A, gH-S47A/T127A, gH-T351A and gH-T751A, but when the gH-HR1 virus
was evaluated, no gH was detected with the SG3 antibody (Figure 6.3A). This is in
agreement with the immunoprecipitation data that suggested the gH-HR1 protein has
lost the SG3 epitope. Typical rosettes, indicative of syncytia formation, were
observed, along with gE (data not shown), demonstrating that virus was present. The
gH292-305 antibody did not recognize the gH-HR1 virus, indicating that the gH-HR1
protein was folded correctly, masking the gH292-305 epitope. Additionally, mAb 206,
178

which recognizes a conformation-dependent epitope on mature, glycosylated gH, and
mAb 258, which recognizes a conformation-dependent epitope on immature,
glycosylated gH, did not detect gH protein in gH-HR1-infected cells (Figure 6.3B). As
demonstrated by deletion of ORF37, gH is an essential protein, and it is therefore
assumed that gH is still produced in the gH-HR1 virus despite the lack of detection by
any available antibodies.
Although gH localization was normal in cells infected with the gH-T751A
virus, syncytia formation was disrupted. Plaque size was not visibly different from that
observed in pOka-infected cells, but when plaques were viewed at a low
magnification, the size of syncytia were noticeably reduced compared to pOka (Figure
6.3C). In pOka-infected cells, typical syncytia involved 30-50 nuclei, with some
smaller syncytia, containing 5-20 nuclei, present on the edges of plaques. In gH-
T751A-infected cells, most syncytia contained 5-20 nuclei, and no 30-50 nuclei
syncytia were observed in any plaques. This indicated that the gH-T751A mutation
disrupted the ability of the virus to induce cell-cell fusion but did not affect virus exit
from infected cells and entry into adjacent cells.
6.4.5. Neutralization of VZV spread by the SG3 antibody
Since SG3, mAb 206 and mAb258 all failed to bind to the gH-HR1 mutant,
and mAb 206 and mAb 258 are known to be neutralizing antibodies, the ability of the
SG3 antibody to neutralize VZV was investigated. HELF cells were infected with
pOka grown in the absence of antibody or in the presence of mAb 206 or SG3 (Figure
6.4). After 96 h of replication, pOka grown in the absence of any antibody had
179

replicated and spread to most cells in the monolayer. Cells treated with mAb 206
showed very few plaques with little evidence of virus spread. Cells treated with the
SG3 antibody also exhibited reduced spread and reduced plaque size. This indicated
that the SG3 antibody was capable of neutralizing VZV pOka spread in a similar
manner as the mAb 206, although it was not as efficient.
6.4.6. Point substitutions and the HR1 mutation do not affect virus replication kinetics
in melanoma cells in vitro
Virus replication kinetics were assayed over six days and compared to wild-
type pOka virus generated using the same method (cosmid or BAC) as the mutant
virus. No statistically significant defect in replication was identified in any point
mutant or in the gH-HR1 virus when compared to wild type pOka (p>0.05; 2-way
ANOVA) (Figure 6.5). In all viruses, virus titers increased approximately 100-fold
during the initial two days following infection. After this increase, titer remained
relative constant for the remaining four days, and by 6 days post infection, most cells
exhibited extensive cytopathic effects or necrosis. Thus, the phenotypes of the point
mutation with respect to glycosylation, gH structure as determined by antibody
epitope detection, and virus-induced cell-cell fusion did not affect the ability of these
viruses to replicate efficiently in vitro.
180

6.4.7. gH-S47A, gH-T751A and gH-HR1 reduce VZV virulence in skin xenografts in
vivo
All gH mutants except for the double S47A/T127A mutant were analyzed for
any impairment of virulence in skin xenografts in the SCID mouse model in vivo.
Inoculum titers were not statistically different (p>0.05; 2-way ANOVA), and the
majority of the point substitution mutants had no effect on virus replication in vivo
(Table 6.2).
The gH-HR1 virus exhibited defective replication in vivo (Table 6.2). Despite a
1-log difference in growth at 10 dpi, titers at this time point were not statistically
different (p>0.05; 2-way ANOVA), probably due to variability in the titers recovered
from the two gH-HR1-infected xenografts. At 21 dpi, only one gH-HR1 xenograft was
positive for virus, and the titer was significantly lower than the pOka titer (p<0.05; 2-
way ANOVA), indicating that mutation of gH-HR1 disrupted an important gH
functional epitope required for efficient virus replication in vivo.
The gH-S47A virus had significantly lower titers recovered from skin
xenografts at 10 dpi (p<0.001; 2-way ANOVA), but not at 21 dpi, indicating that this
point substitution resulted in a delayed replication phenotype in vivo (Table 6.2). The
equal titer recovered at 21 dpi might result if the replication defect was overcome,
allowing the gH-S47A virus to replicate as well as pOka by 21 dpi. It might also result
if pOka infection peaked between 10 and 21 dpi and began to decrease following
extensive tissue necrosis, while the gH-S47A virus continued to replicate and peaked
at a later time point, resulting in an equivalent titer to pOka at 21 dpi. The gH-N45A
virus did not exhibit any replication defects, indicating that the disruption of
181

glycosylation at residue N45 was not responsible for the defective replication of the
gH-S47A virus. The gH-S47T virus also did not exhibit any replication defects,
indicating that the serine/threonine residue at this position might be important for gH
function or maintaining gH structure related to function.
The gH-T751A virus, which had a cell-cell fusion defect in vitro, did not have
any statistically significant effect on replication in vivo (p>0.05; 2-way ANOVA)
(Table 6.2). However, titers recovered from xenografts infected with this virus were
half a log lower than pOka titers at 10 dpi, and half a log higher than pOka at 21 dpi.
This virus might exhibit a very slight delayed replication phenotype, with a peak titer
occurring at a later time point than pOka. This delayed replication might relate to a
disruption in gH function relating to virus-induced cell-cell fusion, but the effect is not
as severe as that observed with the gH-S47A virus.
6.4.8.gH-C540A, gH-C575A, gH-HR2, gH-HR3, gH-αH1, gH-αH2 and gH-FPNG
mutations disrupt gH maturation
A pCDNA3.1 expression vector with ORF37 containing substitution of each of
the seven ectodomain cysteine residues to alanine was created to analyze the
contributions of these residues to gH structure, maturation and localization. Mutations
targeting gH-HR2, gH-HR3, gH-αH1, gH-αH2 or gH-FPNG were lethal, so ORF37
constructs containing each of these mutations were cloned into the pcDNA3.1
expression vector in order to analyze the effect of these mutations on gH. Lysates from
cells transiently expressing mutant gH and gL were analyzed for gH maturation
(Figure 6.6). Immunoprecipitation of the transfected cell lysates with SG3, followed
182

by Western blot detection with SG3, resulted in detection of the same three protein
species identified in immunoprecipitated infected cell lysates. These were the 130 kDa
gH, the mature, glycosylated 118 kDa gH, and the 100 kDa gH. No gH was detected
when vector alone or gH alone was transfected into HEK-293 cells.
The 100 kDa gH was detected in lysates of cells transiently expressing the
cysteine mutants, but the mature, glycosylated 118 kDa form of gH was only detected
in lysates from cells transiently expressing gH-C327A, gH-C647A, gH-C703A, gH-
C724A and gH-C727A (Figure 6.6A). The gH-C540A and gH-C575A proteins were
not detected as 118 kDa protein species, indicating that these mutations disrupt gH
maturation and processing. The gH-647A and gH-703A proteins were much fainter
than the pOka or other cysteine mutants. The 130 kDa gH protein species was very
faint or not present at a high enough level for detection in these two lysates, indicating
that these mutations might have disrupted the processing of gH into the high molecular
weight form. gH-C327A, gH-C724A and gH-C727A had less protein detected
compared to wild type gH, but more protein detected than the gH-C647A and gH-
C703A bands. The rabbit gH292-305 antibody detected protein levels of similar intensity
to the protein levels detected by the SG3 antibody (data not shown), indicating that the
decreased intensity was not due to altered affinity of the SG3 antibody, but more likely
due to lower levels of total gH protein in the transfected cell lysate or lower levels of
gH protein immunoprecipitated by the SG3 antibody.
Lethal mutations gH-HR2, gH-HR3, gH-αH1, gH-αH2, and gH-FPNG were
also tested for maturation (Figure 6.6B). The immature, glycosylated form of gH was
detected in all lysates except the gH-αH2 and gH-FPNG, and no mature, glycosylated
183

118 kDa gH or high Mr gH was detected. This indicates that these gH mutants are
produced as the 100 kDa immature gH protein species but maturation of gH has been
disrupted. The gH-αH2 and gH-FPNG mutants were expressed and detected by the
SG3 antibody when observed by confocal microscopy (see below), and therefore the
lack of detection by immunoprecipitation might have been a result of low transfection
efficiency of these mutants or reduced levels of transiently expressed proteins that
were not sufficient for recovery by immunoprecipitation.
6.4.9. gH-C540A, gH-C575A, gH-HR2, gH-HR3, gH-αH1, gH-αH2 and gH-FPNG
mutations are expressed but are not transported to the cell surface
Localization of gH was examined in melanoma cells transiently expressing gH
and gL (Figure 6.7). Transfection of the vector alone or of gH alone did not result in
any gH detected by SG3. When wild-type gH and gL were transfected together, gH
was detected in permeabilized cells throughout the cytoplasm, colocalizing with a
marker for the TGN, and was also expressed in punctae visible along cell extensions.
In nonpermeabilized cells, indicated by a lack of TGN marker detection, wild-type gH
was detected on the cell membrane. All gH mutants were detected in permeabilized
cells using the SG3 antibody, confirming that all mutants were expressed and that the
SG3 antibody was capable of detecting the mutant gH proteins.
When nonpermeabilized cells were examined for expression of gH mutant
constructs, however, obvious differences existed (Figure 6.7). gH-C327A and
gHC727A were both expressed on the cell surface in a pattern similar to wild type gH.
gH-C647A had a slightly more patchy distribution along the cell surface, and reduced
184

cell-surface expression compared to wild-type gH. gH-C703A and gH-724A were
both expressed on the cell surface, albeit extremely faintly and in a somewhat patchy
distribution. The gH-C647A and gH-C703A mutants also had low levels of gH
immunoprecipitated by the SG3 antibody. gH-C540A and gH-C575A were not
expressed on the cell surface, probably as a direct result of the lack of maturation. The
gH-HR2 and gH-HR3 mutants were expressed on the cell surface but only faintly,
similar to the gH-C703A and gH-C724A mutants. gH-αH1, gH-αH2 and gH-FPNG
were not expressed on the cell surface. This lack of cell surface expression might
result from the lack of maturation of these mutants, but the gH-HR2 and gH-HR3
mutants also failed to produce detectable levels of mature gH, yet were transported to
the cell surface.
185

6.5. Discussion
The conserved glycoprotein gH has been reported to play a role in virus
binding to cell surfaces, entry and fusion in many herpesviruses. The aim of this study
was to investigate the role of potential gH structural and functional motifs by
mutagenesis of ORF37 within the context of the VZV genome. The data presented
here have demonstrated that gH is an essential VZV protein. Point substitutions
demonstrated that the N-terminus of gH contains an important structural domain,
including a glycomoiety, as defined by gH antibody binding, and this domain was
required for efficient gH function and VZV pathogenesis in skin xenografts in vivo. A
substitution near the transmembrane domain demonstrated that this membrane-
proximal region was important for virus-induced cell-cell fusion. Other mutations of
predicted functional domains indicated that heptad repeats and α-helices were
essential for VZV pathogenesis in skin, as was a conserved 781FPNG784 sequence
within the membrane-proximal region. Cysteine residues were important for
maintaining structure related to gH processing, maturation and transport.
This study is the first to demonstrate that gH is an essential VZV protein. gH is
required for virus fusion in both a vaccinia virus fusion assay and a luciferase fusion
assay (66, 218). This role in fusion, probably during entry, is likely to be critical for
VZV pathogenesis. gH has been demonstrated to be an essential protein in HSV and
pseudorabies virus (PRV) (14, 79). In both HSV and PRV, gH is required for fusion of
the virion envelope with the cell plasma membrane but not for attachment. Fusion is a
necessary step to allow herpesvirus entry into cells, and HSV gH plays a role in
186

inducing hemifusion between the two lipid membranes (217). VZV gH might play a
similar role in inducing hemifusion and allowing virus entry into the cell.
This study identified three separate gH protein species from both VZV-
infected and transiently transfected cells. Identification of two of these species was
expected. A 100 kDa form, possibly similar to a previously identified 94 kDa
immature, glycosylated form, and the 118 kDa mature, glycosylated form, also
previously identified, were expected products of ORF37. It has been demonstrated that
the 94/100 kDa form of gH is processed into the 118 kDa form by addition of complex
oligosaccharides during gH transport through the Golgi (166). A new gH protein
species of 130 kDa was also identified in this study, and is likely a highly glycosylated
gH. This 130 kDa gH might result from further processing of the 118 kDa form or
might be a precursor to the 118 kDa form. This protein species could play a role in
VZV pathogenesis, either during replication in the cell or as a component of the
infectious virus particle.
Structural similarities were shown to exist among these three species of gH.
The epitope detected by the SG3 antibody, which has not been mapped, was conserved
among all three gH species. Additionally, the aa 292-305 were masked by protein
folding in all three, based on failure of the antibody to this epitope to recognize any
gH in infected cells. This epitope is identified only under denaturing conditions. One
structural difference between these forms was that even following denaturation, gH292-
305 could not detect aa 292-305 in the 130 kDa gH, which suggests that some
additional glycomoieties are located in the region of aa 292-305 in this form of gH.
187

The data presented here suggests that the SG3 epitope is located in a structural
region of the N-terminus that contains the residues 38LREY41, and contains or is
affected by the proximity of the glycomoiety on N45. Alternatively, these residues
might directly affect the structure that is detected by the SG3 antibody. Based on the
lower affinity of the SG3 antibody for the denatured mutant gH, the neutralization
ability of this antibody, and the fact that the SG3 antibody was raised against gH
expressed in infected cells, it is possible that the SG3 antibody detects a conformation-
dependent epitope. mAb 206 and mAb 258 might also recognize the same epitope
since all three antibodies failed to detect the gH-HR1 mutant and all three are
neutralizing antibodies. A previous study analyzing nine neutralizing antibodies
against gH, not including SG3, mAb 206 or mAb 258, has demonstrated that the nine
antibodies recognized seven different epitopes and that six of these seven epitopes
were near glycomoieties on gH, although the previous study did not map the epitopes
(3). Based on the neutralization ability of these nine antibodies, this previous study
also proposed that the seven epitopes were located on a single gH functional domain
that was required for virus entry and cell-cell spread. The data reported here suggest
that the SG3/mAb 206/mAb 258 epitope involves or is located near the glycomoiety
on N45, and might be the same as one of the seven previously identified epitopes.
The gH N-terminus is an important structural domain, based on evidence that
the SG3/mAb 206/mAb 258 epitope is within this region, and it is an important
functional region as shown by the fact that these antibodies can neutralize VZV
infectivity. The neutralizing activity of antibodies specific for this region points to its
role in gH-dependent fusion. Even though binding of the SG3/mAb 206/mAb 258
188

antibodies is lost, the mutant gH-HR1 protein is compatible with VZV replication in
vitro, indicating that the motif created by the 38LREY41 residues is not absolutely
required for gH function in vitro. However, mutation of 38LREY41, thereby disrupting
the predicted α-helix, appears to significantly alter gH folding and conformation, as
shown by disruption or loss of the structural epitope. This gH mutation resulted in a
delayed growth phenotype in vivo. S47 substituted to alanine also disrupted VZV
pathogenesis in skin xenografts in vivo, while substitution to threonine had no effect,
suggesting that the hydroxyl group present on the serine and threonine residues but
absent on the alanine residue might be involved in amino acid side chain interactions
that are important for gH structure and function. Overall, this indicates that
maintaining the correct structure of the N-terminal region involving the important
residues 38LREY41 and specific side chain interactions involving S47 is important for
gH function and VZV pathogenesis in vivo.
Residues proximal to the transmembrane region are important for a gH role in
virus-induced cell-cell fusion. The gH-T751A substitution disrupted virus-induced
cell-cell fusion and syncytia formation in vitro, and caused a slight delay in VZV
replication in vivo. The 781FPNG784 motif, which is predicted to be part of an α-helix,
was required for VZV pathogenesis. The alanine substitutions, which abolish gH
maturation and cell surface expression, might alter the helical structure, resulting in
lethality based on disruption of gH structure relating to membrane interactions and the
role of gH in fusion. In support of this hypothesis, a corresponding region in HSV gH,
defined as the membrane-proximal region containing aa 761-806, also plays a role in
fusion (86). Mimetic peptides from this region can adopt helical conformations, induce
189

fusion of lipid vesicles, and inhibit HSV infection. In particular, the HSV
779LLLFPNGTV787 motif is required for these peptides to induce fusion, although
glycosylation of the NGT motif is not required for virus pathogenesis, suggesting that
this region plays a role in gH fusion (83, 86). Proline and phenylalanine residues in
this region were thought to be important for peptide insertion into membranes and for
fusion. The VZV gH F781 and P782 residues that were substituted to alanine might be
required for this gH function in VZV pathogenesis.
The T751 residue, which is potentially located structurally near the highly
conserved, essential 781FPNG784 residues, could be involved in interactions important
for maintaining gH structure or function. This would explain the decrease in syncytia
size observed during gH-T751A infection. This reduced cell-cell fusion likely also is
the cause for the slightly delayed growth of this mutant in skin xenografts in vivo,
although this mutation did not cause a defect in virus exit from infected cells and entry
into adjacent cells. This evidence indicates that this region contributes to the role of
gH in virus-induced cell-cell fusion but not in virus egress or entry, suggesting that
regions of gH have distinct roles in gH-induced fusion during entry and gH-induced
fusion during syncytia formation. These data also suggest that virus-induced cell-cell
fusion is not required for spread of the virus. Indeed, cell-cell fusion between a VZV-
infected cell and a neighboring cell does not occur until 9 hpi of the neighboring cell
(199). Additionally, VZV-infected T cells do not under go fusion, indicating that VZV
infection can occur naturally in the absence of virus-induced cell-cell fusion (21).
Syncytia formation may increase the efficiency of VZV cell-cell spread in vivo, as
demonstrated by the very slight delayed-growth phenotype of the gH-T751A virus, but
190

it does not appear to be required for skin infection in vivo. Separation of a gH role in
entry versus cell-cell fusion has also been proposed for HSV, as several residues in the
HSV gH cytoplasmic tail, namely 830SVP832, appear to be required for efficient
syncytia formation but not for virus infection, suggesting these residues are not
important for virus entry (244). For the first time, a point substitution in VZV gH has
demonstrated a similar separation of gH function in entry and syncytia formation.
The predicted heptad repeats gH-HR2 and gH-HR3 are essential for VZV
pathogenesis, indicating that their α-helical structure and potential function, likely in
fusion, are required for VZV replication. gH-HR2 and gH-HR3 were expressed on the
cell surface at low levels, likely indicating that they were mature, although lack of
detection of 118 kDa protein by the SG3 antibody could indicate that only low levels
of mature, glycosylated gH were produced. Another possible explanation is that the
mutations might have resulted in the trafficking of immature gH to the cell surface. If
these gH proteins were properly processed and transported, the lethality of these
mutations might have resulted from disruption of the predicted helices in the two HRs.
The disruption of the helices could prevent gH from undergoing conformational
changes via coiled-coil formation in response to a fusion event, thereby preventing
virus entry and virus-induced cell-cell fusion. Studies are currently underway to
analyze mimetic peptides of these regions to determine if they form α-helical coiled-
coils and whether or not the substitutions disrupt the helical nature of the peptides.
Additionally, the ability of these peptides to interact and any resulting increase in
helicity, indicating the formation of a trimer of coils, is being investigated. Further
evidence for a role of these regions in fusion comes from HSV gH, which has
191

functional heptad repeats that are positionally conserved relative to gH-HR2 and gH-
HR3 (95). Mutations within these HSV heptad repeats disrupted cell-cell fusion and
virus infectivity complementation, similar to the lethality of the VZV substitutions.
HSV peptide studies have also demonstrated that these HSV heptad repeats form
coiled-coils, interact with one another, and can inhibit infection during entry (89, 96).
Peptides corresponding to a predicted heptad repeat in HCMV gH also inhibit virus
entry (151). The VZV peptide studies will shed further light on whether or not the
lethality of these mutations resulted from a disruption in gH structure and function
relating to a role in fusion.
The α-helices gH-αH1 and gH-αH2 might contribute to gH fusion, but the
lethality of the αH1 and αH2 mutations likely directly resulted from a lack of mature
gH on the cell surface. No mature 118 kDa gH was detected in cells transiently
expressing these mutants and gH was not trafficked to the cell surface, indicating that
either maturation has been disrupted or the protein structure has been altered and the
SG3 epitope was disrupted. The double proline substitutions incorporated into these
mutants are predicted to kink the helices formed by the amino acids in these regions,
and the resulting disruption to these helices might alter the overall protein structure,
disrupting either gH/gL interaction, gH maturation, or gH trafficking. Although the
disruption to gH maturation and transport in this study prevents conclusions regarding
VZV gH-αH1 or gH-αH2 function relating to fusion, previous substitution studies
have demonstrated that VZV αH1 is capable of acting as a fusion peptide within HSV
gH (93, 94). αH1 is positionally conserved in all eight human herpesviruses and in a
number of other herpesvirus gH sequences. αH2 is positionally conserved in the
192

human alphaherpesviruses. Substitution of the HSV αH1 sequence with the VZV αH1
sequence resulted in a functional protein but substitution of HSV αH2 with VZV αH1
did not, suggesting both that the VZV αH1 was a functional fusion peptide and that
the αH1 and αH2 sequences might have distinct roles in gH fusion. It was
demonstrated that the HSV α-helices had attributes of fusion peptides: they were
required for efficient cell-cell fusion and virus infectivity complementation, and
mimetic peptides could induce fusion of lipid vesicles and formed helices (84, 85, 93,
94). Further studies of mutations that abolish VZV α-helix function but not gH
maturation and transport, or of mimetic peptide to determine structure and association
with membranes, will be needed to confirm that VZV gH-αH1 and gH-αH2 act as
functional fusion peptides.
gH contains cysteine residues that are important for the overall gH structure,
maturation, and transport. C540 and C575 were required for VZV gH maturation and
transport to the surface, suggesting that these cysteines might be involved in a
disulfide bond or another aspect of gH structure relating to gH/gL interaction or gH
maturation. These cysteines are located in homologous positions to HSV C554 and
C589 (43). Similar results were observed when these HSV cysteines were substituted,
resulting in lower levels of gH protein expression and disrupted gH/gL interaction and
maturation. These cysteines are positionally conserved in numerous alphaherpesvirus
and in all eight human herpesviruses, indicating their potential importance for correct
gH structure. This supports the hypothesis that VZV C540 and C575 are important for
gH structure relating to its interaction with gL or maturation and transport of gH.
193

VZV gH also contains cysteine residues that are important for gH expression
on the cell surface, and possibly gH function. C347A and C727A substitutions did not
appear to affect gH protein expression, maturation or localization, but substitution of
C647, C703 and C724 with alanine resulted in decreased protein levels observed both
by immunoprecipitation and by confocal microscopy analysis of gH cell surface
expression. C647 and C703 are positionally homologous to HSV C652 and C706,
which are important for HSV gH/gL interaction, gL maturation following interaction
with gH, fusion and virus infectivity complementation (43). These two cysteines are
also positionally conserved in numerous alphaherpesvirus and human herpesvirus gH
sequences, signifying that they might be important for gH structure or function
relating to fusion. Since regulation of gH cell surface expression levels by endocytosis
has been suggested as a mechanism by which VZV can modulate virus-induced cell-
cell fusion, the low levels of cell surface expression resulting from the VZV cysteine
substitutions might indicate that these cysteines are important for fusion (185). BAC
constructs containing these point substitutions are being created to analyze the
contribution of these cysteines to VZV pathogenesis.
This report has demonstrated that VZV gH is an essential protein that has
several important functional regions. The structure and function of the N-terminus is
important for efficient VZV pathogenesis in vivo. This region contains the epitope for
the SG3 antibody, as well as mAb 206 and mAb 258, and the requirement of this
region for VZV pathogenesis is consistent with efficient neutralization by antibodies
that bind this region. A hydrophobic membrane-proximal region, predicted to form an
α-helix, is essential for VZV pathogenesis, likely contributing to the gH role in virus-
194

induced cell-cell fusion and syncytia formation. A residue within this region is
important for syncytia formation, but not for a gH role in entry. Predicted helical
structural elements are also essential for VZV pathogenesis, and might compose
heptad repeats and α-helices with characteristics of fusion peptides, indicating that gH
has attributes of a class I fusion protein. Cysteines are important for maintaining gH
structure during maturation and transport. Altogether, these data prove VZV gH is an
essential protein that has multiple functional domains and plays an important role in
VZV fusion and in the pathogenesis of skin infection in vivo.
This work has implications for gH function in both virus entry and in virus-
induced cell-cell fusion, as well as identifying the structure of gH. Furthermore, this
work has possible implications for future VZV vaccine development. The current
VZV vaccine is recommended for use in healthy patients, but because this vaccine is a
live-attenuated vaccine, use in immunocompromised patients is limited due to the
potential for disease development (156). Some of the gH mutations presented in this
study disrupt VZV pathogenicity, and these targeted mutations could be incorporated
into a second-generation VZV vaccine that might have reduced pathogenicity in both
healthy and immunocompromised patients, and furthermore might have reduced
pathogenicity in neuronal tissue, disrupting the ability of the virus to establish latency.
Alternatively, a VZV vaccine with ORF37 deleted could be developed. Although this
vaccine virus would need to be grown on a gH-complementing cell line, it would only
be infectious for one round of replication following administration. This might result
in a protective immune response but a complete absence of disease development, and
possibly also disruption of the establishment of latency. Due to the essential
195

contribution of gH to VZV pathogenesis, incorporating either targeted mutations into
gH or deleting ORF37 altogether could result in a efficient vaccine capable of eliciting
protective immunity but not causing disease.
196

Table 6.1. Primers used for the mutagenesis of ORF37
All primers used for cloning of ORF60 in the pCDNA4.1 expression vector, and all
primers used for mutagenesis and cloning of ORF37 in the pAfl17 cosmid, the self
excisable pOka-DX, the pCR4-TOPO-TA or the pCDNA3.1 expression vector.
Primer Primer use Primer sequence
Cosmid cloning
2149gH[37]-F gH amplification ACAAAGTCAACTGGCGTGAA 133gH[37]-F ACAAAGTTAAAGCCGATCCT gH[37]33-F CCTCTTTGGACCACGGCTAA gH[37]603-F GCCCTTGCCCGACATACTTT gH[37]980-F CGCATATCCCGAAGAGAGTT gH[37]1565-F TTCATCTCGGGTATTAGACG gH[37]2168-F ATGTTTGTATTGCGGAAGTG gH[37]667-R GTATTCTCAACGTTGAGTTGG gH[37]1395-R TGCGGTCTTTAGTTGATCACG gH[37]2168-R TCTTTTAAATTGTCCGGATGG gH[37]4519-R ACAAAGTTAAAGCCGATCCT gH[37]S42A-F S42A substitution ACGAGAATATGCCGACCGTA gH[37]S42A-R TACGGTCGGCATATTCTCGT gH[37]N45A-F N45A substitution CCGACCGTGCTATGTCTCTGA gH[37]N45A-R TCAGAGACATAGCACGGTCGG gH[37]S47A-F S47A substitution GTAATATGTCGCTGAAATTA gH[37]S47A-R TAATTTCAGAGCCATATTAC gH[37]S47T-F S47T substitution CGTAATATGACTCTGAAATTAGAAGC gH[37]S47T-R GCTTCTAATTTCAGAGTCATATTACG gH[37]T351A-F T351A substitution TCTGCGGATGGCAACGAAGG gH[37]T351A-R CCTTCGTTGCCATCCGCAGA gH[37]T751A-F T751A substitution AAGATGCAGAACGACAACTA gH[37]T751A-R TAGTTGTCGTTCTGCATCTT
BAC cloning
gH[37]HR1-F HR1 substitutions AACCCCTGCGACTCGCTCTATCGGACATATGTCTGCTCTTGGACGAGGTGGTTCCGACCGTAATATGTCTAGGATGACGACGATAAGTAGGG
197

gH[37]HR1-R GATAAAAGGCTTCTAATTTCAGAGACATATTACGGTCGGAACCACCTCGTCCAAGAGCAGACATATGTCCCAACCAATTAACCAATTCTGATTAG
gH[37]HR2-F HR2 substitutions AGAATATTTTCTGTTAGATGAGATCGTAGATGTTCAGTATGCAGCAAAATTCCTTAATTACATTAGGATGACGACGATAAGTAGGG
gH[37]HR2-R CTCCTGCTCCTATCCGCATTAAAATGTAATTAAGGAATTTTGCTGCATACTGAACATCTACGATCAACCAATTAACCAATTCTGATTAG
gH[37]HR3-F HR3 substitutions
TCAGGTAAAACCCGCAAATGTCGATTATTTTATTTCATATGCAGCTGCCCGTGATCAAGCAAAGACCGCATACGCGCTTAGGATGACGACGATAAGTAGGG
gH[37]HR3-R
TCACATGGTCTTGACCACGGGAAAGCGCGTATGCGGTCTTTGCTTGATCACGGGCAGCTGCATATGAAATAAAATAATCCAACCAATTAACCAATTCTGATTAG
gH[37]αH1-F αH1 substitutions
CGGATCAACATGACATAAACGAGGAAAGCTATTACCATATCCCGCCTAGAATACCGACATCAATTTTTGCGTTGAGGATGACGACGATAAGTAGGG
gH[37]αH1-R
CTGTGGTACGGCCCATTTCCGACAACGCAAAAATTGATGTCGGTATTCTAGGCGGGATATGGTAATAGCTTTCCCAACCAATTAACCAATTCTGATTAG
gH[37]αH2-F αH2 substitutions GCTTGTGAAGCAAAATTTAAATGCTACAGAGAGGCAGGCTGGACCTCCTGGATCAATGATTTTATTAAATAGGATGACGACGATAAGTAGGG
gH[37]αH2-R AATTTTCTAGTCCTTCGCGGAAATTTAATAAAATCATTGATCCAGGAGGTCCAGCCTGCCTCTCTGTAGCCAACCAATTAACCAATTCTGATTAG
gH[37]FPNG-F FPNG substitutions TCCAGACATGCACGGGGATGACTCTAAGGCTGTGTTGTTGGCTGCTGCCGCTACTGTGGTAACGCTTCTAAGGATGACGACGATAAGTAGGG
gH[37]FPNG-R TGGCTTGTCGTCGTTCGAATCCTAGAAGCGTTACCACAGTAGCGGCAGCAGCCAACAACACAGCCTTAGACAACCAATTAACCAATTCTGATTAG
[D37]-F ORF37 deletion ATAACGTTGCGGTGATATTGTAGCGCAAGTAACAGCGACTTAAAAAACATGTATAATAAGGATGACGACGATAAGTAGGG
[D37]-R GAATACGTTTATAGTGACTTTTTATTATACATGTTTTTTAAGTCGCTGTTACTTGCGCCAACCAATTAACCAATTCTGATTAG
Amplification of cloned BAC products
[37]F72-92 HR1 substitutions AACCCCTGCGACTCGCTCTAT [37]R143-163 GATAAAAGGCTTCTAATTTCA [37]F1155-1175 HR2 substitutions AGAATATTTTCTGTTAGATGA [37]R1220-1240 CTCCTGCTCCTATCCGCATTA [37]F1326-1346 HR3 substitutions TCAGGTAAAACCCGCAAATGT
198
[37]R1406-1426 TCACATGGTCTTGACCACGGG [37]F1061-1081 αH1 substitutions CGGATCAACATGACATAAACG [37]R1136-1156 CTGTGGTACGGCCCATTTCCG [37]F1476-1496 αH2 substitutions GCTTGTGAAGCAAAATTTAAA [37]R1547-1567 AATTTTCTAGTCCTTCGCGGA [37]F2301-2321 FPNG substitutions TCCAGACATGCACGGGGATGA [37]R2372-2392 TGGCTTGTCGTCGTTCGAATC [D37]F65895-65915 ORF37 deletion ATAACGTTGCGGTGATATTGT [D37]R68477-68497 GAATACGTTTATAGTGACTTT [37]F65680-65700 Colony screening ACAACACTTCCTAAATATACC [37]R68697-68717 AACGGCGCTGTGCGTCTATGC P1 (mini-F) BAC screening TTAACTCAGTTTCAATACGGTGCAG P2 (mini-F) TGGGGTTTCTTCTCAGGCTATC P3 (cat) AGGCATTTCAGTCAGTTGCTC P4 (cat) TGCCACTCATCGCAGTACTG
Expression vector cloning
HindIII-Kozak-gH[37]-F gH amplification TTAAAGCTTGCCACCATGTTTGCGCTAGTTTT
AGCG
gH[37]XbaI-BglII-R TCTAGAAGATCTATCATTATGTCAGAGGTATTTTATTATATTC
gH[37]XhoI-R TTACTCGAGTCATTATGTCAGAGGTATTTTATTATAT
gH[37]894BamHI-F CCGGATCCGGGGCCATCTTATCG gH[37]1697AccI-F TACAATACCAAACGTATACAGTCC gH[37]1725AccI-R ACAAGGACTGTATACGTTTGG gH[37]2369BstBI-F AGGATTCGAACGACGACAAGC gH[37]2391BstBI-R GGCTTGTCGTCGTTCGAATCC HindIII-Kozak-gL[60]-F gL amplification TTAAAGCTTGCCACCATGGCATCACATAAATG
GTTAC
gL[60]XhoI-R TTACTCGAGTCATCATTGGCATACGCGTTGGAA
gH[37]C327A-F C327A substitution [Phos]GCCGCATATCCCGAAGAGAGTTTGG gH[37]C327A-R [Phos]TATATCGACCGTAGCATGCTTTG gH[37]C540A-F C540A substitution [Phos]GCTACGGCAGCTCACGCCACGCAAGC gH[37]C540A-R [Phos]CATGGATGTCATTAAAAGCAAAG
gH[37]C575A-AccI-F C575A substitution TACAATACCAAACGTATACAGTCCTGCTATGGGTTCC
gH[37]C647A-F C647A substitution [Phos]GCAGCCGCGCGTAACGGAGAATATG gH[37]C647A-R [Phos]ATCTTGAGCCGTAAATACTTCTGG gH[37]C703A-F C703A substitution [Phos]GAACATGGTGTCATAGAGACG
gH[37]C703A-R [Phos]AGACACGGCAGTATCCTTGCTTAAATAAAC
199

gH[37]C724A-F C724A substitution [Phos]TGCGGAAGTGTTTTTCTTAGG gH[37]C727A-R [Phos]ATACAAAGCTTCTTTTAAATTGTCC gH[37]C727A-F C727A substitution [Phos]GGTATCTAACCACGGGGGCG
gH[37]C727A-R [Phos]TAAGAAAAACACTTCCGGCATACAAACA
gH[37]αH1-R αH1 substitutions [Phos]TGATGTGGGTATTCTTGGGGGGATATGGTAATAGCTTTCC
gH[37]αH1-F [Phos]ATTTTTGCGTTGTCGGAAATGG
gH[37]αH2-R αH2 substitutions [Phos]CATTGAGCCAGGAGGATCAGCCTGCCTCTCTGTAGC
gH[37]αH2-F [Phos]ATTTTATTAAATTTCCGCGAAGG
gH[37]FPNG-BstBI-R FPNG substitutions GTCGTTCGAATCCTAGAAGCGTTACCACAGTTGCGGCTGCAGCCAACAACACAGCCTTAGAG
200

Figure 6.1. Predicted structural and functional motifs in VZV gH and
substitutions used to disrupt them
Predicted structural and functional motifs identified in VZV gH. The signal sequence
and transmembrane region are identified by black boxes. Predicted heptad repeats
(HR) are identified by white boxes. Predicted α-helices are identified by gray boxes.
The location of predicted glycosylation sites, predicted phosphorylation sites, cysteine
residues and a highly conserved hydrophobic region are identified by arrows. The
substitutions indicated were incorporated into the VZV genome.
201

Figure 6.2. Analysis of gH maturation and virus protein in lysates from infected
melanoma cells
Lysates were harvested 48 hpi from melanoma cells infected with wild type or mutant
gH viruses. (A) gH was analyzed by immunoprecipitation with the SG3 antibody, and
then blotted with the SG3 or the gH292-305 antibody. (B) Virus lysates were analyzed
202

for virus gE, IE63 and ORF23, and for cellular α-tubulin. No gH could be detected
directly in non-immunoprecipitated lysates.
203

Figure 6.3. gH localization and syncytia formation in melanoma cells
204

Figure 6.3. gH localization and syncytia formation in melanoma cells
Melanoma cells were mock infected or infected with wild type or mutant gH viruses
and fixed 48 hpi. Cells were stained for gH (red), TGN46 (green; AHP500) and nuclei
(blue; HOECHST 33342). (A) Anti-gH SG3 antibody. (B) Anti-gH mAb 206 and
mAb 258. Inset, uninfected. (C) Anti-gH SG3 antibody. Scale: 25 μm.
205

Figure 6.4. Inhibition of pOka spread by anti-gH mAb 206 and SG3
Inhibition of pOka spread in infected HELF cells by anti-gH mAb 206 and SG3 was
examined. Cells were treated with antibody starting 90 minutes post infection, and
virus was allowed to replicate for 96 h. Virus was stained with polyclonal anti-VZV
serum and visualized with Fast Red substrate. (A) Mock-treated cells. (B) mAb-206-
treated cells. (C) SG3-treated cells.
206

Figure 6.5. pOka and mutant gH virus replication kinetics in melanoma cells in
vitro
Virus replication kinetics were assayed by infecting 1 x 106 melanoma cells with log10
3.0 PFU of virus. Infected cells were harvested and virus was titered in triplicate on
melanoma cells every 24 h over six days.
207
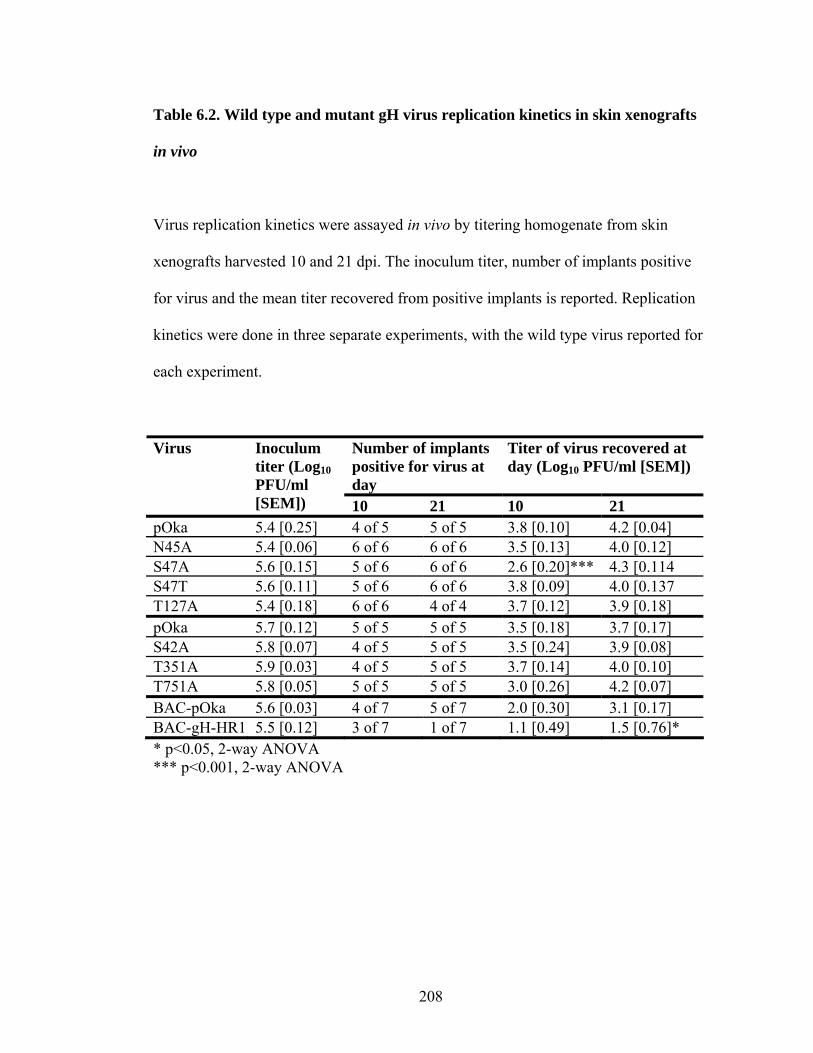
Table 6.2. Wild type and mutant gH virus replication kinetics in skin xenografts
in vivo
Virus replication kinetics were assayed in vivo by titering homogenate from skin
xenografts harvested 10 and 21 dpi. The inoculum titer, number of implants positive
for virus and the mean titer recovered from positive implants is reported. Replication
kinetics were done in three separate experiments, with the wild type virus reported for
each experiment.
Number of implants positive for virus at day
Titer of virus recovered at day (Log10 PFU/ml [SEM])
Virus Inoculum titer (Log10 PFU/ml [SEM]) 10 21 10 21
pOka 5.4 [0.25] 4 of 5 5 of 5 3.8 [0.10] 4.2 [0.04] N45A 5.4 [0.06] 6 of 6 6 of 6 3.5 [0.13] 4.0 [0.12] S47A 5.6 [0.15] 5 of 6 6 of 6 2.6 [0.20]*** 4.3 [0.114 S47T 5.6 [0.11] 5 of 6 6 of 6 3.8 [0.09] 4.0 [0.137 T127A 5.4 [0.18] 6 of 6 4 of 4 3.7 [0.12] 3.9 [0.18] pOka 5.7 [0.12] 5 of 5 5 of 5 3.5 [0.18] 3.7 [0.17] S42A 5.8 [0.07] 4 of 5 5 of 5 3.5 [0.24] 3.9 [0.08] T351A 5.9 [0.03] 4 of 5 5 of 5 3.7 [0.14] 4.0 [0.10] T751A 5.8 [0.05] 5 of 5 5 of 5 3.0 [0.26] 4.2 [0.07] BAC-pOka 5.6 [0.03] 4 of 7 5 of 7 2.0 [0.30] 3.1 [0.17] BAC-gH-HR1 5.5 [0.12] 3 of 7 1 of 7 1.1 [0.49] 1.5 [0.76]* * p<0.05, 2-way ANOVA *** p<0.001, 2-way ANOVA
208

Figure 6.6. Analysis of gH maturation from lysates of transfected HEK-293 cells
HEK-293 cells were transfected with expression plasmids containing wild type gH or
mutant gH and gL. Lysates were harvested 48 h post transfection and gH was
immunoprecipitated and blotted with the SG3 antibody. (A) Expression constructs
containing substitutions of cysteine residues. (B) Expression constructs containing
lethal mutations gH-HR2, gH-HR3, gH-αH1, gH-αH2 and gH-FPNG.
209

Figure 6.7. Localization of transiently expressed gH in HEK-293 cells
210

Figure 6.7. Localization of transiently expressed gH in HEK-293 cells
HEK-293 cells were transfected with expression plasmids containing wild type gH or
mutant gH and gL. Cells were permeabilized (P) to examine total gH expression or not
permeabilized (NP) to examine cell-surface gH expression. Cells were stained for gH
(red; SG3), TGN46 (green; AHP500) and nuclei (blue; HOECHST 33342). No
TGN46 staining was detected in non-permeabilized cells, confirming that the gH
detected is only cell surface gH.
211

CHAPTER VII
SUMMARY
212

Herpesviruses infect nearly all animals, and eight have been identified that
infect humans. Varicella-zoster virus (VZV), which causes chicken pox (varicella) and
shingles (herpes zoster), is the only human herpesviruses for which there is a vaccine.
However, VZV remains a problem for immunocompromised patients, including
cancer patients or those undergoing organ transplant. It is therefore important to
continue studying this virus to determine its mechanisms of pathogenicity.
Glycoprotein H (gH), along with glycoprotein B (gB) and L (gL), are the core
glycoproteins that make up the herpesvirus fusion machinery. The fusion mechanisms
that require multiple viral glycoproteins are not yet well understood. Herpes simplex
virus (HSV) gH plays a role in the induction of hemifusion, while gH and gB is
required for complete fusion (217). gL acts as a chaperone for gH, but it has been
suggested that HSV and Epstein-Barr virus (EBV) gL might also play a role in fusion,
although it has not yet been possible to truly separate the two roles of this protein (67,
142, 178, 195). Aside from fusion, gH has also been demonstrated to play a role in
receptor binding, cell signaling, virus capsid egress from the nucleus, and virus
assembly in some herpesviruses.
Prior to the studies detailed here, the VZV gH protein had been studied but
little was known about VZV gH function. VZV gH is a highly glycosylated protein
that is processed in the endoplasmic reticulum into a 94 kDa protein, then transported
to the Golgi via interactions with gL and processed into the mature 118 kDa protein
(166). Many antibodies targeting gH, including mAb 206, were shown to be
neutralizing, which indicated that gH played an important role in VZV pathogenesis
(166, 219). Four gL cysteines were shown to be important for gH/gL interaction and
213

transport of gH to the Golgi. The only functional study of gH consisted of mutational
analysis of the 835YNKI838 endocytosis motif in the cytoplasmic tail (185, 186).
Mutation or deletion of this motif demonstrated that gH endocytosis served to
downregulate virus-induced fusion. Endocytosis was also shown to be important for
gH targeting to the trans-Golgi network (TGN), the location of virus secondary
envelopment. However, all mutational analysis was done using expression vectors and
no gH mutations were incorporated into ORF37 in the context of the VZV genome.
The first study presented here (see Chapter IV) demonstrated that anti-gH mAb
206 was capable of preventing the establishment of virus infection in vivo, and of
reducing virus replication following the establishment of infection in vivo. When mAb
206 bound to gH, it altered gH trafficking. The antibody-gH complex was
endocytosed, but was not targeted to the TGN, although it trafficked through vesicles
containing markers for early endosomes and the multivesicular body pathway. The
antibody might have interfered with gH interactions that targeted the protein to the
TGN, or might have interacted with Fc receptors to target the gH cargo for
degradation. Interestingly, the antibody also bound gH on virion particles and caused
their uptake into cells, possibly also targeting them for degradation.
The results of this study suggest two points of importance. First, a single
monoclonal antibody to gH was an effective prophylaxis against VZV. With the recent
manufacturing stoppage of Varicella-zoster immunoglobulin (VZIG), used as a
prophylaxis to treat at-risk patients, mAb 206 might be useful as a prophylaxis. Future
studies would need to compare the efficacy of this antibody to other treatments.
214

The second point of importance is that gH is important for VZV pathogenesis,
and thus a vaccine that elicited immune responses specific for VZV gH might be used
in immunocompromised patients. Currently, these patients can be at risk for
developing varicella following vaccine administration. Alternatively, a virus lacking
gH might be an effective as a vaccine that would not cause disease in these
immunocompromised patients. HSV studies have indicated that a vaccine virus
lacking gH is not capable of establishing infection, and suggested that if the virus
establishes latency and then reactivates, the newly produced virus would not be
pathogenic (71). A VZV gH- vaccine strain would have to been grown on a gH-
complementing cell line, and would only be capable of a single round of replication
following administration, but this might be enough to elicit protective immunity.
Further VZV studies will be needed to determine if a VZV strain lacking gH would be
a viable vaccine option.
The second study presented here (see Chapter VI) focused on identifying
important structural and functional domains of gH using mutational analysis in order
to begin to determine the exact role of gH in VZV replication and pathogenesis (see
Table 7.1 for a review). First, this study demonstrated that gH was an essential VZV
protein. It also demonstrated that gH contains an important functional and structural
region in the N-terminus of the protein, and this structure is required for efficient VZV
pathogenesis in vivo. This region also contains the gH residues that are recognized by
at least two neutralizing antibodies, including mAb 206, and a commercially available
anti-gH antibody, which also has some neutralizing properties. Additionally, a region
in the C-terminus, proximal to the transmembrane domain, is required for VZV
215

pathogenesis and contributes to the role of gH in virus-induced cell-cell fusion, and
probably to the formation of syncytia that are characteristic of the vesicular lesions
during skin infection. Important residues that form α-helices, possibly acting as heptad
repeats and fusion peptides, were essential to VZV pathogenesis. The presence of
these structural components indicates that gH might act as a class I fusion protein,
although the requirement for gB in fusion suggests that gH is not capable of acting as
an independent fusion protein. Finally, it was demonstrated that several cysteine
residues were important for gH maturation and transport, and the low cell surface
expression levels associated with mutation of other cysteine residues might indicate
that they are important in gH function in fusion.
Understanding how herpesvirus glycoproteins work in concert to induce fusion
is one of the first necessary steps to understanding how to better prevent herpesvirus
infection. Two examples of VZV fusion assays have been published, but both have
problems. One uses a vaccinia virus infection system, and it is possible that the
vaccinia virus might be contributing to the observed fusion (66, 154). The second, a
luciferase-based assay, requires the use of a mutant form of gH lacking the
endocytosis motif, as well as expression of a receptor found on neuronal cells (218).
No receptor specific to cell types typically used in studies of VZV, such as melanoma
cells or fibroblasts, has been identified. The development of a reliable VZV fusion
assay will contribute greatly to determining how gH functions in fusion. Additionally,
understanding the role that gH plays in fusion might provide evidence for the roles
played by gB and gL, as well as other virus glycoproteins, in herpesvirus fusion.
Mutations in gH that block function or that reduce VZV pathogenesis might contribute
216

to the formation of an attenuated vaccine virus that is not capable of inducing disease
in immunocompromised patients or reactivating to produce shingles. gH is an essential
herpesvirus glycoprotein that plays important roles in pathogenesis, and a better
understanding of how this protein functions will be required in order to better
understand herpesvirus pathogenesis and how to prevent and treat infections.
217
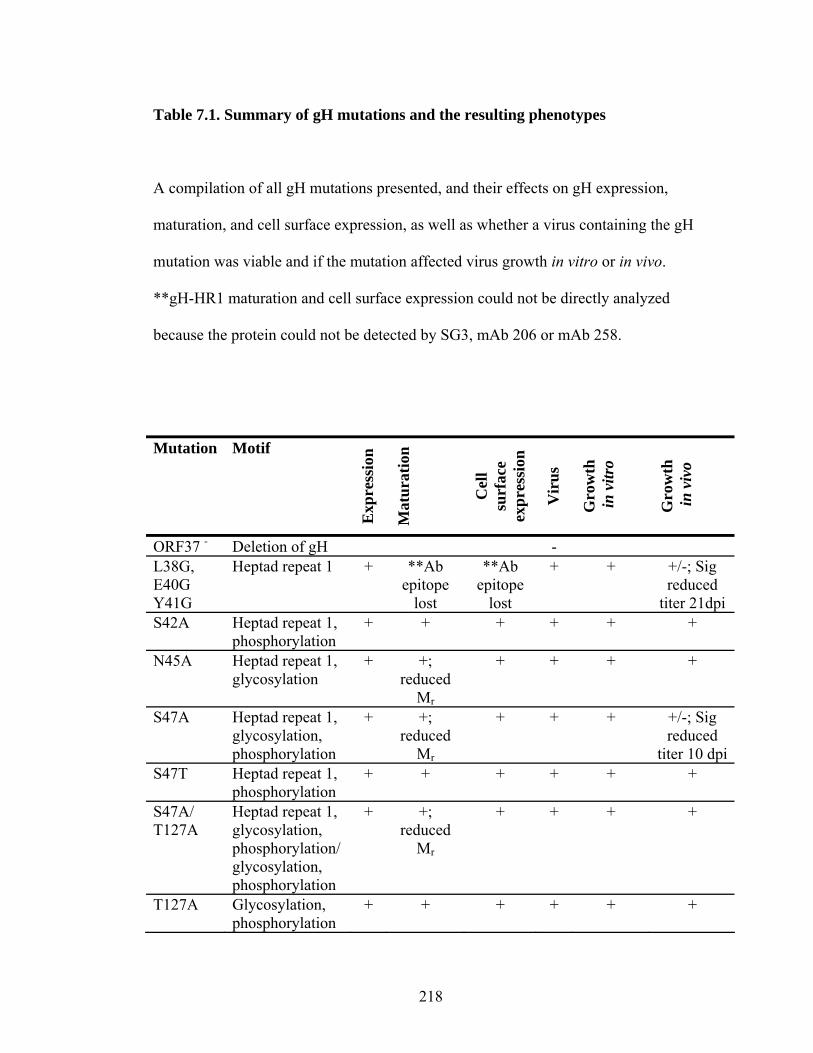
Table 7.1. Summary of gH mutations and the resulting phenotypes
A compilation of all gH mutations presented, and their effects on gH expression,
maturation, and cell surface expression, as well as whether a virus containing the gH
mutation was viable and if the mutation affected virus growth in vitro or in vivo.
**gH-HR1 maturation and cell surface expression could not be directly analyzed
because the protein could not be detected by SG3, mAb 206 or mAb 258.
Mutation Motif
Exp
ress
ion
Mat
urat
ion
Cel
l su
rfac
e ex
pres
sion
Vir
us
Gro
wth
in
vitr
o
Gro
wth
in
viv
o
ORF37 - Deletion of gH - L38G, E40G Y41G
Heptad repeat 1 + **Ab epitope
lost
**Ab epitope
lost
+ + +/-; Sig reduced
titer 21dpi S42A Heptad repeat 1,
phosphorylation + + + + + +
N45A Heptad repeat 1, glycosylation
+ +; reduced
Mr
+ + + +
S47A Heptad repeat 1, glycosylation, phosphorylation
+ +; reduced
Mr
+ + + +/-; Sig reduced
titer 10 dpiS47T Heptad repeat 1,
phosphorylation + + + + + +
S47A/ T127A
Heptad repeat 1, glycosylation, phosphorylation/glycosylation, phosphorylation
+ +; reduced
Mr
+ + + +
T127A Glycosylation, phosphorylation
+ + + + + +
218

Mutation Motif
Exp
ress
ion
Mat
urat
ion
Cel
l su
rfac
e ex
pres
sion
Vir
us
Gro
wth
in
vitr
o
Gro
wth
in
viv
o
A368P, A367P, A372P
α-helix 1 + - - -
Q399A, L400A
Heptad repeat 2 + - Faint -
D456A, E457A, L462A
Heptad repeat 3 + - Faint -
L505D, F506P, F507P, A508D
α-helix 2 + - - -
C327A Cysteine + + + T351A Phosphorylation + + + + + + C540A Cysteine + - - C575A Cysteine + - - C640A Cysteine + + Faint C703A Cysteine + + Faint C724A Cysteine + + Faint C727A Cysteine + + + T751A Phosphorylation + + + + +;
Normal plaque, small
syncytia
+; Delayed growth
(not sig)
F781A, P782A, N783A, G784A
Conserved hydrophobic region
+ - - -
219

CHAPTER VIII
REFERENCES
220

1. Abendroth, A., I. Lin, B. Slobedman, H. Ploegh, and A. M. Arvin. 2001.
Varicella‐zoster virus retains major histocompatibility complex class I protein 888. s in the Golgi compartment of infected cells. J Virol. 75:4878‐4
2. Abendroth, A., B. Slobedman, E. Lee, E. Mellins, M. Wallace, and A. M. Arvin. 2000. Modulation of major histocompatibility class II protein expression by varicella‐zoster virus. J Virol. 74:1900‐1907.
3. Akahori, Y., K. Suzuki, T. Daikoku, M. Iwai, Y. Yoshida, Y. Asano, Y. Kurosawa, and K. Shiraki. 2009. Characterization of neutralizing epitopes of varicella‐zoster virus glycoprotein H. J Virol. 83:2020‐2024.
4. Alconada, A., U. Bauer, and B. Hoflack. 1996. A tyrosine‐based motif and a casein kinase II phosphorylation site regulate the intracellular trafficking of the varicella‐zoster virus glycoprotein I, a protein localized in th ‐e transGolgi network. EMBO J. 15:6096‐6110. Altschul, S. F., W. Gish, W. Miller, E. W. Myers 990. 5. , and D. J. Lipman. 1Basic local alignment search tool. J Mol Biol. 215:403‐410.
6. Anderson, R. A., D. X. Liu, and U. A. Gompels. 1996. Definition of a human herpesvirus‐6 betaherpesvirus‐specific domain in glycoprotein gH that governs interaction with glycoprotein gL: substitution of human cytomega n. lovirus glycoproteins permits group‐specific complex formatioVirology. 217:517‐526. Arvin, A. 20 r 7. 05. Aging, immunity, and the varicella‐zoste virus. N Engl J Med. 352:2266‐2267.
8. Arvin, A. M. 2001. Varicella‐zoster virus, p. 2731‐2768. In D. M. Knipe and P. Howley (ed.), Fields Virology. Lippincott‐Williams & Wilkins, Philadelphia.
9. Arvin, A. M., E. KinneyThomas, K. Shriver, C. Grose, C. M. Koropchak, E. Scranton, A. E. Wittek, and P. S. Diaz. 1986. Immunity to varicella‐zoster viral glycoproteins, gp I (gp 90/58) and gp III (gp 118), and to a nonglyc ‐1351. osylated protein, p 170. J. Immunol. 137:1346
10. Arvin, A. M., C. M. Koropchak, B. R. Williams, F. C. Grumet, and S. K. Foung. 1986. Early immune response in healthy and immunocompromised subjects with primary varicella‐zoster virus infection. J Infect Dis. 154:422‐429.
11. Atanasiu, D., J. C. Whitbeck, M. P. de Leon, H. Lou, B. P. Hannah, G. H. Cohen, and R. J. Eisenberg. 2010. Bimolecular complementation defines functional regions of HSV gB that are involved with gH/gL as necessary steps leading to cell fusion. J Virol.
12. Avitabile, E., C. Forghieri, and G. CampadelliFiume. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 81:11532‐11537.
13. Avitabile, E., C. Forghieri, and G. CampadelliFiume. 2009. Cross talk among the glycoproteins involved in herpes simplex virus entry and
221

fusion: the interaction between gB and gH/gL does not necessarily require gD. J Virol. 83:10752‐10760.
14. Babic, N., B. G. Klupp, B. Makoschey, A. Karger, A. Flamand, and T. C. Mettenleiter. 1996. Glycoprotein gH of pseudorabies virus is essential for penetration and propagation in cell culture and in m of the nervous systemice. J Gen Virol. 77 ( Pt 9):2277‐2285.
15. Babst, M., T. K. Sato, L. M. Banta, and S. D. Emr. 1997. Endosomal transport function in yeast requires a novel AAA‐type ATPase, Vps4p. EMBO J. 16:1820‐1831.
16. Baiker, A., C. Bagowski, H. Ito, M. Sommer, L. Zerboni, K. Fabel, J. Hay, W. Ruyechan, and A. M. Arvin. 2004. The immediate‐early 63 protein of Varicella‐Zoster virus: analysis of functional domains required for replication in vitro and for T‐cell and skin tro pism in the SCIDhu model invivo. J. Virol. 78:1181‐1194.
17. Baldwin, B. R., M. Kleinberg, and S. Keay. 1996. Molecular cloning and expression of receptor peptides that block ell human cytomegalovirus/cfusion. Biochem Biophys Res Commun. 219:668‐673.
18. Baldwin, B. R., C. O. Zhang, and S. Keay. 2000. Cloning and epitope mapping of a functional partial fusion receptor for human cytomegalovirus gH. J. Gen. Virol. 81:27‐35.
19. Bendtsen, J. D., H. Nielsen, G. von Heijne, and S. Brunak. 2004. Improved prediction of signal peptides: Si 3‐gnalP 3.0. J Mol Biol. 340:78795. Berg, J. M., J. L. Tymoczko, and L. Stryer. 2002. Biochemistry. W. H. Freeman an
20. d Company, New York.
21. Besser, J., M. Ikoma, K. Fabel, M. H. Sommer, L. Zerboni, C. Grose, and A. M. Arvin. 2004. Differential requirement for cell fusion and virion formation in the pathogenesis of varicella‐zoster virus infection in skin and T cells. J. Virol. 78:13293‐13305.
22. Blom, N., S. Gammeltoft, and S. Brunak. 1999. Sequence and structure‐based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294:1351‐1362.
23. Blom, N., T. SicheritzPonten, R. Gupta, S. Gammeltoft, and S. Brunak. 2004. Prediction of post‐translational glycosylation and phosphor ylationof proteins from the amino acid sequence. Proteomics. 4:1633‐1649.
24. BoggerGoren, S., J. M. Bernstein, A. A. Gershon, and P. L. Ogra. 1984. Mucosal cell‐mediated immunity to varicella z in oster virus: role protection against disease. J Pediatr. 105:195‐199.
25. Bonifacino, J. S., and L. M. Traub. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 72:395‐447.
26. Bos, C. R., S. L. Shank, and M. D. Snider. 1995. Role of clathrin‐coated vesicles in glycoprotein transport from the cell surface to the Golgi complex. J. Biol. Chem. 270:665‐671.
222

27. sma. 1983. A severe combineimmunodeficiency mutation in the mouse. Nature. 301:527‐530.
28. Browne, H., V. Baxter, and T. Minson. 1993. Analysis of protective immune responses to the glycoprotein H‐glycoprotein L complex of herpes simplex virus type 1. J. Gen.
Bosma, G. C., R. P. Custer, and M. J. Bo d
Virol. 74 ( Pt 12):2813‐2817. 29. Buchbinder, S. P., M. H. Katz, N. A. Hessol, J. Y. Liu, P. M. O'Malley, R.
Underwood, and S. D. Holmberg. 1992. Herpes zoster and human immunodeficiency virus infection. J Infect Dis. 166:1153‐1156. BüchenOsmond, C. (ed.).30. 2006. ICTVdB Management. 00.031.1.01. Simplexvirus. Columbia University, New York, USA. BüchenOsmond, C. (ed.). 01. 31. 2006. ICTVdB Management. 00.031.1.02.0Human herpesvirus 3. Columbia University, New York, USA. BüchenOsmond, C. (ed.). 2. 32. 2006. ICTVdB Management. 00.031.1.0Varicellovirus. Columbia University, New York, USA. BüchenOsmond, C. (ed.).33. 2006. ICTVdB Management. 00.031.1. Alphaherpesvirinae. Columbia University, New York, USA. BüchenOsmond, C. (ed.).34. 2006. ICTVdB Management. 00.031.2.01. Cytomegalovirus. Columbia University, New York, USA. BüchenOsmond, C. (ed.). 3. 35. 2006. ICTVdB Management. 00.031.2.0Roseolovirus. Columbia University, New York, USA. BüchenOsmond, C. (ed.).36. 2006. ICTVdB Management. 00.031.2. Betaherpesvirinae. Columbia University, New York, USA. BüchenOsmond, C. (ed.). . 37. 2006. ICTVdB Management. 00.031.3Gammaherpesvirinae. Columbia University, New York, USA. BüchenOsmond, C. (ed.).38. 2002. ICTVdB Management. 00.031. Herpesviridae. Columbia University, New York, USA. BüchenOsmond, C. (ed.). 2006. Index of Viruse39. s ‐ Herpesviridae. Columbia University, New York, USA.
40. Buckmaster, E. A., U. Gompels, and A. Minson. 1984. Characterisation and physical mapping of an HSV‐1 glycoprotein of approximately 115 X 10(3) molecular weight. Virology. 139:408‐413.
41. Butkinaree, C., K. Park, and G. W. Hart. 2010. O‐linked beta‐N‐acetylglucosamine (O‐GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta. 1800:96‐106.
42. Cairns, T. M., L. S. Friedman, H. Lou, J. C. Whitbeck, M. S. Shaner, G. H. Cohen, and R. J. Eisenberg. 2007. N‐terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J Virol. 81:5102‐5111.
43. Cairns, T. M., D. J. Landsburg, J. C. Whitbeck, R. J. Eisenberg, and G. H. Cohen. 2005. Contribution of cysteine residues to the structure and function of herpes simplex virus gH/gL. Virology. 332:550‐562.
44. Cairns, T. M., R. S. Milne, M. PoncedeLeon, D. K. Tobin, G. H. Cohen, and R. J. Eisenberg. 2003. Structure‐function analysis of herpes simplex
223

virus type 1 gD and gH‐gL: clues from gDgH chimeras. J. Virol. 77:6731‐6742.
45. Calistri, A., P. Sette, C. Salata, E. Cancellotti, C. Forghieri, A. Comin, H. Gottlinger, G. CampadelliFiume, G. Palu, and C. Parolin. 2007. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 81:11468‐11478.
46. Cha, S. C., Y. S. Kim, J. K. Cho, J. Cho, S. Y. Kim, H. Kang, M. H. Cho, and H. H. Lee. 2002. Enhanced protection against HSV lethal challenges in mice by immunization with a combined HSV‐1 glycoprotein B:H:L gene DNAs. Virus Res. 86:21‐31.
47. Chaudhuri, V., M. Sommer, J. Rajamani, L. Zerboni, and A. M. Arvin. 2008. Functions of Varicella‐zoster virus ORF23 capsid protein in viral replication and the pathogenesis of skin infection. J. Virol. 82:10231‐10246.
48. Cheshenko, N., B. Del Rosario, C. Woda, D. Marcellino, L. M. Satlin, and B. C. Herold. 2003. Herpes simplex virus triggers activation of calcium‐signaling pathways. J. Cell Biol. 163:283‐293.
49. Cheshenko, N., W. Liu, L. M. Satlin, and B. C. Herold. 2005. Focal adhesion kinase plays a pivotal role in herpes simplex l virus entry. J BioChem. 280:31116‐31125.
50. Cheshenko, N., W. Liu, L. M. Satlin, and B. C. Herold. 2007. Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol Biol Cell. 18:3119‐3130.
51. Chi, J. H., C. A. Harley, A. Mukhopadhyay, and D. W. Wilson. 2005. The cytoplasmic tail of herpes simplex virus e to nvelope glycoprotein D bindsthe tegument protein VP22 and to capsids. J Gen Virol. 86:253‐261.
52. Cohen, J. I., S. E. Straus, and A. M. Arvin. 2007. Varicella‐zoster virus replication, pathogenesis and management, p. 2773‐2818. In D. M. Knipe and P. Howley (ed.), Fields Virology. Lippincott‐Williams & Wilkins, Philadelphia.
53. Cole, N. L., and C. Grose. 2003. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella‐zoster virus. Rev. Med. Virol. 13:207‐222. Colman, P. M., and M. C. Lawrence. 2003. The struct type I 54. ural biology ofviral membrane fusion. Nat Rev Mol Cell Biol. 4:309‐319.
55. Compton, T., R. R. Nepomuceno, and D. M. Nowlin. 1992. Human cytomegalovirus penetrates host cells by pH‐independent fu t the cell sion asurface. Virology. 191:387‐395.
56. Croen, K. D., J. M. Ostrove, L. J. Dragovic, and S. E. Straus. 1988. Patterns of gene expression and sites of latency in human nerve ganglia are different for varicella‐zoster and herpes simplex viruses. Proc Natl Acad Sci U S A. 85:9773‐9777.
224

57. Danilova, T. A., L. M. Bartova, R. L. Panurina, and I. M. Lyampert. 1981. Studies of Fc receptors of heart valve and joint fibroblasts. Clin. Exp. Immunol. 46:575‐580. Davison, A. J. 1984. Structure of the genome termini of varicella‐zoster virus. J Gen Virol. 65 ( Pt 11):1969‐1977.
58.
59. Davison, A. J., R. Eberle, B. Ehlers, G. S. Hayward, D. J. McGeoch, A. C. Minson, P. E. Pellett, B. Roiz 9. man, M. J. Studdert, and E. Thiry. 200The order Herpesvirales. Arch Virol. 154:171‐177. Davison, A. J., and J. E. Scott. 1986. The complete DNA sequence of 60. varicella‐zoster virus. J. Gen. Virol. 67 ( Pt 9):1759‐1816.
61. Debrus, S., C. SadzotDelvaux, A. F. Nikkels, J. Piette, and B. Rentier. 1995. Varicella‐zoster virus gene 63 encodes an immediate‐early protein that is abundantly expressed during latency. J Virol. 69:3240‐3245.
62. Defechereux, P., S. Debrus, L. Baudoux, B. Rentier, and J. Piette. 1997. Varicella‐zoster virus open reading frame 4 encodes an immediate‐early protein with posttranscriptional regulatory properties. J Virol. 71:7073‐7079.
63. Drew, P. D., M. T. Moss, T. J. Pasieka, C. Grose, W. J. Harris, and A. J. Porter. 2001. Multimeric humanized varicella‐zoster virus antibody fragments to gH neutralize virus while monomeric fragments do not. J. Gen. Virol. 82:1959‐1963.
64. Dubey, L., S. P. Steinberg, P. LaRussa, P. Oh, and A. A. Gershon. 1988. Western blot analysis of antibody to varicella‐zoster virus. J Infect Dis. 157:882‐888.
65. Duus, K. M., and C. Grose. 1996. Multiple regulatory effects of varicella‐zoster virus (VZV) gL on trafficking patterns and fusogenic properties of VZV gH. J. Virol. 70:8961‐8971.
66. Duus, K. M., C. Hatfield, and C. Grose. 1995. Cell surface expression and fusion by the varicella‐zoster vi is rus gH:gL glycoprotein complex: analysby laser scanning confocal microscopy. Virology. 210:429‐440.
67. Fan, Q., E. Lin, and P. G. Spear. 2009. Insertional mutations in herpes simplex virus type 1 gL identify functional domains for association with gH and for membrane fusion. J Virol. 83:11607‐11615.
68. Fariselli, P., M. Finelli, I. Rossi, M. Amico, A. Zauli, P. L. Martelli, and R. Casadio. 2005. TRAMPLE: the transmembrane protein labelling environment. Nucleic Acids Res. 33:W198‐201.
69. Farnsworth, A., T. W. Wisner, and D. C. Johnson. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J Virol. 81:319‐331.
70. Farnsworth, A., T. W. Wisner, M. Webb, R. Roller, G. Cohen, R. Eisenberg, and D. C. Johnson. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. U S A. 104:10187‐10192.
225

71. Farrell, H. E., C. S. McLean, C. Harley, S. Efstathiou, S. Inglis, and A. C. Minson. 1994. Vaccine potential of a herpes simplex virus type 1 mutant with an essential glycoprotein deleted. J. Virol. 68:927‐932.
72. Favoreel, H. W., G. Van Minnebruggen, H. J. Nauwynck, L. W. Enquist, and M. B. Pensaert. 2002. A tyrosine‐based motif in the cytoplasmic tail of pseudorabies virus glycoprotein B is important for both antibody‐induced internalization of viral glycoproteins and efficient cell‐to‐cell spread. J. Virol. 76:6845‐6851. Feldman, S., S. Chaudary 73. , M. Ossi, and E. Epp. 1977. A viremic phase forherpes zoster in children with cancer. J Pediatr. 91:597‐600. Feldman, S., and L. Lott. 1987. Varicella in children with cancer: impact of antiviral therapy and prophyl
74. axis. Pediatrics. 80:465‐472.
75. Ficinska, J., G. Van Minnebruggen, H. J. Nauwynck, K. BienkowskaSzewczyk, and H. W. Favoreel. 2005. Pseudorabies virus glycoprotein gD contains a functional endocytosis motif that acts in concert with an endocytosis motif in gB to drive internalization of antibody‐antige
79:7248‐7254. n
complexes from the surface of infected monocytes. J. Virol. Fleisher, G., W. Henry, M. McSorley, A. Arbeter, and S. Plotkin. 1981
‐899. 76. .
Life‐threatening complications of varicella. Am J Dis Child. 135:89677. Forghani, B., R. Mahalingam, A. Vafai, J. W. Hurst, and K. W. Dupuis.
1990. Monoclonal antibody to immediate early protein encoded by varicella‐zoster virus gene 62. Virus Res. 16:195‐210. Forghani, B., L. Ni, and C. Grose. 1994. Neutralization epitope of the varicella
78. ‐zoster virus gH:gL glycoprotein complex. Virology. 199:458‐462.
79. Forrester, A., H. Farrell, G. Wilkinson, J. Kaye, N. DavisPoynter, and T. Minson. 1992. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341‐348.
80. Forrester, A. J., V. Sullivan, A. Simmons, B. A. Blacklaws, G. L. Smith, A. A. Nash, and A. C. Minson. 1991. Induction of protective immunity with antibody to herpes simplex virus type 1 glycoprotein H (gH) and analysis of the immune response to gH expressed in recombinant vaccinia virus. J. Gen. Virol. 72 ( Pt 2):369‐375.
81. Frey, J., M. Janes, W. Engelhardt, E. G. Afting, C. Geerds, and B. Moller. 1986. Fc gamma‐receptor‐mediated changes in the plasma membrane potential induce prostaglandin release from human fibroblasts. Eur. J. Biochem. 158:85‐89.
82. Fuller, A. O., R. E. Santos, and P. G. Spear. 1989. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 63:3435‐3443.
83. Galdiero, M., A. Whiteley, B. Bruun, S. Bell, T. Minson, and H. Browne. 1997. Site‐directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J. Virol. 71:2163‐2170.
226

84. Galdiero, S., A. Falanga, M. Vitiello, H. Browne, C. Pedone, and M. Galdiero. 2005. Fusogenic domains in herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 280:28632‐28643.
85. Galdiero, S., A. Falanga, M. Vitiello, M. D'Isanto, M. Cantisani, A. Kampanaraki, E. Benedetti, H. Browne, and M. Galdiero. 2008. Peptides containing membrane‐interacting motifs inhibit herpes simplex virus type 1 infectivity. Peptides. 29:1461‐1471.
86. Galdiero, S., A. Falanga, M. Vitiello, M. D'Isanto, C. Collins, V. Orrei, H. Browne, C. Pedone, and M. Galdiero. 2007. Evidence for a role of the membrane‐proximal region of herpes simplex virus Type 1 glycoprotein H in membrane fusion and virus inhibition. Chembiochem. 8:885‐895.
87. Galdiero, S., A. Falanga, M. Vitiello, L. Raiola, R. Fattorusso, H. Browne, C. Pedone, C. Isernia, and M. Galdiero. 2008. Analysis of a membrane interacting region of herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 283:29993‐30009.
88. Galdiero, S., M. Vitiello, M. D'Isanto, E. Di Niola, L. Peluso, K. Raieta, C. Pedone, M. Galdiero, and E. Benedetti. 2004. Induction of signaling pathways by herpes simplex virus type 1 through glycoprotein H peptides. Biopolymers. 76:494‐502.
89. Galdiero, S., M. Vitiello, M. D'Isanto, A. Falanga, C. Collins, K. Raieta, C. Pedone, H. Browne, and M. Galdiero. 2006. Analysis of synthetic peptides from heptad‐repeat domains of herpes simplex virus type 1 glycoproteins H and B. J. Gen. Virol. 87:1085‐1097.
90. Ganem, D. 2007. Kaposi's sarcoma‐associated herpesvirus. In D. M. Knipe and P. Howley (ed.), Fields Virology. Lippincott‐Williams & Wilkins, Philadelphia.
91. Gershon, A. A., D. L. Sherman, Z. Zhu, C. A. Gabel, R. T. Ambron, and M. D. Gershon. 1994. Intracellular transport of newly synthesized varicella‐zoster virus: final envelopment in the trans‐Golgi network. J. Virol. 68:6372‐6390.
92. Gianni, T., M. Amasio, and G. CampadelliFiume. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C‐terminal profusion domain. J Biol Chem. 284:17370‐17382.
93. Gianni, T., R. Fato, C. Bergamini, G. Lenaz, and G. CampadelliFiume. 2006. Hydrophobic {alpha}‐Helices 1 and 2 of Herpes Simplex Virus gH Interact with Lipids, and Their Mimetic Peptides Enhance Virus In fectionand Fusion. J. Virol. 80:8190‐8198.
94. Gianni, T., P. L. Martelli, R. Casadio, and G. CampadelliFiume. 2005. The ectodomain of herpes simplex virus glycoprotein H contains a membrane alpha‐helix with attributes of an interna
l fusion peptide,
positionally conserved in the herpesviridae family. J. Virol. 79:2931‐2940.95. Gianni, T., L. Menotti, and G. CampadelliFiume. 2005. A heptad repeat
in herpes simplex virus 1 gH, located downstream of the alpha‐helix with
227

attributes of a fusion peptide, is critical for virus entry and fusion. J. Virol. 79:7042‐7049.
96. Gianni, T., A. Piccoli, C. Bertucci, and G. CampadelliFiume. 2006. Heptad repeat 2 in herpes simplex virus 1 gH in teracts with heptad repeat1 and is critical for virus entry and fusion. J. Virol. 80:2216‐2224.
97. Glesby, M. J., R. D. Moore, and R. E. Chaisson. 1995. Clinical spectrum of herpes zoster in adults infecte . Clin d with human immunodeficiency virusInfect Dis. 21:370‐375. Gompels, U., and A. Minson. 1986. The properties98. and sequence of glycoprotein H of herpes simplex virus type 1. Virology. 153:230‐247.
99. Gompels, U. A., M. A. Craxton, and R. W. Honess. 1988. Conservation of glycoprotein H (gH) in herpesviruses: nucleotide sequence of the gH gene from herpesvirus saimiri. J Gen Virol. 69 ( Pt 11):2819‐2829.
100. Gould, C. M., F. Diella, A. Via, P. Puntervoll, C. Gemund, S. ChabanisDavidson, S. Michael, A. Sayadi, J. C. Bryne, C. Chica, M. Seiler, N. E. Davey, N. Haslam, R. J. Weatheritt, A. Budd, T. Hughes, J. Pas, L. Rychlewski, G. Trave, R. Aasland, M. HelmerCitterich, R. Linding, and T. J. Gibson. 2010. ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 38:D167‐180.
101. Gray, W. L., B. Starnes, M. W. White, and R. Mahalingam. 2001. The DNA sequ 3‐ence of the simian varicella virus genome. Virology. 284:12130. Grose, C. 1981. Variation on a theme by Fenner: the pathogenesis of chickenpo
102. x. Pediatrics. 68:735‐737.
103. Grose, C., D. P. Edwards, K. A. Weigle, W. E. Friedrichs, and W. L. McGuire. 1984. Varicella‐zoster virus‐specific gp140: a highly immunogenic and disulfide‐linked structural glycoprotein. Virology. 132:138‐146.
104. Gross, S. T., C. A. Harley, and D. W. Wilson. 2003. The cytoplasmic tail of Herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology. 317:1‐12.
105. Guess, H. A., D. D. Broughton, L. J. Melton, 3rd, and L. T. Kurland. 1986. Population‐based studies ‐of varicella complications. Pediatrics. 78:723727.
106. Gupta, R., and S. Brunak. 2002. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput.310‐322.
107. Haddad, R. S., and L. M. HuttFletcher. 1989. Depletion of glycoprotein gp85 from virosomes made with Epstein‐Barr virus proteins abolishes their ability to fuse with virus receptor‐bearing cells. J. Virol. 63:4998‐5005. Hambleton, S., and A. A. Gershon. 2005. Preventing varic108. ella‐zoster disease. Clin Microbiol Rev. 18:70‐80.
109. Hardy, I., A. A. Gershon, S. P. Steinberg, and P. LaRussa. 1991. The incidence of zoster after immunization with live attenuated varicella
228

vaccine. A study in children with leukemi ative a. Varicella Vaccine CollaborStudy Group. N Engl J Med. 325:1545‐1550.
110. Harman, A., H. Browne, and T. Minson. 2002. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein Hplay a role in membrane fusion. J. Virol. 76:10708‐10716.
111. Harpaz, R., I. R. OrtegaSanchez, and J. F. Seward. 2008. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 57:1‐30; quiz CE32‐34.
112. Hata, A., L. Zerboni, M. Sommer, A. A. Kaspar, C. Clayberger, A. M. Krensky, and A. M. Arvin. 2001. Granulysin blocks replication of varicella‐zoster virus and triggers apoptosis of infected cells. Viral Immunol. 14:125‐133. Heineman, T., M. Gong, J. Sample, and E. Kieff. 1988. Identification of 113. the Epstein‐Barr virus gp85 gene. J. Virol. 62:1101‐1107.
114. Hobom, U., W. Brune, M. Messerle, G. Hahn, and U. H. Koszinowski. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes:
galovirus envelope glymutational analysis of human
cytome coprotein genes. J Virol. 74:7720‐7729. Hofmann, K., and W. e‐115. Stoffel. 1993. Presented at the Biol. Chem. HoppSeyler. HopeSimpson, R. E. 1965. The nature of herpes zost116. er: A long‐term study and a new hypothesis. Proc R Soc Med. 58:9‐20.
117. Huang, H. D., T. Y. Lee, S. W. Tzeng, and J. T. Horng. 2005. KinasePhos: a web tool for identifying protein kinase‐specific phosphorylation sites. Nucleic Acids Res. 33:W226‐229.
118. Huber, M. T., and T. Compton. 1998. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H‐glycoprotein L‐containing envelope complex. J. Virol. 72:8191‐8197.
119. Huet, G., V. Gouyer, D. Delacour, C. Richet, J. P. Zanetta, P. Delannoy, and P. Degand. 2003. Involvement of glycosylation in the intracellular trafficking of glycoproteins in polarized epithelial cells. Biochimie. 85:323‐330.
120. Hutchinson, L., H. Browne, V. Wargent, N. DavisPoynter, S. Primorac, K. Goldsmith, A. C. Minson, and D. C. Johnson. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expre 2240‐ssion of gH. J. Virol. 66:2250.
121. Inchauspe, G., S. Nagpal, and J. M. Ostrove. 1989. Mapping of two varicella‐zoster virus‐encoded genes that activate the expression of viral early and late genes. Virology. 173:700‐709.
122. Ito, H., M. H. Sommer, L. Zerboni, A. Baiker, B. Sato, R. Liang, J. Hay, W. Ruyechan, and A. M. Arvin. 2005. Role of the varicella‐zoster virus gene product encoded by open reading frame 35 in viral replication in vitro and in differentiated human skin and T cells in vivo. J. Virol. 79:4819‐4827.
229

123. Jacquet, A., M. Haumont, D. Chellun, M. Massaer, F. Tufaro, A. Bollen, and P. Jacobs. 1998. The varicella zoster virus glycoprotein B (gB) plays a role in virus binding to cell surfac rus Res. 7.
e heparan sulfate proteoglycans. Vi53:197‐20
Jardetzky, T. S., and R. A. Lamb. 2004. Virology: a class act. Nature. 124. 427:307‐308.
125. Jenkins, D. E., R. L. Redman, E. M. Lam, C. Liu, I. Lin, and A. M. Arvin. 1998. Interleukin (IL)‐10, IL‐12, and interferon‐gamma production in primary and memory immun e responses to varicella‐zoster virus. J InfectDis. 178:940‐948.
126. Jones, J. O., and A. M. Arvin. 2006. Inhibition of the NF‐kappaB pathway by varicella‐zoster virus in vitro and in human epidermal cells in vivo. J Virol. 80:5113‐5124.
127. Jones, N. A., and R. J. Geraghty. 2004. Fusion activity of lipid‐anchored envelope glycoproteins of herpes simplex virus type 3‐1. Virology. 324:21228.
128. Julenius, K., A. Molgaard, R. Gupta, and S. Brunak. 2005. Prediction, conservation analysis, and structural characterization of m lian ammamucin‐type O‐glycosylation sites. Glycobiology. 15:153‐164.
129. Kamen, D. E., S. T. Gross, M. E. Girvin, and D. W. Wilson. 2005. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 79:6134‐6141.
130. Kaye, J. F., U. A. Gompels, and A. C. Minson. 1992. Glycoprotein H of human cytomegalovirus (HC MV) forms a stable complex with the HCMVUL115 gene product. J Gen Virol. 73 ( Pt 10):2693‐2698.
131. Keay, S., and B. R. Baldwin. 1996. Evidence for the role of cell protein phosphorylation in human cytomegalovirus/host cell fusion. J Gen Virol. 77 ( Pt 10):2597‐2604.
132. Keay, S., B. R. Baldwin, M. W. Smith, S. S. Wasserman, and W. F. Goldman. 1995. Increases in [Ca tative 2+]i mediated by the 92.5‐kDa pucell membrane receptor for HCMV gp86. Am J Physiol. 269:C11‐21.
133. Kieff, E. D., and A. B. Rickinson. 2007. Epstein‐Barr virus and its replication, p. 2603‐2654. In D. M. Knipe and P. Howley (ed.), Fields Virology. Lippincott‐Williams & Wilkins, Philadelphia.
134. Kirschner, A. N., A. S. Lowrey, R. Longnecker, and T. S. Jardetzky. 2007. Binding‐site interactions between Epstein‐Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B‐cell membrane fusion. J Virol. 81:9216‐9229.
135. Kirschner, A. N., J. Omerovic, B. Popov, R. Longnecker, and T. S. Jardetzky. 2006. Soluble Epstein‐Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B‐cell fusion but not in epithelial cell fusion. J. Virol. 80:9444‐9454.
230

136. Klupp, B., J. Altenschmidt, H. Granzow, W. Fuchs, and T. C. Mettenleiter. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol. 82:6299‐6309.
137. Klupp, B. G., J. Baumeister, A. Karger, N. Visser, and T. C. Mettenleiter. 1994. Identification and characterization of a novel structural glycoprotein in pseudorabies virus, gL. J. Virol. 68:3868‐3878.
138. Klupp, B. G., W. Fuchs, E. Weiland, and T. C. Mettenleiter. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J. Virol. 71:7687‐7695. Klupp, B. G., and T. C. Mettenleiter. 1991. Sequ139. ence and expression of the glycoprotein gH gene of pseudorabies virus. Virology. 182:732‐741. Klupp, B. G., R. Nixdorf, and T. C. Mettenleite s 140. r. 2000. Pseudorabies viruglycoprotein M inhibits membrane fusion. J. Virol. 74:6760‐6768.
141. Klupp, B. G., N. Visser, and T. C. Mettenleiter. 1992. Identification and characterization of pseudorabies virus glycoprotein H. J. Virol. 66:3048‐3055.
142. Klyachkin, Y. M., and R. J. Geraghty. 2008. Mutagenic analysis of herpes simplex virus type 1 glycoprotein L reveals the importance of an arginine‐rich region for function. Virology. 374:23‐32.
143. Klyachkin, Y. M., K. D. Stoops, and R. J. Geraghty. 2006. Herpes simplex virus type 1 glycoprotein L mutants that fail to promote trafficking of glycoprotein H and fail to function in fusion can induce binding of glycoprotein L‐dependent anti‐glycoprotein H antibodies. J. Gen. Virol. 87:759‐767. Krause, P. R., and D. M. Klinman. 1995. Efficacy, immunogenic ety, 144. ity, safand use of live attenuated chickenpox vaccine. J Pediatr. 127:518‐525.
145. Ku, C. C., J. A. Padilla, C. Grose, E. C. Butcher, and A. M. Arvin. 2002. Tropism of varicella‐zoster virus for human tonsillar CD4(+) T lymphocytes that express activation, memory, and skin homing markers. J Virol. 76:11425‐11433.
146. Ku, C. C., L. Zerboni, H. Ito, B. S. Graham, M. Wallace, and A. M. Arvin. 2004. Varicella‐zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon‐alpha. J Exp Med. 200:917‐925.
147. Ku, C. C., L. Zerboni, H. Ito, B. S. Graham, M. Wallace, and A. M. Arvin. 2004. Varicella‐zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon‐alpha. J. Exp. Med. 200:917‐925.
148. Kutinova, L., P. Hainz, V. Ludvikova, L. Maresova, and S. Nemeckova. 2001. Immune response to vaccinia virus recombinants expressing glycoproteins gE, gB, gH, and gL of Varicella‐zoster virus. Virology. 280:211‐220.
231

149. Li, Q., M. A. Ali, and J. I. Cohen. 2006. Insulin degrading enzyme is a cellular receptor mediating varicella‐zoster virus infection and cell‐to‐cellspread. Cell. 127:305‐316.
150. Li, Q., S. M. Turk, and L. M. HuttFletcher. 1995. The Epstein‐Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J. Virol. 69:3987‐3994.
151. Lopper, M., and T. Compton. 2004. Coiled‐coil domains in glycoproteins B and H are involved in human cytome l. galovirus membrane fusion. J. Viro78:8333‐8341. Lupas, A., M. Van Dyke, and J. Stock. 1991. Predicting coiled coils from protein sequences. Science.
152. 252:1162‐1164.
153. Maresova, L., L. Kutinova, V. Ludvikova, R. Zak, M. Mares, and S. Nemeckova. 2000. Characterization of interaction of gH and gL glycoproteins of varicella‐zoster virus: their processing and trafficking. J. Gen. Virol. 81:1545‐1552.
154. Maresova, L., T. J. Pasieka, and C. Grose. 2001. Varicella‐zoster Virus gB and gE coexpression, but not gB or gE alone, leads to abundant fusion and syncytium formation equivalent to those from gH and gL coexpre . ssion. JVirol. 75:9483‐9492.
155. Maresova, L., T. J. Pasieka, E. Homan, E. Gerday, and C. Grose. 2005. Incorporation of three endocytosed varicella‐zoster virus glyco s, proteingE, gH, and gB, into the virion envelope. J. Virol. 79:997‐1007.
156. Marin, M., D. Guris, S. S. Chaves, S. Schmid, and J. F. Seward. 2007. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm. Rep. 56:1‐40. McGeoch, D. J. 1989. The gen : contents, 157. omes of the human herpesvirusesrelationships, and evolution. Annu Rev Microbiol. 43:235‐265.
158. McGeoch, D. J., and S. Cook. 1994. Molecular phylogeny of the alphaherpesvirinae subfamily and a proposed evolutionary timescale. J. Mol. Biol. 238:9‐22.
159. McGeoch, D. J., S. Cook, A. Dolan, F. E. Jamieson, and E. A. Telford. 1995. Molecular phylogeny and ev f olutionary timescale for the family omammalian herpesviruses. J. Mol. Biol. 247:443‐458.
160. McGeoch, D. J., and A. J. Davison. 1986. DNA sequence of the herpes simplex virus type 1 gene encoding glycoprotein gH, and identification of homologues in the genomes of varicella‐zoster virus and Epstein‐Barr virus. Nucleic Acids Res. 14:4281‐4292.
161. Miller, E., J. E. CradockWatson, and M. K. Ridehalgh. 1989. Outcome in newborn babies given anti‐varicella‐zoster immunoglobulin after perinatal maternal infection with va ‐ricella‐zoster virus. Lancet. 2:371373.
162. Miller, N., and L. M. HuttFletcher. 1988. A monoclonal antibody to glycoprotein gp85 inhibits fusion but not attachment of Epstein‐Barr virus. J. Virol. 62:2366‐2372.
232

163. Mocarski, E. S., T. Shenk, and R. F. Pass. 2007. Cytomegaloviruses. In D. M. Knipe and P. Howley (ed.), Fields Virology. Lippincott‐Williams & Wilkins, Philadelphia.
164. Moffat, J. F., M. D. Stein, H. Kaneshima, and A. M. Arvin. 1995. Tropism of varicella‐zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID‐hu mice. J. Virol. 69:5236‐5242.
165. Moffat, J. F., L. Zerboni, P. R. Kinchington, C. Grose, H. Kaneshima, and A. M. Arvin. 1998. Attenuation of the vaccine Oka strain of varicella‐zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID‐hu mouse. J. Virol. 72:965‐974.
166. Montalvo, E. A., and C. Grose. 1986. Neutralization epitope of varicella zoster virus on native viral gly coprotein gp118 (VZV glycoprotein gpIII).Virology. 149:230‐241.
167. Montalvo, E. A., and C. Grose. 1986. Varicella zoster virus glycoprotein gpI is selectively phosphorylated by a virus‐induced protein kinase. Proc. Natl. Acad. Sci. U S A. 83:8967‐8971.
168. Mori, Y., P. Akkapaiboon, X. Yang, and K. Yamanishi. 2003. The human herpesvirus 6 U100 gene product is the third component of the gH‐gL glycoprotein complex on the viral envelope. J. Virol. 77:2452‐2458.
169. Mori, Y., P. Akkapaiboon, S. Yonemoto, M. Koike, M. Takemoto, T. Sadaoka, Y. Sasamoto, S. Konishi, Y. Uchiyama, and K. Yamanishi. 2004. Discovery of a second form of tripartite complex containing f gH‐gL ohuman herpesvirus 6 and observations on CD46. J. Virol. 78:4609‐4616.
170. Mori, Y., X. Yang, P. Akkapaiboon, T. Okuno, and K. Yamanishi. 2003. Human herpesvirus 6 variant A glycoprotein H‐glycoprotein L‐glycoprotein Q complex associates with human CD46. J. Virol. 77:4992‐4999.
171. Moriuchi, H., M. Moriuchi, S. E. Straus, and J. I. Cohen. 1993. Varicella‐zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. J Virol. 67:4290‐4295.
172. Nakatsu, F., and H. Ohno. 2003. Adaptor protein complexes as the key regulators of protein sorting in the post‐Golgi network. Cell Struct Funct. 28:419‐429.
173. Nemeckova, S., V. Ludvikova, L. Maresova, J. Krystofova, P. Hainz, and L. Kutinova. 1996. Induction of varicella‐zoster virus‐neutralizing antibodies in mice by co‐infection with recombinant vaccinia viruses expressing the gH or gL gene. J. Gen. Virol. 77 ( Pt 2 ):211‐215.
174. Nicola, A. V., M. Ponce de Leon, R. Xu, W. Hou, J. C. Whitbeck, C. Krummenacher, R. I. Montgomery, P. G. Spear, R. J. Eisenberg, and G. H. Cohen. 1998. Monoclonal antibodies to distinct sites on herpes simplex virus (HSV) glycoprotein D block HSV binding to HVEM. J. Virol. 72:3595‐3601.
175. Niizuma, T., L. Zerboni, M. H. Sommer, H. Ito, S. Hinchliffe, and A. M. Arvin. 2003. Construction of varicella‐zoster virus recombinants from
233

parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID‐hu mouse model. J. Virol. 77:6062‐6065.
176. Nikkels, A. F., S. Debrus, C. SadzotDelvaux, J. Piette, B. Rentier, and G. E. Pierard. 1995. Localization of varicella‐zoster virus nucleic acids and proteins in human skin. Neurology. 45:S47‐49.
177. Nonoyama, S., F. O. Smith, I. D. Bernstein, and H. D. Ochs. 1993. Strain‐dependent leakiness of mice with severe combined immune deficiency. J. Immunol. 150:3817‐3824.
178. Novotny, M. J., M. L. Parish, and P. G. Spear. 1996. Variability of herpes simplex virus 1 gL and anti‐gL antibodies that inhibit cell fusion but not viral infectivity. Virology. 221:1‐13.
179. O'Regan, K. J., M. A. Bucks, M. A. Murphy, J. W. Wills, and R. J. Courtney. 2007. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE). Virology. 358:192‐200.
180. Ober, B. T., B. Teufel, K. H. Wiesmuller, G. Jung, E. Pfaff, A. Saalmuller, and H. J. Rziha. 2000. The porcine humoral immune response against pseudorabies virus specifically targets attachment sites on glycoprotein gC. J. Virol. 74:1752‐1760.
181. Oliver, S. L., M. Sommer, L. Zerboni, J. Rajamani, C. Grose, and A. M. Arvin. 2009. Mutagenesis of varicella‐zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J Virol.83:7495‐7506.
182. Olson, J. K., and C. Grose. 1997. Endocytosis and recycling of varicella‐zoster virus Fc receptor glycoprotein gE: internalization mediated by a YXXL motif in the cytoplasmic tail. J Virol. 71:4042‐4054.
183. Pagni, M., V. Ioannidis, L. Cerutti, M. ZahnZabal, C. V. Jongeneel, J. Hau, O. Martin, D. Kuznetsov, and L. Falquet. 2007. MyHits: improvements to an interactive resource for analyzing protein sequences. Nucleic Acids Res. 35:W433‐437.
184. Parry, C., S. Bell, T. Minson, and H. Browne. 2005. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J. Gen. Virol. 86:7‐10.
185. Pasieka, T. J., L. Maresova, and C. Grose. 2003. A functional YNKI motif in the short cytoplasmic tail of varicella‐zoster virus glycoprotein gH mediates clathrin‐dependent and antibody‐independent endocytosis. J. Virol. 77:4191‐4204.
186. Pasieka, T. J., L. Maresova, K. Shiraki, and C. Grose. 2004. Regulation of varicella‐zoster virus‐induced cell‐to‐cell fusion by the endocytosis‐competent glycoproteins gH and gE. J. Virol. 78:2884‐2896.
187. Pasieka, T. J., R. F. Woolson, and C. Grose. 2003. Viral induced fusion and syncytium formation: measurement by the Kolmogorov‐Smirnov statistical test. J Virol Methods. 111:157‐161.
234

188. Patrone, M., M. Secchi, E. Bonaparte, G. Milanesi, and A. Gallina. 2007. Cytomegalovirus UL131‐128 products promote gB conformational transition and gB‐gH interaction . during entry into endothelial cells. JVirol. 81:11479‐11488.
189. Pawliczek, T., and C. M. Crump. 2009. Herpes simplex virus type 1 production requires a function of al ESCRT‐III complex but is independent TSG101 and ALIX expression. J Virol. 83:11254‐11264.
190. Pellett, P. E., and B. Roizman. 2001. The Family Herpesviridae: A Brief Introduction, p. 2479‐2499, Fields Virology. Lippincott‐Williams &Wilkins, Philadelphia.
191. Peng, T., M. PoncedeLeon, H. Jiang, G. Dubin, J. M. Lubinski, R. J. Eisenberg, and G. H. Cohen. 1998. The gH‐gL complex of herpes simplex virus (HSV) stimulates neutralizing antibody and protects mice against HSV type 1 challenge. J. Virol. 72:65‐72.
192. Perera, L. P., J. D. Mosca, W. T. Ruyechan, and J. Hay. 1992. Regulation of varicella‐zoster virus gene expression in human T lymphocytes. J Virol. 66:5298‐5304.
193. PerezRomero, P., A. Perez, A. Capul, R. Montgomery, and A. O. Fuller. 2005. Herpes simplex virus entry mediator associates in infected cells in acomplex with viral proteins gD and at least gH. J. Virol. 79:4540‐4544.
194. Pertel, P. E., A. Fridberg, M. L. Parish, and P. G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH‐ ires gL requa gD receptor but not necessarily heparan sulfate. Virology. 279:313‐324.
195. Plate, A. E., J. Smajlovic, T. S. Jardetzky, and R. Longnecker. 2009. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein‐Barr virus‐mediated cell fusion i n ndicates a direct role of gL igB‐induced membrane fusion. J Virol. 83:7678‐7689.
196. Prober, C. G., L. E. Kirk, and R. E. Keeney. 1982. Acyclovir therapy of chickenpox in immunosuppressed children‐‐a collaborative study. J Pediatr. 101:622‐625.
197. Qie, L., D. Marcellino, and B. C. Herold. 1999. Herpes simplex virus entry is associated with tyrosine phospho gy. rylation of cellular proteins. Virolo256:220‐227.
198. Qualtiere, L. F., and G. R. Pearson. 1979. Epstein‐Barr virus‐induced membrane antigens: immunochemical characterization of Triton X‐100 solubilized viral membrane antigens fro . Int m EBV‐superinfected Raji cellsJ Cancer. 23:808‐817.
199. Reichelt, M., J. Brady, and A. M. Arvin. 2009. The replication cycle of varicella‐zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion ass83
embly at the single‐cell level. J. Virol. :3904‐3918.
200. Rickinson, A. B., and E. D. Kieff. 2007. Epstein‐Barr virus, p. 2655‐2700. In D. M. Knipe and P. Howley (ed.), Fields Virology. Lippincott‐Williams & Wilkins, Philadelphia.
235

201. r coated vesicles. Trends CBiol. 14:167‐174.
202. Rodriguez, J. E., T. Moninger, and C. Grose. 1993. Entry and egress of varicella virus blocked by same anti‐gH mono gy.
Robinson, M. S. 2004. Adaptable adaptors fo ell
clonal antibody. Virolo196:840‐844.
203. Roizman, B., D. M. Knipe, and R. J. Whitley. 2007. Herpes simplex viruses, p. 2501‐2601. In D. M. Knipe and P. Howley (ed.), Fields Virology. Lippincott‐Williams & Wilkins, Philadelphia.
204. Rusten, T. E., T. Vaccari, K. Lindmo, L. M. Rodahl, I. P. Nezis, C. SemJacobsen, F. Wendler, J. P. Vincent, A. Brech, D. Bilder, and H. Stenmark. 2007. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol. 17:1817‐1825.
205. Ryckman, B. J., M. C. Chase, and D. C. Johnson. 2008. HCMV gH/gL/UL128‐131 interferes with virus entry into epithelial cells: evidence for cell type‐specific receptors. Proc. N S A. atl. Acad. Sci. U105:14118‐14123.
206. Ryckman, B. J., M. C. Chase, and D. C. Johnson. 2010. Human cytomegalovirus TR strain glycoprotein O acts as a chaperone promoting gH/gL incorporation into virions but is not present in virions. J Virol. 84:2597‐2609.
207. Ryckman, B. J., M. A. Jarvis, D. D. Drummond, J. A. Nelson, and D. C. Johnson. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low‐pH fusion. J Virol. 80:710‐722.
208. Sadaoka, T., K. Yamanishi, and Y. Mori. 2006. Human herpesvirus 7 U47 gene products are glycoproteins expressed in virio e with ns and associatglycoprotein H. J. Gen. Virol. 87:501‐508.
209. Sanavio, B., A. Piccoli, T. Gianni, and C. Bertucci. 2007. Helicity propensity and interaction of synthetic peptides from heptad‐repeat domains of herpes simplex virus 1 glycoprotein H: a circular dichroism study. Biochim. Biophys. Acta. 1774:781‐791.
210. Sanger, C., E. Muhlberger, B. Lotfering, H. D. Klenk, and S. Becker. 2002. The Marburg virus surface protein GP is phosphorylated at its ectodomain. Virology. 295:20‐29.
211. Scanlan, P. M., V. Tiwari, S. Bommireddy, and D. Shukla. 2003. Cellular expression of gH confers resistance to herpes simplex vir y. us type‐1 entrVirology. 312:14‐24.
212. Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment ‐‐> deenvelopment ‐‐> reenvelopment pathway. J Virol. 75:5697‐5702.
213. Sommer, M. H., E. Zagha, O. K. Serrano, C. C. Ku, L. Zerboni, A. Baiker, R. Santos, M. Spengler, J. Lynch, C. Grose, W. Ruyechan, J. Hay, and A. M. Arvin. 2001. Mutational analysis of the repeated open reading frames, ORFs 63 and 70 and ORFs 64 and 69, of varicella‐zoster virus. J. Virol. 75:8224‐8239.
236

214. Spaete, R. R., K. Perot, P. I. Scott, J. A. Nelson, M. F. Stinski, and C. Pachl. 1993. Coexpression of truncated human cytomegalovirus gH with the UL115 gene product or the truncated human fibroblast growth factor receptor results in transport of gH to the cell surface. Virology. 193:853‐861.
215. Spriggs, M. K., R. J. Armitage, M. R. Comeau, L. Strockbine, T. Farrah, B. Macduff, D. Ulrich, M. R. Alderson, J. Mullberg, and J. I. Cohen. 1996. The extracellular domain of the Epstein‐Barr virus BZLF2 protein binds the HLA‐DR beta chain and inh 7‐ibits antigen presentation. J Virol. 70:5555563.
216. Stow, N. D., and A. J. Davison. 1986. Identification of a varicella‐zoster virus origin of DNA replication and its activation by herpes simplex virus type 1 gene products. J Gen Virol. 67 ( Pt 8):1613‐1623.
217. Subramanian, R. P., and R. J. Geraghty. 2007. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. U S A. 104:2903‐2908.
218. Suenaga, T., T. Satoh, P. Somboonthum, Y. Kawaguchi, Y. Mori, and H. Arase. 2010. Myelin‐associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A. 107:866‐871.
219. Sugano, T., T. Tomiyama, Y. Matsumoto, S. Sasaki, T. Kimura, B. Forghani, and Y. Masuho. 1991. A human monoclonal antibody against varicella‐zoster virus glycoprotein III. J Gen Virol. 72 ( Pt 9):2065‐2073. Takahashi, M. 1986. Clinical overview of varicella vaccine: development and early studies. Pediatrics. 78:
220. 736‐741.
221. Takahashi, M. N., W. Jackson, D. T. Laird, T. D. Culp, C. Grose, J. I. Haynes, 2nd, and L. Benetti. 2009. Varicella‐Zoster virus infection induces autoph l. agy in both cultured cells and human skin vesicles. J. Viro
222. Tan, A. Y., C. J. Connett, G. J. Connett, S. C. Quek, H. K. Yap, F. Meurice, and B. W. Lee. 1996. Use of a reformulated Oka strain varicella vaccine (SmithKline Beecham Biologicals/Oka) in healthy children. Eur J Pediatr. 155:706‐711.
223. Teuton, J. R., and C. R. Brandt. 2007. Sialic acid on herpes simplex virus type 1 envelope glycoproteins is required for efficient infection of cells. J Virol. 81:3731‐3739.
224. Tigalonowa, M., J. R. Bjerke, and R. Matre. 1991. Fc gamma‐receptors on Langerhans' cells and keratinocytes in suspension from normal skin characterized using soluble immune complexes and monoclonal antibodies. Acta Derm. Venereol. 71:99‐103.
225. Tischer, B. K., B. B. Kaufer, M. Sommer, F. Wussow, A. M. Arvin, and N. Osterrieder. 2007. A self‐excisable infectious bacterial artificial chromosome clone of varicella‐zoster virus allows analysis of the essential tegument protein encoded by ORF9. J Virol. 81:13200‐13208.
237

226. Turner, A., B. Bruun, T. Minson, and H. Browne. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to m J.
. 72ediate membrane fusion in a Cos cell transfection system.
Virol :873‐875. Tyzzer, E. E. 1906. The Histology of the Skin Lesions in Varicella. J Med Res. 14:361‐392 367
227. .
228. Van de Walle, G. R., H. W. Favoreel, H. J. Nauwynck, P. Van Oostveldt, and M. B. Pensaert. 2001. Involvement of cellular cytoskeleton components in antibody‐induced internalization of viral glycoproteins in pseudorabies virus‐infected monocytes. Virology. 288:129‐138.
229. Vleck, S. E., S. L. Oliver, M. Reichelt, J. Rajamani, L. Zerboni, C. Jones, J. Zehnder, C. Grose, and A. M. Arvin. 2009. Anti‐glycoprotein H antibody impairs the pathogenicity of Varicella‐zoster virus in skin xenografts in the SCID mouse model. J. Virol.
230. Wagenaar, T. R., V. T. Chow, C. Buranathai, P. Thawatsupha, and C. Grose. 2003. The out of Africa model of varicella‐zoster virus evolution: single nucleotide polymorphisms and private alleles distinguish Asian clades from European/North American clades. Vaccine. 21:1072‐1081.
231. Wang, D., and T. Shenk. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tro l. pism. Proc. NatAcad. Sci. U S A. 102:18153‐18158.
232. Wang, X., D. Y. Huang, S. M. Huong, and E. S. Huang. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med. 11:515‐521.
233. Wang, X., S. M. Huong, M. L. Chiu, N. RaabTraub, and E. S. Huang. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature. 424:456‐461.
234. Wang, X., W. J. Kenyon, Q. Li, J. Mullberg, and L. M. HuttFletcher. 1998. Epstein‐Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 72:5552‐5558.
235. Wang, X., W. J. Kenyon, Q. Li, J. Mullberg, and L. M. HuttFletcher. 1998. Epstein‐Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J Virol. 72:5552‐5558.
236. Weigle, K. A., and C. Grose. 1983. Common expression of varicella‐zoster viral glyco icles. protein antigens in vitro and in chickenpox and zoster vesJ. Infect. Dis. 148:630‐638. Weir, J. P. 1998. Genomic organization and evolution of the human herpesviruses. Viru
237. s Genes. 16:85‐93.
238. Westra, D. F., H. B. Kuiperij, G. W. Welling, A. J. Scheffer, T. H. The, and S. WellingWester. 1999. Domains of glycoprotein H of herpes simplex virus type 1 involved in complex formation with glycoprotein L. Virology. 261:96‐105.
239. Westra, D. F., G. M. Verjans, A. D. Osterhaus, A. van Kooij, G. W. Welling, A. J. Scheffer, T. H. The, and S. WellingWester. 2000. Natural infection with herpes simplex virus type 1 (HSV‐1) induces humoral and T
238

239
cell responses to the HSV‐1 glycoprotein H:L complex. J. Gen. Virol. 81:2011‐2015.
240. Whealy, M. E., J. P. Card, R. P. Meade, A. K. Robbins, and L. W. Enquist. 1991. Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J Virol. 65:1066‐1081.
241. Whitbeck, J. C., M. I. Muggeridge, A. H. Rux, W. Hou, C. Krummenacher, H. Lou, A. van Geelen, R. J. Eisenberg, and G. H. Cohen. 1999. The major neutralizing antigenic site on herpes simplex virus glycoprotein D overlaps a receptor‐binding domain. J. Virol. 73:9879‐9890.
242. Whitley, R. J., S. Shukla, and R. J. Crooks. 1998. The identification of risk factors associated with persistent pain following herpes zoster. J Infect Dis. 178 Suppl 1:S71‐75.
243. Wille, P. T., A. J. Knoche, J. A. Nelson, M. A. Jarvis, and D. C. Johnson. 2010. A human cytomegalovirus gO‐null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J Virol. 84:2585‐2596.
244. Wilson, D. W., N. DavisPoynter, and A. C. Minson. 1994. Mutations in the cytoplasmic tail of herpes simplex virus glycoprotein H suppress cell fusion by a syncytial strain. J. Virol. 68:6985‐6993.
245. Yamanishi, K., Y. Mori, and P. E. Pellett. 2007. Human herpesviruses 6 and 7. In D. M. Knipe and P. Howley (ed.), Fields Virology. Lippincott‐Williams & Wilkins, Philadelphia.
246. Yaswen, L. R., E. B. Stephens, L. C. Davenport, and L. M. HuttFletcher. 1993. Epstein‐Barr virus glycoprotein gp85 associates with the BKRF2 gene product and is incompletely processed as a recombinant protein. Virology. 195:387‐396.
247. Yurochko, A. D., E. S. Hwang, L. Rasmussen, S. Keay, L. Pereira, and E. S. Huang. 1997. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF‐kappaB during infection. J. Virol. 71:5051‐5059.
248. Zaia, J. A., M. J. Levin, S. R. Preblud, J. Leszczynski, G. G. Wright, R. J. Ellis, A. C. Curtis, M. A. Valerio, and J. LeGore. 1983. Evaluation of varicella‐zoster immune globulin: protection of immunosuppress
47: ed
children after household exposure to varicella. J. Infect. Dis. 1 737‐743.249. Zerboni, L., C. C. Ku, C. D. Jones, J. L. Zehnder, and A. M. Arvin. 2005.
Varicella‐zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl. Acad. Sci. U S A. 102:6490‐6495.


















